Embed Size (px)
Citation preview

GHTF SG2 - Guidance on Reporting of Medical Device Adverse
Events N54 - SG2 progress report
Dr. Philippe Auclair – Abbott – EUCOMED
Secretary GHTF SG2
"Segundo Foro Internacional y Nacional de Tecnovigilancia"17 y 18 de junio de 2010, Bogotá, D.C. Colombia

Post-Market Surveillance Post-Market Surveillance is the
collection of information on the quality, safety or performance of Medical Devices after they have been placed in the market.
A balanced Post-Market Surveillance system will contain an appropriate mix of proactive and reactive activities.

Post-Market Vigilance
(Post-Market) Vigilance is the reporting and investigation of medical device adverse events and incidents. Both the manufacturer and the Regulatory Authority play major roles.
By its very nature, Vigilance is a REACTIVE activity (the manufacturer or authority receive reports and REACT to them) - this is not intended to be a derogatory statement

A Pictorial view of PMS
Post Market
Surveillance
Vigilance(adverse event
report
investigation)
Post-Market Surveillance
Information is used for:
Injury prevention
Development of standards
Regulatory refinement
Product improvement

SG2 Vision for Harmonization
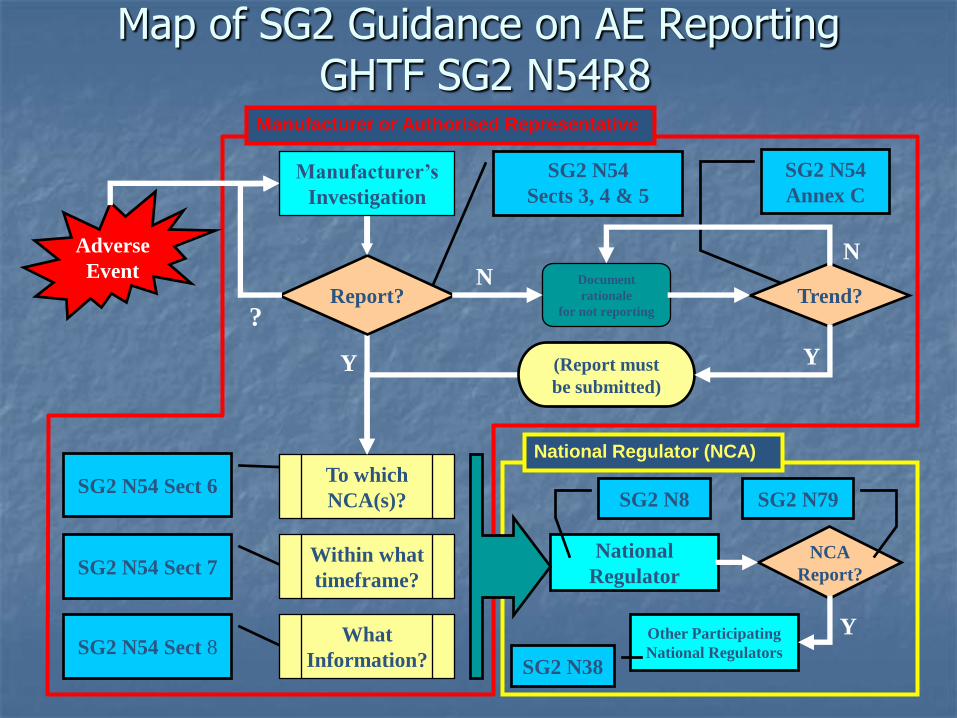
Manufacturer or Authorised Representative
National Regulator (NCA)
Map of SG2 Guidance on AE ReportingGHTF SG2 N54R8
SG2 N54 Sect 8
SG2 N54 Sect 7
SG2 N54 Sect 6
SG2 N54
Sects 3, 4 & 5
SG2 N54
Annex C
To which
NCA(s)?
Within what
timeframe?
What
Information?
Y
?Report?
N
Trend?Document
rationale
for not reporting
N
Y(Report must
be submitted)
National
Regulator
NCA
Report?
Other Participating
National Regulators
Y
SG2 N8 SG2 N79
SG2 N38
Manufacturer’s
Investigation
Adverse
Event

GHTF N54 Section 3.0Three Basic Reporting Criteria
An EVENT must have occurred
AND
The manufacturers device was ASSOCIATEDwith the event
AND
The event led to the death or SERIOUS INJURY of a patient user or other person, ORmight lead to death or serious injury if the event re-occurs

EVENT
Malfunction or deterioration
Inadequate design or manufacture
Inaccuracy in labeling
Significant public health concern
Other information from testing or literature
A change in trend

ASSOCIATION (WITH THE DEVICE)
When the association with the device is difficult to establish, the manufacturer must rely on:
Opinion from healthcare professional
Previous similar events
Other information available to the manufacturer
If there is any doubt, assume that the device was associated with the event.

SERIOUS INJURY
Life threatening illness or injury
Permanent (irreversible) impairment of a body function or permanent damage to a body structure
A condition requiring medical or surgical intervention to prevent permanent impairment of a body function or permanent damage to a body structure

Exemption RulesGHTF N54 Section 4.1- 4.8
1) Deficiency of a new device found by the user prior to its use
2) Adverse event caused by patientconditions
3) Service life or shelf life of the medicaldevice
4) Malfunction protection operated correctly5) Negligible likelihood of occurrence of death or
serious injury6) Expected and foreseeable side effects7) Adverse events described in an advisory notice8) Reporting exemptions granted by NCA

Use Errors reporting N54 Section 5 & Annex D
Use Error: Act, or omission of an act, that has a different result to that intended by the manufacturer or expected by the operator
Use errors related to medical devices, which didresult in death or serious injury or serious public health threat should be reported by the manufacturer to the National Competent Authority
Use errors related to medical devices which did notresult in death or serious injury or serious public health concerns, need not be reported by the manufacturer to the national competent authorities

Abnormal Use reporting N54 Section 5 & Annex D
Abnormal Use:Act, or omission of an act by the operator or user of a medical device as a result of conduct that is beyond any reasonable means of risk control by the manufacturer
Abnormal use need not to be reported by the manufacturer to the national competent authority under adverse event reporting procedure. Abnormal use should be handled by the healthcare facility and appropriate regulatory authorities If manufacturers become aware of instances of abnormal use,
they may bring this to the attention or other appropriate organizations and healthcare facility personnel

The Universe of Device Associated Adverse Events
= Serious Injury
= Death
= Malfunction
/potential Serious injury
RR R
RR NR*
NR*NR*
NR*
R=ReportNR*= No Report*
Manufacturer or Authorised Representative
National Regulator (NCA)
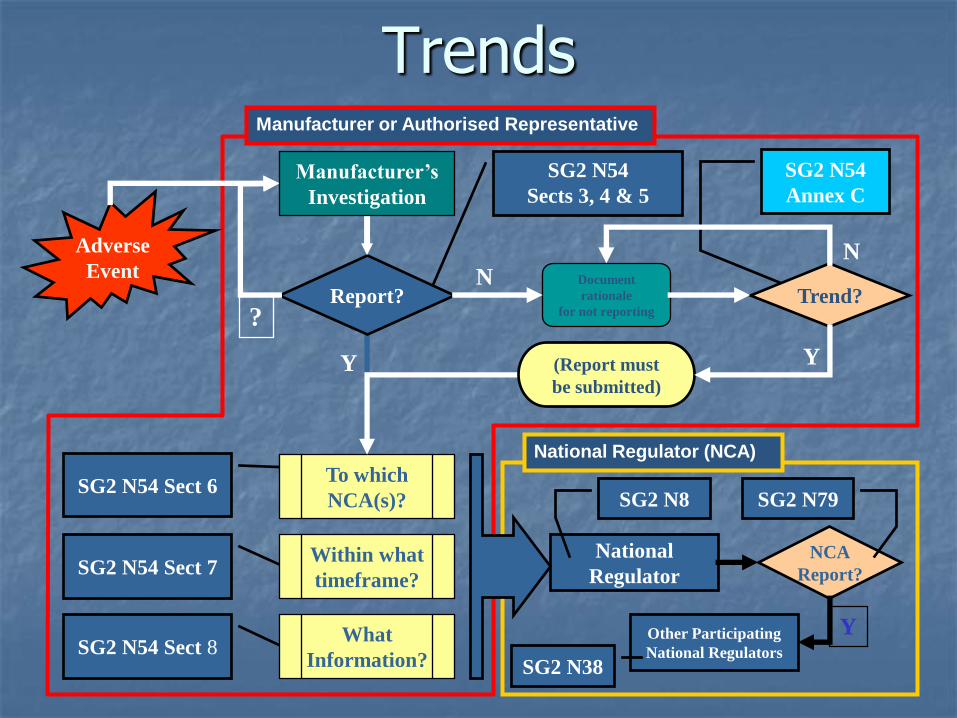
Trends
SG2 N54 Sect 8
SG2 N54 Sect 7
SG2 N54 Sect 6
SG2 N54
Sects 3, 4 & 5
SG2 N54
Annex C
To which
NCA(s)?
Within what
timeframe?
What
Information?
Y
?Report?
N
Trend?Document
rationale
for not reporting
N
Y(Report must
be submitted)
National
Regulator
NCA
Report?
Other Participating
National Regulators
Y
SG2 N8 SG2 N79
SG2 N38
Manufacturer’s
Investigation
Adverse
Event

AE Trend Reporting
Adverse events specifically exempted from reporting become reportable, if there is a change in trend (usually an increase in frequency) or pattern is identified.
The SG2 document on trend reporting describes the criteria for identifying a significant increase in the rate of adverse events.

AE Trend Reporting
Example of an upward shift in trend
time
IB baseline
Incidence of events (i)
IT threshold
RoV* new baseline RoV*
* normal Range of Variance
Report
new threshold
Manufacturer or Authorised Representative
National Regulator (NCA)
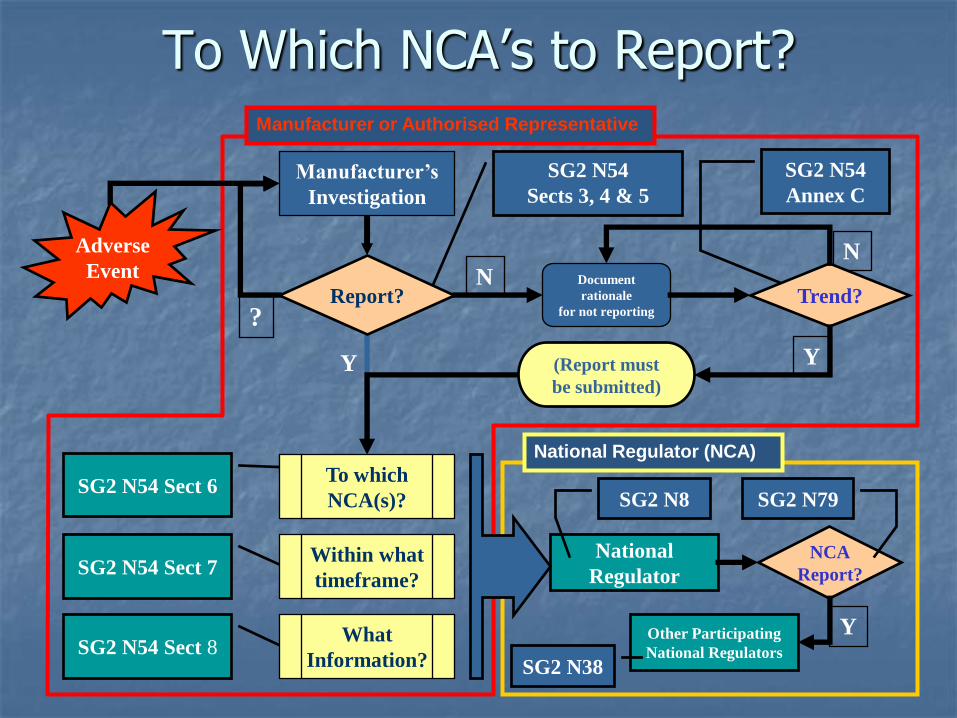
To Which NCA’s to Report?
SG2 N54 Sect 8
SG2 N54 Sect 7
SG2 N54 Sect 6
SG2 N54
Sects 3, 4 & 5
SG2 N54
Annex C
To which
NCA(s)?
Within what
timeframe?
What
Information?
Y
?Report?
N
Trend?Document
rationale
for not reporting
N
Y(Report must
be submitted)
National
Regulator
NCA
Report?
Other Participating
National Regulators
Y
SG2 N8 SG2 N79
SG2 N38
Manufacturer’s
Investigation
Adverse
Event

To Whom to ReportN54 Section 6
Adverse Events must be reported to a National Competent Authority (NCA) according to applicable requirements in each jurisdiction. NCA’s should provide a contact point to manufacturer from reporting.
SG2 considered several options that might resolve this situation, including the establishment of a global database for submission of adverse event reports.
Manufacturer or Authorised Representative
National Regulator (NCA)
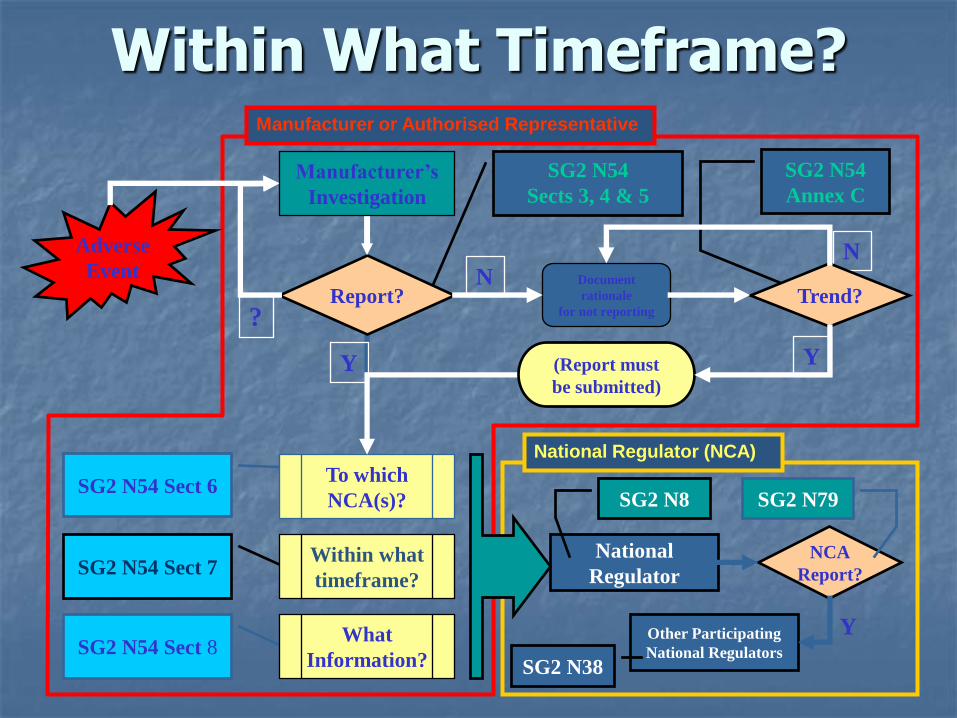
Within What Timeframe?
SG2 N54 Sect 8
SG2 N54 Sect 7
SG2 N54 Sect 6
SG2 N54
Sects 3, 4 & 5
SG2 N54
Annex C
To which
NCA(s)?
Within what
timeframe?
What
Information?
Y
?Report?
N
Trend?Document
rationale
for not reporting
N
Y(Report must
be submitted)
National
Regulator
NCA
Report?
Other Participating
National Regulators
Y
SG2 N8 SG2 N79
SG2 N38
Manufacturer’s
Investigation
Adverse
Event

Reporting TimeframesN54 Section 7 & Annex B
Adverse events that result in unanticipated death or unanticipated serious injury or represent a serious public health threat must be reported immediately by the manufacturer.
All other reportable events must be reported as soon as possible by the manufacturer, but not later than 30-elapsed calendar days following the date of awareness of the event.

Reporting Timeframes
Immediately: For purposes of adverse event reporting, immediately means as soon as possible, but not later than 10 elapsed calendar days following the date of awareness of the event.
Serious public heath threat: Any event type, which results in imminent risk of death, serious injury, or serious illness that may require prompt remedial action.
Manufacturer or Authorised Representative
National Regulator (NCA)
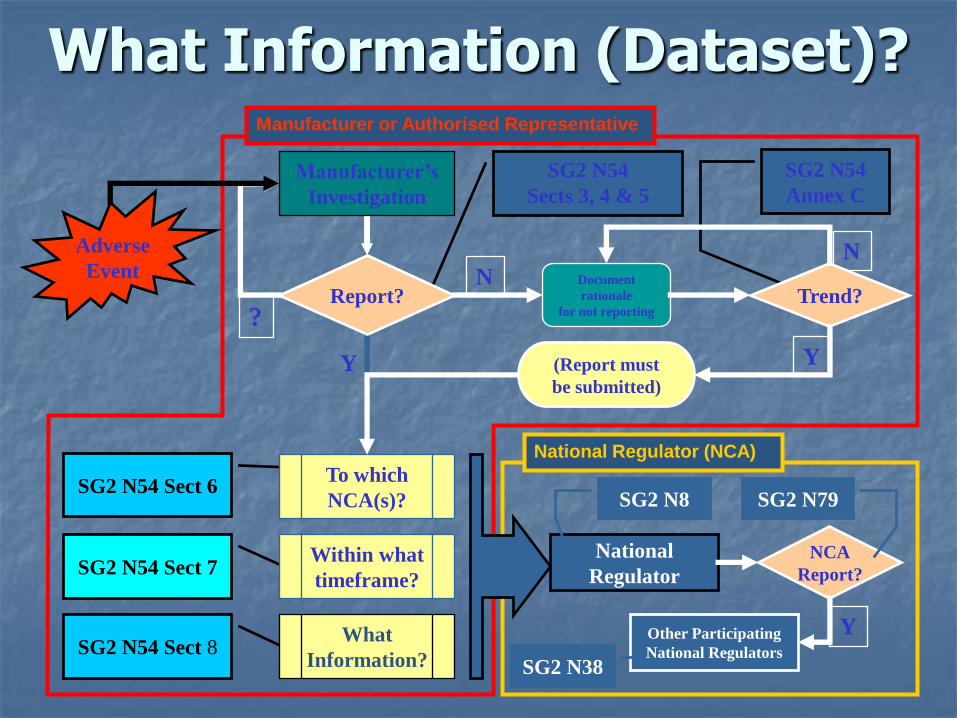
What Information (Dataset)?
SG2 N54 Sect 8
SG2 N54 Sect 7
SG2 N54 Sect 6
SG2 N54
Sects 3, 4 & 5
SG2 N54
Annex C
To which
NCA(s)?
Within what
timeframe?
What
Information?
Y
?Report?
N
Trend?Document
rationale
for not reporting
N
Y(Report must
be submitted)
National
Regulator
NCA
Report?
Other Participating
National Regulators
Y
SG2 N8 SG2 N79
SG2 N38
Manufacturer’s
Investigation
Adverse
Event

Report Data Set
Event information: Dates, Reporter details, Healthcare facility details, Patient details, Event type and description, Notified CA’s, Resolution description
Device Information: Manufacturer, Generic device group, Disposition, Results of analysis, Corrective action taken.
Other: Comments, Notified Body details, CA’s notified of Corrective action

Courtesy of Isabelle Demade, Principal Administrator, European Commission, DG Health and
Consumers, Unit B2, Cosmetics and Medical Devices ,
Chair GHTF SG2
SG2 STATUS REPORT- May 2010

SG2 Main Tasks
Updating/developing guidance on A/E reporting and other PM surveillance activities, when appropriate.
Improving/developing/maintaining the NCAR Exchange Programme
Providing training on SG2 Guidance and NCAR Exchange Program upon request

SG2 2009 Main Achievements
Updating and improving guidance on NCAR Exchange Program: N38:2009;N79:2009
Establishing closer links with AHWP WG2

SG2 guidance in progress (1)
Definition and Classification of FSCA, incl. Recalls
Reason: term ‘recall’ not uniformally applied. Has impeded communication between NCA’s+ led to anxieties among general public. Recall classification equally disparate.
Expected completion: Q4 2010/ Q1 2011

SG2 guidance in progress (2)
Severe Adverse Events REPORTINGDURING CLINICAL INVESTIGATION
Reason: C.I. conducted globally
Joint SG2-SG5 TF
Draft: along lines GHTF SG2 N54 R8
Expected completion : Q4 - 2010

Training SG2 Activities
Ongoing:
- Centralizing training requests
- Developing/updating training slide presentations
Request: development of SG2 Training
Documents
Scope: trainers qualified, type of training sessions, feedback on training.

GHTF/SC/N2R12:2010 :GHTF Roles and Responsibilities
GHTF Membership Update

Rules on participation to GHTF (SG2)GHTF/SC/N2R12:2010 :GHTF Roles
and Responsibilities
GHTF - voluntary international group of medical device regulatory authorities, and medical device trade associations grouped into three geographical areas, namely Europe, North America and Asia-Pacific
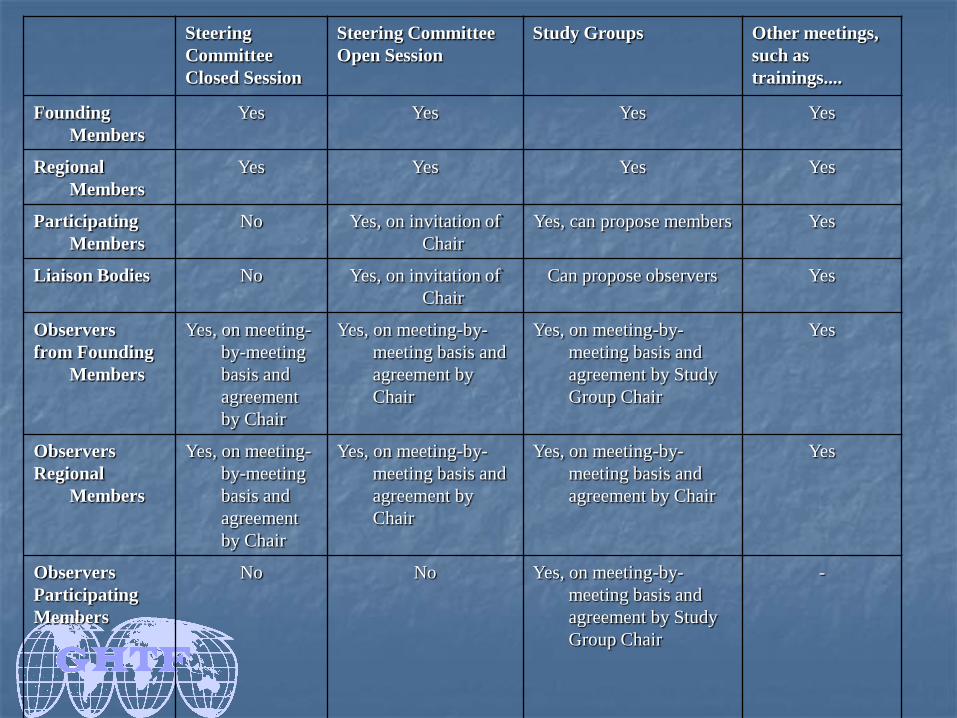
Different categories of membership – Founding Members, Regional Members, Participating Members and Liaison Bodies.

Founding Members : Regulatory authorities or industry representatives from Europe, the United States of America (USA), Canada, Japan and Australia.
Regional members: Members represent the regulatory authorities and industry representatives in a formal regional grouping of economies, not otherwise represented by a Founding Member.
Participating members : representatives of medical device regulatory authorities or medical device trade associations of countries other than the Founding Members
Liaison bodies: public health organizations, international standard-setting bodies or other groups who can contribute to or benefit from participation in GHTF.
GHTF Member Categories
Steering
Committee
Closed Session
Steering Committee
Open Session
Study Groups Other meetings,
such as
trainings....
Founding
Members
Yes Yes Yes Yes
Regional
Members
Yes Yes Yes Yes
Participating
Members
No Yes, on invitation of
Chair
Yes, can propose members Yes
Liaison Bodies No Yes, on invitation of
Chair
Can propose observers Yes
Observers
from Founding
Members
Yes, on meeting-
by-meeting
basis and
agreement
by Chair
Yes, on meeting-by-
meeting basis and
agreement by
Chair
Yes, on meeting-by-
meeting basis and
agreement by Study
Group Chair
Yes
Observers
Regional
Members
Yes, on meeting-
by-meeting
basis and
agreement
by Chair
Yes, on meeting-by-
meeting basis and
agreement by
Chair
Yes, on meeting-by-
meeting basis and
agreement by Chair
Yes
Observers
Participating
Members
No No Yes, on meeting-by-
meeting basis and
agreement by Study
Group Chair
-

SG2 Vision for Harmonization
Thanks for your attention
Questions and suggestions
welcome !





























