Embed Size (px)
Citation preview

R E S E A R C H A R T I C L E
Gerstmann-Sträussler-Scheinker Syndrome with VariablePhenotype in a New Kindred with PRNP-P102L MutationMiguel A. Riudavets1; María Alejandra Sraka2; Marcelo Schultz1; Estefanía Rojas1; Horacio Martinetto1;Christian Begué1; Inés Noher de Halac3; Anna Poleggi4; Michele Equestre4; Maurizio Pocchiari4;Gustavo Sevlever1; Ana Lía Taratuto1
1 Department of Neuropathology, Institute for Neurological Research, FLENI, Buenos Aires, Argentina.2 Department of Neurology, Hospital Larrain de Berisso, La Plata, Argentina.3 Department of Internal Medicine, University of Córdoba, Córdoba, Argentina.4 Department of Cell Biology and Neurosciences, Istituto Superiore di Sanitá, Rome, Italy.
Keywords
Argentina, ataxia, Gerstmann-Sträussler-Scheinker, P102L, variable phenotype.
Corresponding author:
Gustavo Sevlever, MD, PhD, Department ofNeuropathology, Institute for NeurologicalResearch, FLENI, Montañeses 2325,Buenos Aires 1428, Argentina(E-mail: [email protected])
Received 14 February 2013Accepted 8 August 2013Published Online Article Accepted 15August 2013
Conflict of interest: All authors declare noconflict of interest.
doi:10.1111/bpa.12083
AbstractGerstmann-Sträussler-Scheinker syndrome (GSS) is a dominantly inherited disorderbelonging to the group of transmissible human spongiform encephalopathies or priondiseases. Several families affected by GSS with patients carrying mutations in the prionprotein gene have been described worldwide. We report clinical, genealogical, neuropa-thology and molecular study results from two members of the first Argentine kindredaffected by GSS. Both family members presented a frontotemporal-like syndrome, onewith and the other without ataxia, with different lesions on neuropathology. A Pro to Leupoint mutation at codon 102 (P102L) of the prion protein gene was detected in one of thesubjects studied. The pathogenic basis of phenotypic variability observed in this familyremains unclear, but resembles that observed in other P102L GSS patients from the samefamily.
INTRODUCTIONTransmissible human spongiform encephalopathies or prion dis-eases encompass a spectrum of disorders including sporadic,variant and genetic Creutzfeldt-Jakob disease (CJD), sporadic andfamilial fatal insomnia, kuru and Gerstmann-Sträussler-Scheinkersyndrome (GSS). These disorders are characterized by the depo-sition, mainly in the brain, of an abnormal isoform of the hostprion protein (PrP) (24). About 10–15% of human prion diseasesare dominantly inherited, including 100% of GSS cases andapproximately 10–15% of CJD cases (19).
As GSS was first described in an Austrian family in 1936 (6, 10,11,), families suffering from the disorder have been identified in 10different countries worldwide (12, 18, 26). All GSS cases arelinked to mutations in the prion protein gene (PRNP), with theproline to leucine substitution at residue 102 (P102L) (15) beingthe most common one (5) among others previously described (21).
GSS patients develop the disease relatively early [mean age51.6 years, standard deviation (SD) 12.8 years] with a long clinicalduration (mean 40 years, SD 25 months) (16), characterized inmost cases by progressive cerebellar ataxia accompanied by
cognitive decline (9). However, GSS patients may have differentclinical phenotypes ranging from almost pure cerebellar signswithout dementia, psychiatric manifestations at onset followed byataxia (3), slowly progressive dementia (frontotemporal-like) (29),or with clinical features resembling sporadic CJD (2).
Neuropathological examination reveals the presence of abun-dant multicentric amyloid plaques in the cerebellar and cerebrumcortices, with variable amount of spongiform changes, rangingfrom complete absence to severe spongiform changes (2, 9).
This study reports an Argentine family whose members carrythe P102L mutation of the PRNP gene. This is the first GSSkindred observed, to our knowledge, in a Latin American country.
MATERIALS AND METHODS
Family history and clinical assessment
Clinical histories and examination details were obtained frommedical notes completed at the time of clinical review. Familyhistory and complementary clinical information were obtainedfrom alive and healthy members of the kindred (Figure 1).
Brain Pathology ISSN 1015-6305
142 Brain Pathology 24 (2014) 142–147
© 2013 International Society of Neuropathology

Informed consent for genetic analyses and brain autopsy wereobtained from the next of kin. This study was approved by theEthical Committee of FLENI, Buenos Aires, Argentina.
Neuropathological assessment
The purpose and procedures of the autopsy were explained andobjectives discussed with the relatives. We performed the autopsyon the proband (subject IV-7). Cerebrum, brain stem and cerebel-lum were available. A piece of frontal cortex was taken fresh andwas frozen in −80°C.
The brain was fixed in 10% formaldehyde, and cut 2 weeks afterfixation. Samples of fixed tissue were obtained from the frontal,temporal, parietal and occipital cortices, as well as from thehippocampal formation, basal ganglia, thalamus, cerebellum andbrain stem.
The cerebrum from subject IV-5 was received as a consult.Samples were obtained from the frontal, temporal, parietal andoccipital cortices, as well as from the hippocampal formation,basal ganglia and thalamus. The tissue was processed for paraffinembedding and 10-μm thick sections stained with hematoxylinand eosin (H&E), and immunostained for the pathological prionprotein (PrPTSE) (3F4. 1:500 after 88% formic acid treatment).
PRNP gene sequencing
DNA was extracted from frozen brain tissue (proband, IV-7) usingthe Wizard Genomic DNA Purification kit (Promega Corporation,Fitchburg, WI, USA) and from paraffin embedded/formalin fixedbrain tissue (patient IV-5) using the DNeasy Blood and Tissue Kit(Qiagen, Venlo, Limburg, Netherlands). We genotyped the P102Lmutation by direct Sanger sequencing as previously described(19). To verify the presence of the mutation on the Met allele, wehave first cloned the fragment by thymine-adenine cloning(Invitrogen, Carlsbad, CA, USA) and then sequenced clones.
RESULTS
Family description
The family (Figure 1) was originally from a small village in Sicily(San Piero, Messina), who later left Italy to settle in Argentina(near Buenos Aires) in the early XXth century.
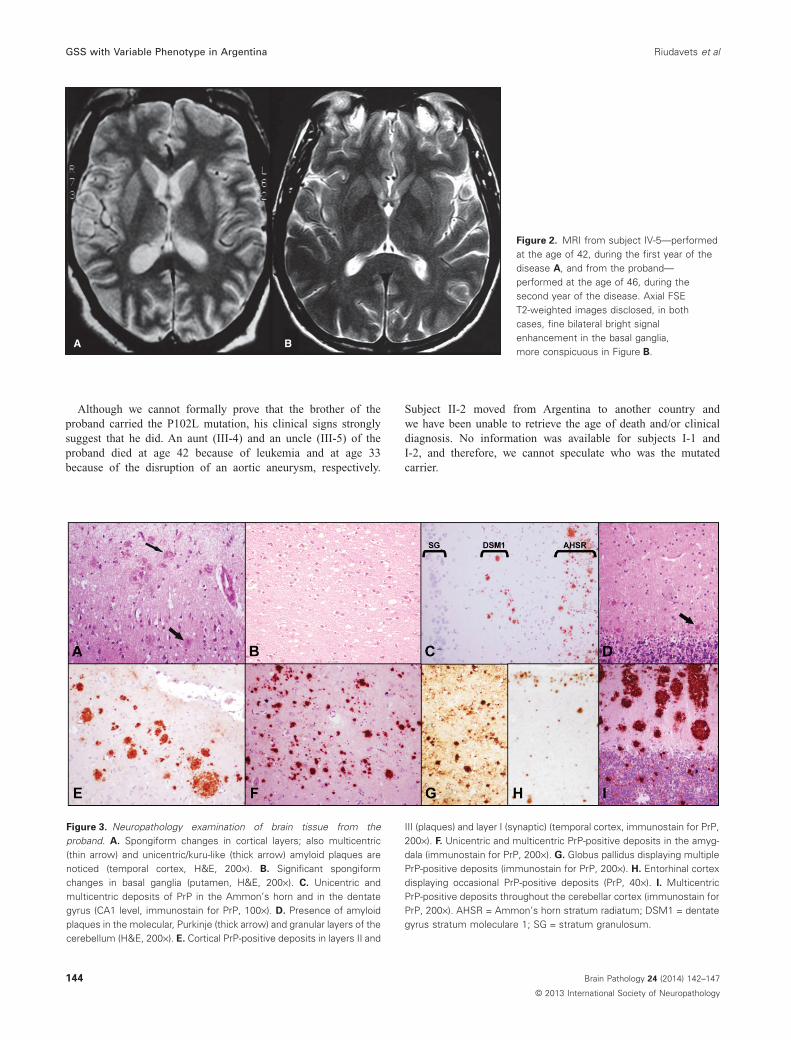
The proband (IV-7, Figure 1) was a 50-year-old physician who,in 2001 at age 44, referred abnormal gait suggestive of ataxia. In2002, a neuropsychological examination was within normal limits;however, electromyography (EMG) results reported an axonalpattern, and brain magnetic resonance imaging (MRI) indicatedhigh signal intensity in the basal ganglia (Figure 2B). Somemonths later, the patient progressed with cognitive declinemainly with a frontotemporal pattern consisting of disinhibited,repetitive and compulsive behaviors. Over the following years, itevolved with aphasia and added agnosia, sphincter incontinence,amyotrophy, apraxia, difficulty in swallowing and finally broncho-pneumonia and sepsis. The patient died in early 2007 and anautopsy was performed in our unit.
Subject IV-5: In 2000, this 41-year-old computer technicianexperienced neurological impairment mainly with a fronto-temporal pattern imparted by the presence of cognitive declinewith disinhibited behavior and attentional deficits. At that time,the mini-mental state examination score was 23/30. During theensuing years, the patient developed passivity, aphasia, memoryloss and agnosia, although on neurological examination, noataxia or pyramidal/extrapyramidal signs were evident. EMGperformed in 2001 showed no relevant abnormalities. BrainMRI indicated high signal intensity in the basal ganglia butno further alterations in cortical or subcortical regions(Figure 2A). The patient finally died in 2004 with a clinical diag-nosis of Pick’s disease at the age of 45 in his death certificate.Autopsy was performed, and the brain, without the cerebellumand the brain stem, was referred to our brain bank in BuenosAires.
Scant clinical information is available for the other members ofthe family. However, according to reports from relatives, the father(III-1) of the GSS patient IV-5 died in 1978 with a clinical diag-nosis of dementia with motoneuron signs, and his father (II-1) hadin turn been wheelchair-bound. As both of them were obligatecarriers, it is likely that he died of GSS.
On the other side of the family, the brother (IV-6) of the probanddied in 2002 at 52 years with a clinical diagnosis of frontotemporaldementia with motoneuron signs, the father (III-3) had died in1976 at age 54 with a clinical diagnosis of dementia with moto-neuron signs, while their grandmother (II-3) in 1956 at age 52 witha clinical diagnosis of Pick’s disease. The father and the grand-mother were obligate carriers and it is conceivable that both ofthem were affected by GSS.
Figure 1. Family pedigree. Solid symbolsindicate individuals with autopsy; strippedsymbols reflect individuals affected by aneurological disorder; and diagonal lines,those who are deceased. The arrow singlesout the proband.
Riudavets et al GSS with Variable Phenotype in Argentina
143Brain Pathology 24 (2014) 142–147
© 2013 International Society of Neuropathology
Although we cannot formally prove that the brother of theproband carried the P102L mutation, his clinical signs stronglysuggest that he did. An aunt (III-4) and an uncle (III-5) of theproband died at age 42 because of leukemia and at age 33because of the disruption of an aortic aneurysm, respectively.
Subject II-2 moved from Argentina to another country andwe have been unable to retrieve the age of death and/or clinicaldiagnosis. No information was available for subjects I-1 andI-2, and therefore, we cannot speculate who was the mutatedcarrier.
A B
Figure 2. MRI from subject IV-5—performedat the age of 42, during the first year of thedisease A, and from the proband—performed at the age of 46, during thesecond year of the disease. Axial FSET2-weighted images disclosed, in bothcases, fine bilateral bright signalenhancement in the basal ganglia,more conspicuous in Figure B.
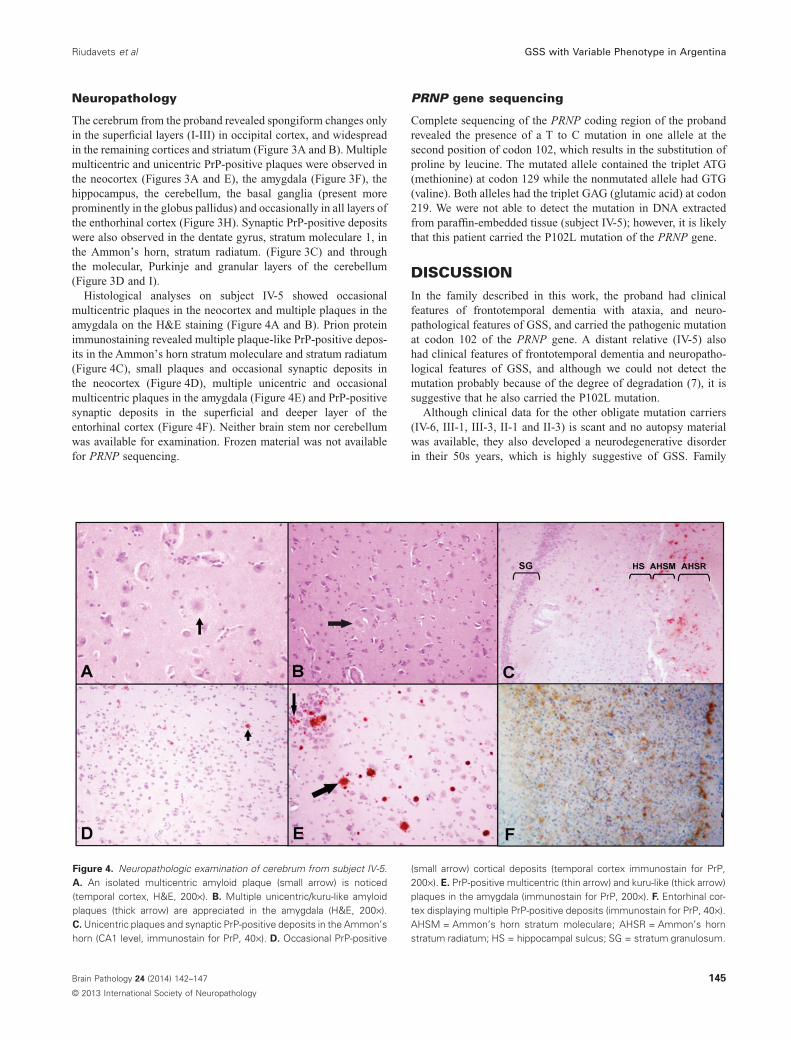
Figure 3. Neuropathology examination of brain tissue from theproband. A. Spongiform changes in cortical layers; also multicentric(thin arrow) and unicentric/kuru-like (thick arrow) amyloid plaques arenoticed (temporal cortex, H&E, 200×). B. Significant spongiformchanges in basal ganglia (putamen, H&E, 200×). C. Unicentric andmulticentric deposits of PrP in the Ammon’s horn and in the dentategyrus (CA1 level, immunostain for PrP, 100×). D. Presence of amyloidplaques in the molecular, Purkinje (thick arrow) and granular layers of thecerebellum (H&E, 200×). E. Cortical PrP-positive deposits in layers II and
III (plaques) and layer I (synaptic) (temporal cortex, immunostain for PrP,200×). F. Unicentric and multicentric PrP-positive deposits in the amyg-dala (immunostain for PrP, 200×). G. Globus pallidus displaying multiplePrP-positive deposits (immunostain for PrP, 200×). H. Entorhinal cortexdisplaying occasional PrP-positive deposits (PrP, 40×). I. MulticentricPrP-positive deposits throughout the cerebellar cortex (immunostain forPrP, 200×). AHSR = Ammon’s horn stratum radiatum; DSM1 = dentategyrus stratum moleculare 1; SG = stratum granulosum.
GSS with Variable Phenotype in Argentina Riudavets et al
144 Brain Pathology 24 (2014) 142–147
© 2013 International Society of Neuropathology
Neuropathology
The cerebrum from the proband revealed spongiform changes onlyin the superficial layers (I-III) in occipital cortex, and widespreadin the remaining cortices and striatum (Figure 3A and B). Multiplemulticentric and unicentric PrP-positive plaques were observed inthe neocortex (Figures 3A and E), the amygdala (Figure 3F), thehippocampus, the cerebellum, the basal ganglia (present moreprominently in the globus pallidus) and occasionally in all layers ofthe enthorhinal cortex (Figure 3H). Synaptic PrP-positive depositswere also observed in the dentate gyrus, stratum moleculare 1, inthe Ammon’s horn, stratum radiatum. (Figure 3C) and throughthe molecular, Purkinje and granular layers of the cerebellum(Figure 3D and I).
Histological analyses on subject IV-5 showed occasionalmulticentric plaques in the neocortex and multiple plaques in theamygdala on the H&E staining (Figure 4A and B). Prion proteinimmunostaining revealed multiple plaque-like PrP-positive depos-its in the Ammon’s horn stratum moleculare and stratum radiatum(Figure 4C), small plaques and occasional synaptic deposits inthe neocortex (Figure 4D), multiple unicentric and occasionalmulticentric plaques in the amygdala (Figure 4E) and PrP-positivesynaptic deposits in the superficial and deeper layer of theentorhinal cortex (Figure 4F). Neither brain stem nor cerebellumwas available for examination. Frozen material was not availablefor PRNP sequencing.
PRNP gene sequencing
Complete sequencing of the PRNP coding region of the probandrevealed the presence of a T to C mutation in one allele at thesecond position of codon 102, which results in the substitution ofproline by leucine. The mutated allele contained the triplet ATG(methionine) at codon 129 while the nonmutated allele had GTG(valine). Both alleles had the triplet GAG (glutamic acid) at codon219. We were not able to detect the mutation in DNA extractedfrom paraffin-embedded tissue (subject IV-5); however, it is likelythat this patient carried the P102L mutation of the PRNP gene.
DISCUSSIONIn the family described in this work, the proband had clinicalfeatures of frontotemporal dementia with ataxia, and neuro-pathological features of GSS, and carried the pathogenic mutationat codon 102 of the PRNP gene. A distant relative (IV-5) alsohad clinical features of frontotemporal dementia and neuropatho-logical features of GSS, and although we could not detect themutation probably because of the degree of degradation (7), it issuggestive that he also carried the P102L mutation.
Although clinical data for the other obligate mutation carriers(IV-6, III-1, III-3, II-1 and II-3) is scant and no autopsy materialwas available, they also developed a neurodegenerative disorderin their 50s years, which is highly suggestive of GSS. Family
Figure 4. Neuropathologic examination of cerebrum from subject IV-5.A. An isolated multicentric amyloid plaque (small arrow) is noticed(temporal cortex, H&E, 200×). B. Multiple unicentric/kuru-like amyloidplaques (thick arrow) are appreciated in the amygdala (H&E, 200×).C. Unicentric plaques and synaptic PrP-positive deposits in the Ammon’shorn (CA1 level, immunostain for PrP, 40×). D. Occasional PrP-positive
(small arrow) cortical deposits (temporal cortex immunostain for PrP,200×). E. PrP-positive multicentric (thin arrow) and kuru-like (thick arrow)plaques in the amygdala (immunostain for PrP, 200×). F. Entorhinal cor-tex displaying multiple PrP-positive deposits (immunostain for PrP, 40×).AHSM = Ammon’s horn stratum moleculare; AHSR = Ammon’s hornstratum radiatum; HS = hippocampal sulcus; SG = stratum granulosum.
Riudavets et al GSS with Variable Phenotype in Argentina
145Brain Pathology 24 (2014) 142–147
© 2013 International Society of Neuropathology

members of generations I and II were born in Southern Italy(a small town in the province of Messina, Sicily) where severalapparently unrelated families experienced GSS in carriers with theP102L mutation (2, 3, 8, 13, 17, 19). It is therefore likely that thisfamily is related to one of the previously described GSS-affectedfamilies in Sicily and work is in progress to identify a possiblecommon ancestral origins of the P102L mutation-associated chro-mosomes in these families through genealogical (4) and geneticstudies (20).
Both the proband (IV-7) and IV-5 had clinical signs resemblingFTD but the former showed also ataxia while IV-5 did not. Differ-ences in clinical signs and disease duration of P102L-GSS patientshave been previously observed, even within the same family, wherecases might resemble sporadic CJD, have pure cerebellar signswith no dementia or psychiatric disturbances (1, 2, 3, 14, 22, 27,28). The proband and IV-5 also had different neuropathologicalpatterns. Spongiform changes ranged from moderate to prominentin the proband while were almost absent in subject IV-5. PrPTSE
staining pattern also showed marked differences in these twoautopsied family members (see Figures 3 and 4). The reason forthis variability remains poorly understood and it is still controver-sial whether it depends on the presence of methionine or valine inthe polymorphic codon 129 of the nonmutated PrP allotype (2, 27),different patterns of truncated prion protein fragments (22) orprion protein heterogeneity (23, 25).
Once clinical suspicion arises, cases potentially considered to becaused by prions are normally referred to the Centre for HumanTransmissible Spongiform Encephalopathies (HTSE), functioningat our institution since 1983, where patients are studied exten-sively, with workup including electroencephalography (EEG),MRI and 14-3-3 protein WB in CSF. However, on a limited numberof occasions, a few prion-related cases presenting clinically withsymptoms of more common neurodegenerative disorders mayescape this tight surveillance system and be handled at autopsyas “standard” cases. The material will therefore be sent to thebrain bank instead of the Centre for HTSE. Within the group ofprion-related diseases, this holds true especially for Gerstmann-Sträussler-Scheinker syndrome.
It is important to acknowledge that the identification of thiskindred would have been missed by routine clinical and laboratoryinvestigations suggesting that PRNP genetic analyses and post-mortem examination should be recommended in all cases with afamily history of any type of neuropsychiatric syndrome (27), evenwhen they are not associated with cognitive signs.
In conclusion, we report the first Argentine kindred affectedby Gerstmann-Sträussler-Scheinker disease associated with theP102L mutation of the PRNP gene, with possible intrafamilialclinical variability and Italian origins.
ACKNOWLEDGMENTSWe are grateful to the members of this family, whose effort tofight against this tremendous disease makes this work worth to bepresented. We are also indebted to Dr Lina Nuñez for helping uson the preparation of the manuscript. This work was supportedby the Department of Teaching and Research (FLENI), theFLENI SECyT BID 1728/OC-AR and PID 351/2003, the IstitutoSuperiore di Sanita (ISS)-NIH research program “Rare Diseases
2006” and the Italian Ministry of Health (CJD Registry). Weacknowledge the support of the Research Council of Argentina(CONICET).
REFERENCES1. Arata H, Takashima H, Hirano R, Tomimitsu H, Machigashira K,
Izumi K et al (2006) Early clinical signs and imaging findings inGerstmann–Sträussler–Scheinker syndrome (Pro102Leu). Neurology66:1672–1678.
2. Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F, MarasB et al (1996) Polymorphism at codon 129 or codon 219 of PRNPand clinical heterogeneity in a previously unreported family withGerstmann-Sträussler-Scheinker disease (PrP-P102L mutation).Neurology 47:734–741.
3. Bianca M, Bianca S, Vecchio I, Raffaele R, Ingegnosi C, Nicoletti F(2003) Gerstmann-Sträussler-Scheinker disease with P102L-V129mutation: a case with psychiatric manifestations at onset. Ann Genet46:467–469.
4. Bruni A, Montesi M, Rainero I, Ferini-Strambi L, Macciardi F,Pinessi L et al (1993) The power of systematic genealogicalstudy in familial Alzheimer disease. Ital J Neurol Sci14:239–244.
5. Collins S, Lawson VA, Masters CL (2004) Transmissiblespongiform encephalopathies. Lancet 363:51–61.
6. Dimitz L (1913) Bericht des Vereines fur Psychiatrie undNeurologie in Wein. Jahrb Psychiatr Neurol 34:384.
7. Ferrer I, Armstrong J, Capellari S, Parchi P, Arzberger T, Bell Jet al (2007) Effects of formalin fixation, paraffin embedding, andtime of storage on DNA preservation in brain tissue: a BrainNetEurope study. Brain Pathol 17:297–303.
8. Galatioto S, Ruggeri D, Gullotta F (1995) Gerstmann-Sträussler-Scheinker syndrome in a Sicilian patient. Neuropathological aspects.Pathologica 87:659–665.
9. Gambetti P, Parchi P, Chen SG (2003) Hereditary Creutzfeldt-Jakobdisease and fatal familial insomnia. Clin Lab Med 23:43–64.
10. Gerstmann J (1928) Über ein noch nicht beschriebenesReflexphänomen bei einer Erkrankung des zerebellären Systems.Wien Med Wochenschr 78:906–908.
11. Gerstmann J, Straussler E, Scheinker J (1936) Über eine eigenartigehereditär-familiäre Erkrankung des Zentralnervensystems. Z Neurol154:736–762.
12. Ghetti B, Dlouhy SR, Giaccone G, Bugiani O, Frangione B, FarlowMR, Tagliavini F (1995) Gerstmann-Sträussler-Scheinker diseaseand the Indiana kindred. Brain Pathol 5:61–75.
13. Giovagnoli A, Di Fede G, Aresi A, Reati F, Rossi G, Tagliavini F(2008) Atypical frontotemporal dementia as a new clinicalphenotype of Gerstmann-Sträussler-Scheinker disease with thePrP-P102L mutation. Description of a previously unreported Italianfamily. Neurol Sci 29:405–410.
14. Hainfellner JA, Brantner Inthaler S, Cervenakova L,Brown P, Kitamoto T, Tateishi J et al (1995) The originalGerstmann-Sträussler-Scheinker family of Austria: divergentclinicopathological phenotypes but constant PrP genotype. BrainPathol 5:201–211.
15. Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JDet al (1989) Linkage of a prion protein missense variant toGerstmann-Straussler syndrome. Nature 338:342–345.
16. Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H,Van Duijn C et al (2005) Genetic prion disease: the EUROCJDexperience. Hum Genet 118:166–174.
17. Kretzschmar HA, Kufer P, Riethmuller G, DeArmond S, PrusinerSB, Schiffer D (1992) Prion protein mutation at codon 102 in an
GSS with Variable Phenotype in Argentina Riudavets et al
146 Brain Pathology 24 (2014) 142–147
© 2013 International Society of Neuropathology

Italian family with Gerstmann-Sträussler-Scheinker syndrome.Neurology 42:809–810.
18. Kulczycki J, Collinge J, Lojkowska W, Parnowski T,Wierzba-Bobrowicz T (2001) Report on the first polish case of theGerstmann-Sträussler-Scheinker syndrome. Folia Neuropathol39:27–31.
19. Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins Set al (2005) Mortality from Creutzfeldt-Jakob disease and relateddisorders in Europe, Australia, and Canada. Neurology64:1586–1591.
20. Lee HS, Sambuughin N, Cervenakova L, Chapman J, Pocchiari M,Litvak S et al (1999) Ancestral origins and worldwide distributionof the PRNP 200K mutation causing familial Creutzfeldt-Jakobdisease. Am J Hum Genet 64:1063–1070.
21. Liberski PP (2012) Gerstmann-Sträussler-Scheinker disease. AdvExp Med Biol 724:128–137.
22. Parchi P, Chen S, Brown P, Zou W, Capellari S, Budka H et al(1998) Different patterns of truncated prion protein fragmentscorrelate with distinct phenotypes in P102L Gerstmann–Sträussler–Scheinker disease. Proc Natl Acad Sci U S A95:8322–8327.
23. Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD,Nochlin D et al (1998) Phenotypic variability of Gerstmann-
Sträussler-Scheinker disease is associated with prion proteinheterogeneity. J Neuropathol Exp Neurol 57:979–988.
24. Prusiner SB (1991) Molecular biology of prion diseases. Science252:1515–1522.
25. Wadsworth J, Joiner S, Linehan JM, Cooper S, Powell S, MallinsonG et al (2006) Phenotypic heterogeneity in inherited prion disease(P102L) is associated with differential propagation ofprotease-resistant wild-type and mutant prion protein. Brain129:1557–1569.
26. Wang Y, Qiao X, Zhao C, Gao X, Yao Z, Qi L, Lu C (2006) Reporton the first Chinese family with Gerstmann-Sträussler-Scheinkerdisease manifesting the codon 102 mutation in the prion proteingene. Neuropathology 26:429–432.
27. Webb T, Poulter M, Beck J, Uphill J, Adamson G, Campbell T et al(2008) Phenotypic heterogeneity and genetic modification of P102Linherited prion disease in an international series. Brain131:2632–2646.
28. Worrall B, Rowland L, Chin S, Mastrianni JA (2000) amyotrophy inprion diseases. Arch Neurol 57:33–38.
29. Woulfe J, Kertesz A, Frohn I, Bauer S, St. George-Hyslop P,Bergeron C (2005) Gerstmann-Sträussler-Scheinker disease with theQ217R mutation mimicking frontotemporal dementia. Acta Neurol110:317–319.
Riudavets et al GSS with Variable Phenotype in Argentina
147Brain Pathology 24 (2014) 142–147
© 2013 International Society of Neuropathology



![KM C364e-20190813142255 · Voc [VI Vmp [VI Imax [Adc Isc Adc Prnp [W] Maximum system voltage [V] Maximum Over-Current Protection Ratin [A Page 2 of 3 xxx= 380 to 425 in step of 5](https://img.pdfslide.us/doc/110x75/5fd27ac4724ba838c266a207/km-c364e-20190813142255-voc-vi-vmp-vi-imax-adc-isc-adc-prnp-w-maximum-system.jpg)
















