Embed Size (px)
Citation preview

Plant Gene 6 (2016) 1–12
Contents lists available at ScienceDirect
Plant Gene
j ourna l homepage: www.e lsev ie r .com/ locate /p lantgene
Genome-wide analysis of the SPL family transcription factors and theirresponses to abiotic stresses in maize
Hu-De Mao a,b,⁎, Li-Juan Yu b, Zhan-Jie Li b, Yan Yan b, Ran Han b, Hui Liu b, Meng Ma b
a State Key Laboratory of Crop Stress Biology for Arid Areas and College of Plant Protection, Northwest A&F University, Yangling, Shaanxi 712100, Chinab College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China
⁎ Corresponding author.E-mail address: [email protected] (H.-D. Mao).
http://dx.doi.org/10.1016/j.plgene.2016.03.0032352-4073/© 2016 The Authors. Published by Elsevier B.V
a b s t r a c t
a r t i c l e i n f oArticle history:Received 9 January 2016Received in revised form 20 February 2016Accepted 1 March 2016Available online 4 March 2016
SQUAMOSA promoter binding protein-like (SPL) transcription factors (TFs) are plant-specific and play vital reg-ulatory roles in plant growth and development. Even though they are one of the unique groups of TFs in plants,their characteristics, evolutionary relationships and expression patterns are largely unknown inmaize, an impor-tant food crop worldwide. In this study, we identified 31 SPL genes (ZmSPLs) in the maize B73 genome. A phylo-genetic analysis showed that these genes were divided into six groups (Groups 1–6) and members within thesame group shared conserved exon/intron distributions and motif compositions, implying their functional re-dundancy. The 31 ZmSPL genes were distributed unevenly on 9 of the 10 chromosomes, with 10 segmental du-plication events, suggesting that the expansion of the ZmSPL genes occurred due to segmental duplication.Analysis of the Ka/Ks ratios showed that the duplicated ZmSPL genes had primarily undergone strong purifyingselection. In addition, 19 of the 31 ZmSPLs, belonging to Groups 1, 2 and 3, were targets of microRNAmiR156, in-dicating of the miR156-mediated posttranscriptional regulation of these ZmSPL genes. Expression analysis of theZmSPLs in various tissues at different development stages revealed distinct spatiotemporal patterns. Moreover,quantitative real-time PCR analysis identified several ZmSPL genes that were potentially involved in responseto abiotic stresses. Our results present a comprehensive overview of the maize SPL gene family and provide animportant foundation for further uncovering the biological functions of ZmSPLs in the growth and developmentof maize.
© 2016 The Authors. Published by Elsevier B.V.
Keywords:MaizeGenome-wide analysisSPL genesPosttranscriptional regulationExpression patternAbiotic stress response
1. Introduction
Transcription factors (TFs) are a large class of regulators that controlgene expression at the transcriptional level and often serve as on–offswitches in the developmental processes of eukaryotic organisms(Sun and Oberley, 1996). SQUAMOSA promoter binding protein-like(SPL) TFs are specific to plants and have a highly conserved SQUAMOSApromoter binding protein (SBP) domain, with approximately 78 aminoacid residues. This domain contains three functionally importantmotifs,including two zinc-binding sites, Cys–Cys–Cys–His (Zn1) and Cys–Cys–His–Cys (Zn2), and a nuclear localization signal (NLS) that partiallyoverlaps with the second Zn-finger, located at the C-terminal of theSBP domain (Yamasaki et al., 2004; Birkenbihl et al., 2005). Genesencoding SPLs were first identified for SBP1 and SBP2 in Antirrhinummajus (Klein et al., 1996). These genes have recently been identified insingle-celled green algae, mosses and gymnosperms, as well as angio-sperms (Jin et al., 2014). SPLs are encoded by a large gene family inplants. For instance, there are 16 SPL genes in Arabidopsis thaliana
.
(Cardon et al., 1999), 19 in rice (Xie et al., 2006), and 28 in Populustrichocarpa (Li and Lu, 2014).
Recent studies in various species have suggested that SPL genes af-fect a broad range of developmental processes. To date, sixteen SPLgenes have been identified in the Arabidopsis genome (Cardon et al.,1999), and many of them have been found responsible for a diversenumber of developmental process events, including embryogenesis(Unte et al., 2003), shoot and leaf development (Wu and Poethig,2006; Schwarz et al., 2008), flowering (Gandikota et al., 2007), vegeta-tive and reproductive phase transitions (Jung et al., 2011), plastochronformation (Wang et al., 2008), fertility (Xing et al., 2010), copper ho-meostasis (Yamasaki et al., 2009) and plant hormone signaling (Zhanget al., 2007). Besides Arabidopsis, knowledge of the functions of SPLgenes in other plant species, especially important agricultural and eco-nomical crops, has also begun to accumulate, highlighting the diverseroles of the SPL proteins in plant development. For example, OsSPL14,also known as ideal plant architecture1 (IPA1), is related to plant archi-tecture and substantially enhances grain yield (Jiao et al., 2010; Miuraet al., 2010; Lu et al., 2013); OsSPL16 promotes grain quality and yield(Wang et al., 2012); andOsLG1 regulates a closed panicle trait in domes-ticated rice (Ishii et al., 2013). Epigenetic mutation analysis of the toma-to colorless non-ripening (cnr) mutant has demonstrated that one of the

2 H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
tomato SPL genes is vital for tomato fruit ripening (Manning et al.,2006). In maize, liguleless1, which contains the SBP domain, regulatesligule and auricle formation (Moreno et al., 1997), and tasselsheath4(tsh4) is an SPL gene that regulates bract development, a necessity inbranch meristem initiation and maintenance (Chuck et al., 2010).
Along with the deepening of research, an increasing number ofmicroRNAs (miRNAs) have been found to play a crucial role in theregulation of gene function in plants (Chen and Rajewsky, 2007;Lauressergues et al., 2015). MiRNAs are small RNA molecules (20–24nucleotides in length) that can cause the degradation of mRNA, or re-press translation by binding to the transcripts of their target genesand forming an RNA-induced silencing complex; approximately half oftheir target genes encode transcription factors (Rhoades et al., 2002;Bartel, 2004). As a gene family that encodes transcription factors,more than half of the SPL genes identified to date have been found tobe targeted by miR156/157. In Arabidopsis, for instance, 10 of the 16SPL genes are putative targets of AtmiR156 (Wu and Poethig, 2006;Gandikota et al., 2007; Addo-Quaye et al., 2008), while 11 of the 19SPL genes in rice have been revealed to be putative targets ofOsmiR156 (Xie et al., 2006). The target sites are located both in the cod-ing region and in the 3′-untranslated region (3′ UTR) (Xie et al., 2006).
Unfortunately, despite an increasing body of physiological and bio-chemical data, the biological role of the SPL gene family remains elusive.Although regulatory and functional data of the SPL genes are increasing,the biological role of many members in this family still needs to beclarified. Maize as one of most important cropworldwide, it is thereforeintriguing for us to do a genomewide analysis of SPL family in this spe-cies after its full genome sequence released (Schnable et al., 2009), eventhough some early work has been done (Hultquist and Dorweiler,2008). In order to characterize the whole SPL gene family in maize, wesearched the maize genome assembly which identified 31 full-lengthZmSPLs. This current study aimed to systematically analyze phylogenet-ic relationships, gene structures, conserved proteinmotifs, chromosom-al locations, gene duplications and expression patterns of all theidentified ZmSPLs. Additionally, we investigated the miR156-mediatedposttranscriptional regulation of ZmSPLs, providing useful informationto elucidate further the biological functions of SPL genes in maize.
2. Materials and methods
2.1. Identification of maize SPL genes
Different approaches were applied to identify the putative SPL pro-teins from Zea mays L. Initially, amino acid sequences encoding SPL pro-teins from Zea mayswere retrieved from the Plant Transcription FactorDatabase 3.0 (http://planttfdb.cbi.pku. edu.cn/index.php) (Jin et al.,2014). Then, genes in themaize genome annotatedwith a Pfam SBP do-main (PF03110) were retrieved from Phytozome v10.0 (http://www.phytozome.net/eucalyptus.php). In addition, BLAST searches wereperformed against the maize genome using 16 known ArabidopsisSPL proteins, to identify any additional SPL members. All but the lon-gest splice variants were removed and redundant sequences werealso removed using the decrease redundancy tool (web.expasy.org/decrease_redundancy). The presence of the SBP domain in the pro-teins was evaluated using the Pfam (http://pfam.sanger.ac.uk/search)(Finn et al., 2006) and SMART (http://smart.embl-heidelberg.de/)tools (Letunic et al., 2004). Arabidopsis SPL proteins sequences weredownloaded from the Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org/). Rice and sorghum SPL sequences weredownloaded from the Plant Transcription Factor Database 3.0 (http://planttfdb.cbi.pku.edu.cn/index.php) (Jin et al., 2014).
Information on the ZmSPL genes, including the number of aminoacids, coding sequence (CDS), open reading frame (ORF) lengths andlocation coordinates were acquired from the Phytozome database.Physical parameters, including the molecular mass and isoelectric
point (pI) of the deduced proteins were generated by ExPASy (http://web.expasy.org/protparam/).
2.2. Multiple sequence alignments and phylogenetic analysis
To reveal the phylogenetic relationships among the ZmSPL proteinsand their orthologs in rice, sorghum and Arabidopsis, a neighbor-joining(NJ) phylogenetic tree was constructed for 31 ZmSPLs, 16 AtSPLs, 19OsSPLs and 18 SbSPLs using MEGA5.0 (Tamura et al., 2011). The boot-strap value was set to 1000 replicates and the pairwise deletion optionwas used. Protein sequences used for the phylogenetic tree are listed inSupplementary File A1.
2.3. Gene structure and conserved motif analysis
To illustrate the exon–intron structures of the individual ZmSPLgenes, a comparison of the genomic sequences and their correspondingcoding sequences (CDS) was operated using GSDS (Gene Structure Dis-play Server) (http://gsds.cbi.pku.edu.cn/) (Guo et al., 2007). Conservedmotif of ZmSPL proteins was identified using the online MEME (Multi-ple Expectation Maximization for Motif Elicitation) (http://meme.nbcr.net/meme/cgibin/meme.cgi) (Bailey and Elkan, 1995). The parameterswere adopted as follows: optimum motif width was set to between 6and 150 residues;maximumnumber ofmotifs was set to 15. Each struc-tural motif annotation was performed using the Pfam (http://pfam.sanger.ac.uk/search) and SMART (http://smart.embl-heidelberg.de/)tools. The 78 amino acids of the SBP domain were aligned usingclustalW. Sequence logos were generated using the weblogo platform(http://weblogo.berkeley.edu/).
2.4. Chromosome location and gene duplication
Chromosome locations of the ZmSPL genes were determined by aBLAST analysis of the ZmSPLs against the maize genome fromPhytozome v10.0 (http://www. phytozome.net/eucalyptus.php). Thetandem duplicates were identified, according to Hanada et al. (2008),as pairs of ZmSPL genes within 100 kb of each other that had 10 orfewer nonhomologous genes between them. Segmental duplicategene pairs were analyzed on the Plant Genome Duplication Databaseserver (http://chibba.agtec.uga.edu/duplication/index/locus), with adisplay range of 100 kb. The nonsynonymous rates (Ka), synonymousrates (Ks) and evolutionary constraints (Ka/Ks) between the duplicatedpairs of ZmSPLs were calculated using the CODEML program in PAML(Yang, 2007). The approximate date of the duplication events was cal-culated using T = Ks / 2λ × 10−6 million years ago (Mya), based onthe clock-like rates (λ) in grasses of 6.5 × 10−9 (Gaut et al., 1996).
2.5. Prediction of ZmSPLs targeted by miR156
The sequences of maize miR156a–miR156l were obtained from themiRBase (Kozomara and Griffiths-Jones, 2011) (http://www.mirbase.org/). ZmSPLs targeted bymiR156were predicted by searching the cod-ing regions and 3′ UTRs of all the ZmSPL genes for complementary se-quences of maize miR156a–miR156l on the psRNATarget server(http://plantgrn.noble.org/psRNATarget/?function=3), using defaultparameters (Dai and Zhao, 2011).
2.6. Microarray analysis
The published transcriptome data of the genome-wide gene expres-sion atlas, of the maize inbred line B73, provided a useful complementto understand the expression patterns of the ZmSPLs during differentdevelopmental stages (Sekhon et al., 2011). Normalized gene expres-sion values, expressed as the number of fragments per kilobase ofexon per million fragments mapped (FPKM), were transformed usinglog2 (FPKM+ 1), and then used for further expression analysis.

3H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
2.7. Plant materials and growth conditions
Seeds of the maize inbred line B73 were surface-sterilized in 1‰(v/v) Topsin-M (Rotam Crop Sciences Ltd.) for 10 min. Then theywere washed in deionized water and germinated on wet filter paperat 28 °C for 3 days. The germinated seeds were placed in a nutrient so-lution (0.75mMK2SO4, 0.1mMKCl, 0.25mMKH2PO4, 0.65mMMgSO4,0.1 mM EDTA–Fe, 2 mM Ca(NO3)2, 1.0 mM MnSO4, 1.0 mM ZnSO4,0.1 mM CuSO4, 0.005 mM (NH4)6Mo7O24) for hydroponic cultivationwith a 16-h light/8-h dark cycle at 28 °C. Four kinds of treatments:drought, low temperature (10 °C), NaCl (200 mmol/L), and abscisicacid (ABA; 100 mmol/L) were separately applied to three-leaf stageB73 seedlings. The salt and ABA treatments were conducted by cultur-ing the seedlings in 200mmol/L of NaCl and 100mmol/L of ABA culturesolutions, respectively. For drought treatment, the seedlings wereplaced on a clean bench and subjected to dehydration (28 °C, relativehumidity of 40–60%). Leaves from a minimum of three seedlings werecollected after 5 h treatment, and then immediately frozen into liquidnitrogen and stored at−80 °C for RNA extraction.
2.8. RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was isolated from the collected samples using TRIZOLreagent (Biotopped, China) according to the manufacturer's instruc-tions. The RNA was subsequently treated with RNase-free DNase I(Takara, China) to remove genomic DNA contamination. First-strand cDNA was synthesized from 1 μg of total RNA using Recombi-nant M-MLV reverse transcriptase (Promega, USA). Quantitative realtime-PCR (qRT-PCR) was performed in optical 48-well plates using theABI7300 Thermo-cycler (Applied Biosystems, USA). Reactionswere car-ried out in a 10-μl volume containing 1 μl diluted cDNA, 200 nM gene-specific primers, and 5 μl SYBR Premix Ex Taq II (Takara, China) withthe following conditions: 10 min at 95 °C, 40 cycles of 15 s at 95 °C,
Table 1The 31 ZmSPL genes identified in maize and their sequence characteristics.
Gene name Sequenced IDProtein
Length (aa) Mass (Da)
ZmSPL1 GRMZM2G160917 403 41,667.07ZmSPL2 GRMZM2G169270 1106 121,171.61ZmSPL3 GRMZM2G156756 972 105,912.24ZmSPL4 GRMZM2G036297 399 43,359.10ZmSPL5 GRMZM2G126018 383 40,199.40ZmSPL6 GRMZM2G061734 429 44,959.34ZmSPL7 GRMZM2G113779 99 11,294.57ZmSPL8 GRMZM2G133646 881 98,614.88ZmSPL9 GRMZM5G878561 447 46,723.30ZmSPL10 GRMZM2G106798 325 34,384.04ZmSPL11 GRMZM2G101511 464 45,168.34ZmSPL12 AC233751.1_FG002 588 62,871.73ZmSPL13 GRMZM2G460544 408 41,901.31ZmSPL14 GRMZM2G098557 1112 121,807.20ZmSPL15 GRMZM2G168229 406 42,601.38ZmSPL16 GRMZM2G163813 331 36,182.26ZmSPL17 GRMZM2G065451 482 50,723.66ZmSPL18 GRMZM2G138421 961 105,083.27ZmSPL19 GRMZM2G414805 440 47,112.40ZmSPL20 GRMZM2G097275 479 50,856.67ZmSPL21 GRMZM2G126827 332 36,514.70ZmSPL22 GRMZM2G111136 441 46,001.58ZmSPL23 GRMZM2G156621 332 36,514.70ZmSPL24 GRMZM2G101499 434 46,035.69ZmSPL25 GRMZM2G307588 378 39,599.11ZmSPL26 GRMZM2G067624 206 21,040.19ZmSPL27 GRMZM2G081127 861 96,563.72ZmSPL28 GRMZM2G109354 850 94,022.87ZmSPL29 GRMZM2G371033 309 33,655.13ZmSPL30 GRMZM2G148467 450 47,007.47ZmSPL31 GRMZM2G058588 396 43,268.96
and 30 s at 60 °C. The specificity of the amplicon for each primer pairwas verified by melting curve analysis. The expression of ZmUbi-2(UniProtKB/TrEMBL; ACC:Q42415) was used as an internal control.The quantification method (2−ΔΔCt) (Livak and Schmittgen, 2001)was used and the variation in expression was estimated from thethree biological replicates. The primer pairs used for the qRT-PCR anal-ysis of the ZmSPL genes are listed in Supplementary File A2.
3. Results
3.1. Identification of SPL family genes in maize
Gene models in the maize genome containing a SBP domain wereidentified using a superfamily search (see the Materials and Methodssection). The candidates were then examined by Pfam and SMART toconfirm the presence of the SBP domain. After removing the redundantsequences, 31 non-redundant SPL genes were identified. The 31 SPLgenes were designated as ZmSPL, followed by the Arabic numbers1–31, according to their position (from top to bottom) on chromosomes1–10 (Table 1).
The physical parameters of each SPL protein were calculated usingExPASy server. As shown in Table 1, the full-length coding sequences(CDS) of the ZmSPL genes ranged from 300 bp (ZmSPL7) to 3339 bp(ZmSPL14) with the deduced proteins of 99–1112 amino acids. Further-more, the computed molecular weights of these SPL proteins rangedfrom 11.2 to 121.9 kDa. The theoretical pI of the deduced ZmSPL pro-teins ranged from 5.41 to 10.05.
3.2. Phylogenetic analysis of SPLs
To better understand themolecular evolution and phylogenetic rela-tionship among SPLs in plants, we constructed a phylogenetic treebased on multiple sequence alignments of maize, sorghum, rice, and
ORF GRASSIUS
PI Chr. length (bp) Exons TF name
8.73 1 1212 3 ZmSBP86.97 1 3321 10 –5.43 1 2919 11 –7.45 2 1200 3 ZmSBP158.64 2 1152 3 ZmSBP236.73 2 1290 3 –
10.05 2 300 2 ZmSBP137.27 3 2646 11 –9.21 3 1344 4 –8.70 4 978 3 ZmSBP37.18 4 1395 3 –8.64 4 1767 6 –8.73 4 1227 3 ZmSBP307.54 4 3339 10 –8.88 4 1221 3 –8.98 4 996 4 ZmSBP199.01 4 1449 4 ZmSBP205.78 5 2886 11 ZmSBP69.10 5 1323 4 ZmSBP259.15 5 1440 4 –8.96 5 999 4 ZmSBP126.70 5 1326 3 ZmSBP108.96 5 999 4 ZmSBP319.14 6 1305 3 –9.33 7 1137 3 ZmSBP29.75 7 621 2 ZmSBP297.48 8 2586 12 –5.41 8 2553 10 –8.84 8 930 3 –9.31 10 1353 3 ZmSBP217.81 10 1191 3 ZmSBP28
4 H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
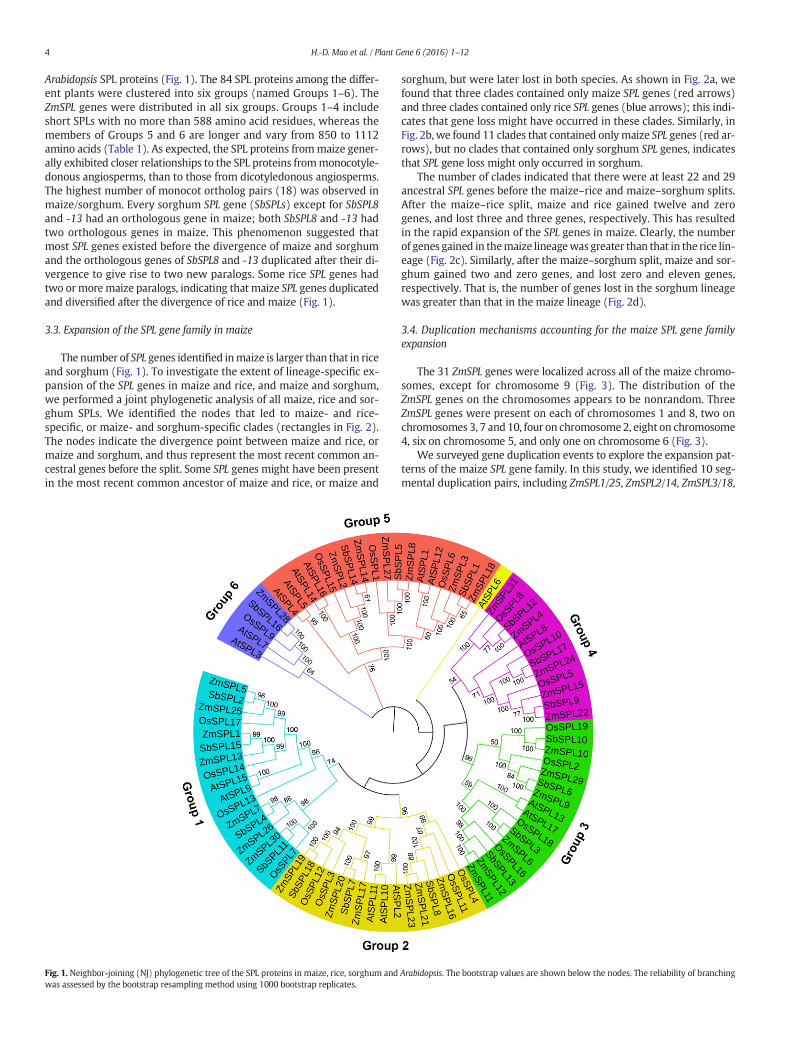
Arabidopsis SPL proteins (Fig. 1). The 84 SPL proteins among the differ-ent plants were clustered into six groups (named Groups 1–6). TheZmSPL genes were distributed in all six groups. Groups 1–4 includeshort SPLs with no more than 588 amino acid residues, whereas themembers of Groups 5 and 6 are longer and vary from 850 to 1112amino acids (Table 1). As expected, the SPL proteins frommaize gener-ally exhibited closer relationships to the SPL proteins frommonocotyle-donous angiosperms, than to those from dicotyledonous angiosperms.The highest number of monocot ortholog pairs (18) was observed inmaize/sorghum. Every sorghum SPL gene (SbSPLs) except for SbSPL8and -13 had an orthologous gene in maize; both SbSPL8 and -13 hadtwo orthologous genes in maize. This phenomenon suggested thatmost SPL genes existed before the divergence of maize and sorghumand the orthologous genes of SbSPL8 and -13 duplicated after their di-vergence to give rise to two new paralogs. Some rice SPL genes hadtwo ormoremaize paralogs, indicating that maize SPL genes duplicatedand diversified after the divergence of rice and maize (Fig. 1).
3.3. Expansion of the SPL gene family in maize
The number of SPL genes identified inmaize is larger than that in riceand sorghum (Fig. 1). To investigate the extent of lineage-specific ex-pansion of the SPL genes in maize and rice, and maize and sorghum,we performed a joint phylogenetic analysis of all maize, rice and sor-ghum SPLs. We identified the nodes that led to maize- and rice-specific, or maize- and sorghum-specific clades (rectangles in Fig. 2).The nodes indicate the divergence point between maize and rice, ormaize and sorghum, and thus represent the most recent common an-cestral genes before the split. Some SPL genes might have been presentin the most recent common ancestor of maize and rice, or maize and
Fig. 1. Neighbor-joining (NJ) phylogenetic tree of the SPL proteins in maize, rice, sorghum andwas assessed by the bootstrap resampling method using 1000 bootstrap replicates.
sorghum, but were later lost in both species. As shown in Fig. 2a, wefound that three clades contained only maize SPL genes (red arrows)and three clades contained only rice SPL genes (blue arrows); this indi-cates that gene loss might have occurred in these clades. Similarly, inFig. 2b, we found 11 clades that contained onlymaize SPL genes (red ar-rows), but no clades that contained only sorghum SPL genes, indicatesthat SPL gene loss might only occurred in sorghum.
The number of clades indicated that there were at least 22 and 29ancestral SPL genes before the maize–rice and maize–sorghum splits.After the maize–rice split, maize and rice gained twelve and zerogenes, and lost three and three genes, respectively. This has resultedin the rapid expansion of the SPL genes in maize. Clearly, the numberof genes gained in themaize lineagewas greater than that in the rice lin-eage (Fig. 2c). Similarly, after the maize–sorghum split, maize and sor-ghum gained two and zero genes, and lost zero and eleven genes,respectively. That is, the number of genes lost in the sorghum lineagewas greater than that in the maize lineage (Fig. 2d).
3.4. Duplication mechanisms accounting for the maize SPL gene familyexpansion
The 31 ZmSPL genes were localized across all of the maize chromo-somes, except for chromosome 9 (Fig. 3). The distribution of theZmSPL genes on the chromosomes appears to be nonrandom. ThreeZmSPL genes were present on each of chromosomes 1 and 8, two onchromosomes 3, 7 and 10, four on chromosome2, eight on chromosome4, six on chromosome 5, and only one on chromosome 6 (Fig. 3).
We surveyed gene duplication events to explore the expansion pat-terns of the maize SPL gene family. In this study, we identified 10 seg-mental duplication pairs, including ZmSPL1/25, ZmSPL2/14, ZmSPL3/18,
Arabidopsis. The bootstrap values are shown below the nodes. The reliability of branching

Fig. 2. Phylogenetic tree and copy number changes of themaize and rice (a), andmaize and sorghum (b) SPL genes. The numbers on the branches indicate the bootstrap percentage valuescalculated from 1000 replicates; only values higher than 50% are shown. The nodes that represent themost recent common ancestral genes before themaize–rice (a) andmaize–sorghum(b) splits are indicated by the red rectangles (bootstrap support N50%). Clades that contain onlymaize or rice SPLs are indicated by the red and blue arrows, respectively. The copy numberchanges in the maize and rice (c), and maize and sorghum (d) SPL genes are shown; the numbers in the circles and rectangles represent the numbers of SPL genes in the extant andancestral species, respectively, while the numbers on the branches (with plus and minus symbols, respectively) represent the numbers of gene gains and losses.
5H.-D. Mao et al. / Plant Gene 6 (2016) 1–12

Fig. 3.Genomic localization of the ZmSPL and Zm-miR156 genes. Regions that are assumed to correspond to homologous genome blocks are connected by gray lines. The paralogous ZmSPLgenes are indicated by the red lines located within the gray trapezoids.
6 H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
ZmSPL4/31, ZmSPL5/25, ZmSPL7/26, ZmSPL8/27, ZmSPL9/29, ZmSPL16/21and ZmSPL22/24, and each of them were located in a pair of paralogousblocks (Fig. 3; Table 2). In addition, no ZmSPL genes were arranged intandem repeats. The results provide strong evidence that segmental du-plication has made an important contribution to maize SPL gene familyexpansion.
Further, the selective constraints of duplicated ZmSPL geneswere ex-plored by calculating the ratio of nonsynonymous substitutions (Ka) tosynonymous substitutions (Ks) of each duplicated pairs. Generally, du-plicated genes with a high Ka/Ks ratio (N1) are deemed to be evolvingunder positive selection, Ka/Ks = 1 indicates neutral selection, whileKa/Ks b 1 indicates negative or purifying selection (Juretic et al.,2005). A summary of Ka/Ks for 10 ZmSPL duplicated pairs is shown inTable 2. The ratios of duplicated ZmSPL gene pairs were all less than0.7, which implied that the ZmSPL genes have mainly undergone strongpurifying selection after the duplication events with limited functional
Table 2The Ka/Ks ratios and estimated divergence times for the duplicated ZmSPL proteins.
Paralogous pairs Ka Ks Ka/Ks Duplication date (MY)
ZmSPL1-ZmSPL25 0.33 0.74 0.45 60.66ZmSPL2-ZmSPL14 0.05 0.24 0.21 19.67ZmSPL3-ZmSPL18 0.05 0.19 0.26 15.57ZmSPL4-ZmSPL31 0.08 0.25 0.32 20.49ZmSPL5-ZmSPL25 0.12 0.32 0.38 26.23ZmSPL7-ZmSPL26 0.13 0.21 0.62 17.21ZmSPL8-ZmSPL27 0.09 0.23 0.39 18.85ZmSPL9-ZmSPL29 0.09 0.23 0.39 18.85ZmSPL16-ZmSPL21 0.07 0.19 0.37 15.57ZmSPL22-ZmSPL24 0.44 0.74 0.59 60.66
divergence. We also estimated the evolutionary timescale based on asubstitution rate of 6.5 × 10−9 substitutions per synonymous site peryear. As shown in Table 2, the divergence time of duplicated ZmSPLgene pairs ranged from 15.57 to 60.66 million years.
3.5. Gene structure and conserved motif analysis of ZmSPLs
To better examine the structural diversity of the ZmSPL genes, weperformed an exon/intron analysis (Fig. 4). The schematic structuressuggest that introns existed in the coding sequences of all the ZmSPLgenes and the number of exons ranged from2 to 12. In addition, thema-jority of ZmSPL genes in the same group (Fig. 1) bore similar numbers ofexons. For example, all genes within Group 2 were made up of fourexons. However, genes in Group 5 contained either ten or eleven exons.
Analysis the conserved domains of ZmSPL proteins showed that allof the 31 ZmSPLs contained an SBP domain, which was located in a re-gion close to the N-terminus and was encoded by the first two exonsof the ZmSPL genes (Fig. 4). Sequence analysis of SBP domains revealedthat the conserved zinc-binding sites, Zn1 and Zn2, also existed in theSBP domain of the ZmSPL proteins. Zn1 contains the CX4CX16CX2H(CCCH) or CX4CX16CX2C (CCCC) signature sequence, whereas Zn2 con-tains the CX2CX3HX11C (CCHC) signature (Supplementary File A3). Inaddition to Zn1 and Zn2, the SBP domain contains a conserved nuclearlocation signal (NLS) in the C-terminus of the SBP domains, with theconsensus sequence of KRX11RRRK (Supplementary File A3). Moreover,six ZmSPL proteins (ZmSPL2, ZmSPL3, ZmSPL8, ZmSPL14, ZmSPL18 andZmSPL27) belonging to Group 5 contained an ANK or Ank-2 domain,with three or four ankyrin repeats (Supplementary File A4), indicatingthat these ZmSPL proteins may function by interacting with other pro-teins in plant cells.

Fig. 4. Gene structures of the ZmSPLs. Exons, introns, SQUAMOSA promoter binding protein (SBP) domains, intron phases and miR156/157 target sites are shown.
7H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
In addition to the conserved domains, other conserved motifscould also be important for the function of SPLs (Xie et al., 2006;Guo et al., 2008). We identified 15 motifs for the 31 ZmSPLs (Fig. 5;Supplementary File A5). Motifs 1, 2, 5 and 6 make up the SBP domain(Supplementary File A3), and Motif 4 corresponds to the ANK do-main. However, the biological annotation of the other putative mo-tifs remain unclear. Moreover, the number of motifs in each SPLvaried from 3 to 12, and the ZmSPLs within a same subfamily sharedcommon motif compositions (Fig. 5), indicating functional similari-ties among these proteins.
3.6. MiR156-mediated posttranscriptional regulation of ZmSPLs
According to the available annotation information (Zhang et al.,2009), the twelve maize miR156 genes (Zm-miR156a–Zm-miR156l)were distributed on 7 of the tenmaize chromosomes: twowere presenton each of chromosomes 2, 3, 4, 5 and 7, and onewas present on each ofchromosomes 6 and 10 (Fig. 3). In order to understand the miR156-mediated posttranscriptional regulation of the ZmSPL genes, wesearched the coding regions and 3′ UTRs of all ZmSPLs for the targetsof maize miR156a–miR156l. The results showed that 19 ZmSPL genes,belonged to Group 1–3 were targets of miR156. The miR156-targetsites in 17 of these ZmSPL genes present in the last exon, and encodedthe conserved peptide ALSLLS. The target sites for the other twoZmSPL genes were located in the 3′ UTRs, close to the stop codons(Fig. 4; Fig. 6). ThemiR156-targeted SPL genes consistently included se-quences from rice and Arabidopsis and were distributed into only three
of the subgroups (Groups 1, 2 and 3). This suggests that miR156-mediated posttranscriptional regulation of the SPLs is conserved inplants.
3.7. Expression profiles of ZmSPLs in different tissues
The global transcriptome data provide us valuable information topredict the gene functions. In this study, we focus on the temporal andspatial expression patterns of ZmSPL genes to reveal their roles in devel-opmental regulation. For this purpose, we conducted a comprehensiveexpression analysis of ZmSPL genes using the publicly availablemicroar-ray data (Sekhon et al., 2011). It can be seen from the heatmap that theexpression patterns of different ZmSPL genes varied greatly (Fig. 7). Thetranscripts of ZmSPL3, -8, -9, -12, -14, and -18 were constitutivelyexpressed in the various tissues. ZmSPL3, -8, -14, -18, -19, -27, and -28exhibit relatively high expression levels in most of the developmentalstages, while the transcripts of ZmSPL5, -9, -12, -15, -22, -24, and -30showed relatively low expression levels. In addition, some tissue- ororgan-specific genes were discovered. For example, ZmSPL4wasmainlyexpressed in the husk, while ZmSPL6 and ZmSPL11 were expressed athigh levels in the cob (Fig. 7). Moreover, there tended to be little or novariation in the expression (among the tissues tested) of the ZmSPLgenes that lacked an miR156 target site, including ZmSPL2, -3, -4, -8,-14, -15, -18, -22, -24, -27, -28, and -31. In contrast, the expression levelsof ZmSPL genes containing amiR156 target site varied among the differ-ent tissues, except for ZmSPL5, -9, -12, and -30 (Fig. 7). The results indi-cate that the ZmSPLsmay play important roles in maize development.

Fig. 5. Distribution of conserved motifs in ZmSPL proteins. The conserved motifs were identified through MEME; different motifs are represented by the different colored boxes. Thelocation of each motif can be estimated using the scale at the bottom.
8 H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
3.8. Expression profiles of ZmSPLs in response to abiotic stresses
In order to decipher the role of the ZmSPL genes in response to abiot-ic stresses, the expression of 31 ZmSPLs was analyzed in response toacute dehydration, salinity, cold and ABA exposures (Fig. 8; Supplemen-tary File A6). The heat map of the expression levels is shown in Fig. 8a.Overall, the qRT-PCR analysis demonstrates that all of the genesdisplayed variations in their expression behavior in response to one ormore stresses. Among the four treatments, cold stress induced relativelymore dramatic changes in the transcript abundances of the ZmSPL genesthan dehydration, salinity and ABA. Some genes were differentiallyexpressed in response to a specific stress. For example, ZmSPL11 andZmSPL21 were only induced by ABA, while ZmSPL10 was only inducedby the salinity treatment. Some genes were co-regulated by two ormore stresses. For example, transcripts of ZmSPL2 and ZmSPL20 accu-mulated during the cold and drought treatments; ZmSPL4, ZmSPL19and ZmSPL24 were induced by the drought, salinity and ABA treat-ments; and ZmSPL1, -9, -12, -13, -15, -22, -26, and -29were induced byall four stresses (Fig. 8). The results suggest that these ZmSPL genesmay involve in regulating abiotic stresses in maize.
4. Discussion
SPL genes are plant-specific transcription factors that are broadlyexist in photosynthetic organisms (Cardon et al., 1999; Guo et al.,2008; Jin et al., 2014), and are reported to participate in many crucial
biological processes in plants. Recently, SPL genes were identified in di-verse plant species, and the number of SPL genes varies among landplants. In this study, three methods were used to identify all SPL genesin maize, and result the different numbers of SPLs, respectively. For ex-ample, there are 65 maize SPL members in Phytozome 10 (https://phytozome.jgi.doe.gov/pz/ portal.html), while only 55 members inPlantTFDB 3.0 (http://planttfdb.cbi.pku.edu. cn/). After removed all re-dundant and splice variant sequences, 31 non-redundant ZmSPLgenes, and distributed on nine chromosomes were identified in themaize genome (Table 1; Fig. 3). Phylogenetic analyzes demonstratedthat all the SPL genes were clustered into six groups (Group 1–6)(Fig. 1), consistent with previous findings (Guo et al., 2008). In addition,this classification was consistent with gene structure and motif compo-sition. Geneswithin a group shared a similar length, structure,motif dis-tribution, and miR156 target site location (Fig. 4; Fig. 5). The resultsindicated that the classification and evolution of SPL genes might beclosely related to their structural divergence and diversification.
Our analysis of the conserved protein motifs in themaize SPL family(using MEME) showed that the majority of the ZmSPL proteins in thesame group shared similar motif distributions. However, between dif-ferent groups, there was a high divergence in the motif patterns(Fig. 5). For example, motif 10 was more specific to Group 2, whereasmotifs 4, 7, 8 and 13 were only found in Group 5; these results indicatethe intricate nature of the function of SPL proteins in maize. In addition,we found some motifs, like motifs 1, 2, 5 and 6, were more conservedand appeared inmany ZmSPL proteins (Fig. 5). Thesemotifs could be es-sential components that determine the commonmolecular functions of

Fig. 6. ZmSPLs targeted by miR156. The green lines represent the open reading frames (ORFs). The lines flanking the ORFs represent 3′-UTR. The red lines represent the SQUAMOSApromoter binding protein (SBP)-domain. The miRNA complementary sites (black) with the nucleotide positions of the ZmSPL cDNAs are indicated. The RNA sequence of eachcomplementary site, from 5′ to 3′, and the predicted miRNA sequence, from 3′ to 5′, are shown in the expanded regions.
9H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
the SBP domain among the different familymembers. The differences inmotif patterns among the different subfamilies of the ZmSPL genes arethe structural foundations for the diversity in the gene functions.
To illuminate the phylogenetic relationship of the SPL genes, a com-parative genomic analysis of the plant SPL members from monocots
(maize, rice and sorghum) and dicotyledons (Arabidopsis) was per-formed. Orthologs are generally defined as genes from different ge-nomes that were derived from a single ancestral gene and may havethe same function; in contrast, paralogs are genes that originated froma single genewithin a genome,were created by gene duplication events,

Fig. 7.Hierarchical clustering of expression profiles of ZmSPL genes in 16 tissues. The colorscale on the bottom of the figure represents the RPKM-normalized log2-transformedexpression values, based on transcriptome data. Key: Red, high expression; white,intermediate expression; green, low expression. The tissues from the differentdevelopmental stages are denoted at the top of the heat map. ZmSPL genes that containmiR156/157 target sites are indicated by red color.
10 H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
and often display different functions (Sonnhammer and Koonin, 2002).Therefore, the analysis has allowed us to understand how the diversefunctions of the SPL gene family members evolved; the phylogenetictree indicated that all the SPL genes from the four higher plants were di-vided into six well-supported groups (Fig. 1). The SPL proteins withinthe same group seemed to be more closely related to each other thanto those from the same species, but in different groups. Thus, numerousorthologous (i.e., ZmSPL5/SbSPL2, ZmSPL7/SbSPL4, ZmSPL16/OsSPL11,ZmSPL29/OsSPL2, AtSPL7/OsSPL9) genes were identified (Fig. 1). The ex-istence of orthologous genes between the monocot and dicot SPL genessuggest that some ancestral SPL genes may have existed before thedicot–monocot split.
The number of SPL genes found inmaize (31)wasmuch greater thanthat in Arabidopsis (16), rice (19), and sorghum (18), and similar to thenumber in Populus (28) (Cardon et al., 1999; Xie et al., 2006; Jin et al.,2014; Li and Lu, 2014). Phylogenetic analysis indicated that the maizegained twelve genes after themaize and rice plants split, demonstratingthe rapid expansion of SPL genes in maize (Fig. 2). In this study, ten sis-ter pairs of close paralogs were found to be involved in segmental
duplications among the 31 ZmSPL genes (Table 2; Fig. 3). These genesrepresented about 64% of the ZmSPL genes that evolved from duplicatedchromosomal regions. The results indicate that segmental duplicationplayed a predominant role in the expansion of the maize SPL genes.Gene duplication has been reported for many plant TF gene families,such as AP2, MADS, and DOF, among others (Zahn et al., 2005;Moreno-Risueno et al., 2007; Shigyo et al., 2007). Duplicated SPL genepairs have been identified in Arabidopsis (AtSPL10/11, AtSPL4/5 andAtSPL1/12) and rice (OsSPL2/19, OsSPL3/12, OsSPL4/11, OsSPL5/10 andOsSPL16/18) (Yang et al., 2008). However, the number of duplicatedZmSPL gene pairs identified was obviously greater than that inArabidopsis and rice, indicating that (1) more segment duplicationevents happened in maize, and (2) most SPL genes in Arabidopsis, riceand maize expanded in a species-specific manner.
To further investigate the possible functions of the ZmSPL genes inplant growth and development, expression patterns of the 31 ZmSPLsin 16 different tissues were detected, based on the transcriptomedata (Fig. 7). The heat map of the ZmSPLs expression patternsshowed that some genes, like ZmSPL9 and ZmSPL12, appeared to beconsistently expressed at low levels among all tissues. In contrast,ZmSPL3 and ZmSPL18 exhibited high expression levels in all tissues(Fig. 7). The consistent gene expression levels across all tissues canbe considered constitutive expression. The low-level expression ofmany ZmSPL genes observed indicates that these genes may worksynergistically with other proteins during plant growth and develop-ment. In addition, the three groups of ZmSPL genes discussed above(Groups 1, 2 and 3) all contained a miR156 target site (Fig. 4;Fig. 6). The expression levels of these genes were varied in the differ-ent tissues. In contrast, the genes in Groups 4, 5 and 6 did not containan miR156 target site and were all expressed ubiquitously and con-stitutively, with little or no variation in any of the tissues analyzed(Fig. 7). These results indicate that maize genes from these twogroups may have functions that are distinct from the miR156-targeted SPL genes in Groups 1, 2 and 3.
To date, several important and divergent biological processes regu-lated by SPL genes have been reported; however, only a small numberof these genes have been shown to play a role in response to stress.For example, in Arabidopsis, the expression of an SPL gene respondedto various types of biotic and abiotic stresses, through interactionswith genes involved in the defense response pathway (Wang et al.,2009). In addition, AtSPL14 was found to be involved in programmedcell death and plays a role in sensitivity to fumonisin B1 (Stone et al.,2005). In our study, a preliminary expression profiling of the 31 ZmSPLgenes showed that they were influenced by several environmentalstimuli, including drought, cold, salinity and ABA exposure (Fig. 8);this indicates their role in abiotic stress responses. The differential ex-pression profiles of the ZmSPL genes observed in this study underscorethe daunting task of comprehending the global milieu associated witha stress response. However, an important outcome could be the com-parison of their expressions patterns tomultiple environmental stimuli,for the accurate identification of prospective candidate genes.
In conclusion, a total of 31 full-length SPLs were identified in maizegenome. These proteins harbor sufficient structural diversities, in termsof their exon/intron distributions and motif compositions that may berelated to their diverse functions. The expression patterns of the 31ZmSPLs in different tissues further prove the presence of functional di-versification of this family genes. The different expression patterns ofmiR156-targeted and -nontargeted genes indicate that miR156 playsimportant roles in maize development. In addition, the change in ex-pression of ZmSPL genes to various abiotic stresses indicated thatthese genes may play important roles in responding to abiotic stress.These preliminary results of the SPL family genes in maize provide afoundation for further studies on the physiological and biochemicalfunctions of SPL proteins.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.plgene.2016.03.003.

Fig. 8. Expression profile of the ZmSPL genes in response to various abiotic stresses. (a) Hierarchical clustering of the differential gene expression, in response to dehydration, salinity, coldand abscisic acid (ABA) stress. The heat map was generated based on the fold-change values in the treated samples, when compared with unstressed control samples. The color scale forthe fold-change values is shown at the bottom. (b and c) Venn diagram showing stress-specific distribution of the ZmSPL genes into two categories: (b) up-regulated, and (c) down-regulated. The genes regulated by two or more stresses are shown in the overlapping areas.
11H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
Author contribution
Conceived and designed the experiments: HDM. Performed the ex-periments: HDM, LJY, ZJL. Analyzed the data: HDM, LJY, ZJL, YY, RH,HL, MM. Wrote the paper: HDM, LJY. All authors read and approvedthe final manuscript and have no conflicts of interest with regard tothis research or its funding.
Conflicts of interest
The authors declare that there is no conflict of interest.
Acknowledgments
We thank all our collaboratorswhomade thework on SPL transcrip-tion factors a rewarding experience. This work was supported by theNational Natural Science Foundation of China (31471505).
References
Addo-Quaye, C., Eshoo, T.W., Bartel, D.P., Axtell, M.J., 2008. Endogenous siRNA andmiRNAtargets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 18,758–762.
Bailey, T.L., Elkan, C., 1995. The value of prior knowledge in discovering motifs withMEME. Proc. Int. Conf. Intell. Syst. Mol. Biol. 3, 21–29.
Bartel, D.P., 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116,281–297.
Birkenbihl, R.P., Jach, G., Saedler, H., Huijser, P., 2005. Functional dissection of the plant-specific SBP-domain: overlap of the DNA-binding and nuclear localization domains.J. Mol. Biol. 352, 585–596.
Cardon, G., Höhmann, S., Klein, J., Nettesheim, K., Saedler, H., Huijser, P., 1999. Molecularcharacterisation of the Arabidopsis SBP-box genes. Gene 237, 91–104.
Chen, K., Rajewsky, N., 2007. The evolution of gene regulation by transcription factors andmicroRNAs. Nat. Rev. Genet. 8, 93–103.
Chuck, G., Whipple, C., Jackson, D., Hake, S., 2010. The maize SBP-box transcription factorencoded by tasselsheath4 regulates bract development and the establishment of mer-istem boundaries. Development 137, 1243–1250.
Dai, X., Zhao, P.X., 2011. psRNATarget: a plant small RNA target analysis server. NucleicAcids Res. 39, W155–W159.
Finn, R.D., Mistry, J., Schuster-Böckler, B., Griffiths-Jones, S., Hollich, V., Lassmann, T.,Moxon, S., Marshall, M., Khanna, A., Durbin, R., Eddy, S.R., Sonnhammer, E.L.,Bateman, A., 2006. Pfam: clans, web tools and services. Nucleic Acids Res. 34,D247–D251.
Gandikota, M., Birkenbihl, R.P., Höhmann, S., Cardon, G.H., Saedler, H., Huijser, P., 2007.The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP boxgene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J.49, 683–693.
Gaut, B.S., Morton, B.R., McCaig, B.C., Clegg, M.T., 1996. Substitution rate comparisons be-tween grasses and palms: synonymous rate differences at the nuclear gene Adh par-allel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. U. S. A. 93,10274–10279.
Guo, A.Y., Zhu, Q.H., Chen, X., Luo, J.C., 2007. GSDS: a gene structure display server. YiChuan 29, 1023–1026.
Guo, A.Y., Zhu, Q.H., Gu, X., Ge, S., Yang, J., Luo, J., 2008. Genome-wide identification andevolutionary analysis of the plant specific SBP-box transcription factor family. Gene418, 1–8.
Hanada, K., Zou, C., Lehti-Shiu, M.D., Shinozaki, K., Shiu, S.H., 2008. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environ-mental stimuli. Plant Physiol. 148, 993–1003.
Hultquist, J.F., Dorweiler, J.E., 2008. Feminized tassels of maize mop1 and ts1 mutants ex-hibit altered levels of miR156 and specific SBP-box genes. Planta 229, 99–113.

12 H.-D. Mao et al. / Plant Gene 6 (2016) 1–12
Ishii, T., Numaguchi, K., Miura, K., Yoshida, K., Thanh, P.T., Htun, T.M., Yamasaki, M.,Komeda, N., Matsumoto, T., Terauchi, R., Ishikawa, R., Ashikari, M., 2013. OsLG1 regu-lates a closed panicle trait in domesticated rice. Nat. Genet. 45, 462–465.
Jiao, Y., Wang, Y., Xue, D., Wang, J., Yan, M., Liu, G., Dong, G., Zeng, D., Lu, Z., Zhu, X., Qian,Q., Li, J., 2010. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture inrice. Nat. Genet. 42, 541–544.
Jin, J., Zhang, H., Kong, L., Gao, G., Luo, J., 2014. PlantTFDB 3.0: a portal for the functionaland evolutionary study of plant transcription factors. Nucleic Acids Res. 42,D1182–D1187.
Jung, J.H., Seo, P.J., Kang, S.K., Park, C.M., 2011. miR172 signals are incorporated into themiR156 signaling pathway at the SPL3/4/5 genes in Arabidopsis developmental tran-sitions. Plant Mol. Biol. 76, 35–45.
Juretic, N., Hoen, D.R., Huynh, M.L., Harrison, P.M., Bureau, T.E., 2005. The evolutionaryfate of MULE-mediated duplications of host gene fragments in rice. Genome Res.15, 1292–1297.
Klein, J., Saedler, H., Huijser, P., 1996. A new family of DNA binding proteins includes pu-tative transcriptional regulators of the Antirrhinum majus floral meristem identitygene SQUAMOSA. Mol. Gen. Genet. 250, 7–16.
Kozomara, A., Griffiths-Jones, S., 2011. miRBase: integrating microRNA annotation anddeep-sequencing data. Nucleic Acids Res. 39, D152–D157.
Lauressergues, D., Couzigou, J.M., Clemente, H.S., Martinez, Y., Dunand, C., Bécard, G.,Combier, J.P., 2015. Primary transcripts of microRNAs encode regulatory peptides.Nature 520, 90–93.
Letunic, I., Copley, R.R., Schmidt, S., Ciccarelli, F.D., Doerks, T., Schultz, J., Ponting, C.P., Bork,P., 2004. SMART 4.0: towards genomic data integration. Nucleic Acids Res. 32,D142–D144.
Li, C., Lu, S., 2014. Molecular characterization of the SPL gene family in Populus trichocarpa.BMC Plant Biol. 14, 131.
Livak, K.J., Schmittgen, T.D., 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408.
Lu, Z., Yu, H., Xiong, G., Wang, J., Jiao, Y., Liu, G., Jing, Y., Meng, X., Hu, X., Qian, Q., Fu, X.,Wang, Y., Li, J., 2013. Genome-wide binding analysis of the transcription activatorideal plant architecture1 reveals a complex network regulating rice plant architec-ture. Plant Cell 25, 3743–3759.
Manning, K., Tör, M., Poole, M., Hong, Y., Thompson, A.J., King, G.J., Giovannoni, J.J.,Seymour, G.B., 2006. A naturally occurring epigenetic mutation in a gene encodingan SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38,948–952.
Miura, K., Ikeda, M., Matsubara, A., Song, X.J., Ito, M., Asano, K., Matsuoka, M., Kitano, H.,Ashikari, M., 2010. OsSPL14 promotes panicle branching and higher grain productiv-ity in rice. Nat. Genet. 42, 545–549.
Moreno, M.A., Harper, L.C., Krueger, R.W., Dellaporta, S.L., Freeling,M., 1997. liguleless1 en-codes a nuclear-localized protein required for induction of ligules and auricles duringmaize leaf organogenesis. Genes Dev. 11, 616–628.
Moreno-Risueno, M.A., Martínez, M., Vicente-Carbajosa, J., Carbonero, P., 2007. The familyof DOF transcription factors: from green unicellular algae to vascular plants. Mol. Gen.Genomics. 277, 379–390.
Rhoades, M.W., Reinhart, B.J., Lim, L.P., Burge, C.B., Bartel, B., Bartel, D.P., 2002. Predictionof plant microRNA targets. Cell 110, 513–520.
Schnable, P.S., Ware, D., Fulton, R.S., Stein, J.C., 2009. The B73 maize genome: complexity,diversity, and dynamics. Science 326, 1112–1115.
Schwarz, S., Grande, A.V., Bujdoso, N., Saedler, H., Huijser, P., 2008. ThemicroRNA regulat-ed SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol.Biol. 67, 183–195.
Sekhon, R.S., Lin, H., Childs, K.L., Hansey, C.N., Buell, C.R., de Leon, N., Kaeppler, S.M., 2011.Genome-wide atlas of transcription during maize development. Plant J. 66, 553–563.
Shigyo, M., Tabei, N., Yoneyama, T., Yanagisawa, S., 2007. Evolutionary processes duringthe formation of the plant-specific Dof transcription factor family. Plant Cell Physiol.48, 179–185.
Sonnhammer, E.L., Koonin, E.V., 2002. Orthology, paralogy and proposed classification forparalog subtypes. Trends Genet. 18, 619–620.
Stone, J.M., Liang, X.W., Nekl, E.R., Stiers, J.J., 2005. Arabidopsis AtSPL14, a plants pecificSBP-domain transcription factor, participates in plant development and sensitivityto fumonisin B1. Plant J. 41, 744–754.
Sun, Y., Oberley, L.W., 1996. Redox regulation of transcriptional activators. Free Radic. Biol.Med. 21, 335–348.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S., 2011. MEGA5: molec-ular evolutionary genetics analysis using maximum likelihood, evolutionary distance,and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Unte, U.S., Sorensen, A.M., Pesaresi, P., Gandikota, M., Leister, D., Saedler, H., Huijser, P.,2003. SPL8, an SBP-box gene that affects pollen sac development in Arabidopsis.Plant Cell 15, 1009–1019.
Wang, J.W., Schwab, R., Czech, B., Mica, E., Weigel, D., 2008. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size inArabidopsis thaliana. Plant Cell 20, 1231–1243.
Wang, Y., Hu, Z., Yang, Y., Chen, X., Chen, G., 2009. Function annotation of an SBP-box genein Arabidopsis based on analysis of co-expression networks and promoters. Int. J. Mol.Sci. 10, 116–132.
Wang, S., Wu, K., Yuan, Q., Liu, X., Liu, Z., Lin, X., Zeng, R., Zhu, H., Dong, G., Qian, Q., Zhang,G., Fu, X., 2012. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet.44, 950–954.
Wu, G., Poethig, R.S., 2006. Temporal regulation of shoot development in Arabidopsisthaliana by miR156 and its target SPL3. Development 133, 3539–3547.
Xie, K., Wu, C., Xiong, L., 2006. Genomic organization, differential expression, and interac-tion of SQUAMOSA promoter-binding-like transcription factors and microRNA156 inrice. Plant Physiol. 142, 280–293.
Xing, S., Salinas, M., Höhmann, S., Berndtgen, R., Huijser, P., 2010. miR156-targeted andnontargeted SBP-box transcription factors act in concert to secure male fertility inArabidopsis. Plant Cell 22, 935–3950.
Yamasaki, K., Kigawa, T., Inoue, M., Tateno, M., Yamasaki, T., Yabuki, T., Aoki, M., Seki, E.,Matsuda, T., Nunokawa, E., Ishizuka, Y., Terada, T., Shirouzu, M., Osanai, T., Tanaka,A., Seki, M., Shinozaki, K., Yokoyama, S., 2004. A novel zinc-binding motif revealedby solution structures of DNA-binding domains of Arabidopsis SBP-family transcrip-tion factors. J. Mol. Biol. 337, 49–63.
Yamasaki, H., Hayashi, M., Fukazawa,M., Kobayashi, Y., Shikanai, T., 2009. SQUAMOSA pro-moter binding protein-like7 is a central regulator for copper homeostasis inArabidopsis. Plant Cell 21, 347–361.
Yang, Z.H., 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol.24, 1586–1591.
Yang, Z., Wang, X., Gu, S., Hu, Z., Xu, H., Xu, C., 2008. Comparative study of SBP-box genefamily in Arabidopsis and rice. Gene 407, 1–11.
Zahn, L.M., Kong, H., Leebens-Mack, J.H., Kim, S., Soltis, P.S., Landherr, L.L., Soltis, D.E.,Depamphilis, C.W., Ma, H., 2005. The evolution of the SEPALLATA subfamily ofMADS-box genes: a preangiosperm origin with multiple duplications throughout an-giosperm history. Genetics 169, 2209–2223.
Zhang, Y., Schwarz, S., Saedler, H., Huijser, P., 2007. SPL8, a local regulator in a subset ofgibberellin-mediated developmental processes in Arabidopsis. Plant Mol. Biol. 63,429–439.
Zhang, L., Chia, J.M., Kumari, S., Stein, J.C., Liu, Z., Narechania, A., Maher, C.A., Guill, K.,McMullen, M.D., Ware, D., 2009. A genome-wide characterization of microRNAgenes in maize. PLoS Genet. 5, e1000716.





























