Embed Size (px)
Citation preview

J. Pathol. 189: 496–503 (1999)
GENETIC ALTERATIONS IN ‘NORMAL’ LUMINALAND MYOEPITHELIAL CELLS OF THE BREAST
. 1*, 2, 3, 1, 4, 2, . 4 . ’3
1Department of Histopathology, University College London Medical School, Rockefeller Building, University Street,London WC1E 6JJ, U.K.
2The Ludwig Institute for Cancer Research, Courtauld Building, 91 Riding House Street, London W1P 8BT, U.K.3The LICR/UCL Breast Cancer Laboratory, Department of Surgery, University College London Medical School,
Charles Bell House, 67–73 Riding House Street, London W1P 7LD, U.K.4Section of Cancer Genetics, Institute of Cancer Research, 15 Cotswold Road, Sutton, Surrey SM2 5NG, U.K.
SUMMARY
Chromosomal loci exhibiting loss of heterozygosity (LOH) at high frequency in invasive breast cancer have been investigated in‘normal’ breast tissue from patients with carcinoma and from reduction mammoplasty specimens. Duct-lobular units dissected fromparaffin-embedded tissues and 485 ‘normal’ luminal and myoepithelial cell clones were studied. Overall, LOH was found in normal cellsin 5/10 breast cancer cases and 1/3 reduction mammoplasty specimens. LOH was identified in normal cells adjacent to and distant fromthe tumour. In one case, all luminal and myoepithelial samples exhibited loss of the same allele on chromosome 13q. One case in whichthe patient had a germline truncating mutation in the BRCA1 gene exhibited LOH on 17q in 3/33 normal clones. One of these clonesshowed loss of wild-type allele indicating gene inactivation. This sample also had LOH at markers on chromosomes 11p and 13q. Oneof 93 clones from three reduction mammoplasties showed allele loss at a locus on chromosome 13q. The identification of LOH in breastlobules suggests that they may be clonal. The demonstration of genetic alteration in luminal and myoepithelial cells provides evidencefor the presence of a common stem cell for the two epithelial cell types. LOH has been demonstrated in normal tissues near and awayfrom the carcinoma, suggesting that genetic alterations are likely to be more heterogeneous and widespread than is currently envisaged,and probably occur very early in breast development. Homozygous deletion of BRCA1 per se does not appear to provide clonaladvantage. Copyright � 1999 John Wiley & Sons, Ltd.
KEY WORDS—loss of heterozygosity; breast; luminal; epithelium; myoepithelium; genetic alteration; normal tissue
INTRODUCTION
The multistep model for breast carcinogenesis sug-gests that invasive carcinoma arises via a series ofintermediate ‘hyperplastic’ and neoplastic stages. Theevidence that these are precursors of invasive cancer isderived from animal studies and from a review of humanhistopathological material. When mice are infected withthe murine mammary tumour virus, breast epitheliumundergoes nodular proliferation to form ‘hyperplasticalveolar nodules’ (HANs). When HANs are trans-planted into cleared mammary fat pads, they developinto tumours more frequently than normal breast tis-sue.1,2 Transformation is not inevitable. Evidence fromhuman studies is, however, indirect. Invasive carcinomais often associated with in situ carcinoma and hyperplas-tic lesions, suggesting that they may represent precursorlesions. Studies carried out by Dupont and Page3 showan increased risk of carcinoma in women with hyper-plasia, particularly when this proliferation is atypical.The risk is doubled in the presence of a positive familyhistory. Together, these data support the hypothesis that‘hyperplastic’ lesions, with and without atypia, may beprecursors of invasive carcinoma.
CCC 0022–3417/99/130496–08$17.50Copyright � 1999 John Wiley & Sons, Ltd.
We have previously demonstrated that loss of hetero-zygosity (LOH) identified at various chromosomal lociat high frequency in invasive cancer is already present inin situ carcinoma,4,5 atypical ductal hyperplasia,6 andnon-atypical hyperplasia of the breast.7 It would there-fore clearly be of interest to know whether LOH is alsopresent in normal tissues. Deng et al.8 reported thatLOH identified in invasive carcinoma is indeed presentin morphologically normal breast lobules adjacent tocarcinomas, but not away from the tumour. Since theirstudies were carried out on microdissected tissues fromparaffin-embedded sections, the possibility that the LOHwas a result of tumour cells migrating to the lobularunits via pagetoid spread cannot be entirely excluded.Furthermore, it was not possible for them to study theluminal and myoepithelial cells separately. Their findingof LOH in normal lobules could therefore be accountedfor by one of three hypotheses: first, that the LOHidentified is actually due to the presence of tumour cellswhich have migrated into the normal lobule from thenearby invasive carcinoma; second, that the LOH waspresent in normal cells, which would mean that both‘normal’ luminal and myoepithelial cell types must showan identical abnormality; or third, that the proportion ofmyoepithelial cells in the dissected sample was small andhence did not act as a contaminant to mask LOHpresent in ‘normal’ luminal cells.
Although the first hypothesis appears to be moreplausible, if the conclusions were correct, they suggest
*Correspondence to: Dr S. R. Lakhani, Department of Histo-pathology, University College London Medical School, RockefellerBuilding, University Street, London WC1E 6JJ, U.K.E-mail: [email protected]
Received 19 February 1999Revised 21 May 1999
Accepted 28 July 1999

497GENETIC ALTERATIONS IN NORMAL BREAST TISSUE
that the normal lobule is clonal, which raises questionsabout the existence or otherwise of a common stem cellfor these two breast epithelial cell types. It also raisesquestions about whether such alterations could accountfor some cases of local recurrence after ‘completeexcision’ of the tumour.
Using an in vitro cell cloning technique, we haveattempted to address these issues by looking for LOH insamples free of contamination from tumour cells andhave examined LOH independently in the luminal andmyoepithelial cells of the breast.
MATERIALS AND METHODS
Collection of tissue samples
All tissues were collected from resection specimensreceived within the Department of Histopathology,UCL, London. We collected ‘normal’ tissues fromten cases with carcinoma and from three reductionmammoplasties Apart from two cases, the resectionswere from patients without a family history of breastdisease. One of these two patients had a known germlinetruncating mutation in the BRCA1 gene. The specimensarrived unfixed and on ice within 15 min of resection andwere examined by an experienced breast pathologist(SRL). A piece of tumour with some adjacent normaltissue was sectioned using a clean blade and used forthe in vitro cloning of epithelial cells. The rest ofthe specimen was examined and processed routinely.Criteria for the histopathological diagnosis were thoseadopted for the U.K. National Breast ScreeningProgramme.9
Cloning of ‘normal’ cells from breast tissue samples
Fresh tissue samples were obtained both from thetumour and, when available, from more distant non-involved areas of the surgical specimen. After dicing into�1–2 mm3 fragments using a scalpel, the tissues wereincubated at 37�C for 4–6 h in 20 ml of a solution of 0·25per cent (w/v) collagenase (Type 1, Sigma, Poole, DorsetU.K.) in Leibovitz L-15 medium plus 10 per cent fetalcalf serum. Samples, in 30 ml Universal tubes, wererotated on a Spiramix until the tissue fragments weredisaggregated. Remaining large fragments were removedby filtration though a 100 �m nylon mesh. In cases inwhich sufficient tumour tissue was available, the reactivestromal cells present in the filtrate were labelled usingthe F-19 monoclonal antibody against FAP (fibroblastactivation protein)10 and removed immunomagneticallyusing anti-mouse Dynabeads. Remaining epithelialtissue was washed twice in 0·02 per cent EDTA andincubated for 10 min at 37�C in 5 ml of a solution of1 mg/ml trypsin (porcine pancreatic Type III, Sigma) in0·02 per cent EDTA with vigorous shaking at intervalsto release disaggregated cells. Trypsinization wasstopped by diluting the sample in 20 ml of L-15/FCS andfiltering it though a 30 �m nylon mesh to give a pre-dominantly single cell suspension. These cells wereplated, at different dilutions, in a series of 75 cm2 culture
Copyright � 1999 John Wiley & Sons, Ltd.
flasks which had been pre-seeded with 2�106 lethallyirradiated (40 Gy) mouse 3T6 fibroblasts. The culturemedium was Ham’s F-12 (Imperial Laboratories,Andover, Hampshire, U.K.) plus 10 per cent FCS,1 �g/ml hydrocortisone, 5 �g/ml insulin, 20 ng/ml chol-era toxin, and 10 ng/ml recombinant human EGF (allculture additives from Sigma). These conditions havebeen previously shown to result in the clonal growth ofboth luminal epithelial and myoepithelial cells fromcosmetic surgery-derived normal breast tissues.11 Theydo not cause bona fide malignant tumour cells to growin this manner, as shown by failure to derive any cloneswhen secondary breast cancers such as lymph nodemetastases are cultured in this manner.12
After 10–14 days, clones of epithelial cells consistingof 1000–5000 cells were identified and marked. Theirphase-contrast morphology corresponded to either lumi-nal or myoepithelial clones as seen when normal tissue iscloned and analysed for cell type-specific characteris-tics.11 In cases in which no immunomagnetic separationof reactive stromal cells was attempted, some fibroblastcolonies were also seen.
After rinsing the cultures in 0·02 per cent (w/v)EDTA, luminal, myoepithelial, and fibroblast cloneswere individually harvested, using trypsinization, byconventional ring-cloning techniques. Each clonesample was then removed using a separate glass pipette,diluted in L-15/FCS to inactivate trypsin, and spundown in a 0·5 ml Eppendorf tube. After removing thesupernatant and resuspending the sample in phosphatebuffered saline (PBS), each was spun down again, thePBS aspirated, and the pellet flash-frozen in liquid N2and stored at �80�C to await analysis.
LOH analysis
The tumour and normal duct-lobular units were dis-sected using the fine point of a glass pipette. The point ofthe pipette was ‘pulled’ by heating and stretching. Adrop of 50 per cent alcohol was placed onto the lesion.After separating the cells of interest, the sample wassucked back into the pipette and transferred into anEppendorf tube. DNA extraction procedures were asdescribed previously.7 LOH was investigated by ampli-fication of polymorphic microsatellite markers usingfluorescence tagged primers and the polymerase chainreaction (PCR). A touchdown protocol was used for theexperiment.7 Seven dinucleotide repeats were used forthis study. These were D3s1597 (3p24.2), D3s1578(3p21.1), D11s902 (11p15), D13s267 (13q13), D16s413(16q24.3), D17s796 (17p13.2), and D17s250 (17q12).These lie within genomic regions which exhibit LOH athigh frequency in invasive carcinoma. D3s1597 lieswithin the region identified by Deng et al.8 as showinghigh frequency of LOH in normal lobules. D17s796 liesin the vicinity of the TP53 gene, D17s250 in the regionof the BRCA1 gene, and D13s267 in the region of theBRCA2 gene. All the markers apart from D17s25013
were identified from the second-generation linkage mapconstructed by the Genethon group.14 One primer ofeach pair was fluorescently labelled using either FAMdye, HEX dye or TET dye. The primers were also
J. Pathol. 189: 496–503 (1999)
498 S. R. LAKHANI ET AL.
purified using standard HPLC. The fluorescent gel datacollected during the run were automatically analysed bythe GeneScan Analysis Software (version 2.02) (AppliedBiosystems, Foster City, CA, U.S.A.) at the end of therun. LOH was assessed by comparing tumour:normalallele intensities.7 We assigned a value of 0·5 or less asindicative of LOH.
Comparative genomic hybridization analysis (CGH)
CGH analysis was carried out on case 2. The invasivetumour and two clones (one luminal and one myoepi-thelial) exhibiting LOH of the same allele were analysed.Following DNA extraction, the sample was subjected toPCR using degenerate primers, DOP-PCR. Additionalrounds of amplification were required in some cases toobtain visible amounts of DNA as assessed on anagarose gel. Direct labelling with fluorescein-12-dUTPor rhodamine-12-dUTP (FluoroGreen, FluoroRed,Amersham International plc, Amersham, U.K.) wascarried out by incorporation during further rounds ofDOP-PCR. For all CGH preparations, 250–500 ng ofdifferentially labelled test and control DNA plus15–25 �g of CotI DNA (BRL Gibco, Paisley, Scotland)was co-hybridized to normal denatured metaphases for48 h at 37oC before washing and mounting in antifadewith 0·1 �g/ml DAPI as a counterstain. Images werecaptured using a cooled CCD camera (Photometrics,Munich, Germany) with SmartCapture software(Digital Scientific, Cambridge, U.K.). CGH analysis wascarried out using software package and independentlychecked. At least five representative images were fullyanalysed and the results from these were studied separ-ately and also combined to produce an average fluor-escence ratio for each chromosome. A copy numberchange was recorded when the average fluorescenceratio lay outside the normal range and the normal rangewas determined in CGH experiments using only controlDNA.
Copyright � 1999 John Wiley & Sons, Ltd.
Sequence analysis in one patient with a BRCA1mutation
One of the patients analysed had a known germlinemutation in the BRCA1 gene (5382insC). Exon 20 of thegene was amplified using PCR with the following primerpair: forward—cac tgt gcc tgg cct gat ac and reverse—atg tta aat tca aag tct cta. Amplification was carried outusing a touchdown PCR protocol and 50 ng of DNA ina 100 �l reaction. The product was cleaned up usingsodium acetate/ethanol and precipitated. It was resus-pended in 20 �l of water. The product (2–5 �l) wassequenced in both directions (same primers as for PCR)using an ABI prism Taq FS thermocycling sequencingkit. Excess dye was removed by sodium acetate/ethanolprecipitation. The products were run on an ABI 377automated sequencer.
RESULTS
A total of 485 ‘normal’ clones (392 from malignantbreasts and 93 from reduction mammoplasties) wereanalysed from the 13 cases studied. Together withtumour and normal tissue microdissected from paraffin-embedded sections, the analysis comprised more than500 samples studied at seven microsatellite markers.
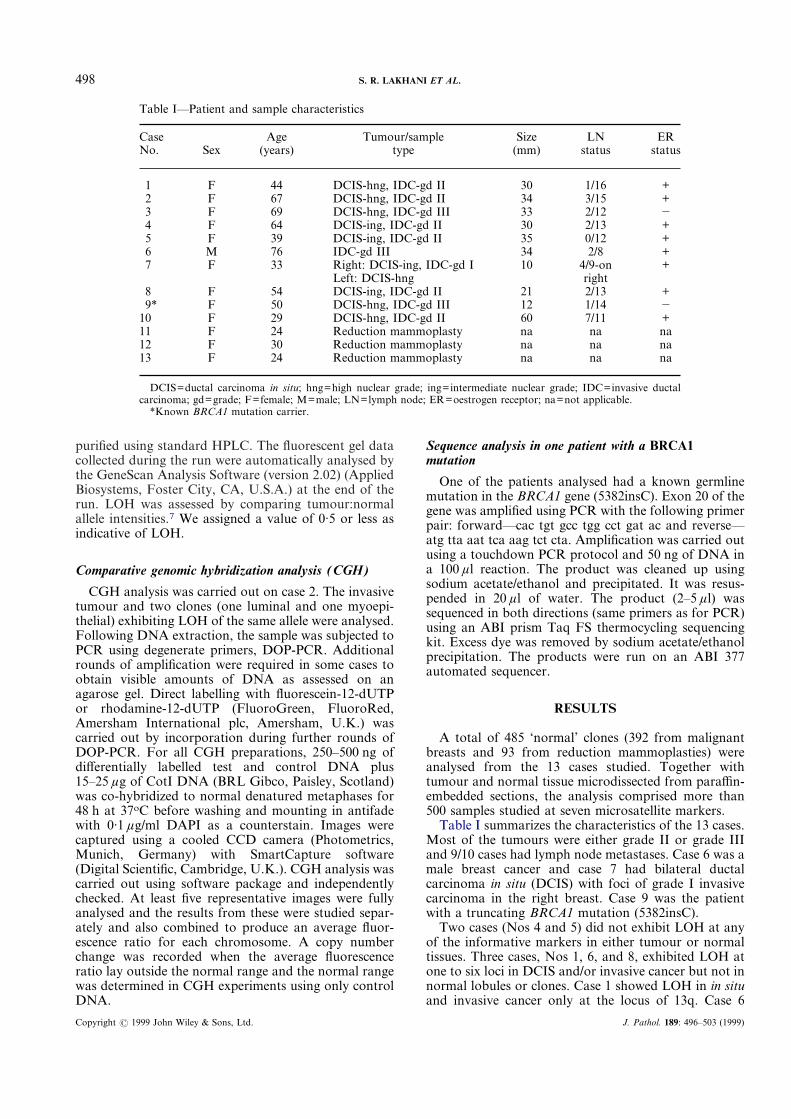
Table I summarizes the characteristics of the 13 cases.Most of the tumours were either grade II or grade IIIand 9/10 cases had lymph node metastases. Case 6 was amale breast cancer and case 7 had bilateral ductalcarcinoma in situ (DCIS) with foci of grade I invasivecarcinoma in the right breast. Case 9 was the patientwith a truncating BRCA1 mutation (5382insC).
Two cases (Nos 4 and 5) did not exhibit LOH at anyof the informative markers in either tumour or normaltissues. Three cases, Nos 1, 6, and 8, exhibited LOH atone to six loci in DCIS and/or invasive cancer but not innormal lobules or clones. Case 1 showed LOH in in situand invasive cancer only at the locus of 13q. Case 6
Table I—Patient and sample characteristics
CaseNo. Sex
Age(years)
Tumour/sampletype
Size(mm)
LNstatus
ERstatus
1 F 44 DCIS-hng, IDC-gd II 30 1/16 +2 F 67 DCIS-hng, IDC-gd II 34 3/15 +3 F 69 DCIS-hng, IDC-gd III 33 2/12 �4 F 64 DCIS-ing, IDC-gd II 30 2/13 +5 F 39 DCIS-ing, IDC-gd II 35 0/12 +6 M 76 IDC-gd III 34 2/8 +7 F 33 Right: DCIS-ing, IDC-gd I 10 4/9-on +
Left: DCIS-hng right8 F 54 DCIS-ing, IDC-gd II 21 2/13 +9* F 50 DCIS-hng, IDC-gd III 12 1/14 �
10 F 29 DCIS-hng, IDC-gd II 60 7/11 +11 F 24 Reduction mammoplasty na na na12 F 30 Reduction mammoplasty na na na13 F 24 Reduction mammoplasty na na na
DCIS=ductal carcinoma in situ; hng=high nuclear grade; ing=intermediate nuclear grade; IDC=invasive ductalcarcinoma; gd=grade; F=female; M=male; LN=lymph node; ER=oestrogen receptor; na=not applicable.
*Known BRCA1 mutation carrier.
J. Pathol. 189: 496–503 (1999)

499GENETIC ALTERATIONS IN NORMAL BREAST TISSUE
showed LOH in both DCIS and invasive cancer atthree loci on 3p (two loci) and 11p. Case 8 showed LOHat six loci. The two loci on 3p showed LOH in bothDCIS and invasive carcinoma, while the four loci on13q, 17p, 17q, and 16q exhibited LOH only in invasivecancer.
Five cases (Nos 2, 3, 7, 9, and 10) showed LOH innormal tissue. In case 3, LOH was only seen in a normallobule dissected from a paraffin section. The tissueadjacent to the tumour did not generate any normalepithelial clones, but reactive fibroblast colonies wereharvested and analysed. None of 12 fibroblast coloniesexhibited LOH at any locus. Clones derived from nor-mal breast away from the tumour were also negativewith all the informative markers. In the other four cases,LOH was demonstrated in ‘normal’ clones. Three pat-terns of LOH were identified in these cases: (a) LOH intumour and normal cells from tissue adjacent to thetumour involving the same allele; (b) LOH in tumourand normal cells from sites distant from the tumour; and(c) LOH in normal tissue but not in the adjacenttumour.
Of the three reduction mammoplasty cases, one caseshowed LOH in a single myoepithelial clone (1/93clones).
LOH in tumour and normal cells from tissue adjacent tothe carcinoma
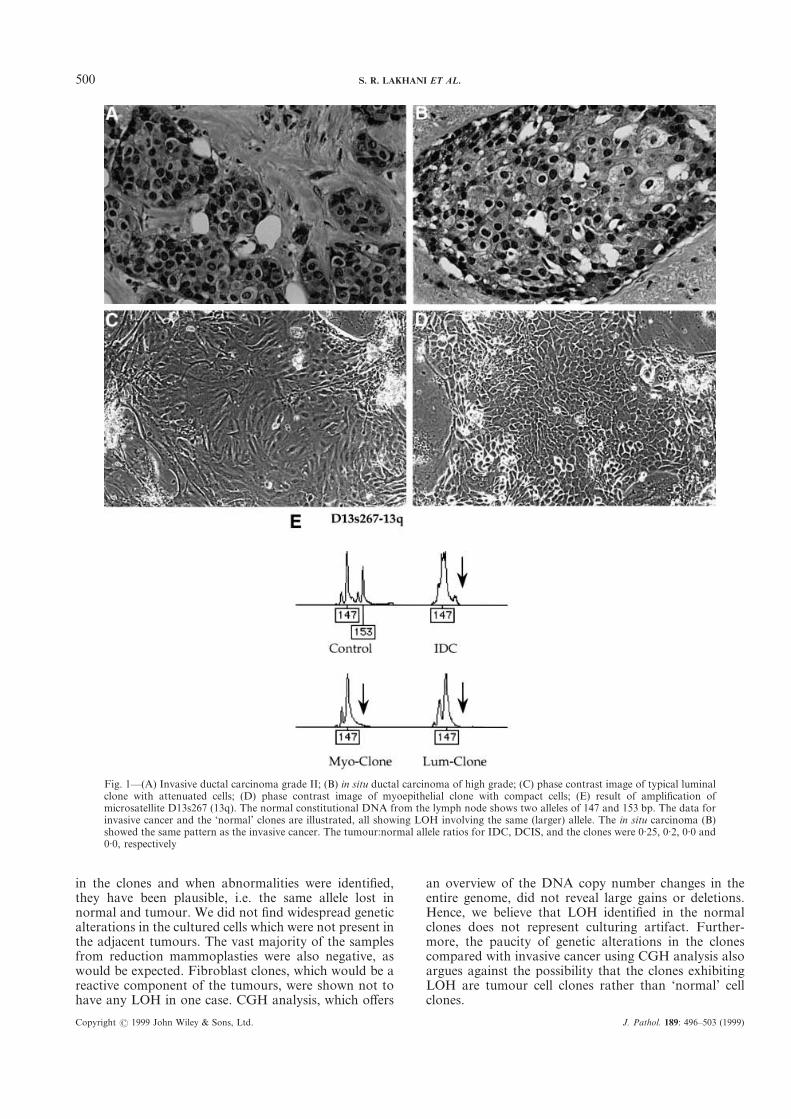
Figure 1 (case 2) illustrates the morphology of theinvasive carcinoma, ductal carcinoma in situ, and theluminal and myoepithelial clones. The adjacent graphillustrates the LOH identified at microsatellite D13s267,a marker on chromosome 13q, which is in the vicinity ofthe BRCA2 gene. The 153 bp allele is lost from all thetumour and normal samples. Table II summarizes thedata for all seven microsatellites analysed from the case.Five markers were informative, of which only the 13qlocus exhibited LOH.
LOH in tumour and normal cells cloned from sitesdistant from the tumour
Table III summarizes the data from case 9 (BRCA1mutation). All seven markers were informative, of whichthree [on chromosomal arms 3p(1597), 13q and 17q]showed LOH in invasive cancer as well as DCIS. LOHwas identified in ‘normal’ clones derived from tissuedistant from the tumour, but not in clones derived fromthe vicinity of the tumour. One clone showed LOH atthree markers on chromosomes 11p, 13q, and 17q. Oneclone showed LOH only at 13q, and two clones only at17q. Hence overall, 4/33 clones exhibited LOH at one ormore locus. Of the three clones exhibiting LOH at 17q(in the vicinity of the BRCA1 gene), two exhibited LOHinvolving the 150 bp allele, while one clone exhibitedLOH involving the 154 bp allele. The invasive cancershowed LOH of the 154 bp allele. Most patients withmutations in the BRCA1 gene exhibit LOH in thetumour and this usually involves loss of the wild-typeallele.15 This would indicate that the 154 bp allele wasthe wild type and the 150 bp allele was on the arm
Copyright � 1999 John Wiley & Sons, Ltd.
containing the truncating BRCA1 mutation. DNA forsequencing was available from normal lymph node andfrom one clone exhibiting LOH of the 150 bp allele andthis confirmed deletion of the mutant allele in this cellclone. Sequencing of this clone revealed a normal, wildtype allele only. Hence, the tumour and one clone hadlost the 154 bp wild-type allele. It was this clone thatalso showed allele loss at markers on chromosomes 11pand 13q.
Case 10 exhibited LOH at 13q in invasive and in situcarcinoma and at 11p only in DCIS. In this case, 1/91myoepithelial clones derived from tissue away from thetumour showed LOH at 11p, but not at any othermarker. None of 46 ‘normal’ clones from near thetumour showed any genetic alterations.
LOH in normal tissue but not in the adjacent tumour
Case 7 had bilateral DCIS with foci of invasive cancerin the right breast. One of 12 myoepithelial clones fromthe left breast exhibited LOH at a locus on 13q. NoLOH was identified in any other clones from eitherbreast, or from the foci of DCIS in the two breasts(Table IV).
LOH in reduction mammoplasty specimens
Fourteen luminal and 79 myoepithelial clones werestudied from three benign reduction mammoplastyspecimens. One of 28 myoepithelial clones from case 1(overall 1/93 ‘normal’ clones) exhibited LOH at D13s267(13q). All other clones from case 1 and all clones fromcases 2 and 3 were negative at the informative markers.
CGH analysis
One invasive carcinoma and two clones (one myoepi-thelial and one luminal) from the same case were sub-jected to CGH analysis to exclude the possibility thatthese ‘normal’ clones represented a rare event of estab-lishing tumour colonies by the in vitro method describedabove. The tumour sample showed widespread DNAcopy number changes as has been described previouslyin invasive breast cancers.16 In contrast, the ‘normal’clones showed either no changes or small deletions andgains, particularly near the centromere or telomere,regions known to produce artifacts in CGH analysis.
DISCUSSION
Until the recent development of laser capture micro-dissection, it has not been possible to study LOHindependently in both the luminal and the myoepithelialcells, and hence we used an in vitro cloning technique toisolate and clone the two cell types.
The conditions used for the in vitro cell culture do notallow tumour cells to proliferate, but do allow theexpansion of ‘normal’ epithelial cells. A potentialcriticism levelled at such in vitro techniques is thatculturing itself may lead to genetic alterations. In thepresent study, relatively few abnormalities were detected
J. Pathol. 189: 496–503 (1999)
500 S. R. LAKHANI ET AL.
in the clones and when abnormalities were identified,they have been plausible, i.e. the same allele lost innormal and tumour. We did not find widespread geneticalterations in the cultured cells which were not present inthe adjacent tumours. The vast majority of the samplesfrom reduction mammoplasties were also negative, aswould be expected. Fibroblast clones, which would be areactive component of the tumours, were shown not tohave any LOH in one case. CGH analysis, which offers
Copyright � 1999 John Wiley & Sons, Ltd.
an overview of the DNA copy number changes in theentire genome, did not reveal large gains or deletions.Hence, we believe that LOH identified in the normalclones does not represent culturing artifact. Further-more, the paucity of genetic alterations in the clonescompared with invasive cancer using CGH analysis alsoargues against the possibility that the clones exhibitingLOH are tumour cell clones rather than ‘normal’ cellclones.
Fig. 1—(A) Invasive ductal carcinoma grade II; (B) in situ ductal carcinoma of high grade; (C) phase contrast image of typical luminalclone with attenuated cells; (D) phase contrast image of myoepithelial clone with compact cells; (E) result of amplification ofmicrosatellite D13s267 (13q). The normal constitutional DNA from the lymph node shows two alleles of 147 and 153 bp. The data forinvasive cancer and the ‘normal’ clones are illustrated, all showing LOH involving the same (larger) allele. The in situ carcinoma (B)showed the same pattern as the invasive cancer. The tumour:normal allele ratios for IDC, DCIS, and the clones were 0·25, 0·2, 0·0 and0·0, respectively
J. Pathol. 189: 496–503 (1999)
501GENETIC ALTERATIONS IN NORMAL BREAST TISSUE
We confirmed the result of Deng et al.,8 that LOHidentified in invasive carcinoma is present in normaltissues adjacent to tumour. By the use of an in vitrotechnique, we were able to demonstrate unequivocallythat the LOH identified was not a spurious result due topagetoid spread of tumour cells into the adjacent normallobules. Further confirmation stems from a recent find-ing of LOH in lobules dissected from paraffin-embeddedtissues from reduction mammoplasties.17 We extendedthis observation to show that LOH is detected indepen-dently in both the luminal and the myoepithelial celltypes. Taking the data of Deng et al.,8 and the observa-tions from our own study together, the results imply thatthe normal breast lobule is likely to be clonal in nature.The observation of LOH in both luminal and myo-epithelial cells provides evidence for the existence of a
Copyright � 1999 John Wiley & Sons, Ltd.
common stem cell that gives rise to the two breast celltypes by differentiation. When normal luminal andmyoepithelial cells are cloned,11 each retains all theappropriate cell type-specific markers and mixed clonesare not seen. In the present study, all clones were clearlyof luminal and myoepithelial origin as demonstrated bytheir distinct morphology (Fig. 1). Further evidence ofthe ‘non-malignant’ origin of tumour-derived clones isprovided by the observation of Smith et al.18,19 that suchclones are predominantly diploid. Their interpretationthat such cells are tumour ‘stem cells’ is not borne out bythe observation that no such clones occur when second-ary tumours are cultured,12 that they correspond mor-phologically to luminal and myoepithelial clones derivedfrom cosmetic surgery samples,11 and that CGH doesnot show widespread genetic alterations. These results
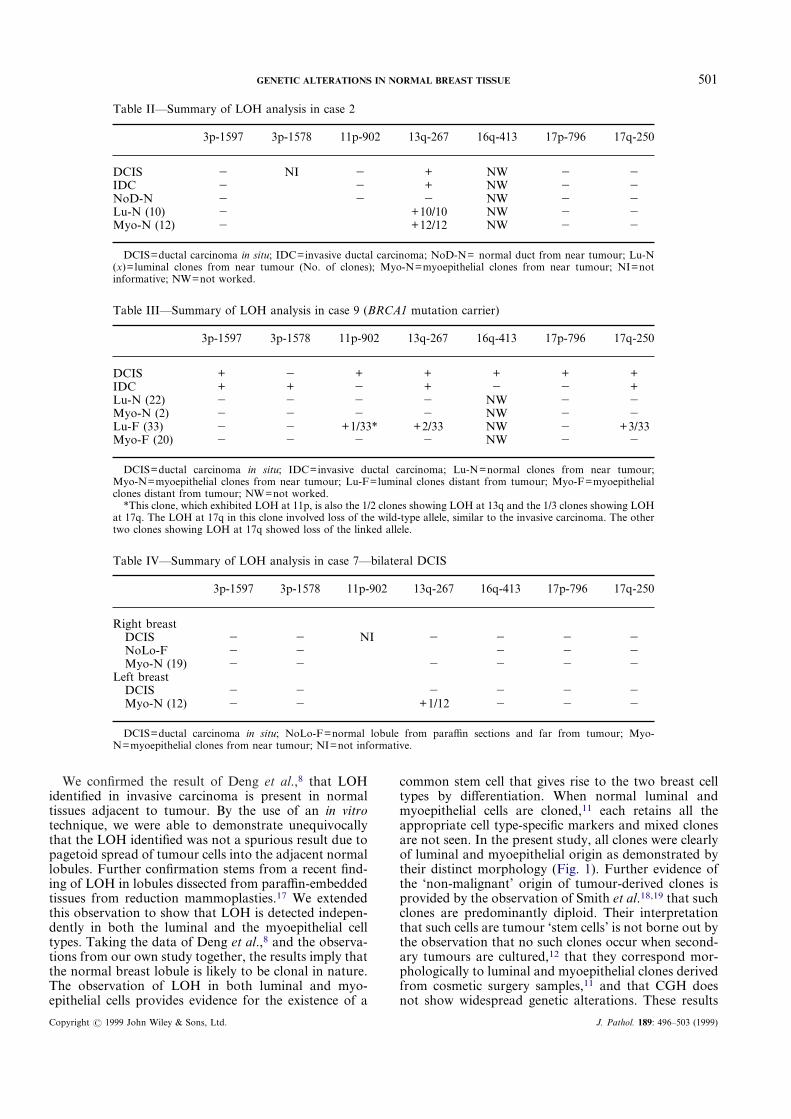
Table II—Summary of LOH analysis in case 2
3p-1597 3p-1578 11p-902 13q-267 16q-413 17p-796 17q-250
DCIS � NI � + NW � �IDC � � + NW � �NoD-N � � � NW � �Lu-N (10) � +10/10 NW � �Myo-N (12) � +12/12 NW � �
DCIS=ductal carcinoma in situ; IDC=invasive ductal carcinoma; NoD-N= normal duct from near tumour; Lu-N(x)=luminal clones from near tumour (No. of clones); Myo-N=myoepithelial clones from near tumour; NI=notinformative; NW=not worked.
Table III—Summary of LOH analysis in case 9 (BRCA1 mutation carrier)
3p-1597 3p-1578 11p-902 13q-267 16q-413 17p-796 17q-250
DCIS + � + + + + +IDC + + � + � � +Lu-N (22) � � � � NW � �Myo-N (2) � � � � NW � �Lu-F (33) � � +1/33* +2/33 NW � +3/33Myo-F (20) � � � � NW � �
DCIS=ductal carcinoma in situ; IDC=invasive ductal carcinoma; Lu-N=normal clones from near tumour;Myo-N=myoepithelial clones from near tumour; Lu-F=luminal clones distant from tumour; Myo-F=myoepithelialclones distant from tumour; NW=not worked.
*This clone, which exhibited LOH at 11p, is also the 1/2 clones showing LOH at 13q and the 1/3 clones showing LOHat 17q. The LOH at 17q in this clone involved loss of the wild-type allele, similar to the invasive carcinoma. The othertwo clones showing LOH at 17q showed loss of the linked allele.
Table IV—Summary of LOH analysis in case 7—bilateral DCIS
3p-1597 3p-1578 11p-902 13q-267 16q-413 17p-796 17q-250
Right breastDCIS � � NI � � � �NoLo-F � � � � �Myo-N (19) � � � � � �
Left breastDCIS � � � � � �Myo-N (12) � � +1/12 � � �
DCIS=ductal carcinoma in situ; NoLo-F=normal lobule from paraffin sections and far from tumour; Myo-N=myoepithelial clones from near tumour; NI=not informative.
J. Pathol. 189: 496–503 (1999)

502 S. R. LAKHANI ET AL.
indicate that multipotential stem cells must constitute avery small proportion of the total.
In contrast to Deng et al.,8 our data show that LOHmay be present in ‘normal’ tissues well away from thetumour and that genetic alterations in the normal breastmay be more heterogeneous and widespread than iscurrently envisaged. We examined only a limitedamount of non-tumoural breast tissue and we did notattempt to characterize the topographical relationship tothe tumour; hence the extent of breast tissues involvedby the LOH is unknown. It is likely that genetic altera-tions in ‘normal’ tissues occur very early in breastdevelopment, but the delineation of when the first eventsoccur would require extensive LOH analysis of differenttopographical regions of the breast. This type of analysismay have implications for patient management. Ifpatients with breast cancer have widespread geneticinstability in ‘normal’ tissues within the breast, this maybe one reason why some patients with apparent ‘com-plete excision’ of their tumour present with recurrentdisease. We also know that the extent of malignantdisease is often underestimated using conventionalmammography and we have demonstrated that foci oftumour away from the index quadrant can be identifiedusing MRI imaging.20 Although, at present, there is noevidence that these further foci are important in predict-ing local recurrence of carcinoma, the delineation ofgenetic alteration in tissues away from the index quad-rant warrants further investigation.
One patient had a known truncating mutation in thepredisposition gene BRCA1. We were able to demon-strate that LOH identified in one normal cloneinvolved loss of the wild-type allele, indicating that thegene was inactivated. This clone also exhibited furthergenetic alterations involving chromosomal arms 11pand 13q. Although the sample size here is clearly small,there does not appear to be a preponderance of normalcells showing loss of the wild-type allele. This suggeststhat inactivation of the BRCA1 gene does not confer aclonal advantage on the cells. The combination of adefect in DNA repair and other genetic alterations (inthis case, LOH at 13q, 11p) is probably required toconfer genetic instability and clonal advantage fortumour growth. This hypothesis is consistent with datafrom BRCA2 knockout mice21 which suggest thathomozygous deletion of BRCA2 per se is not sufficientfor tumour formation and that loss of the p53 check-point is essential for tumour progression. Two furtherclones from this patient showed LOH involving thelinked (mutated) allele, hence leaving the wild-typeallele only. The effect of the mutation is thereforeneutralized. The significance of this finding remainsunclear at present and a larger study will be needed toaddress this issue.
The third pattern of LOH in normal tissues but not inthe adjacent tumour is also intriguing. In this situation,this particular genetic alteration is clearly not patho-genetically significant for the development of thetumour. This is further borne out by the demonstrationof LOH in ‘normal’ clones from benign breasts removedpurely for cosmetic reasons. At what frequency thesegenetic alterations occur in the population and what
Copyright � 1999 John Wiley & Sons, Ltd.
combination of events triggers proliferation remainimportant unanswered questions.
The breast is not the only organ in which geneticalterations have been detected in ‘normal’ tissue.Cytogenetic abnormalities of chromosome 7 have beendetected in normal bronchial epithelium from patientswith lung carcinoma22 and t(14;18) translocation, usu-ally seen in follicular lymphomas, has been reported inreactive lymphoid follicles.23 TEL (ETV6)–AML1(CBFA2) gene fusion due to chromosomal rearrange-ment occurs in approximately 25 per cent of commonacute lymphoblastic leukaemia. Ford et al.24 reportedthat twins with acute lymphoblastic leukaemia shared anidentical clonotypic TEL–AML1 genomic sequence.This suggests that the tumours in both babies arose froma single cell in utero. Since few studies of this type havebeen carried out to date, and none in the breast using invitro cloning techniques, such changes in ‘normal’ tissueare likely to be underestimated.
Finally, although the finding of LOH in normal breastepithelial and myoepithelial cells does not prove atransition from normal to malignancy, it does addweight to the multistep hypothesis for breast carcinogen-esis. Since the LOH probably occurs in stem cells, whichthen differentiate into myoepithelial and luminal cells, itraises questions as to why myoepithelial tumours of thebreast are so rare. Further studies of the myoepithelialcell, which appears to be resistant to transformation,might also yield important clues about the mechanismsof breast carcinogenesis, for example in relation topatterns of tumour suppressor gene activity.25
ACKNOWLEDGEMENTS
We are grateful to Professor A. Munro Neville for hisinterest, enthusiasm and encouragement for this projectand to Dr Betsy Richards, LICR—New York Branch,for the gift of F19 antibody. We would also like toacknowledge the support from Professor I. Taylor,Professor M. Baum, and Dr T. Davidson fromthe Department of Surgery, UCL and Professor M.Waterfield, Director, LICR—UCL Branch.
REFERENCES
1. Cardiff RD, Muller WJ. Transgenic mouse models of mammary tumori-genesis. Cancer Surv 1993; 16: 97–113.
2. Morris DW, Cardiff RD. The multistep model of mouse mammary tumourdevelopment. Adv Virol Oncol 1987; 7: 123–140.
3. Dupont WD, Page DL. Risk factors for breast cancer in women withproliferative breast disease. N Engl J Med 1985; 312: 146–151.
4. Stratton MR, Collins N, Lakhani SR, Sloane JP. Loss of heterozygosity inductal carcinoma in situ of the breast. J Pathol 1995; 175: 195–201.
5. Lakhani S, Collins N, Sloane J, Stratton M. Loss of heterozygosity inlobular carcinoma in situ of the breast. J Clin Pathol: Mol Pathol 1995; 48:M74–M78.
6. Lakhani SR, Collins N, Stratton MR, Sloane JP. Atypical ductal hyper-plasia of the breast: clonal proliferation with loss of heterozygosity onchromosomes 16q and 17p. J Clin Pathol 1995; 48: 611–615.
7. Lakhani S, Slack D, Hamoudi R, Collins N, Stratton M, Sloane J.Detection of alleleic imbalance indicates that a proportion of mammaryhyperplasia of usual type are clonal, neoplastic proliferations. Lab Invest1996; 74: 129–135.
8. Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS. Loss of heterozygosity innormal tissue adjacent to breast carcinomas. Science 1996; 274: 2057–2059.
J. Pathol. 189: 496–503 (1999)

503GENETIC ALTERATIONS IN NORMAL BREAST TISSUE
9. National Coordinating Group for Breast Screening Pathology. PathologyReporting in Breast Cancer Screening (2nd edn). NHSBSP PublicationsNo. 3. Sheffield, UK 1995.
10. Welt S, Divgi CR, Scott AM, et al. Antibody targeting in metastatic coloncancer: a phase I study of monoclonal antibody F19 against a cell-surfaceprotein of reactive tumor stromal fibroblasts. J Clin Oncol 1994; 12:1193–1203.
11. O’Hare M, Ormerod MG, Monaghan P, Lane EB, Gusterson BA. Charac-terization in vitro of luminal and myoepithelial cells isolated from thehuman mammary gland by cell sorting. Differentiation 1991; 46: 209–221.
12. O’Hare M. Breast cancer. In Human Cancer in Primary Culture. A Hand-book, Masters J (ed.). Kluwer: Dordrecht, 1990; 271–286.
13. Weber J, Kwitek AE, May PE, Wallace MR, Collins FS, Ledbetter DH.Dinucleotide repeat polymorphisms at the D17S250 and D17S261 loci.Nuceicl Acid Res 1990; 18: 4640.
14. Weissenbach J, Gyapay G, Dib C, et al. A second generation linkage map ofthe human genome. Nature 1992; 359: 794–797.
15. Merajver SD, Frank TS, Xu J, et al. Germline BRCA1 mutations and lossof the wild-type allele in tumors from families with early onset breast andovarian cancer. Clin Cancer Res 1995; 1: 539–544.
16. Tirkkonen M, Tanner M, Karhu R, Kallioniemi A, Isola J, Kallioniemi OP.Molecular cytogenetics of primary breast cancer by CGH. Genes Chromo-somes Cancer 1998; 21: 177–184.
17. Larson PS, de las Morenas A, Cupples LA, Huang K, Rosenberg CL.Genetically abnormal clones in histologically normal breast tissue. Am JPathol 1998; 152: 1591–1598.
Copyright � 1999 John Wiley & Sons, Ltd.
18. Smith HS, Lan S, Ceriani R, Hackett AJ, Stampfer MR. Clonal prolifer-ation of cultured nonmalignant and malignant human breast epithelia.Cancer Res 1981; 41: 4637–4643.
19. Wolman SR, Smith HS, Stampfer M, Hackett AJ. Growth of diploid cellsfrom breast cancers. Cancer Genet Cytogenet 1985; 16: 49–64.
20. Douek M, Vaidya JS, Lakhani SR, Hall-Craggs MA, Baum M, Taylor I.Can magnetic-resonance imaging help elucidate natural history of breastcancer multicentricity? Lancet 1998; 351: 801–802.
21. Connor F, Bertwistle D, Mee PJ, et al. Tumorigenesis and a DNA repairdefect in mice with a truncating BRCA2 mutation. Nature Genet 1997; 17:423–430.
22. Sozzi G, Miozzo M, Tagliabue E, et al. Cytogenetic abnormalities andoverexpression of receptors for growth factors in normal bronchial epi-thelium and tumor samples of lung cancer patients. Cancer Res 1991; 51:400–404.
23. Limpens J, de Jong D, van Krieken JH, et al. Bcl-2/JH rearrangements inbenign lymphoid tissues with follicular hyperplasia. Oncogene 1991; 6:2271–2276.
24. Ford AM, Bennett CA, Price CM, Bruin MCA, Van Wering ER, GreavesM. Fetal origins of the TEL–AML1 fusion gene in identical twins withleukemia. Proc Natl Acad Sci U S A 1998; 95: 4584–4588.
25. Sager R. Expression genetics in cancer: shifting the focus from DNA toRNA. Proc Natl Acad Sci U S A 1997; 94: 952–955.
J. Pathol. 189: 496–503 (1999)







![Transcriptomic response of goat mammary epithelial cells ...€¦ · previously [Ogorevc et al. 2009b]. Luminal, myoepithelial and fibroblast cells were characterized using antibodies](https://img.pdfslide.us/doc/110x75/605fe504ab02910182502932/transcriptomic-response-of-goat-mammary-epithelial-cells-previously-ogorevc.jpg)





















