Embed Size (px)
Citation preview

TECHNOLOGY REPORT
Generation of a Conditional Disruption of the Tsc2 GeneOmar Hernandez, Sharon Way, James McKenna III, and Michael J. Gambello*Department of Pediatrics, Division of Medical Genetics, University of Texas Health Science Center, Houston, Texas
Received 13 September 2006; Revised 11 December 2006; Accepted 17 December 2006
Summary: Tuberous sclerosis complex (TSC) is an auto-somal dominant disorder caused by mutations in theTSC1 or TSC2 gene. Patients afflicted with TSC developtumors in various organ systems, but cerebral pathologyis particularly severe. Conventional gene disruptionof the Tsc1 or Tsc2 gene in mice cause limited centralnervous system pathology. Homozygous deletion ofeither gene causes midgestation lethality. To circumventthe homozygous lethality of the conventional Tsc2knockout we have generated a conditional allele of theTsc2 gene by homologous recombination in mouse EScells. The homozygous Tsc2flox/flox mice are identical towildtype in many organs typically affected by TSC, espe-cially the brain. Using this Tsc2flox allele we have gener-ated a null allele using Cre recombination. This allele willbe useful in investigating TSC pathology with appropri-ate cell and organ specific Cre-transgenic mice. genesis45:101–106, 2007. VVC 2007 Wiley-Liss, Inc.
Key words: Tsc2; tuberin; Cre-loxP; tuberous sclerosis;conditional allele
Tuberous Sclerosis Complex (TSC) is an autosomal domi-nant hamartomatous disorder with an incidence ofapproximately 1/6,000 (Hyman and Whittemore, 2000).Benign and occasionally malignant tumors occur in anyorgan, but predominantly the brain, kidney, heart, lung,and skin (Gomez, 1999). Mutations in either TSC1or TSC2, encoding hamartin and tuberin, respectively,are the principle cause of TSC (Consortium, 1993; vanSlegtenhorst et al., 1997). Although any organ can beaffected, brain pathology, often as cortical tubersand subependymal nodules, is particularly debilitating.Significant morbidity and mortality result from epi-lepsy, autism, and other developmental and behavioraldisabilities.
TSC1 and TSC2 control cell growth, division, adhesion,and vesicular traffic by modulating the insulin signaling/AKT/mTOR, Wnt/Disheveled/b-catenin, and Rho-GTP/p27Kip1 pathways (Au et al., 2004). How perturbationsin these pathways lead to pathology is unclear. Conven-tional targeted disruptions of the Tsc1 and Tsc2 genes inthe mouse have been generated to model and study TSC(Kobayashi et al., 1999, 2001; Kwiatkowski et al., 2002;Onda et al., 1999; Wilson et al., 2005). Homozygous
disruption of either gene causes embryonic lethality.Heterozygous mice eventually develop some aspects ofTSC (renal tumors), but have not been demonstrated tohave any major central nervous system pathology. Toaddress CNS pathology, a conditional disruption of theTsc1 gene has been crossed to a glial-specific Cre trans-genic mouse (Jansen et al., 2005; Uhlmann et al., 2002).All of the progeny develop seizures after 1 month of ageand demonstrate increased numbers of astrocytes aswell as hippocampal disorganization. These Tsc1-nullmice may be a good model for the neuropathology andseizures associated with TSC. To further study the CNSpathophysiology of TSC and better define the roles ofTsc2 in normal brain physiology, we have generated andcharacterized a conditional disruption of the Tsc2 geneusing the Cre-loxP and Flp-frt systems.
To generate a floxed Tsc2 allele, we made a Tsc2neo
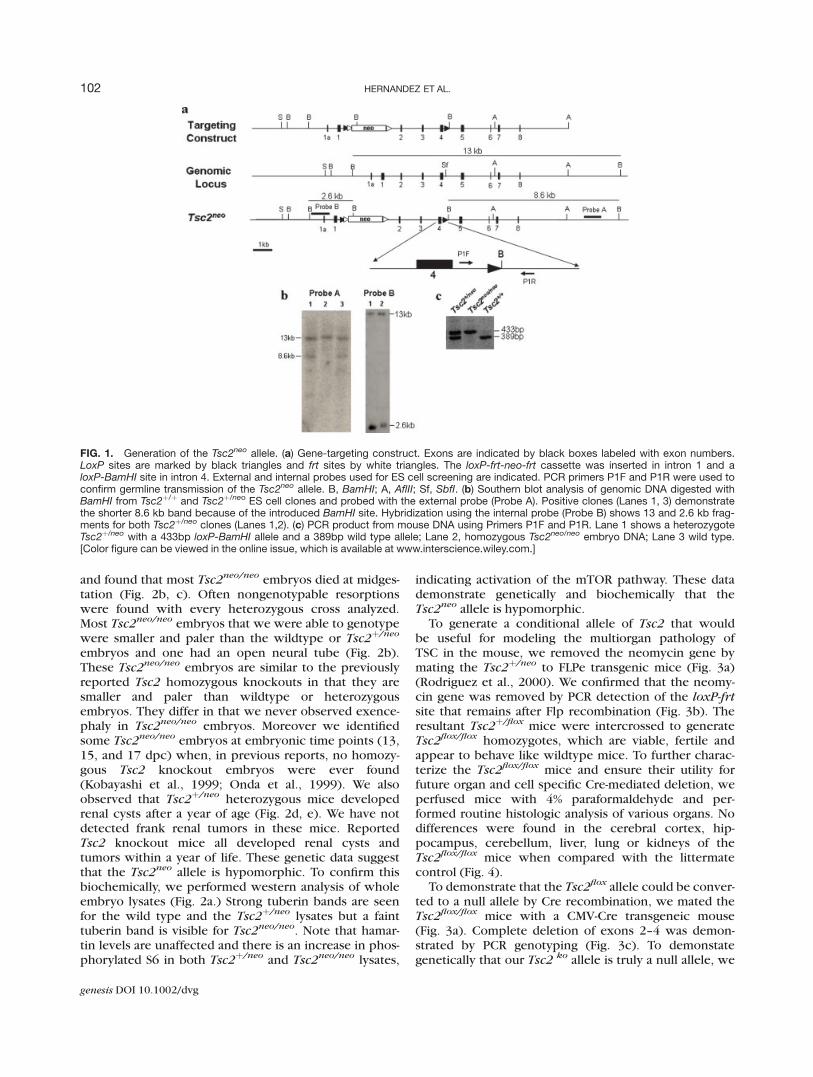
targeting vector by inserting a loxP-BamHI site intoexon 4 and a loxP-frt-neo-frt cassette into exon 1(Fig. 1a). To screen for neomycin resistant ES cells weused the 30 external and 50 internal probes indicated.Seven of 180 clones demonstrated 13 kb wildtype and8.6 kb mutant bands on Southern blot when the genomicDNA was digested with BamH1 and probed with theexternal probe (Fig 1b). After Southern analysis ofBamHI digested DNA using the internal probe demon-strated the correct 13 kb wildtype and the 2.6 kb mutantbands (Fig. 1b), two of the clones were selected forblastocyst injection. Only one chimera demonstratedgermline transmission as demonstrated by PCR genotyp-ing for the presence of the loxP-BamH1 site (Fig. 1c).
Breeding of Tsc2þ/neo heterozygotes did not generateany live born Tsc2neo/neo homozygotes, suggesting thatthe neomycin gene interfered with the expression ofTsc2. We dissected pregnant females from heterozygousTsc2þ/neo crosses at different embryonic time points
* Correspondence to: Michael J. Gambello, Department of Pediatrics,
Division of Medical Genetics, University of Texas Health Science Center,
MSB 3.144, 6431 Fannin, Houston, TX 77030.
E-mail: [email protected]
Contract grant sponsor: Tuberous Sclerosis AlliancePublished online in
Wiley InterScience (www.interscience.wiley.com).
DOI: 10.1002/dvg.20271
' 2007 Wiley-Liss, Inc. genesis 45:101–106 (2007)
and found that most Tsc2neo/neo embryos died at midges-tation (Fig. 2b, c). Often nongenotypable resorptionswere found with every heterozygous cross analyzed.Most Tsc2neo/neo embryos that we were able to genotypewere smaller and paler than the wildtype or Tsc2þ/neo
embryos and one had an open neural tube (Fig. 2b).These Tsc2neo/neo embryos are similar to the previouslyreported Tsc2 homozygous knockouts in that they aresmaller and paler than wildtype or heterozygousembryos. They differ in that we never observed exence-phaly in Tsc2neo/neo embryos. Moreover we identifiedsome Tsc2neo/neo embryos at embryonic time points (13,15, and 17 dpc) when, in previous reports, no homozy-gous Tsc2 knockout embryos were ever found(Kobayashi et al., 1999; Onda et al., 1999). We alsoobserved that Tsc2þ/neo heterozygous mice developedrenal cysts after a year of age (Fig. 2d, e). We have notdetected frank renal tumors in these mice. ReportedTsc2 knockout mice all developed renal cysts andtumors within a year of life. These genetic data suggestthat the Tsc2neo allele is hypomorphic. To confirm thisbiochemically, we performed western analysis of wholeembryo lysates (Fig. 2a.) Strong tuberin bands are seenfor the wild type and the Tsc2þ/neo lysates but a fainttuberin band is visible for Tsc2neo/neo. Note that hamar-tin levels are unaffected and there is an increase in phos-phorylated S6 in both Tsc2þ/neo and Tsc2neo/neo lysates,
indicating activation of the mTOR pathway. These datademonstrate genetically and biochemically that theTsc2neo allele is hypomorphic.
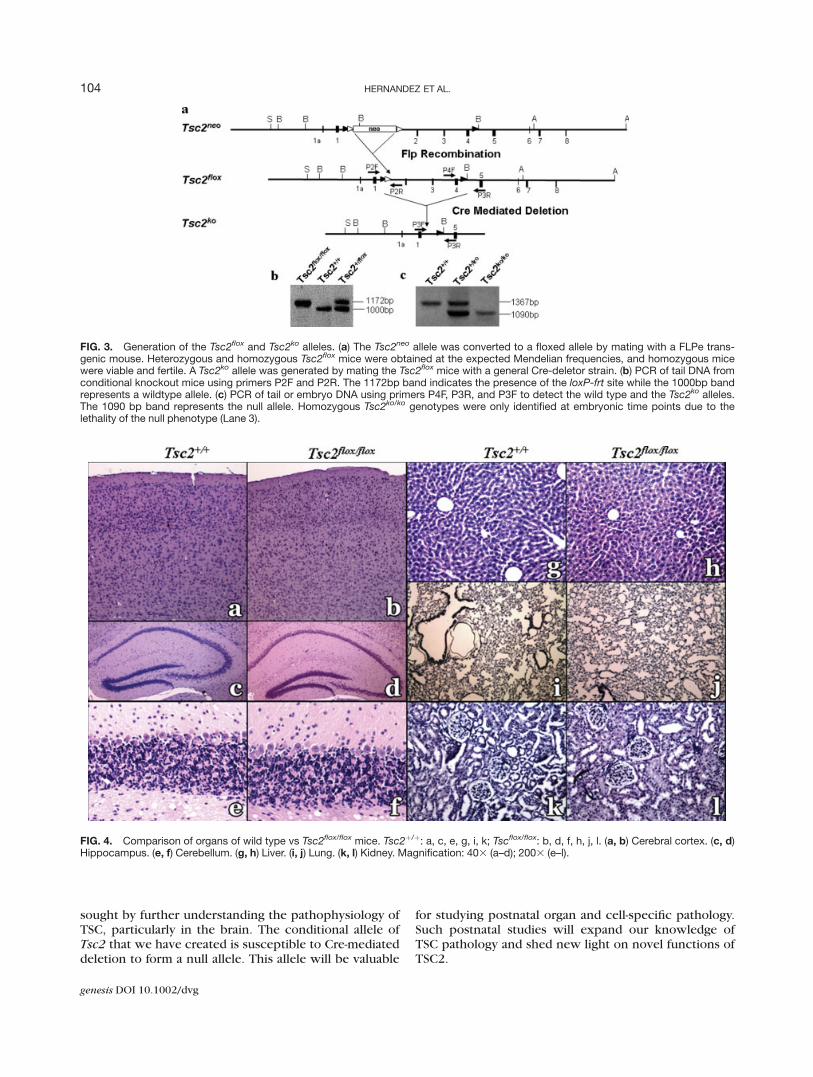
To generate a conditional allele of Tsc2 that wouldbe useful for modeling the multiorgan pathology ofTSC in the mouse, we removed the neomycin gene bymating the Tsc2þ/neo to FLPe transgenic mice (Fig. 3a)(Rodriguez et al., 2000). We confirmed that the neomy-cin gene was removed by PCR detection of the loxP-frtsite that remains after Flp recombination (Fig. 3b). Theresultant Tsc2þ/flox mice were intercrossed to generateTsc2flox/flox homozygotes, which are viable, fertile andappear to behave like wildtype mice. To further charac-terize the Tsc2flox/flox mice and ensure their utility forfuture organ and cell specific Cre-mediated deletion, weperfused mice with 4% paraformaldehyde and per-formed routine histologic analysis of various organs. Nodifferences were found in the cerebral cortex, hip-pocampus, cerebellum, liver, lung or kidneys of theTsc2flox/flox mice when compared with the littermatecontrol (Fig. 4).
To demonstrate that the Tsc2flox allele could be conver-ted to a null allele by Cre recombination, we mated theTsc2flox/flox mice with a CMV-Cre transgeneic mouse(Fig. 3a). Complete deletion of exons 2–4 was demon-strated by PCR genotyping (Fig. 3c). To demonstategenetically that our Tsc2 ko allele is truly a null allele, we
FIG. 1. Generation of the Tsc2neo allele. (a) Gene-targeting construct. Exons are indicated by black boxes labeled with exon numbers.LoxP sites are marked by black triangles and frt sites by white triangles. The loxP-frt-neo-frt cassette was inserted in intron 1 and aloxP-BamHI site in intron 4. External and internal probes used for ES cell screening are indicated. PCR primers P1F and P1R were used toconfirm germline transmission of the Tsc2neo allele. B, BamHI; A, AflII; Sf, SbfI. (b) Southern blot analysis of genomic DNA digested withBamHI from Tsc2þ/þ and Tsc2þ/neo ES cell clones and probed with the external probe (Probe A). Positive clones (Lanes 1, 3) demonstratethe shorter 8.6 kb band because of the introduced BamHI site. Hybridization using the internal probe (Probe B) shows 13 and 2.6 kb frag-ments for both Tsc2þ/neo clones (Lanes 1,2). (c) PCR product from mouse DNA using Primers P1F and P1R. Lane 1 shows a heterozygoteTsc2þ/neo with a 433bp loxP-BamHI allele and a 389bp wild type allele; Lane 2, homozygous Tsc2neo/neo embryo DNA; Lane 3 wild type.[Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
102 HERNANDEZ ET AL.
genesis DOI 10.1002/dvg

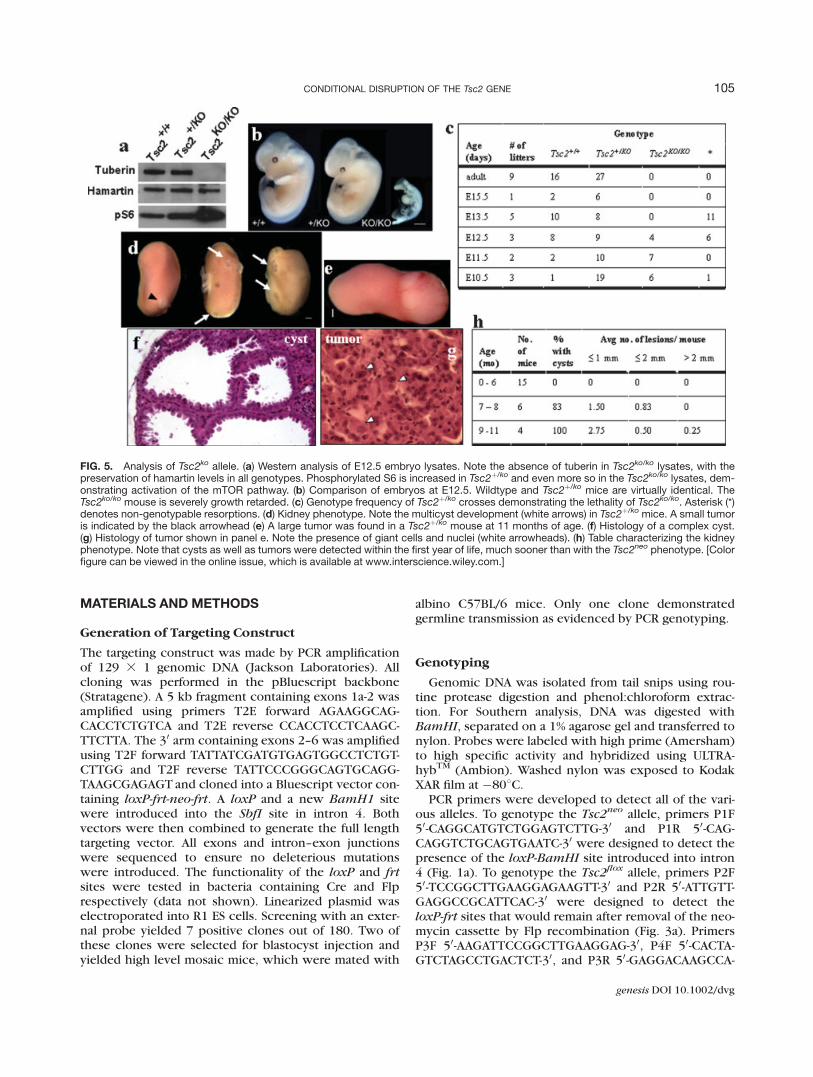
performed heterozygous crosses to determine if ourhomozygous knockout Tsc2ko/ko die at midgestation aspreviously reported. No adult homozygous knockoutTsc2 mice were detected (Fig. 5c). Embryonic analysis atmidgestation demonstrated midgestation lethality similarto previously published reports. Severe growth retarda-tion was noted in the Tsc2ko/ko mice (Figure 5b). West-ern analysis of whole embryo lysates demonstrates thecomplete absence of tuberin antigen in the Tsc2ko/ko
embryos, with unaffected hamartin levels (Fig. 5a). Notein particular that phosphorylated S6 is increased in bothTsc2þ/ko and Tsc2ko/ko embryos, demonstrating the acti-vation of the mTOR pathway. The kidney phenotype inour Tsc2þ/ko mice is also similar to previously publishedreports. Lesions are detected within the first year of life
(Fig. 5h) and are comprised of cysts and frank tumors(Fig. 5d,e). Histology demonstrates cystadenomas andrenal tumors with abnormal giant cells (Fig. 5f,g).
Though TSC is complex, efforts to understand thepathophysiology of this disease have been rather fruitful.TSC2 occupies a central role in inhibiting mTOR activity(Au et al., 2004). Loss of tuberin results in increasedmTOR activity with consequent increases in translation,cell growth, and proliferation. Rapamycin, a well knowninhibitor of mTOR, has been used in trials to replace theTSC2 inhibition lost in TSC patients. Animal and humantrials have been very encouraging (Franz et al., 2006;Kenerson et al., 2005; Lee et al., 2006). Nonetheless,tumors can often develop resistance to one chemothera-peutic agent. Consequently other targets should be
FIG. 2. Analysis of Tsc2neo allele (a) Western analysis of lysates from E12.5 embryos. Note the low level of tuberin antigen in the Tsc2neo/neo
lane. Hamartin levels are unaffected. Phosphorylated S6 is increased in both Tsc2þ/neo and Tsc2neo/neo lysates. (b) Comparison of embryosat E12.5. Tsc2neo/neo embryos were slightly smaller than wildtype and were distinctly underdeveloped and pale. Most noticeable is the lackof digitation of paws and the head size. Note the open neural tube indicated by the arrows. (c) Genotype frequency of Tsc2þ/neo crossesdemonstrating the lethality of Tsc2neo/neo embryos. Asterisk (*) denotes nongenotypable resorptions. (d) Kidneys from Tsc2þ/neo mice atvarious ages demonstrating cysts, and an H and E histology of a simple cyst, demonstrating renal cyst. Bar represents 1 mm for grosskidney photographs. (e) Table characterizing the kidney cyst phenotype. Note that no cysts developed in the first year of life, most cystswere 1 mm or less. No tumors were detected. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
103CONDITIONAL DISRUPTION OF THE Tsc2 GENE
genesis DOI 10.1002/dvg
sought by further understanding the pathophysiology ofTSC, particularly in the brain. The conditional allele ofTsc2 that we have created is susceptible to Cre-mediateddeletion to form a null allele. This allele will be valuable
for studying postnatal organ and cell-specific pathology.Such postnatal studies will expand our knowledge ofTSC pathology and shed new light on novel functions ofTSC2.
FIG. 3. Generation of the Tsc2flox and Tsc2ko alleles. (a) The Tsc2neo allele was converted to a floxed allele by mating with a FLPe trans-genic mouse. Heterozygous and homozygous Tsc2flox mice were obtained at the expected Mendelian frequencies, and homozygous micewere viable and fertile. A Tsc2ko allele was generated by mating the Tsc2flox mice with a general Cre-deletor strain. (b) PCR of tail DNA fromconditional knockout mice using primers P2F and P2R. The 1172bp band indicates the presence of the loxP-frt site while the 1000bp bandrepresents a wildtype allele. (c) PCR of tail or embryo DNA using primers P4F, P3R, and P3F to detect the wild type and the Tsc2ko alleles.The 1090 bp band represents the null allele. Homozygous Tsc2ko/ko genotypes were only identified at embryonic time points due to thelethality of the null phenotype (Lane 3).
FIG. 4. Comparison of organs of wild type vs Tsc2flox/flox mice. Tsc2þ/þ: a, c, e, g, i, k; Tscflox/flox: b, d, f, h, j, l. (a, b) Cerebral cortex. (c, d)Hippocampus. (e, f) Cerebellum. (g, h) Liver. (i, j) Lung. (k, l) Kidney. Magnification: 403 (a–d); 2003 (e–l).
104 HERNANDEZ ET AL.
genesis DOI 10.1002/dvg
MATERIALS AND METHODS
Generation of Targeting Construct
The targeting construct was made by PCR amplificationof 129 3 1 genomic DNA (Jackson Laboratories). Allcloning was performed in the pBluescript backbone(Stratagene). A 5 kb fragment containing exons 1a-2 wasamplified using primers T2E forward AGAAGGCAG-CACCTCTGTCA and T2E reverse CCACCTCCTCAAGC-TTCTTA. The 30 arm containing exons 2–6 was amplifiedusing T2F forward TATTATCGATGTGAGTGGCCTCTGT-CTTGG and T2F reverse TATTCCCGGGCAGTGCAGG-TAAGCGAGAGT and cloned into a Bluescript vector con-taining loxP-frt-neo-frt. A loxP and a new BamH1 sitewere introduced into the SbfI site in intron 4. Bothvectors were then combined to generate the full lengthtargeting vector. All exons and intron–exon junctionswere sequenced to ensure no deleterious mutationswere introduced. The functionality of the loxP and frtsites were tested in bacteria containing Cre and Flprespectively (data not shown). Linearized plasmid waselectroporated into R1 ES cells. Screening with an exter-nal probe yielded 7 positive clones out of 180. Two ofthese clones were selected for blastocyst injection andyielded high level mosaic mice, which were mated with
albino C57BL/6 mice. Only one clone demonstratedgermline transmission as evidenced by PCR genotyping.
Genotyping
Genomic DNA was isolated from tail snips using rou-tine protease digestion and phenol:chloroform extrac-tion. For Southern analysis, DNA was digested withBamHI, separated on a 1% agarose gel and transferred tonylon. Probes were labeled with high prime (Amersham)to high specific activity and hybridized using ULTRA-hybTM (Ambion). Washed nylon was exposed to KodakXAR film at �808C.
PCR primers were developed to detect all of the vari-ous alleles. To genotype the Tsc2neo allele, primers P1F50-CAGGCATGTCTGGAGTCTTG-30 and P1R 50-CAG-CAGGTCTGCAGTGAATC-30 were designed to detect thepresence of the loxP-BamHI site introduced into intron4 (Fig. 1a). To genotype the Tsc2flox allele, primers P2F50-TCCGGCTTGAAGGAGAAGTT-30 and P2R 50-ATTGTT-GAGGCCGCATTCAC-30 were designed to detect theloxP-frt sites that would remain after removal of the neo-mycin cassette by Flp recombination (Fig. 3a). PrimersP3F 50-AAGATTCCGGCTTGAAGGAG-30, P4F 50-CACTA-GTCTAGCCTGACTCT-30, and P3R 50-GAGGACAAGCCA-
FIG. 5. Analysis of Tsc2ko allele. (a) Western analysis of E12.5 embryo lysates. Note the absence of tuberin in Tsc2ko/ko lysates, with thepreservation of hamartin levels in all genotypes. Phosphorylated S6 is increased in Tsc2þ/ko and even more so in the Tsc2ko/ko lysates, dem-onstrating activation of the mTOR pathway. (b) Comparison of embryos at E12.5. Wildtype and Tsc2þ/ko mice are virtually identical. TheTsc2ko/ko mouse is severely growth retarded. (c) Genotype frequency of Tsc2þ/ko crosses demonstrating the lethality of Tsc2ko/ko. Asterisk (*)denotes non-genotypable resorptions. (d) Kidney phenotype. Note the multicyst development (white arrows) in Tsc2þ/ko mice. A small tumoris indicated by the black arrowhead (e) A large tumor was found in a Tsc2þ/ko mouse at 11 months of age. (f) Histology of a complex cyst.(g) Histology of tumor shown in panel e. Note the presence of giant cells and nuclei (white arrowheads). (h) Table characterizing the kidneyphenotype. Note that cysts as well as tumors were detected within the first year of life, much sooner than with the Tsc2neo phenotype. [Colorfigure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
105CONDITIONAL DISRUPTION OF THE Tsc2 GENE
genesis DOI 10.1002/dvg

ACATCCAT-30 were designed to detect the null allele aswell as the wildtype allele (Fig. 3a).
Protein Analysis
Whole cell lysates were made from E12.5 embryosthat were quick frozen in liquid nitrogen. After genotyp-ing a small piece of each embryo, samples were pooledby genotype and homogenized in a dounce homogenizerwith 10 volumes of Ripa buffer with protease inhibitorcocktail (Sigma). Lysates were centrifuged at 48C,sonicated and frozen until use. Protein concentrationswere determined with a BCA reagent kit (Pierce). Equalamounts of protein were separated on a denaturing4–12% gradient gel (Invitrogen) and transferred to nitro-cellulose. The same membrane was probed with threedifferent antibodies using a stripping procedure aftereach experiment. The membrane was first probed withtuberin antibody (1:1,000, Cell Signaling), then withhamartin (1:1,000, Santa Cruz), and lastly with phospho-rylated (Ser 240/244) S6 (1:2,000, Cell Signaling).Secondary antibodies were horseradish peroxidase con-jugated. Visualization was done with an enhanced chem-iluminescence kit (Amersham).
Embryo and Organ Analysis
The day of the vaginal plug was 0.5. Mice were thenanesthetized with avertin and killed by cervical disloca-tion before embryos were dissected into cold PBS. Theyolk sac or a small piece of the embryo was used for gen-otyping. For organ analysis, mice were anesthetized withavertin and transcardially perfused with 4% paraformal-dehyde. Organs were postfixed in paraformaldehydeovernight and then dehydrated and embedded in paraf-fin. Five micron microtome sections were cut. Slideswere then rehydrated, stained with routine hematoxylinand eosin, and coverslipped. Embryo and tissue imageswere captured with a SPOT RT digital camera.
ACKNOWLEDGMENTS
We thank Gail Martin for the loxp-frt-Neo-frt construct.Special thanks to Jan Parker-Thornberg and RichardBehringer for reagents and use of the MD Anderson Corefacility.
LITERATURE CITED
Au K-S, Williams A, Gambello M, Northrup H. 2004. Molecular geneticbasis of tuberous sclerosis complex: From bench to bedside.J Child Neurol 19:699–709.
Consortium ECTS. 1993. Identification and characterization of thetuberous sclerosis gene on chromosome 16. Cell 75:1305–1315.
Franz D, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopou-los A, Thomas G, Crone K. 2006. Rapamycin causes regression ofastrocytomas in tuberous sclerosis complex. Ann Neurol 59:490–498.
Gomez MR, editor. 1999. Tuberous sclerosis complex, 3rd ed. NewYork: Oxford University Press.
Hyman M, Whittemore V. 2000. National Institutes of Health ConsensusConference: The tuberous sclerosis complex. Arch Neurol 57:662–665.
Jansen L, Uhlmann E, Crino P, Gutmann D, Wong M. 2005. Epileptogen-esis and reduced inward rectifier potassium current in tuberoussclerosis complex-1-deficient astrocytes. Epilepsia 46:1871–1880.
Kenerson H, Dundon T, Yeung R. 2005. Effects of rapamycin in theEker rat model of tuberous sclerosis complex. Pediatr Res 57:67–75.
Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T. 1999. Renalcarcinogenesis, hepatic hemangiomatosis, and embryonic lethalitycaused by a germ-line Tsc2 mutation in mice. Cancer Res 59:1206–1211.
Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E,Noda T, Hino O. 2001. A germ-line Tsc1 mutation causes tumordevelopment and embryonic lethality that are similar, but notidentical to, those caused by Tsc2 mutation in mice. Proc NatlAcad Sci USA 98:8762–8767.
Kwiatkowski D, Zhang H, Bandura J, Heiberger K, Glogauer M,el-Hashemite N, Onda H. 2002. A mouse model of Tsc1 revealssex-dependent lethality from liver hemangiomas, and up-regula-tion of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet11:525–534.
Lee L, Sudentas P, Dabora S. 2006. Combination of rapamycin analog(CCI-779) and interferon-gamma is more effective than singleagents in treating a mouse model of tuberous sclerosis. GenesChromosomes Cancer 45:933–944.
Onda H, Lueck A, Marks P, Warren H. 1999. Tsc2þ/� mice developtumors in multiple sites that express gelsolin and are influencedby genetic background. J Clin Invest 104:687–695.
Rodriguez C, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R,Stewart A, Dymecki S. 2000. High-efficiency deleter mice showthat FLPe is an alternative to Cre-loxP. Nat Genet 25:139–140.
Uhlmann E, Wong M, Baldwin R, Bajenaru M, Onda H, Kwiatkowski D,Yamada K, Gutmann D. 2002. Astrocyte-specific TSC1 conditionalknockout mice exhibit abnormal neuronal organization andseizures. Ann Neurol 52:285–296.
van Slegtenhorst M, deHoogt R, Hermans C, Nellist M, Janssen B,Verhoef S, Lindhout D, Ouweland A, Halley D, Young J, Burley M,Jeremiah S, Woodward K, Nahmias J, Fox M, Ekong R, Osborne J,Wolfe J, Povey S, Snell RG, Cheadle JP, Jones AC, Tachataki M,Ravine D, Sampson JR, Reeve MP, Richardson P, Wilmer F, MunroC, Hawkins TL, Sepp T, Ali JB, Ward S, Green J, Yates JR,Kwiatkowska J, Henske EP, Short MP, Haines JH, Jozwiak S, Kwiat-kowski DJ. 1997. Identification of the tuberous sclerosis geneTSC1 on chromosome 9q34. Science 77:805–808.
Wilson C, Idziaszczyk S, Parry L, Griffiths D, Lazda E, Bayne R, Smith A,Sampson J, Cheadle J. 2005. A mouse model of tuberous sclerosis1 showing background specific early post-natal mortality and met-astatic renal cell carcinoma. Hum Mol Genet 14:1839–1850.
106 HERNANDEZ ET AL.
genesis DOI 10.1002/dvg





























