Embed Size (px)
Citation preview

Methods 56 (2012) 452–460
Contents lists available at SciVerse ScienceDirect
Methods
journal homepage: www.elsevier .com/locate /ymeth
Generation and genetic modification of 3D cultures of human dopaminergicneurons derived from neural progenitor cells
Catarina Brito a,b, Daniel Simão a,b, Inês Costa a,b, Rita Malpique a,b, Cristina I. Pereira a,b,Paulo Fernandes a,b, Margarida Serra a,b, Sigrid C. Schwarz c, Johannes Schwarz c, Eric J. Kremer d,Paula M. Alves a,b,⇑a iBET, Instituto de Biologia Experimental e Tecnológica, Apartado 12, 2780-901 Oeiras, Portugalb Instituto de Tecnologia Química e Biológica–Universidade Nova de Lisboa (ITQB–UNL), Avenida da República, Estação Agronómica Nacional, 2780-157 Oeiras, Portugalc Department of Neurology, University of Leipzig, Liebigstr. 20, 04103 Leipzig, Germanyd Institut de Génétique Moléculaire de Montpellier, CNRS UMR 5535, Universities of Montpellier I and II, 34293 Montpellier, France
a r t i c l e i n f o
Article history:Available online 14 March 2012
Keywords:Human neural stem cellsNeurospheresNeuronal differentiationDopaminergic neuronsStirred culture systems3D cell models
1046-2023/$ - see front matter � 2012 Elsevier Inc. Ahttp://dx.doi.org/10.1016/j.ymeth.2012.03.005
Abbreviations: b3-tub, b3-tubulin; CNS, central nepidermal growth factor; EM, expansion medium; AMGFP, enhanced green fluorescent protein; hmNPC, humhuman pluripotent stem; hpt, hours post-transductidisease; PI, propidium iodide; PLOF, poly-L-ornithine-⇑ Corresponding author at: iBET, Instituto de Biolog
E-mail address: [email protected] (P.M. Alves).
a b s t r a c t
Central nervous system (CNS) disorders remain a formidable challenge for the development of efficienttherapies. Cell and gene therapy approaches are promising alternatives that can have a tremendousimpact by treating the causes of the disease rather than the symptoms, providing specific targetingand prolonged duration of action. Hampering translation of gene-based therapeutic treatments of neuro-degenerative diseases from experimental to clinical gene therapy is the lack of valid and reliable pre-clin-ical models that can contribute to evaluate feasibility and safety.
Herein we describe a robust and reproducible methodology for the generation of 3D in vitro models ofthe human CNS following a systematic technological approach based on stirred culture systems. We tookadvantage of human midbrain-derived neural progenitor cells (hmNPC) capability to differentiate intothe various neural phenotypes and of their commitment to the dopaminergic lineage to generate differ-entiated neurospheres enriched in dopaminergic neurons. Furthermore, we describe a protocol for effi-cient gene transfer into differentiated neurospheres using CAV-2 viral vectors and stable expression ofthe transgene for at least 10 days. CAV-2 vectors, derived from canine adenovirus type 2, are promisingtools to understand and treat neurodegenerative diseases, in particular Parkinson’s disease. CAV-2 vec-tors preferentially transduce neurons and have an impressive level of axonal retrograde transport in vivo.
Our model provides a practical and versatile in vitro approach to study the CNS in a 3D cellular context.With the successful differentiation and subsequent genetic modification of neurospheres we are increas-ing the collection of tools available for neuroscience research and contributing for the implementation andwidespread utilization of 3D cellular CNS models. These can be applied to study neurodegenerative dis-eases such as Parkinson’s disease; to study the interaction of viral vectors of therapeutic potential withinhuman neural cell populations, thus enabling the introduction of specific therapeutic genes for treatmentof CNS pathologies; to study the fate and effect of delivered therapeutic genes; to study toxicologicaleffects. Furthermore these methodologies may be extended to other sources of human neural stem cells,such as human pluripotent stem cells, including patient-derived induced pluripotent stem cells.
� 2012 Elsevier Inc. All rights reserved.
1. Introduction multipotent ability to generate at least the three neural lineages
Neural stem cells (NSCs) are self-renewing, exist in specific re-gions of the developing and adult mammalian CNS, and have the
ll rights reserved.
ervous system; CAV-2, canine ad, aggregation medium; FDA, fluoresan midbrain-derived neural proge
on; ip, infectious particles; MOI, mfibronectin; RPL22, ribosomal protia Experimental e Tecnológica, Ap
– neurons, astrocytes and oligodendrocytes. Neural progenitorcells (NPC) have a limited capacity for self-renewal and may retainmultipotency or present reduced differentiation potential. For the
enovirus type 2; DM, differentiation medium; DN, dopaminergic neurons; EGF,cein diacetate; FGF-2, fibroblast growth factor-2; GFAP, glial fibrillary acidic protein;nitor cell; hiPS, human induce pluripotent stem; hNSC, human neural stem cell; hPS,
ultiplicity of infection; PCNA, proliferating cell nuclear antigen; PD, Parkinson’sein L22; TH, tyrosine hydroxylase; vg, viral genomes.artado 12, 2780-901 Oeiras, Portugal. Fax: +351 21 4421161.

C. Brito et al. / Methods 56 (2012) 452–460 453
last two decades multipotent NSC/NPC have been isolated frommultiple brain regions and more recently, protocols have beendeveloped for the in vitro derivation of human NSC (hNSC) fromhuman pluripotent stem (hPS) cells, both embryonic and induced(iPS) [1–3]. The possibility of stable expansion, in vitro differentia-tion into neural phenotypes and genetic modification makes hNSC/NPC an attractive cell source for cell therapy as well as diseasemodeling of a wide variety of nervous system diseases, rangingfrom Parkinson’s disease (PD) to stroke and brain trauma, withapplication in basic cell biology, drug discovery and toxicology[1]. Therefore, despite the significant body of research on hNSC/NPC, the field is still gaining momentum. To address specific ques-tions in biomedical research or to generate therapeutically relevanteffector cells for cell therapy, technologies for the culture, differen-tiation and modification of hNSC/NPC and derivatives are required.
Two well-described culture systems commonly used to expandhNSC/NPC include the adherent monolayer and the neurosphereculture system, where cells are cultured as free-floating aggre-gates. The neurosphere culture system, developed in the earlynineties to identify neural stem cells, has been widely used forthe isolation and expansion of embryonic and adult CNS stem cell.It is based on the ability of NSC/NPC to aggregate and proliferateunder serum-free media conditions, in the presence of epidermalgrowth factor (EGF) and/or fibroblast growth factor-2 (FGF2) [4].Numerous bioprocesses for the large-scale production of NSC/NPC isolated from different regions of murine and human brainhave been developed based on both the classic and on the stirredsuspension neurosphere culture systems [5–7]. The utility of theneurosphere formation assay as quantitative in vitro method formeasuring NSC frequency was challenged by the discovery thatthe assay conditions allow the expansion of multipotent NSC/NPCbut also of more committed progenitors, with the production of aheterogeneous cell population [4,8]. More recently, it has been ar-gued that adherent monolayer culture conditions, using laminin orfibronectin as adherent substrate, in defined serum free media andexposure to EGF and FGF2 are better suited for long-term NSCexpansion; these conditions allow cells to divide symmetricallyretaining their tripotent neurogenic potential [1]. Concerning NSCin vitro differentiation, typically cells are plated on adhesive sub-tracts, such as laminin, EGF and FGF2 are withdrawn and neurotro-phic factors added. Differentiation depends not only on the appliedprotocol but also on the developmental stage and region of the tis-sue of origin [9,10].
Several methods for the genetic modification of hNSC/NPC havebeen applied [11], including non-viral [12] and viral gene transfer.The transduction of proliferative neurospheres with human adeno-virus vectors [13], was optimal only after dissociation of the neur-ospheres. Likewise, techniques for gene transfer into differentiated2D cultures are abundant in the literature [14].
Amongst the viral vectors available, CAV-2 vectors, which arederived from canine adenovirus type 2, are promising tools forgene transfer due to the lack of immunological memory, long-termepisomal expression and high cloning capacity [15]. Moreover,both in rat brain and in vitro 2D neural cultures, CAV-2 vectorspreferentially transduce neurons and undergo efficient long-dis-tance targeting via axonal transport [16], making them promisingtools for the treatment of degenerative diseases, such as PD as wellas for the development of robust human CNS cell models.
2. Materials and methods
2.1. Overview
In this study, we provide protocols for the generation of 3Din vitro models of the human CNS following an approach based
on stirred culture systems (Fig. 1). As cell source we used humanmidbrain-derived neural progenitor cells (hmNPC) as these canbe expanded in vitro and differentiated into tyrosine hydroxilase(TH)-positive cells, in 2D culture conditions [17]. In addition tothe expression of TH, the key enzyme for dopamine synthesis,these neurons exhibited morphological and functional propertiesof dopaminergic neurons in culture, such as dopamine productionand release [3]. We describe the generation of differentiated neur-ospheres enriched in TH-positive neurons. Furthermore, we estab-lished a protocol for efficient gene transfer of differentiatedneurospheres using CAV-2 vectors that allowed long-term trans-gene expression.
We took advantage of a well-described 2D protocol for long-term proliferation of hmNPC based on the use of O2 levels closeto physiological conditions (3%) and serum-free media [17]. Theseconditions led to the expansion of EGF/FGF-2 responding cells formore than 1 year, while retaining their tripotency and the abilityto differentiate into dopaminergic neurons [3]. This expanded pop-ulation of hmNPC was used as starting point for a robust and repro-ducible methodology based on stirred culture systems fordifferentiation and transduction.
2.2. Cell isolation and expansion
All tissue procurement was performed with mother’s consentand in accordance with the Ethics Committee of the University ofLeipzig and with all German state and federal laws. Human neuralprogenitor cells derived from aborted fetal brain tissue 12–14 weeks post-fertilization were isolated as described previously[17–20]. In brief, prior to trituration, the tissue was incubated in100 lg/mL Papain (Roche Diagnostics) and 10 lg/mL DNase inphosphate-buffered saline (PBS), for 30 min at 37 �C, followed bywashing with PBS, and incubation with antipain (50 lg/mL; Roche)for 30 min at 37 �C.
Expansion of hmNPC was performed under 3% O2, on poly-L-ornithine-fibronectin (PLOF)-coated surfaces and serum-free med-ium, as described previously [17,19,20]. Expansion medium (EM)was composed of Dulbecco’s modified Eagle medium (DMEM)and Ham’s F12 Nutrient Mix (both from Invitrogen) in a 1:1 ratio,2% B27 supplement (Invitrogen) 20 ng/mL rhu-FGF2 and rhu-EGF(both from PrepoTech), 1 lg/mL Tocopherol (Fluka), 1 lg/mLTocopherol Acetate (Sigma) and 10 lg/mL Gentamycin (Invitogen).Cells were split, typically every 10–14 days, at 90–100% confluency(Fig. 2), corresponding to a 3- to 5-fold increase in cellconcentration.
For splitting or collecting cells, the monolayer was incubatedwith Accutase� (Sigma) up to 30 min at 37 �C and cells detachedby gentle mechanical dislodgment using a cell scrapper. Cells werecollected with PBS and sedimented by centrifugation at 300g,5 min, with no brake setting. After resuspension in a small volumeof EM (approximately 1 mL/T150 flask collected), a 1 mL pipette ora glass Pasteur pipette was used to obtain a homogeneous cell sus-pension, thus avoiding air bubbles. Viability was determined byTrypan blue exclusion assay: after incubation with 0.1% (v/v) Try-pan blue (Invitrogen) in PBS, colorless (viable) and blue (unviable)cells were counted using a Fuchs–Rosenthal haemocytometerchamber.
To avoid cell death and spontaneous differentiation (i) mediaexchange should be performed every 3–4 days or whenever a dropin pH is detectable by visual inspection in the media (the use phe-nol red containing media is recommended unless it interferes withsubsequent assays); (ii) avoid overgrowing the monolayers, toavoid cell migration, culture polarization and consequent sponta-neous differentiation.
Using this protocol, hmNPC were stably expanded for long peri-ods of time, >20 passages (at least 10–12 population doublings), in

Fig. 1. Schematic workflow of expansion, 3D differentiation and transduction of human midbrain derived neural progenitor cell (hmNPC) cultures using stirredculture systems and low oxygen concentrations. Expansion of hmNPC was performed using 2D culture systems, on poly-L-ornithine-fibronectin (PLOF)-surfaces, inexpansion medium (EM). Differentiation in 3D culture systems was performed using shake-flasks: cells are inoculated in aggregation medium (AM). Afterneurosphere formation differentiation was induced with differentiation media (DM). Differentiated neurospheres can be kept in long-term cultures and be transducedwith CAVGFP vectors.
Fig. 2. 2D expansion cultures of human midbrain derived neural progenitor cells (hmNPC). Phase contrast images of hmNPC expansion cultures 1 and 14 days after seeding,representing successful adherence and confluence phase (A), respectively. Immunofluorescence microscopy of hmNPC: nestin (red), Ki67 (green) and DAPI (blue) (B); nestin(red), b3-tubulin (green) and DAPI (blue) (C). Scale bars – 100 lm.
454 C. Brito et al. / Methods 56 (2012) 452–460

C. Brito et al. / Methods 56 (2012) 452–460 455
a multi-gas cell incubator (Sanyo), at 37 �C, in a humidified atmo-sphere with 5% CO2 and 3% O2 [17,19,20].
2.3. Cell cryopreservation
Expanded hmNPC were cryopreserved as single cell suspensionsin CryoStor-CS10 (BioLife Solutions), at 5 � 106 viable cell/mL.Cryovials (Nunc) filled with 1 mL of cell suspension were held at4 �C for 20 min for cryoprotectant equilibration, frozen to �80 �Cin an isopropanol-based freezing system, ‘‘Mr. Frosty’’ (Nalgene),which allows for a cooling rate close to 1 �C/min (accordingly tothe manufacture’s specifications). Long-term storage was per-formed in the gas phase of a liquid nitrogen storage reservoir.Thawing was performed in a water bath at 37 �C and immediatelyafter thawing a three-step drop-wise dilution (1:2, 1:4, 1:10) inpre-warmed EM was performed, with 2 and 5 min equilibrationsteps between additions, respectively. The stepwise addition ofthe EM during thawing was critical to avoid cell injury by the os-motic imbalance caused by the hypertonic DMSO present in Cryo-Stor-CS10. Cells were collected by centrifugation at 300g, 5 min (nobrake setting), resuspended in EM and seeded on PLOF-coated sur-faces at 5–8 � 104 cell/cm2.
Viabilities of 85–90% are usually attained with this cryopreser-vation-thawing protocol. A media exchange should be performed48 h after thawing, particularly if viability after thawing is lowerthan the expected range.
2.4. 2D differentiation
Differentiation of hmNPC in 2D cultures was performed asdescribed previously [17,19,20]. hmNPC were cultured on PLOF-coated surfaces, using the expansion protocol described inSection 2.2. Once cells attained confluency (Fig. 2), EM wasexchanged to differentiation medium (DM), composed of Neurobasalmedium (Invitrogen), supplemented with 2% B27 (Invitrogen),2 mM Glutamax (Invitrogen), 100 lM dibutyryl c-AMP (Sigma),10 lM forskolin (Sigma–Aldrich) and 10 lg/mL Gentamycin (Invi-togen) for at least 7 days. Total media exchange was performedevery 3–4 days.
2.5. 3D neurosphere differentiation
hmNPC were cultured as neurospheres in stirred systems withorbital shaking, using a multi-gas cell incubator (Sanyo), in ahumidified atmosphere with 5% CO2 and 3% O2, at 37 �C. Typicallycells were cultured in 125 mL shake flasks (Corning) with a work-ing volume of 10–25 mL and a stirring rate of 100 rpm.
After cell expansion in 2D monolayers, obtaining a single-cellsuspension (see Section 2.2) was particularly important to achievehomogeneous aggregation and uniform aggregate size in 3D cul-tures. Therefore, the cell suspension was filtered through a70 lm nylon strainer (Millipore) prior to inoculation in shakeflasks at 2 � 105 cell/mL in aggregation medium (AM). This mediahad the same composition of EM, except for reduced mitogen con-centration (5 ng/mL of both FGF2 and EGF). The aggregation stepallowed the neurospheres to form and grow up to 150–200 lm(5–7 days), with 50% media exchange at day 3–4. Neurosphereswere not allowed to reach diameters above 200–350 lm to avoidthe formation of necrotic centers due to limitations in nutrient dif-fusion and O2 uptake in the center of the aggregate [27]. Differen-tiation was induced by removal of mitogens and culture for at least10 days in DM. A 50% media exchange was performed every 3–4 days.
For media exchange, neurospheres were allowed to settle bygravity before removing the medium and not centrifuged to avoidaltering the shape and inducing fusion of neurospheres. The cul-
tures were kept outside the incubator for the minimum time re-quired for manipulation. Typically, when using manipulationtimes of 10–15 min and fresh media equilibrated at 37 �C and 3%O2, which reduce even further the perturbations in dissolved O2,no significant differences in proliferation and differentiation effi-ciency were observed. Importantly, repeated opening of the incu-bator was avoided to guarantee a stable 3% O2 level throughoutthe culture time.
Differentiation was analyzed by fluorescence microscopy andqRT-PCR tools to confirm differentiation into the 3 neural lineages(see Section 2.7).
2.6. Transduction of differentiated neurospheres
2.6.1. Viral stock titrationThe quality of the vector stock plays a key role in the efficacy
and reproducibility of the transduction protocol. Non-purified viralvectors have been successfully used but presented significant var-iability between experiments.
We routinely produced CAVGFP (an E1-deleted CAV-2 vectorexpressing GFP) [21] stocks by infection of E1-complementingdog kidney cells (DKZeo) [22] maintained in DMEM (Invitrogen)with 10% (v/v) FBS. Forty hours post-infection (hpi) cells were col-lected and lysed with 0.1% (v/v) of TritonX-100 (Sigma–Aldrich) inPBS. The cleared lysate was purified by CsCl gradient and the puri-fied vector stored in PBS with 10% (v/v) glycerol at �85 �C [23].CAVGFP was quantified using qPCR of viral genomes (vg)/mL andan infectivity assay – number of infectious particles/mL (ip/mL)[23,24]. The CAVGFP preps had a vg/ip unit ratio of 20:1. In thisstudy, multiplicity of infection (MOI) is the number of infectiousviral particles per cell.
2.6.2. Cell concentration determinationTo determine the optimal amount of vector particles, cell con-
centration must be assessed initially. Proliferative neurospheresand 2D cultures were easily dissociated with Accutase� (Sigma),without significant loss of cell viability, which readily allowed cellconcentration quantification using Trypan blue exclusion assay(Section 2.2). Differentiated neurospheres, however, were difficultto dissociate without significant losses in cell viability due to theincreased mechanical susceptibility of differentiated cells, andthe intricate net of neurites formed. Thus, we assessed viabilityof differentiated neurospheres using a fluorescence microscopybased method, the fluoresceine diacetate (FDA)/propidium iodide(PI) assay [25]. Subsequently, neurospheres were disrupted in0.1 M citric acid with 1% TritonX-100, at 37 �C, overnight, and nu-clei were stained with 0.1% crystal violet [26] and counted in aFuchs–Rosenthal haemocytometer chamber. A sample size of atleast 1 mL of culture was used to reliably determine cellconcentration.
2.6.3. TransductionTransduction of neurospheres was carried out in 50% reduction
of the working volume, making sure that the minimum workingvolumes of the shake flasks were respected (see Section 2.5) andmaintaining the remaining culture conditions as described in Sec-tion 2.5; CAVGFP (see Section 2.6.1) was added to the culture,according to the intended MOI. For 2D controls, the supernatantwas discarded and vector stock diluted in fresh DM (according tothe MOI) added; 2D transduction was performed in static condi-tions. Four hours post-transduction (hpt) the medium was eithercompletely replaced or the initially working volume restored byadding fresh DM and changed 72 h post-transduction. Transduc-tion efficacy as a function of time was evaluated by fluorescencemicroscopy and qRT-PCR tools and, for 2D cultures, flow cytometry

456 C. Brito et al. / Methods 56 (2012) 452–460
(see Section 2.7). Our low and high MOI conditions correspondedto approximately 100 and 500 ip/cell, respectively.
2.7. Analysis of differentiation and transduction
2.7.1. Culture samplingSampling was performed with a 5 or 10 mL pipette. For qRT-PCR
and microscopy, 8–12 aggregates were collected. Neurosphereswere sedimented by gravity; centrifugation was used only whencells were processed for mRNA or protein analysis.
2.7.2. Viability assayThe qualitative assessment of cell viability was performed using
FDA, which is converted to fluorescein by intracellular esterasesand retained within cells with intact membranes and the mem-brane-impermeable DNA-dye PI. Neurospheres were incubatedwith 20 lg/mL FDA and 10 lg/mL PI in PBS for 5 min, washed withPBS and observed using fluorescence microscopy (DMI6000, Leica).
2.7.3. qRT-PCRNeurospheres were sedimented by centrifugation at 500g for
5 min, washed with PBS and the dry pellet snap-freezed by immer-sion in liquid nitrogen. Samples were stored at �80 �C until RNAextraction. Total RNA was extracted with High Pure RNA IsolationKit (Roche), which included a DNase digestion step, according tothe manufacturer instructions. RNA was eluted with 50 lL ofsterile deionized water, quantified in a NanoDrop 2000c (ThermoScientific) and used directly for cDNA synthesis or stored at�80 �C.
Reverse transcription was performed with High Fidelity cDNASynthesis Kit (Roche), using Anchored-oligo(dT)18 Primer (Roche).Firstly, the concentrations of all RNA samples were normalizedby dilution in sterile deionized water (until 10.4 lL, the maximumvolume recommended per reaction in cDNA synthesis kit). Theresulting cDNA was quantified and used directly for qPCR or storedat �20 �C. qPCR was performed in triplicates according to Light-Cycler� 480 SYBR Green I Master Kit (Roche), in 20 lL reactions
Table 1List of primers used in real-time quantitative PCR.
Gene Primers forward (top) and reverse (bottom) Product size (bp)
RPL22 CACGAAGGAGGAGTGACTGGTGTGGCACACCACTGACATT
116
PCNA CGGAGTGAAATTTTCTGCAAGTTCAGGTACCTCAGTGCAAAAG
144
TH AGCCCTACCAAGACCAGACGGCGTGTACGGGTCGAACTT
132
GFP CAGAAGAACGGCATCAAGGTCTGGGTGCTCAGGTAGTGG
143
Table 2Thermal parameters used in real-time quantitative PCR.
Cycles Analysis mode Temp (�C
Denaturation 1 None 95Amplification 45 Quantification
956272
Melting 1 Melting Curves956097
Cooling 1 None 40
with 1:2 diluted cDNA template and 5 lM primers (listed in Ta-ble 1). For each pair of primers, calibration curves with serial dilu-tions of cDNA were performed in order to determine the amount ofcDNA to be used in the qPCR reaction. The samples were loaded inLightCycler� 480 Multiwell Plate 96 (Roche), always maintainingreagents and well-plates on ice. The thermal parameters used aredescribed in Table 2; the reactions were performed using Light-Cycler� 480 Instrument II 96-well block (Roche). Cycles threshold(Ct’s) and melting curves were determined using LightCycler�
D480 Software version 1.5 (Roche). All data was analyzed usingthe 2�DDCt method for relative gene expression analysis [27]. Thechanges in gene expression (PCNA, proliferating cell nuclear anti-gen; TH, tyrosine hydroxylase; GFP, enhanced green fluorescentprotein) were normalized using the housekeeping gene RPL22(ribosomal protein L22) as internal control. Statistical analysiswas carried out using GraphPad Prism 5 software.
2.7.4. Fluorescence microscopyProtocols for cryosectioning and immunocytochemistry of
neurospheres have been described in detail elsewhere [28–30].Briefly, neurospheres were frozen at �80 �C in Tissue Teck OCT™Compound (Sakura) and sectioned in a cryostat (Leica) (10–15 lm sections). Cryosections and intact neurospheres for wholemount microscopy were fixed in PFA 4 + 4% sucrose and processedfor immunostaining as described [25,31]. The antibodies used forpopulation characterization, as well as the secondary antibodiesare described in Table 3; cell nuclei were counterstained with DAPI(Invitrogen).
Samples were visualized using fluorescence (DMI6000, Leica),spinning disk (Nikon Eclipse Ti-E, confocal scanner: YokogawaCSU-x1) and point scan confocal (SP5, Leica) microscopes. Duringconfocal image acquisition, each fluorophore used was detectedseparately to avoid channel bleed-through. For each channel, pho-tomultiplier gains and offsets were adjusted to use full image dy-namic range and for each image, focus intervals were set to meetthe demands of the Nyquist sampling criteria [32]. Merge betweenchannels, maximum z-projections and orthogonal projections, aswell as linear brightness and contrast adjustments of the images,were created using the open source ImageJ software version1.43 m (http://rsbweb.nih.gov/ij/).
2.7.5. Flow cytometryFlow cytometry of 2D cultures was performed essentially as de-
scribed [33]. Cells were detached by incubation of the monolayerwith Accutase� (Sigma) and processed as described in Section 2.2.The single cell suspension was resuspended in PBS with 2% FBScells were analyzed in a CyFlowH space (Partec) instrument as re-ported elsewhere [30]. Ten thousand events were registered persample.
) Time Ramp rate Acquisition mode
00:10:00 4.4 None
00:00:10 4.4 None00:00:10 2.2 None00:00:15 4.4 Single
00:00:05 4.4 None00:01:00 2.2 None00:00:00 0.11 Continuous00:00:30 2.2 None

Table 3List of antibodies used for immunofluorescence microscopy.
Antibody Marker Concentration Manufacturer
Anti-ß3-tubulin Neurons 1:200 Millipore, cat. No. MAB1637Anti-nestin Neural progenitors 1:200 Millipore, cat. No. AB5922Anti-Ki67-FITC Proliferative cells 1:200 Abcam, cat. No. ab27619Anti-GFAP Astrocytes 1:200 Millipore, cat. No. AB5804Anti-O4 Olygodendrocytes 1:200 Millipore, cat. No. MAB345Anti-mouse Alexa Fluor 488 (Secondary antibody) 1:500 Invitrogen, cat. No. A-11001Anti-rabbit Alexa Fluor 594 (Secondary antibody) 1:500 Invitrogen, cat. No. A-11012
Fig. 3. Characterization of cell population dynamics of neurospheres during differentiation. Immunofluorescence microscopy of cryosections of neurospheres, scale bars –50 lm: day 7 of culture – nestin (red) and DAPI (blue) (A), Ki67 (green) and DAPI (blue) (B); day 21 of culture – GFAP (red), O4 (green), DAPI (blue) (C). Spinning disk confocalmicroscopy of whole neurospheres (day 21 of culture): b3-tubulin (green); DAPI (blue), maximum intensity z-projections of 40–80 optical sections of 0.8 lm (D and E).Confocal microscopy of cryosections of neurospheres (21 days of culture): b3-tubulin (green), maximum intensity z-projections of 30 optical sections of 0.5 lm; arrowsindicate b-tubulin-positive cell bodies in the inner part of the neurosphere (F); qRT-PCR analysis of PCNA (proliferating cell nuclear antigen) and TH (tyrosine hydroxylase)gene expression: fold increase in gene expression of neurospheres with 21 days of culture relatively to proliferative cells expanded in 2D; data are mean ± SEM of threeindependent cultures (G).
C. Brito et al. / Methods 56 (2012) 452–460 457

458 C. Brito et al. / Methods 56 (2012) 452–460
3. Results and discussion
3.1. 3D neurosphere differentiation
Typically, neurosphere aggregation occurred within 24 h afterinoculation. During the first 7 days of culture, neurosphereswere mainly progenitor cells positive for nestin, although cellsprogressively lost the proliferation marker ki67 (Fig. 3A and B).After 14 days in DM, nestin-positive cells were distributed inthe inner part of the aggregate together with glial fibrillaryacidic protein (GFAP)-positive cells, whereas cells immunoreac-tive for b3-tubulin (b3-tub) were concentrated at the surfaceof the differentiated neurosphere, where a few O4-positiveoligodendrocytes were also detected (Fig. 3C and D). Althoughb3-tub-positive neurons presented an intricate network of neu-rites that covered most of the neurosphere surface and spannedinto the core (Fig. 3E), b3-tub-positive cell bodies were also de-tected in the inner part of the aggregate (Fig. 3F, arrows). qRT-PCR analysis revealed that there was a pronounced decrease inthe expression of the proliferation marker PCNA (proliferatingcell nuclear antigen), reaching almost undetectable levels after2 weeks in DM (Fig. 3G), which suggests that most of the neuralprogenitors present in the neurospheres reached a final asym-
Fig. 4. Characterization of transduction of differentiated neurospheres with CAVGFP. Fltransduction time: 4 h, 5 days post-transduction (dpt) (A). qRT-PCR analysis of effectexpression of transduced differentiated neurospheres relatively to transduced differentithree independent cultures and transduction experiments. Asterisks indicate significant dhoc multiple comparison test (B). Spinning disk confocal microscopy analysis of transprojections of 180–240 optical sections of 0.35 lm (C); viability of transduced 3D differinto the DNA of cells with damaged cell membrane; GFP (green) (D).
metric division and that the culture remained morphologicallystable over time [1]. Most importantly, an increased expressionof the dopaminergic marker tyrosine hydroxylase (TH) (Fig. 3G)indicated that neurons derived from hmNPC acquired a dopami-nergic phenotype. When performing 3D neurosphere differentia-tion, we consistently obtained at least 3-fold increase in THexpression, quantified by qRT-PCR, compared to the standard2D differentiation protocol (data not shown).
These results showed that the protocol described herein was arobust methodology for the differentiation of neurospheres de-rived from hmNPC. A consistent differentiated phenotype was ob-tained within 3 weeks of 3D culture (2 weeks in differentiationmedia).
3.2. Transduction of differentiated neurospheres
Typically, when performing transduction of hmNPC differenti-ated in 2D with CAVGFP (MOI: 100 ip/cell, transduction time:4 h), we attained approximately 30% of GFP-positive cells(Fig. 4A). For the 3D culture system flow cytometry analysiswas not possible to perform due to the complex cell-cell interac-tions and dense networks of cell projections established whichpreclude an efficient dissociation of the differentiated neuro-
ow cytometry analysis of transduced 2D differentiated cultures – MOI 100 ip/cell,of MOI and transduction time on GFP expression at 5 dpt. Fold increase in gene
ated 2D cultures (MOI: 100 ip/cell; transduction time: 4 h); data are mean ± SEM ofifference (⁄P < 0.05; ⁄⁄⁄P < 0.001) by a one-way ANOVA analysis with a Tukey’s post
duced differentiated neurospheres at 5 dpt – GFP (green), maximum intensity z-entiated neurospheres at 5 dpt, assessed by propidium iodide (PI, red) intercalation
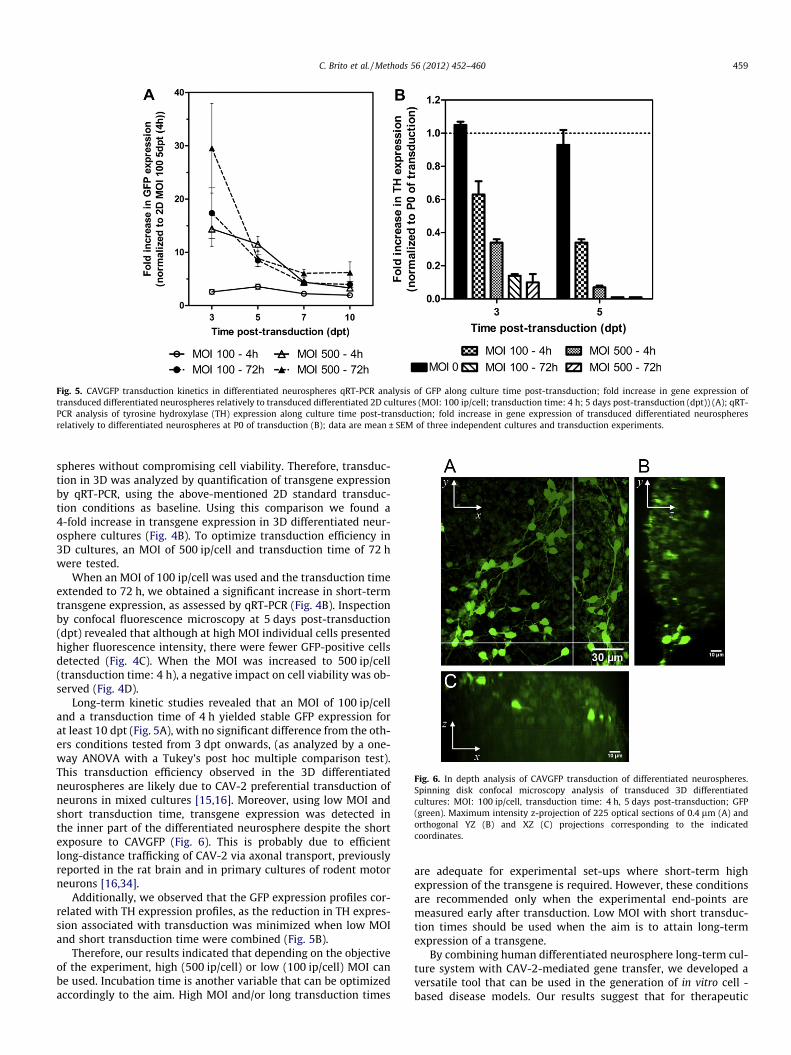
Fig. 5. CAVGFP transduction kinetics in differentiated neurospheres qRT-PCR analysis of GFP along culture time post-transduction; fold increase in gene expression oftransduced differentiated neurospheres relatively to transduced differentiated 2D cultures (MOI: 100 ip/cell; transduction time: 4 h; 5 days post-transduction (dpt)) (A); qRT-PCR analysis of tyrosine hydroxylase (TH) expression along culture time post-transduction; fold increase in gene expression of transduced differentiated neurospheresrelatively to differentiated neurospheres at P0 of transduction (B); data are mean ± SEM of three independent cultures and transduction experiments.
Fig. 6. In depth analysis of CAVGFP transduction of differentiated neurospheres.Spinning disk confocal microscopy analysis of transduced 3D differentiatedcultures: MOI: 100 ip/cell, transduction time: 4 h, 5 days post-transduction; GFP(green). Maximum intensity z-projection of 225 optical sections of 0.4 lm (A) andorthogonal YZ (B) and XZ (C) projections corresponding to the indicatedcoordinates.
C. Brito et al. / Methods 56 (2012) 452–460 459
spheres without compromising cell viability. Therefore, transduc-tion in 3D was analyzed by quantification of transgene expressionby qRT-PCR, using the above-mentioned 2D standard transduc-tion conditions as baseline. Using this comparison we found a4-fold increase in transgene expression in 3D differentiated neur-osphere cultures (Fig. 4B). To optimize transduction efficiency in3D cultures, an MOI of 500 ip/cell and transduction time of 72 hwere tested.
When an MOI of 100 ip/cell was used and the transduction timeextended to 72 h, we obtained a significant increase in short-termtransgene expression, as assessed by qRT-PCR (Fig. 4B). Inspectionby confocal fluorescence microscopy at 5 days post-transduction(dpt) revealed that although at high MOI individual cells presentedhigher fluorescence intensity, there were fewer GFP-positive cellsdetected (Fig. 4C). When the MOI was increased to 500 ip/cell(transduction time: 4 h), a negative impact on cell viability was ob-served (Fig. 4D).
Long-term kinetic studies revealed that an MOI of 100 ip/celland a transduction time of 4 h yielded stable GFP expression forat least 10 dpt (Fig. 5A), with no significant difference from the oth-ers conditions tested from 3 dpt onwards, (as analyzed by a one-way ANOVA with a Tukey’s post hoc multiple comparison test).This transduction efficiency observed in the 3D differentiatedneurospheres are likely due to CAV-2 preferential transduction ofneurons in mixed cultures [15,16]. Moreover, using low MOI andshort transduction time, transgene expression was detected inthe inner part of the differentiated neurosphere despite the shortexposure to CAVGFP (Fig. 6). This is probably due to efficientlong-distance trafficking of CAV-2 via axonal transport, previouslyreported in the rat brain and in primary cultures of rodent motorneurons [16,34].
Additionally, we observed that the GFP expression profiles cor-related with TH expression profiles, as the reduction in TH expres-sion associated with transduction was minimized when low MOIand short transduction time were combined (Fig. 5B).
Therefore, our results indicated that depending on the objectiveof the experiment, high (500 ip/cell) or low (100 ip/cell) MOI canbe used. Incubation time is another variable that can be optimizedaccordingly to the aim. High MOI and/or long transduction times
are adequate for experimental set-ups where short-term highexpression of the transgene is required. However, these conditionsare recommended only when the experimental end-points aremeasured early after transduction. Low MOI with short transduc-tion times should be used when the aim is to attain long-termexpression of a transgene.
By combining human differentiated neurosphere long-term cul-ture system with CAV-2-mediated gene transfer, we developed aversatile tool that can be used in the generation of in vitro cell -based disease models. Our results suggest that for therapeutic

460 C. Brito et al. / Methods 56 (2012) 452–460
applications, low CAV-2 dosage should probably be pursued toachieve therapeutic efficiency while minimizing inherent toxicitywith any vector platform and side effects such as loss of TH-posi-tive dopaminergic neurons.
4. Conclusions
Herein we describe a robust and amenable methodology for theproduction of human differentiated neurospheres, using stirredculture systems. This system allows culture and handling of differ-entiated neurospheres for long periods of time, including geneticmanipulation using viral vectors. The use of hmNPC, with a pro-pensity for dopaminergic differentiation, makes this culture sys-tem appropriate for PD modeling. Nevertheless, the protocol canbe easily adapted to other hNPC sources and specific differentiationprotocols.
The particular features of CAV-2 vectors, namely the preferen-tial tropism towards neurons and the efficient long-distance tar-geting were explored for the transduction of the differentiated3D model. Transduction efficiency was optimized to (1) allowshort-term expression of the transgene at very high levels (MOI:500 ip/cell; time of transduction: 4 h); (2) attain constant levelsof long-term expression of the transgene (MOI: 100 ip/cell; timeof transduction: 4 h).
The requirement for multi-gas cell incubators and the handlingof cells under low O2 conditions, as well as the requirement forstirring devices can be daunting for some laboratories. However,multiple solution for cell culture at low O2 concentrations arebecoming widely available, from multi-gas incubators to sealedchambers that can be placed inside an regular incubator or evencontrolled bioreactors or complete hypoxic workstations. Althoughsome of these options are quite expensive, a multi-gas incubatorwith an orbital stirring plate is relatively accessible. Cell handlingcan be performed in a regular laminar flow without major pertur-bation of oxygen tensions, as oxygen solubility in the culture med-ium is limited, taking several hours [35], whereas culturemanipulation outside the incubator requires only few minutes.
The 3D cell models generated herein have a wide range of appli-cations, from disease modeling and analysis of viral vector interac-tion with human neural cell populations, to efficacy studies ofspecific therapeutic genes for treatment of CNS pathologies andtoxicological studies. Furthermore these methodologies may be ex-tended to other sources of hNSC, such as hPS cells, including pa-tient-derived induced iPS cells, broadening even further theirapplicability.
Acknowledgments
We gratefully acknowledge Dr. Giampietro Schiavo, Cancer Re-search UK, for fruitful discussion and helpful advice. Rui Tostões isacknowledged for support on 3D culture systems and confocalmicroscopy and Ana Amaral for revision of the manuscript. Thiswork was supported by BrainCAV (FP7-222992), funded by theEU, and PTDC/EBB-BIO/112786/2009, funded by Fundação para a
Ciência e Tecnologia. Daniel Simão and Paulo Fernandes wererecipients of PhD fellowship from FCT, Portugal.
References
[1] L. Conti, E. Cattaneo, Nat. Rev. Neurosci. 11 (2010) 176–187.[2] P.H. Schwartz, D.J. Brick, A.E. Stover, J.F. Loring, F.J. Muller, Methods 45 (2008)
142–158.[3] S.C. Schwarz, J. Schwarz, Transl. Res. 156 (2010) 155–160.[4] B.A. Reynolds, R.L. Rietze, Nat. Methods 2 (2005) 333–336.[5] A. Sen, M.S. Kallos, L.A. Behie, Bioprocess Engineering of Neural Stem Cells,
Encyclopedia of Industrial Biotechnology, in: M.C. Flickinger (Ed.), John Wiley& Sons, 2010, ISBN: 9-471-79930-6.
[6] S. Ahmed, J. Cell. Biochem. 106 (2009) 1–6.[7] C.A. Rodrigues, T.G. Fernandes, M.M. Diogo, C.L. da Silva, J.M. Cabral,
Biotechnol. Adv. (2011).[8] E. Pastrana, V. Silva-Vargas, F. Doetsch, Cell Stem Cell 8 (2011) 486–498.[9] M. Sabolek, B. Baumann, M. Heinrich, A.K. Meyer, A. Herborg, S. Liebau, M.
Maisel, A. Hermann, K. Ventz, J. Schwarz, T. Wirth, A. Storch, Stem Cells 27(2009) 2009–2021.
[10] M.R. Costa, R. Jagasia, B. Berninger, in: L.C. Doering (Ed.), Protocols for NeuralCell Culture, Springer, 2009, pp. 29–49.
[11] R. Jandial, I. Singec, C.P. Ames, E.Y. Snyder, Mol. Ther. 16 (2008) 450–457.[12] M.T. Dieterlen, F. Wegner, S.C. Schwarz, J. Milosevic, B. Schneider, M. Busch, U.
Romuss, A. Brandt, A. Storch, J. Schwarz, J. Neurosci. Methods 178 (2009) 15–23.
[13] C.M. Bertram, S.M. Hawes, S. Egli, S.L. Peh, M. Dottori, U.R. Kees, P.B. Dallas,Stem Cells Dev. 19 (2010) 569–578.
[14] P. Alberts, R. Rudge, I. Hinners, A. Muzerelle, S. Martinez-Arca, T. Irinopoulou,V. Marthiens, S. Tooze, F. Rathjen, P. Gaspar, T. Galli, Mol. Biol. Cell 14 (2003)4207–4220.
[15] C. Soudais, N. Skander, E.J. Kremer, FASEB J. 18 (2004) 391–393.[16] T. Bru, S. Salinas, E.J. Kremer, Viruses 2 (2010) 2134–2153.[17] A. Storch, G. Paul, M. Csete, B.O. Boehm, P.M. Carvey, A. Kupsch, J. Schwarz,
Exp. Neurol. 170 (2001) 317–325.[18] G. Schaarschmidt, F. Wegner, S.C. Schwarz, H. Schmidt, J. Schwarz, PLoS ONE 4
(2009) e6168.[19] J. Milosevic, A. Brandt, U. Roemuss, A. Arnold, F. Wegner, S.C. Schwarz, A.
Storch, H. Zimmermann, J. Schwarz, J. Neurochem. 99 (2006) 913–923.[20] J. Milosevic, M. Maisel, F. Wegner, J. Leuchtenberger, R.H. Wenger, M. Gerlach,
A. Storch, J. Schwarz, J. Neurosci. 27 (2007) 412–421.[21] E.J. Kremer, S. Boutin, M. Chillon, O. Danos, J. Virol. 74 (2000) 505–512.[22] C. Soudais, C. Laplace-Builhe, K. Kissa, E.J. Kremer, FASEB J. 15 (2001) 2283–
2285.[23] P. Fernandes, V. Santiago, N. Viana, E.J. Kremer, A.S. Coroadinha, P.M. Alves,
N. Jenkins, N. Barron, P. Alves, vol. 5, Springer, Netherlands, 2012, pp. 671–674.
[24] T.B. Ferreira, R. Perdigao, A.C. Silva, C. Zhang, J.G. Aunins, M.J. Carrondo, P.M.Alves, Biotechnol. Prog. 25 (2009) 235–243.
[25] M. Serra, C. Correia, R. Malpique, C. Brito, J. Jensen, P. Bjorquist, M.J. Carrondo,P.M. Alves, PLoS ONE 6 (2011) e23212.
[26] S. Sá Santos, L.L. Fonseca, M.A. Monteiro, M.J. Carrondo, P.M. Alves, J. Neurosci.Res. 79 (2005) 26–32.
[27] K.J. Livak, T.D. Schmittgen, Methods 25 (2001) 402–408.[28] G.A. Jacqueline, T. Kasia, P.K.K. Laura, D.C. Laurie, S. Shelley, Protoc. Exch.
(2006).[29] R. Malpique, L.M. Osorio, D.S. Ferreira, F. Ehrhart, C. Brito, H. Zimmermann,
P.M. Alves, Tissue Eng. C Methods 16 (2010) 965–977.[30] M. Serra, C. Brito, E.M. Costa, M.F. Sousa, P.M. Alves, BMC Biotechnol. 9 (2009)
82.[31] M. Serra, C. Brito, S.B. Leite, E. Gorjup, H. von Briesen, M.J. Carrondo, P.M. Alves,
Ann. Anat. 191 (2009) 104–115.[32] J.B. Pawley, J.B. Pawley, Springer, US, 2006.[33] M. Serra, C. Brito, M.F. Sousa, J. Jensen, R. Tostoes, J. Clemente, R. Strehl, J.
Hyllner, M.J. Carrondo, P.M. Alves, J. Biotechnol. 148 (2010) 208–215.[34] S. Salinas, L.G. Bilsland, D. Henaff, A.E. Weston, A. Keriel, G. Schiavo, E.J.
Kremer, PLoS Pathog. 5 (2009) e1000442.[35] D. Wion, T. Christen, E.L. Barbier, J.A. Coles, Cell Stem Cell 5 (2009) 242–
243.





























