Embed Size (px)
DESCRIPTION
Gene Therapy & Molecular Biology Volume 13 Issue A
Citation preview

GENE THERAPY
&
MOLECULAR BIOLOGY
Volume 13
Number 1
June 2009
Published by Gene Therapy Press
ISSN 1529-9120

Instructions to authors:
Gene Therapy and Molecular Biology (GTMB) OPEN ACCESS www.gtmb.org
Scope
Gene Therapy and Molecular Biology, bridging various fields is one of the most rapid with free access
at gtmb.org.
The scope of Gene Therapy and Molecular Biology is to promote interaction between researchers in
the fields of Gene Therapy and Molecular Biology providing rapid publication of review articles and
research papers. Articles (both invited and submitted) review or report novel findings of importance to
a general audience in gene therapy, molecular medicine, gene discovery, and molecular biology with
emphasis to molecular mechanisms. The journal will accept papers on all aspects of gene therapy,
including gene delivery systems, gene therapy of cancer and other diseases (e.g. CFTR, hemophilia,
AIDS, restenosis) at the clinical, preclinical or cell culture stage, gene discovery, cancer
immunotherapy, DNA vaccines, use of DNA regulatory elements in gene transfer, cell therapy and
transplantation, arraying technologies & DNA chips, peptide libraries and drug discovery related to
gene therapy, cell targeting, gene targeting, therapy with oligonucleotides (antisense, ribozymes,
triplex). The authors are encouraged to elaborate on the molecular mechanisms that govern a gene
therapy approach. Gene Therapy and Molecular Biology will also publish articles on, transcription
factors, DNA replication, recombination, repair, chromatin, nuclear matrix, DNA regulatory regions,
locus control regions, protein phosphorylation, signal transduction, development, and on molecular
mechanism of human disease. To make the publication attractive authors are encouraged to
include color figures.
Type of articles
Both review articles and original research articles will be considered. In addition, short 1-2 page news
& views will also be considered for publication. Original research articles should contain a generous
introduction in addition to experimental data. The articles contain information important to a general
audience as the volume is also addressed to researches outside the field. There is no limit on the length
of the articles provided that the subject is interesting to a general audience and covers exhaustively a
field. The typical length of each manuscript is a approximately 4-20 printed page including Figures
and Tables. This is 12-60 manuscript pages.
Charges, Complimentary reprints & Subscriptions
There are no charges for color figures or page numbers. Corresponding authors get a one-year free
subscription (hard copy) plus 25 reprints free of charge. The free subscription can be renewed for
additional years by having one paper per year accepted for publication.
The free electronic access to articles published in " Gene Therapy and Molecular Biology " to a big
general audience, the attractive journal title, the speed of the reviewing process, the no-charges for
page numbers or color figure reproduction, the 25 complimentary reprints, the rapid electronic
publication, the embracing of many fields in gene therapy (from molecular mechanisms to clinical
trials), the high quality in depth reviews and first rate research articles and most important, the
eminent members of the Editorial Board being assembled are prognostic factors of a big success for
GTMB.

Sections of the manuscript
Each manuscript should have a Title, Authors, Affiliation, Corresponding Author (with Tel, Fax, and
E-mail), Summary, key words , running title and Introduction; review articles are subdivided into
headings I, II, III, etc. (starting with I. Introduction) subdivided into A, B, C, and further subdivided
using 1, 2, 3, etc. You can further subdivide into 1, 2, 3, etc. Research articles are divided into
Summary; I. Introduction; II. Materials and Methods III. Results; IV. Discussion; Acknowledgments;
and References. Please include in your text citations the name of authors and year in parenthesis; for
three or more authors use: (name of first author et al, with year); for two authors please use both
names. Please delete hidden text for references. In the reference list, please, type references with year
and Journal in boldface and provide full title of the article such as:
Buschle M, Schmidt W, Berger M, Schaffner G, Kurzbauer R, Killisch I, Tiedemann J-K, Trska B,
Kirlappos H, Mechtler K, Schilcher F, Gabler C, and Birnstiel ML (1998) Chemically defined, cell-
free cancer vaccines: use of tumor antigen-derived peptides or polyepitope proteins for vaccination.
Gene Ther Mol Biol 1, 309-321.
To avoid delays it is essential to submit an electronic and a hard copy version of your manuscript via
e-mail and mail in a floppy, CD-ROM or ZIP, containing the manuscript that will be used to typeset
the paper. Please include in the digital media: Tables, if any, (preferably as a Microsoft Word text) and
Figure legends. Please use Microsoft Word, font “Times” (Mac users) or “Times New Roman” (PC
users) and insert Greek or other characters using the “Insert/Symbol” function in the Microsoft Word
rather than simple conversion to font “Symbol”. Please boldface Figure 1, 2, 3 etc. as well as Table 1,
2, etc. throughout the text. Please provide the highest quality of prints of your Figures; whenever
possible, please provide in addition an electronic version of your figures.
Article contributors are kindly requested to provide a color (or black/white) photo of themselves
(preferably 4x5 cm or any size) or a group photo of the authors, as we shall include these in the
publication
Submission and reviewing
Peer reviewing is by members of the Editorial Board and external referees. Please suggest 2-3
reviewers providing their electronic addresses, mailing addresses and telephone/fax numbers. Authors
are sent page proofs.
Gene Therapy and Molecular Biology is published in on high quality paper, hardbound, and with
excellent reproduction of color figures.
Reviewing is completed within 5-15 days from receiving the manuscript.
Articles accepted without revisions (i.e., review articles) will be published online (www.gtmb.org) in
approximately 1 month following submission.
Please submit an electronic version of full text and figures preferably in jpeg format. The electronic
version of the figures will be used for the rapid reviewing process. High quality prints or photograph
of the figures and the original with one copy should be sent via express mail to the Editorial Office.
Citation in MedLine
Articles accepted for publication by GTMB or Cancer Therapy can be included in MedLine (PubMed)
as full articles upon the request of authors provided that the authors have completed their published
work under a government grant by NIH (or EU/Japan government grant). If this is you case, please
consult the NIH Manuscript Submission System http://www.nihms.nih.gov/.

Editorial Office
Teni Boulikas, Ph.D./ Maria Koutoudi, B.A. , M.A.
Gregoriou Afxentiou 7
Alimos, Athens 17455
Greece
Tel: +30-210-985-8454
Fax: +30-210-985-8453
and electronically to
The free electronic access to articles published in "GTMB" to a big general audience, the attractive
journal title, the speed of the reviewing process, the no-charges for page numbers or color figure
reproduction, the 25 complimentary reprints, the rapid electronic publication, the embracing of many
fields in cancer, the anticipated high quality in depth reviews and first rate research articles and most
important, the eminent members of the Editorial Board being assembled are prognostic factors of a big
success for the newly established journal.

Gene Therapy and Molecular Biology (GTMB) is
covered in the following Thomson Scientific
services:
! Science Citation Index Expanded (also known as
SciSearch")
! Biotechnology Citation Index"
Journals Citation Reports/Science Edition

Gene Therapy & Molecular Biology is acknowledged by the National
Library of Medicine
http://www.ncbi.nlm.nih.gov/sites/entrez search field: Gene ther mol biol
search: journals http://www.ncbi.nlm.nih.gov/sites/entrez?Db=nlmcatalog&doptcmdl=Expanded&cmd=search&Term=9815849[NlmId]

Table of contents
Gene Therapy and Molecular Biology
Vol 13 Number 1, June 2009
Pages Type of
Article Article title Authors (corresponding author is in
boldface)
1-9 Review
Article
New trends in aptamer-based
electrochemical biosensors
Maria N. Velasco-Garcia, Sotiris
Missailidis
10-14
Research
Article
Mapping of MHC class binding
nonamers from lipid binding protein of
Ascaridia galli
Virendra S Gomase, Somnath B
Waghmare, Baba Jadhav, Karbhari V
Kale
15-19 Review
Article
Perspectives in vector development for
systemic cancer gene therapy
Arash Hatefi, Brenda F. Canine
20-25 Research
Article
Curcumin is not a ligand for
peroxisome proliferator-activated
receptor-!
Venkata R. Narala, Monica R. Smith,
Ravi K. Adapala, Rajesh Ranga, Kalpana
Panati, Bethany B. Moore, Todd Leff,
Vudem D. Reddy, Anand K. Kondapi,
Raju C. Reddy
26-35 Review
Article
FAK as a target for cancer therapy
Steven N. Hochwald, Vita M.
Golubovskaya
36-52 Review
Article
Combination of immunotherapy with
anaerobic bacteria for immunogene
therapy of solid tumours
Jian Xu, Xiao Song Liu, Shu-Feng
Zhou, Ming Q Wei
53-63 Review
Article
Non-viral and local gene medicine for
improvement of cutaneous wound
healing
Markus Rimann, Heike Hall

GENE THERAPY & MOLECULAR BIOLOGY Addresses of Members of the Editorial Board
OPEN ACCESS www.gtmb.org
Missailidis, Sotiris, DPhil
(York)
Lecturer in Chemistry and
Analytical Sciences, The Open
University, UK
Roberts, Michael, Ph.D.,
Regulon A.E., Athens Greece
Magos, Alexandros D. Ph.D.
Chemist, Nanotechnology
Formulations
Regulon A.E., Athens Greece
Rossi, John, Ph.D., Beckman
Research Institute of the City of
Hope, USA
Crooke, Stanley, M.D., Ph.D.,
ISIS Pharmaceuticals, Inc,
USA
Shen, James, Ph.D., Institute of
Molecular Biology, Academia
Sinica, Taipei, Taiwan, Republic
of China & University of
California at Davis, USA.
Gronemeyer, Hinrich, Ph.D.
I.N.S.E.R.M., IGBMC, France
Webb, David, Ph.D., Celgene
Corporation, USA
Aguilar-Cordova, Estuardo,
Ph.D., AdvantaGene, Inc.,
USA
Berezney, Ronald, Ph.D., State
University of New York at
Buffalo, USA
Editor
Editor Assistants
Boulikas, Teni,
Ph.D.
Chairman of the
Board, Regulon, Inc.
Mt View CA 94043
and Regulon AE,
Athens, Greece
Koutoudi, Maria M.A.
Vougiouka, Maria, B.Sc.
Kruit, Adrian, Ph.D.
Bellimezi, M., Ph.D
Katsoupi J, Mph,
Tsogas I., Ph.D,
Magkos, A., Ph.D,
Christofis Petros., Ph.D,
Leto Tziveleka., Ph.D
Associate Editors

Editorial Board Members
Akporiaye, Emmanuel,
Ph.D., Arizona Cancer Center,
USA
Baldwin, H. Scott, M.D
Vanderbilt University Medical
Center, USA
Amiji, Mansoor M. Ph.D.,
Professor of Pharmaceutical
Sciences
Northeastern University,
Boston, MA
Anson, Donald S., Ph.D.,
Women's and Children's
Hospital, Australia
Barranger, John, MD, Ph.D.,
University of Pittsburgh, USA
Ariga, Hiroyoshi, Ph.D.,
Hokkaido University, Japan
Black, Keith L. M.D., Maxine
Dunitz Neurosurgical Institute,
Cedars-Sinai Medical Center,
USA
Blum, Kenneth, Ph.D., Wake
Forest University School of
Medicine, USA
Eckstein, Jens W., Ph.D.,
Akikoa Pharmaceuticals Inc,
USA
Bode, Jürgen, Gesellschaft für
Biotechnologische Forschung
m.b.H., Germany
Fisher, Paul A. Ph.D., State
University of New York, USA
Bohn, Martha C., Ph.D., The
Feinberg School of Medicine,
Northwestern University, USA
Georgiev, Georgii, Ph.D.,
Russian Academy of Sciences,
USA
Bresnick, Emery, Ph.D.,
University of Wisconsin
Medical School, USA
Getzenberg, Robert, Ph.D.,
Institute Shadyside Medical
Center, USA
Caiafa, Paola, Ph.D.,
Università di Roma “La
Sapienza”, Italy
Ghosh, Sankar Ph.D., Yale
University School of Medicine,
USA

Cheng, Seng H. Ph.D.,
Genzyme Corporation, USA
Gojobori, Takashi, Ph.D.,
Center for Information Biology,
National Institute of Genetics,
Japan
Cole, David J. M.D., Medical
University of South Carolina,
USA
Harris David T., Ph.D., Cord
Blood Bank, University of
Arizona, USA
Crooke, Stanley, M.D.,
Ph.D. ISIS
Pharmaceuticals, Inc. USA
Heldin, Paraskevi Ph.D.,
Uppsala Universitet, Sweden
Davie, James R, Ph.D.,
Manitoba Institute of Cell
Biology, USA
Hesdorffer, Charles S., M.D.,
Columbia University, USA
DePamphilis, Melvin L,
Ph.D., National Institute of
Child Health and Human,
National Institutes of Health,
USA
Hoekstra, Merl F, Ph.D.,
Epoch Biosciences, Inc., USA
Hung, Mien-Chie, Ph.D., The
University of Texas, USA
Kuroki, Masahide, M.D.,
Ph.D., Fukuoka University
School of Medicine, Japan
Johnston, Brian, Ph.D.,
Somagenics, Inc, USA
Lai, Mei T. Ph.D., Lilly
Research Laboratories USA
Jolly, Douglas J, Ph.D.,
Advantagene, Inc.,USA
Latchman, David S., PhD,
Dsc, MRCPath
University of London, UK
Joshi, Sadhna, Ph.D., D.Sc.,
University of Toronto
Canada
Lavin, Martin F, Ph.D., The
Queensland Cancer Fund
Research Unit, The Queensland
Institute of Medical Research,
Australia
Kiyama, Ryoiti, Ph.D.,
National Institute of
Bioscience and Human-
Technology, Japan
Lebkowski, Jane S., Ph.D.,
GERON Corporation, USA

Kotoku Kurachi, Ph.D.,
University of Michigan
Medical School, USA
Li, Liangping Ph.D., Max-
Delbrück-Center for Molecular
Medicine, Germany
Kottaridis, Stavros D.,
Ph.D. Regulon Inc. USA
Lu, Yi, Ph.D., University of
Tennessee Health Science
Center, USA
Krawetz, Stephen A., Ph.D.,
Wayne State University School
of Medicine. USA
Lundstrom Kenneth, Ph.D.,
Bioxtal/Regulon, Inc.
Switzerland
Kruse, Carol A., Ph.D.,
Sidney Kimmel Cancer
Center. USA
MacDougald, Ormond A,
Ph.D., University of Michigan
Medical School, USA
Kuo, Tien, Ph.D., The
University of Texas M. D.
Anderson Cancer USA
Mirkin, Sergei, M. Ph.D.,
University of Illinois at
Chicago, USA
Malone, Robert W., M.D.,
Aeras Global TB Vaccine
Foundation, USA
Noteborn, Mathieu, Ph.D.,
Leiden University, The
Netherlands
Royer, Hans-Dieter, M.D.,
(CAESAR), Germany
Paleos, Constantinos M.,
Ph.D.
Institute of Physical
Chemistry Demokritos.
Greece
Rubin, Joseph, M.D., Mayo
Medical School
Mayo Clinic, USA
Perry, George , Ph.D.
Dean and Professor
College of Sciences
University of Texas at San
Antonio
Saenko Evgueni L., Ph.D.,
University of Maryland School
of Medicine Center for
Vascular and Inflammatory
Diseases, USA
Pomerantz, Roger, J., M.D.,
Tibotec, Inc., USA
Santoro, M. Gabriella, Ph.D.,
University of Rome Tor
Vergata, Italy

Raizada, Mohan K., Ph.D.,
University of Florida, USA
Salmons, Brian, Ph.D., (FSG-
Biotechnologie GmbH), Austria
Razin, Sergey, Ph.D., Institute
of Gene Biology
Russian Academy of Sciences,
USA
Sharrocks, Andrew, D.,
Ph.D., University of
Manchester, UK
Robbins, Paul, D, Ph.D.,
University of Pittsburgh, USA
Smythe Roy W., M.D., Texas
A&M University Health
Sciences Center, USA
Rosenblatt, Joseph, D., M.D,
University of Miami School of
Medicine, USA
Srivastava, Arun Ph.D.,
University of Florida College of
Medicine, USA
Rosner, Marsha, R., Ph.D.,
Ben May Institute for Cancer
Research, University of
Chicago, USA
Steiner, Mitchell, M.D.,
University of Tennessee, USA
Tainsky, Michael A., Ph.D.,
Karmanos Cancer Institute,
Wayne State University, USA
White, Robert, J., University
of Glasgow, UK
Taira, Kazunari, Ph.D., The
University of Tokyo, Japan
White-Scharf, Mary, Ph.D.,
Biotransplant, Inc., USA
Thierry, Alain, Ph.D.,
National Cancer Institute,
National Institutes of Health,
France
Wiginton, Dan, A., Ph.D.,
Children's Hospital Research
Foundation, CHRF , USA
Trifonov, Edward, N. Ph.D.,
University of Haifa, Israel
Yung, Alfred, M.D.,
University of Texas, USA

Van Dyke, Michael, W.,
Ph.D., The University of Texas
M. D. Anderson Cancer
Center, USA
Zannis-Hadjopoulos, Maria
Ph.D., McGill Cancer Centre,
Canada
Vournakis, John N., Ph.D.
Medical University of
South Carolina, USA
Zorbas, Haralabos, Ph.D.,
BioM AG Team, Germany
Chi-Un Pae, MD, PhD,
Associate Professor,
Department of Psychiatry
The Catholic Universoty
of Korea College of
Medicine
Sikorska, Marianna Ph.D.
Neurogenesis and Brain Repair,
Institute for Biological
Sciences, National Research
Council Canada,
Ottawa, Ontario, Canada
Associate Board Members
Falasca, Marco, M.D.,
University College
London, UK
Hiroki, Maruyama, M.D.,
Ph.D., Niigata University
Graduate School of Medical
and Dental Sciences, Japan
Gao, Shou-Jiang, Ph.D.,
The University of Texas
Health Science Center at
San Antonio, USA
Kazunori, Aoki, M.D., Ph.D.,
National Cancer Center
Research Institute, Japan
Gibson, Spencer Bruce,
Ph.D., University of
Manitoba, USA
Rigoutsos, Isidore, Ph.D.,
Thomas J. Watson Research
Center, USA
Gu, Baohua, Ph.D., The
Jefferson Center, USA
Priya, Aggarwal Ph.D.,
University of Pennsylvania
Morris, Kevin Vance, Assistant Professor,
The Scripps Research
Institute, La Jolla, CA
W. Todd Penberthy, PH.D.,
Assistant Professor,
Department of Molecular
Genetics,Biochemistry, and
Microbiology,

Romano, Gaetano
Ph.D.
Research Associate
Professor; Temple
University,
Philadelphia, U.S.A.
Yuefei Yu Ph.D.
Texas Tech University Health
Science Center. Research
Scientist. Head of the research
group.
Hongying Hao
M.D./Ph.D.,
Instructor, Department
of Surgery
School of Medicine
University of
Louisville
U.S.A.
Robert Harrod, Ph.D.
Associate Professor
Department of Biological
Sciences
Southern Methodist
University
Dallas
Prof. Emo Chiellini
Department of
Chemistry & Industrial
Chemistry
University of Pisa
Pisa (Italy)
Chittaranjan Patra
Assistant Professor,
Department of Biochemistry
and Molecular Biology,
Mayo Clinic Cancer Center,
Rochester, MN, USA.
Natesan Pushparaj,
Peter, Ph.D Research Scientist
Glasgow Biomedical
Research Centre,
University of Glasgow
Raju Reddy, M.D.
Assistant Professor of
Medicine
University of Michigan,
Ann Arbor
Hossam M Ashour,
Ph.D
Department of
Microbiology and
Immunology
Faculty of Pharmacy
Cairo University, Egypt
Arash Hatefi (Ph.D.,
Pharm.D.)
Assistant Professor
Department of Pharmaceutical
Sciences, Center for Integrated
Biotechnology,
Washington State
University
Selvarangan
Ponnazhagan, Ph.D.
Professor Department
of Pathology
The University of
Alabama at
Birmingham
Ekaterina Breous, Ph.D
Postdoctoral fellow, University
of Pennsylvania,
Philadelphia, USA

Gene Therapy and Molecular Biology Vol 13, page 1
1
Gene Ther Mol Biol Vol 13, 1-9, 2009
New trends in aptamer-based electrochemical
biosensors Review Article
Maria N. Velasco-Garcia*, Sotiris Missailidis Department of Chemistry and Analytical Sciences, Faculty of Science, The Open University, Walton Hall, Milton Keynes,
United Kingdom, MK7 6AA
__________________________________________________________________________________
*Correspondence: Maria N. Velasco-Garcia, Department of Chemistry and Analytical Sciences, Faculty of Science, The Open
University, Walton Hall, Milton Keynes, United Kingdom, MK7 6AA; e-mail: [email protected]
Sotiris Missailidis, Department of Chemistry and Analytical Sciences, Faculty of Science, The Open University, Walton Hall, Milton
Keynes, United Kingdom, MK7 6AA; e-mail: [email protected]
Key words: Aptamer, Biosensor, Aptasensor, Electrochemical detection, SELEX
Abbreviations: Platelet-derived growth factor BB (PDGF-BB); reverse-transcription PCR (RT PCR); self-assembled monolayers
(SAMs); Systematic Evolution of Ligands by EXponential enrichment, (SELEX)
Received: 28 January 2009; Revised: 6 February 2009
Accepted: 6 February 2009; electronically published: 8 February 2009
Summary The analytical characteristics of aptamers are comparable with those of antibodies for the development of biosensor
technology. However, aptamers offer some crucial advantages over antibodies such as selection capability for a
variety of targets, easy synthesis, improved reproducibility and stability, simple modification for immobilization to
solid supports and enhanced selectivity. This article reviews aptamer technology as well as aptamer-based assay
configurations and goes on to explore reported applications in electrochemical aptasensors.
I. Introduction Biosensor technology holds a great promise for the
healthcare market, the security sector, the food industry,
environmental and veterinary diagnostic; harnessing the
specificity and sensitivity of biological-based assays
packaged into portable and low cost devices which allow
for rapid analysis of complex samples in out-of-laboratory
environments. However the application of biosensors lags
far behind the fundamental research; the challenges facing
this basic technology are associated with sensitive
detection of specific molecules in samples, stability issues,
quality assurance, instrumentation design and cost
considerations (Velasco-Garcia and Tottram, 2003).
The main biological sensing materials used in
biosensor development are the couples enzyme/substrate
and antibody/antigen. These are limited by temperature,
sensitivity, stability, batch-to-batch variation, large size
and difficulty in production. Recent advances and
developments in the aptamer area offer a powerful
alternative approach involving the use of small RNA or
DNA molecules that bind to specific targets with very high
affinity and specificity. Aptamer receptors are a novel
entity of undeniable potential in analytical applications
and can complement or substitute antibodies or offer
applications where the later are not compatible (Tombelli
et al, 2005, 2007).
Despite the fact that development of aptasensors has
been boosted by using optical and acoustic transducers,
this review summarizes the recent developments in the
design of electrochemical aptamer-based affinity sensors.
In comparison with other detection systems, the
electrochemical detection combines a high sensitivity,
direct electronic signal production, fast response,
robustness, low cost, the possibility of miniaturization and
simultaneous multianalyte detection.
II. Aptamers As aptamers approach 20 years since they were
originally described (Ellington and Szostak, 1990; Tuerk
and Gold, 1990), they are currently receiving a wider
recognition in the literature as research reagents,
inhibitors, imaging or diagnostic agents (Luzi et al, 2003;
Hamula et al, 2006). Aptamers are short, single stranded
oligonucleotides, which inherently adopt stable three
dimensional sequence-dependent structures. This intrinsic
property makes them efficient binding molecules, capable
of binding to an array of molecular targets ranging from
small ions and organic molecules to large glycoproteins

Velasco-Garcia and Missailidis: New trends in aptamer-based electrochemical biosensors
2
and mucins (Ferreira et al, 2006). Aptamers are a novel
and particularly interesting targeting modality, with the
ability to bind to a variety of targets including proteins,
peptides, enzymes, antibodies and cell surface receptors,
as well as small molecules ranging from glucose and
caffeine, to steroids to TNT. Aptamers are single stranded
oligonucleotides that vary in size between 25-90 bases
long and adopt complex secondary and tertiary structures,
which facilitate specific interactions with other molecules.
They are derived from vast combinatorial libraries through
selective targeting and competitive binding. There are two
different configurations of aptamers: (i) linear and (ii)
molecular beacon. Aptamers with a linear configuration
maintain in certain physicochemical conditions a typical 3-
D conformation with specific binding sites for the target
molecule. On the other hand aptamers with a molecular
beacon configuration initially form a loop that changes
conformation following binding to the analyte of interest.
Aptamers offer unique benefits compared to other
targeting agents; not only they bind specific ligands with
high affinity and selectivity, but aptamers can be easily
selected using in vitro techniques and are chemically
synthesized, overcoming the use of animal for their
production. In comparison to antibodies, aptamers are
purified to a very high degree of purity, which eliminates
the batch-to-batch variation found in antibodies. Aptamers
have higher temperature stability (stable at room
temperature) and because of their small size, denser
receptor layers could be generated. The animal-free
production of aptamers is especially advantageous in cases
where the immune response can fail when the target
molecule (e.g. a protein) has a structure similar to
endogenous proteins or when the antigen consists of toxic
or non-immunogenic compounds. Aptamers are relatively
stable under a wide range of buffer conditions and
resistant to chemical degradation, although, due to their
DNA or RNA constitution, they are sensitive to hydrolytic
digestion by nucleases. Aptamers have been modified into
nuclease-resistant moieties by modification of the ribose
ring at the 2’-position or by the specific modification of
the pyrimidine nucleotide (Pieken et al, 1991; Heidenreich
and Eckstein, 1992; Kusser, 2000). It is also possible to
chemically modify aptamers to facilitate covalent
conjugation to reporters and nanoparticles with 5’ or 3’
amino, biotin or thiol groups. These characteristics make
them extremely attractive as alternatives to antibodies and
peptides for use in assays, or as diagnostic agents.
A. The SELEX process Aptamers are typically isolated from combinatorial
libraries by a process of in vitro evolution, termed SELEX
(Systematic Evolution of Ligands by EXponential
enrichment). This procedure is an in vitro evolutionary
selection process that allows the isolation of aptamer(s),
with unique binding properties, from a large library of
oligonucleotides through iterative cycles of (i) interaction
of a large library of aptamers with the target molecule, (ii)
separation of bound from unbound aptamer species, (iii)
elution of bound aptamers and (iv) PCR amplification of
the binding aptamers for further selection rounds (Figure
1 for an example of the process).
An aptamer library usually consists of a variable
region (20-40 nucleotides) flanked by known primer
sequences on either end for the amplification during the
SELEX procedure. The variable region makes up to 1015
different sequences which, combined with the innate
ability of oligonucleotides to form stable sequence-
Figure 1. The SELEX process

Gene Therapy and Molecular Biology Vol 13, page 3
3
dependent structures, provide an array of molecular
shapes available for the selection process (Khan and
Missailidis, 2008). In the selection steps, the library is
incubated with the immobilised target. Unbound or weak-
binding species are removed and bound aptamers are
eluted using high salt, temperature, chaotropic agents or
other such conditions that would affect molecular structure
or disrupt molecular interactions. Eluted aptamers are
subsequently amplified by PCR (DNA) or reverse-
transcription PCR (RT PCR) using primers
complementary to the flanking sequences in the aptamer
library. The enriched pool of binding species forms the
pool for the next round of selection. Repeated selection
and amplification steps allow identification of the highest
binding species, through competitive binding. The
selection and amplification step constitutes one round or
cycle in a typical SELEX procedure, with anything
between 1 and 15 cycles often described in the literature.
Counter- or negative selection steps can ensure that the
finally selected aptamers are very specific for their target
and do not interact with homologous proteins or
chemically closely-related molecular targets (Missailidis,
2008).
Selected aptamers are subsequently cloned and
sequenced to identify the sequence of the binding species
and their interactions are usually characterised by a variety
of analytical methodologies, prior to move into the various
applications they were originally destined for. Selected
aptamer can be easily produced by solid phase synthesis
and appropriate modifications can be introduced at this
stage to confer additional properties to the selected
aptamers, such as nuclease resistance (Figure 2), cross-
linking ability or improved pharmacokinetic properties.
Although SELEX has been the initial methodology
associated with aptamer selection and has remained a
robust and powerful technique, which has been adapted to
various systems and targets, a number of other
methodologies have also emerged for the selection of
aptamers. Such “non-SELEX” based methods for the
selection of aptamers include capillary electrophoresis
methodologies (Berezovski et al, 2005; Drabovich et al,
2005), isolation of aptamers with predefined kinetic and
thermodynamic properties of their interaction with the
target, without the need for amplification, allowing the use
of libraries which are difficult or cannot be amplified, or
computational methods, which are particularly important
in selecting aptamers with inhibitory activities or
sequences that undergo ligand dependent conformational
changes (Ikebukuro et al, 2005).
The SELEX procedure and subsequent technologies for
aptamer selection have offered the tools for the designing
of aptamers that have found a range of diagnostic
applications (Khan and Missailidis, 2008). Such
applications include Photo-SELEX (www.somalogic.com)
and SELEX NADIR (Winters-Hilt, 2006) using optical
probe reporting or nanopore reporting mechanisms
respectively, aptamer microarrays (Cho et al., 2005),
currently in the market by LC Sciences
(www.lcsciences.com), fluorescent aptamers in chips and
microspheres (Kirby et al, 2004; Potyrailo et al., 1998),
fluorescent sensors for small molecule recognition (Ozaki
et al, 2006; Yamana et al, 2003), quantum dots (Liu et al,
2007; Levy et al, 2005; Choi et al, 2006; Ivanovic et al,
2007), colorimetric detection (Liu and Lu, 2004; Cho et al,
2006; Liu and Lu, 2006), electrochemical detection (Lai et
al, 2007; Xiao et al, 2005; Papamichael et al, 2007; Mir et
al, 2006) and piezoelectric quartz crystal sensors (Bini et
al, 2007).
The above methods, fluorescent, electrochemical and
colorimetric detection, have also been used in molecular
switch type sensors or modular sensor assemblies, where
the aptamers usually change conformation upon binding to
either emit a fluorescent signal based on an aptamer
beacon on sensor, or through non-covalent interaction with
the fluorescent label, triggering an electrochemical sensor
or leading to change of colour (Stojanovic and
Kolpashchikov, 2004; Stojanovic et al, 2001; Baker et al,
2006; Zuo et al, 2007; Stojanovic and Landry, 2002;
Frauendorf and Jaschke, 2001), with particular
sensitivities in the recognition of small analytes.
Aptamers have also been used in enzymatic sensing,
without the use of any label or signal related directly to the
aptamer. These applications remain based on changes in
the conformation of bifunctional aptamers that recognise
the target ligand and an enzyme or ribosome. The binding
of the aptamer to the ligand results in conformational
changes that affect enzymatic activity or protein
expression, and it is the later that is subsequently
measured (Ogawa and Maeda, 2007; Yoshida et al, 2006;
Yoshida et al, 2006) or utilises an enzyme to ligate
proximally bound aptamers to large protein targets and
allow their subsequent PCR amplification (Fredriksson et
al, 2002).
III. Aptamer immobilisation Aptamers can certainly be used as molecular
recognition elements in affinity sensing. The small size of
aptamers provides advantages over antibodies: (i) a greater
Figure 2. An amino or fluoro modification at the 2’ position of
the sugar can confer the oligonucleotide aptamer stability against
nuclease degradation. An alternative to using modifications at the
2’ of the sugar (whether at the 3’ or 5’ end of the aptamer, or
both) for nuclease resistance is to use a flipped base added to the
end of the aptamer.

Velasco-Garcia and Missailidis: New trends in aptamer-based electrochemical biosensors
4
surface density of receptors and (ii) multiple binding to
target molecules for sandwich assays.
The method of immobilization of aptamers to a solid
support affects the sensitivity of the aptamer to the target
molecule. Thus, the selected method should maintain the
binding affinity and selectivity that the aptamers display in
solution (Balamurugan et al, 2008).
Aptamers can be attached to the solid support at
either the 5’-end or the 3’ end. Both positions have been
reported as being used for aptasensor development.
However, there are very few studies looking at the effect
of the two types of end attachment. Recent work suggests
that it depends on the particular aptamer (Cho et al, 2006),
although for biological targeting it may be that the 3’ end
is more suitable, since the 3’ end is the primary target for
exonucleases, and thus its coupling to the solid support
would simultaneously confer resistance to nucleases.
Gold is used for many electrochemical
measurements. Direct attachment of aptamers to gold
surfaces could be achieved by using a thiol-alkane linked
to the aptamer sequence. The gold surface could also be
functionalized and the type of chemistry selected is
dependent on what type of terminal functional group is
linked to the aptamer (amine, thiol or biotin termini;
Figure 3).
Gold surfaces functionalized with self-assembled
monolayers (SAMs) can address the nonspecific
adsorption of aptamer to the surface, which is a particular
problem for long oligonucleotides with larger numbers of
amine groups. Avidin-biotin technology has also been
exploited for aptamer immobilization. Strepavidin can be
physically adsorbed or covalently immobilized onto the
support and the method mainly requires incubation of the
biotin-tethered aptamer with the modified substrate.
Studies of the anti-thrombin aptamer revealed this
biocoating method gives best results regarding sensitivity
compared to other immobilization strategies (Hianik et al,
2007).
IV. Electrochemical assays In principle, aptamers can be selected for any given
target, ranging from small molecules to large proteins and
even cells. When aptamers bind small molecular targets,
these get incorporated into the nucleic acid structure,
buried within the binding pockets of aptamer structures.
On the other hand, large molecules (e.g. proteins) are
structurally more complicated, allowing aptamer
interactions at various sites via hydrogen bonding,
electrostatic interactions and shape complementarity. The
use of aptamers as bio-recognition elements for small
molecules has not been reported as extensively as for
protein targets.
Mainly two different assay configurations have been
reported to transduce these target-binding aptamer events:
(i) single-site binding and (ii) dual-site binding (Song et al,
2008). Small molecules are often assayed using the single-
site binding configuration. Protein targets can be assayed
via both single-site and dual-site binding. The dual-site
binding assay is commonly known as the sandwich assay.
Normally, the target molecule is sandwiched between a
pair of aptamers that bind to different regions of the large
Figure 3. Standard nucleic acid modifications used for aptamer
immobilisation. Most of the common modifications are linked
via the phosphate group of the oligonucleotide aptamer. Various
lengths carbon chains are used that can offer higher or lower
flexibility.
molecule. One aptamer is immobilized on a suitable solid
support to capture the target while the other aptamer for
detection is conjugated to a catalytic label. Enzymes,
inorganic or organic catalysts or nanoparticles are often
used for electrochemical detection. In some cases, when
there is only one aptamer for the molecule of interest,
antibodies have been reported to be used instead of the
second aptamer (Ferreira et al, 2008). If the target protein
contains two identical binding sites, the selection of a
single aptamer still allows the development of a sandwich
assay.
Displacement assays have been also proposed to
overcome the more challenging detection of small
molecules. Affinity interactions between aptamers and
small ligands are weaker than interaction with large
molecules (with dissociation constants in the µM range, in
comparison with constants for large molecules that are in
the pM-nM range). The presence of the small target could
induce the separation of two strands of a duplex nucleic
acid (one strand being the aptamer immobilised to a solid
support). Another strategy could rely on the displacement
of the aptamer from its complex with the immobilised
target molecule when the molecule is present in solution
(De-los-Santos-Alvarez et al, 2008).
Induced-fit conformational changes of the aptamer
after binding to the target molecule can also be used to
monitor a bio-recognition event by tagging the aptamer
(Figure 4). The use of labels requires precise knowledge
of the aptamer folding mechanism after binding to the
target and the binding sites. In the case of a redox active
marker, the accessibility of the label to the conducting
support is associated with the tertiary structure of the
aptamer before and after the binding event. However, for
small molecules, this strategy is not always viable,

Gene Therapy and Molecular Biology Vol 13, page 5
5
because the aptamer 3D structure could only be slightly
perturbed after the ligand interaction.
Redox-active reporting labels could not be covalently
tethered to aptamers. Methylene blue has been intercalated
into the double-stranded DNA domain of a hairpin
configuration aptamer. The binding of the target with the
aptamer opens the hairpin structure and releases the
intercalated methylene blue. As a result, the amperometric
response decreased with the addition of the analyte. This
approach is known as “label-free” method (Figure 5).
Related approaches use cationic redox-active reporting
units bound to the electrode via electrostatic interactions
with the DNA aptamer phosphate backbone. The binding
of the target molecule with the aptamer blocked the
binding of the cationic reporting units and the
electrochemical response decreased. The main
disadvantage of these latter approaches is a negative
detection signal.
Recently, nanomaterials are also providing novel
electrochemical sensing approaches. Single-walled carbon
nanotube field-effect transistor sensors were developed to
monitor aptamer-protein binding studies. Aptamers are
well suited for FET sensing due to their small size (1-2
nm) and recognition occurs inside the electrical double-
layer associated with the gate (within the Debye length).
The single-walled carbon nanotubes were assembled
between source and drain electrodes and the aptamers
were immobilized to these nanomaterials. In this label-free
approach, the binding of the target molecule to the
aptamers altered conductance through the device. The ease
of miniaturization of these sensing devices opens up the
feasibility of high-throughput assays in microarrays.
Nanoparticles have also been reported as catalytic
labels, instead of enzymes, and carriers for ultrasensitive
electrochemical detection; because one nanoparticle
contains a large number of aptamers, the target binding
process is amplified.
Impedance spectroscopy has been the most
frequently used electrochemical method in the
development of electrochemical aptasensors and has
shown excellent sensitivity, achieving limit of detection of
fM. However, despite the fact that the analytical technique
is simple to perform, the data fitting remains a bit
complicated. Easier data processing and faster response
could be achieved with chonoamperometry, but the limit
of detection will be higher and in the nM range.
IV. Applications of electrochemical
aptasensors Aptamer publications have now appeared in the
literature using most of the electrochemical transducers.
The majority of aptamer work on electrochemical sensors
is focused on amperometric transducers, but there have
been references on aptamers used in impedimetric, FET
and recently potentiometric sensors. Furthermore, a lot of
the work on the aptamers in electrochemical sensors has
been on the model protein, thrombin, which is one of the
best characterised complexes in the aptamer literature.
Figure 4. Assays based on induced-fit conformational changes of aptamers.
Figure 5. Label-free electrochemical assays based on: (A) methylene blue intercalated into the DNA aptamer and (B) cationic redox-
active reporting units bound to DNA aptamer phosphate backbone.

Velasco-Garcia and Missailidis: New trends in aptamer-based electrochemical biosensors
6
These have provided proof of principle concepts as to
how aptamers could be developed in novel sensors.
However, a number of other systems have also now been
described, which will be presented in this review.
A. Electrochemical aptasensors for the model
protein The thrombin-binding aptamer (15-mer, 5’-
GGTTGGTGTGGTTGG-3’) was the first one selected in
1992 by Block and colleagues and its structure has been
well characterized and studied. The folded structure in
solution is composed of two guanine quartets connected
by two T-T loops spanning the narrow grooves at one end
and a T-G-T loop spanning a wide groove at the other end
(known as the G-quartet structure). This anti-thrombin
aptamer has been extensively used as the model
oligonucleotide by many researchers to demonstrate the
wide applicability of aptamers as bio-recognition elements
in biosensors.
In the literature, many different electrochemical
aptasensors for thrombin detection have been reported.
The most straightforward configuration is based on the
immobilization of a thiol terminated aptamer on a gold
electrode. The aptamer-thrombin interaction is transduced
by the electrochemical quantification of p-nitroaniline
produced by the thrombin’s enzymatic reaction. Thrombin
has two electropositive exosites both capable of binding
the aptamer, allowing the development of an
electrochemical sensor system in a sandwich manner. The
thiolated aptamer was immobilized on a gold electrode
and, after incubation with the thrombin, a second
incubation step with an HRP labelled aptamer took place.
Electrochemical detection of HRP was performed using
H2O2 and a diffusional osmium based mediator. A similar
aptasensor system in sandwich manner for thrombin was
developed based on the aptamer for detection, labelled
with pyrroquinoline quinine glucose dehydrogenase, and
the electric current generated from glucose addition after
the formation of the complex on a gold electrode
(Ikebukuro et al, 2005). Another strategy for the thrombin
sensing is the direct immobilization of the protein on the
electrode surface. After the incubation with biotin-labelled
aptamer and then with streptavidin-HRP, the
electrochemical detection is performed using H2O2 and a
diffusional osmium-based mediator. The latter approach
achieved the lower limit of detection, 3.5 nM (Mir et al,
2006).
Mir and colleagues also developed in 2008 a
chronoamperometric beacon biosensor based on a
ferrocene-labelled thiol-aptamer. The aptamer adopts a 3-
D conformational change after binding the thrombin,
allowing the ferrocene label to approach to the gold
electrode. The interaction is detected via a
microperoxidase mediated electron transfer between the
label and the electrode surface. The system was
demonstrated with impedance spectroscopy and
chronoamperometry measurements, achieving a limit of
detection of 30 fM with the impedance spectroscopy (Mir
et al, 2008).
Methylene blue has also been used as an
electrochemical marker. The beacon aptamer surface was
prepared following formation of 11-mercaptoundecanoic
acid self-assembled monolayer on gold electrode.
Methylene blue was intercalated on the aptamer by the
interaction with two guanine bases. Binding of the
thrombin is correlated with the decrease in electrical current intensity in voltammetry. The estimated detection
limit of the target thrombin was 11 nM (Bang et al, 2005).
The modification of antibodies is difficult, costly and
time consuming; however researchers have been using
conventional polyclonal antibodies as a capturing probe
and labelled-aptamers as the detection probe in new
sandwich approaches for protein detection. Kang and
colleagues reported in 2008 a modified electrochemical
sandwich model for thrombin, based on capturing
antibody immobilized onto glassy carbon electrodes with
nanogold-chitosan composite film and Methylene blue
labelled aptamer as the electrochemical detection probe.
Lu and colleagues described in 2008 an
electrochemical aptasensor for thrombin that is not based
on the target binding-induced conformational change of
aptamers. The thrombin-binding aptamer is first assembled
onto a gold electrode and then hybridized with a ferrocene
labelled short aptamer-complementary DNA
oligonucleotide. The binding of the thrombin to the
aptamer destroys the double-stranded DNA
oligonucleotide and leads to the dissociation of the label
short complementary DNA oligonucleotide from the
electrode surface, resulting in a decrease in the differential
pulse voltammetry responses at the electrode (Lu et al,
2008). This strategy is based on the stronger binding
affinity of the aptamers towards their targets rather than to
the short aptamer-complementary DNA oligonucleotide
labelled with electroactive moieties.
The majority of the work performed on aptamer-
based electrochemical biosensors is based on aptamers
labelled using redox compounds, such as methylene blue,
and catalysts such as horseradish peroxidase. However,
nanoparticle-based materials offer excellent prospects for
a new signal amplification strategy for ultrasensitive
electrochemical aptasensing. Platinum nanoparticles have
been reported as catalytic labels when linked to a thiolated
aptamer. The nanoparticles catalysed the electrochemical
reduction of H2O2 and the resulting current enabled the
amplified detection of thrombin sandwiched between the
aptamer on the electrode surface and the aptamer labelled
with the nanoparticles (Polsky et al, 2006). Gold
nanoparticles offer several advantages such as electrical
conductivity, biocompatibility, ease of self-assembly
through a thiol group, increase electrode surface area and
amount of immobilized capturing probe. Gold
nanoparticles have been used as an electrochemical
sensing platform for direct detection of thrombine. The
aptamer was immobilised on a screen-printed electrode
modified with gold-nanoparticles by avidin-biotin
technology. The gold-nanoparticles surface status is
evaluated by the Au/Au oxide film formation with cyclic
and stripping voltammetry. Gold nanoparticles signal
changed with the deposition of biolayers due to
differences in electron transfer efficacy and availability of
buffer oxygen. Aptamers prefer to adopt the G-quarter
structure when binding with thrombin and the

Gene Therapy and Molecular Biology Vol 13, page 7
7
conformational changes made double strand DNA zones
appear and facilitated the electron transfer from solution to
the electrode surface, based on the double stranded DNA’s
ability to transport charge along the nucleotide stacking
(Suprun et al, 2008). The detection limit of this novel
approach is in the nM range. However, the aptasensor
measured directly binding events and opened 4 orders of
magnitude the operating range of protein concentration.
Assays coupling aptamers with magnetic beads for
the aptamer or target immobilisation before the
electrochemical transduction have also been proposed
(Centi et al, 2008). The use of magnetic beads improved
the assay kinetics due to the beads being in suspension and
also minimized matrix effect because of better washing
and separation steps.
An ultrasensitive electrochemical aptasensor for
thrombin in a sandwich format of magnetic nanoparicle-
immobilized aptamer, thrombin and gold nanoparticle-
labelled aptamer was reported by Zheng and colleagues in
2007. The magnetic nanoparticle-immobilized aptamer
was used for capturing and separating the target protein.
The gold nanoparticle-labelled aptamer offered the
electrochemical signal transduction. The signal was
amplified by forming a network like thiocyanuric acid/
gold nanoparticles to cap more nanoparticles per assay,
lowering the detection limit to the aM range
B. Other targets Aptamer have been selected against a wide range of
targets with typical binding affinities in the nanomolar to
picomolar range. Recently, electrochemical aptasensors
have been reported to detect proteins, hormones and drugs.
Papamichael and colleagues described in 2007 a
disposable electrochemical aptasensor for
Immunoglobulin E, a key marker of atopic diseases (such
as asthma, dermatitis and pollenosis). The sensor
incorporates a competitive format for IgE detection using
a biotinylated form of the aptamer. A standard, indirect
method was used where competition between surface-
bound IgE and IgE in solution proceeded for the aptamer.
The electrochemical detection is achieved by the use of an
extravidin-alkaline phosphatase label. After careful
optimization of conditions (buffer pH, ionic strength,
additional ions and proteins), the aptasensor was
performing at levels suitable for human testing (>300ng
ml-1).
Platelet-derived growth factor BB (PDGF-BB) is one
important cytokine involved in neural inflammation and
was selected as target for the development of an
electrochemical aptasensor based on capacitance change
induced by aptamer-protein specific binding, measured by
non-faradic impedance spectroscopy. The biosensor
detection limit was 40 nM. Electrochemical impedance
spectroscopy is a very attractive method for in vivo
diagnostics, due to its high sensitivity and label free
characteristics (Liao and Cui, 2007). A similar
electrochemical detection was also reported to a
tuberculosis-related cytokine, the interferon-!. The
aptamer-based electrochemical impedance biosensor
successfully detected interferon-! to a level of 100 fM
with an RNA aptamer and 1 pM with a DNA aptamer
probe (Min et al, 2008).
Electrochemical aptasensors for 17-" estradiol have
also been reported. The selected biotinylated DNA
aptamer was immobilized on a streptavidin-modified gold
electrode. The chemical binding of the hormone to the
aptamer was monitored by cyclic and square wave
voltammetry. When the 17-" estradiol interacted with the
aptamer, the current decreased due to the interference of
the bound target molecule with the electron flow produced
by a redox reaction between ferrocyanide (the mediator)
and ferricyanide. The linear range of this aptasensing
device was 1-0.01 nM of 17-" estradiol (Kim et al, 2007).
Cocaine has been detected by an electrochemical
aptasensor incorporating gold nanoparticles onto the
surface of a gold electrode. The thiol-derivative aptamer
was self-assembled onto the gold nanoparticles. The
aptamer was also functionalized at the other termini of the
strand with a redox-active ferrocene moiety. The cocaine
binding to the aptamer induces the conformational change
of the aptamer, bringing the redox tag in close proximity
to the electrode, leading to an increase in the current (Li et
al, 2008). Methylene blue tagged aptamer has been also
explored for the detection of cocaine (Baker et al, 2006).
A novel adenosine aptasensor was reported based on
the structure change of an aptamer probe immobilized on a
gold electrode. After the binding aptamer-target
nucleoside, a higher surface charge density and an
increasing steric hindrance were obtained that reduce the
diffusion of [Fe(CN)6]3-/[Fe(CN)6]
4- towards the electrode
surface, resulting in a decrease of the current. The
biosensing surface was easily regenerated and the
aptasensor limit of detection was 10 nM (Zheng et al,
2008).
C. Aptasensor arrays Some of the aptamer-based biosensor technology
described in this review could be transferred from single-
analyte devices to electrochemical methods offering the
possibility of simultaneous measurements of a panel of
targets. Wang reviewed the use of metal nanoparticles as
tracers for the analysis of nucleic acid hybridization.
Magnetic nanoparticles were linked to different probe
DNAs and incubated with samples containing different
DNA targets. Semiconductor quantum dots were
functionalized each with different nucleic acids
complementary to the free chain of the target DNA. After
dissolution of the metal nanoparticles, the identification of
the metal ions by stripping voltammetry enabled the
analysis of the different DNA targets (Wang, 2003).
Thrombin and lysozyme were detected in parallel
using a competitive assay in which thrombin and
lysozyme were modified with different semiconductor
quantum dots (Hansen et al, 2006). Specific aptamers were
immobilized on a gold electrode and bound to the
respective labelled protein. In the presence of unlabelled
protein in the sample, the quantum-dot functionalized
protein is displaced from the electrode into solution. The
dissolution of the remaining metal ions on the surface and
the electrochemical detection of the released ions enabled
the quantitative detection of the proteins.

Velasco-Garcia and Missailidis: New trends in aptamer-based electrochemical biosensors
8
IV. Conclusions Aptamers have been widely used in a variety of
diagnostic and sensor applications, offering a variety of
possibilities for aptamer-based sensors in early disease
diagnosis and prognosis, substance control, environmental
measurements or national security applications on
measurements of explosives or potential infectious agents.
Yet, despite the advances and the huge body of literature
documenting the success of the technology, the
commercial application of aptamers in the field of
diagnostics remains relatively undeveloped, not least due
to the exclusive IP portfolio, and the fact that there is a
vast antibody-based diagnostic market and a certain degree
of hesitation to move to a new type of product, unless
aptamers offer verifiably significant improvements on
current technologies that warrant substitution of antibodies
in some current assay formats. In this review, different
types of electrochemical aptamer-based biosensors have
been discussed. Although the optical and mass-sensitive aptasensors have been the most commonly described in
the literature, electrochemical transducers have enormous
potential and offer simple, rapid, cost-effective and easy to
miniaturize sensing in many diagnostic fields. Emerging
nanomaterials have also brought new possibilities for
developing novel ultrasensitive electrochemical
aptasensors.
References Baker BR, Lai RY, Wood MCS, Doctor EH, Heeger AJ, Plaxco
KW (2006) An electronic, aptamer-based small-molecule
sensor for the rapid label-free detection of cocaine in
adulterated samples and biological fluids. J Am Chem Soc
128, 3138-3139.
Balamurugan S, Obubuafo A, Soper SA, Spivak DA (2008)
Surface immobilization methods for aptamer diagnostic
applications. Anal Bioanal Chem 390, 1009-1021.
Bang GS, Cho S, Kim BG (2005) A novel electrochemical
detection method for aptamer biosensors. Biosens Bioelec
21, 863-870.
Berezovski M, Drabovich A, Krylova SM, Musheev M,
Okhonin V, Petrov A, Krylov SN (2005) Nonequilibrium
Capillary Electrophoresis of Equilibrium Mixtures: A
Universal Tool for Development of Aptamers. J Am Chem
Soc 127, 3165-3171.
Bini A, Minunni M, Tombelli S, Centi S, Mascini M (2007)
Analytical performances of aptamer-based sensing for
thrombin detection. Anal Chem 79, 3016-3019.
Centi S, Messina G, Tombelli S, Palchetti I, Mascini M (2008)
Different approaches for the detection of thrombin by an
electrochemical aptamer-based assay coupled to magnetic
beads. Biosens Bioelec 23, 1602-1609.
Cho EJ, Collett JR, Szafranska AE, Ellington AD (2006)
Optimization of aptamer microarray technology for multiple
protein targets. Anal Chim Acta 564, 82-90.
Cho S, Kim JE, Lee BR, Kim JH, Kim BG (2005) Bis-aptazyme
sensors for hepatitis C virus replicase and helicase without
blank signal. Nucleic Acids Res 33, e177.
Choi JH, Chen KH, Strano MS (2006) Aptamer-Capped
Nanocrystal Quantum Dots: A New Method for Label-Free
Protein Detection. J Am Chem Soc 128, 15584-15585.
De-los-Santos-Alvarez N, Lobo-Castañón MJ, Miranda-
Ordieres AJ, Tuñón-Blanco P (2008) Aptamers as
recognition elements for label-free analytical devices.
Trends Anal Chem 27, 437-446.
Drabovich A, Berezovski M, Krylov SN (2005) Selection of
Smart Aptamers by Equilibrium Capillary Electrophoresis of
Equilibrium Mixtures (ECEEM). J Am Chem Soc 127,
11224-11225.
Ellington AD, Szostak JW (1990) In vitro selection of RNA
molecules that bind specific ligands. Nature 346, 818-822.
Ferreira CSM, Matthews CS, Missailidis S (2006) DNA
aptamers that bind to MUC1 tumour marker: Design and
characterization of MUC1-binding single stranded DNA
aptamers. Tumor Biol 27, 289-301.
Ferreira CSM, Papamichael K, Guilbault G, Schwarzacher T,
Gariepy J, Missailidis S (2008) Design of aptamer-antibody
sandwich ELISA for early tumour diagnosis. Anal Bioanal
Chem 390, 1039-1050.
Frauendorf C, Jaschke A (2001) Detection of small organic
analytes by fluorescing molecular switches. Bioorg Med
Chem 9, 2521-2524.
Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K,
Gustafsdottir SM, Ostman A, Landegren U (2002) Protein
detection using proximity-dependent DNA ligation assays.
Nature Biotechnol 20, 473-477.
Hamula CLA, Guthrie JW, Zhang H, Li XF, Le XC (2006)
Selection and analytical applications of aptamers. Trends
Anal Chem 25, 681-691.
Hansen JA, Wang J, Kawde AN, Xiang Y, Gothelf KV, Collins
G (2006) Quantum-dot/aptamer-based ultrasensitive multi-
analyte electrochemical biosensor. J Am Chem Soc 128,
2228-2229.
Heidenreich O, Eckstein F (1992) Hammerhead ribozyme-
mediated cleavage of the long terminal repeat RNA of
human immunodeficiency virus type 1. J Biol Chem 267,
1904-1909.
Hianik T, Ostatna V, Sonlajtnerova M, Grman I (2007)
Influence of ionic strength, pH and aptamer configuration for
binding affinity to thrombin. Bioelectrochemistry 70, 127-
133.
Ikanovic M, Rudzinski WE, Bruno JG, Allman A, Carrillo MP,
Dwarakanath S, Bhahdigadi S, Rao P, Kiel JL, Andrews CJ
(2007) Fluorescence Assay Based on Aptamer-Quantum Dot
Binding to Bacillus thuringiensis Spores. J Fluoresc 17, 193-
199.
Ikebukuro K, Kiyohara C, Sode K (2005) Novel electrochemical
sensor system for protein using the aptamers in sandwich
manner. Biosens Bioelec 20, 2168-2172.
Ikebukuro K, Okumura Y, Sumikura K, Karube I (2005) A
novel method of screening thrombin-inhibiting DNA
aptamers using an evolution-mimicking algorithm. Nucleic
Acids Res 33, e108.
Kang Y, Feng KJ, Chen JW, Jiang JH, Shen GL, Yu RQ (2008)
Electrochemical detection of thrombin by sandwich approach
using antibody and aptamer. Bioelectrochemistry 73, 76-81.
Khan H, Missailidis S (2008) Aptamers in oncology: a
diagnostic perspective. Gene Ther Mol Biol 12, 111-128.
Kim YS, Jung HS, Matsuura T, Lee HY, Kawai T, Gu MB
(2007) Electrochemical detection of 17beta-estradiol using
DNA aptamer immobilized gold electrode chip. Biosens
Bioelec 22, 2525-2531.
Kirby R, Cho EJ, Gehrke B, Bayer T, Park YS, Neikirk DP,
McDevitt JT, Ellington AD (2004) Aptamer-based sensor
arrays for the detection and quantification of proteins. Anal
Chem 76, 4066-4075.
Kusser W (2000) Chemically modified nucleic acid aptamers
for in vitro selections: evolving evolution. Rev Mol
Biotechnol 74, 27-38.
Lai RY, Plaxco KW, Heeger AJ (2007) Aptamer-based
electrochemical detection of picomolar platelet-derived
growth factor directly in blood serum. Anal Chem 79, 229-
233.

Gene Therapy and Molecular Biology Vol 13, page 9
9
Levy M, Cater SF, Ellington AD (2005) Quantum-Dot Aptamer
Beacons for the Detection of Proteins. ChemBioChem 6,
2163-2166.
Li X, Qi H, Shen L, Gao Q, Zhang C (2008) Electrochemical
Aptasensor for the Determination of Cocaine Incorporating
Gold Nanoparticles Modification. Electroanalysis 20, 1475-
1482.
Liao W, Cui XT (2007) Reagentless aptamer based impedance
biosensor for monitoring a neuro-inflammatory cytokine
PDGF. Biosens Bioelec 23, 218-224.
Liu J, Lee JH, Lu Y (2007) Quantum Dot Encoding of Aptamer-
Linked Nanostructures for One-Pot Simultaneous Detection
of Multiple Analytes. Anal Chem 79, 4120-4125.
Liu J, Lu Y (2004) Adenosine-dependent assembly of
aptazyme-functionalized gold nanoparticles and its
application as a colorimetric biosensor. Anal Chem 76,
1627-1632.
Liu J, Lu Y (2006) Fast colorimetric sensing of adenosine and
cocaine based on a general sensor design involving aptamers
and nanoparticles. Angew Chem Int Ed 45, 90-94.
Lu Y, Zhu N, Yu P, Mao L (2008) Aptamer-based
electrochemical sensors that are not based on the target
binding-induced conformational change of aptamers.
Analyst 133, 1256-1260.
Luzi E, Minunni M, Tombelli S, Mascini M (2003) New trends
in affinity sensing: aptamers for ligand binding. Trends
Anal Chem 22, 810-818.
Min K, Cho M, Han SY, Shim YB, Ku J, Ban C (2008) A
simple and direct electrochemical detection of interfero-
gamma using its RNA and DNA aptamers. Biosens Bioelec
23, 1819-1824.
Mir M, Vreeke M, Katakis I (2006) Different strategies to
develop an electrochemical thrombin aptasensor.
Electrochem Comm 8, 505-511.
Mir M, Vreeke M, Katakis I (2008) Ultrasensitive detection
based on an aptamer beacon electron transfer chain.
Electrochem Comm 10, 1533-1536.
Missailidis S (2008) Combinatorial Approach to Anticancer
Drug Design, In-Anticancer Therapeutics (Eds: S.
Missailidis), Wiley & Sons Ltd.
Ogawa A, Maeda M (2007) Aptazyme-based riboswitches as
label-free and detector-free sensors for cofactors. Bioorg
Med Chem Letters 17, 3156-3160.
Ozaki H, Nishihira A, Wakabayashi M, Kuwahara M, Sawai H
(2006) Biomolecular sensor based on fluorescence-labeled
aptamer. Bioorg Med Chem Letters 16, 4381-4384.
Papamichael KI, Kreuzer MP, Guilbault GG (2007) Viability of
allergy (IgE) detection using an alternative aptamer receptor
and electrochemical means. Sens Actuat B-Chem 121, 178-
186.
Pieken W, Olsen DB, Benseler F, Aurup H, Eckstein HF (1991)
Kinetic characterization of ribonuclease-resistant 2’-modified
hammerhead ribozymes. Science 253, 314-317.
Polsky R, Gill R, Willner I (2006) Nucleic Acid-Functionalized
Pt Nanoparticles: Catalytic Labels for the Amplified
Electrochemical Detection of Biomolecules. Anal Chem 78,
2268-2271.
Potyrailo RA, Conrad RC, Ellington AD, Hieftje GM (1998)
Adapting selected nucleic acid ligands (aptamers) to
biosensors. Anal Chem 70, 3419-3425.
Song S, Wang L, Li J, Zhao J, Fan C (2008) Aptamer-based
biosensors. Trends Anal Chem 27, 108-117.
Stojanovic MN, Kolpashchikov DM (2004) Modular aptameric
sensors. J Am Chem Soc 126, 9266-9270.
Stojanovic MN, Landry DW (2002) Aptamer-based colorimetric
probe for cocaine. J Am Chem Soc 124, 9678-9679.
Stojanovic MN, Prada PD, Landry DW (2001) Aptamer-based
folding fluorescent sensor for cocaine. J Am Chem Soc 123,
4928-4931.
Suprun E, Shumyantseva V, Bulko T, Rachmetova S, Rad’ko S,
Bodeoev N, Archakov A (2008) Au-nanoparticles as an
electrochemical sensing platform for aptamer-thrombin
interaction. Biosens Bioelec 24, 831-836.
Tombelli S, Minunni M, Mascini M (2005) Analytical
applications of aptamers. Biosens Bioelec 20, 2424-2434.
Tombelli S, Minunni M, Mascini M (2007) Aptamers- based
assays for diagnostics, environmental and food analysis.
Biomol Eng 24, 191-200.
Tuerk C, Gold L (1990) Systematic evolution of ligands by
exponential enrichment - RNA ligands to bacteriophage-T4
DNA-polymerase. Science 249, 505-510.
Velasco-Garcia MN, Mottram TT (2003) Biosensor Technology
addressing Agricultural Problems Biosys Eng 84, 1-12.
Wang J (2003) Nanoparticle-based electrochemical DNA
detection. Anal Chim Acta 500, 247-257.
Winters-Hilt S (2006) Nanopore Detector based analysis of
single-molecule conformational kinetics and binding
interactions. BMC Bioinformatics 7, S21.
www.lcsciences.com , accessed January 2009.
www.somalogic.com, accessed January 2009.
Xiao Y, Lubin AA, Heeger AJ, Plaxco KW (2005) Label-free
electronic detection of thrombin in blood serum by using an
aptamer-based sensor. Angew Chem Int Ed 44, 5456-5459.
Yamana K, Ohtani Y, Nakano H, Saito I (2003) Bis-pyrene
labeled DNA aptamer as an intelligent fluorescent biosensor.
Bioorg Med Chem Letters 13, 4329-4331.
Yoshida W, Sode K, Idebukuro K (2006b) Homogeneous DNA
sensing using enzyme-inhibiting DNA aptamers. Biochem
Biophys Res Comm 348, 245-252.
Yoshida W, Sode K, Ikebukuro K (2006a) Aptameric Enzyme
Subunit for Biosensing Based on Enzymatic Activity
Measurement. Anal Chem 78, 3296-3303.
Zheng F, Wu ZS, Zhang SB, Guo MM, Chen CR, Shen GL, Yu
RQ (2008) Aptamer-based Electrochemical Biosensors for
Highly Selective and Quantitative Detection of Adenosine.
Chem Res Chinese Universities 24, 138-142.
Zheng J, Feng W, Lin L, Zhang F, Cheng G, He P, Fang Y
(2007) A new amplification strategy for ultrasensitive
electrochemical aptasensor with network-like thiocyanuric
acid/gold nanoparticles. Biosens Bioelec 23, 341-347.
Zuo X, Song S, Zhang J, Pan D, Wang L, Fan C (2007) A
target-responsive electrochemical aptamer switch (TREAS)
for reagentless detection of nanomolar ATP. J Am Chem
Soc 129, 1042-1043.
Maria N. Velasco-Garcia and Sotiris Missailidis

Gomase et al: Mapping of MHC class binding nonamers from lipid binding protein of Ascaridia galli
10
Gene Ther Mol Biol Vol 13, 10-14, 2009
Mapping of MHC class binding nonamers from lipid
binding protein of Ascaridia galli Research Article
Virendra S Gomase1,*, Somnath B Waghmare2, Baba Jadhav2, Karbhari V Kale1 1Department of Computer Science and Information Technology, Dr. Babasaheb Ambedkar Marathwada University,
Aurangabad, 431004, (MS), India 2Department of Zoology, Dr. Babasaheb Ambedkar Marathwada University, Aurangabad, 431004, (MS), India
__________________________________________________________________________________
*Correspondence: Virendra S Gomase, Department of Computer Science and Information Technology, Dr. Babasaheb Ambedkar
Marathwada University, Aurangabad, 431004 (MS), India; Mobile- +91-9987770696; e-mail: [email protected]
Key words: lipid binding protein, MHC, epitope, solvent accessibility, peptide vaccine
Abbreviations: Major histocompatibility complex (MHC); Position Specific Scoring Matrices, (PSSMs); Support Vector Machine,
(SVM)
Received: 3 March 2009; Revised: 12 March 2009
Accepted: 16 March 2009; electronically published: March 2009
Summary
Ascaridia galli involved multiple antigenic components to direct and empower the immune system to protect the
host from infection. MHC molecules are cell surface proteins, which take active part in host immune reactions and
involvement of MHC class in response to almost all antigens and it give effects on specific sites. Predicted MHC
binding regions acts like red flags for antigen specific and generate immune response against the parent antigen. So
a small fragment of antigen can induce immune response against whole antigen. This theme is implemented in
designing subunit and synthetic peptide vaccines. In this study, we analyzed lipid-binding protein of Ascaridia galli
and is allows potential drug targets to identify active sites, which form antibodies against or infection. The method
integrates prediction of peptide MHC class binding; proteosomal C terminal cleavage and TAP transport efficiency.
Antigenic epitopes of lipid binding protein are important antigenic determinants against the various toxic reactions
and infections.
I. Introduction Ascaridia galli parasitic nematodes produce at least
two structurally novel classes of small helix-rich retinol-
and fatty-acid-binding proteins that have no counterparts
in their plant or animal hosts and thus represent potential
targets for new nematicides. Nematode-specific fatty-acid
family of proteins localises to the surface of the organism,
placing it in a strategic position for interaction with the
host. Their function as a broad-spectrum and it is thought
that it is involved in the evasion of primary host plant
defence systems. Prediction of peptide fragments from
lipid binding protein of Ascaridia galli involved multiple
antigenic components to direct and empower the immune
system to protect the host from infection (Timanova et al,
1999; Jordanova et al, 2005a,b). Major histocompatibility
complex (MHC) molecules are cell surface proteins,
which take active part in host immune reactions and
involvement of MHC class-I & II in response to almost all
antigens. The predicted binding affinity is normalized by
the 1% fractil. The MHC peptide binding is predicted
using neural networks trained on C terminals of known
epitopes. In analysis predicted MHC/peptide binding is a
log-transformed value related to the IC50 values in nM
units (Gomase et al, 2008b). This approach is based on the
phenomenon of cross-protection, whereby a host infected
with a Ascaridia galli is protected against a more severe
strain of the same lipid binding protein of Ascaridia galli.
The phenotype of the resistant transgenic hosts includes
fewer centers of initial infection, a delay in symptom
development, and low accumulation. Lipid binding protein
of Ascaridia galli is necessary for new paradigm of
synthetic vaccine development and target validation
(Gomase, 2008a,b).
II. Methodology Antigenic epitopes of lipid binding protein of Ascaridia
galli is determined using the Gomase in 2007, Welling, Parker
antigenicity methods (Gomase et al, 2007a, b). We also found the
Abraham & Leo hydrophobicity, Bull & Breese hydrophobicity,
Guy hydrophobicity, Miyazawa hydrophobicity, Roseman
hydrophobicity, Wolfenden hydrophobicity, scales. Theses scales

Gene Therapy and Molecular Biology Vol 13, page 11
11
are essentially a hydrophilic index, with polar residues assigned
negative values (Gomase et al, 2008a). The MHC peptide
binding of lipid binding protein is predicted using neural
networks trained on C terminals of known epitopes. In analysis
predicted MHC/peptide binding of lipid binding protein is a log-
transformed value related to the IC50 values in nM units.
MHC2Pred predicts peptide binders to MHCI and MHCII
molecules from protein sequences or sequence alignments using
Position Specific Scoring Matrices (PSSMs). Support Vector
Machine (SVM) based method for prediction of promiscuous
MHC class II binding peptides. SVM has been trained on the
binary input of single amino acid sequence (Reche et al, 2002;
Buus et al, 2003; Nielsen et al, 2003; Bhasin and Raghava,
2005). In addition, we predict those MHC ligands from whose C-
terminal end is likely to be the result of proteosomal cleavage.
III. Results and Interpretation Lipid binding protein is 508 residues long, having
antigenic MHC binding peptides. MHC molecules are cell
surface glycoproteins, which take active part in host
immune reactions and involvement of MHC class-I and
MHC II in response to almost all antigens. BepiPrep
Server antigenicity determinant shows epitopes present in
the Ascaridia galli the desired immune response. PSSM
based server predict the peptide binders to MHCI
molecules of lipid binding protein to MHCII molecules of
lipid binding protein sequence as H2_Db, I_Ab, I_Ag7,
I_Ad, analysis found antigenic epitopes region in lipid
binding protein (Tables 1, 2). We also found the SVM
based MHCII-IAb; MHCII-IAd; MHCII-IAg7 and
MHCII- RT1.B peptide regions, which represented
predicted binders from lipid binding protein. The predicted
binding affinity is normalized by the 1% fractil. We
describe an improved method for predicting linear
epitopes (Table 2). The region of maximal hydrophilicity
is likely to be an antigenic site, having hydrophobic
characteristics, because terminal regions of lipid binding
protein is solvent accessible and unstructured, antibodies
against those regions are also likely to recognize the native
protein (Figures 1-4). It was shown that lipid binding
protein is hydrophobic in nature and contains segments of
low complexity and high-predicted flexibility (Figures 5-
8). Predicted antigenic fragments can bind to MHC
molecule is the first bottlenecks in vaccine design.
IV. Conclusion Lipid binding protein of Ascaridia galli peptide
nonamers are from a set of aligned peptides known to bind
to a given MHC molecule as the predictor of MHC-
peptide binding. MHCII molecules bind peptides in
similar yet different modes and alignments of MHCII-
ligands were obtained to be consistent with the binding
mode of the peptides to their MHC class, this means the
increase in affinity of MHC binding peptides may result in
enhancement of immunogenicity of lipid binding protein.
These predicted of lipid binding protein antigenic peptides
to MHC class molecules are important in vaccine
development from Ascaridia galli.
Table 1. PSSM based prediction of MHC ligands, from whose C-terminal ends are proteosomal cleavage sites.
MHC-I POS. N Sequence C MW (Da)
8mer_H2_Db 254 NLR SEENAISL VNG 843.9
8mer_H2_Db 277 QSS SYASWDTL IAS 900.98
8mer_H2_Db 64 LLE KSPEKMDI MML 929.1
8mer_H2_Db 117 KAL SKGSHPTK EEM 822.91
8mer_H2_Db 338 LSE DEHSKHDI DAA 961.99
9mer_H2_Db 253 ENL RSEENAISL VNG 1000.09
9mer_H2_Db 53 RDP MLYDNVTKL LEK 1078.28
9mer_H2_Db 447 HKT VTFPNALHL IQR 993.17
9mer_H2_Db 157 HSY LKDENIHAL QEV 1034.18
9mer_H2_Db 276 KQS SSYASWDTL IAS 988.06
9mer_H2_Db 408 LKE VKAKNEKLY YIL 1074.28
9mer_H2_Db 260 NAI SLVNGFTEV CKA 947.05
9mer_H2_Db 420 YIL FLINDHVAM LRR 1041.23
9mer_H2_Db 37 IAK KKARSFAHV LSK 1025.22
9mer_H2_Db 427 DHV AMLRRYNEL SDP 1147.37
10mer_H2_Db 125 PTK EEMTNLAKEL SAK 1159.32
10mer_H2_Db 370 KII SSMSFYSECI ITP 1135.29
10mer_H2_Db 457 HLI QRYANTTEEY HHQ 1256.3
10mer_H2_Db 276 KQS SSYASWDTLI ASL 1101.22
10mer_H2_Db 148 ELI NALFAGHSYL KDE 1074.21
10mer_H2_Db 253 ENL RSEENAISLV NGF 1099.22
10mer_H2_Db 259 ENA ISLVNGFTEV CKA 1060.21
11mer_H2_Db 369 KKI ISSMSFYSECI ITP 1248.45
11mer_H2_Db 419 YYI LFLINDHVAML RRY 1267.55
11mer_H2_Db 445 FFH KTVTFPNALHL IQR 1222.44
11mer_H2_Db 291 LEK APRSHARAVIL RDI 1172.41
11mer_H2_Db 325 KAM SAILGLLKVML SED 1139.5
11mer_H2_Db 298 HAR AVILRDIHRCL VKK 1290.6
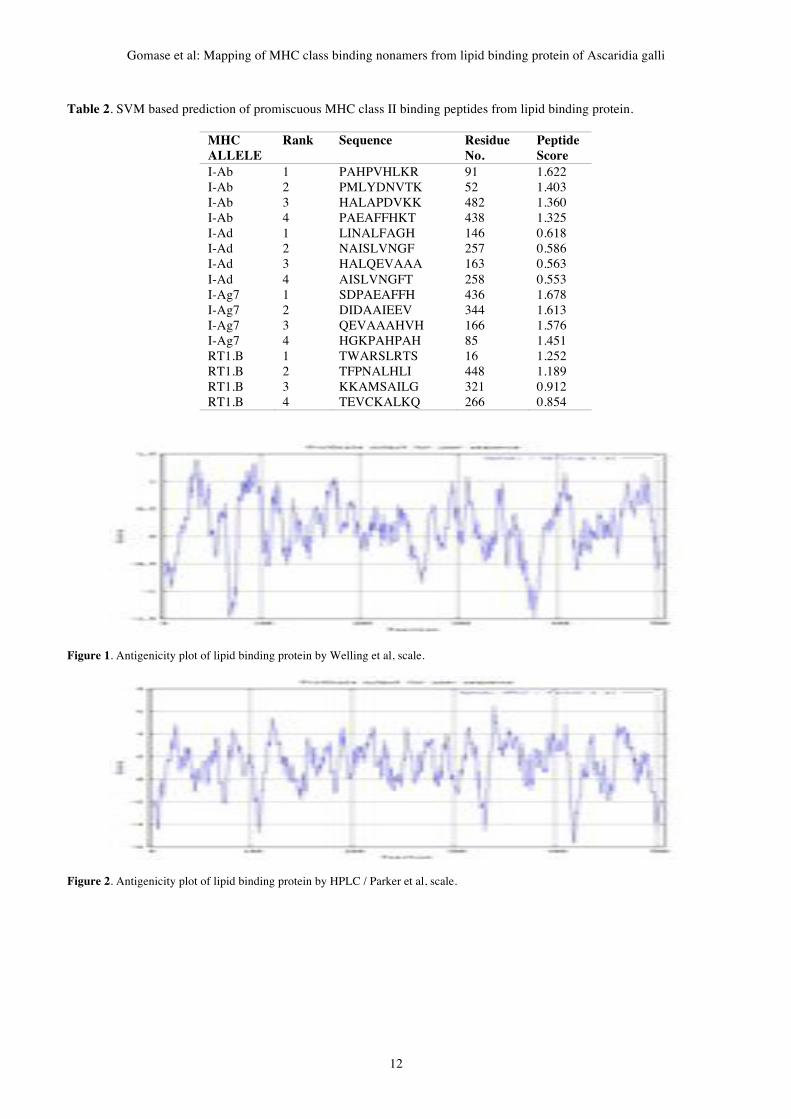
Gomase et al: Mapping of MHC class binding nonamers from lipid binding protein of Ascaridia galli
12
Table 2. SVM based prediction of promiscuous MHC class II binding peptides from lipid binding protein.
MHC
ALLELE
Rank Sequence Residue
No.
Peptide
Score
I-Ab 1 PAHPVHLKR 91 1.622
I-Ab 2 PMLYDNVTK 52 1.403
I-Ab 3 HALAPDVKK 482 1.360
I-Ab 4 PAEAFFHKT 438 1.325
I-Ad 1 LINALFAGH 146 0.618
I-Ad 2 NAISLVNGF 257 0.586
I-Ad 3 HALQEVAAA 163 0.563
I-Ad 4 AISLVNGFT 258 0.553
I-Ag7 1 SDPAEAFFH 436 1.678
I-Ag7 2 DIDAAIEEV 344 1.613
I-Ag7 3 QEVAAAHVH 166 1.576
I-Ag7 4 HGKPAHPAH 85 1.451
RT1.B 1 TWARSLRTS 16 1.252
RT1.B 2 TFPNALHLI 448 1.189
RT1.B 3 KKAMSAILG 321 0.912
RT1.B 4 TEVCKALKQ 266 0.854
Figure 1. Antigenicity plot of lipid binding protein by Welling et al, scale.
Figure 2. Antigenicity plot of lipid binding protein by HPLC / Parker et al, scale.
Gene Therapy and Molecular Biology Vol 13, page 13
13
Figure 3. Hydrophobicity plot of lipid binding protein by Wolfenden et al, scale.
Figure 4. Hydrophobicity plot of lipid binding protein by Bull and Breese scale.
Figure 5. Hydrophobicity plot of lipid binding protein by Gut scale.
Figure 6. Hydrophobicity plot of lipid binding protein by Miyazawa et al, scale.
Gomase et al: Mapping of MHC class binding nonamers from lipid binding protein of Ascaridia galli
14
Figure 7. Hydrophobicity plot of lipid binding protein by Roseman scale.
Figure 8. Hydrophobicity plot of lipid binding protein by Abraham and Leo scale.
References Bhasin M, Raghava GP (2005) P cleavage: an SVM based
method for prediction of constitutive proteasome and
immunoproteasome cleavage sites in antigenic sequences.
Nucleic Acids Res 33, W202-207.
Buus S, Lauemøller SL, Worning P, Kesmir C, Frimurer T,
Corbet S, Fomsgaard A, Hilden J, Holm A, Brunak S. (2003)
Sensitive quantitative predictions of peptide-MHC binding
by a 'Query by Committee' artificial neural network
approach. Tissue Antigens 62, 378-384.
Gomase VS, Kale KV, Shyamkumar K, Shankar S (2008a)
Computer Aided Multi Parameter Antigen Design: Impact of
Synthetic Peptide Vaccines from Soybean Mosaic Virus.
ICETET 2008, IEEE Computer Society in IEEE Xplore, Los
Alamitos, California, 629-634.
Gomase VS, Tagore S, Shyamkumar K (2008b) Prediction of
antigenic binders from c-terminal domain Human
papillomavirus oncoprotein e7. Gene Ther Mol Biol 12,
147-166.
Gomase VS (2008a) Computer aided multi parameter antigen
design: Impact of synthetic peptide vaccines from
Latrodectus tredecimguttatus. Int J Bioinformatics 1, 53-54.
Gomase VS (2008b) In silico prediction of antigenic epitope of
neurotoxin M14 from Buthus eupeus. Int J Bioinformatics
1, 47-51.
Gomase VS, Kale KV, Jyotiraj A, Vasanthi R (2007a)
Identification of mhc ligands from alfalfa mosaic virus. Med
Chem Res 15, 160.
Gomase VS, Kale KV, Chikhale NJ, Changbhale SS (2007b)
Prediction of MHC Binding Peptides and Epitopes from
Alfalfa mosaic virus. Curr Drug Discov Technol 4, 117-
1215.
Jordanova R, Radoslavov G, Fischer P, Liebau E, Walter RD,
Bankov I, Boteva R (2005a) Conformational and functional
analysis of the lipid binding protein Ag-NPA-1 from the
parasitic nematode Ascaridia galli. FEBS J 272, 180-9.
Jordanova R, Radoslavov G, Fischer P, Torda A, Lottspeich F,
Boteva R, Walter RD, Bankov I, Liebau E (2005b) The
highly abundant protein Ag-lbp55 from Ascaridia galli
represents a novel type of lipid-binding proteins. J Biol
Chem 280, 41429-38.
Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth
K, Buus S, Brunak S, Lund O (2003) Reliable prediction of
T-cell epitopes using neural networks with novel sequence
representations. Protein Sci 12, 1007-1017.
Reche PA, Glutting JP, Reinherz EL (2002) Prediction of MHC
class I binding peptides using profile motifs. Hum Immunol
63, 701-709.
Timanova A, Müller S, Marti T, Bankov I, Walter RD (1999)
Ascaridia galli fatty acid-binding protein, a member of the
nematode polyprotein allergens family. Eur J Biochem 261,
569-76.
Virendra Gomase and Somnath B Waghmare

Gene Therapy and Molecular Biology Vol 13, page 15
15
Gene Ther Mol Biol Vol 13, 15-19, 2009
Perspectives in vector development for systemic cancer gene therapy Review Article
Arash Hatefi*, Brenda F. Canine
Department of Pharmaceutical Sciences, Center for Integrated Biotechnology, Washington State University, Pullman, WA,
USA
__________________________________________________________________________________
*Correspondence: Arash Hatefi, Department of Pharmaceutical Sciences, Center for Integrated Biotechnology, Washington State
University, P.O. Box 646534, Pullman, WA, 99164, USA; Tel: 509-335-6253; Fax: 509-335-5902; e-mail: [email protected]
Key words: non-viral vectors, cancer gene therapy, vector development, viral vectors
Abbreviations: adenovirus, (Ad); coxsackievirus and adenovirus receptor, (CAR); fibroblast growth factor 2, (FGF2); fibroblast growth
factor receptor, (FGFR); herpes simplex virus, (HSV); multiplicity of infection, (MOI); ornithine transcarbamylase, (OTC)
Received: 10 February 2009; Revised: 25 March 2009 Accepted: 26 March 2009; electronically published: April 2009
Summary Gene therapy is perceived as a revolutionary technology with the promise to cure almost any disease, provided that
we understand its genetic basis. However, enthusiasm has rapidly abated as multiple clinical trials have failed to
show efficacy. The limiting factor seems to be the lack of a suitable delivery system to carry the therapeutic genes to
the target tissue safely and efficiently. Therefore, advancements in cancer gene therapy in general depend on the
development of novel vectors with maximum therapeutic efficacy at the target site and minimal toxicity to normal
tissues. This mini-review highlights both the major fortes and the unique challenges associated with the state-of–the-
art gene carriers currently being used in cancer gene therapy.
I. Introduction Gene therapy is perceived as a ground-breaking
technology with the promise to cure almost any disease,
provided that we understand the genetic and molecular
basis of the malady being treated. However, enthusiasm
has rapidly abated as multiple clinical trials have failed to
show efficacy. The limiting factor seems to be the lack of
a suitable delivery system to carry the therapeutic genes
safely and efficiently to the target tissue (Louise, 2006).
Gene-transfer technology is still in a nascent stage owing
to several inherent limitations in the existing delivery
methods. While lipid-based vectors (liposomes) provide
high transfection efficiency, their large scale production,
reproducibility and cytotoxicity remain a major concern
(Lv et al, 2006). On the other hand, cationic polymers are
robust and relatively biocompatible, but they suffer from
poor gene-transfer efficiency (Pack et al, 2005).
Adenoviruses are the vehicles of choice for cancer gene
therapy at this point particularly due to their ability to
overcome the intracellular barriers and the enormous
possibility for recombinant engineering. However, non-
specific binding to all cells that over-express
coxsackievirus and adenovirus receptor (CAR), potential
immunogenicity, high costs of production, and the fact
that the majority of cancer cells do not express CAR has
limited their use for cancer gene therapy (Thomas et al,
2003; Shen and Nemunaitis, 2006). What has been long
desired is a technology which combines the
biocompatibility, efficiency and the ability to engineer an
effective gene-transfer technology. Since internalization of
both viral and non-viral vectors is the first step in their
transfection pathway, knowledge and understanding of
their entry mechanisms is of major importance for the
design of efficient viral and non-viral vehicles for cancer
gene therapy.
II. Strengths and weaknesses of
current vectors A. Viral vectors for systemic cancer gene
therapy Viruses have evolved to efficiently infect their host,
overcome the cellular barriers and transfer their genetic
material into the cell’s nucleus. One viral vector that has
received considerable attention in cancer gene therapy is
adenovirus. The basic elements of the trafficking pathway
for adenovirus include high affinity binding of the capsid
to receptors on the cell surface, internalization by
endocytosis, lysis of the endosomal membrane resulting in
escape to the cytosol, trafficking along microtubules,
binding to the nuclear envelope, and insertion of the viral

Hatefi and Canine: Perspectives in vector development for systemic cancer gene therapy
16
genome through the nuclear pore (Leopold and Crystal,
2007).
Adenoviruses have high affinity for the CAR and use
it to enter the cells. Although they are highly efficient in
transducing cells that over-express CAR on their surface,
they are considered poor gene delivery systems in cells
that have low expression of CAR (Li et al, 1999). In
addition, CAR is expressed on many normal cells which
undermines the ability of this vector to specifically reach
target cancer cells when administered systemically. Thus,
adenovirus is not considered a universal efficient vehicle
for cancer gene therapy as the majority of cancer cells do
not over-express CAR (Shen and Nemunaitis, 2006).
Another virus, Herpes Simplex Virus overcomes this
deficiency by utilizing a different receptor to enter cancer
cells. The initial attachment of HSV involves the
interaction of viral envelope glycoproteins with the
glycosaminoglycan moieties of cell surface heparan
sulfates (Spear et al, 1992). However, like CAR,
expression of heparin sulfates is not unique to cancer cells
and can be found routinely in normal cells. As a result,
systemic administration of HSV could also be
problematic.
Attachment of a targeting ligand to the viral capsid
has been used as a means to make adenovirus specifically
bind cancer cells and internalize via receptor mediated
endocytosis. One example is attachment of the ligand,
fibroblast growth factor 2 (FGF2) which has affinity for
the basic fibroblast growth factor receptor (FGFR) (Green
et al, 2008) (Figure 1). This receptor is over-expressed in
subpopulations of lung, prostate and breast cancer
(Chandler et al, 1999). While promising, the attachment of
the ligand to the virus capsid involves chemical
conjugation during which a significant portion of viruses
could become inactive. As a result, obtaining high titers of
active virus for delivery becomes expensive. Alternatively,
retargeted viruses can be genetically engineered through
the abrogation of CAR binding (e.g., Y477A mutation in
adenoviral fiber protein) and insertion of a receptor-
specific binding peptide in the HI loop of the fiber protein
(Piao et al, 2009). In this approach, no chemical
conjugation step is involved. However, one potential
problem with this approach is that targeting peptides with
considerable 3D structure could interfere with the proper
packaging of the viral capsid proteins and result in reduced
transduction efficiency. Furthermore, such alterations in
receptor targeting could impact transduction efficiency of
viruses due to the change in trafficking routes and
internalization pathways (Varga et al, 2000).
B. Are viral vectors highly immunogenic? There are five main classes of viral vectors which
can be categorized into two groups (Table 1) according to
whether their genomes integrate into host cellular
chromatin (oncoretroviruses and lentiviruses) or persist in
the cell nucleus predominantly as extrachromosomal
episomes (AAVs, adenoviruses and herpes viruses).
Figure 1. Schematic representation of cell transfection by
adenoviruses (Ad). While CAR represents coxsackie adenovirus
receptor, FGFR represents fibroblast growth factor receptor
(FGFR).
Table 1. Characteristics of major classes of viral vectors.
Vector Immunogenic
Potential Specificity Limitation Major Advantage
Integrated
Retrovirus Low Dividing Cells only Integration may induce
oncogenesis
Persistent gene
transfer in dividing
cells
Lentivirus Low Broad Integration may induce
oncogenesis
Persistent gene
transfer in most
cells
Episomal
AAV Low Broad Small packaging
capacity
Non-inflammatory
and non-pathogenic
Herpes
Simplex Virus High High in neurons
Transient expression in
some non-neuronal cells
Large packaging
capacity
Adenovirus High Broad (CAR
receptor)
Capsid may induce
inflammatory response
Efficient
transduction of
most cells

Gene Therapy and Molecular Biology Vol 13, page 17
17
Out of these five, only herpes simplex virus (HSV) and
adenovirus (Ad) have been shown to be highly
immunogenic. In general, introduction of any non-self
molecule, including viruses, into the body has the potential
to trigger an immune response. However, the level of
immune response to the foreign entity is dependent on the
dose, the structure and any previous exposures. For
example, a patient (Jesse Gelsinger) who suffered from a
partial deficiency of ornithine transcarbamylase (OTC)
took part in a gene therapy clinical trial conducted at the
University of Pennsylvania in 1999. OTC is a liver
enzyme that is required for the safe removal of excessive
nitrogen from amino acids and proteins. Gelsinger
received the highest dose of vector in the trial (3.8 ! 1013
particles). After 4 hours of treatment Gelsinger developed
a high fever and within four days of treatment he died
from multiorgan failure. A female patient who received a
similar dose (3.6 ! 1013 particles) experienced no
unexpected side effects. It has been speculated that
previous exposure to a wild-type virus infection might
have sensitized Gelsinger’s immune system to the vector
(Bostanci, 2002). If a lowered dose of the adenovirus was
administered, Gelsinger’s symptoms may not have been as
catastrophic. Therefore, drawing a firm conclusion that
viral vectors are highly immunogenic and deadly is
premature.
C. Are non-viral vectors biocompatible? Polymeric or liposomal based non-viral vectors are
utilized to complex plasmid DNA forming stable
nanoparticles. This complexation protects the DNA from
serum endonucleases and also condenses the DNA into
nanosize particles suitable for cellular uptake. Non-viral
polymeric vectors are generally believed to be non-
immunogenic mostly due to their lack of structural
hierarchy. Although there has been reports on the toxicity
of such vectors (e.g., PEI or liposomes) (Lv, Zhang et al,
2006), in general they are assumed to have low
immunogenic potential. Polymers such as poly (ethylene
glycol), for example, have been utilized to sterically
stabilize the surface of particles reducing the interaction of
particles with the elements of the immune system
(Chekhonin et al, 2005). However, two separate groups
recently reported that repeated injection of PEGylated
liposomes in rats and mice elicits PEG-specific IgM/IgG
(Ishida et al, 2006; Judge et al, 2006). These studies
highlight the potential that even a presumably safe
polymer such as PEG can invoke an immune response if
injected in high doses and repeatedly. This in turn may
undermine the ability of PEG to be used as surface
stabilizer in drug delivery systems that need multiple
injections to achieve significant therapeutic response. As a
result, drawing a general conclusion that non-viral vectors
are less immunogenic than viral vectors is also premature
at this point. Therefore, there is a continuing need for the
development of more biocompatible and bio-interactive
polymers that can reduce immunogenicity. This in turn
enhances blood circulation time of drug delivery systems
maximizing their therapeutic efficacy at the target site.
D. Are viral vectors more efficient than
non-viral vectors? 1. Viral vectors versus targeted non-viral
vectors From the available literature, it is apparent that the
efficiency of non-viral vectors is usually compared with
the adenoviral vector which arguably is the most efficient
viral vector (Thomas et al, 2003). As a result of this
comparison, it is generally believed that non-viral vectors
are less efficient. This comparison may not be completely
reliable in all situations as adenoviral vectors are targeted
systems which utilize abundant CAR receptors to enter the
cells (Wickham et al. 1993). When CAR receptors are not
abundant the transfection levels are markedly decreased
(Li et al, 1999). Targeted non-viral vectors are usually
equipped with ligands that are intended to bind to over-
expressed receptors. These include growth factor receptors
(e.g., FGFR and HER2) and transferrin. The abundance of
these receptors on the surface of the cells and their
affinities towards their corresponding ligands may not be
as high as CAR. Therefore, non-viral vectors that could be
as efficient as adenoviruses in trasfecting dividing cells
will show less efficiency when internalizing through
receptors because of the difference in receptor number and
binding affinity. The question then is how viral and
targeted non-viral vectors can be fairly compared in terms
of gene transfer efficiency. One potential solution would
be evaluation of transfection efficiency normalized to the
abundance of the receptor being utilized. This is to remove
the bias associated with the receptor numbers. Another
answer could be as simple as comparison of FGF2 targeted
non-viral vector with FGF2 retargeted adenovirus. In this
approach, the bias associated with receptor binding
affinity and internalization pathway can be eliminated.
Alternatively, adenovirus can be compared with non-viral
vectors that are equipped with CAR ligands to target cells.
In this way the bias associated with the number of entry
gates as well as receptor binding affinity will be
eliminated. It is also noteworthy that the number of viral
or non-viral particles delivered needs to be kept equal to
achieve the same multiplicity of infection (MOI). To date
no study has been reported that has considered the
abovementioned factors in order to appropriately compare
viral versus targeted non-viral vectors.
2. Viral vectors versus non-targeted non-viral
vectors For non-targeted non-viral vectors, the surface
charge of the nanoparticles usually dictates the binding
efficiency to the surface of the cells. Once complexed with
pDNA, the nanoparticles are formulated to have a slight
positive surface charge (e.g., 10-40 mV). This facilitates
binding to the negatively charged phosphate groups on the
surface of the cell membranes resulting in internalization
via caveolae or clathrin mediated endocytosis (Midoux et
al, 2008). In this scenario, comparison of viral with non-
targeted non-viral vectors would not be appropriate as they
utilize entirely different internalization pathways.
Transfection efficiency, in this case, will be dependent on
the cell type not the vector. In one cell line (e.g., CAR

Hatefi and Canine: Perspectives in vector development for systemic cancer gene therapy
18
positive), the viral vector will be more efficient than the
non-viral vector, while in another cell line (e.g., CAR
negative), the non-viral vector will show higher efficiency.
Therefore, drawing any conclusion regarding the
efficiency of viral vectors versus non-targeted non-viral
vector may not be appropriate.
III. Emerging new technologies In recent years there has been a great deal of interest
on the development of recombinant polymers
(biopolymers) with applications in tissue engineering, drug
delivery and gene therapy (Dreher et al, 2006; Furgeson et
al, 2006; Hatefi et al, 2006, 2007; Canine et al, 2008;
Nettles et al, 2008). The major advantage of the polymers
that are genetically engineered versus chemical synthetic
methods is the homogeneity, control over sterotacticity
and full control over the architecture (Urry, 1997). These
biopolymers bear the potential to hybridize the strengths
of both viral and non-viral vectors in order to overcome
the extra- and intracellular barriers to efficient, safe and
cost-effective gene delivery. This is due to their versatility,
flexibility, unlimited capacity and most importantly ability
to bioengineer at the molecular level.
In addition to genetically engineered polymers with
well-defined architecture, synthetic inorganic gene carriers
(e.g., nano- rods and tubes) are exciting, emerging
technologies that would allow precise control of
composition, size and multifunctionality of the delivery
system (Krajcik et al, 2008; Liu et al, 2008). For example,
Leong’s group recently reported a non-viral gene-delivery
system based on multi-segment bimetallic nanorods with
the ability to simultaneously bind condensed plasmid
DNA and targeting ligands in a spatially defined manner
(Salem et al, 2003). Although promising, there are some
concerns related to the toxicity and pharmacological fate
of inorganic nanocarriers (Lacerda et al, 2006).
Nonetheless, synthetic inorganic gene carriers have great
potential to make a significant impact on the science of
cancer gene therapy.
IV. Conclusion Lack of an efficient, non-toxic and non-immunogenic
gene delivery system remains the major limiting factor to
advancements in cancer gene therapy. Adenovirus while
efficient in some cell lines (CAR positive) raises concerns
about safety as well as targetability. Non-viral vectors
while potentially less immunogenic than viral vectors have
not been studied thoroughly enough to reliably state that
they do not trigger major immune responses. Further
studies need to be done in terms of long term
administration, dose scheduling, and treatment thresholds
to examine these effects. The efficiency of non-viral
vectors also needs to be reinvestigated taking into account
the model system being used before blanket comparisons
between non-viral and viral efficiency levels can be made.
In both non-specific viral and non-viral vectors the use of
targeting ligands is an attractive alternative to non-specific
delivery particularly in cancer therapy. No matter which
system, viral or non-viral, improvements in current
technologies continue to be needed.
Acknowledgement This work was supported in part by the NIH
biotechnology training fellowship (GM008336) to Canine
and Reeves
References Bostanci A (2002) Gene therapy. Blood test flags agent in death
of Penn subject. Science 295, 604-5.
Canine BF, Wang Y, Hatefi A (2008) Evaluation of the effect of
vector architecture on DNA condensation and gene transfer
efficiency. J Control Release 129, 117-123.
Chandler LA, Sosnowski BA, Greenlees L, Aukerman SL, Baird
A, Pierce GF (1999) Prevalent expression of fibroblast
growth factor (FGF) receptors and FGF2 in human tumor
cell lines. Int J Cancer 81, 451-8.
Chekhonin VP, Zhirkov YA, Gurina OI, Ryabukhin IA, Lebedev
SV, Kashparov IA, Dmitriyeva TB (2005) PEGylated
immunoliposomes directed against brain astrocytes. Drug
Deliv 12, 1-6.
Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F,
Chilkoti A (2006) Tumor vascular permeability,
accumulation, and penetration of macromolecular drug
carriers. J Natl Cancer Inst 98, 335-44.
Furgeson DY, Dreher MR, Chilkoti A (2006) Structural
optimization of a "smart" doxorubicin-polypeptide conjugate
for thermally targeted delivery to solid tumors. J Control
Release 110, 362-9.
Green NK, Morrison J, Hale S, Briggs SS, Stevenson M, Subr V,
Ulbrich K, Chandler L, Mautner V, Seymour LW, Fisher KD
(2008) Retargeting polymer-coated adenovirus to the FGF
receptor allows productive infection and mediates efficacy in
a peritoneal model of human ovarian cancer. J Gene Med
10, 280-9.
Hatefi A, Cappello J, Ghandehari H (2007) Adenoviral gene
delivery to solid tumors by recombinant silk-elastinlike
protein polymers. Pharm Res 24, 773-9.
Hatefi A, Megeed Z, Ghandehari H (2006) Recombinant
polymer-protein fusion: a promising approach towards
efficient and targeted gene delivery. J Gene Med 8, 468-76.
Ishida T, Ichihara M, Wang X, Yamamoto K, Kimura J, Majima
E, Kiwada H (2006) Injection of PEGylated liposomes in rats
elicits PEG-specific IgM, which is responsible for rapid
elimination of a second dose of PEGylated liposomes. J
Control Release 112, 15-25.
Judge A, McClintock K, Phelps JR, Maclachlan I (2006)
Hypersensitivity and loss of disease site targeting caused by
antibody responses to PEGylated liposomes. Mol Ther 13,
328-37.
Krajcik R, Jung A, Hirsch A, Neuhuber W, Zolk O (2008)
Functionalization of carbon nanotubes enables non-covalent
binding and intracellular delivery of small interfering RNA
for efficient knock-down of genes. Biochem Biophys Res
Commun 369, 595-602.
Lacerda L, Bianco A, Prato M, Kostarelos K (2006) Carbon
nanotubes as nanomedicines: from toxicology to
pharmacology. Adv Drug Deliv Rev 58, 1460-70.
Leopold PL, Crystal RG (2007) Intracellular trafficking of
adenovirus: many means to many ends. Adv Drug Deliv Rev
59, 810-21.
Li D, Duan L, Freimuth P, O'Malley BW Jr (1999) Variability of
adenovirus receptor density influences gene transfer
efficiency and therapeutic response in head and neck cancer.
Clin Cancer Res 5, 4175-81.
Liu Y, Yu ZL, Zhang YM, Guo DS, Liu YP (2008)
Supramolecular architectures of beta-cyclodextrin-modified
chitosan and pyrene derivatives mediated by carbon

Gene Therapy and Molecular Biology Vol 13, page 19
19
nanotubes and their DNA condensation. J Am Chem Soc
130, 10431-9.
Louise C (2006) Nonviral vectors. Methods Mol Biol 333, 201-
26.
Lv H, Zhang S, Wang B, Cui S, Yan J (2006) Toxicity of
cationic lipids and cationic polymers in gene delivery. J
Control Release 114, 100-9.
Midoux P, Breuzard G, Gomez JP, Pichon C (2008) Polymer-
based gene delivery: a current review on the uptake and
intracellular trafficking of polyplexes. Curr Gene Ther 8,
335-52.
Nettles DL, Kitaoka K, Hanson NA, Flahiff CM, Mata BA, Hsu
EW, Chilkoti A, Setton LA (2008) In situ crosslinking
elastin-like polypeptide gels for application to articular
cartilage repair in a goat osteochondral defect model. Tissue
Eng Part A 14, 1133-40.
Pack DW, Hoffman AS, Pun S, Stayton PS (2005) Design and
development of polymers for gene delivery. Nat Rev Drug
Discov 4, 581-93.
Piao Y, Jiang H, Alemany R, Krasnykh V, Marini FC, Xu J,
Alonso MM, Conrad CA, Aldape KD, Gomez-Manzano C,
Fueyo J (2009) Oncolytic adenovirus retargeted to Delta-
EGFR induces selective antiglioma activity. Cancer Gene
Ther 16, 256-65.
Salem AK, Searson PC, Leong KW (2003) Multifunctional
nanorods for gene delivery. Nat Mater 2, 668-71.
Shen Y, Nemunaitis J (2006) Herpes simplex virus 1 (HSV-1)
for cancer treatment. Cancer Gene Ther 13, 975-92.
Spear PG, Shieh MT, Herold BC, WuDunn D, Koshy TI (1992)
Heparan sulfate glycosaminoglycans as primary cell surface
receptors for herpes simplex virus. Adv Exp Med Biol 313,
341-53.
Thomas CE, Ehrhardt A, Kay MA (2003) Progress and problems
with the use of viral vectors for gene therapy. Nat Rev
Genet 4, 346-58.
Urry DW (1997) Physical chemistry of biological free energy
transduction as demonstrated by elastic protein-based
polymers. J Phys Chem B 101, 11007-11028.
Varga CM, Wickham TJ, Lauffenburger DA (2000) Receptor-
mediated targeting of gene delivery vectors: insights from
molecular mechanisms for improved vehicle design.
Biotechnol Bioeng 70, 593-605.
Wickham TJ, Filardo EJ, Cheresh DA, Nemerow GWickham TJ,
Filardo EJ, Cheresh DA, Nemerow GR (1993) Integrins
alpha v beta 3 and alpha v beta 5 promote adenovirus
internalization but not virus attachment. Cell 73, 309-19.
Arash Hatefi

Narala et al: Curcumin is not a ligand for PPAR-!
20
Gene Ther Mol Biol Vol 13, 20-25, 2009
Curcumin is not a ligand for peroxisome
proliferator-activated receptor-! Research Article
Venkata R. Narala1, Monica R. Smith1, Ravi K. Adapala1, Rajesh Ranga1, Kalpana
Panati2, Bethany B. Moore1, Todd Leff3, Vudem D. Reddy2, Anand K. Kondapi4,
Raju C. Reddy1,* 1Department of Internal Medicine, Division of Pulmonary and Critical Care Medicine, University of Michigan Medical
Center, Ann Arbor, MI 48109, USA 2Center for Plant Molecular Biology, Osmania University, Hyderabad 500 007, India 3Center for Integrative Metabolic and Endocrine Research, Wayne State University School of Medicine, Detroit, MI
48201, USA 4Department of Biotechnology, School of Life Sciences, University of Hyderabad, Hyderabad 500 046, India
__________________________________________________________________________________
*Correspondence: Raju C. Reddy M.D., University of Michigan, Division of Pulmonary and Critical Care Medicine, 109 Zina Pitcher
Place, 4062 BSRB, Ann Arbor, MI 48109-2200, USA; Tel: (734) 615-2871; Fax: (734) 615-2111; e-mail: [email protected]
Key words: PPAR-!, TGF-", rosiglitazone, ciglitazone, PPRE, preadipocyte, fibroblast, turmeric, peroxisome, curcumin
Abbreviations: dithiothreitol, (DTT); glutathione-S-transferase, (GST); glyceraldehyde-3-phosphate dehydrogenase, (GAPDH);
isopropyl-1-"-D-galactopyranoside, (IPTG); peroxisome proliferator response element, (PPRE); peroxisome proliferator-activated
receptor-!, (PPAR-!); #-smooth muscle actin, (#-SMA)
Received: 24 February 2009; Revised: 14 March 2009
Accepted: 16 March 2009; electronically published: April 2009
Summary
Curcumin, a compound found in the spice turmeric, has been shown to possess a number of beneficial biological
activities exerted through a variety of different mechanisms. Some curcumin effects have been reported to involve
activation of the nuclear transcription factor peroxisome proliferator-activated receptor-! (PPAR-!), but the
concept that curcumin might be a PPAR-! ligand remains controversial. Results reported here demonstrate that, in
contrast to the PPAR-! ligands ciglitazone and rosiglitazone, curcumin is inactive in five different reporter or DNA-
binding assays, does not displace [3H]rosiglitazone from the PPAR-! ligand-binding site, and does not induce
PPAR-!-dependent differentiation of preadipocytes, while its ability to inhibit fibroblast-to-myofibroblast
differentiation is not affected by any of four PPAR-! antagonists. These multiple lines of evidence conclusively
demonstrate that curcumin is not a PPAR-! ligand and indicate the need for further investigation of the
mechanisms through which the compound acts.
I. Introduction The polyphenol curcumin (diferuloylmethane; 1,7-
bis(4-hydroxy-3-methoxy-phenyl)1,6-heptadiene-3,5-
dione) is an orange-yellow compound with limited water
solubility that is obtained from the turmeric plant,
Curcuma longa. Curcumin has been shown to exhibit a
variety of biological effects (Maheshwari et al, 2006) such
as anti-oxidant, anti-inflammatory, anti-tumor and wound-
healing properties (Srivastava et al, 1995). These activities
are exerted through an equally wide variety of signaling
pathways, which may involve either inhibition (Chen and
Tan, 1998; Gaedeke et al, 2004; Zhou et al, 2007) or
activation (Hu et al, 2005) of specific intracellular
signaling pathways. These varied beneficial effects have
led to investigation of curcumin as a potential therapeutic
agent in a number of disease conditions (Reddy et al,
2005; Thangapazham et al, 2006; Aggarwal et al, 2007).
Peroxisome proliferator-activated receptor-! (PPAR-
!) is a member of the nuclear receptor family of
transcription factors, a large group of proteins that mediate
ligand-dependent transcriptional activation and
transrepression (Issemann and Green, 1990). PPAR-! is
highly expressed in adipose tissue and plays a crucial role
in adipocyte differentiation (Lemberger et al, 1996). It is
also expressed in a variety of other tissue and cell types,
where it plays key roles in the regulation of metabolism
and inflammation. Ligands for PPAR-! include a variety

Gene Therapy and Molecular Biology Vol 13, page 21
21
of natural and synthetic compounds. Most of the natural
ligands are fatty acids or fatty acid derivatives. Synthetic
ligands include the thiazolidinediones, which are used as
insulin sensitizing agents for treatment of type 2 diabetes
(Berger and Moller, 2002).
Curcumin has been reported to activate PPAR-! (Xu
et al, 2003; Zheng and Chen, 2004; Chen and Xu, 2005;
Lin and Chen, 2008). It remains unclear, however,
whether this activation reflects curcumin binding to the
receptor, as has been suggested (Chen and Xu, 2005;
Jacob et al, 2007), or is entirely the result of indirect
effects. The present study, utilizing multiple molecular and
cellular assays, is the first to directly investigate the ability
of curcumin to act as a PPAR-!-activating ligand.
II. Material and Methods
A. Reagents DMEM and DMEM/F12 were purchased from Gibco-BRL
Life Technologies (Grand Island, NY). High purity curcumin
was obtained from Sigma Chemical Co. (St. Louis, MO),
Bioprex (Pune, Maharashtra, India), and Alfa Aesar (Ward Hill,
MA); all experiments were repeated using each formulation.
Fetal bovine serum (FBS) was obtained from HyClone (Logan,
UT). PPAR-! antagonists GW9662 and BADGE were purchased
from Cayman Chemical (Ann Arbor, MI), while PPAR-!
Antagonist III (G3335), and T0070907 were purchased from
Calbiochem (La Jolla, CA). The PPAR-! agonists ciglitazone
and rosiglitazone were purchased from Cayman. Aliquots of
agonists and antagonists were dissolved in
DMSO (Sigma-
Aldrich, St. Louis, MO) at 100 mM and stored
at -30°C until use.
[3H]rosiglitazone was obtained from American Radiolabeled
Chemicals (St. Louis, MO). Anti-glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) mouse monoclonal antibody was
obtained from Abcam (Cambridge, UK), while anti-#-smooth
muscle actin (#-SMA) mouse antibody,
clone 1A4, was obtained
from Dako Automation (Carpentaria, CA), and TGF-"1 was
obtained from R&D Systems (Minneapolis, MN). GAL4-PPAR-
! plasmid was a kind gift from YE Chen, University of
Michigan, Ann Arbor. The aP2-luc plasmid (Camp et al, 2001)
and the FATP-PPRE-luc plasmid (Monajemi et al, 2007) were
constructed as previously described.
B. Cell culture and transfection CV-1 and 3T3-L1 cells were obtained from American
Type Culture Collection (Manassas, VA). IMR-90 cells were
obtained from the Coriell Institute for Medical Research
(Camden, NJ). CV-1 cells were grown to 70% confluence in
DMEM/F12 supplemented with 10% FBS and 1% penicillin-
streptomycin. Cells were then transiently co-transfected with
pRL-SV40 and a PPAR-dependent luciferase reporter, pFATP-
luc. In separate experiments, cells were co-transfected with pRL-
SV40 plus a luciferase gene under the control of four Gal4 DNA-
binding elements (UASG $ 4 TK-luciferase) and a plasmid
containing the ligand-binding domain for PPAR-! fused to the
Gal4 DNA-binding domain. All transfections were performed
using Lipofectamine 2000 (Invitrogen) according to the
manufacturer’s instructions. Twenty-four h after transfection,
cells were treated with test compounds and incubated for an
additional 24 h in medium with 10% FBS. The resulting
luciferase activity was measured with reporter luciferase assay
kits (Promega; Madison, WI) employing a Modulus 9201
luminometer (Turner Biosystems; Sunnydale, CA) and
normalized by comparison to Renilla luciferase.
C. Nuclear protein preparation and PPAR-!-
DNA binding assay CV-1 and IMR-90 cells were plated in 100 mm dishes at
70% confluence. The cells were treated with curcumin or
rosiglitazone for 3 h, after which nuclear protein was isolated
(Cayman nuclear protein extraction kit). Protein concentrations
were estimated using the Bio-Rad (Hercules, CA) DC protein
assay. PPAR-! DNA-binding activity in the nuclear protein was
detected by an ELISA-based PPAR-! transcription factor assay
(Cayman) that detects PPAR-! bound to PPRE-containing
double-stranded DNA sequences.
D. Ligand binding by PPAR-!-GST The ligand binding domain of PPAR-! was introduced into
the pGEX-2T bacterial expression vector (Amersham Pharmacia;
Buckinghamshire, UK). Expression of glutathione-S-transferase
(GST)-tagged PPAR-! in Escherichia coli strain BL21-DE3
(Novagen; San Diego, CA) was induced by the addition of 1 mM
isopropyl-1-"-D-galactopyranoside (IPTG) to the growth
medium. Bacterial extracts were prepared using standard
methods and the fusion proteins were purified using Glutathione
Sepharose 4B (GE Healthcare; Piscataway, NJ). GST-PPAR-!
protein induction and receptor binding was assessed as described
(Fu et al, 2003). Briefly, 5 %g of GST-PPAR-! protein,
[3H]rosiglitazone (specific activity, 5 Ci/mmol), and various
concentrations of curcumin or unlabeled rosiglitazone were
incubated for 2 h at 25°C in a buffer containing 10 mM Tris HCl
(pH 8.0), 50 mM KCl, and 10 mM dithiothreitol (DTT). Bound
[3H]rosiglitazone was separated from free [3H]rosiglitazone by
centrifugation at 8000 rpm for 1 min. The radioactivity of the
bound [3H]rosiglitazone fraction was determined by liquid
scintillation counting.
E. 3T3-L1 differentiation and Oil Red O
staining
3T3-L1
preadipocytes were grown and maintained in
DMEM containing
10% FBS. Differentiation of preadipocytes
was studied in cells 2 days following confluence (designated day
0). These cells were cultured for 14 d in DMEM containing 10%
FBS
and either curcumin or rosiglitazone. The medium was
changed every
2 d. The differentiated
adipocytes were stained by
Oil Red O (Sigma) as described previously (Song et al, 2007).
Briefly, cells were washed with PBS and fixed in 4%
paraformaldehyde for 1 h, followed by rinsing with PBS and
with water. After the rinsing, cells were stained with 0.1% Oil
Red O for 1 h. Plates were rinsed with water and images of cells
on the plate were taken in water.
F. RNA isolation and real-time PCR Total RNA was extracted using TRI-Reagent (Sigma)
according to the manufacturer’s instructions. cDNA was
generated from 1 %g of total RNA and real-time quantitative PCR
was performed using Sybr Green PCR Master Mix (Applied
Biosystems; Foster City, CA) according to the manufacturer’s
protocol. Quantitative changes were expressed relative to "-actin.
Primers used were:
PPAR-!: (F) 5'-ATTCTGGCCCACCAACTTCGG-3'
(R) 5'-TGGAAGCCTGATGCTTTATCCCCA-3'
"-actin: (F) 5'-GTGGGGCGCCCCCAGGCACCA-3'
(R) 5'-GCTCGGCCGTGGTGGTGAAGC-3'
G. Western immunoblotting Cells were lysed in radioimmunoprecipitation (RIPA)
buffer and whole-cell protein was quantified. Ten %g of protein
was subjected to 12% Tris-glycine SDS-PAGE (Invitrogen).
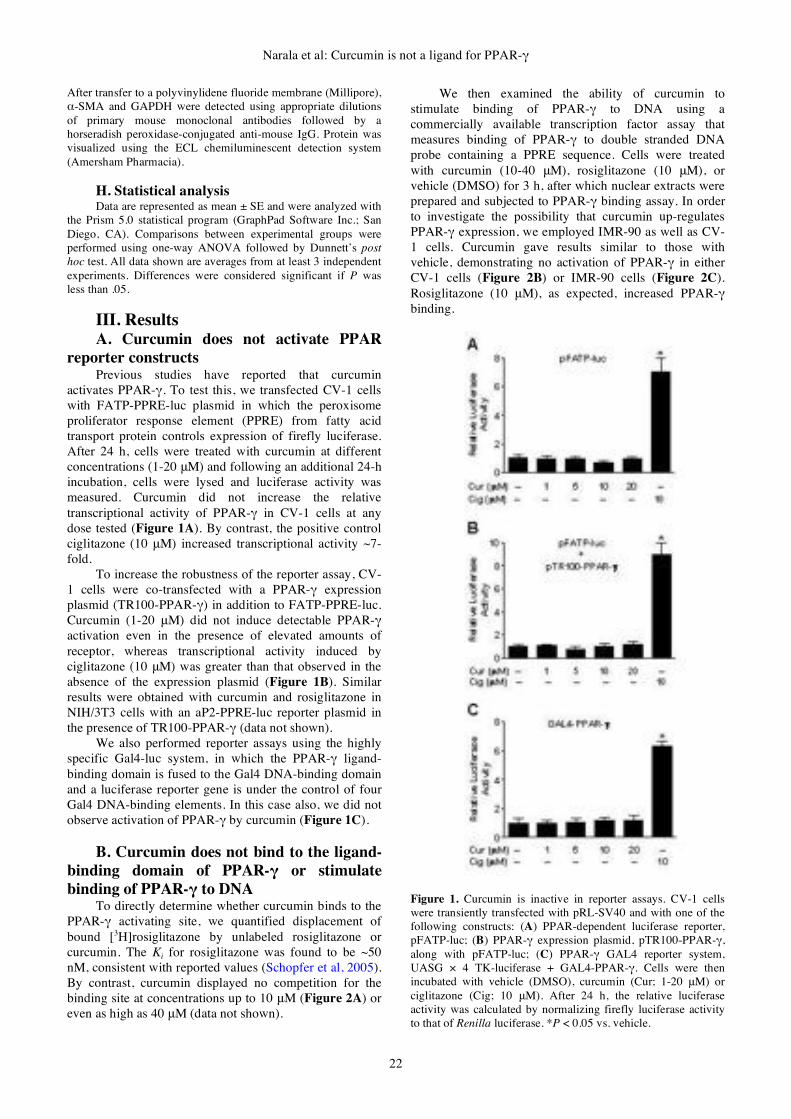
Narala et al: Curcumin is not a ligand for PPAR-!
22
After transfer to a polyvinylidene fluoride membrane (Millipore),
#-SMA and GAPDH were detected using appropriate dilutions
of primary mouse monoclonal antibodies followed by a
horseradish peroxidase-conjugated anti-mouse IgG. Protein was
visualized using the ECL chemiluminescent detection system
(Amersham Pharmacia).
H. Statistical analysis Data are represented as mean ± SE and were analyzed with
the Prism 5.0 statistical program (GraphPad Software Inc.; San
Diego, CA). Comparisons between experimental groups were
performed using one-way ANOVA followed by Dunnett’s post
hoc test. All data shown are averages from at least 3 independent
experiments. Differences were considered significant if P was
less than .05.
III. Results A. Curcumin does not activate PPAR
reporter constructs Previous studies have reported that curcumin
activates PPAR-!. To test this, we transfected CV-1 cells
with FATP-PPRE-luc plasmid in which the peroxisome
proliferator response element (PPRE) from fatty acid
transport protein controls expression of firefly luciferase.
After 24 h, cells were treated with curcumin at different
concentrations (1-20 %M) and following an additional 24-h
incubation, cells were lysed and luciferase activity was
measured. Curcumin did not increase the relative
transcriptional activity of PPAR-! in CV-1 cells at any
dose tested (Figure 1A). By contrast, the positive control
ciglitazone (10 %M) increased transcriptional activity ~7-
fold.
To increase the robustness of the reporter assay, CV-
1 cells were co-transfected with a PPAR-! expression
plasmid (TR100-PPAR-!) in addition to FATP-PPRE-luc.
Curcumin (1-20 %M) did not induce detectable PPAR-!
activation even in the presence of elevated amounts of
receptor, whereas transcriptional activity induced by
ciglitazone (10 %M) was greater than that observed in the
absence of the expression plasmid (Figure 1B). Similar
results were obtained with curcumin and rosiglitazone in
NIH/3T3 cells with an aP2-PPRE-luc reporter plasmid in
the presence of TR100-PPAR-! (data not shown).
We also performed reporter assays using the highly
specific Gal4-luc system, in which the PPAR-! ligand-
binding domain is fused to the Gal4 DNA-binding domain
and a luciferase reporter gene is under the control of four
Gal4 DNA-binding elements. In this case also, we did not
observe activation of PPAR-! by curcumin (Figure 1C).
B. Curcumin does not bind to the ligand-
binding domain of PPAR-! or stimulate
binding of PPAR-! to DNA To directly determine whether curcumin binds to the
PPAR-! activating site, we quantified displacement of
bound [3H]rosiglitazone by unlabeled rosiglitazone or
curcumin. The Ki for rosiglitazone was found to be ~50
nM, consistent with reported values (Schopfer et al, 2005).
By contrast, curcumin displayed no competition for the
binding site at concentrations up to 10 %M (Figure 2A) or
even as high as 40 %M (data not shown).
We then examined the ability of curcumin to
stimulate binding of PPAR-! to DNA using a
commercially available transcription factor assay that
measures binding of PPAR-! to double stranded DNA
probe containing a PPRE sequence. Cells were treated
with curcumin (10-40 %M), rosiglitazone (10 %M), or
vehicle (DMSO) for 3 h, after which nuclear extracts were
prepared and subjected to PPAR-! binding assay. In order
to investigate the possibility that curcumin up-regulates
PPAR-! expression, we employed IMR-90 as well as CV-
1 cells. Curcumin gave results similar to those with
vehicle, demonstrating no activation of PPAR-! in either
CV-1 cells (Figure 2B) or IMR-90 cells (Figure 2C).
Rosiglitazone (10 %M), as expected, increased PPAR-!
binding.
Figure 1. Curcumin is inactive in reporter assays. CV-1 cells
were transiently transfected with pRL-SV40 and with one of the
following constructs: (A) PPAR-dependent luciferase reporter,
pFATP-luc; (B) PPAR-! expression plasmid, pTR100-PPAR-!,
along with pFATP-luc; (C) PPAR-! GAL4 reporter system,
UASG $ 4 TK-luciferase + GAL4-PPAR-!. Cells were then
incubated with vehicle (DMSO), curcumin (Cur; 1-20 %M) or
ciglitazone (Cig; 10 %M). After 24 h, the relative luciferase
activity was calculated by normalizing firefly luciferase activity
to that of Renilla luciferase. *P < 0.05 vs. vehicle.
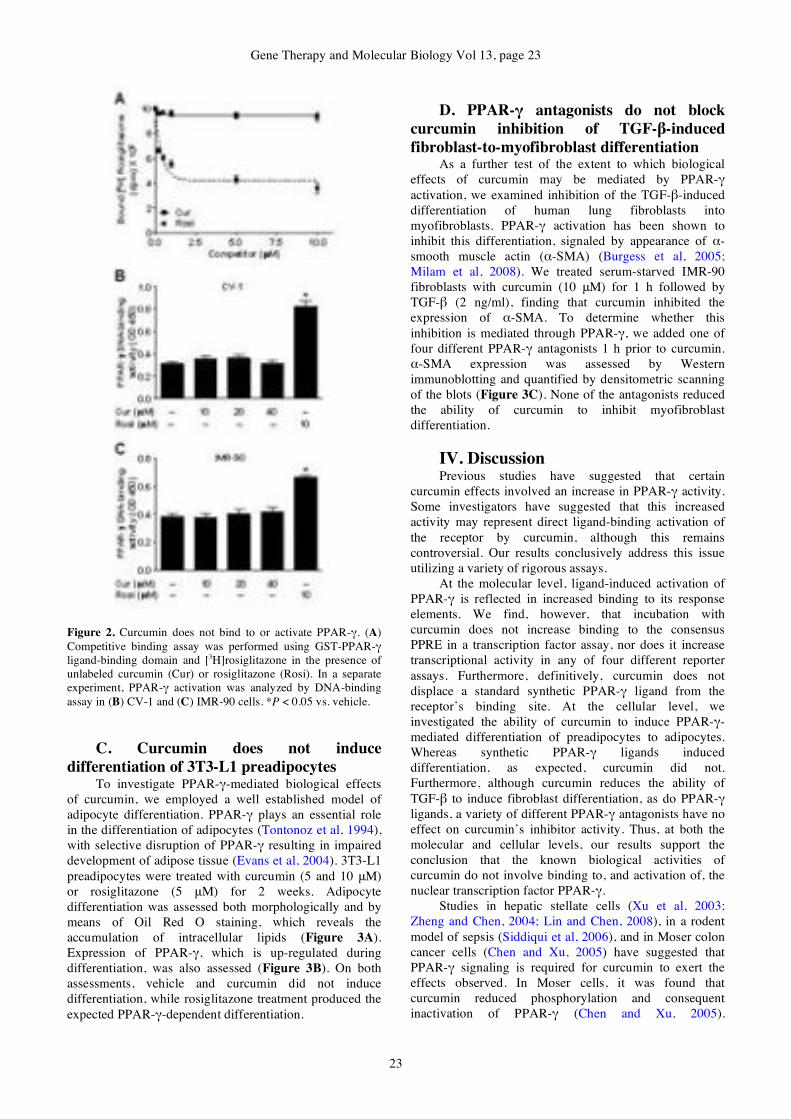
Gene Therapy and Molecular Biology Vol 13, page 23
23
Figure 2. Curcumin does not bind to or activate PPAR-!. (A)
Competitive binding assay was performed using GST-PPAR-!
ligand-binding domain and [3H]rosiglitazone in the presence of
unlabeled curcumin (Cur) or rosiglitazone (Rosi). In a separate
experiment, PPAR-! activation was analyzed by DNA-binding
assay in (B) CV-1 and (C) IMR-90 cells. *P < 0.05 vs. vehicle.
C. Curcumin does not induce
differentiation of 3T3-L1 preadipocytes To investigate PPAR-!-mediated biological effects
of curcumin, we employed a well established model of
adipocyte differentiation. PPAR-! plays an essential role
in the differentiation of adipocytes (Tontonoz et al, 1994),
with selective disruption of PPAR-! resulting in impaired
development of adipose tissue (Evans et al, 2004). 3T3-L1
preadipocytes were treated with curcumin (5 and 10 %M)
or rosiglitazone (5 %M) for 2 weeks. Adipocyte
differentiation was assessed both morphologically and by
means of Oil Red O staining, which reveals the
accumulation of intracellular lipids (Figure 3A).
Expression of PPAR-!, which is up-regulated during
differentiation, was also assessed (Figure 3B). On both
assessments, vehicle and curcumin did not induce
differentiation, while rosiglitazone treatment produced the
expected PPAR-!-dependent differentiation.
D. PPAR-! antagonists do not block
curcumin inhibition of TGF-"-induced
fibroblast-to-myofibroblast differentiation As a further test of the extent to which biological
effects of curcumin may be mediated by PPAR-!
activation, we examined inhibition of the TGF-"-induced
differentiation of human lung fibroblasts into
myofibroblasts. PPAR-! activation has been shown to
inhibit this differentiation, signaled by appearance of #-
smooth muscle actin (#-SMA) (Burgess et al, 2005;
Milam et al, 2008). We treated serum-starved IMR-90
fibroblasts with curcumin (10 %M) for 1 h followed by
TGF-" (2 ng/ml), finding that curcumin inhibited the
expression of #-SMA. To determine whether this
inhibition is mediated through PPAR-!, we added one of
four different PPAR-! antagonists 1 h prior to curcumin.
#-SMA expression was assessed by Western
immunoblotting and quantified by densitometric scanning
of the blots (Figure 3C). None of the antagonists reduced
the ability of curcumin to inhibit myofibroblast
differentiation.
IV. Discussion Previous studies have suggested that certain
curcumin effects involved an increase in PPAR-! activity.
Some investigators have suggested that this increased
activity may represent direct ligand-binding activation of
the receptor by curcumin, although this remains
controversial. Our results conclusively address this issue
utilizing a variety of rigorous assays.
At the molecular level, ligand-induced activation of
PPAR-! is reflected in increased binding to its response
elements. We find, however, that incubation with
curcumin does not increase binding to the consensus
PPRE in a transcription factor assay, nor does it increase
transcriptional activity in any of four different reporter
assays. Furthermore, definitively, curcumin does not
displace a standard synthetic PPAR-! ligand from the
receptor’s binding site. At the cellular level, we
investigated the ability of curcumin to induce PPAR-!-
mediated differentiation of preadipocytes to adipocytes.
Whereas synthetic PPAR-! ligands induced
differentiation, as expected, curcumin did not.
Furthermore, although curcumin reduces the ability of
TGF-" to induce fibroblast differentiation, as do PPAR-!
ligands, a variety of different PPAR-! antagonists have no
effect on curcumin’s inhibitor activity. Thus, at both the
molecular and cellular levels, our results support the
conclusion that the known biological activities of
curcumin do not involve binding to, and activation of, the
nuclear transcription factor PPAR-!.
Studies in hepatic stellate cells (Xu et al, 2003;
Zheng and Chen, 2004; Lin and Chen, 2008), in a rodent
model of sepsis (Siddiqui et al, 2006), and in Moser colon
cancer cells (Chen and Xu, 2005) have suggested that
PPAR-! signaling is required for curcumin to exert the
effects observed. In Moser cells, it was found that
curcumin reduced phosphorylation and consequent
inactivation of PPAR-! (Chen and Xu, 2005).
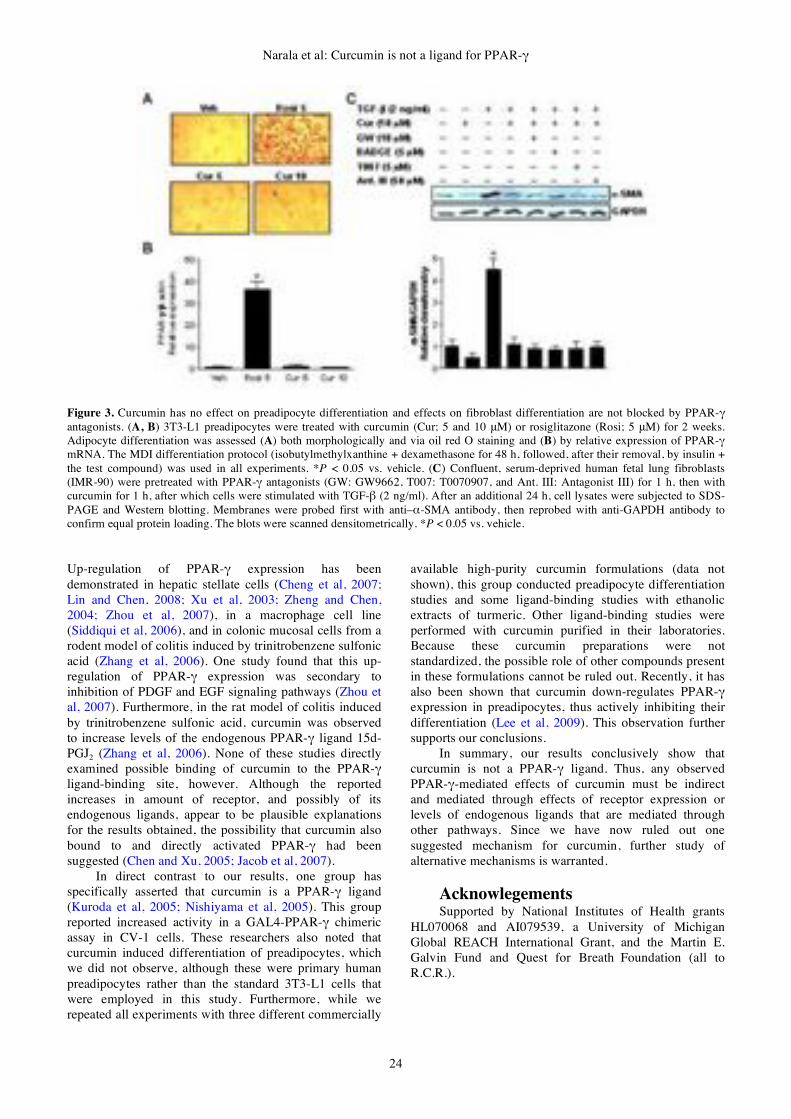
Narala et al: Curcumin is not a ligand for PPAR-!
24
Figure 3. Curcumin has no effect on preadipocyte differentiation and effects on fibroblast differentiation are not blocked by PPAR-! antagonists. (A, B) 3T3-L1 preadipocytes were treated with curcumin (Cur; 5 and 10 %M) or rosiglitazone (Rosi; 5 %M) for 2 weeks.
Adipocyte differentiation was assessed (A) both morphologically and via oil red O staining and (B) by relative expression of PPAR-!
mRNA. The MDI differentiation protocol (isobutylmethylxanthine + dexamethasone for 48 h, followed, after their removal, by insulin +
the test compound) was used in all experiments. *P < 0.05 vs. vehicle. (C) Confluent, serum-deprived human fetal lung fibroblasts
(IMR-90) were pretreated with PPAR-! antagonists (GW: GW9662, T007: T0070907, and Ant. III: Antagonist III) for 1 h, then with
curcumin for 1 h, after which cells were stimulated with TGF-" (2 ng/ml). After an additional 24 h, cell lysates were subjected to SDS-
PAGE and Western blotting. Membranes were probed first with anti–#-SMA antibody, then reprobed with anti-GAPDH antibody to
confirm equal protein loading. The blots were scanned densitometrically. *P < 0.05 vs. vehicle.
Up-regulation of PPAR-! expression has been
demonstrated in hepatic stellate cells (Cheng et al, 2007;
Lin and Chen, 2008; Xu et al, 2003; Zheng and Chen,
2004; Zhou et al, 2007), in a macrophage cell line
(Siddiqui et al, 2006), and in colonic mucosal cells from a
rodent model of colitis induced by trinitrobenzene sulfonic
acid (Zhang et al, 2006). One study found that this up-
regulation of PPAR-! expression was secondary to
inhibition of PDGF and EGF signaling pathways (Zhou et
al, 2007). Furthermore, in the rat model of colitis induced
by trinitrobenzene sulfonic acid, curcumin was observed
to increase levels of the endogenous PPAR-! ligand 15d-
PGJ2 (Zhang et al, 2006). None of these studies directly
examined possible binding of curcumin to the PPAR-!
ligand-binding site, however. Although the reported
increases in amount of receptor, and possibly of its
endogenous ligands, appear to be plausible explanations
for the results obtained, the possibility that curcumin also
bound to and directly activated PPAR-! had been
suggested (Chen and Xu, 2005; Jacob et al, 2007).
In direct contrast to our results, one group has
specifically asserted that curcumin is a PPAR-! ligand
(Kuroda et al, 2005; Nishiyama et al, 2005). This group
reported increased activity in a GAL4-PPAR-! chimeric
assay in CV-1 cells. These researchers also noted that
curcumin induced differentiation of preadipocytes, which
we did not observe, although these were primary human
preadipocytes rather than the standard 3T3-L1 cells that
were employed in this study. Furthermore, while we
repeated all experiments with three different commercially
available high-purity curcumin formulations (data not
shown), this group conducted preadipocyte differentiation
studies and some ligand-binding studies with ethanolic
extracts of turmeric. Other ligand-binding studies were
performed with curcumin purified in their laboratories.
Because these curcumin preparations were not
standardized, the possible role of other compounds present
in these formulations cannot be ruled out. Recently, it has
also been shown that curcumin down-regulates PPAR-!
expression in preadipocytes, thus actively inhibiting their
differentiation (Lee et al, 2009). This observation further
supports our conclusions.
In summary, our results conclusively show that
curcumin is not a PPAR-! ligand. Thus, any observed
PPAR-!-mediated effects of curcumin must be indirect
and mediated through effects of receptor expression or
levels of endogenous ligands that are mediated through
other pathways. Since we have now ruled out one
suggested mechanism for curcumin, further study of
alternative mechanisms is warranted.
Acknowlegements Supported by National Institutes of Health grants
HL070068 and AI079539, a University of Michigan
Global REACH International Grant, and the Martin E.
Galvin Fund and Quest for Breath
Foundation (all to
R.C.R.).

Gene Therapy and Molecular Biology Vol 13, page 25
25
References Aggarwal BB, Sundaram C, Malani N, Ichikawa H (2007)
Curcumin: the Indian solid gold. Adv Exp Med Biol 595, 1-
75.
Berger J, Moller DE (2002) The mechanisms of action of
PPARs. Annu Rev Med 53, 409-435.
Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM,
Redonnet M, Phipps RP, Sime PJ (2005) PPARgamma
agonists inhibit TGF-beta induced pulmonary myofibroblast
differentiation and collagen production: implications for
therapy of lung fibrosis. Am J Physiol Lung Cell Mol
Physiol 288, L1146-1153.
Camp HS, Chaudhry A, Leff T (2001) A novel potent antagonist
of peroxisome proliferator-activated receptor gamma blocks
adipocyte differentiation but does not revert the phenotype of
terminally differentiated adipocytes. Endocrinology 142,
3207-3213.
Chen A, Xu J (2005) Activation of PPAR! by curcumin inhibits
Moser cell growth and mediates suppression of gene
expression of cyclin D1 and EGFR. Am J Physiol
Gastrointest Liver Physiol 288, G447-456.
Chen Y-R, Tan T-H (1998) Inhibition of the c-Jun N-terminal
kinase (JNK) signaling pathway by curcumin. Oncogene 17,
173-178.
Cheng Y, Ping J, Xu LM (2007) Effects of curcumin on
peroxisome proliferator-activated receptor ! expression and
nuclear translocation/redistribution in culture-activated rat
hepatic stellate cells. Chin Med J (Engl) 120, 794-801.
Evans RM, Barish GD, Wang YX (2004) PPARs and the
complex journey to obesity. Nat Med 10, 355-361.
Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De
Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia
G, Piomelli D (2003) Oleylethanolamide regulates feeding
and body weight through activation of the nuclear receptor
PPAR-alpha. Nature 425, 90-93.
Gaedeke J, Noble NA, Border WA (2004) Curcumin blocks
multiple sites of the TGF-" signaling cascade in renal cells.
Kidney Int 66, 112-120.
Hu M, Du Q, Vancurova I, Lin X, Miller EJ, Simms HH, Wang
P (2005) Proapoptotic effect of curcumin on human
neutrophils: activation of the p38 mitogen-activated protein
kinase pathway. Crit Care Med. 33, 2571-2578.
Issemann I, Green S (1990) Activation of a member of the
steroid hormone receptor superfamily by peroxisome
proliferators. Nature 347, 645-650.
Jacob A, Wu R, Zhou M, Wang P (2007) Mechanism of the
Anti-inflammatory Effect of Curcumin: PPAR-gamma
Activation. PPAR Res 2007, 89369.
Kuroda M, Mimaki Y, Nishiyama T, Mae T, Kishida H,
Tsukagawa M, Takahashi K, Kawada T, Nakagawa K,
Kitahara M (2005) Hypoglycemic effects of turmeric
(Curcuma longa L. rhizomes) on genetically diabetic KK-Ay
mice. Biol Pharm Bull 28, 937-939.
Lee YK, Lee WS, Hwang JT, Kwon DY, Surh YJ, Park OJ
(2009) Curcumin exerts antidifferentiation effect through
AMPKalpha-PPAR-gamma in 3T3-L1 adipocytes and
antiproliferatory effect through AMPKalpha-COX-2 in
cancer cells. J Agric Food Chem. 57, 305-310.
Lemberger T, Desvergne B, Wahli W (1996) Peroxisome
proliferator-activated receptors: a nuclear receptor signaling
pathway in lipid physiology. Annu Rev Cell Dev Biol 12,
335-363.
Lin J, Chen A (2008) Activation of peroxisome proliferator-
activated receptor-! by curcumin blocks the signaling
pathways for PDGF and EGF in hepatic stellate cells. Lab
Invest 88, 529-540.
Maheshwari RK, Singh AK, Gaddipati J, Srimal RC (2006)
Multiple biological activities of curcumin: a short review.
Life Sci 78, 2081-2087.
Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR,
Hogaboam CM, Standiford TJ, Thannickal VJ, Reddy RC
(2008) PPAR-gamma agonists inhibit profibrotic phenotypes
in human lung fibroblasts and bleomycin-induced pulmonary
fibrosis. Am J Physiol Lung Cell Mol Physiol 294, L891-
901.
Monajemi H, Zhang L, Li G, Jeninga EH, Cao H, Maas M,
Brouwer CB, Kalkhoven E, Stroes E, Hegele RA, Leff T
(2007) Familial partial lipodystrophy phenotype resulting
from a single-base mutation in deoxyribonucleic acid-
binding domain of peroxisome proliferator-activated
receptor-gamma. J Clin Endocrinol Metab 92, 1606-1612.
Nishiyama T, Mae T, Kishida H, Tsukagawa M, Mimaki Y,
Kuroda M, Sashida Y, Takahashi K, Kawada T, Nakagawa
K, Kitahara M (2005) Curcuminoids and sesquiterpenoids in
turmeric (Curcuma longa L.) suppress an increase in blood
glucose level in type 2 diabetic KK-Ay mice. J Agric Food
Chem. 53, 959-963.
Reddy RC, Vatsala PG, Keshamouni VG, Padmanaban G,
Rangarajan PN (2005) Curcumin for malaria therapy.
Biochem Biophys Res Commun 326, 472-474.
Schopfer FJ, Lin Y, Baker PRS, Cui T, Garcia-Barrio M, Zhang
J, Chen K, Chen YE, Freeman BA (2005) Nitrolinoleic acid:
an endogenous peroxisome proliferator-activated receptor !
ligand. Proc Natl Acad Sci U S A 102, 2340-2345.
Siddiqui AM, Cui X, Wu R, Dong W, Zhou M, Hu M, Simms
HH, Wang P (2006) The anti-inflammatory effect of
curcumin in an experimental model of sepsis is mediated by
up-regulation of peroxisome proliferator-activated receptor-
!. Crit Care Med 34, 1874-1882.
Song DH, Getty-Kaushik L, Tseng E, Simon J, Corkey BE,
Wolfe MM (2007) Glucose-dependent insulinotropic
polypeptide enhances adipocyte development and glucose
uptake in part through Akt activation. Gastroenterology
133, 1796-1805.
Srivastava KC, Bordia A, Verma SK (1995) Curcumin, a major
component of food spice turmeric (Curcuma longa) inhibits
aggregation and alters eicosanoid metabolism in human
blood platelets. Prostaglandins Leukot Essent Fatty Acids
52, 223-227.
Thangapazham RL, Sharma A, Maheshwari RK (2006) Multiple
molecular targets in cancer chemoprevention by curcumin.
AAPS J 8, E443-449.
Tontonoz P, Hu E, Spiegelman BM (1994) Stimulation of
adipogenesis in fibroblasts by PPAR gamma 2, a lipid-
activated transcription factor. Cell 79, 1147-1156.
Xu J, Fu Y, Chen A (2003) Activation of peroxisome
proliferator-activated receptor-! contributes to the inhibitory
effects of curcumin on rat hepatic stellate cell growth. Am J
Physiol Gastrointest Liver Physiol 285, G20-30.
Zhang M, Deng C, Zheng J, Xia J, Sheng D (2006) Curcumin
inhibits trinitrobenzene sulphonic acid-induced colitis in rats
by activation of peroxisome proliferator-activated receptor !.
Int Immunopharmacol 6, 1233-1242.
Zheng S, Chen A (2004) Activation of PPAR! is required for
curcumin to induce apoptosis and to inhibit the expression of
extracellular matrix genes in hepatic stellate cells in vitro.
Biochem J 384, 149-157.
Zhou Y, Zheng S, Lin J, Zhang QJ, Chen A (2007) The
interruption of the PDGF and EGF signaling pathways by
curcumin stimulates gene expression of PPAR! in rat
activated hepatic stellate cell in vitro. Lab Invest 87, 488-
498.

Hochwald and Golubovskaya: FAK and cancer therapy
26
Gene Ther Mol Biol Vol 13, 26-35, 2009
FAK as a target for cancer therapy Review Article
Steven N. Hochwald*, Vita M. Golubovskaya
Department of Surgery, University of Florida College of Medicine, Gainesville, Florida
__________________________________________________________________________________
*Correspondence: Steven N. Hochwald MD, Department of Surgery, University of Florida College of Medicine, 1600 SW Archer
Road, P.O. Box 100109, Gainesville, FL 32609, USA; Tel: 352-265-0761, Fax: 352-265-0262, e-mail:
Key words: ocal Adhesion Kinase; malignancy; cancer; Y15
Abbreviations: FAK, (Focal Adhesion Kinase); FERM, (Focal Adhesion Kinase Ezrin/Radixin/Moesin; FRNK, (FAK-related non
kinase)
This work was supported by the following NIH grant: CA113766 (S.N.H.)
Received: 20 March 2009; Accepted: 24 March 2009; electronically published: April 2009
Summary
We have learned that malignant cells are similar to normal cells in the signaling pathways that they use. However,
cancer cells acquire aberrations that favor their growth in the complex environments of living tissues. This includes
their ability to invade and metastasize and their ability to grow and divide indefinitely. The progression of human
cancer is characterized by a process of tumor cell motility, invasion, and metastasis to distant sites, requiring the
cancer cells to be able to survive the apoptotic pressures of anchorage-independent conditions. One of the main
tyrosine kinases that are linked to this malignant phenotype is the Focal Adhesion Kinase (FAK). FAK is
overexpressed in many types of tumors and recently has been proposed to be a target for anti-cancer therapy. In
this review, we will review the FAK structure, its role in signaling, and FAK targeted therapy approaches in
malignancy.
I. Introduction Despite recent advances in surgery, chemotherapy
and radiation treatment, survival of patients with advanced
malignancy remains suboptimal. Fortunately, our
understanding of the origins of cancer has changed
dramatically over the last twenty-five years, owing in large
part to the revolution in molecular biology that has
changed all biomedical research. Powerful experimental
tools are available to cancer biologists and have made it
possible to uncover and dissect the complex molecular
machinery operating inside normal and malignant cells. In
addition, these tools have allowed researchers to pinpoint
the defects that cause cancer cells to signal and proliferate
abnormally.
Focal Adhesion Kinase (FAK) was discovered about
15 years ago as a tyrosine phosphorylated protein kinase.
Investigations in several laboratories have shown that this
protein plays a critical role in intracellular processes of
cell adhesion, motility, survival, and cell cycle
progression. The FAK gene encodes a non-receptor
tyrosine kinase that localizes at contact points of cells with
extracellular matrix and is activated by integrin (cell
surface receptor) signaling. The FAK gene was first
isolated from chicken embryo fibroblasts transformed by
v-src (Schaller et al, 1992). Subsequently, the FAK gene
was identified in human tumors, and FAK mRNA has
been shown to be up-regulated in invasive and metastatic
human breast and colon cancer samples as compared to
normal tissues (Weiner et al, 1993). This was the first
evidence that FAK might be regulated at the level of gene
transcription. Subsequently, up-regulation of FAK has
been demonstrated at the protein level in a wide variety of
human tumors, including breast cancer, colon cancer,
ovarian cancer, thyroid cancer, melanoma, and sarcoma
(Owens et al, 1995, 1996; Judson et al, 1999; Cance et al,
2000). Recently, the regulatory promoter region of the
FAK gene was cloned and confirmed transcriptional up-
regulation in cancer cell lines (Golubovskaya et al, 2004).
II. Molecular structure of focal
adhesion kinase The human FAK (also known as PTK2a) gene has
been mapped to chromosome 8 (Fiedorek, Jr. and Kay,
1995; Agochiya et al, 1999), and there appears to be a high

Gene Therapy and Molecular Biology Vol 13, page 27
27
degree of homology between vertebrate species. Human
complete FAK mRNA sequence (NCBI Accession
number: L13616) is 3791 bases long and includes a 5’-
untranslated 233 base pair region (Whitney et al, 1993).
Human FAK cDNA was first isolated from primary
sarcoma tissue and increased FAKmRNA was seen in
tumor samples compared with normal tissue samples
(Weiner et al, 1993). Subsequently, Xenopus laevis FAK
cDNA (Zhang et al, 1995) and rat FAK cDNA (Burgaya
and Girault, 1996) were identified. Recently, Drosophila
FAK cDNA (Dfak56) was isolated (Fujimoto et al, 1999).
FAK cDNA is closely related to the homologous proline-
rich calcium dependent tyrosine kinase (45% amino-acid
identity) that is also located on human chromosome 8,
locus p21.1, named PYK2 (RAFTK (related adhesion
focal tyrosine kinase), CADTK (calcium-dependent
tyrosine kinase), CAK (cell adhesion kinase) b, PTK 2b
(protein tyrosine kinase 2b) (Avraham et al, 1995; Lev et
al, 1995; Sasaki et al, 1995).
The gene coding FAK contains 34 exons (NCBI
Gene ID: 5747), and genomic sequence spans 230 kb
(Corsi et al, 2006). The FAK gene contains four 5’ non-
coding exons and 34 coding exons and has been shown to
have multiple alternatively spliced forms. Comparison of
the mouse and human FAK genes detected conservative
and non-conservative 5’-untranslated exons that suggests a
complex regulation of FAK expression. Exons (Sasaki et
al, 1995; Burgaya and Girault, 1996; Fujimoto et al, 1999;
Golubovskaya et al, 2002) are highly conserved among
vertebrate species, suggesting their critical function in
gene regulation (Corsi et al, 2006).
It is known that alternative splicing often occurs and
plays an important role in cancer (Caballero et al, 2001;
Venables, 2006). Alternative splicing most often results
from different exon inclusion, but can also occur from
intron retention or alternative choice between two splice
sites leading to changes in protein localization, structure,
removal of phosphorylation sites, or proteasomal
degradation (Venables, 2006). There were several cases of
alternatively spliced genes that are involved in invasion
and metastasis (Rac 1, !-catenin, Crk) or angiogenesis
(VEGFR-2, VEGFR-3 (Flt-4)). Thus, detailed study of
alternatively spliced forms of FAK that are overexpressed
in pre- and metastatic cancers will be critical for
understanding mechanisms and regulation of FAK
expression in carcinogenesis, either by changes in mRNA,
by changes in the coding sequence (exon
inclusion/exclusion), or by changes in protein levels
(stability, etc.).
The human FAK promoter regulating FAK
expression contains 600 base pairs and includes many
transcription binding sites, such as AP-1, AP-2, SP-1,
PU.1, GCF, TCF-1, EGR-1, NF-!B and p53
(Golubovskaya et al, 2004). Interestingly, two
transcription binding sites for p53 have been identified in
the FAK promoter, and p53 can block FAK promoter
activity (Golubovskaya et al, 2004). Recently, the mouse
promoter has been cloned and found to be highly
homologous to the human promoter and contains the same
binding sites (Corsi et al, 2006). In addition, the FAK gene
has an internal FRNK promoter or C-terminal, FAK-CD
promoter that has been recently cloned by Parsons group
(Hayasaka et al, 2005), regulating expression of
autonomously expressed FRNK protein.
A. FAK protein structure The FAK protein is a 125 kDa tyrosine kinase
(p125FAK) with a large amino-N-terminal domain,
exhibiting homology with a FERM (protein 4.1, ezrin,
radixin and moesin) domain with an autophosphorylation
site (Y-397), a central catalytic domain, and a large
carboxy-C-terminal domain that contains a number of
potential protein interacting sites, including two proline-
rich domains and FAT domain (Schaller and Parsons,
1994; Schaller et al, 1994; Hanks and Polte, 1997) (Figure
1).
B. The kinase domain The central kinase domain of FAK (amino acids 424-
676) contains the Y576 and Y577, major phosphorylation
sites, and also K454, which is the ATP binding site
(Figure 1). Phosphorylation of FAK by Src on Y576 and
Y577 is an important step in the formation of an active
signaling complex and is required for maximal FAK
enzymatic activity (Calalb et al, 1995). The crystal
structure of the FAK kinase domain reveals an open
conformation similar to other kinases (Nowakowski et al,
2002). The FAK kinase domain structure has an unusual
bisulphite bond between the conserved cysteines 456 and
459, suggesting a possible role in protein-protein
interactions and kinase function (Nowakowski et al, 2002).
The ATP binding site of protein kinases is the most
common target for the small-molecule inhibitors, although
the design and specificity of these inhibitors can be
complicated by structural similarities between kinase
domains. Thus, finding small structural differences
between the ATP binding site of kinases is crucial in the
design of small molecule kinase inhibitors. For example,
the side chain of glutamic acid, E506 forms a bifurcated
hydrogen bond to the 2’ and 3’ hydroxyl groups of the
ribose (Nowakowski et al, 2002). The corresponding side
chains in EphA2 and Aurora-A kinases are smaller and do
not contact with sugar (Nowakowski et al, 2002).
C. The N-terminal domain The first function of the N-terminal, homologous to
FERM domain was linked to the binding of integrins, via
their ! subunits (Schaller et al, 1995). The N-terminal
domain of FAK protein contains the major
autophosphorylation site Y397-tyrosine, that in its
phosphorylated form becomes a binding site of the SH-2
domain of Src, leading to its conformational changes and
activation (Hanks and Polte, 1997). Tyrosine
phosphorylation of FAK and binding of Src leads to
tyrosine phosphorylation of other tyrosine phosphorylation
sites of FAK: Y407; Y576,Y577- major phosphorylation
sites in the catalytic domain of FAK; Y861 and Y925
(Hanks and Polte, 1997; McLean et al, 2005), and to
phosphorylation of FAK binding proteins, such as paxillin
and Cas (Schaller et al, 1999). This leads to subsequent
cytoskeletal changes and activation of RAS-MAPK
(mitogen-activated protein kinase) signaling pathways
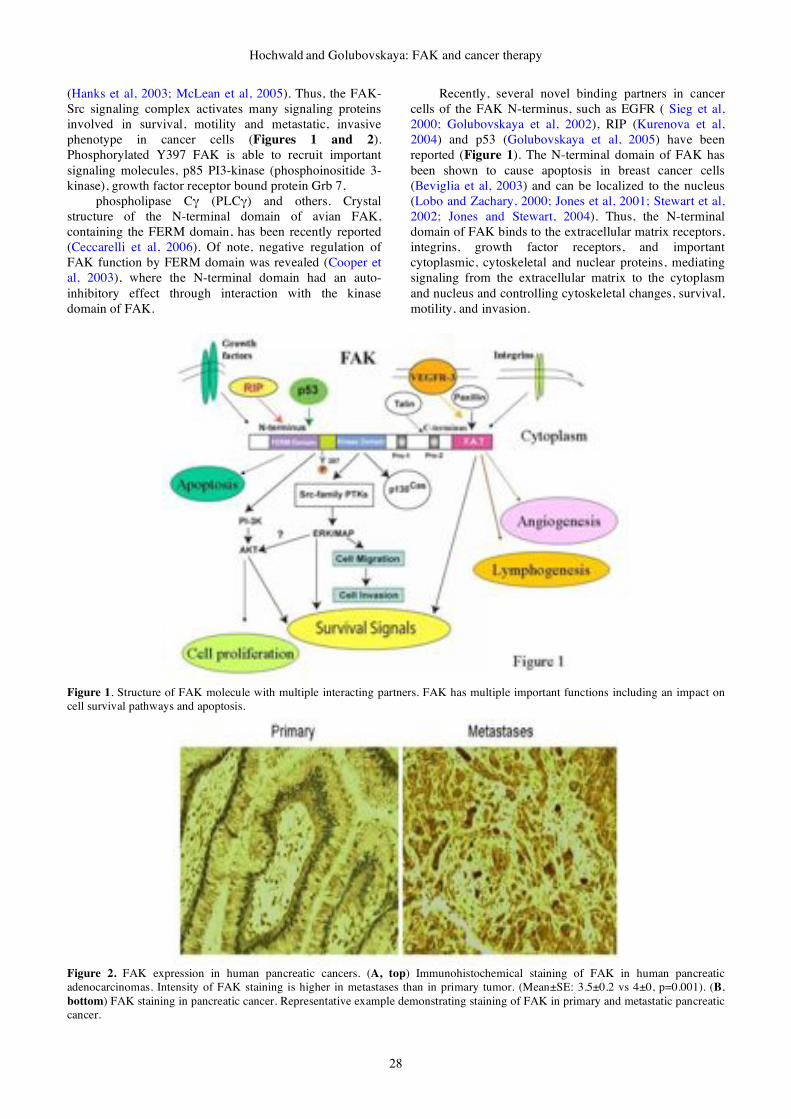
Hochwald and Golubovskaya: FAK and cancer therapy
28
(Hanks et al, 2003; McLean et al, 2005). Thus, the FAK-
Src signaling complex activates many signaling proteins
involved in survival, motility and metastatic, invasive
phenotype in cancer cells (Figures 1 and 2).
Phosphorylated Y397 FAK is able to recruit important
signaling molecules, p85 PI3-kinase (phosphoinositide 3-
kinase), growth factor receptor bound protein Grb 7,
phospholipase C" (PLC") and others. Crystal
structure of the N-terminal domain of avian FAK,
containing the FERM domain, has been recently reported
(Ceccarelli et al, 2006). Of note, negative regulation of
FAK function by FERM domain was revealed (Cooper et
al, 2003), where the N-terminal domain had an auto-
inhibitory effect through interaction with the kinase
domain of FAK.
Recently, several novel binding partners in cancer
cells of the FAK N-terminus, such as EGFR ( Sieg et al,
2000; Golubovskaya et al, 2002), RIP (Kurenova et al,
2004) and p53 (Golubovskaya et al, 2005) have been
reported (Figure 1). The N-terminal domain of FAK has
been shown to cause apoptosis in breast cancer cells
(Beviglia et al, 2003) and can be localized to the nucleus
(Lobo and Zachary, 2000; Jones et al, 2001; Stewart et al,
2002; Jones and Stewart, 2004). Thus, the N-terminal
domain of FAK binds to the extracellular matrix receptors,
integrins, growth factor receptors, and important
cytoplasmic, cytoskeletal and nuclear proteins, mediating
signaling from the extracellular matrix to the cytoplasm
and nucleus and controlling cytoskeletal changes, survival,
motility, and invasion.
Figure 1. Structure of FAK molecule with multiple interacting partners. FAK has multiple important functions including an impact on
cell survival pathways and apoptosis.
Figure 2. FAK expression in human pancreatic cancers. (A, top) Immunohistochemical staining of FAK in human pancreatic
adenocarcinomas. Intensity of FAK staining is higher in metastases than in primary tumor. (Mean±SE: 3.5±0.2 vs 4±0, p=0.001). (B,
bottom) FAK staining in pancreatic cancer. Representative example demonstrating staining of FAK in primary and metastatic pancreatic
cancer.

Gene Therapy and Molecular Biology Vol 13, page 29
29
D. The C-terminal domain Different proteins can bind to the C-terminal domain
of FAK (amino acids 677-1052), including paxillin,
p130cas, PI3-kinase, and GTP-ase-activating protein Graf,
leading to changes in the cytoskeleton and to activation of
the Ras-MAP kinase pathway (Schaller and Parsons, 1994;
Windham et al, 2002; Hanks et al, 2003; Parsons, 2003).
The carboxy-terminal domain of FAK contains sequences
responsible for its targeting to focal adhesions, also known
as the FAT domain. Alternative splicing of FAK results in
autonomous expression of the C-terminal part of FAK,
FAK-related non-kinase (FRNK) (Richardson and
Parsons, 1995). The crystal structure of the C-terminal
domain of FAK, FAT, has been determined recently by
several groups (Hayashi et al, 2002; Prutzman et al, 2004)
and structure analysis demonstrates that it can exist as a
dimer or monomer, allowing binding of several binding
partners.
E. Post-translational protein
modifications FAK function is altered by post-translational
modifications including phosphorylation of tyrosines or
serines. FAK has numerous tyrosine phosphorylated sites:
Y397, Y407, Y576/Y577, Y861 and Y925.
Phosphorylation of Y397, creates a binding site for Src,
PI3K, PLC-g, Grb-7 and Grb-2-SOS. Phosphorylation of
tyrosine 407, as well as Y397, correlated with
differentiation and with the level of gastrin-releasing
peptide and its receptor in colon cancer cells (Matkowskyj
et al, 2003). Phosphorylation of Y576 and Y577 correlated
with maximal activity of FAK (Calalb et al, 1995). Src-
dependent phosphorylation of Y861 was induced by
VEGFR in HUVEC endothelial cells (bu-Ghazaleh et al,
2001). FAT domain mediates signaling through Grb-2
binding to Y925 site of FAK (Arold et al, 2002).
Inhibition of FAK that resulted in decreased Y925
phosphorylation of FAK resulted in decreased FAK-Grb2-
MAPK signaling and VEGFR-induced tumor growth of
4T1 breast carcinoma cells (Mitra et al, 2006).
In addition to tyrosine phosphorylation, several
serine phosphorylation sites have been reported to play a
major role in FAK function, such as serines 722, 732, 843
and 910. The role of serine phosphorylation is less
described than phosphorylation of tyrosines but was
suggested to play a role in binding/stability of proteins
(Parsons, 2003).
In addition, recent mass spectrometry analysis of
chicken FAK revealed 19 new sites of phosphorylation
with some sites reported before: 15 serine, 5 threonine,
and 5 tyrosine residues (Grigera et al, 2005). The authors
suggested that coordinated phosphorylation of FAK by
tyrosine and serine/threonine-specific kinases may be
critical a step in regulation of FAK function (Grigera et al,
2005). Some of the sites were present only in chicken
FAK, such as S386, T388 and T393, but several chicken
phosphorylation sites were conserved in human, mouse,
and frog species, such as S29, Y155, S390, S392, T394,
Y397, T406, Y407, Y570, T700, S708, S722, S725, S726,
S732, S766, S845 (S843 in human), S894, Y899 and S911
(S910 in human and mouse) (Grigera et al, 2005). Thus,
now there are total of 30 sites of phosphorylation of FAK,
including those reported before, requiring detailed analysis
of their biological functioning in vivo.
III. FAK functioning in cells Attachment to the underlying extracellular matrix
provides cells with both a means of anchorage needed for
traction during migration via a link to the actin
cytoskeleton and also with intracellular structures that
house membrane-associated signaling proteins. This leads
to the transmission of biochemical signals into the cell
interior to induce multiple biological responses. Loss of
regulation of the process of adhesion formation or
turnover, or of downstream signaling is likely to contribute
to primary tumor development and/or tumor
dissemination. Signaling via adhesion-associated kinases
controls the changes that are necessary for cell migration
including regulation of cell-matrix adhesion turnover and
coordination of remodeling of the actin cytoskeleton
network (Cance et al, 2000). FAK has numerous functions
in cell survival, motility, metastasis, invasion, and
angiogenesis. FAK has also been shown to be important
for cell motility (Hauck et al, 2001; Schaller, 2001; Hanks
et al, 2003; Schlaepfer and Mitra, 2004). FAK-null
embryos exhibit decreased motility in vitro (Ilic et al,
1995). Furthermore, forced expression of FAK stimulated
cell migration (Hildebrand et al, 1993; Sieg et al, 1999).
Cell migration is initiated by protrusion at the leading edge
of the cell, by the formation of peripheral adhesions,
exertion of force on these adhesions, and then the release
of the adhesions at the rear of the cell (Tilghman et al,
2005). FAK has been shown to be required for the
organization of the leading edge in migrating cells by
coordinating integrin signaling in order to direct the
correct activation of membrane protrusion (Tilghman et al,
2005). SH2 domain of Src, targeting Src to focal adhesions
and Y397 activity has been shown to be important for
motility (Yeo et al, 2006). PI3 kinase has been also shown
to be critical for FAK-mediated motility in Chinese
hamster ovary (CHO) cells (Reiske et al, 1999). Tumor
suppressor gene PTEN, encoding phosphatase has been
shown to interact with FAK, causing its dephosphorylation
and blocked motility (Tamura et al, 1998). Moreover,
Y397FAK was important for PTEN interaction with FAK
(Tamura et al, 1999). Overexpression of FAK reversed the
inhibitory effect of PTEN on cell migration (Tamura et al,
1998).
Activation of FAK is linked to invasion and
metastasis signaling pathways. FAK was important in Erb-
2/Erb3-induced oncogenic transformation and invasion
(Benlimame et al, 2005). Inhibition of FAK in FAK-
proficient invasive cancer cells prevented cell invasion and
metastasis processes (Benlimame et al, 2005). In addition,
FAK has been shown to be activated in invading
fibrosarcoma and regulated metastasis (Hanada et al,
2005). Inhibition of FAK with dominant-negative FAK-
CD disrupted invasion of cancer cells (Hauck et al, 2001).
We have also shown high FAK expression in breast
cancers associated with an aggressive tumor phenotype
(Lark et al, 2005). Subsequently, we analyzed FAK
expression in pre-invasive ductal carcinoma in situ, DCIS

Hochwald and Golubovskaya: FAK and cancer therapy
30
tumors and detected protein overexpression in preinvasive
tumors (Lightfoot, Jr. et al, 2004), suggesting that FAK
survival function occurs as an early event in breast
tumorigenesis.
FAK plays a major role in survival signaling and has
been linked to detachment-induced apoptosis or anoikis
(Frisch et al, 1996). It has been shown that constitutively
activated forms of FAK rescued epithelial cells from
anoikis, suggesting that FAK can regulate this process
(Frisch et al, 1996; Frisch and Ruoslahti, 1997; Frisch,
1999; Frisch and Screaton, 2001; Windham et al, 2002).
Similarly, both FAK antisense oligonucleotides (Xu et al,
1996; Smith et al, 2005), as well as dominant-negative
FAK protein (FAK-CD), caused cell detachment and
apoptosis in tumor cells (Xu et al, 1996, 1998, 2000; van
de et al, 2001; Golubovskaya et al, 2002, 2003; Beviglia et
al, 2003; Gabarra-Niecko et al, 2003; Park et al, 2004b).
The anti-apoptotic role of FAK was also demonstrated in
FAK-transfected FAK/HL60 cells that were highly
resistant to apoptosis induced with etoposide and hydrogen
peroxide compared with the parental HL-60 cells or the
vector-transfected cells (Sonoda et al, 2000; Kasahara et
al, 2002). HL-60/FAK cells activated the AKT pathway
and NF-!B survival pathways with the induction of
inhibitor-of-apoptosis proteins, IAPs (Sonoda et al, 2000).
We have demonstrated that EGFR and Src signaling
cooperate with FAK survival signaling in colon and breast
cancer cells (Golubovskaya et al, 2002, 2003; Park et al,
2004a,b). We have also demonstrated that simultaneous
inhibition of Src and FAK or EGFR and FAK pathways
was able to increase apoptosis in cancer cells
(Golubovskaya et al, 2002, 2003). Thus, cancer cells use
the cooperative function of kinases and growth factor
receptor signaling to increase survival.
Vascular endothelial growth factor (VEGF) is one of
the known angiogenic growth factors, stimulating
formation of new blood vessels or angiogenesis. FAK has
been shown to play a major role in vasculogenesis. It has
been shown that VEGF induced tyrosine phosphorylation
of FAK in human umbilical vein endothelial cells
(HUVEC) and other endothelial cell lines (Abedi and
Zachary, 1997). VEGF-induced stimulation of FAK
phosphorylation was also demonstrated in cultured rat
cardiac myocytes that was accompanied by subcellular
translocation of FAK from perinuclear sites to the focal
adhesions and increased association with the adaptor
proteins Shc, Grb-2 and c-Src (Takahashi et al, 1999).
VEGF-induced phosphorylation of FAK was inhibited by
the tyrosine kinase inhibitors tyrphostin and genistein
(Takahashi et al, 1999). VEGF-induced phosphorylation
of FAK was induced in human brain microvascular
endothelial cell (HBMEC) (Avraham et al, 2003).
Furthermore, inhibition of FAK with the dominant-
negative inhibitor FRNK (FAK-related non-kinase) or the
C-terminal FAK (FAK-CD) significantly decreased
HBMEC spreading and migration (Avraham et al, 2003,
2004). In addition, angiogenic inhibitor endostatin blocked
VEGF-induced activation of FAK (Kim et al, 2002).
Recently, we have shown that FAK binds to VEGFR-3
(Flt-4) protein in cancer cell lines (Garces et al, 2006),
suggesting an important role of FAK in lymphogenesis in
addition to angiogenesis. We have shown that the C-
terminal domain of FAK binds to VEGFR-3. Disruption of
this binding with VEGFR peptides caused apoptosis in
breast cancer cells, allowing novel therapeutic approaches
in breast tumors (Garces et al, 2006). The detailed
interaction of FAK and VEGFR signaling and its
mechanisms remain to be discovered in the future.
IV. FAK as a target for therapy Recently, several reports describe the properties of
FAK inhibitors in vitro and in vivo. FAK has been
proposed to be a new therapeutic target (McLean et al,
2005). Initial studies which evaluated the effects of FAK
inhibition in preclinical models focused on dominant
negative mutants of FAK, antisense oligonucleotides and
siRNAs (Parsons et al, 2008). More recently, scientists at
Novartis Pharmaceuticals designed and synthesized a
series of 2-amino-9-aryl-7H-pyrrolo[2,3-d]pyrimidines to
inhibit FAK using molecular modeling in conjunction with
a co-crystal structure (Choi et al, 2006). Chemistry was
developed to introduce functionality onto the 9-aryl ring,
which resulted in the identification of potent FAK
inhibitors. We and others have published reports on the
use of such FAK inhibitors that have targeted the ATP
binding site in the kinase domain. In human pancreatic
cancer, we have shown widespread expression of FAK in
primary pancreatic adenocarcinoma. In addition, we have
shown significant upregulation of FAK protein expression
in metastatic lesions (Figure 2, unpublished data). In
human pancreatic cancer cells, we have identified that the
FAK kinase inhibitor, TAE226, decreases viability,
increases cell detachment and increases apoptosis (Liu et
al, 2008). Other studies have shown that TAE226 readily
induced apoptosis in human breast cancer cells with
overexpressed Src or EGFR. Of note, these cells were
resistant to adenoviral FAK dominant negative treatment,
indicating that kinase inhibition was important for
downregulation of FAK function and the observed
phenotypic changes (Golubovskaya et al, 2008b).
Subsequent studies have studied the in vivo effects of
TAE226. The expression status of FAK in Barrett’s
esophageal adenocarcinoma has been recently reported.
FAK expression was studied in frank adenocarcinoma,
areas of Barrett’s epithelia, squamous epithelia, and gastric
epithelia. FAK expression was increased in cancerous
parts compared to non-cancerous areas and strong
expression (>50% positive staining cells per area) were
observed in 94% of Barrett’s esophageal adenocarcinoma
compared with 18% of Barrett’s epithelia. In a
subcutaneous model of human esophageal cancer,
TAE226 given orally at 30 mg/kg significantly decreased
tumor volume and weight compared with placebo
(Watanabe et al, 2008). Similar results from in vivo studies
have confirmed the ability of TAE226 to decrease the
growth of ovarian and glioma xenografts (Shi et al, 2007).
While initial results with kinase inhibition of FAK
has shown anti-neoplastic effects, TAE226 has been
shown to also inhibit the activity of IGF-1R at nanomolar
concentrations (Liu et al, 2007). Therefore, the activities
against multiple tumor types likely reflect its dual
inhibition of adhesion and growth promoting pathways.
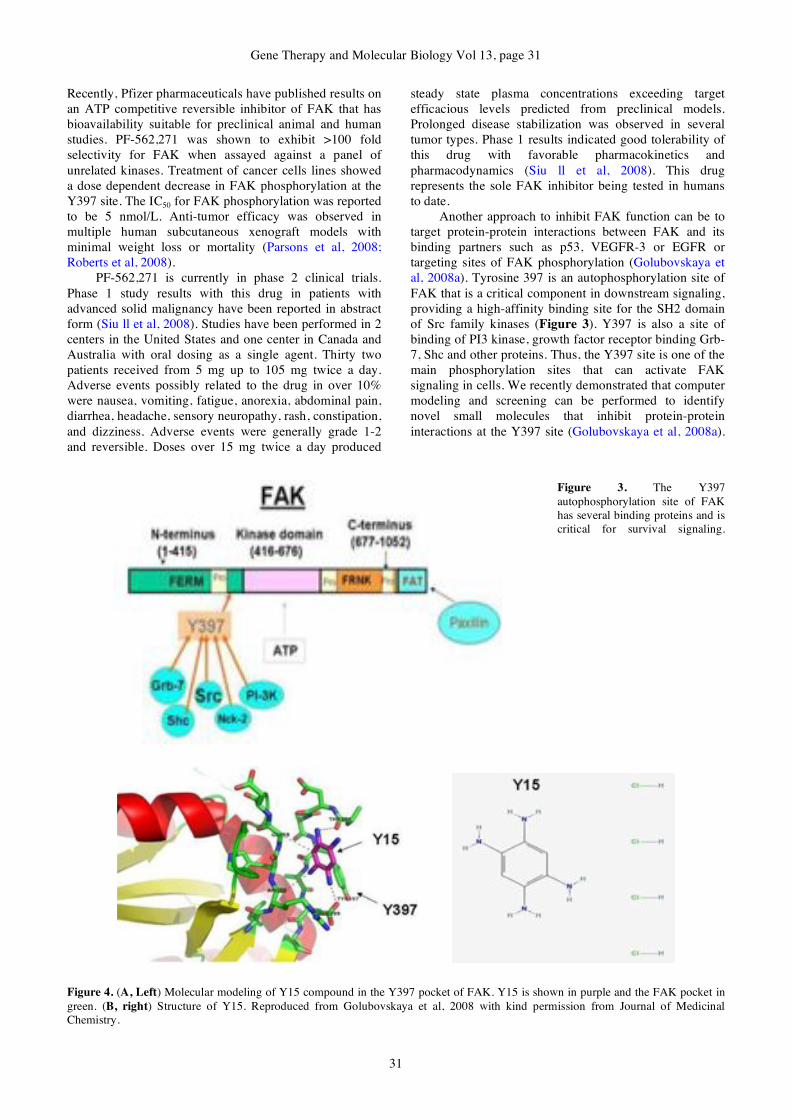
Gene Therapy and Molecular Biology Vol 13, page 31
31
Recently, Pfizer pharmaceuticals have published results on
an ATP competitive reversible inhibitor of FAK that has
bioavailability suitable for preclinical animal and human
studies. PF-562,271 was shown to exhibit >100 fold
selectivity for FAK when assayed against a panel of
unrelated kinases. Treatment of cancer cells lines showed
a dose dependent decrease in FAK phosphorylation at the
Y397 site. The IC50 for FAK phosphorylation was reported
to be 5 nmol/L. Anti-tumor efficacy was observed in
multiple human subcutaneous xenograft models with
minimal weight loss or mortality (Parsons et al, 2008;
Roberts et al, 2008).
PF-562,271 is currently in phase 2 clinical trials.
Phase 1 study results with this drug in patients with
advanced solid malignancy have been reported in abstract
form (Siu ll et al, 2008). Studies have been performed in 2
centers in the United States and one center in Canada and
Australia with oral dosing as a single agent. Thirty two
patients received from 5 mg up to 105 mg twice a day.
Adverse events possibly related to the drug in over 10%
were nausea, vomiting, fatigue, anorexia, abdominal pain,
diarrhea, headache, sensory neuropathy, rash, constipation,
and dizziness. Adverse events were generally grade 1-2
and reversible. Doses over 15 mg twice a day produced
steady state plasma concentrations exceeding target
efficacious levels predicted from preclinical models.
Prolonged disease stabilization was observed in several
tumor types. Phase 1 results indicated good tolerability of
this drug with favorable pharmacokinetics and
pharmacodynamics (Siu ll et al, 2008). This drug
represents the sole FAK inhibitor being tested in humans
to date.
Another approach to inhibit FAK function can be to
target protein-protein interactions between FAK and its
binding partners such as p53, VEGFR-3 or EGFR or
targeting sites of FAK phosphorylation (Golubovskaya et
al, 2008a). Tyrosine 397 is an autophosphorylation site of
FAK that is a critical component in downstream signaling,
providing a high-affinity binding site for the SH2 domain
of Src family kinases (Figure 3). Y397 is also a site of
binding of PI3 kinase, growth factor receptor binding Grb-
7, Shc and other proteins. Thus, the Y397 site is one of the
main phosphorylation sites that can activate FAK
signaling in cells. We recently demonstrated that computer
modeling and screening can be performed to identify
novel small molecules that inhibit protein-protein
interactions at the Y397 site (Golubovskaya et al, 2008a).
Figure 3. The Y397
autophosphorylation site of FAK
has several binding proteins and is
critical for survival signaling.
Figure 4. (A, Left) Molecular modeling of Y15 compound in the Y397 pocket of FAK. Y15 is shown in purple and the FAK pocket in
green. (B, right) Structure of Y15. Reproduced from Golubovskaya et al, 2008 with kind permission from Journal of Medicinal
Chemistry.
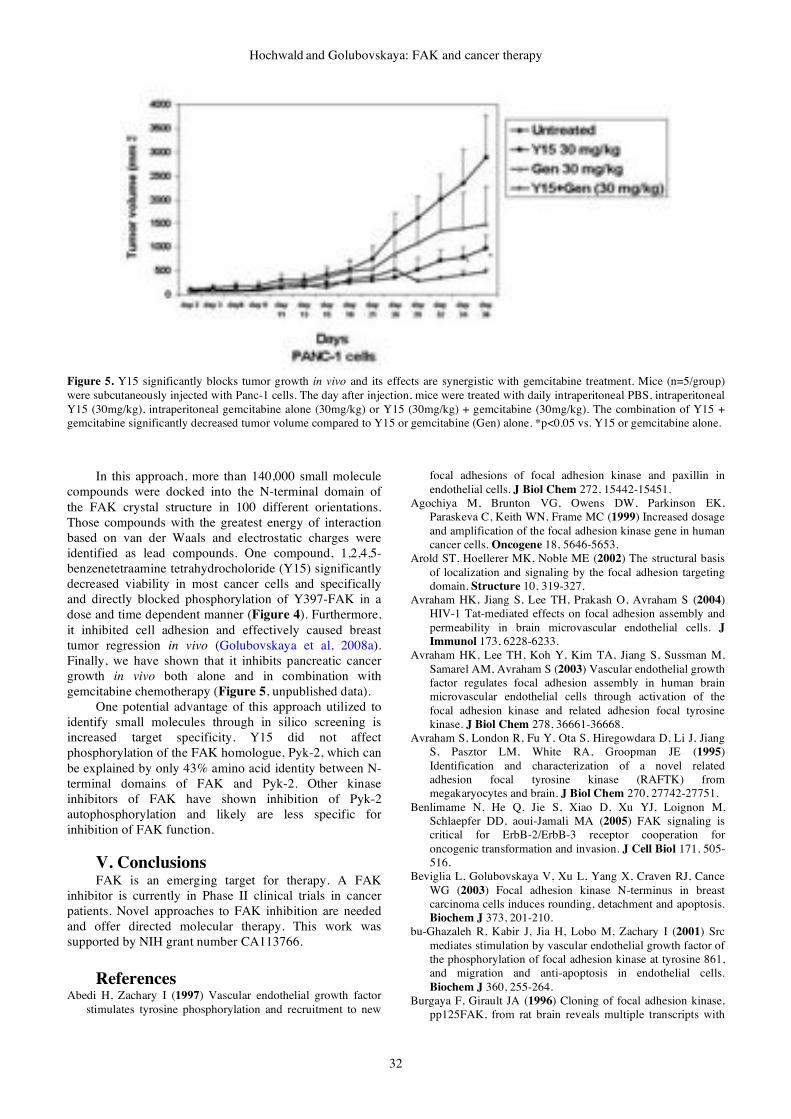
Hochwald and Golubovskaya: FAK and cancer therapy
32
Figure 5. Y15 significantly blocks tumor growth in vivo and its effects are synergistic with gemcitabine treatment. Mice (n=5/group)
were subcutaneously injected with Panc-1 cells. The day after injection, mice were treated with daily intraperitoneal PBS, intraperitoneal
Y15 (30mg/kg), intraperitoneal gemcitabine alone (30mg/kg) or Y15 (30mg/kg) + gemcitabine (30mg/kg). The combination of Y15 +
gemcitabine significantly decreased tumor volume compared to Y15 or gemcitabine (Gen) alone. *p<0.05 vs. Y15 or gemcitabine alone.
In this approach, more than 140,000 small molecule
compounds were docked into the N-terminal domain of
the FAK crystal structure in 100 different orientations.
Those compounds with the greatest energy of interaction
based on van der Waals and electrostatic charges were
identified as lead compounds. One compound, 1,2,4,5-
benzenetetraamine tetrahydrocholoride (Y15) significantly
decreased viability in most cancer cells and specifically
and directly blocked phosphorylation of Y397-FAK in a
dose and time dependent manner (Figure 4). Furthermore,
it inhibited cell adhesion and effectively caused breast
tumor regression in vivo (Golubovskaya et al, 2008a).
Finally, we have shown that it inhibits pancreatic cancer
growth in vivo both alone and in combination with
gemcitabine chemotherapy (Figure 5, unpublished data).
One potential advantage of this approach utilized to
identify small molecules through in silico screening is
increased target specificity. Y15 did not affect
phosphorylation of the FAK homologue, Pyk-2, which can
be explained by only 43% amino acid identity between N-
terminal domains of FAK and Pyk-2. Other kinase
inhibitors of FAK have shown inhibition of Pyk-2
autophosphorylation and likely are less specific for
inhibition of FAK function.
V. Conclusions FAK is an emerging target for therapy. A FAK
inhibitor is currently in Phase II clinical trials in cancer
patients. Novel approaches to FAK inhibition are needed
and offer directed molecular therapy. This work was
supported by NIH grant number CA113766.
References Abedi H, Zachary I (1997) Vascular endothelial growth factor
stimulates tyrosine phosphorylation and recruitment to new
focal adhesions of focal adhesion kinase and paxillin in
endothelial cells. J Biol Chem 272, 15442-15451.
Agochiya M, Brunton VG, Owens DW, Parkinson EK,
Paraskeva C, Keith WN, Frame MC (1999) Increased dosage
and amplification of the focal adhesion kinase gene in human
cancer cells. Oncogene 18, 5646-5653.
Arold ST, Hoellerer MK, Noble ME (2002) The structural basis
of localization and signaling by the focal adhesion targeting
domain. Structure 10, 319-327.
Avraham HK, Jiang S, Lee TH, Prakash O, Avraham S (2004)
HIV-1 Tat-mediated effects on focal adhesion assembly and
permeability in brain microvascular endothelial cells. J
Immunol 173, 6228-6233.
Avraham HK, Lee TH, Koh Y, Kim TA, Jiang S, Sussman M,
Samarel AM, Avraham S (2003) Vascular endothelial growth
factor regulates focal adhesion assembly in human brain
microvascular endothelial cells through activation of the
focal adhesion kinase and related adhesion focal tyrosine
kinase. J Biol Chem 278, 36661-36668.
Avraham S, London R, Fu Y, Ota S, Hiregowdara D, Li J, Jiang
S, Pasztor LM, White RA, Groopman JE (1995)
Identification and characterization of a novel related
adhesion focal tyrosine kinase (RAFTK) from
megakaryocytes and brain. J Biol Chem 270, 27742-27751.
Benlimame N, He Q, Jie S, Xiao D, Xu YJ, Loignon M,
Schlaepfer DD, aoui-Jamali MA (2005) FAK signaling is
critical for ErbB-2/ErbB-3 receptor cooperation for
oncogenic transformation and invasion. J Cell Biol 171, 505-
516.
Beviglia L, Golubovskaya V, Xu L, Yang X, Craven RJ, Cance
WG (2003) Focal adhesion kinase N-terminus in breast
carcinoma cells induces rounding, detachment and apoptosis.
Biochem J 373, 201-210.
bu-Ghazaleh R, Kabir J, Jia H, Lobo M, Zachary I (2001) Src
mediates stimulation by vascular endothelial growth factor of
the phosphorylation of focal adhesion kinase at tyrosine 861,
and migration and anti-apoptosis in endothelial cells.
Biochem J 360, 255-264.
Burgaya F, Girault JA (1996) Cloning of focal adhesion kinase,
pp125FAK, from rat brain reveals multiple transcripts with

Gene Therapy and Molecular Biology Vol 13, page 33
33
different patterns of expression. Brain Res Mol Brain Res
37, 63-73.
Caballero OL, de Souza SJ, Brentani RR, Simpson AJ (2001)
Alternative spliced transcripts as cancer markers. Dis
Markers 17, 67-75.
Calalb MB, Polte TR, Hanks SK (1995) Tyrosine
phosphorylation of focal adhesion kinase at sites in the
catalytic domain regulates kinase activity: a role for Src
family kinases. Mol Cell Biol 15, 954-963.
Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J,
Simkins S, Xu L (2000) Immunohistochemical analyses of
focal adhesion kinase expression in benign and malignant
human breast and colon tissues: correlation with preinvasive
and invasive phenotypes. Clin Cancer Res 6, 2417-2423.
Ceccarelli DF, Song HK, Poy F, Schaller MD, Eck MJ (2006)
Crystal structure of the FERM domain of focal adhesion
kinase. J Biol Chem 281, 252-259.
Choi HS, Wang Z, Richmond W, He X, Yang K, Jiang T,
Karanewsky D, Gu XJ, Zhou V, Liu Y, Che J, Lee CC,
Caldwell J, Kanazawa T, Umemura I, Matsuura N, Ohmori
O, Honda T, Gray N, He Y (2006) Design and synthesis of
7H-pyrrolo[2,3-d]pyrimidines as focal adhesion kinase
inhibitors. Part 2. Bioorg Med Chem Lett 16, 2689-2692.
Cooper LA, Shen TL, Guan JL (2003) Regulation of focal
adhesion kinase by its amino-terminal domain through an
autoinhibitory interaction. Mol Cell Biol 23, 8030-8041.
Corsi JM, Rouer E, Girault JA, Enslen H (2006) Organization
and post-transcriptional processing of focal adhesion kinase
gene. BMC Genomics 7, 198.
Fiedorek FT, Jr., Kay ES (1995) Mapping of the focal adhesion
kinase (Fadk) gene to mouse chromosome 15 and human
chromosome 8. Mamm Genome 6, 123-126.
Frisch SM (1999) Evidence for a function of death-receptor-
related, death-domain-containing proteins in anoikis. Curr
Biol 9, 1047-1049.
Frisch SM, Ruoslahti E (1997) Integrins and anoikis. Curr Opin
Cell Biol 9, 701-706.
Frisch SM, Screaton RA (2001) Anoikis mechanisms. Curr
Opin Cell Biol 13, 555-562.
Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY (1996) Control
of adhesion-dependent cell survival by focal adhesion kinase.
J Cell Biol 134, 793-799.
Fujimoto J, Sawamoto K, Okabe M, Takagi Y, Tezuka T,
Yoshikawa S, Ryo H, Okano H, Yamamoto T (1999)
Cloning and characterization of Dfak56, a homolog of focal
adhesion kinase, in Drosophila melanogaster. J Biol Chem
274, 29196-29201.
Gabarra-Niecko V, Schaller MD, Dunty JM (2003) FAK
regulates biological processes important for the pathogenesis
of cancer. Cancer Metastasis Rev 22, 359-374.
Garces CA, Kurenova EV, Golubovskaya VM, Cance WG
(2006) Vascular endothelial growth factor receptor-3 and
focal adhesion kinase bind and suppress apoptosis in breast
cancer cells. Cancer Res 66, 1446-1454.
Golubovskaya V, Beviglia L, Xu LH, Earp HS, III, Craven R,
Cance W (2002) Dual inhibition of focal adhesion kinase and
epidermal growth factor receptor pathways cooperatively
induces death receptor-mediated apoptosis in human breast
cancer cells. J Biol Chem 277, 38978-38987.
Golubovskaya V, Kaur A, Cance W (2004) Cloning and
characterization of the promoter region of human focal
adhesion kinase gene: nuclear factor kB and p53 binding
sites. Biochim Biophys Acta 1678, 111-125.
Golubovskaya VM, Finch R, Cance WG (2005) Direct
interaction of the N-terminal domain of focal adhesion kinase
with the N-terminal transactivation domain of p53. J Biol
Chem 280, 25008-25021.
Golubovskaya VM, Gross S, Kaur AS, Wilson RI, Xu LH, Yang
XH, Cance WG (2003) Simultaneous inhibition of focal
adhesion kinase and SRC enhances detachment and
apoptosis in colon cancer cell lines. Mol Cancer Res 1, 755-
764.
Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A,
Ostrov D, Cance WG (2008a) A small molecule inhibitor,
1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the
y397 site of focal adhesion kinase decreases tumor growth. J
Med Chem 51, 7405-7416.
Golubovskaya VM, Virnig C, Cance WG (2008b) TAE226-
induced apoptosis in breast cancer cells with overexpressed
Src or EGFR. Mol Carcinog 47, 222-234.
Grigera PR, Jeffery ED, Martin KH, Shabanowitz J, Hunt DF,
Parsons JT (2005) FAK phosphorylation sites mapped by
mass spectrometry. J Cell Sci 118, 4931-4935.
Hanada M, Tanaka K, Matsumoto Y, Nakatani F, Sakimura R,
Matsunobu T, Li X, Okada T, Nakamura T, Takasaki M,
Iwamoto Y (2005) Focal adhesion kinase is activated in
invading fibrosarcoma cells and regulates metastasis. Clin
Exp Metastasis 22, 485-494.
Hanks SK, Polte TR (1997) Signaling through focal adhesion
kinase. Bioessays 19, 137-145.
Hanks SK, Ryzhova L, Shin NY, Brabek J (2003) Focal adhesion
kinase signaling activities and their implications in the
control of cell survival and motility. Front Biosci 8, d982-
d996.
Hauck CR, Sieg DJ, Hsia DA, Loftus JC, Gaarde WA, Monia
BP, Schlaepfer DD (2001) Inhibition of focal adhesion
kinase expression or activity disrupts epidermal growth
factor-stimulated signaling promoting the migration of
invasive human carcinoma cells. Cancer Res 61, 7079-7090.
Hayasaka H, Simon K, Hershey ED, Masumoto KH, Parsons JT
(2005) FRNK, the autonomously expressed C-terminal
region of focal adhesion kinase, is uniquely regulated in
vascular smooth muscle: analysis of expression in transgenic
mice. J Cell Biochem 95, 1248-1263.
Hayashi I, Vuori K, Liddington RC (2002) The focal adhesion
targeting (FAT) region of focal adhesion kinase is a four-
helix bundle that binds paxillin. Nat Struct Biol 9, 101-106.
Hildebrand JD, Schaller MD, Parsons JT (1993) Identification of
sequences required for the efficient localization of the focal
adhesion kinase, pp125FAK, to cellular focal adhesions. J
Cell Biol 123, 993-1005.
Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N,
Nomura S, Fujimoto J, Okada M, Yamamoto T (1995)
Reduced cell motility and enhanced focal adhesion contact
formation in cells from FAK-deficient mice. Nature 377,
539-544.
Jones G, Machado J, Jr., Merlo A (2001) Loss of focal adhesion
kinase (FAK) inhibits epidermal growth factor receptor-
dependent migration and induces aggregation of nh(2)-
terminal FAK in the nuclei of apoptotic glioblastoma cells.
Cancer Res 61, 4978-4981.
Jones G, Stewart G (2004) Nuclear import of N-terminal FAK by
activation of the FcepsilonRI receptor in RBL-2H3 cells.
Biochem Biophys Res Commun 314, 39-45.
Judson PL, He X, Cance WG, Van LL (1999) Overexpression of
focal adhesion kinase, a protein tyrosine kinase, in ovarian
carcinoma. Cancer 86, 1551-1556.
Kasahara T, Koguchi E, Funakoshi M, izu-Yokota E, Sonoda Y
(2002) Antiapoptotic action of focal adhesion kinase (FAK)
against ionizing radiation. Antioxid Redox Signal 4, 491-
499.
Kim YM, Hwang S, Kim YM, Pyun BJ, Kim TY, Lee ST, Gho
YS, Kwon YG (2002) Endostatin blocks vascular endothelial
growth factor-mediated signaling via direct interaction with
KDR/Flk-1. J Biol Chem 277, 27872-27879.

Hochwald and Golubovskaya: FAK and cancer therapy
34
Kurenova E, Xu LH, Yang X, Baldwin AS, Jr., Craven RJ,
Hanks SK, Liu ZG, Cance WG (2004) Focal adhesion kinase
suppresses apoptosis by binding to the death domain of
receptor-interacting protein. Mol Cell Biol 24, 4361-4371.
Lark AL, Livasy CA, Dressler L, Moore DT, Millikan RC,
Geradts J, Iacocca M, Cowan D, Little D, Craven RJ, Cance
W (2005) High focal adhesion kinase expression in invasive
breast carcinomas is associated with an aggressive
phenotype. Mod Pathol 18, 1289-1294.
Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM,
Plowman GD, Rudy B, Schlessinger J (1995) Protein
tyrosine kinase PYK2 involved in Ca(2+)-induced regulation
of ion channel and MAP kinase functions. Nature 376, 737-
745.
Lightfoot HM, Jr., Lark A, Livasy CA, Moore DT, Cowan D,
Dressler L, Craven RJ, Cance WG (2004) Upregulation of
focal adhesion kinase (FAK) expression in ductal carcinoma
in situ (DCIS) is an early event in breast tumorigenesis.
Breast Cancer Res Treat 88, 109-116.
Liu TJ, LaFortune T, Honda T, Ohmori O, Hatakeyama S, Meyer
T, Jackson D, de GJ, Yung WK (2007) Inhibition of both
focal adhesion kinase and insulin-like growth factor-I
receptor kinase suppresses glioma proliferation in vitro and
in vivo. Mol Cancer Ther 6, 1357-1367.
Liu W, Bloom DA, Cance WG, Kurenova EV, Golubovskaya
VM, Hochwald SN (2008) FAK and IGF-IR interact to
provide survival signals in human pancreatic
adenocarcinoma cells. Carcinogenesis 29, 1096-1107.
Lobo M, Zachary I (2000) Nuclear localization and apoptotic
regulation of an amino-terminal domain focal adhesion
kinase fragment in endothelial cells. Biochem Biophys Res
Commun 276, 1068-1074.
Matkowskyj KA, Keller K, Glover S, Kornberg L, Tran-Son-Tay
R, Benya RV (2003) Expression of GRP and its receptor in
well-differentiated colon cancer cells correlates with the
presence of focal adhesion kinase phosphorylated at
tyrosines 397 and 407. J Histochem Cytochem 51, 1041-
1048.
McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton
VG, Frame MC (2005) The role of focal-adhesion kinase in
cancer - a new therapeutic opportunity. Nat Rev Cancer 5,
505-515.
Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A,
Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, Cheresh
DA, Schlaepfer DD (2006) Intrinsic FAK activity and Y925
phosphorylation facilitate an angiogenic switch in tumors.
Oncogene 25, 5969-5984.
Nowakowski J, Cronin CN, McRee DE, Knuth MW, Nelson CG,
Pavletich NP, Rogers J, Sang BC, Scheibe DN, Swanson RV,
Thompson DA (2002) Structures of the cancer-related
Aurora-A, FAK, and EphA2 protein kinases from
nanovolume crystallography. Structure 10, 1659-1667.
Owens LV, Xu L, Craven RJ, Dent GA, Weiner TM, Kornberg
L, Liu ET, Cance WG (1995) Overexpression of the focal
adhesion kinase (p125FAK) in invasive human tumors.
Cancer Res 55, 2752-2755.
Owens LV, Xu L, Dent GA, Yang X, Sturge GC, Craven RJ,
Cance WG (1996) Focal adhesion kinase as a marker of
invasive potential in differentiated human thyroid cancer.
Ann Surg Oncol 3, 100-105.
Park HB, Golubovskaya V, Xu L, Yang X, Lee JW, Scully S,
Craven RJ, Cance WG (2004b) Activated Src increases
adhesion, survival and alpha2-integrin expression in human
breast cancer cells. Biochem J 378, 559-567.
Park HB, Golubovskaya V, Xu L, Yang X, Lee JW, Scully S,
Craven RJ, Cance WG (2004a) Activated Src increases
adhesion, survival and alpha2-integrin expression in human
breast cancer cells. Biochem J 378, 559-567.
Parsons JT (2003) Focal adhesion kinase: the first ten years. J
Cell Sci 116, 1409-1416.
Parsons JT, Slack-Davis J, Tilghman R, Roberts WG (2008)
Focal adhesion kinase: targeting adhesion signaling pathways
for therapeutic intervention. Clin Cancer Res 14, 627-632.
Prutzman KC, Gao G, King ML, Iyer VV, Mueller GA, Schaller
MD, Campbell SL (2004) The focal adhesion targeting
domain of focal adhesion kinase contains a hinge region that
modulates tyrosine 926 phosphorylation. Structure 12, 881-
891.
Reiske HR, Kao SC, Cary LA, Guan JL, Lai JF, Chen HC (1999)
Requirement of phosphatidylinositol 3-kinase in focal
adhesion kinase-promoted cell migration. J Biol Chem 274,
12361-12366.
Richardson A, Parsons JT (1995) Signal transduction through
integrins: a central role for focal adhesion kinase? Bioessays
17, 229-236.
Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C,
Richter D, Emerson E, Lin J, Kath J, Coleman K, Yao L,
Martinez-Alsina L, Lorenzen M, Berliner M, Luzzio M, Patel
N, Schmitt E, LaGreca S, Jani J, Wessel M, Marr E, Griffor
M, Vajdos F (2008) Antitumor activity and pharmacology of
a selective focal adhesion kinase inhibitor, PF-562,271.
Cancer Res 68, 1935-1944.
Sasaki H, Nagura K, Ishino M, Tobioka H, Kotani K, Sasaki T
(1995) Cloning and characterization of cell adhesion kinase
beta, a novel protein-tyrosine kinase of the focal adhesion
kinase subfamily. J Biol Chem 270, 21206-21219.
Schaller MD (2001) Biochemical signals and biological
responses elicited by the focal adhesion kinase. Biochim
Biophys Acta 1540, 1-21.
Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB,
Parsons JT (1992) pp125FAK a structurally distinctive
protein-tyrosine kinase associated with focal adhesions. Proc
Natl Acad Sci U S A 89, 5192-5196.
Schaller MD, Hildebrand JD, Parsons JT (1999) Complex
formation with focal adhesion kinase: A mechanism to
regulate activity and subcellular localization of Src kinases.
Mol Biol Cell 10, 3489-3505.
Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR,
Parsons JT (1994) Autophosphorylation of the focal adhesion
kinase, pp125FAK, directs SH2-dependent binding of
pp60src. Mol Cell Biol 14, 1680-1688.
Schaller MD, Otey CA, Hildebrand JD, Parsons JT (1995) Focal
adhesion kinase and paxillin bind to peptides mimicking beta
integrin cytoplasmic domains. J Cell Biol 130, 1181-1187.
Schaller MD, Parsons JT (1994) Focal adhesion kinase and
associated proteins. Curr Opin Cell Biol 6, 705-710.
Schlaepfer DD, Mitra SK (2004) Multiple connections link FAK
to cell motility and invasion. Curr Opin Genet Dev 14, 92-
101.
Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson
D, Ohmori O, Bigner DD, Friedman HS, Rich JN (2007) A
novel low-molecular weight inhibitor of focal adhesion
kinase, TAE226, inhibits glioma growth. Mol Carcinog 46,
488-496.
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky
CH, Schlaepfer DD (2000) FAK integrates growth-factor and
integrin signals to promote cell migration. Nat Cell Biol 2,
249-256.
Sieg DJ, Hauck CR, Schlaepfer DD (1999) Required role of focal
adhesion kinase (FAK) for integrin-stimulated cell migration.
J Cell Sci 112 ( Pt 16), 2677-2691.
Siu ll, Burris HA, Mileshkin L, Camidge DR, Rishchin D, Chen
EX, Jones S, Tin D, Fingert H (2008) Journal of Clinical
Oncology, 2007 ASCO Annual Meeting Proceedings Part 1.
J Clin Oncol 25, 3527.

Gene Therapy and Molecular Biology Vol 13, page 35
35
Smith CS, Golubovskaya VM, Peck E, Xu LH, Monia BP, Yang
X, Cance WG (2005) Effect of focal adhesion kinase (FAK)
downregulation with FAK antisense oligonucleotides and 5-
fluorouracil on the viability of melanoma cell lines.
Melanoma Res 15, 357-362.
Sonoda Y, Matsumoto Y, Funakoshi M, Yamamoto D, Hanks
SK, Kasahara T (2000) Anti-apoptotic role of focal adhesion
kinase (FAK). Induction of inhibitor-of-apoptosis proteins
and apoptosis suppression by the overexpression of FAK in a
human leukemic cell line, HL-60. J Biol Chem 275, 16309-
16315.
Stewart A, Ham C, Zachary I (2002) The focal adhesion kinase
amino-terminal domain localises to nuclei and intercellular
junctions in HEK 293 and MDCK cells independently of
tyrosine 397 and the carboxy-terminal domain. Biochem
Biophys Res Commun 299, 62-73.
Takahashi N, Seko Y, Noiri E, Tobe K, Kadowaki T, Sabe H,
Yazaki Y (1999) Vascular endothelial growth factor induces
activation and subcellular translocation of focal adhesion
kinase (p125FAK) in cultured rat cardiac myocytes. Circ
Res 84, 1194-1202.
Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada
KM (1999) PTEN interactions with focal adhesion kinase
and suppression of the extracellular matrix-dependent
phosphatidylinositol 3-kinase/Akt cell survival pathway. J
Biol Chem 274, 20693-20703.
Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM
(1998) Inhibition of cell migration, spreading, and focal
adhesions by tumor suppressor PTEN. Science 280, 1614-
1617.
Tilghman RW, Slack-Davis JK, Sergina N, Martin KH, Iwanicki
M, Hershey ED, Beggs HE, Reichardt LF, Parsons JT (2005)
Focal adhesion kinase is required for the spatial organization
of the leading edge in migrating cells. J Cell Sci 118, 2613-
2623.
van de WB, Houtepen F, Huigsloot M, Tijdens IB (2001)
Suppression of chemically induced apoptosis but not necrosis
of renal proximal tubular epithelial (LLC-PK1) cells by focal
adhesion kinase (FAK). Role of FAK in maintaining focal
adhesion organization after acute renal cell injury. J Biol
Chem 276, 36183-36193.
Venables JP (2006) Unbalanced alternative splicing and its
significance in cancer. Bioessays 28, 378-386.
Watanabe N, Takaoka M, Sakurama K, Tomono Y, Hatakeyama
S, Ohmori O, Motoki T, Shirakawa Y, Yamatsuji T, Haisa
M, Matsuoka J, Beer DG, Nagatsuka H, Tanaka N, Naomoto
Y (2008) Dual tyrosine kinase inhibitor for focal adhesion
kinase and insulin-like growth factor-I receptor exhibits
anticancer effect in esophageal adenocarcinoma in vitro and
in vivo. Clin Cancer Res 14, 4631-4639.
Weiner TM, Liu ET, Craven RJ, Cance WG (1993) Expression
of focal adhesion kinase gene and invasive cancer. Lancet
342, 1024-1025.
Whitney GS, Chan PY, Blake J, Cosand WL, Neubauer MG,
Aruffo A, Kanner SB (1993) Human T and B lymphocytes
express a structurally conserved focal adhesion kinase,
pp125FAK. DNA Cell Biol 12, 823-830.
Windham TC, Parikh NU, Siwak DR, Summy JM, McConkey
DJ, Kraker AJ, Gallick GE (2002) Src activation regulates
anoikis in human colon tumor cell lines. Oncogene 21, 7797-
7807.
Xu LH, Owens LV, Sturge GC, Yang X, Liu ET, Craven RJ,
Cance WG (1996) Attenuation of the expression of the focal
adhesion kinase induces apoptosis in tumor cells. Cell
Growth Differ 7, 413-418.
Xu LH, Yang X, Bradham CA, Brenner DA, Baldwin AS, Jr.,
Craven RJ, Cance WG (2000) The focal adhesion kinase
suppresses transformation-associated, anchorage-
independent apoptosis in human breast cancer cells.
Involvement of death receptor-related signaling pathways. J
Biol Chem 275, 30597-30604.
Xu LH, Yang X, Craven RJ, Cance WG (1998) The COOH-
terminal domain of the focal adhesion kinase induces loss of
adhesion and cell death in human tumor cells. Cell Growth
Differ 9, 999-1005.
Yeo MG, Partridge MA, Ezratty EJ, Shen Q, Gundersen GG,
Marcantonio EE (2006) Src SH2 arginine 175 is required for
cell motility: specific focal adhesion kinase targeting and
focal adhesion assembly function. Mol Cell Biol 26, 4399-
4409.
Zhang X, Wright CV, Hanks SK (1995) Cloning of a Xenopus
laevis cDNA encoding focal adhesion kinase (FAK) and
expression during early development. Gene 160, 219-222.
Steven N. Hochwald and Vita M. Golubovskaya

Gene Therapy and Molecular Biology Vol 13, page 36
36
Gene Ther Mol Biol Vol 13, 36-52, 2009
Combination of immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours Review Article
Jian Xu1, Xiao Song Liu 2*, Shu-Feng Zhou3, Ming Q Wei1*
1Division of Molecular and Gene Therapies, Griffith Institute for Health and Medical research, School of Medical Science, Griffith University, Gold Coast campus, Southport, Queensland 4215 2Diamantina Institute for Cancer, Immunology and Metabolic Medicine, University of Queensland, Princess Alexandra Hospital, Wollongabba, Queensland 410 3School of Health Sciences, RMIT, Victoria 3083, Australia
*Correspondence: A/Prof Ming Q Wei, Director of Division of Molecular and Gene Therapies, Griffith Institute for Health and
Medical research, School of Medical Science, Griffith University, Gold Coast campus, Qld 4215, Australia. Tel: 617 5678 0745;
Mobile: 61 422888780; Email: [email protected]
Dr Xiao Song Liu, Diamantina Institute for Cancer, Immunology and Metabolic Medicine, University of Queensland, Princess
Alexandra, Hospital, Wollongabba, Qld 4102, Australia, Email: [email protected]
Key words: Tumour microenvironment, Immunotherapy, Anaerobic bacteria, Hypoxia, Clostridial spores
Received: 16 December 2009; Revised 2009;
Accepted: 14 April 2009; electronically published: 26 April 2009
Summary
Solid tumours possess unique microenvironment characterised by defective vessels, heterogeneous tumour cell,
hypoxic regions, and anaerobic metabolisms. These often become intrinsic and acquired barriers to current
therapeutical approaches, but they also create an ideal condition for the growth of anaerobic bacteria, which have
shown specificity in their germination and multiplication. Spores from the strictly anaerobic clostridial had
demonstrated ability in tumour specific colonisation and induction of tumour lysis following intravenous delivery.
Clostridial strains genetically modified to act as “Trojan horse” gene therapy vectors have been developed.
Similarly, recent development in immunotherapy strategies for cancer also utilizes gene transfer to facilitate a
dormant host immune response directed against the tumour. Combination of anaerobic bacteria for cancer gene
therapies with immunotherapy will probably be the most promising approach that can potentially generate a
prolonged anti-tumour effect beyond the immediate treatment period of gene therapy, allowing for treatment of
advanced primary tumours and disseminated disease. In this review, we introduce the recent understanding of
tumour microenvironment and detail the advances in the use of anaerobic bacteria for cancer gene therapies and
recent studies in immuno therapy for cancers. We believe that the use of combined treatment modalities of such will
provide a rational paradigm to improve upon the clinical efficacy of cancer therapy.
I. Introduction
Cancer is one of the major health problems of
mankind, accounting for 7.6 million of death world wide.
Cancer mortality is expected to increase further, with an
estimated 9 million people dying from cancer in 2015.
This figure will rise to 11.4 million in 2030 (WHO 2006)
(Cho, 2007).
Of all cancer diagnosed, 90% of these are solid
tumours. As they do not have particular noticeable
symptom or signs for early detection, a significant
percentage of the patients with newly diagnosed disease
have regional or advanced, inoperable disease, especially
in developing countries where diagnostic facilities are
suboptimal. Conventional therapies include surgical
operation, radiation and chemotherapy. Single or a
combination of methods may be used, depending on
various factors such as the type and location of the cancer.
Unfortunately, current cancer treatments are limited to
effect. Furthermore they also cause severe side effects.
The search for new cancer therapies is one of the most
pressing tasks of medical science.
Cancer development results from constant battle
between tumour cells and host defence system. Once it
establish by itself. Its microenvironments are hostile to
therapeutic including immunotherapy as well as gene

Xu J et al: immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours
37
therapy. In this paper, we review current understanding of
tumour microenvironments and recent advances in therapy
of solid tumour and explore potential combinations of
immunization and anaerobic bacteria for cancer
management.
II. The unique microenvironment of solid tumours
A. Overview
All solid tumours, when they grow more than 2 mm
diameter in size, undergo angiogenesis that results in
biological changes and adaptive metabolisms, i.e.:
formation of defective vessels, appearance of hypoxic
areas, and emergence of heterogeneous tumour cell
population. Thus, solid tumours are organ-like structures
that are heterogeneous and structurally complex,
consisting cancer cells and stromal cells (i.e., fibroblasts
and inflammatory cells) that are embedded in an
extracellular matrix and nourished by a vascular network;
each of these components may vary from one location to
another in the same tumour. Compared with normal
tissues, the tumour stroma is associated with an altered
extracellular matrix and an increased number of stromal
that synthesize growth factors, chemokines, and adhesion
molecules (Aznavoorian et al, 1990). The extracellular
matrix can vary greatly among tumours, both in amount
and in composition (Ohtani, 1998). Also the tumour
stroma can influence malignant transformation (Tlsty
2001) plays an important role in the ability of tumours to
invade and metastasize, and affects the sensitivity of
tumour cells to drug treatment. The amount composition
and structure of stromal components in tumours also
contribute to an increase in interstitial fluid pressure,
which hinders the penetration of macromolecules through
tissue (Croker, 2008). Also, the three-dimensional
structure of tissue itself can influence the sensitivity of
constituent cells to both radiation and chemotherapy
(Shicang 2007).
B. Tumour vasculature and blood flow
Solid tumours at advanced stages have abnormal
vasculature, which influences the sensitivity of the tumour
to therapies. Anticancer drugs gain access to tumours via
the blood and limited supply of nutrients in tumours leads
to metabolic changes (including hypoxia) and gradients of
cell proliferation that influence drug sensitivity (Tatum et
al, 2006). Also, blood vessels in tumours are often dilated
and convoluted. Compared with normal tissues, tumour
blood vessels have branching patterns that feature
excessive loops and arteriolar–venous shunts, in some
tumours they are not organized into arterioles, capillaries,
and venules but instead share features of all of these
structures. The walls of tumour vessels may have
fenestrations, discontinuous or absent basement
membranes that may lack perivascular smooth muscle
(Hallmann et al, 2005) and fewer pericytes than walls of
normal vessels. In addition, cancer cells may be integrated
into the vessel wall. These abnormalities tend to make
tumour vessels leaky, although their permeability varies
both within and among tumours.
C. Tumour hypoxia and acidity
Most solid tumours contain regions of hypoxia (Wu
et al, 2006). The limited vasculature of tumours results in
insufficient blood supply and chronic or diffusion-limited
hypoxia. Tumour cells in hypoxic regions may be viable,
but they are often adjacent to regions of necrosis. Tumour
cells in regions proximal to blood vessels can migrate into
hypoxic areas and become necrotic, presumably because of
nutrient deprivation. If cells close to blood vessels are
killed by treatment, the nutrient supply to previously
hypoxic cells may improve, allowing those cells to survive
and regenerate the tumour (Trédan et al, 2007). Transient
hypoxia is also common in tumours and results from the
temporary shutdown of blood vessels. Hypoxic regions of
tumours are likely to have a decreased supply of nutrients
such as glucose and essential amino acids (Pouysségur et
al, 2006). The presence of hypoxia in tumours is known to
lead to the activation of genes associated with
angiogenesis and cell survival that is mediated by the
transcription factor hypoxia-inducible factor 1(Bos R et al,
2004). Expression of these genes may result in the
expansion of populations of cells with altered biochemical
pathways that may have a drug-resistant phenotype.
Transient hypoxia has been reported to cause amplification
and increased expression of the genes encoding P-
glycoprotein and dihydrofolate reductase, which induce
drug resistance to substrates of P-glycoprotein and to
folate antagonists, respectively. Transient hypoxia that is
associated with glucose deprivation can also disrupt
protein folding in the endoplasmic reticulum; this effect
may confer resistance to topoisomerase II–targeted drugs
and enhance P-glycoprotein expression and multidrug
resistance (Chen et al, 2003).
The pH in the tumour microenvironment can
influence the cytotoxicity of anticancer drugs (Philip et al,
2005). Molecules diffuse passively across the cell
membrane most efficiently in the uncharged form. The
extracellular pH in tumours is low and the intracellular pH
of tumour cells is neutral to alkaline, weakly basic drugs
that have an acid dissociation constant of 7.5–9.5 are
protonated and display decreased cellular uptake.
Alkalinization of the extracellular environment enhances
the uptake and cytotoxicity of some of these drugs (Trédan
et al, 2007). By contrast, weakly acidic drugs concentrate
some in the relatively neutral intracellular space. The
acidic microenvironment may also inhibit active transport
of some drugs (Mahoney et al, 2003).
D. Tumour immunosuppression
During the constant battle between tumour and
immune system, tumour cells developed multiple ways to
fight back the immune system.
1. Avoidance of effectors T cell killing

Gene Therapy and Molecular Biology Vol 13, page 38
38
One of well established strategies is down regulation
of antigen presentation by tumour cells, especially through
MHC class I restricted antigen presentation pathway.
Tumour cells can down regulation, even loss of MHC
class I molecules on their cell surface (Frey, 2006),
mutation of proteins associated with this pathway, such as
TAP and LMP2 and LMP7.
Tumour or stromal cells also secrete factors that
damp immune responses. TGF (tumour growth factor), IL-
10 are two cytokines with immune suppressive functions
usually found with high levels within tumour. TGF levels
are associated with poor prognoses of cancers including
prostate, gastric and bladder carcinoma (Biswas et al,
2007). TGF inhibits T cell activation and differention of
cytotoxic T cells and promotes NKT cells mediated
inhibition of CTL responses together with IL-13 (Biswas
et al, 2007). IL-10 down regulate antigen presentation by
dendritic cells and promote the generation of Tr1
regulatory T cell generation (Suciu-Foca et al, 2003) and
Inhibit CTL response in antigen experienced host (Tamada
et al, 2002). High levels of prostaglandin E2 (PGE2) have
been shown in colorectal, lung and bladder cancer
(Akasaki et al, 2006). It has been demonstrated that PGE2
promotes the generation of IL-10 secreting CD4 T cells
through the induction of IL-10 secreting dendritic cells
(Cools et al, 2007).
Different tumour types have also been expressed PD-
L1, an immune suppressive molecule. Tissue histology
study showed that freshly isolated carcinomas of human
lung, ovarian, colon, melanoma, head and neck cancers,
and breast cancers can express PD-L123. PD-L1 a
suppressive molecule, engagement of PD-L1 with PD-1 of
effector T cells causes T cell apoptosis (Yang et al, 2008).
B7-H1 positive melanoma cells were also more resistant to
specific CTL, while nearly all B7-H1 negative tumour
cells were eliminated in the cultures (Dong and Chen,
2003), these results suggest that expression of suppressive
molecule is another strategy used by tumour cells to avoid
from killing by effector cells.
2. Regulation of immunoresponses by
regulatory T cells
Regulatory T cells are groups of T cells that
regulatory immune response, different compartments of T
regulatory cells including CD4+, CD8+ and NKT cells
have been identified. CD4+CD25+ Foxp3+ thymus
derived T regulatory cells and antigen induced IL-10
secreting CD4 T cells are the 2 main types identified.
NKT cells have also been shown to have regulatory
function during tumour development (Berzofsky et al,
2008). However, the number of T regulatory cells with
human ovary cancer is related to poor prognosis of cancer
(Koido et al, 2005). Also, it has been shown that myeloma
cells promote the generation of IL-10 secreting Tr1 T cells
(Battaglia et al, 2006). Tr1 cells can be isolated from
tumour infiltrating lymphocytes in B16 tumour model
(Seo et al, 2001). Human bladder cancer tissues contain
high number of Foxp3+ cells and mRNA level of IL-10
(Petrulio et al, 2006). It is not clear whether the T
regulatory cells were boosted from existing T regulatory
cells or vaccine induced.
However, immunotherapy has shown to amplify
tumour specific T regulatory cells, thus impede effective
immunotherapy in a mouse tumour model (Reilly et al,
2000); moreover, similar results were also observed
clinically. Patients with resected HPV16-positive cervical
cancer were vaccinated with an overlapping set of long
peptides comprising the sequences of the HPV16 E6 and
E7 oncoproteins emulsified in Montanide ISA-51. The
vaccine-induced responses were dominated by effector
type CD4(+)CD25(+)Foxp3(-) type 1 cytokine IFN
gamma-producing T cells but also included the expansion
of T cells with a CD4(+)CD25(+)Foxp3(+) phenotype
(Welters et al, 2008).
3. Abnormal antigen presentation cells
Antigen presentation cells include dendritic cells
(DC), macropaghes and B cells. Matured DCs play key
roles for the priming of naive T cells, including CD8+ T
cells, which is critical for the killing of tumour cells.
Tumour microenvironments usually have less functional
competent matured but more immature DCs, which can
not effectively activate T cells. Furthermore, it has been
reported that in tumour tissues, there are subset of DCs
that suppress T cell function. This T cell suppression has
been shown in cancer patients as well as animal tumour
models.
Immune cells in the tumour microenvironment are
dysfunctional, generally fail to control tumour growth and
may even promote its progression. Molecular mechanisms
responsible for tumour-induced local and systemic
immune suppression are currently under intense discussed.
It appears that tumours can deregulate recruitment,
effector functions and survival of immune cells,
interfering with all stages of antitumour response.
Suppressive mechanisms targeting key signalling
pathways in immune cells have been identified. Strategies
for reversal of tumour-mediated immunsuppression are
being developed. Confirmation of multiple and varied
mechanisms used by tumours to escape immune
surveillance is crucial for the future design in antitumour
therapies.
III. Current cancer gene therapy and immunotherapy approaches
A. Current development in gene therapy
of solid tumour
Cancer is, at present, the disease most frequently
targeted by gene therapy because its promise of potential
for selective potency. To achieve this aim, cancer gene
therapy strategies attempt to exploit the biological
uniqueness of each particular tumour. Cancer gene therapy
may be defined as the transfer of recombinant DNA into
human cells to achieve an anti-tumour effect. Gene
therapy will have a major impact on the healthcare of our
population only when vectors are developed that can

Xu J et al: immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours
39
safely and efficiently be injected directly into patients as
drugs. One of the most strategies of vector development is
that of non-viral vectors, which consist of liposomes,
molecular conjugates, and naked DNA delivered by
mechanical methods. The modifying viral vectors should
be focused to reduce toxicity and immunogenic, increasing
the transduction efficiency of non-viral vectors, enhancing
vector targeting and specificity, regulating gene
expression, and identifying synergies between gene-based
agents and other cancer therapeutics. A universal gene
delivery system has yet to be identified, but the further
optimization of each of these vectors should result in each
having a unique application.
1. Pro-Drug activation vectors
Several experimental models relying on pro-drug
activation vectors (Kanai et al, 2008). One such a model
involves local injection of gene therapy vectors into
tumour sites. This model may benefit from the so-called
"bystander effect," a reflection of the biological
observation that pro-drug activation to 5-fluorocysteine (5-
FU) releases this chemotherapeutic not just in the tumour
cells, but in the surrounding cell environment as well. In
fact, using in vitro systems, it has been found that only 5%
of tumour cells need to be infected by the delivery vector
for anti-tumour effect to be seen throughout the whole
tumour cell population. An adenovirus vector expressing
the cytosine deaminase enzyme will be injected into the
prostate bed using similar techniques as those now used
for radiation implants. These patients will then be given
the pro-drug, which in principle will be activated to 5-FU
in the prostate gland. This should allow localized
cytotoxic therapy to the prostate and possible synergistic
benefit between 5-FU and the concurrent radiation
therapy.
The other model system which is used in clinical
trials deals with autologous transplantation for metastatic
breast cancer. In this system, harvested bone marrow is
exposed to the viral vector, which infects the epithelial
tumour cells efficiently, but normal marrow stem cells less
efficiently. After intensive chemotherapy, patients are then
given this modified marrow population. Once engrafted,
patients are treated with the pro-drug 5-FC, which in
principle should be toxic only to the infected tumour cells.
This trial is open to women with known marrow involvement by tumour cells, and who are
therefore not candidates for standard high-dose
therapy.
2. Tumour-specific gene promoters
The L-plastin gene (Akbulut et al, 2003), as another
means of conferring tumour-specific expression which
encodes an actin-binding protein, show the new vector
model with a tumour specific gene promoter. The
estrogen-dependent tissues such as ovary and breast were
selectively expressed in ovarian and breast cancer. The
promoter for this gene is added to the adenoviral vector,
and a reporter enzyme, such as beta-galactosidase, is
linked to the promoter to allow for assessment of
expression. In preliminary experiments, this vector was
able to transfect ovarian cancer cells isolated from ascites
fluid, and confer tumour-specific expression of beta-
galactosidase. This method creates the possibility of
targeting expression of certain genes in specific tissues
3. Herpes simplex virus thymidine kinase
gene
To broaden the effect of gene therapy, vectors
employing both the thymidine kinase gene and the genes
for immunomodulatory cytokines such as IL-2 or
granulocyte-macrophage colony-stimulating factor (GM-
CSF) have been developed (Iwadate et al, 1997). In mice,
injection of these vectors into tumours and treatment with
ganciclovir had both a direct anti-tumour effect in the
liver, as well as a systemic effect in generating tumour-
specific immune responses. As a result, these mice are
resistant to subsequent tumour challenge. This system
establishes the principle that localized gene therapy might
ultimately have systemic protective or therapeutic effect
by stimulating immune mechanisms which can act
throughout the organism. A phase I trial for patients that
would include treatment with a thymidine kinase and
cytokine (IL-2) vector is being planned. The principle
endpoint of the study will be the determination of an anti-
tumour immune response.
4. Dendritic cells as targets for cancer gene
therapy
DCs are the most potent APCs in the immune system
and are central to the success of these genetically
engineered tumour vaccine strategies. Activated DCs can
present prostate tumour vaccine-associated antigens; they
have processed to both CD4 (helper) and CD8 (cytolytic)
T cells in the draining lymph node of the vaccination sites,
activating a systemic tumouricidal immune response. The
possibility of obtaining large numbers of DCs in vitro has
boosted research on their ontogeny and functions. The
unique ability of DCs to take up, process, and present
antigens, and to activate naive CD4+ and CD8+ T cells,
makes them appropriate candidates for the
immunotherapeutic approach.
In a mouse model, DCs are harvested and then
transfected with adenoviral vectors. These vectors
expressed a foreign protein, beta-galactosidase. The
dendritic cells were then injected into mice, and served to
prime an immune response against that protein. This ex
vivo gene therapy has many potential human applications.
Three major myeloid DC populations have been identified
in vivo: (1) epidermal Langerhans’ cells (LC); (2)
interstitial (or dermal) immature DC; and (3) mature
interdigitating DC, found in secondary lymphoid organs.
In the early stages of DC research, the limited accessibility
of these cells in vivo as well as their difficult ex vivo
culture hampered attempts to study this particular cell type
in more detail. In the 1990s, this problem was solved by
the efforts of various research teams which revealed the
hematopoietic lineages through which DC differentiate,

Gene Therapy and Molecular Biology Vol 13, page 40
40
and established in vitro expansion protocols to obtain
sufficient quantities of DC for clinical use (Caux et al,
1992; Sallusto, 1994). The unique ability of DC to
stimulate primary immune responses stems from several
factors. The immature DC type uses elegant systems,
including macropinocytosis, mannose receptor-mediated
uptake, Fcg receptor III (FcgRIII)-mediated uptake and
phagocytosis to efficiently take up exogenous antigens,
either self or non-self, from the periphery (Steinman et al,
1999). After antigen capture, DC leaves the peripheral
tissue and migrates via blood or lymphatic vessels to the
draining lymph nodes where they activate T cells Given
their central role in controlling immunity and their link
with the innate immune system, DC are often called
nature’s adjuvant. Therefore, DC is logical targets for
immunotherapy of cancer. The fact that tumours do not
elicit a therapeutic T cell response may be due to the
absence of competent DC at the tumour site.
B. Cancer gene therapy existing problems
Currently, there are many different approaches to
fight cancer with gene therapy. Morgan et al report has
revealed encouraging results for the use of gene therapy as
a treatment for cancer (Morgan et al, 2006). However; two
principal obstacles continue to limit further advances in
gene therapy. The first is a technical problem, the
development of an appropriate delivery system -- a
reliable, safe, and effective means for introducing genetic
material into the target cells or tissues. The second
problem is a biological one -- developing an understanding
of the molecular basis underlying cancer in order to
determine where single alterations in genetic expression
might allow effective anti-cancer therapy. In viral vector,
the efficiency of transduction is not sufficient for
therapeutic measures (Marina et al, 2003). One important
parameter is whether the genetic alteration has to be
lasting or temporary (stable or transient transfection). Of
overall importance is the question of biological safety,
which means that the vector itself does not create a novel
threat to the patient's health. The key to a successful gene
therapy is the vector system. Various vectors have been
developed with unique features, including viral and non-
viral based therapy systems (Wagner, 2007). However,
due to the complex nature of cancers, these vectors suffer
from several deficiencies: firstly the majority of vectors
currently in use require intratumoural injection to elicit an
effect, far from ideal as many tumours are inaccessible and
spread to other areas of the body making them difficult to
locate and treat. Second, most vectors do not have the
capacity to efficiently enter and kill every tumour cell.
The emerging challenges of cancer gene therapy: i)
which better route of administration is best for improving
gene delivery; iii) optimizing new vector best suited to the
target type of tissue and reducing toxicity, Although as
with many gene-therapy approaches, considerable barriers
will need to be overcome to make the technique more
reliable and widely applicable - achieving long-term
expression of therapeutic genes is a particular problem -
these results are nevertheless a heartening 'proof-of-
principle' demonstration of the potential power of gene
therapy to combat cancers.
To establish efficient and safe gene delivery in vivo,
a number of new techniques and concepts have been
introduced with improvements in targeted or controlled
delivery of genes. But we have come a long way in
understanding the cellular barriers which prevent proper
delivery of DNA or viral vectors. Cancer gene therapy has
still a long way to go in the basic and clinical sciences.
C. Anaerobic bacteria for cancer
treatment
Interest in microbe-based approaches to cancer
therapy has recently re-emerged with the development of
methods to genetically engineer bacteria, reducing their
toxicity and arming them with genes encoding pro drug-
metabolizing enzymes.
1. Anaerobic bacteria as tumour target
vector
The unique solid tumour micro-milieu, though,
provides a haven for anaerobic bacteria. Anaerobic and
facultative anaerobes tested so far fell into three classes.
(1) the lactic acid, Gram-positive anaerobic bacteria; (2)
the intracellular, Gram-negative facultative anaerobes, and
(3) the strictly anaerobic, Gram-positive saccharolytic/
proteolytic Clostridia. At the molecular level, bacterial
infections like those of Clostridia novyi (C. novyi) are
associated with the release of pathogen-associated
molecular patterns (PAMPs) from bacteria and Hsp70
from necrotic cells (Gelman, 2003). Hsp70 induces
maturation of DCs, professional antigen-presenting cells
that are essential for the production of potent immune
responses. PAMPs interact with Toll-like receptors,
leading to up-regulation of costimulatory molecules such
as CD40 and proinflammatory cytokines such as IL-12.
These in turn induce the production of IFN-! and initiate a
Th1-dependent cell-mediated response, primarily affected
by CD8+ cytolytic T cells (Kay, 2001). The demonstration
that CD8+ T cells from C. novyi-NT-cured mice can confer
adoptive immunity in a tumour-specific fashion is
consistent.
Clostridium is strictly anaerobic, sporulating Gram-
positive bacteria. This genus is one of the largest genera
comprising of about 80 species. Up to 10 species of
Clostridia have been studied and as strictly anaerobic
bacteria they all require an anaerobic environment to grow
but their oxygen tolerance and biochemical profile varies
considerably among different species. Clostridial spores
had been used to induce tumour lysis following
intravenous delivery and shown a distinct advantage over
Bifidobacterium and Salmonella in terms of easy
production, hardy storage and impressive oncolytic
effects. Both proteolytic and saccharolytic Clostridia have
been tested for cancer therapy. When C. novyi-NT spores
are injected intravenously into immunodeficient mice
bearing human xenografts, the spores quickly germinate
within necrotic regions of the tumours. Hypoxic and

Xu J et al: immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours
41
necrotic regions are generally localized within the central
parts of tumours, with well perfused tumour cells
occupying the rim. Because of the exquisite sensitivity of
C. novyi-NT to oxygen (Dang et al, 2001), bacterial
germination and spread halt when the bacteria reach the
well oxygenated rim. It was shown that conventional
chemotherapy and radiation therapy could be used to
destroy the well oxygenated cells in this rim, and that the
combination of C. novyi-NT provided substantial
antitumour activity in several xenograft models.
2. Anaerobic bacteria and immune response
C. novyi is well known for its capacity to induce
massive leukocytosis and inflammation (Agrawal et al,
2004), whereas many other species of Clostridia do not
induce this level of response. The inflammatory reaction
is classic in many ways, including the observed increase in
neutrophil-directed cytokines in serum and the cellular
nature and time course of the infiltrate. The antitumour
effects of inflammation are well documented. Systemically
administered C. novyi-NT spores are distributed
throughout the body, but due to their strict anaerobic
growth requirements, germinate only within anoxic or
markedly hypoxic regions of tumours. Once germinated,
the bacteria destroy adjacent cancer cells through the
secretion of lipases, proteases, and other degradative
enzymes. At the same time, the host reacts to this localized
infection, producing cytokines such as IL-6, MIP-2, G-
CSF, TIMP-1, and KC that attract a massive influx of
inflammatory cells, initiated largely by neutrophils and
followed within a few days by monocyte and lymphocyte
infiltration. The inflammatory reaction restrains the spread
of the bacterial infection, providing a second layer of
control in addition to that provided by the requisite
anaerobic environment. The inflammation may also
directly contribute to the destruction of tumour cells
through the production of reactive oxygen species,
proteases, and other degradative enzymes. Moreover, it
stimulates a potent cellular immune response that can
subsequently destroy residual tumour cells not lysed by the
bacteria. The cure rate is determined by the balance
between bacteriolysis, angiogenesis, regrowth of residual
tumour cells, and the rate of development of the immune
response.
During these years, bacteriological research on
tumour associated anaerobic spore forming bacteria has
accumulated a considerable amount of information and a
variety of new concepts in experimental and clinical
oncology (Agrawal et al, 2004). Of great importance was
the systematic elucidation which convincingly
demonstrated that the growth of anaerobes can be strictly
interconnected with tumour growth. A whole series of
experimental studies have been performed to elucidate the
mechanisms which governed the selective, temporarily
unrestricted clostridial growth and which formed the basis
for the liquefaction of tumour tissue. Since tumour lysis
with Clostridium oncolyticum spores is incomplete and,
possibly, subject to non-specific systemic incompatibility
[‘acute tumour lysis syndrome’]. Clostridia became
significant in pursuing the concept of engineered
Clostridia to produce anti-cancer drugs (Jennifer et al,
2006). The strictly anaerobic clostridia, on the other hand,
have been shown to selectively colonise in solid tumours
when delivered systemically and has resulted in high
percentage of "cures" of experimental tumours. A phase I
clinical trial combining spores of a non toxic strain (C.
novyi-NT) with an antimicrotubuli agent has been
initiated.
The recombinant DNA technology reignited the
field, enabling genetic improvement of Clostridia’s innate
oncolytic capability. It provides a possible alternative to
overcome the hitch of using wild type strains Anaerobic
bacteria, such as Clostridia have now been convincingly
shown to selectively colonise and regerminate in the
hypoxic/necrotic regions of solid tumours and can be
delivered systemically. Furthermore, existing plasmid-
based gene modification strategy harbours several safety
concerns regarding possible horizontal plasmid transfer
and spread of plasmid-associated antibiotic resistant
genes.
IV. Current approaches for immunotherapy of cancer
A. Overview
The aim of cancer immunotherapy is to activate
patient’s immune system to eradiate tumour cells. It was
expected that when appropriately primed, the activated
host immune cells, especially tumour antigen specific
CD4+ and CD8+ T cells, can specifically kill tumour cells.
Tumour antigens are usually self antigens, both
central and peripheral tolerance apply to tumour antigens.
Central tolerance occurs in the thymus, T cells with strong
self reactivity are eliminated. Peripheral tolerance make
tumour specific T cells anergy or suppressive. Cancer
vaccine will activate T cells purged of strong activity and
influenced by different peripheral tolerance mechanisms.
Different approaches have been employed to overcome the
tolerance, in order to achieve better T cell responses,
including immunization with different routs and with
different adjuvant, providing co-stimulating signals while
inhibiting signals such as CTLA-4. Neutralizing IL-10 at
the same time of immunization has been show to generate
better CTL response in antigen experienced host, which is
important for cancer immunotherapy; as patients with
cancer are tumour antigens experienced.
B. Combining immunostimulation with gene-silencing by siRNA
The innate immune system recognizes pathogens by
means of germ line-encoded pattern recognition receptors
(PRRs) (Gro F, 2006). A subfamily of PRRs is the Toll-
like receptors (TLRs), which is important for initiation of
an immune response. siRNAs can activate innate
immunity through the activation of Toll-like receptor
(Sioud et al, 2007). These findings suggest potential
prophylactic and therapeutic use of immunostimulatory
siRNAs as adjuvant. In addition, to immune stimulation,

Gene Therapy and Molecular Biology Vol 13, page 42
42
gene-silencing through RNAi is another potency of
immunostimulatory siRNAs. RNAi is a widely conserved
post-transcriptional gene-silencing mechanism where
double-stranded (ds) RNAs trigger the degradation of
homologous mRNA sequences and certain siRNA
sequences can activate immune cells to secrete
proinflammatory cytokines and type I interferons in
immune cells. As a consequence of these findings any
therapeutic siRNA should be tested in human blood cells
prior to use in (Gelman, 2003). However, if we view the
activation of innate immunity by siRNAs as beneficial for
cancer therapy and infectious diseases, then
immuostimulatory siRNAs could emerge as useful agents
to knockdown gene expression and activate innate and
adaptive immunity against tumour cells. This observation
prompted us to design bifunctional siRNAs, which
combine gene-silencing and immunostimulation in one
single siRNA molecule (Gro F, 2006).
C. Development of strategies to promote effector cell recruitment into tumour
One strategy is to promote effector cell recruitment
into metastases when it fails spontaneously (Shakhar,
2003). Intratumoural introduction of chemokines through
the use of viral vectors would serve as a proof of concept.
Transduction of tumour cells to express specific
chemokines has shown benefit in some experimental
murine models. Similarly, introduction of the TNF
superfamily member LIGHT (homologous to
lymphotoxins, inducible expression, competes with HSV
glycoprotein D for HVEM, a receptor expressed on T
lymphocytes) has been expressed at tumour sites with
dramatic results (Kunz M et al, 1999). However, direct
intratumoural injection of recombinant viral vectors will
only serve as a proof of concept, and development of
agents that can be delivered systemically yet target tumour
metastases would have to be pursued for practical
application.
D. Modulating tumour cell biology to alter the tumour microenvironment
Once the oncogenic signals present in tumour cells
that determine the nature of the tumour microenvironment
are defined, then it should be possible to target those
pathways directly to eliminate the underlying basis for
immunosuppression at tumour sites. For example, Stat can
drive the expression of vascular endothelial growth factor
(VEGF) (Burdelya et al, 2005), which in addition to
promoting neoangiogenesis has been reported to be
inhibitory for dendritic cell generation in vivo (Della et al,
2005). The interface between tumour biology and the
creation of the immunosuppressive tumour
microenvironment is an area ripe for additional research.
Another strategy in the immunotherapy of tumours is
the use of mRNA-encoding tumour antigens to induce T-
and B-cell immunity to the encoded antigens. In vivo
application of mRNA induced cytotoxic T-cell activity
and specific antibodies in mice. Furthermore, human DCs
transfected ex vivo with mRNA induced an antigen-
specific immune response both in vitro to a viral antigen
and in vivo to a tumour-associated antigen in patients with
cancer.
Current efforts in cancer immune therapy and
bacteria therapy are largely aimed at stimulating anti-
tumour immune responses by using various tumour
antigens and adjuvants. The involvement of TLR-activated
pathways in immune response is supported by the
induction of DC maturation and secretion of various
cytokines (Palucka et al, 2007), leading to the induction of
innate and adaptive immunity.
E. Targeting cancer stem/progenitor cells
for anticancer therapy
The cancer recurrence phenomenon has been
associated with the accumulating genetic or epigenic
alterations in cancer cells which may contribute to their
uncontrolled growth, survival and invasion as well as their
intrinsic or acquired resistance to clinical treatments
(Lowenberg et al, 2003; Mimeault et al, 2005). Recent
investigations have revealed that the most aggressive
cancers may originate from the malignant transformation
of embryonic or adult stem/progenitor cells into cancer
progenitor cells (Mimeault, 2006). The cancer progenitor
cells can provide critical functions in cancer initiation and
progression into metastatic and recurrent disease states.
Numerous investigations have provided evidence that the
genetic and/or epigenic alterations occurring in the multi-
potent tissue-specific adult stem cells, the most cancers
may arise from the malignant transformation of multi-
potent tissue-specific adult stem cells and/or their early
progenitors into cancer progenitor cells, the accumulation
of different genetic and/or epigenic alterations in cancer
progenitor cells during cancer progression also seems to
be associated with the occurrence of highly aggressive
cancer subtypes. The functional properties of cancer
progenitor cells may be influenced through external
signals mediated by other further differentiated cancer
cells and host stromal cells including activated fibroblasts
and infiltrating immune cells, such as macrophages and
endothelial cells (Kopp et al, 2006).
Among the diverse growth factors, chemokines and
angiogenic substances released by stromal cells (Kopp,
2006). All these soluble factors can influence, of autocrine
or paracrine manner, the tumour cell behaviour and
neovascularization process during cancer progression. The
intrinsic or acquired resistance of poorly differentiated and
tumourigenic cancer progenitor cells to current clinical
therapies may lead to their persistence in primary and
secondary neoplasms after treatments, and thereby
contribute to cancer recurrence (Mimeault, 2007; de
Jonge-Peeters et al, 2007). The cancer stem/progenitor cell
model of carcinogenesis may also explain the differences
of response of distinct cancer subtypes to current therapies
as well as the dormancy phenomenon and disease relapse,
which may be associated with a higher resistance of
cancer progenitor cells to conventional therapies under
specific conditions prevalent in primary and/or secondary
neoplasms relative to their further differentiated progeny

Xu J et al: immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours
43
(Mimeault, 2007). Based on these observations, the new
cancer therapeutic strategies should be based on targeting
of different oncogenic cascades activated in tumourigenic
cancer progenitor cells, and which must now be
considered for improving the current therapeutic
treatments. The molecular targeting of tumourigenic
cancer progenitor cells must be considered for improving
the efficacy of the current cancer therapies.
F. Gene-based tumour immunization
For any gene therapy application including genetic
immunization, the goal is to deliver genes into
therapeutically-relevant cells while avoiding other cells
that cannot contribute to immunization or therapeutic
effects. While this is the goal, particularly for in vivo gene
therapy, current gene delivery vectors cannot specifically
deliver genes to the cells we want and frequently deliver
genes into non-target tissues reducing therapy and
increasing dangerous side effects.
Generally, the level of gene transfer into tumour cells
and immune effector cells determined the level of
immunogenetics, they have been shown to be limited, and
this has been thought to account for the poor results
obtained by cancer gene immunotherapy. Therefore,
vector design is one of the most critical areas for future
research (Logan et al, 2002). Gene delivery vectors thus
are required fall into three areas: 1) identification of cell-
targeting ligands using random peptide-presenting phage
libraries; 2) engineering viral and non-viral gene delivery
vectors to accept cell-targeting ligands; and 3) developing
effective methods to image gene and vector delivery in
vivo to determine the efficacy of targeted vectors in the
complex tumour environment. The different vector
systems can have strengths or weaknesses, depending on
their use. For ex vivo gene delivery and clinical use in
cancer protocols, design of optimized transduction
protocols and development of improved vectors,
exhibiting improved gene transfer efficiency and stability
for large-scale production, have just begun to be
evaluated. Nonviral gene delivery systems are cost- and
time-effective and large-scale manufacturing of clinical-
grade plasmid vectors is logistically simple. The major
disadvantages are the low transfection efficiency and the
transient expression in target cells. As already mentioned
earlier, one of the attractive features of immunological
gene therapy approaches is that they capitalize on the
ability to amplify the outcome of the gene transfer
(‘genetic immunopotentiation’). Consequently, high
efficiency gene transfer may not be an essential
requirement in these protocols. Given this problem, we are
interested in developing gene delivery with recombinant
engineer bacteria vectors that can be tuned to target
specific cells in vivo for gene therapy and immunization
applications. As recombinant engineer bacteria are so far
the best characterized bacteria vectors, they are most
frequently used vectors for immuno-gene therapy of
cancer.
Immunogene therapies have the theoretical
advantage of inducing a systemic anti-tumour response
associated with immunologic memory. Such a response
potentially allows for treatment of disseminated disease
and a prolonged anti-tumour effect that persists beyond the
immediate treatment period. Immunogene therapy
strategies involve both ex vivo and in vivo approaches
(Glick et al, 2006). Increasing the capacity of the immune
system to mediate tumour regression has been a major
goal for tumour immunologists. Progress towards tumour
vaccines has been recently made by the molecular
identification of novel tumour-associated antigens (TAA)
and by a better understanding of cellular signals required
for efficient T cell activation (Pule et al, 2002). Cancer
vaccination is of therapeutic rather than prophylactic
nature, involving attempts to activate immune responses
against TAA to which the immune system has already
been exposed. To date, advances in gene delivery
technology have led to the development of immuno-gene
therapy strategies to augment host-immune responses to
tumours. These approaches include (1) the use of tumour
cells genetically modified with genes encoding
costimulatory ligands, cytokines or HLA molecules to
enhance their immunogenicity and (2) the genetic
modification of immune-competent cells with TAA in
order to enhance their anti-tumour response.
Despite the continuous increase in clinical gene
therapy protocols for immunotherapy of cancer, many
aspects of gene transfer are still far from ideal. A basic
requirement, not yet adequately and routinely fulfilled, is
to introduce the gene of interest with sufficient efficiency
into the target cells in order to achieve therapeutic benefit
in cancer patients.
G. Breakdown of immune tolerance to
tumours
The current rationale lies in the local recruitment of
inflammatory cells that can destroy a fraction of the
tumour cells directly or indirectly, thereby releasing
tumour antigens. These antigens can be taken up in the
form of peptides, proteins or apoptotic bodies by
professional antigenpresenting cells (APC) by a process
known as cross-priming (i.e. indirect presentation of
tumour antigens to the immune system by a host-derived
APC), that travel to the draining lymph nodes where they
will activate naive antigen-specific T cells and initiate a
primary cellular immune response. The new approach
enlists the help of the immune system to target and kill
tumour blood vessel cells, through an unprecedented
recruitment of the immune system; they were able to
generate a strong anti-tumour effect by targeting the
central component of what tumours need most-a blood
supply (Niethammer et al, 2002).
According to the classical paradigm in tumour
immunology, immune responses are believed to follow a
model of discrimination between self and non-self.
Consequently, tumours should be considered as non-self,
like viruses or bacteria. Therefore, an important task of the
immune system is to search for and destroy tumour cells
as they arise, in concordance with the original proposals of
Burnet’s immunological surveillance hypothesis.

Gene Therapy and Molecular Biology Vol 13, page 44
44
However, the limited successes of cancer immunotherapy
approaches based on these concepts, prompted a revision
of tumour immunology (Luis et al, 2005). Ultimately, it
appears that the immune response at the T cell level is
based on the presence of the appropriate costimulatory
molecules on APC that promote T cell activation. DCs
(DC) form a complex network of antigen-capturing and -
presenting cells (APC) defined by morphological,
phenotypical and functional criteria which distinguish
them from monocytes and macrophages (Elke et al, 2002).
Immunity against cancer is necessary if gene transfer
is going to be applied in a clinically relevant way. Instead
of exploiting the increasing knowledge on cytokines and
their plethora of actions in the immune response,
immunology may provide a more fundamental mechanism
to explain the immunological unresponsiveness to cancer
than the classical self/non-self paradigm. At a later stage,
we will focus on a new gene-based tumour immunization
that seems to fit within this conceptual framework.
H. Stimulation to illicit an active immunoresponse in a solid tumour
environment
Van Pel and Boon (1982) demonstrated that a
protective immune response could be generated against a
‘non-immunogenic’ murine tumour, providing the first
experimental evidence that the lack of tumour immunity
was not due to the absence of TAA but rather to the
inability to stimulate the immune system. Factors that can
explain the failure of the immune system in tumour-
bearing hosts are numerous, and it is not clear which of
them are critical in the clinical context. We all know that
tumour cells are poor stimulators of immune responses
and capable of inducing immune tolerance. Alternatively,
it may well be that the lack of costimulatory molecules
(e.g. CD80, CD86) on the surface of tumour cells accounts
for the immune tolerance which keeps the tumour from
being rejected. Deficiency of the immune system could be
responsible for the lack of immunity and induction of T
cell tolerance (von Euler et al, 2008). In this case; the
tumour actively suppresses host antigen presentation and
immune effect or functions by expression of a variety of
local inhibitory molecules, such as VEGF and IL-10,
especially when large tumour burdens are involved.
Antigen-specific cytotoxic cells that do specifically
recognize tumour cells can be generated by cell cloning
techniques ex vivo or can be genetically engineered by the
stable transfection of a TCR that specifically recognizes a
certain MHC-tumour antigen complex (Keith et al, 2002).
This has been made possible by the use of defined tumour
antigens to stimulate lymphocytes in vitro, and the ability
to clone lymphocytes derived from a single, antigen-
specific T cell (Pule et al, 2002). Adoptive transfer of
clonally expanded lymphocytes to lymphopenic hosts after
nonmyeloablative conditioning chemotherapy has resulted
in cell proliferation and persistent clonal repopulation
correlated with tumour regressions in patients with
melanoma (Keith et al, 2002). Ex vivo–expanded clonal
populations of tumour antigen–specific lymphocytes can
be derived from a natural or genetically engineered
initiating cell. Moreover, the TCR of cytotoxic T cells can
be substituted with an immunoglobulin-like surface
molecule, which allows the binding to tumour-specific
surface molecules not presented by MHC molecules (Keith
et al, 2002). These more elaborate forms of adoptive
transfer of killer cells are being studied in ongoing clinical
trials. A second approach in preclinical development
involves genetic modification of DCs with the gene for
interleukin-7 (IL-7). IL-7 stimulates cytotoxic T-
lymphocyte responses and down-regulates tumour
production of the immunosuppressive growth factor, TGF-
!.
V. Cancer vaccine
A. Overview
In the past two decades, adoptive immunotherapy,
based on tumour-infiltrating lymphocytes or lymphokine-
activated killer cells, has been used in clinical trials
(Rosenberg et al, 1986; Rosenberg et al, 1987). These
early results gave first evidence that the manipulation of
the immune system represents a promising tool in cancer
immunotherapy. The main rationale of genetic
immunopotentiation protocols is the possibility of
enlisting the immune system for a potentially vast
amplification of gene therapy, thereby enhancing
therapeutic benefit. The recognition that most tumours
encode TAA and are capable of inducing protective
immunity in preclinical models has reinvigorated the field
of cancer immunotherapy (Pule et al, 2002). It has been
hypothesized that the immune system of tumour patients,
characterized by tolerance, can be modified to mount an
immunological response against the tumour and thus
facilitate tumour rejection. This ‘cancer vaccination’ is to
be accomplished through exposure of TAA in a more
favourable context to the immune system (Christian et al,
2006). Despite ongoing efforts to define and characterize
TAA and, more importantly, clinically relevant TAA, little
is known about TAA for the majority of human cancers
and the largest part of clinical experience with tumour
vaccines has been obtained in melanoma patients.
Therefore, most cancer vaccines, to date, use tumour cells
as a source of TAA. The molecular identification of
antigens expressed by tumour cells that can be recognized
by specific CD8+ cytotoxic T lymphocytes (CTLs) has
provided a means by which to explore anti-tumour T-cell
parameters in patients and also to develop antigen-specific
immunotherapies.
B. Current vaccines
1. Antigen Presentation to the Immune
System
The immune system responds to intracellular events
in target cells by the recognition of intracellularly derived
protein fragments presented on the cell surface by major
histocompatibility complex (MHC) molecules. Circulating
T lymphocytes can potentially engage these peptide-MHC
complexes through their T-cell receptors (TCR). This

Xu J et al: immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours
45
mechanism allows the immune system to differentiate
abnormal intracellular processes from normally
functioning cells expressing so-called self proteins. The
key steps in the generation of an immune response to
cancer cells include loading of tumour antigens onto
antigen-present cells in vitro or in vivo (Figure 1).
2. Intratumoral bacillus Calmette-Guérin
(BCG)
This strategy may be one of the earliest forms of
cellular immunotherapy tested by the Intratumoral
injection of the BCG in cancer (Mathe et al, 1973). The
immunologic basis is that BCG generates an inflammatory
process ideal for the attraction of APCs, which pick up
tumour antigens released by the tumour cells, damaged by
the bacterial infection and cross-present them in a so-
called danger environment. This form of treatment
generates occasional antitumor immune responses.
3. Intratumoral HLA-B7
The intratumoral injection of BCG, the recognition of
a powerful alloantigen by cells with NK activity allows
the recruitment of APCs, among other inflammatory cells,
which will pick up tumour antigens released by the HLA-
B7–transfected cells and cross-present them to cytotoxic
effector cells. These tumours antigen-specific CD8+ CTLs
would then be permitted to attack other tumour cells
without the requirement of the presence of the alloantigen
HLA-B7 on tumour cells.
4. Whole-cell tumour vaccines
Whole-cell autologous tumour vaccines are
personalized vaccines, and it can be assumed that they
contain the relevant tumour antigens; however, the logistic
drawback is that it is difficult to obtain and individually
prepare vaccines for each patient. To avoid this problem,
other tumour cell vaccines have been formulated as lysates
of allogeneic laboratory cell lines containing shared
tumour antigens (Sondak et al, 2002).
5. Naked DNA and gene-modified tumour
vaccines
Intramuscular injection of naked DNA sequences
results in gene expression and the generation of immune
responses (Wolff et al, 1990; Kumar et al, 1996). These
DNA plasmids, which consist of an antigen gene regulated
by a promoter with constitutive activity can be conjugated
with gold particles and propelled into the skin using a
helium gas gene gun. The protein antigen produced by the
target cells is taken up by host APCs, processed, and cross-
presented to the immune system in the draining lymph
nodes.
Gene-modified tumour vaccines have been tested in
clinical trials for many years, the paracrine expression of
cytokines such as IL-2 or IFN!, would allow the tumour
cell to provide all of the signals for direct cytotoxic T cell
activation, bypassing the need for host APCs and CD4+ T
lymphocyte assist (Fearon et al, 1990). However,
comparison of the antitumor capacity of gene-modified
tumour vaccines in preclinical models was surprising in
that the introduction of GM-CSF into tumour cells
produced the most active vaccine (Dranoff et al, 1993).
Bone marrow chimeras were used to show that the GM-
CSF gene-modified tumour vaccines attracted host APCs,
which picked up tumour antigens and cross-presented
them to the host immune system (Huang et al, 1994).
Figure1: Cross-presentation of tumour antigens derived from cancer vaccines.
Several immunologic manipulations lead to a common pathway of cross presentation of proteins derived from tumour antigens. a) in vivo
APC-Based Vaccines; b) ex Vivo APC-Based Vaccines; c) augment the number of APC; d) non-T cell-DC. These host antigen-presenting
cells (APCs), the most powerful of which are the DCs, circulate through the afferent lymphatic vessels to the T-cell areas of lymph nodes.
There they cross-present the tumour antigen to T lymphocytes.

Gene Therapy and Molecular Biology Vol 13, page 46
46
6. Microbe-based vaccines
A variety of microbiology vectors have been adapted
to cancer immunotherapy. Tumour antigen DNA
sequences can be inserted into attenuated pox viruses that
are unable to replicate in mammalian hosts or tumour
antigen gene segments have been introduced into bacteria
such as Salmonella and Listeria, resulting in protective
immunity in animal models (Huang et al, 1994). Other
vectors include recombinant replication-incompetent viral
vectors (adenovirus, retrovirus, lentivirus), which are
modified viruses that have been specifically mutated to be
incapable of self-replication into infectious progeny
virions after infection of a single target cell, but that
efficiently express the foreign gene inserted in the vector.
This form of genetic immunization has also resulted in
weak immunologic responses in humans (Rosenberg et al,
1998), enhancing the immune potency of viral vector.
Immunization can be achieved by the coexpression of
cytokines or costimulatory molecules in the viral vector
because these viral vectors usually have a large capacity to
carry and express multiple genes (Rosenberg et al, 1998).
Several anaerobic bacteria vectors are testing in lab now.
Advantages may include the ability to use the oral route
for immunization and the strong inflammatory milieu
created by bacterial products, leading to the attraction of
APCs, and a preferential Th1 cytokine polarizing pattern
stimulated by certain bacteria such as Listeria.
7. The prime-boost strategy
The sequential administration of naked DNA and a
viral vector has resulted in synergistic immune activation;
it is a potent method of generating immune responses to
tumour antigens in what is now known as the prime-boost
strategy. The initial injection of a plasmid allows the
activation of infrequent T cells without other immune cells
competing for the antigen because the naked DNA has a
limited inflammatory potential. After a rest period, these
antigen-specific high-avidity lymphocytes are boosted by
the re-exposure to the same antigen, now in a more
inflammatory milieu generated by the highly
immunogenic viral proteins from the recombinant viral
vector. Preclinical murine and primate models have shown
that this heterologous prime-boost regimen induces 10- to
100-fold higher frequencies of T cells than do naked DNA
or recombinant viral vectors alone (Ramshaw et al, 2000).
A modification of this strategy is the sequential
administration of two different viral vectors carrying the
same tumour antigen gene, which bypasses the limitation
of the development of neutralizing antibodies to the viral
backbone by boosting with a different vector without
shared viral epitopes (Mincheff et al, 2000; Marshall et al,
2000). These strategies, which avoid the need of cell
culture common to the majority of highly immunologically
active vaccine strategies, are rapidly undergoing clinical
testing for infectious disease and cancer.
8. Augmentation of the number of APCs
As can be noted by the mechanism of action of most
of the prior immunologic maneuvers, the common
pathway of anticancer immune activation is the
recruitment and activation of host APCs to cross-present
tumour antigens to effector CD8+ cytotoxic T cells
(Figure1). Cytokines such as GM-CSF have been used as
vaccine adjuvants with the hope of attracting and
activating DCs locally at the site of vaccination. Other
strategies are aimed at systemically expanding the
dendritic cell pool in the hosts, which may be achieved by
the administration of cytokines such as the combination of
GM-CSF and IL-4 (Roth et al, 2000). In retrospective
studies of tumour biopsies, a greater number of APCs
infiltrating the cancer have been correlated with
improvements in survival of patients (Lotze, 1997). This
increase in the availability of intratumoral APCs may
allow more efficient cross-presentation of tumour antigens.
C. Ex vivo APC-based vaccines
1. DCs and exosomes
The crucial role of DCs was discovered for the
induction of primary T-cell–dependent immune responses.
DCs are now considered to be the best adjuvant for
antitumor immunity. Different antigen loading procedures
have been used for dendritic cell antigen presentation. For
well-characterized antigens, synthetic HLA-binding
peptide epitopes or the complete DNA sequence in a viral
vector can be used to load the dendritic cell vaccines. DCs
pulsed with peptide epitopes and genetically-modified with
recombinant viral or bacteria vectors are conceptually
similar to the vaccination with peptides in immunologic
adjuvants or the direct administration of recombinant
viruses, respectively, in which the DCs should be
perceived as powerful immunologic adjuvants for the
tumour antigen. Also, DCs can be loaded with defined
antigens to take advantage of antigen uptake surface
receptors, such as FC receptors to take up immune
complexes carrying a tumour antigen (Rafiq et al, 2002).
The nanometer vesicles derived from late endosomes
are released differentiated in vitro by DCs , which contain
most of the appropriate molecules to adequately present
MHC-antigen complexes to the immune system (Wolfers
et al, 2001; Zitvogel et al, 1998). These exosomes can be
isolated by filtration of dendritic cell culture media and
then loaded with custom antigens. Their use alone as
vaccines or as vehicles to transfer back preassembled
MHC-peptide complexes to DCs is under clinical
investigation
2. Non–T-cell–directed cancer vaccines
Monoclonal antibodies to surface receptors, such as
trastuzumab or rituximab, have complex mechanisms of
action leading to effective tumour regressions. One such
mechanism is the stimulation of antibody-dependent cell-
mediated cytotoxicity. This immune-based effect, together
with the recognized ability of immune complexes to allow
antigen cross-presentation in DCs (Clynes et al, 2000),
may contribute to their antitumour effects by a coordinated
humoral and cellular response. Several other cancer
vaccines are in different phases of clinical testing. Most of

Xu J et al: immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours
47
these strategies rely on the activation of humoral
(antibody) responses to a peptide or nonpeptide antigen.
Resultant tumour cell damage and cross-presentation of
antigen by host APCs may allow the transfer of the
immunologic stimulus to cellular immune responses.
Advances in the understanding of the mechanisms of
action of cellular antitumour immune responses have
allowed the development of new generations of cancer
vaccines, in which the key step is the recognition of the
need for professional APCs to cross-present the antigen to
the host immune system. The most immunologically active
vaccines usually require costly and laborious ex vivo
cellular cultures, whereas the cell-free vaccines that can be
directly administered from an easily stored and transported
vial are usually less immunologically active but more
suitable for widespread clinical testing. New advances in
the formulation of cancer vaccines brought by a more
precise knowledge of the requirements for the generation
of cellular immune responses to tumour antigens, together
with the current ability to closely monitor cellular immune
responses, will likely provide powerful, nonindividualized,
cell-free vaccines in the near future.
VI. Combined multi-modality therapy: immunization with anaerobic
bacteria therapy for tumour
Immunotherapy strategies for cancer gene
therapy utilize gene transfer to facilitate a dormant
host immune response directed against the tumour.
Evasion of autologous host cellular immunity is a
common feature of tumour cell neoantigens. Tumour
cells are poor antigen presenting cells. ‘Cancer
vaccine’ strategies are based on optimization of the
context in which tumour antigens or tissue-specific
antigens are presented to the host immune system
(Sobol et al, 1995). Utilizing gene therapy to
optimize tumourantigen presentation is through the
targeted expression of cytokines in tumour cells.
Targeted paracrine expression eliminates the
toxicities associated with systemic cytokine
administration. The transduced cytokines result in a
combination of improved tumour cell vaccine
antigen presentation, and activation of APCs, both
essential for effective priming of the cellular immune
response.
The vector-induced inflammatory/immune
response functions as an adjuvant to the transduced
antigen, resulting in local release of cytokines and
influx of APCs to the vaccine site. The
immunotherapy of cancer is now being assessed in
the clinics. An immune response has a potentially
long-term clinical impact on the course of the disease
by stabilising the condition and thus prolonging
survival rather than by performing massive tumour
elimination, those with minor tumour burden or
patients who have had their tumour surgically
removed but who have a high risk of relapse. In these
categories of patients, disease stabilisation, frequency
of relapse, time-span to relapse and length of survival
are the most rational parameters for evaluating
cancer immunization effectiveness. Even if optimal
gene delivery is achieved, the success of gene
therapy, like conventional therapy, may be impeded
by tumour cell resistance and intratumoural cell
heterogeneity. The use of combined treatment
modalities provides a rational paradigm to improve
upon the clinical efficacy of cancer gene therapy
(Klencke et al, 2002). Within the modality of gene
therapy itself, multiple therapies may be combined in
an attempt to benefit from additive or synergistic
efficacy. Multi-gene therapy approaches already
under evaluation include the transduction of dual
immunostimulatory molecules for immunotherapy,
and anaerobic bacteria therapy (Figure 2).
A major limitation in the use of gene therapy in
solid tumours in vivo is the diffusion-limited tissue
penetration into the target tissue. The ability of
immunotherapy and anaerobic bacteria therapy has
been observed in vitro and in vivo. The effects we
observed in animals are contingent on both
bacteriolysis and immunity. There are three reasons
to believe that systemic injection of Clostridium.
Novyi-NT (C. novyi-NT) into humans would lead to
bacteriolysis of tumours. First, C. novyi-NT
germinates within the tumours of all three species
tested (rabbits, rats, and mice), whether the tumours
are s.c., intramuscular, or intrahepatic. Second, C.
novyi-NT can germinate within human tumour
xenografts in the nude mouse host (although
complete regressions and cures are not generally
observed as there is minimal T cell-mediated
immunity). And third, there are many case reports of
C. novyi germination and gangrene developing in
penetrating wounds or after illicit drug injection.
These reports demonstrate that the parental strain of
C. novyi, differing from C. novyi-NT only in that the
latter is devoid of the lethal "-toxin gene, can
proliferate within hypoxic regions in humans.
C. novyi-NT infection of cancers in humans will
induce tumour immunity is more difficult to predict
(Dang et al, 2004). There are many studies indicating
that human tumours are immunogenic, as assessed by
the presence of specific antibodies or reactive T cells
in untreated patients. Furthermore, it has been shown
that stronger immune responses can be elicited
through the administration of various vaccines in
several clinical trials. But there are also many studies
indicating that human tumour cells can protect
themselves against potential immune responses
through a variety of direct and indirect mechanisms.

Gene Therapy and Molecular Biology Vol 13, page 48
48
Figure 2: Anaerobic bacteria-mediated immunologic therapy for solid tumour
Anaerobic bacteria therapy has been observed these effects in treatment of solid tumour: a) nonspecific immunologic therapy which the
characterization of cytokines produced by immune system cells and their production by genetic recombinant techniques, such as IL-2 and IFN,
the significant toxicity of high-dose systemic cytokine therapy is the major drawback; b) specific immunisation represent which allow the
stimulation of an immune response while avoiding the high toxicity of systemic administration of recombinant anaerobic bacteria vectors and
gene modification of tumour cells, which allows an initial direct cytotoxic effect on the cancer cell by antibody dependent cellular cytotoxicity,
thereby releasing tumour antigens; c) the adoptive transfer of immune effector cells from the immune system, T cell, DCs pulsed with
genetically-modified with recombinant anaerobic bacteria vectors are conceptually similar to the vaccination with peptides in immunologic
adjuvant.
As similar observations, both with respect to the
potential of tumours to elicit an immune response and
their ability to evade such responses, have been
recorded in animals, there is reason to hope that the
immune therapeutic effects stimulated by C. novyi-
NT germination might be obtainable in carefully
selected patients.
In experimental setting, the strictly anaerobic
Clostridia have demonstrated several advantages
over others as clostridial spores specifically colonise
and germinate into vegetative cells in the hypoxic
regions of solid tumours, causing tumour lysis and
destruction. Early trials in the 70's of non pathogenic
strains in human had shown plausible safety (Carey
et al, 1976).
VII. Conclusions
Current innovative approaches for cancer therapy
hold significant potentials for effective cancer
management; bacteria therapies and immunotherapies will
probably be the most promising, especially when genetic
manipulation of bacteria to improve its potential have
applied. Recent understanding of tumour
microenvironment, detailed characterization of tumour
antigens and the increased revealing of the immunological
pathways involved in tumour immunity have paved the
way for the design of gene-immune therapies (Ribas et al,
2000). To this end, three cellular sources can be
envisaged for genetic modification: tumour cells, effector
T cells and DCs. However, before ex vivo immuno-gene
therapy can become a realistic treatment modality for
cancer, several barriers have yet to be overcome. First,
improved (bacteria) vectors should lead to higher gene
delivery rates and transgene expression. Therefore,
carefully designed clinical studies are necessary to assess
gene transfer efficiency, safety and toxicity, and
eventually to establish the clinical efficacy of the tumour
immunization. With regard to gene-modified tumour
cells, another major issue still unsolved at the clinical level
is to determine what is the best cytokine the tumour cells
to release in order to recruit the immune system. Second, it
will be imperative to break down the immunological
tolerance against the tumour through reversal of T cell
ignorance, anergy or tumour-induced immunosuppression
in order to achieve a therapeutic outcome. Use of DCs,
whether gene-modified or not, in the context of danger
signals could provide a means to initiate a cellular immune
response against the tumour. An additional general feature
to be considered when designing immuno-gene therapy of
cancer is the complex redundancy of the immune system.
Its effectiveness in protecting the body from harmful
infections demands a sophisticated network to control the
pathways of activation and termination of an immune
response, as well as maintenance of life-long tolerance.
This suggests that a combination of multiple strategies,

Xu J et al: immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours
49
gene-based or not, acting at different levels may be
advantageous to boost the immune system against the
tumour. Moreover, it is believed that the breakdown of
tolerance to tumours will require, in addition to the
strategies discussed in this review, complementary
strategies that specifically counteract the active tumour-
induced immunosuppression.
VIII. Future directions
The challenges facing the implementation of
successful gene therapeutic strategies will be better
understood as the early clinical trials for cancer gene
therapy begin to return more results. Vector development
with increased transgene size capacity, optimized
immunogenic properties, and improved gene transfer
efficiency and targeting will facilitate the next generation
of gene therapy strategies (Kanai et al, 1998). The
burgeoning field of genomics provides an exciting new
resource for the design of tumour-specific gene therapy
strategies. Harnessing these tumour gene products and
others for use as immunization offers exciting prospects
for a whole new class of cancer gene therapy strategies.
As the diversity of molecular lesions underlying
tumourigenesis is better characterized, new targets for
corrective and cytoreductive approaches will emerge.
Effective anticancer gene therapy may ultimately require
individualized molecular profiles. Solid tumours meet
their demands for nascent blood vessels and increased
glycolysis, to combat hypoxia, by activating multiple
genes involved in angiogenesis and glucose metabolism.
Hypoxia inducible factor-1(HIF-1) is a constitutively
expressed basic helix-loop-helix transcription factor,
formed by the assembly of HIF-1alpha and HIF-1beta,
which is stabilized in response to hypoxia, and rapidly
degraded under normoxic conditions (Kanai et al, 1998). It
activates the transcription of genes important for
maintaining oxygen homeostasis but failed to stimulate
systemic T-cell-mediated antitumour immunity, and
synergized with B7-1-mediated immunotherapy. This
approach holds promise to form the foundation for the
transition between the traditional anticancer therapies and
the molecular antineoplastic gene therapy of the future.
Other approaches are to develop new gene therapy vectors
whose expression is selectively activated by hypoxia
(Rosenberg et al, 1998). As VEGF is upregulated by
hypoxia, such regulatory mechanisms would enable us to
achieve hypoxia-inducible expression of therapeutic
genes. The unique pathophysiology of solid tumours
presents a huge problem for the conventional therapies.
Thus, the outcomes of current therapies are so far
disappointing. Several new approaches aiming at
developing effective treatments are on the horizon. These
include a variety of bacteria-based therapy systems.
Amongst all these, anaerobic bacteria vector-mediated
cancer therapy is most promising and expected to generate
new data and new protocols for cancer gene therapy.
Acknowledgements
This work is partly supported by a project grant from
the NHMRC/Cancer Council Queensland (Grant ID No.
401681) and the Dr. Jian Zhou smart state fellowship from
the State Government of Queensland to MQW.
References
Agrawal N, Bettegowda C, Cheong I, et al (2004). Bacteriolytic
therapy can generate a potent immune response against
experimental tumors. Proc Natl Acad Sci U S A 101, 15172-7.
Akasaki Y, Liu G, Matundan HH, et al (2006). A peroxisome
proliferator-activated receptor-gamma agonist, troglitazone,
facilitates caspase-8 and -9 activities by increasing the enzymatic
activity of protein-tyrosine phosphatase-1B on human glioma
cells. J Biol Chem 281, 6165-74.
Aznavoorian S, Stracke ML, Krutzsch H, Schiffmann E, Liotta LA
(1990). Signal transduction for chemotaxis and haptotaxis by
matrix molecules in tumor cells. J Cell Biol; 110, 1427-38.
Akbulut L, Zhang Y, Deisseroth A (2003). Cytotoxic effect of
replication-competent adenoviral vectors carrying L-plastin
promoter regulated E1A and cytosine deaminase genes in
cancers of the breast, ovary and colon, Cancer Gene Ther 10,
388–95.
Bubenik J, Vonka V (2003). MHC class I status of tumours and
design of immunotherapeutic strategies. Immunol Lett 90, 177-
8.
Biswas S, Guix M, Rinehart C, et al (2007). Inhibition of TGF-beta
with neutralizing antibodies prevents radiation-induced
acceleration of metastatic cancer progression. J Clin Invest 117,
1305-13.
Bos R vDP, van der Groep P, Shvarts A, Greijer AE, van der Wall E
(2004). Expression of hypoxia-inducible factor-1alpha and cell
cycle proteins in invasive breast cancer are estrogen receptor
related. Breast Cancer Res 6: R450-9.
Burdelya L, Kujawski M, Niu G, et al (2005). Stat3 activity in
melanoma cells affects migration of immune effector cells and
nitric oxide-mediated antitumor effects. J Immunol 174, 3925-
31.
Berzofsky JA, Terabe M (2008). NKT cells in tumor immunity:
opposing subsets define a new immunoregulatory axis. J
Immunol 180, 3627-35.
Battaglia M, Gregori S, Bacchetta R, Roncarolo MG (2006). Tr1
cells: from discovery to their clinical application. Semin
Immunol 18, 120-7.
Bonizzi RP (2007). Welfare and immune response. Veterinary
Research Communications 31, 97-102.
Cho W. Contribution of oncoproteomics to cancer biomarker
discovery (2007). Mol Cancer 6, 25-9.
Croker AK, Allan AL (2008). Cancer stem cells: implications for
the progression and treatment of metastatic disease. J Cell Mol
Med 12, 374-90.
Chen ZS, Robey RW, Belinsky MG, et al (2003). Transport of
methotrexate, methotrexate polyglutamates, and 17beta-estradiol
17-(beta-D-glucuronide) by ABCG2: effects of acquired
mutations at R482 on methotrexate transport. Cancer Res 63,
4048-54.
Cools N, Ponsaerts P, Van Tendeloo VF, Berneman ZN (2007).
Balancing between immunity and tolerance: an interplay between
dendritic cells, regulatory T cells, and effector T cells. J Leukoc
Biol 82, 1365-74.
Caux CD, Schmitt D, Banchereau J (1992). GM-CSF and TNF-
alpha cooperate in the generation of dendritic Langerhans cells.
Nature 360, 258-61.

Gene Therapy and Molecular Biology Vol 13, page 50
50
Clynes RA, Towers TL, Presta LG, et al (2000). Inhibitory Fc
receptors modulate in vivo cytoxicity against tumor targets. Nat
Med 6, 443–6,
Carey RW, Holland JF, Whang HY, Neter E (1967), Bryant B:
Clostridial oncolysis in man. Euro J Can 3, 37-46.
Christian A Petrulio SK-S, Howard L Kaufman (2006). The tumour
microenvironment and implications for cancer immunotherapy.
Expert Opinion on Biological Therapy 6, 671-84.
Dong H, Chen L (2003). B7-H1 pathway and its role in the evasion
of tumor immunity. J Mol Med 81, 281-7.
Della Porta M, Danova M, Rigolin GM, et al (2005). Dendritic cells
and vascular endothelial growth factor in colorectal cancer:
correlations with clinicobiological findings. oncology 68, 276-
84.
Dean M, Fojo T, Bates S (2005). Tumour stem cells and drug
resistance. Nat Rev Cancer 5, 275-84
Dranoff G, Jaffee E, Lazenby A, et al (1993). Vaccination with
irradiated tumor cells engineered to secrete murine granulocyte-
macrophage colony-stimulating factor stimulates potent, specific,
and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S
A 90, 3539–43.
Dang LH, Bettegowda C, Huso DL, Kinzler KW, Vogelstein B
(2001). Combination bacteriolytic therapy for the treatment of
experimental tumors. Proc Natl Acad Sci U S A 98, 15155-60.
Dang LH, Bettegowda C, Agrawal N, et al. (2004). Targeting
vascular and avascular compartments of tumors with C. novyi-
NT and anti-microtubule agents. Cancer Biol. Ther. 3, 326-337
de Jonge-Peeters SD, Kuipers F, de Vries EG, Vellenga E (2007).
ABC transporter expression in hematopoietic stem cells and the
role in AML drug resistance. Crit Rev Oncol Hematol 62, 214-
26.
Elke Jäger DJ, Alexander Knuth (2002). Clinical cancer vaccine
trials. Current Opinion in immunology 14, 178-82.
Frey AB (2006). Myeloid suppressor cells regulate the adaptive
immune response to cancer. J Clin Invest 116, 2587-90.
Fearon ER, Pardoll DM, Itaya T, et al (1990). Interleukin-2
production by tumour cells bypasses T helper function in the
generation of an antitumor response. Cell 60, 397–403.
Gro F MS (2006). Design of bifunctional siRNAs: Combining
immunostimulation and gene-silencing in one single siRNA
molecule. Biochemical and Biophysical Research
Communications 352, 642-9.
Ganss R (2006). Tumor stroma fosters neovascularization by
recruitment of progenitor cells into the tumor bed. J Cell Mol
Med 10, 857-65.
Glick RP LT, Lin H, Tarlock K, Cohen EP (2006). Immunogene
therapy as a treatment for malignant brain tumors in young mice.
J Neurosurg 105, 65-70.
Gelman AE (2003). Autoimmunity heats up. Nat Med 9, 1465-6.
Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM
(2005). Expression and function of laminins in the embryonic
and mature vasculature. Physiol Rev 85,979-1000.
Huang AY, Golumbek P, Ahmadzadeh M, et al (1994). Role of
bone marrow-derived cells in presenting MHC class I-restricted
tumor antigens. Science 264, 961–5.
Iwadate Y, Namba H, Tagawa M, et al (1997). Induction of
acquired immunity in rats that have eliminated intracranial
gliosarcoma cell by the expression of herpes simplex virus
thymidine kinase gene and ganciclovir administration. Oncology
54, 329-34
Jennifer A L, Philipp K (2006). Recent developments in
antibacterial drug discovery: microbe-derived natural products –
from collection to the clinic. Expert Opinion on Investigational
Drugs 15, 211-26.
Koido S, Nikrui N, Ohana M, et al (2005). Assessment of fusion
cells from patient-derived ovarian carcinoma cells and dendritic
cells as a vaccine for clinical use. Gynecol Oncol 99, 462-71.
Kay A (2001). Allergy and allergic diseases. First of two parts. N
Engl J Med 344, 30-7.
Keith LK BA, David A M, Kathy S, Mary LD (2002). Adoptive T-
cell therapy for the treatment of solid tumours. Expert Opinion
on Biological Therapy 2, 55-66.
Klencke B, Matijevic M, Urban RG, et al (2002). Encapsulated
plasmid DNA treatment for human papillomavirus 16-associated
anal dysplasia: A phase I study of ZYC101. Clin Cancer Res 8,
1028–37,
Kanai F, Kawakami T, Hamada H, et al (1998). Adenovirus-
mediated transduction of Escherichia coli uracil
phosphoribosyltransferase gene sensitizes cancer cells to low
concentrations of 5-fluorouracil. Cancer Res 58, 1946-51.
Kunz M TA, Goebeler M, Engelhardt E, Bröcker E, Gillitzer R
(1999). Strong expression of the lymphoattractant C-X-C
chemokine Mig is associated with heavy infiltration of T cells in
human malignant melanoma. J Pathol 189, 552-8.
Kopp HG, Ramos CA, Rafii S (2006). Contribution of endothelial
progenitors and proangiogenic hematopoietic cells to
vascularization of tumor and ischemic tissue. Curr Opin
Hematol 13, 175-81.
Kumar V, Sercarz E (1996). Genetic vaccination: The advantages of
going naked. Nat Med 2, 857–9.
Logan AC, Lutzko C, Kohn DB (2002). Advances in lentiviral
vector design for gene-modification of hematopoietic stem cells.
Curr Opin Biotechnol 13, 429-36.
Lotze MT (1997). Getting to the source: Dendritic cells as
therapeutic reagents for the treatment of patients with cancer.
Ann Surg 226, 1–5,
Lowenberg B, Griffin JD, Tallman MS (2003). Acute myeloid
leukemia and acute promyelocytic leukemia. Hematology Am
Soc Hematol Educ Program 82-101.
Luis SF, Eckhard RP (2005). Lung Cancer Immunotherapy. Clin
Med Res 3, 221-28.
Mahoney BP, Baggett B, Gillies RJ (2003). Tumor acidity, ion
trapping and chemotherapeutics. I. Acid pH affects the
distribution of chemotherapeutic agents in vitro. Biochem
Pharmacol 66, 1207-18.
Morgan RA, Dudley ME, Wunderlich JR (2006). Cancer regression
in patients after transfer of genetically engineered lymphocytes.
Science. 314(5796), 126-9
Marina Mata JCG, David J. Fink (2003). Gene Transfer to the
Nervous System: Prospects for Novel Treatments Directed at
Diseases of the Aging Nervous System. The Journals of
Gerontology Series A: Biological Sciences and Medical
Sciences 58, M1111-18
Mimeault M, Batra SK (2006). Concise review: recent advances on
the significance of stem cells in tissue regeneration and cancer
therapies. Stem Cells 24, 2319-45.
Mimeault M, Batra SK (2007). Interplay of distinct growth factors
during epithelial mesenchymal transition of cancer progenitor
cells and molecular targeting as novel cancer therapies. Ann
Oncol 18, 1605-19.
Mathe G, Halle-Pannenko O, Bourut C (1973). BCG in cancer
immunotherapy: Results obtained with various BCG preparations
in a screening study for systemic adjuvants applicable to cancer
immunoprophylaxis or immunotherapy. Natl Cancer Inst
Monogr 39, 107–13.

Xu J et al: immunotherapy with anaerobic bacteria for immunogene therapy of solid tumours
51
Martin-Orozco N, Dong C (2006). New battle fields for
costimulation. J Exp Med 203, 817-20.
Mimeault M, Brand RE, Sasson AA, Batra SK(2005). Recent
advances on the molecular mechanisms involved in pancreatic
cancer progression and therapies. Pancreas 31, 301-16.
Mimeault M, Batra SK (2006). Recent advances on multiple
tumorigenic cascades involved in prostatic cancer progression
and targeting therapies. Carcinogenesis 27, 1-22.
Marshall JL, Hoyer RJ, Toomey MA, et al (2000). Phase I study in
advanced cancer patients of a diversified prime-and-boost
vaccination protocol using recombinant vaccinia virus and
recombinant nonreplicating avipox virus to elicit anti-
carcinoembryonic antigen immune responses. J Clin Oncol 18,
3964–73,
Mincheff M, Tchakarov S, Zoubak S, et al (2000). Naked DNA and
adenoviral immunizations for immunotherapy of prostate cancer:
A phase I/II clinical trial. Eur Urol 38, 208–17.
Mabjeesh N J, Simons J W (2002). Gene therapy of prostate cancer:
current and future directions. Endocrine-Related Cancer 9,
115-39.
Nuyts S VML, Theys J, Landuyt W, Lambin P, Anné J (2002).
Clostridium spores for tumor-specific drug delivery. Anticancer
Drugs 13, 115-25.
Niethammer AG, Xiang R, Becker JC, et al (2002). A DNA vaccine
against VEGF receptor 2 prevents effective angiogenesis and
inhibits tumor growth. Nat Med 8, 1369-75.
Ohtani H (1998). Stromal reaction in cancer tissue:
pathophysiologic significance of the expression of matrix-
degrading enzymes in relation to matrix turnover and
immune/inflammatory reactions. Pathol Int 48,1-9.
Pouysségur J DF, Mazure NM (2006). Hypoxia signalling in cancer
and approaches to enforce tumour regression. Nature 441, 437-
43.
Petrulio CA, Kim-Schulze S, Kaufman HL (2006). The tumour
microenvironment and implications for cancer immunotherapy.
Expert Opin Biol Ther 6, 671-84.
Pule BC, Heslop HE (2002). Genetically engineered T-cells for
adoptive immunotherapy. Curr Opin Mol Ther ; 4, 467-75.
Philip W, Wong CL, Ian F (2005). Tannock Reduction of
Intracellular pH as a Strategy to Enhance the pH-Dependent
Cytotoxic Effects of Melphalan for Human Breast Cancer Cells
Clinical Cancer Research 11, 3553-7.
Palucka AK, Joseph W F, Jacques B (2007). Taming cancer by
inducing immunity via dendritic cells. Immunological Reviews
220, 129-50.
Roth MD, Gitlitz BJ, Kiertscher SM, et al (2000). Granulocyte
macrophage colony-stimulating factor and interleukin 4 enhance
the number and antigen-presenting activity of circulating CD14+
and CD83+ cells in cancer patients. Cancer Res 60, 1934–41.
Rafiq K, Bergtold A, Clynes R (2002). Immune complex-mediated
antigen presentation induces tumor immunity. J Clin Invest
110, 71–79.
Reilly RT, Gottlieb MB, Ercolini AM, et al (2000). HER-2/neu is a
tumor rejection target in tolerized HER-2/neu transgenic mice.
Cancer Res 60, 3569-76.
Rosenberg SA, Zhai Y, Yang JC, et al (1998). Immunizing patients
with metastatic melanoma using recombinant adenoviruses
encoding MART-1 or gp100 melanoma antigens. J Natl Cancer
Inst 90, 1894–1900.
Ramshaw IA, Ramsay AJ (2000). The prime-boost strategy:
Exciting prospects for improved vaccination. Immunol Today
21, 163–5.
Rosenberg SA, Spiess P, Lafreniere R (1986). A new approach to
the adoptive immunotherapy of cancer with tumor-infiltrating
lymphocytes. Science 233, 1318–1321.
Rosenberg SA, Lotze MT, Muul LM, et al (1987). A progress report
on the treatment of 157 patients with advanced cancer using
lymphokine-activated killer cells and interleukin-2 or high-dose
interleukin-2 alone. N Engl J Med 316, 889–97.
Ribas A, Butterfield LH, Economou JS (2000). Genetic
immunotherapy for cancer. Oncologist 5, 87–98.
Shicang Y, Guijun H, Guisheng Q, Yuying L, Guoming W, Ruiling
G (2007). Efficacy of chemotherapeutic agents under hypoxic
conditions in pulmonary adenocarcinoma multidrug resistant cell
line. J Chemother 19, 203-11.
Suciu-Foca N, Manavalan JS, Cortesini R (2003). Generation and
function of antigen-specific suppressor and regulatory T cells.
Transpl Immunol 11, 235-44.
Sallusto FA (1994). Efficient presentation of soluble antigen by
cultured human dendritic cells is maintained by
granulocyte/macrophage colony-stimulating factor plus
interleukin 4 and downregulated by tumor necrosis factor alpha.
J Exp Med 179, 1109-18.
Steinman RM, Turley S, Pierre P, Mellman I (1999). Antigen
capture, processing, and presentation by dendritic cells: recent
cell biological studies. Hum Immunol 60,562-7.
Seo N, Hayakawa S, Takigawa M, Tokura Y (2001). Interleukin-10
expressed at early tumour sites induces subsequent generation of
CD4(+) T-regulatory cells and systemic collapse of antitumour
immunity. Immunology 103, 449-57.
Sioud M FG, Cekaite L (2007). Suppression of immunostimulatory
siRNA-driven innate immune activation by 2'-modified RNAs.
Biochem Biophys Res Commun 361, 122-6.
Shakhar Guy B-ES (2003). Potential Prophylactic Measures Against
Postoperative Immunosuppression: Could They Reduce
Recurrence Rates in Oncological Patients? . Annals of Surgical
Oncology 10, 972-92.
Sondak VK, Liu PY, Tuthill RJ, et al (2002). Adjuvant
immunotherapy of resected, intermediate-thickness, node-
negative melanoma with an allogeneic tumour vaccine: Overall
results of a randomized trial of the Southwest Oncology Group. J
Clin Oncol 20, 2058–66,
Sobol RE, Fakhrai H, Shawler D, et al (1995). Interleukin-2 gene
therapy in a patient with glioblastoma. Gene Ther 2, 164–7.
Shibata T GAJ, Brown J M (2000). Development of a hypoxia-
responsive vector for tumor-specific gene therapy. Gene therapy
7, 493-8.
Tatum JL, Kelloff GJ, Gillies RJ, et al (2006). Hypoxia: importance
in tumor biology, noninvasive measurement by imaging, and
value of its measurement in the management of cancer therapy.
Int J Radiat Biol 82, 699-757.
Tlsty TD, Hein PW (2001). Know thy neighbor: stromal cells can
contribute oncogenic signals. Curr Opin Genet Dev 11, 54-9.
Trédan O GC, Patel K, Tannock IF (2007). Drug resistance and the
solid tumor microenvironment. J Natl Cancer Inst 99, 1441-
54.
Tamada K, Tamura H, Flies D, et al (2002). Blockade of
LIGHT/LTbeta and CD40 signaling induces allospecific T cell
anergy, preventing graft-versus-host disease. J Clin Invest
109,549-57.
Van Pel A, Boon T (1982). Protection against a nonimmunogenic
mouse leukemia by an immunogenic variant obtained by
mutagenesis. Proc Natl Acad Sci USA 79, 4718-22

Gene Therapy and Molecular Biology Vol 13, page 52
52
von Euler H, Sadeghi A, Carlsson B, et al (2008). Efficient
adenovector CD40 ligand immunotherapy of canine malignant
melanoma. J Immunother 31, 377-84.
Wu W, Luo Y, Sun C, et al (2006). Targeting cell-impermeable
prodrug activation to tumor microenvironment eradicates
multiple drug-resistant neoplasms. Cancer Res 66:970-80.
Welters MJ, Kenter GG, Piersma SJ, et al (2008). Induction of
tumor-specific CD4+ and CD8+ T-cell immunity in cervical
cancer patients by a human papillomavirus type 16 E6 and E7
long peptides vaccine. Clin Cancer Res 14, 178-87.
Wagner E (2007). Converging Paths of Viral and Non-viral Vector
Engineering. Molecular Therapy 16, 1-2.
Wolff JA, Malone RW, Williams P, et al (1990). Direct gene
transfer into mouse muscle in vivo. Science 247, 1465–8,.
Wolfers J, Lozier A, Raposo G, et al (2001). Tumor-derived
exosomes are a source of shared tumor rejection antigens for
CTL cross-priming. Nat Med 7, 297–303.
Yang W, Chen PW, Li H, Alizadeh H, Niederkorn JY (2008). PD-
L1: PD-1 Interaction Contributes to the Functional Suppression
of T-Cell Responses to Human Uveal Melanoma Cells In Vitro.
Invest Ophthalmol Vis Sci 49, 2518-25.
Zitvogel L, Regnault A, Lozier A, et al (1998). Eradication of
established murine tumors using a novel cell-free vaccine:
Dendritic cell-derived exosomes. Nat Med 4, 594–600.

Gene Therapy and Molecular Biology Vol 13, page 53
53
Gene Ther Mol Biol Vol 13, 53-63, 2009
Non-viral and local gene medicine for improvement of cutaneous wound healing Review Article
Markus Rimann1, Heike Hall1* 1Cells and BioMaterials, Department of Materials, ETH Zurich, Zurich, Switzerland
__________________________________________________________________________________
*Correspondence: Heike Hall, ETH Zurich, Department of Materials, HCI E415, Cells and BioMaterials, Wolfgang-Pauli-Strasse 10,
CH-8093 Zurich, Switzerland; Tel: +41 44 633 69 75; Fax: +41 44 632 10 73; email: [email protected]
Key words: wound healing, local gene therapy, gene medicine, non-viral gene delivery systems, matrix-mediated gene delivery, PLL-g-
PEG nanoparticles
Abbreviations: adeno-associated viruses, (AAV); early endosome antigen-1, (EEA-1); extracellular matrix, (ECM); hypoxia-inducible
factor, (HIF); HIF-1! lacking the oxygen-sensitive degradation domain (HIF-1!!ODD); Low-level laser therapy, (LLLT); matrix
metalloproteinases, (MMPs); negative pressure wound therapy, (NPWT); platelet-derived growth factor, (PDGF); poly(ethylene glycol),
(PEG); poly(lactide-co-glycolide), (PLGA); polyethylenimine, (PEI); poly-L-lysine, (PLL); transferrin receptor, (TFR); US Food and
Drug Administration, (FDA); vacuum-assisted closure, (VAC); vascular endothelial growth factor-A, (VEGF-A)
Received: 23 March 2009; Revised: 01 April 2009
Accepted: 03 April 2009; electronically published: April 2009
Summary
Deficient vascularisation is a major clinical incidence and affects wound healing especially in elderly people as well
as in diabetes patients. Many studies and different technologies aim to locally increase blood perfusion and improve
the endogenous wound healing capacity and thereby ameliorate the patient’s life quality. Gene therapy has gained a
lot of attention for treatment of chronic diseases, cancer and genetic disorders. It is also considered as a valuable
alternative for conventional protein therapy, since it overcomes inherent problems that are associated with
administration of protein drugs in terms of bioavailability, systemic toxicity, in vivo clearance rate and
manufacturing costs. For this reason safe and efficient delivery systems for therapeutic DNA are developed.
Polycationic substances have been shown to form complexes with DNA and are widely used as an attractive
alternative to viral vectors in gene therapy. One promising approach consists in the usage of grafted copolymers of
poly-L-lysine (PLL) and poly(ethylene glycol) (PEG) that forms stable complexes with plasmid DNA, which are
highly transfection-efficient and are suitable to deliver DNA from 3D-fibrin wound healing matrices. A gene of
interest to be delivered should stimulate endogenous wound healing and may consist of a stabilized form of hypoxia-
inducible factor-1! (HIF-1!!ODD), a transcription factor that ultimately leads to the increase in vascular
endothelial growth factor-A (VEGF-A) expression that in turn activates angiogenesis followed by wound healing.
Local administration of a matrix-mediated DNA delivery system on cutaneous wounds will be a big step in the
direction of specific gene medicine and might represent a powerful tool in clinical wound therapy.
I. Introduction For most people, wound healing is a natural process
of repair, which follows injuries of the skin and other soft
tissues. For diseased individuals, however, it becomes a
complex medical problem requiring specialized treatment
and care. Together with many local factors that impede the
healing process such as trauma, edema and infections,
many systemic factors also contribute to impair wound
healing processes. Among them are age, chronic diseases,
such as diabetes mellitus, vascular insufficiencies,
immunosuppressant and radiation therapy (Gosain and
DiPietro, 2004; Hausman and Rinker, 2004; Jeffecoate et
al, 2004; Anscher and Vujaskovic, 2005), (for review:
Branski et al, 2007; Eming et al, 2007; Jensen, 2007).
Since these risk factors affect large proportions of the
aging population, the need of an adequate approach to
treat impaired wound healing e.g. by locally increasing the
blood perfusion seems essential. Worldwide
approximately twenty million people suffer from chronic
wounds caused by diabetes (alone > 7 million diabetic
ulcers), circulatory problems and many other conditions
such as surgical site infections that generate huge demands
on the health care systems (http://www.prlog.org
/10076809-wound-types-and-advanced-wound-products-

Rimann and Hall: Gene therapy in wound healing
54
market-worldwide). In Europe only, diabetic patients
exceed 30 million people and cause 5-10 % of the total
health care costs (www.idf-europe.org). Therefore,
therapeutic improvements of wound healing especially by
increasing the patients’ endogenous wound healing
potentials are highly appreciated by the patients
themselves and by the entire society.
II. Wound healing Wound healing is a highly dynamic process related to
growth and tissue regeneration and involves complex
interactions of extracellular matrix (ECM) molecules,
soluble mediators, various resident cells and infiltrating
cells to reachieve tissue integrity (Singer and Clark, 1999;
Baum and Arpey, 2005; Gurtner et al, 2008). Wound
healing comprises four overlapping phases: hemostasis
and inflammation, migration, proliferation and maturation
(Gurtner et al, 2008). Details about the overlapping phases
of wound healing are available in excellent recent reviews
(Baum and Arpey, 2005; Barrientos et al, 2008; Gurtner et
al, 2008) and will not be repeated here. The focus of this
review will be on elucidating impaired wound healing that
results when the well-orchestrated sequence of events is
disturbed or stopped and non-healing or chronic wounds
develop.
A. Impaired wound healing Wounds can be categorized into two different types
distinguished by their healing properties: i) The acute
wound follows the well-orchestrated phases of
inflammation, new tissue formation and remodeling
leading to tissue repair and scar formation, whereas ii)
chronic wounds fail to heal within the expected time
frame, which arises from the disruption of the orderly
sequence of events at one or more stages in the wound
healing process. In order to ensure an effective wound
repair, interfering factors such as diseases (e.g. diabetes
mellitus), drug therapies (e.g. growth factor delivery) and
patient circumstances (e.g. pressure sores because of
neuropathy), wounds in immunocompromised people
(after systemic chemotherapy and/or radiation therapy,
chronic steroid use) must be all taken into considerations
(Boateng et al, 2008). In addition, aged people often show
slowed or impaired wound healing even without an
underlying disease (Swift et al, 2001). On the molecular
level chronic wounds display a deficiency of endogenous
growth factors (Pierce et al, 1995; Jeffecoate et al, 2004;
Whitney, 2005) or an excessive production of exudate and
expression of high levels of tissue-degrading proteases
creating a destructive non-healing-promoting wound
environment (Fahey et al, 1991; Loots et al, 1998). Often
prolonged inflammation, impaired neovascularization,
decreased synthesis of collagen, increased levels of
proteases and defective macrophage function are observed
(Fahey et al, 1991; Loots et al, 1998; Branski et al, 2007;
Bao et al, 2008). In the case of prolonged inflammation
the upregulation of neutrophils leads to increased secretion
of matrix metalloproteinases (MMPs) that are imbalanced
because of the lack of their natural inhibitors. Furthermore
the mitogenic activity of cells is suppressed because of
missing growth factors that promote proliferation such as
platelet-derived growth factor (PDGF). Therefore the
chronic wound displays a destructive environment that is
not favorable for wound healing.
III. Treatment of chronic wounds Treatment of wounds can be divided into physical
and biological methods. The physical treatments include
surgical debridement, vacuum-assisted closure (VAC)
therapy and low level laser treatments. Surgical
debridement involves the removal of necrotic tissue out of
the wound bed. This may eventually lead to a reset of the
disturbed sequence of wound healing processes (Falanga,
2004, 2005). Clinical success of these methods is assigned
to a reduction of excess of wound fluid, edema and
exudate. Furthermore the putative bacterial burden and
phenotypically abnormal cells are removed. In vacuum-
assisted closure therapy also termed negative pressure
wound therapy (NPWT), a controlled level of negative
pressure of -80 to -125 mmHg is applied on the wounds
leading to accelerated debridement and promotion of
healing in many different types of wounds (Saxena et al,
2004; Lindstedt et al, 2006; Jones et al, 2005; Kanakaris et
al, 2007; Körber et al, 2008; Labanaris et al, 2008). The
underlying mechanisms of NPWT suggest mechanical
deformation of cells in and around the wound resulting in
increased matrix synthesis, which ultimately leads to an
improved wound healing (Saxena et al, 2004; Wilkes et al,
2007; Eneroth and van Houtum, 2008; Ennis et al, 2008;
Jacobs et al, 2008). Low-level laser therapy (LLLT) has
been introduced by Mester and colleagues in 1968 and
uses a single, coherent, monochromatic wavelength of
light. The power varies from 5 to 500 mW and the
emission wavelength is between 600 to 1000 nm. It has
been shown that LLLT led to increased production of
procollagen by human skin fibroblasts, increased
fibroblast and keratinocyte proliferation, increased
angiogenesis, tension resistance of scars and improved
epithelialization (Sobanko and Alster, 2008).
Another way to treat wounds and improve their
healing capacities is to use specialized and bioactive
wound dressings either made of molecules from the ECM
or of synthetic polymers. Many dressings are already
commercially available and are composed of collagen,
hyaluronic acid, amelogenins, chondroitin-6-sulphate and
fibrin. The compositions, functionalities and their
applications have been recently reviewed in (Agren and
Werthen, 2007). Some of the dressings combine ECM
molecules with exogenously applied cells, such as human
fibroblasts, autologous human keratinocytes and allogenic
human fibroblasts. These bioactive dressings have been
recently reviewed in (Boateng et al, 2008). Today more
and more dressings are composed of polymeric molecules
either artificial or of natural origin. The idea is to simulate
the native ECM of the wound site and adjacent tissue
using water swollen, gas permeable and fibrillar polymer
structures that allow gas and nutrient exchange. Often used
polymers are poly(lactide-co-glycolide) (PLGA),
poly(vinyl pyrrolidone), poly(vinyl alcohol), polyurethane
foams, hydrocolloid and alginate dressings (reviewed in
(Boateng et al, 2008)). Other hydrogel dressings are made
of native polymers such as hyaluronic acid, collagen,

Gene Therapy and Molecular Biology Vol 13, page 55
55
chitosan and fibrin (reviewed in (Boateng et al, 2008)).
Hydrogel dressings can be loaded with therapeutically-
active substances in order to achieve a controlled and
sustained release thereby avoiding multiple interventions
by changing the wound dressing several times. Commonly
used are bactericides such as silver ions, antibiotics, or
antimicrobial peptides and different growth factors. In
order to support physical wound therapies significant
efforts have been made to develop protein growth factors
as wound healing therapeutics. First clinical trials were
performed with exogenous application of growth factors
like platelet-derived growth factor (PDGF) and others
(Robson et al, 2001; Steed, 2006; Viswanathan et al,
2006). So far only PDGF-BB has received approval by the
US Food and Drug Administration (FDA) but solely for
the treatment of diabetic foot ulcers (Margolis et al, 2004;
Robson et al, 2001; Steed, 2006; Viswanathan et al, 2006).
Unfortunately, these efforts have not produced clinically
significant improvements. The overall lack of success with
protein growth factors has been attributed in part to short
persistence of the growth factor in the protease-rich
environment of the wound bed as only 1-9 % of the
applied growth factor dose reached a depth of 1-3 mm
(Trengove et al, 1999; Yager and Nwomeh, 1999). This
required repeated applications of growth factors that are
very costly to produce. Further difficulties are associated
with the wound healing process itself as it is very complex
and involves many different growth factors acting in
concert and need to succeed one after the other thus being
very difficult to simulate by application of single
therapeutics.
IV. Gene therapy/gene medicine to
improve wound healing In recent years, gene therapy has been evaluated as
an alternative approach in wound therapy (Chandler et al,
2000; Petrie et al, 2003; Eriksson and Vranckx, 2004;
Keswani et al, 2004; Theopold et al, 2004; Yla-Herttuala
et al, 2004; Glover et al, 2005). Two different strategies
are distinguished concerning the introduction and time of
foreign gene expression: gene therapy that refers to the
permanent substitution of a defect or missing gene
whereas gene medicine leads to transient transformation
and short term expression of a gene product (Morgan and
Anderson, 1993; Khavari et al, 2002). As genes encoding
for a growth factor or a defective protein could be placed
into the wound milieu (reviewed by: (Hirsch et al, 2007;
Davidson, 2008)), sustained local production of the growth
factor might yield improvements over purely protein-
based therapies. Advantages of gene-based as compared to
protein-based therapies are longer life times of applied
genes and therefore prolonged expression of the
therapeutic protein, immunological tolerance and faster
and easier production and storage of the components that
might finally lead to a reduction in costs for the health care
systems. Gene therapy and gene medicine use a number of
different DNA delivery systems that can be divided into
two major groups: namely viral and non-viral delivery
systems.
A. Gene delivery systems Viral gene delivery systems use recombinant viruses,
such as retroviruses (including lentiviruses), adenoviruses
and adeno-associated viruses (AAV) containing
therapeutic DNA (Breckpot et al, 2007; Flotte, 2007;
Stender et al, 2007). Due to their inherent cell infection
ability these gene delivery systems are very efficient and
transduce dividing and some of them also non-dividing
cells. Thus most of the clinical trials used viral delivery
systems for gene therapy
(http://www.wiley.co.uk/genetherapy/clinical/). Recent
reviews summarize applications of viral vectors for
cutaneous wound healing in animal and human studies
(Branski et al, 2007; Eming et al, 2007; Jensen, 2007).
Unfortunately viruses have several drawbacks such as high
immunogenicity, packaging size limitations and some of
them show random integration into the host genome,
which leads to non-controllable side effects (Wu and
Burgess, 2004).
Therefore, many non-viral gene delivery vehicles
have been designed to overcome the inherent limitations
of viral vectors. Non-viral gene delivery systems may
consist of naked DNA transfer, lipid-mediated, peptide-
mediated and polymer-mediated condensation of
therapeutic DNA that lead to an improved cellular uptake
(Panyam and Labhasetwar, 2003; Wells, 2004; Trentin et
al, 2005; Park et al, 2006; Jeon et al, 2006; Gao et al,
2007; Shigeta et al, 2007). The major limits of these non-
viral vectors are their poor in vivo transfection efficiencies
resulting in low protein production, as well as their
transient gene expression profile, which in some cases
such as in wound healing, is desirable. The benefits of
non-viral gene delivery vehicles are their safety and their
unlimited gene size transportation capacity (Tal, 2000).
Currently three different strategies for non-viral
applications of therapeutic DNA are pursued. The simplest
way is the use of naked DNA, which is either injected
directly into the target tissue (Liu et al, 2007), applied via
electroporation or ultrasound (Kusumanto et al, 2007) or
loaded onto nano-sized particles of heavy metal and
brought into the cell by gene gun applications (Kuriyama
et al, 2000). Alternatively, microseeding delivers DNA
directly into target cells by solid microneedles (Eriksson et
al, 1998). However, enzymatic degradation of the
unprotected DNA and poor cell transfection efficiencies
are the major drawbacks (Liu et al, 2007). Another
approach is the use of lipoplexes that are lipid-based DNA
vehicles, entering the cytoplasm by cell membrane fusion
(Felgner et al, 1987; Lv et al, 2006). Many different
modifications for specific cell targeting and intracellular
routing have been developed (Kawakami et al, 2000;
Vandenbroucke et al, 2007). However, the primary
drawback of lipid-based DNA delivery systems is their
rapid clearance from the blood stream and their short-term
stability (Lai and van Zanten, 2002).
Another group of DNA-complexing substances
consists of polycationic molecules such as PLL, poly-L-
ornithine or polyethylenimine (PEI) that have been
previously demonstrated to be used as gene delivery
vehicles (Ramsay et al, 2000; Pichon et al, 2001; Davis,
2002; Zaitsev et al, 2004) for review see: (Park et al,
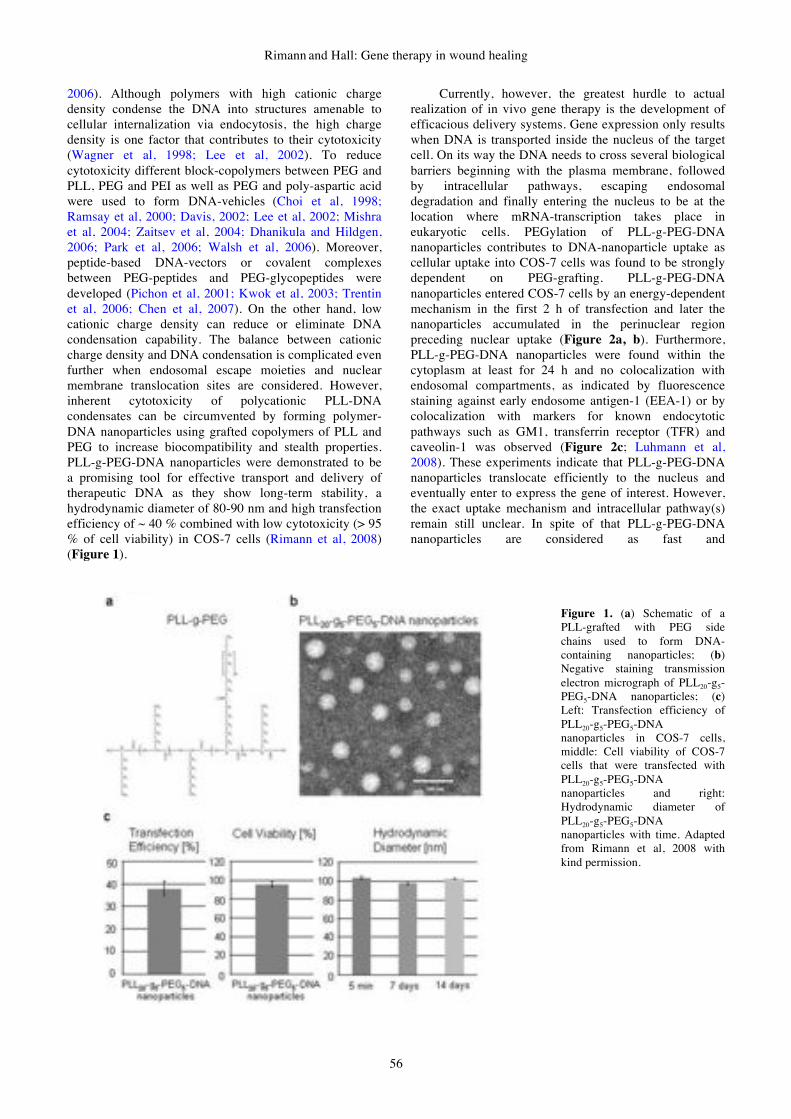
Rimann and Hall: Gene therapy in wound healing
56
2006). Although polymers with high cationic charge
density condense the DNA into structures amenable to
cellular internalization via endocytosis, the high charge
density is one factor that contributes to their cytotoxicity
(Wagner et al, 1998; Lee et al, 2002). To reduce
cytotoxicity different block-copolymers between PEG and
PLL, PEG and PEI as well as PEG and poly-aspartic acid
were used to form DNA-vehicles (Choi et al, 1998;
Ramsay et al, 2000; Davis, 2002; Lee et al, 2002; Mishra
et al, 2004; Zaitsev et al, 2004; Dhanikula and Hildgen,
2006; Park et al, 2006; Walsh et al, 2006). Moreover,
peptide-based DNA-vectors or covalent complexes
between PEG-peptides and PEG-glycopeptides were
developed (Pichon et al, 2001; Kwok et al, 2003; Trentin
et al, 2006; Chen et al, 2007). On the other hand, low
cationic charge density can reduce or eliminate DNA
condensation capability. The balance between cationic
charge density and DNA condensation is complicated even
further when endosomal escape moieties and nuclear
membrane translocation sites are considered. However,
inherent cytotoxicity of polycationic PLL-DNA
condensates can be circumvented by forming polymer-
DNA nanoparticles using grafted copolymers of PLL and
PEG to increase biocompatibility and stealth properties.
PLL-g-PEG-DNA nanoparticles were demonstrated to be
a promising tool for effective transport and delivery of
therapeutic DNA as they show long-term stability, a
hydrodynamic diameter of 80-90 nm and high transfection
efficiency of ~ 40 % combined with low cytotoxicity (> 95
% of cell viability) in COS-7 cells (Rimann et al, 2008)
(Figure 1).
Currently, however, the greatest hurdle to actual
realization of in vivo gene therapy is the development of
efficacious delivery systems. Gene expression only results
when DNA is transported inside the nucleus of the target
cell. On its way the DNA needs to cross several biological
barriers beginning with the plasma membrane, followed
by intracellular pathways, escaping endosomal
degradation and finally entering the nucleus to be at the
location where mRNA-transcription takes place in
eukaryotic cells. PEGylation of PLL-g-PEG-DNA
nanoparticles contributes to DNA-nanoparticle uptake as
cellular uptake into COS-7 cells was found to be strongly
dependent on PEG-grafting. PLL-g-PEG-DNA
nanoparticles entered COS-7 cells by an energy-dependent
mechanism in the first 2 h of transfection and later the
nanoparticles accumulated in the perinuclear region
preceding nuclear uptake (Figure 2a, b). Furthermore,
PLL-g-PEG-DNA nanoparticles were found within the
cytoplasm at least for 24 h and no colocalization with
endosomal compartments, as indicated by fluorescence
staining against early endosome antigen-1 (EEA-1) or by
colocalization with markers for known endocytotic
pathways such as GM1, transferrin receptor (TFR) and
caveolin-1 was observed (Figure 2c; Luhmann et al,
2008). These experiments indicate that PLL-g-PEG-DNA
nanoparticles translocate efficiently to the nucleus and
eventually enter to express the gene of interest. However,
the exact uptake mechanism and intracellular pathway(s)
remain still unclear. In spite of that PLL-g-PEG-DNA
nanoparticles are considered as fast and
Figure 1. (a) Schematic of a
PLL-grafted with PEG side
chains used to form DNA-
containing nanoparticles; (b)
Negative staining transmission
electron micrograph of PLL20-g5-
PEG5-DNA nanoparticles; (c)
Left: Transfection efficiency of
PLL20-g5-PEG5-DNA
nanoparticles in COS-7 cells,
middle: Cell viability of COS-7
cells that were transfected with
PLL20-g5-PEG5-DNA
nanoparticles and right:
Hydrodynamic diameter of
PLL20-g5-PEG5-DNA
nanoparticles with time. Adapted
from Rimann et al, 2008 with
kind permission.
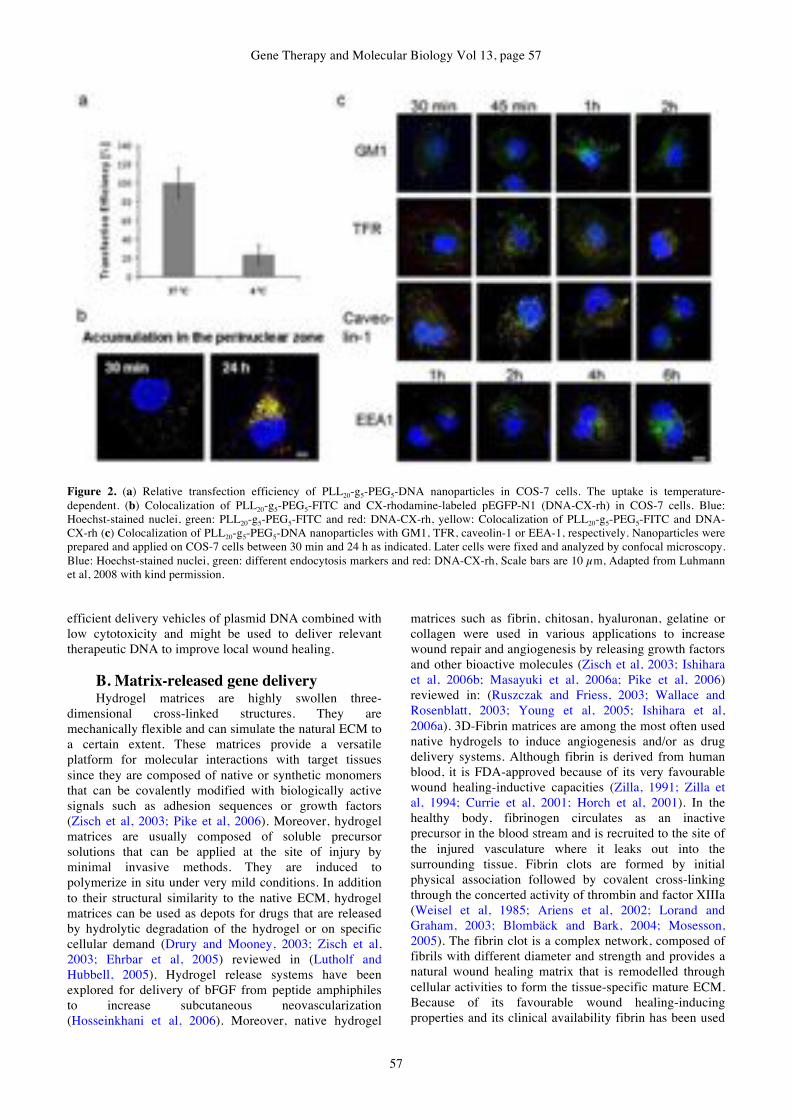
Gene Therapy and Molecular Biology Vol 13, page 57
57
Figure 2. (a) Relative transfection efficiency of PLL20-g5-PEG5-DNA nanoparticles in COS-7 cells. The uptake is temperature-
dependent. (b) Colocalization of PLL20-g5-PEG5-FITC and CX-rhodamine-labeled pEGFP-N1 (DNA-CX-rh) in COS-7 cells. Blue:
Hoechst-stained nuclei, green: PLL20-g5-PEG5-FITC and red: DNA-CX-rh, yellow: Colocalization of PLL20-g5-PEG5-FITC and DNA-
CX-rh (c) Colocalization of PLL20-g5-PEG5-DNA nanoparticles with GM1, TFR, caveolin-1 or EEA-1, respectively. Nanoparticles were
prepared and applied on COS-7 cells between 30 min and 24 h as indicated. Later cells were fixed and analyzed by confocal microscopy.
Blue: Hoechst-stained nuclei, green: different endocytosis markers and red: DNA-CX-rh, Scale bars are 10 µm, Adapted from Luhmann
et al, 2008 with kind permission.
efficient delivery vehicles of plasmid DNA combined with
low cytotoxicity and might be used to deliver relevant
therapeutic DNA to improve local wound healing.
B. Matrix-released gene delivery Hydrogel matrices are highly swollen three-
dimensional cross-linked structures. They are
mechanically flexible and can simulate the natural ECM to
a certain extent. These matrices provide a versatile
platform for molecular interactions with target tissues
since they are composed of native or synthetic monomers
that can be covalently modified with biologically active
signals such as adhesion sequences or growth factors
(Zisch et al, 2003; Pike et al, 2006). Moreover, hydrogel
matrices are usually composed of soluble precursor
solutions that can be applied at the site of injury by
minimal invasive methods. They are induced to
polymerize in situ under very mild conditions. In addition
to their structural similarity to the native ECM, hydrogel
matrices can be used as depots for drugs that are released
by hydrolytic degradation of the hydrogel or on specific
cellular demand (Drury and Mooney, 2003; Zisch et al,
2003; Ehrbar et al, 2005) reviewed in (Lutholf and
Hubbell, 2005). Hydrogel release systems have been
explored for delivery of bFGF from peptide amphiphiles
to increase subcutaneous neovascularization
(Hosseinkhani et al, 2006). Moreover, native hydrogel
matrices such as fibrin, chitosan, hyaluronan, gelatine or
collagen were used in various applications to increase
wound repair and angiogenesis by releasing growth factors
and other bioactive molecules (Zisch et al, 2003; Ishihara
et al, 2006b; Masayuki et al, 2006a; Pike et al, 2006)
reviewed in: (Ruszczak and Friess, 2003; Wallace and
Rosenblatt, 2003; Young et al, 2005; Ishihara et al,
2006a). 3D-Fibrin matrices are among the most often used
native hydrogels to induce angiogenesis and/or as drug
delivery systems. Although fibrin is derived from human
blood, it is FDA-approved because of its very favourable
wound healing-inductive capacities (Zilla, 1991; Zilla et
al, 1994; Currie et al, 2001; Horch et al, 2001). In the
healthy body, fibrinogen circulates as an inactive
precursor in the blood stream and is recruited to the site of
the injured vasculature where it leaks out into the
surrounding tissue. Fibrin clots are formed by initial
physical association followed by covalent cross-linking
through the concerted activity of thrombin and factor XIIIa
(Weisel et al, 1985; Ariens et al, 2002; Lorand and
Graham, 2003; Blombäck and Bark, 2004; Mosesson,
2005). The fibrin clot is a complex network, composed of
fibrils with different diameter and strength and provides a
natural wound healing matrix that is remodelled through
cellular activities to form the tissue-specific mature ECM.
Because of its favourable wound healing-inducing
properties and its clinical availability fibrin has been used
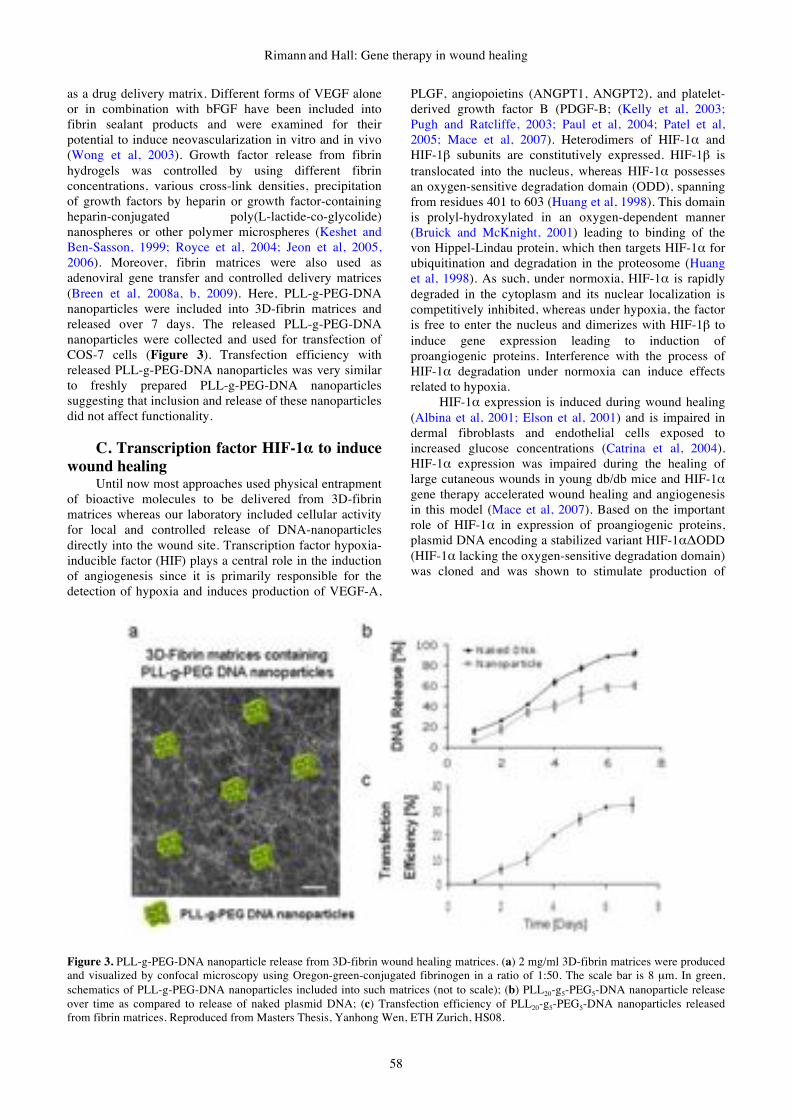
Rimann and Hall: Gene therapy in wound healing
58
as a drug delivery matrix. Different forms of VEGF alone
or in combination with bFGF have been included into
fibrin sealant products and were examined for their
potential to induce neovascularization in vitro and in vivo
(Wong et al, 2003). Growth factor release from fibrin
hydrogels was controlled by using different fibrin
concentrations, various cross-link densities, precipitation
of growth factors by heparin or growth factor-containing
heparin-conjugated poly(L-lactide-co-glycolide)
nanospheres or other polymer microspheres (Keshet and
Ben-Sasson, 1999; Royce et al, 2004; Jeon et al, 2005,
2006). Moreover, fibrin matrices were also used as
adenoviral gene transfer and controlled delivery matrices
(Breen et al, 2008a, b, 2009). Here, PLL-g-PEG-DNA
nanoparticles were included into 3D-fibrin matrices and
released over 7 days. The released PLL-g-PEG-DNA
nanoparticles were collected and used for transfection of
COS-7 cells (Figure 3). Transfection efficiency with
released PLL-g-PEG-DNA nanoparticles was very similar
to freshly prepared PLL-g-PEG-DNA nanoparticles
suggesting that inclusion and release of these nanoparticles
did not affect functionality.
C. Transcription factor HIF-1! to induce
wound healing Until now most approaches used physical entrapment
of bioactive molecules to be delivered from 3D-fibrin
matrices whereas our laboratory included cellular activity
for local and controlled release of DNA-nanoparticles
directly into the wound site. Transcription factor hypoxia-
inducible factor (HIF) plays a central role in the induction
of angiogenesis since it is primarily responsible for the
detection of hypoxia and induces production of VEGF-A,
PLGF, angiopoietins (ANGPT1, ANGPT2), and platelet-
derived growth factor B (PDGF-B; (Kelly et al, 2003;
Pugh and Ratcliffe, 2003; Paul et al, 2004; Patel et al,
2005; Mace et al, 2007). Heterodimers of HIF-1! and
HIF-1" subunits are constitutively expressed. HIF-1" is
translocated into the nucleus, whereas HIF-1! possesses
an oxygen-sensitive degradation domain (ODD), spanning
from residues 401 to 603 (Huang et al, 1998). This domain
is prolyl-hydroxylated in an oxygen-dependent manner
(Bruick and McKnight, 2001) leading to binding of the
von Hippel-Lindau protein, which then targets HIF-1! for
ubiquitination and degradation in the proteosome (Huang
et al, 1998). As such, under normoxia, HIF-1! is rapidly
degraded in the cytoplasm and its nuclear localization is
competitively inhibited, whereas under hypoxia, the factor
is free to enter the nucleus and dimerizes with HIF-1" to
induce gene expression leading to induction of
proangiogenic proteins. Interference with the process of
HIF-1! degradation under normoxia can induce effects
related to hypoxia.
HIF-1! expression is induced during wound healing
(Albina et al, 2001; Elson et al, 2001) and is impaired in
dermal fibroblasts and endothelial cells exposed to
increased glucose concentrations (Catrina et al, 2004).
HIF-1! expression was impaired during the healing of
large cutaneous wounds in young db/db mice and HIF-1!
gene therapy accelerated wound healing and angiogenesis
in this model (Mace et al, 2007). Based on the important
role of HIF-1! in expression of proangiogenic proteins,
plasmid DNA encoding a stabilized variant HIF-1!"ODD
(HIF-1! lacking the oxygen-sensitive degradation domain)
was cloned and was shown to stimulate production of
Figure 3. PLL-g-PEG-DNA nanoparticle release from 3D-fibrin wound healing matrices. (a) 2 mg/ml 3D-fibrin matrices were produced
and visualized by confocal microscopy using Oregon-green-conjugated fibrinogen in a ratio of 1:50. The scale bar is 8 µm. In green,
schematics of PLL-g-PEG-DNA nanoparticles included into such matrices (not to scale); (b) PLL20-g5-PEG5-DNA nanoparticle release
over time as compared to release of naked plasmid DNA; (c) Transfection efficiency of PLL20-g5-PEG5-DNA nanoparticles released
from fibrin matrices. Reproduced from Masters Thesis, Yanhong Wen, ETH Zurich, HS08.
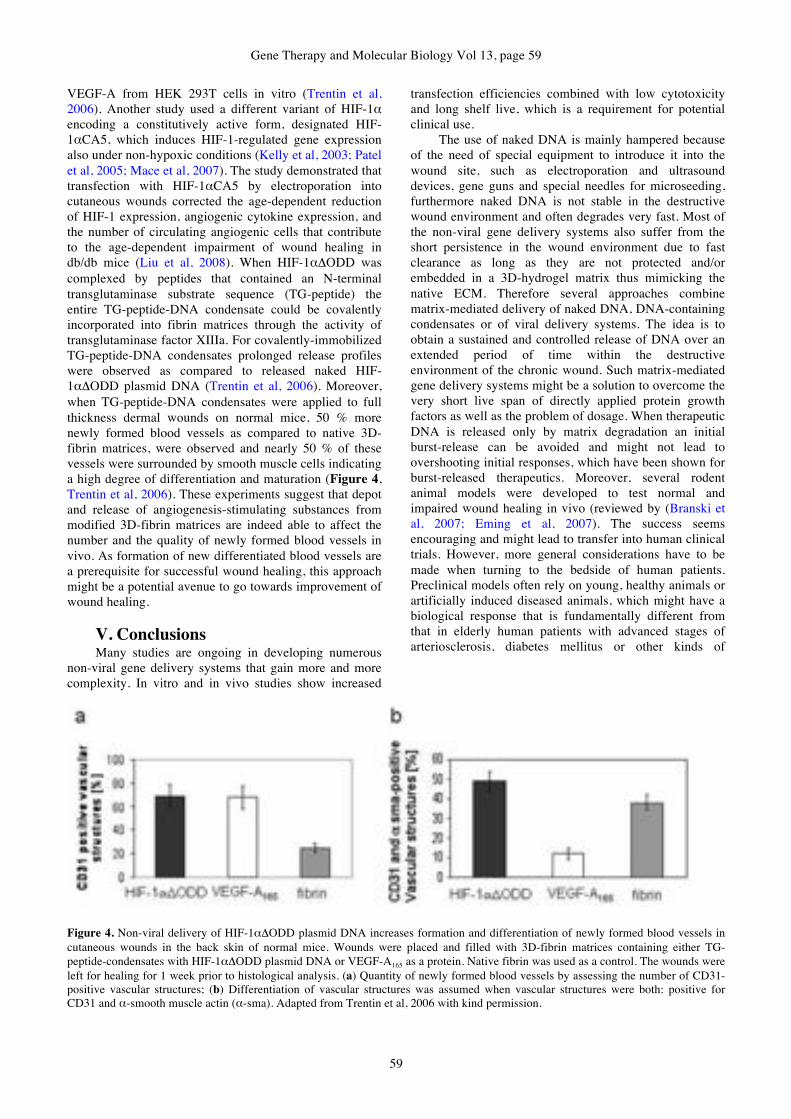
Gene Therapy and Molecular Biology Vol 13, page 59
59
VEGF-A from HEK 293T cells in vitro (Trentin et al,
2006). Another study used a different variant of HIF-1!
encoding a constitutively active form, designated HIF-
1!CA5, which induces HIF-1-regulated gene expression
also under non-hypoxic conditions (Kelly et al, 2003; Patel
et al, 2005; Mace et al, 2007). The study demonstrated that
transfection with HIF-1!CA5 by electroporation into
cutaneous wounds corrected the age-dependent reduction
of HIF-1 expression, angiogenic cytokine expression, and
the number of circulating angiogenic cells that contribute
to the age-dependent impairment of wound healing in
db/db mice (Liu et al, 2008). When HIF-1!!ODD was
complexed by peptides that contained an N-terminal
transglutaminase substrate sequence (TG-peptide) the
entire TG-peptide-DNA condensate could be covalently
incorporated into fibrin matrices through the activity of
transglutaminase factor XIIIa. For covalently-immobilized
TG-peptide-DNA condensates prolonged release profiles
were observed as compared to released naked HIF-
1!!ODD plasmid DNA (Trentin et al, 2006). Moreover,
when TG-peptide-DNA condensates were applied to full
thickness dermal wounds on normal mice, 50 % more
newly formed blood vessels as compared to native 3D-
fibrin matrices, were observed and nearly 50 % of these
vessels were surrounded by smooth muscle cells indicating
a high degree of differentiation and maturation (Figure 4,
Trentin et al, 2006). These experiments suggest that depot
and release of angiogenesis-stimulating substances from
modified 3D-fibrin matrices are indeed able to affect the
number and the quality of newly formed blood vessels in
vivo. As formation of new differentiated blood vessels are
a prerequisite for successful wound healing, this approach
might be a potential avenue to go towards improvement of
wound healing.
V. Conclusions Many studies are ongoing in developing numerous
non-viral gene delivery systems that gain more and more
complexity. In vitro and in vivo studies show increased
transfection efficiencies combined with low cytotoxicity
and long shelf live, which is a requirement for potential
clinical use.
The use of naked DNA is mainly hampered because
of the need of special equipment to introduce it into the
wound site, such as electroporation and ultrasound
devices, gene guns and special needles for microseeding,
furthermore naked DNA is not stable in the destructive
wound environment and often degrades very fast. Most of
the non-viral gene delivery systems also suffer from the
short persistence in the wound environment due to fast
clearance as long as they are not protected and/or
embedded in a 3D-hydrogel matrix thus mimicking the
native ECM. Therefore several approaches combine
matrix-mediated delivery of naked DNA, DNA-containing
condensates or of viral delivery systems. The idea is to
obtain a sustained and controlled release of DNA over an
extended period of time within the destructive
environment of the chronic wound. Such matrix-mediated
gene delivery systems might be a solution to overcome the
very short live span of directly applied protein growth
factors as well as the problem of dosage. When therapeutic
DNA is released only by matrix degradation an initial
burst-release can be avoided and might not lead to
overshooting initial responses, which have been shown for
burst-released therapeutics. Moreover, several rodent
animal models were developed to test normal and
impaired wound healing in vivo (reviewed by (Branski et
al, 2007; Eming et al, 2007). The success seems
encouraging and might lead to transfer into human clinical
trials. However, more general considerations have to be
made when turning to the bedside of human patients.
Preclinical models often rely on young, healthy animals or
artificially induced diseased animals, which might have a
biological response that is fundamentally different from
that in elderly human patients with advanced stages of
arteriosclerosis, diabetes mellitus or other kinds of
Figure 4. Non-viral delivery of HIF-1#!ODD plasmid DNA increases formation and differentiation of newly formed blood vessels in
cutaneous wounds in the back skin of normal mice. Wounds were placed and filled with 3D-fibrin matrices containing either TG-
peptide-condensates with HIF-1#!ODD plasmid DNA or VEGF-A165 as a protein. Native fibrin was used as a control. The wounds were
left for healing for 1 week prior to histological analysis. (a) Quantity of newly formed blood vessels by assessing the number of CD31-
positive vascular structures; (b) Differentiation of vascular structures was assumed when vascular structures were both: positive for
CD31 and !-smooth muscle actin (!-sma). Adapted from Trentin et al, 2006 with kind permission.

Rimann and Hall: Gene therapy in wound healing
60
underlying systemic diseases. In addition, the question
remains whether in vitro models can be compared with in
vivo experiments in rodent animal models and if the
results obtained can then be transferred to human patients.
When a comparison between different DNA delivery
vehicles in vitro and in vivo was performed,
Lipofectamine 2000 and DOTAP/Chol lipoplexes showed
significantly enhanced gene transfer in vitro, whereas no
transfection was detected for naked DNA. In contrast,
naked DNA was found to be most efficient in gene
transfer in experimental burn wounds in rats
(Steinstraesser et al, 2007). Therefore, it has to be taken
into consideration that in vitro test systems offer very
limited predictability for subsequent in vivo gene
therapy/gene medicine approaches especially when
diseased human tissues are addressed. Moreover, transfer
from small animal models where cardiovascular diseases
such as diabetes mellitus or old age can only be simulated
by genetic manipulation, medication or specific inbreeding
of several strains, the etiology of impaired wound healing
might look comparable to human wounds but the
underlying mechanisms can not be compared so easily.
VI. Future perspectives It is well understood that one single growth factor
gene therapy/gene medicine cannot stimulate all
interlinked phases of wound healing in an orchestrated
manner as required. In order to address the complexity of
succeeding active factors (growth factors, cytokines and
enzymes) acting in normal wound healing processes the
strategies must go towards the direction of multiple gene
delivery or address key control genes that stimulate entire
cascades of complex processes. It was demonstrated in a
partial thickness wound healing model that the
combination of PDGF and IGF-I was more effective than
either growth factor alone (Lynch et al, 1987). Moreover a
combination of PDGF and FGF-2 increased the DNA
content of wounds in the rat more than any single growth
factor alone (Sprugel et al, 1987) and transfection of KGF
cDNA in combination with IGF-I cDNA compared to the
same genes individually seems to be more efficient than
the single genes (Jeschke and Klein, 2004). Alternatively,
it was described that specific and local transfection of
single key-transcription factor genes such as HIF-1! at
strategic time points of wound healing might substitute a
sequential growth factor therapy (Trentin et al, 2006; Liu
et al, 2008). This single transcription factor might be able
to switch on the entire cascade of angiogenesis followed
by proper wound healing such that a self-regulating
system is activated. Upon such stimulation ideally the
patients' endogenous regulatory system would take over
and a natural healing response would follow. Another very
important aspect for future gene therapy and gene
medicine will be to formulate safe and efficient delivery
systems that can be controlled by endogenous regulation
such as by cellular demand, in addition it might be
therapeutically interesting to be able to support the release
of therapeutic DNA by external stimuli e.g. by slight
temperature or pH change or light of specific wavelengths.
However, in order to define such key-genes a lot of
basic research is still necessary to find out which
regulatory factors need to be activated at what time.
Moreover, it requires joining forces from interdisciplinary
researchers coming from medicine, life sciences,
pharmaceutical sciences and engineering.
Acknowledgments
This study was supported by Gebert Rüf Foundation
(GRS-053/05), Switzerland.
References Agren MS, Werthen M (2007) The extracellular matrix in wound
healing: a closer look at therapeutics for chronic wounds. Int
J Low Extrem Wounds 6, 82-97.
Albina JE, Mastrofrancesco B, Vessella JA, Louis CA, Henry
WL, Jr., Reichner JS (2001) HIF-1 expression in healing
wounds: HIF-1alpha induction in primary inflammatory cells
by TNF-alpha. Am J Physiol Cell Physiol 281, C1971-1977.
Anscher MS, Vujaskovic Z (2005) Mechanisms and potential
targets for prevention and treatment of normal tissue injury
after radiation therapy. Semin Oncol 32, S86-91.
Ariens RAS, LAi TS, Weisel JW, Greenberg CS, Grant PJ
(2002) Role of factor XIII in fibrin clot formation and effects
of genetic polymorphisms. Blood 100, 743-754.
Bao P, Kodra A, Tomic-Canic M, Gelinko MS, Ehrlich HP,
Brem H (2008) The role of vascular endothelial growth
factor in wound healing. J Surg Res, in press.
Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-
Canic M (2008) Growth factors and cytokines in wound
healing. Wound Repair Regen 16, 585-601.
Baum CL, Arpey CJ (2005) Normal cutaneous wound healing:
clinical correlation with cellular and molecular events.
Dermatol Surg 31, 674-686; discussion 686.
Blombäck B, Bark N (2004) Fibrinopeptides and fibrin gel
structure. Biophys Chem 112, 147-151.
Boateng JS, Matthews KH, Stevens HN, Eccleston GM (2008)
Wound healing dressings and drug delivery systems: a
review. J Pharm Sci 97, 2892-2923.
Branski LK, Pereira CT, Herndon DN, Jeschke MG (2007) Gene
therapy in wound healing: present status and future
directions. Gene Ther 14, 1-10.
Breckpot K, Aerts JL, Thielemans K (2007) Lentiviral vectors
for cancer immunotherapy: transforming infectious particles
into therapeutics. Gene Ther 14, 847-862.
Breen A, Dockery P, O'Brien T, Pandit A (2008a) Fibrin scaffold
promotes adenoviral gene transfer and controlled vector
delivery. J Biomed Mater Res A, in press.
Breen A, O'Brien T, Pandit A (2009) Fibrin as a Delivery System
for Therapeutic Drugs and Biomolecules. Tissue Eng Part B
Rev, in press.
Breen AM, Dockery P, O'Brien T, Pandit AS (2008b) The use of
therapeutic gene eNOS delivered via a fibrin scaffold
enhances wound healing in a compromised wound model.
Biomaterials 29, 3143-3151.
Bruick RK, McKnight SL (2001) A conserved family of prolyl-
4-hydroxylases that modify HIF. Science 294, 1337-1340.
Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L
(2004) Hyperglycemia regulates hypoxia-inducible factor-
1alpha protein stability and function. Diabetes 53, 3226-
3232.
Chandler LA, Gu DL, Ma C, Gonzalez AM, Doukas J, Nguyen
T, Pierce GF, Phillips ML (2000) Matrix-enabled gene
transfer for cutaneous wound repair. Wound Repair Regen
8, 473-479.

Gene Therapy and Molecular Biology Vol 13, page 61
61
Chen CP, Kim JS, Liu D, Rettig GR, McAnuff MA, Martin ME,
Rice KG (2007) Synthetic PEGylated glycoproteins and their
utility in gene delivery. Bioconjug Chem 18, 371-378.
Choi YH, Liu F, Kim JS, Choi YK, Park JS, Kim SW (1998)
Polyethylene glycol-grafted poly-L-lysine as polymeric gene
carrier. J Control Release 54, 39-48.
Currie LJ, Sharpe JR, Martin R (2001) The use of fibrin glue in
skin grafts and tissue-engineered skin replacements: a
review. Plast Reconstr Surg 108, 1713-1726.
Davidson JM (2008) First class delivery: getting growth factors
to their destination. J Invest Dermatol 128, 1360-1362.
Davis ME (2002) Non-viral gene delivery systems. Curr Opin
Biotechnol 13, 128-131.
Dhanikula RS, Hildgen P (2006) Synthesis and evaluation of
novel dendrimers with a hydrophilic interior as nanocarriers
for drug delivery. Bioconjug Chem 17, 29-41.
Drury JL, Mooney DJ (2003) Hydrogels for tissue engineering:
scaffold design variables and applications. Biomaterials 24,
4337-4351.
Ehrbar M, Metters A, Zammaretti P, Hubbell JA, Zisch AH
(2005) Endothelial cell proliferation and progenitor
maturation by fibrin-bound VEGF variants with differential
susceptibilities to local cellular activity. J Control Release
101, 93-109.
Elson DA, Thurston G, Huang LE, Ginzinger DG, McDonald
DM, Johnson RS, Arbeit JM (2001) Induction of
hypervascularity without leakage or inflammation in
transgenic mice overexpressing hypoxia-inducible factor-
1alpha. Genes Dev 15, 2520-2532.
Eming SA, Krieg T, Davidson JM (2007) Gene therapy and
wound healing. Clin Dermatol 25, 79-92.
Eneroth M, van Houtum WH (2008) The value of debridement
and Vacuum-Assisted Closure (V.A.C.) Therapy in diabetic
foot ulcers. Diabetes Metab Res Rev 24 Suppl 1, S76-80.
Ennis WJ, Meneses P, Borani M (2008) Push-pull therory: using
mechanotransduction to achieve tissue perfusion and wound
healing in complex cases. Gynecol Oncol 111, S81-S86.
Eriksson E, Vranckx J (2004) Wet wound healing: from
laboratory to patients to gene therapy. Am J Surg 188, 36-
41.
Eriksson E, Yao F, Svensjo T, Winkler T, Slama J, Macklin MD,
Andree C, McGregor M, Hinshaw V, Swain WF (1998) In
vivo gene transfer to skin and wound by microseeding. J
Surg Res 78, 85-91.
Fahey TJ, 3rd, Sadaty A, Jones WG, 2nd, Barber A, Smoller B,
Shires GT (1991) Diabetes impairs the late inflammatory
response to wound healing. J Surg Res 50, 308-313.
Falanga V (2004) The chronic wound: impaired healing and
solutions in the context of wound bed preparation. Blood
Cells Mol Dis 32, 88-94.
Falanga V (2005) Wound healing and its impairment in the
diabetic foot. Lancet 366, 1736-1743.
Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M,
Northrop JP, Ringold GM, Danielsen M (1987) Lipofection:
a highly efficient, lipid-mediated DNA-transfection
procedure. Proc Natl Acad Sci U S A. 84, 7413-7417.
Flotte TR (2007) Gene therapy: The first two decades and the
current state-of-the-art. J Cell Physiol 213, 301-305.
Gao X, Kim KS, Liu D (2007) Nonviral gene delivery: what we
know and what is next. AAPS J 9, E92-104.
Glover DJ, Lipps HJ, Jans DA (2005) Towards safe, non-viral
therapeutic gene expression in humans. Nat Rev Genet 6,
299-310.
Gosain A, DiPietro LA (2004) Aging and wound healing. World
J Surg 28, 321-326.
Gurtner GC, Werner S, Barrandon Y, Longaker MT (2008)
Wound repair and regeneration. Nature 453, 314-321.
Hausman MR, Rinker BD (2004) Intractable wounds and
infections: the role of impaired vascularity and advanced
surgical methods for treatment. Am J Surg 187, 44S-55S.
Hirsch T, Spielmann M, Yao F, Eriksson E (2007) Gene therapy
in cutaneous wound healing. Front Biosci 12, 2507-2518.
Horch RE, Bannasch H, Stark GB (2001) Transplantation of
cultured autologous keratinocytes in fibrin sealant biomatrix
to resurface chronic wounds. Transplant Proc 33, 642-644.
Hosseinkhani H, Hosseinkhani M, Khademhosseini A,
Kobayashi H, Tabata Y (2006) Enhanced angiogenesis
through controlled release of basic fibroblast growth factor
from peptide amphiphile for tissue regeneration.
Biomaterials 27, 5836-5844.
Huang LE, Gu J, Schau M, Bunn HF (1998) Regulation of
hypoxia-inducible factor 1alpha is mediated by an O2-
dependent degradation domain via the ubiquitin-proteasome
pathway. Proc Natl Acad Sci U S A 95, 7987-7992.
Ishihara M, Kiyohaya O, Nakamura SMF, Masuoka K, Kanatani
H, Morimoto Y, Ishihara M, Machara T, Kikuchi M (2006a)
Chitosan hydrogel as a drug delivery carrier to control
angioenesis. J Artif Organs 9, 8-16.
Ishihara M, Masanori F, Kiyohaya O, Hattori H, Kikuchi M,
Maehara T (2006b) Controlled release of FGF-2 and
paclitaxol from chitosan hdrogels and their subsequent
effects on wound repair, angiogenesis, tumor growth. Curr
Drug Deliv 3, 351-358.
Jacobs S, Simhaee DA, Marsano A, Fomovsky GM, Niedt G,
Wu JK (2008) Efficacy and mechanisms of vacuum-assisted
closure (VAC) therapy in promoting wound healing: a rodent
model. J Plast Reconstr Aesthet Surg, in press.
Jeffecoate WJ, Price P, Harding KG (2004) Wound healing and
treatments for people with diabetic foot ulcers. Diabetes
Metab Res Rev 20(suppl. 1), S79-89.
Jensen TG (2007) Cutaneous gene therapy. Ann Med 39, 108-
115.
Jeon O, Kang SW, Lim HW, Chung JH, Kim BS (2006) Long-
term and zero order release of basic fibroblast growth factor
from heparin-conjugated poly(L-lactide-co-glycolide)
nanospheres and fibrin gel. Biomaterials 27, 1598-1607.
Jeon O, Ryu SH, Chung JH, Kim BS (2005) Control of basic
fibroblast growth factor release from fibrin gel with heparin
and concentrations of fibrinogen and thrombin. J Contr Rel
105, 249-259.
Jeschke MG, Klein D (2004) Liposomal gene transfer of multiple
genes is more effective than gene transfer of a single gene.
Gene Ther 11, 847-855.
Jones SM, Banwell PE, Shakespeare PG (2005) Advances in
wound healing: topical negative pressure therapy. Postgrad
Med J 81, 353-357.
Kanakaris NK, Thanasas C, Keramaris N, Kontakis G, Granick
MS, Giannoudis PV (2007) The efficacy of negative pressure
wound therapy in the management of lower extremity
trauma: Review of clinical evidence. Injury, Int J Care
Injured 38S, S8-S17.
Kawakami S, Sato A, Nishikawa M, Yamashita F, Hashida M
(2000) Mannose receptor-mediated gene transfer into
macrophages using novel mannosylated cationic liposomes.
Gene Ther 7, 292-299.
Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon
S, Rowan A, Yan Z, Campochiaro PA, Semenza GL (2003)
Cell type-specific regulation of angiogenic growth factor
gene expression and induction of angiogenesis in
nonischemic tissue by a constitutively active form of
hypoxia-inducible factor 1. Circ Res 93, 1074-1081.
Keshet E, Ben-Sasson SA (1999) Anticancer drug targets:
approaching angiogenesis. J Clin Invest 104, 1497-1501.
Keswani SG, Katz AB, Lim FY, Zoltick P, Radu A, Alaee D,
Herlyn M, Crombleholme TM (2004) Adenoviral mediated

Rimann and Hall: Gene therapy in wound healing
62
gene transfer of PDGF-B enhances wound healing in type I
and type II diabetic wounds. Wound Repair Regen 12, 497-
504.
Khavari PA, Rollman O, Vahlquist A (2002) Cutaneous gene
transfer for skin and systemic diseases. J Intern Med 252, 1-
10.
Körber A, Franckson T, Grabbe S, Dissemond J (2008) Vacuum
Assisted Closure Device Improves the Take of Mesh Grafts
in Chronic Leg Ulcer Patients. Dermatology 218, 250-256.
Kuriyama S, Mitoro A, Tsujinoue H, Nakatani T, Yoshiji H,
Tsujimoto T, Yamazaki M, Fukui H (2000) Particle-
mediated gene transfer into murine livers using a newly
developed gene gun. Gene Ther 7, 1132-1136.
Kusumanto YH, Mulder NH, Dam WA, Losen MH, Meijer C,
Hospers GA (2007) Improvement of In vivo Transfer of
Plasmid DNA in Muscle: Comparison of Electroporation
versus Ultrasound. Drug Deliv 14, 273-277.
Kwok KY, Park Y, Yang Y, McKenzie DL, Liu Y, Rice KG
(2003) In vivo gene transfer using sulfhydryl cross-linked
PEG-peptide/glycopeptide DNA co-condensates. J Pharm
Sci 92, 1174-1185.
Labanaris AP, Polykandriotis E, Horch RE (2008) The effect of
vacuum-assisted closure on lymph vessels in chronic
wounds. J Plast Reconstr Aesthet Surg, in press.
Lai E, van Zanten JH (2002) Evidence of lipoplex dissociation in
liquid formulations. J Pharm Sci 91, 1225-1232.
Lee H, Jeong JH, Park TG (2002) PEG grafted polylysine with
fusogenic peptide for gene delivery: high transfection
efficiency with low cytotoxicity. J Control Release 79, 283-
291.
Lindstedt S, Malmsjö M, Gesslein G, Ingemansson R (2006)
Topical negative pressure effects on coronary blood flow in a
sternal wound model. Int Wound J 5, 503-509.
Liu F, Shollenberger LM, Conwell CC, Yuan X, Huang L (2007)
Mechanism of naked DNA clearance after intravenous
injection. J Gene Med 9, 613-619.
Liu L, Marti GP, Wei X, Zhang Z, Zhang H, Liu YV, Nastai M,
Semenza GL, Harmon JW (2008) Age-Dependent
Impairment of HIF-1! Expression in Diabetic Mice:
Correction With Electroporation-Facilitated Gene Therapy
Increases Wound Healing, Angiogenesis, Circulating
Angiogenic Cells. J Cell Physiol217, 319-327.
Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD,
Middelkoop E (1998) Differences in cellular infiltrate and
extracellular matrix of chronic diabetic and venous ulcers
versus acute wounds. J Invest Dermatol 111, 850-857.
Lorand L, Graham RM (2003) Transglutaminases: crosslinking
enzymes with pleiotropic functions. Nat Rev Mol Cell Biol
4, 140-156.
Luhmann T, Rimann M, Bittermann AG, Hall H (2008) Cellular
uptake and intracellular pathways of PLL-g-PEG-DNA
nanoparticles. Bioconjug Chem 19, 1907-1916.
Lutholf MP, Hubbell JA (2005) Synthetic biomaterials as
instructive extracellular microenvironments for
morphogenesis in tissue engineering. Nat Biotechnol 23, 47-
53.
Lv H, Zhang S, Wang B, Cui S, Yan J (2006) Toxicity of
cationic lipids and cationic polymers in gene delivery. J
Control Release 114, 100-109.
Lynch SE, Nixon JC, Colvin RB, Antoniades HN (1987) Role of
platelet-derived growth factor in wound healing: synergistic
effects with other growth factors. Proc Natl Acad Sci USA
84, 7696-700.
Mace KA, Yu DH, Paydar KZ, Boudreau N, Young DM (2007)
Sustained expression of Hif-1alpha in the diabetic
environment promotes angiogenesis and cutaneous wound
repair. Wound Repair Regen 15, 636-645.
Margolis DJ, Cromblehome T, Herlyn M, Cross P, Weinberg L,
Filip J, Propert K (2004) Clinical protocol. Phase I trial to
evaluate the safety of H5.020CMV.PDGF-b and limb
compression bandage for the treatment of venous leg ulcer:
trial A. Hum Gene Ther 15, 1003-1019.
Masayuki I, Masanori F, Kiyohaya O, Hidemi H, Shigo N,
Masaki N, Tomoharu K, Yasuhiro K, Bonpei T, Makoto K,
Tadaaki M (2006a) Controlled release of FGF-2 and
paclitaxol from chitosam hydrogels and their subsequent
effects on wound repair, angiogenesis, tumor growth. Curr
Drug Del 2, 351-358.
Mester E, Juhasz J, Varga P, Karika G (1968) Lasers in clinical
practice. Acta Chir Acad Sci Hung 9, 349-357.
Mishra S, Webster P, Davis ME (2004) PEGylation significantly
affects cellular uptake and intracellular trafficking of non-
viral gene delivery particles. Eur J Cell Biol 83, 97-111.
Morgan RA, Anderson WF (1993) Human gene therapy. Annu
Rev Biochem 62, 191-217.
Mosesson MW (2005) Fibrinogen and fibrin structure and
functions. J Thromb Haemost 3, 1894-1904.
Panyam J, Labhasetwar V (2003) Biodegradable nanoparticles
for drug and gene delivery to cells and tissue. Adv Drug
Deliv Rev 55, 329-347.
Park TG, Jeong JH, Kim SW (2006) Current status of polymeric
gene delivery systems. Adv Drug Deliv Rev 58, 467-486.
Patel TH, Kimura H, Weiss CR, Semenza GL, Hofmann LV
(2005) Constitutively active HIF-1alpha improves perfusion
and arterial remodeling in an endovascular model of limb
ischemia. Cardiovasc Res 68, 144-154.
Paul SAM, Simons JW, Mabjeesh NJ (2004) HIF at the
crossroads between ischemia and carcinogenesis. J Cell
Physiol 200, 20-30.
Petrie NC, Yao F, Eriksson E (2003) Gene therapy in wound
healing. Surg Clin North Am 83, 597-616.
Pichon C, Goncalves C, Midoux P (2001) Histidine-rich peptides
and polymers for nucleic acids delivery. Adv Drug Deliv
Rev 53, 75-94.
Pierce GF, Tarpley JE, Tseng J, Bready J, Chang D, Kenney
WC, Rudolph R, Robson MC, Vande Berg J, Reid P,
Kaufman S, Farrell CL (1995) Detection of platelet-derived
growth factor (PDGF)-AA in actively healing human wounds
treated with recombinant PDGF-BB and absence of PDGF in
chronic nonhealing wounds. J Clin Invest 96, 1336-1350.
Pike DB, Cai S, Pomraning KR, Firpo MA, Fisher RJ, Shu X, Z.,
Prestwich GD, Peattie RA (2006) Heparin-regulated release
of growth factors in vitro and angiogenic response in vivo to
implanted hyaluronic hydrogels containing VEGF and bFGF.
Biomaterials 27, 5242-5251.
Pugh CW, Ratcliffe PJ (2003) Regulation of angiogenesis by
hypoxia: role of the HIF system. Nat Med 9, 677-684.
Ramsay E, Hadgraft J, Birchall J, Gumbleton M (2000)
Examination of the biophysical interaction between plasmid
DNA and the polycations, polylysine and polyornithine, as a
basis for their differential gene transfection in-vitro. Int J
Pharm 210, 97-107.
Rimann M, Luhmann T, Textor M, Guerino B, Ogier J, Hall H
(2008) Characterization of PLL-g-PEG-DNA nanoparticles
for the delivery of therapeutic DNA. Bioconjug Chem 19,
548-557.
Robson MC, Phillips TJ, Falanga V, Odenheimer DJ, Parish LC,
Jensen JL, Steed DL (2001) Randomized trial of topically
applied repifermin (recombinant human keratinocyte growth
factor-2) to accelerate wound healing in venous ulcers.
Wound Repair Regen 9, 347-352.
Royce SM, Askari M, Marra KG (2004) Incorporation of
polymer microspheres within fibrin scaffolds for the
controlled delivery of FGF-1. J Biomater Sci Polymer Edn
15, 1327-1336.

Gene Therapy and Molecular Biology Vol 13, page 63
63
Ruszczak R, Friess W (2003) Collagen as a carrier for on-site
delivery of antibacterial drugs. Adv Drug Delivery Rev 55,
1679-1689.
Saxena V, Hwang C-W, Huang S, Eichbaum Q, Ingber D, Orgill
DP (2004) Vacuum-Assisted Closure: Microdeformations of
Wounds and Cell Proliferation. Plast Reconstr Surg 114,
1086-1096.
Shigeta K, Kawakami S, Higuchi Y, Okuda T, Yagi H,
Yamashita F, Hashida M (2007) Novel histidine-conjugated
galactosylated cationic liposomes for efficient hepatocyte-
selective gene transfer in human hepatoma HepG2 cells. J
Control Release 118, 262-270.
Singer AJ, Clark RA (1999) Cutaneous wound healing. N Engl J
Med 341, 738-746.
Sobanko JF, Alster TS (2008) Efficacy of low-level laser therapy
for chronic cutaneous ulceration in humans: a review and
discussion. Dermatol Surg 34, 991-1000.
Sprugel KH, McPherson JM, Clowes AW, Ross R (1987) Effects
of growth factors in vivo. I. Cell ingrowth into porous
subcutaneous chambers. Am J Pathol 129, 601-613.
Steed DL (2006) Clinical evaluation of recombinant human
platelet-derived growth factor for the treatment of lower
extremity ulcers. Plast Reconstr Surg 117, 143S-149S.
Steinstraesser L, Hirsch T, Beller J, Mittler D, Sorkin M,
Pazdierny G, Jacobsen F, Eriksson E, Steinau HU (2007)
Transient non-viral cutaneous gene delivery in burn wounds.
J Gene Med 9, 949-955.
Stender S, Murphy M, O'Brien T, Stengaard C, Ulrich-Vinther
M, Soballe K, Barry F (2007) Adeno-associated viral vector
transduction of human mesenchymal stem cells. Eur Cell
Mater 13, 93-99.
Swift ME, Burns AL, Gray KL, DiPietro LA (2001) Age-related
alterations in the inflammatory response to dermal injury. J
Invest Dermatol 117, 1027-1035.
Tal J (2000) Adeno-associated virus-based vectors in gene
therapy. J Biomed Sci 7, 279-291.
Tal R, Shaish A, Bangio L, Peled M, Breitbart E, Harats D
(2008) Activation of C-transactivation domain is essential for
optimal HIF-1 alpha-mediated transcriptional and angiogenic
effects. Microvasc Res 76, 1-6.
Theopold C, Yao F, Eriksson E (2004) Gene therapy in the
treatment of lower extremity wounds. Int J Low Extrem
Wounds 3, 69-79.
Trengove NJ, Stacey MC, MacAuley S, Bennett N, Gibson J,
Burslem F, Murphy G, Schultz G (1999) Analysis of the
acute and chronic wound environments: the role of proteases
and their inhibitors. Wound Repair Regen. 7, 442-452.
Trentin D, Hall H, Wechsler S, Hubbell JA (2006) Peptide-
matrix-mediated gene transfer of an oxygen-insensitive
hypoxia-inducible factor-1alpha variant for local induction of
angiogenesis. Proc Natl Acad Sci U S A. 103, 2506-2511.
Trentin D, Hubbell J, Hall H (2005) Non-viral gene delivery for
local and controlled DNA release. J Control Release 102,
263-275.
Vandenbroucke RE, De Smedt SC, Demeester J, Sanders NN
(2007) Cellular entry pathway and gene transfer capacity of
TAT-modified lipoplexes. Biochim Biophys Acta 1768,
571-579.
Viswanathan V, Pendsey S, Sekar N, Murthy GSR (2006) A
phase III study to evaluate the safety and efficacy of
recombinant human epidermal growth factor (REGEN-D™
150) in healing diabetic foot ulcers. Wounds 18, 186-196.
Wagner E, Ogris M, Zauner W (1998) Polylysine-based
transfection systems utilizing receptor-mediated delivery.
Adv Drug Deliv Rev 30, 97-113.
Wallace DG, Rosenblatt J (2003) Collagen gel systems for
sustained delivery and tissue engineering. Adv Drug
Delivery Rev 55, 1631-1649.
Walsh M, Tangney M, O'Neill MJ, Larkin JO, Soden DM,
McKenna SL, Darcy R, O'Sullivan GC, O'Driscoll CM
(2006) Evaluation of cellular uptake and gene transfer
efficiency of pegylated poly-L-lysine compacted DNA:
implications for cancer gene therapy. Mol Pharm 3, 644-
653.
Weisel JW, Stauffacher CV, Bullitt E, Cohen C (1985) A model
for fibrinogen: domains and sequence. Science 230, 1388-
1391.
Wells DJ (2004) Gene therapy progress and prospects:
electroporation and other physical methods. Gene Ther 11,
1363-1369.
Whitney JD (2005) Overview: acute and chronic wounds. Nurs
Clin North Am 40, 191-205.
Wilkes RP, McNulty AK, Feeley TD, Schmidt MA, Kieswetter
K (2007) Bioreactor for Application of Subatmospheric
Pressure to Three-Dimensional Cell Culture. Tissue Eng 13,
3003-3010.
Wong C, Imman E, Spaethe R, Helgerson S (2003) Fibrin-based
biomaterials to deliver human growth factors. Thromb
Haemost 89, 573-582.
Wu X, Burgess SM (2004) Integration target site selection for
retroviruses and transposable elements. Cell Mol Life Sci 61,
2588-2596.
Yager DR, Nwomeh BC (1999) The proteolytic environment of
chronic wounds. Wound Repair Regen 7, 433-441.
Yla-Herttuala S, Markkanen JE, Rissanen TT (2004) Gene
therapy for ischemic cardiovascular diseases: some lessons
learned from the first clinical trials. Trends Cardiovasc
Med 14, 295-300.
Young S, Wong M, Tabata Y, Mikos AG (2005) Gelatin as a
delivery vehicle for the controlled release of bioactive
molecules. J Control Release 109, 256-274.
Zaitsev S, Cartier R, Vyborov O, Sukhorukov G, Paulke BR,
Haberland A, Parfyonova Y, Tkachuk V, Bottger M (2004)
Polyelectrolyte nanoparticles mediate vascular gene delivery.
Pharm Res 21, 1656-1661.
Zilla P (1991) Endothelialization of vascular grafts. Curr Opin
Cardiol 6, 877-886.
Zilla P, Deutsch M, Meinhart J, Puschmann R, Eberl T, Minar E,
Dudczak R, Lugmaier H, Schmidt P, Noszian I, Fischlein T
(1994) Clinical in vitro endothelialization of femoropopliteal
bypass grafts: an actuarial follow-up over three years. J Vasc
Surg 19, 540-548.
Zisch AH, Lutolf MP, Hubbell JA (2003) Biopolymeric delivery
matrices for angiogenic growth factors. Cardiovasc Pathol
12, 295-310.
Heike Hall and Markus Rimann





























