Embed Size (px)
Citation preview

ABCDEFG
UNIVERS ITY OF OULU P.O.B . 7500 F I -90014 UNIVERS ITY OF OULU F INLAND
A C T A U N I V E R S I T A T I S O U L U E N S I S
S E R I E S E D I T O R S
SCIENTIAE RERUM NATURALIUM
HUMANIORA
TECHNICA
MEDICA
SCIENTIAE RERUM SOCIALIUM
SCRIPTA ACADEMICA
OECONOMICA
EDITOR IN CHIEF
PUBLICATIONS EDITOR
Senior Assistant Jorma Arhippainen
University Lecturer Santeri Palviainen
Professor Hannu Heusala
Professor Olli Vuolteenaho
University Lecturer Hannu Heikkinen
Director Sinikka Eskelinen
Professor Jari Juga
Professor Olli Vuolteenaho
Publications Editor Kirsti Nurkkala
ISBN 978-952-62-0062-0 (Paperback)ISBN 978-952-62-0063-7 (PDF)ISSN 0355-3221 (Print)ISSN 1796-2234 (Online)
U N I V E R S I TAT I S O U L U E N S I S
MEDICA
ACTAD
D 1194
ACTA
Mika N
evalainen
OULU 2012
D 1194
Mika Nevalainen
GENE PRODUCT TARGETING INTO AND MEMBRANE TRAFFICKING FROMTHE ENDOPLASMIC/SARCOPLASMIC RETICULUM IN SKELETAL MYOFIBERS
UNIVERSITY OF OULU GRADUATE SCHOOL;UNIVERSITY OF OULU,FACULTY OF MEDICINE,INSTITUTE OF BIOMEDICINE,DEPARTMENT OF ANATOMY AND CELL BIOLOGY


A C T A U N I V E R S I T A T I S O U L U E N S I SD M e d i c a 1 1 9 4
MIKA NEVALAINEN
GENE PRODUCT TARGETING INTO AND MEMBRANE TRAFFICKING FROM THE ENDOPLASMIC/SARCOPLASMIC RETICULUMIN SKELETAL MYOFIBERS
Academic dissertation to be presented with the assentof the Doctoral Training Committee of Health andBiosciences of the University of Oulu for public defencein Auditorium A101 of the Department of Anatomy andCell Biology (Aapistie 7 A), on 25 January 2013, at 12noon
UNIVERSITY OF OULU, OULU 2012

Copyright © 2012Acta Univ. Oul. D 1194, 2012
Supervised byDocent Kalervo Metsikkö
Reviewed byDocent Pasi TaviDocent Tiina Laitala-Leinonen
ISBN 978-952-62-0062-0 (Paperback)ISBN 978-952-62-0063-7 (PDF)
ISSN 0355-3221 (Printed)ISSN 1796-2234 (Online)
Cover DesignRaimo Ahonen
JUVENES PRINTTAMPERE 2012

Nevalainen, Mika, Gene product targeting into and membrane trafficking from theendoplasmic/sarcoplasmic reticulum in skeletal myofibers. University of Oulu Graduate School; University of Oulu, Faculty of Medicine, Institute ofBiomedicine, Department of Anatomy and Cell Biology, P.O. Box 5000, FI-90014 University ofOulu, FinlandActa Univ. Oul. D 1194, 2012Oulu, Finland
Abstract
Skeletal muscle cells (myofibers) are huge multinucleated cells responsible for muscle contractionand hence for the everyday movements of the joints. The structure of these voluminous cellsdiffers greatly from that of the mononucleated cells – the characteristic features of the myofibersinclude dozens of peripherally located nuclei, tightly packed contractile apparatus and asophisticatedly organized endomembrane system. The basic physiology involving myofibers isquite well known, but scarce data exist on the membrane biology of the myofibers.
The purpose of this study was to examine the localization of mRNA and the site of proteinsynthesis in the myofibers. The characterization of the membrane dynamics in muscle cells wasalso performed.
In this study we utilized a primary cell culture model obtained from the rat flexor digitorumbrevis (FDB) muscle. Also frozen sections from the rat extensor digitorum longus muscle wereused. The precursor cells of the myofibers – myoblasts and myotubes – were also utilized in someexperiments. Furthermore, methods of immunohistochemistry and molecular biology wereapplied extensively in this study.
We found that in FDB myofibers the mRNA lies just under the plasma membrane. Proteinsynthesis seemed to be concentrated in the vicinity of nuclei locating beneath the plasmamembrane but also in interfibrillar dot-like structures. Protein products moved hundreds ofmicrometers away from the nuclei of origin. Moreover, there were no barriers for proteinmovement into the core regions of the myofibers. Movement of proteins was found to be rapid inthe cytosol and in the endomembrane system, too. Interestingly, when examining exocytictrafficking we observed that ER-to-Golgi trafficking significantly differed from that ofmononucleated cells. Finally, myofibers were found to be able to generate lipid bodies under stressconditions. The dynamics of lipid bodies seemed to deviate from the dynamics found in other cellstypes.
Nowadays not much muscle research with primary myofibers is done worldwide, and thereforedilemmas involving myofibers such as insulin resistance and myotoxicity of statins are mostlyunresolved. The knowledge gained from this study may be used in the future to solve clinicalproblems related to the cell biology of the myofibers.
Keywords: endoplasmic reticulum, Golgi apparatus, lipid body, mRNA, proteintransport, sarcoplasmic reticulum, skeletal muscle fiber


Nevalainen, Mika, Geenituotteiden kohdentaminen ja kalvostoliikenneendoplasmisessa/sarkoplasmisessa kalvostossa luurankolihassoluissa. Oulun yliopiston tutkijakoulu; Oulun yliopisto, Lääketieteellinen tiedekunta, Biolääketieteenlaitos, Anatomia ja solubiologia, PL 5000, 90014 Oulun yliopistoActa Univ. Oul. D 1194, 2012Oulu
Tiivistelmä
Luurankolihassolut eli myofiiberit ovat jättimäisiä monitumaisia soluja, jotka vastaavat lihassu-pistuksen aikaansaamisesta ja siten mahdollistavat jokapäiväisen liikkumisemme. Näiden suur-ten solujen rakenne poikkeaa selkeästi yksitumaisten solujen rakenteesta: myofiiberien tunnus-omaisia piirteitä ovat kymmenet solun reunoille sijoittuneet tumat, tiiviisti pakkautunut supistu-miskoneisto ja monimutkaisesti järjestynyt solukalvostojärjestelmä. Vaikka myofiiberien perus-fysiologia tunnetaankin hyvin, niin tiedetään itse myofiiberien kalvostobiologiasta sangenvähän.
Kokonaisuutena tämän tutkimuksen tarkoituksena oli tarkastella mRNA:n ja proteiinisyntee-sin sijaintia myofiibereissä. Lisäksi selvitimme lihassolujen kalvostodynamiikkaa.
Tässä tutkimuksessa käytimme rotan flexor digitorum brevis (FDB) -lihaksesta saatua pri-määristä soluviljelymallia. Lisäksi hyödynsimme rotan extensor digitorum longus -lihaksestahankittuja jääleikkeitä. Joissakin kokeissa käytimme myös myofiiberien esiastesoluja (myoblas-teja ja myotuubeja). Immunohistokemian ja molekyylibiologian menetelmiä sovellettiin tutki-muksessa laajasti.
Havaitsimme, että FDB –myofiibereissä mRNA sijaitsee aivan solukalvon alla. Proteiinisyn-teesi vaikutti olevan keskittynyt solukalvon alla sijaitsevien tumien ympärille, mutta myössolunsisäisiin pistemäisiin rakenteisiin. Proteiinituotteet ylsivät satojen mikrometrien päähänalkuperäisestä tumastaan. Lisäksi proteiineille ei ilmennyt leviämisestettä myofiiberin sisäosiin.Leviämisen havaittiin olevan nopeaa sekä solulimassa että solulimakalvostoissa. Tutkiessammesolun eritystoimintaa huomasimme, että kuljetus ER:stä Golgin laitteeseen eroaa huomattavastiyksitumaisten solujen vastaavasta kuljetuksesta. Lopuksi havaitsimme myofiiberien pystyvänmuodostamaan rasvapisaroita rasitusolosuhteissa. Rasvapisaroiden käyttäytyminen näytti myöspoikkeavan siitä, mitä muissa soluissa on havaittu.
Nykyään lihastutkimusta primäärisoluilla ei juuri tehdä maailmalla, minkä vuoksi myofiibe-reihin liittyvät lääketieteelliset pulmat kuten insuliiniresistenssi ja statiinien lihashaitat ovat suu-relta osin ratkaisematta. Tästä tutkimuksesta saatuja tuloksia voitaneen jatkossa käyttää myofii-berien solubiologiaan liittyvien kliinisten ongelmien selvittämiseen.
Asiasanat: Golgin laite, luurankolihassolu, lähetti-RNA, proteiinikuljetus, rasvapisara,sarkoplasmakalvosto, solulimakalvosto


Omnia praeclara rara

8

9
Acknowledgements
This work was carried out at the Department of Anatomy and Cell Biology,
Institute of Biomedicine, University of Oulu during 2007–2012. The work was
financially supported by the Finnish Medical Society Duodecim, the Finnish-
Norwegian Medical Society, the Emil Aaltonen Foundation, the Oulu University
Scholarship Foundation and the Orion-Farmos Research Foundation.
I wish to express my deepest gratitude to my supervisor docent Kalervo
Metsikkö for his unwavering guidance and support during this work. Kalervo’s
door has always been open for me. I deeply appreciate his vast experience on cell
biology and his practical laboratory skills. His logical approach to science is also
one thing I sincerely admire.
I am grateful to Professor Juha Tuukkanen, the head of the department, for
providing the facilities to do research. His supportive and encouraging attitude
towards my work is also acknowledged. Professor Petri Lehenkari is
acknowledged for giving different perspectives in the field of science. For his
collegial attitude towards my research I am also grateful. Docent Ulla Petäjä-
Repo I want to thank for making me give presentations at our seminar series.
Giving speeches and showing your results is a crucial part of doing science. I
wish to thank Docent Pasi Tavi and Docent Tiina Laitala-Leinonen for their
critical reading of my thesis and for their valuable comments. Additionally,
biostatistician Risto Bloigu is acknowledged for the valuable statistical advice
given to me. My dear friend M.Sc. Jukka Ahola is warmly thanked for thorough
and efficient revision of the language.
I would also like to thank my co-authors and colleagues. Their experience
and support has considerably helped me to understand science. All the fellow
researchers and friends at the department are also warmly thanked. Especially
Ph.D. Tuula Kaisto, who was my first mentor, deserves my gratitude. My former
roommate, brother-in-arms, Ph.D. Mika Kaakinen, is also acknowledged for his
vast knowledge in cell biology. The environment of our room is not the same
without you. Additionally, M.D., Ph.D. Paula Kuvaja is thanked for sharing
thoughts and working space with me. M.D. Riina Myllylä also deserves my
gratitude. Special thanks go also to the lab technicians Paula Salmela and Marja
Paloniemi for their invaluable practical help during this work.
My friends, both old ones and new ones, have always been important for me.
Your friendship and support is very much appreciated. Especially, I would like to
thank Jukka Ahola for intellectual and competitive atmosphere back in Lyseo and

10
nowadays too. All my physician friends are also acknowledged. Special thanks go
to our 100 kg from bench –club and its members and newcomers. M.D. Tapio
Räihä is thanked for being a great man. It is invaluable to have you as a friend.
Finally, I wish express my thanks to my parents Paula and Markku
Nevalainen. You have always encouraged me to push forward. Additionally, you
have always been there for me. Thank you for that. My brother Olli-Pekka is
thanked for being a little brother and sharing thoughts every now and then.
Ultimately, my deepest thanks go to my beloved girlfriend Jaana Marttala. Thank
you for being there – in life, in science, and in love.
Oulu, December 2012 Mika Nevalainen

11
Abbreviations
ADRP adipose differentiation related protein
BrU bromouridine
CAV3 caveolin 3 protein
CLQ calsequestrin
CLN calnexin
cDNA complementary deoxyribonucleic acid
COPI coat protein I
COPII coat protein II
DHPR dihydropyridine receptor
EDL extensor digitorum longus
ER endoplasmic reticulum
ERES ER exit site
FDB flexor digitorum brevis
FRAP fluorescence recovery after photobleach
GFP green fluorescent protein
LB lipid body
MHC myosin heavy chain
mRNA messenger ribonucleic acid
mRNP messenger ribonucleoprotein
MTJ myotendinous junction
NMJ neuromuscular junction
NPC nuclear pore complex
OST oligosaccharyltransferase
PDI protein disulfide isomerise
PM plasma membrane
POL II RNA polymerase II
RBP RNA binding protein
SERCA sarco/endoplasmic reticulum Ca2+ -ATPase
SFV Semliki Forest Virus
SNARE N-ethylmaleimide sensitive factor attachment protein receptor
SR sarcoplasmic reticulum
SREBP sterol regulatory element binding protein
recSFV recombinant SFV
RER rough endoplasmic reticulum
rRNA ribosomal RNA

12
RYR ryanodine receptor
TGN trans-Golgi network
tRNA transfer RNA
tsG temperature sensitive G-protein
UTR untranslated region
VSV vesicular stomatitis virus
VTC vesicular tubular complex
YFP yellow fluorescent protein

13
List of original publications
This thesis is based on the following publications, which are referred to in the text
by their Roman numerals:
I Nevalainen M, Kaakinen M & Metsikkö K (2012) Localization of mRNA
transcripts and translation activity in skeletal myofibers. Manuscript.
II Nevalainen M, Nissinen M, Kaakinen M & Metsikkö K (2010) Influenza
virus infection in multinucleated skeletal myofibers. Exp Cell Res 316: 1784–
1794.
III Nevalainen M, Kaisto T & Metsikkö K (2010) Mobile ER-to-Golgi but not
post-Golgi membrane transport carriers disappear during the terminal
myogenic differentiation. Cell Tissue Res 342: 107–116
IV Nevalainen M, Kaakinen M, Rahkila P & Metsikkö K (2012) Reversible
stress-induced lipid body formation in fast twitch rat myofibers. Exp Cell Res
318: 2191–2199.

14

15
Contents
Abstract
Tiivistelmä
Acknowledgements 9 Abbreviations 11 List of original publications 13 Contents 15 1 Introduction 17 2 Review of literature 19
2.1 Logistics of gene products ...................................................................... 19 2.1.1 mRNA synthesis ........................................................................... 20 2.1.2 mRNA trafficking and degradation .............................................. 21 2.1.3 Protein synthesis ........................................................................... 23 2.1.4 Protein trafficking and degradation .............................................. 25
2.2 Exocytic trafficking ................................................................................. 27 2.2.1 ER-to-Golgi trafficking ................................................................ 28 2.2.2 Post-Golgi trafficking ................................................................... 29
2.3 Skeletal muscle cells ............................................................................... 32 2.3.1 Myogenesis ................................................................................... 32 2.3.2 Architecture of myofibers ............................................................. 33 2.3.3 Plasma membrane and its specialized macrodomains .................. 34 2.3.4 SR, ER and the Golgi apparatus ................................................... 36 2.3.5 Ribosomes and mRNA ................................................................. 38 2.3.6 Studying the trafficking of gene products .................................... 39
2.4 LBs in mammalian cells .......................................................................... 41 2.4.1 LBs in skeletal muscle and insulin resistance............................... 42
3 Aims of the study 43 4 Materials and methods 45
4.1 Cell culture of L6 myoblasts, myotubes and BHK cells (II) ................... 45 4.1.1 Isolation of myofibers (I, II, III, IV) ............................................. 45
4.2 Recombinant mammalian expression plasmids (I, IV) ........................... 45 4.2.1 In vivo electroporation of recombinant plasmids (I, IV) .............. 46
4.3 Preparation of recombinant SFV (I, II, III, IV) ....................................... 46 4.3.1 Other viruses used (II) .................................................................. 46
4.4 Immunofluorescence studies (I, II, III, IV) ............................................. 47 4.5 Live cell microscopy and FRAP (I, III, IV) ............................................ 48

16
4.6 In situ hybridization (I, II) ....................................................................... 48 4.7 Pulse labeling and SDS/PAGE (III) ........................................................ 49 4.8 Western blotting (I, III, IV) ..................................................................... 49 4.9 Statistical testing (III) .............................................................................. 49
5 Results 51 5.1 The localization of mRNA and translation in myofibers (I) .................... 51
5.1.1 Poly-A tails of endogenous mRNAs and ribosomes
localize subsarcolemmally and perinuclearly ............................... 51 5.1.2 Bromouridine labeling also marks myonuclei and
subsarcolemmal region ................................................................. 51 5.1.3 Translation is concentrated in the vicinity of myonuclei
and in interfibrillar spots .............................................................. 52 5.2 Dynamics of proteins in myofibers is versatile (I, II, III) ....................... 53
5.2.1 The Influenza A viral proteins travel a long distance from
the nucleus of origin, whereas the viral mRNA does not ............. 53 5.2.2 Rapid transverse movement of cytoplasmic and membrane
proteins occurs in myofibers ........................................................ 54 5.2.3 Cellular subcompartments, where mobility is restricted,
exist in myofibers ......................................................................... 55 5.3 Exocytic membrane trafficking in myofibers (III) .................................. 56
5.3.1 Adult myofibers lack ER-to-Golgi transport carriers ................... 56 5.3.2 Post-Golgi vesicles are present in skeletal muscle cells ............... 58
5.4 LB formation in myofibers is inducible (IV) .......................................... 59 5.4.1 LBs are immobile structures without a connection to the
ER ................................................................................................. 59 5.4.2 ER stress induces LBs accumulation in myofibers ....................... 60
6 Discussion 63 6.1 mRNA localization differs from the site of translation in
myofibers ................................................................................................ 63 6.2 Protein mobility and its efficacy in vast myofibers ................................. 65 6.3 Regression of Golgi associated trafficking in myofibers ........................ 66 6.4 ER stress, LBs and insulin resistance in myofibers ................................. 68
7 Conclusions 71 References 73 Original publications 85

17
1 Introduction
In a eukaryotic cell, gene products – namely messenger RNA (mRNA) and
proteins – are the outcomes of the sophisticated reading of DNA within the
nucleus. Subsequently, the targeting and trafficking of gene products forms the
very basis of a functional cell. However, the logistics of gene products has been
studied almost exclusively in mononucleated cells and profound understanding of
it is still somewhat elusive. Eukaryotic cells contain many complex membrane
structures, which are responsible for various vital functions of the cell.
Accordingly, membrane trafficking is a term used to describe the flow of
membrane material between endomembrane systems and the plasma membrane
(PM), and it is crucial for the transport of proteins and other macromolecules to
various destinations inside and outside the cell. Membrane trafficking is also
closely linked with the cell’s fundamental need to maintain cellular homeostasis.
In mononucleated mammalian cells, as well as in plant and yeast cells, the
intracellular membrane trafficking is fairly well characterized, whereas larger and
more differentiated cells have not been studied so extensively in this respect.
Multinucleated skeletal muscle cells, also known as myofibers, are colossal
cells containing an elaborate endomembrane system as well as the contraction
machinery, the myofibrils. Dozens of peripheral nuclei located under the PM are
also characteristic to myofibers. In an adult muscle cell, all this is wrapped into a
tight cylindrical package to accomplish the functions of a fully differentiated
force generating cell. The squeezed architecture of the myofibers poses many
challenges, and consequently little knowledge exists on the logistics of gene
products or on membrane dynamics in myofibers.
This work was carried out to investigate the localization of mRNA, and the
site of translation in myofibers. Additionally, membrane trafficking within and
from the endomembrane system of the myofibers was analyzed. The insights
gained from this research may be used in the future to solve clinical problems
related to muscle cells.

18

19
2 Review of literature
2.1 Logistics of gene products
In a eukaryotic cell a segment of DNA encoding for either RNA or protein as a
final gene product, is called a gene. The synthesis, trafficking and targeting of
gene products in mononucleated cell models is fairly well understood, but in more
differentiated and complex cells, such as multinucleated muscle cells, the
understanding of the logistics of gene products is still quite vague.
RNA, like DNA, is basicly a linear polymer consisting of nucleotides. In
RNA each nucleotide contains a nucleobase (adenine, cytosine, guanine or uracil),
a ribose sugar and a phosphate group. DNA, on the contrary, contains a
deoxyribose sugar and instead of uracil uses a thymine nucleobase. DNA is also
double-stranded, whereas most RNAs are single-stranded and can have complex
three-dimensional secondary structure. This sequence of nucleotides allows RNA
to transmit genetic information. There are many different classes of RNAs in
eukaryotic cells, for example messenger RNA (mRNA), transfer RNA (tRNA),
ribosomal RNA (rRNA) and non-coding RNA such as microRNA, small
interfering RNA and small nuclear RNA. RNAs have many functional properties
in cells; they catalyze translation and splicing, control gene expression, modify
other RNAs and function in various other cellular processes.
Perhaps the most important task of RNAs is the protein synthesis, where
mRNA guides the assembly of proteins in ribosomes. The process involves tRNA
to deliver amino acids to the ribosome, in which rRNA fuses the incoming amino
acid into a polypeptide chain to make the protein product. The mammalian
proteins consist of 20 different amino acid joined together as long polypeptide
chains varying in length. Lastly, proteins are then further processed to achieve
their final state and three-dimensional conformation.
The logistics of gene products – especially mRNA – have been studied
eagerly during the last few years (Donnelly et al. 2010, St Johnston 2005, Weis et al. 2012) and many new techniques are available nowadays to track gene products
even in live cells (Armitage 2011, Konig et al. 2012, Yamada et al. 2011). Since
the 1970s it was thought that the main way of trafficking of gene products is the
targeting of the proteins, but nowadays it is even more widely recognized that
mRNA targeting also plays a crucial role (Weis et al. 2012). The trafficking of

20
gene products involves many complex steps, some of which are understood well,
while others are yet to be explained by the scientists.
2.1.1 mRNA synthesis
In eukaryotic cells the mRNA synthesis (transcription) occurs in the nucleus. A
tiny fraction of mRNA is also synthesized in the mitochondria for their own use.
The transcription begins with the opening and unwinding of a small local portion
of the DNA double helix. Either of the exposed strands then serves as a template
for the synthesis of RNA. The nucleotide sequence of the synthesized RNA is
exactly complementary the strand of DNA used as template. However, the length
of mRNA is considerably shorter than that of the DNA molecule: only a few
thousand nucleotides in mammals.
In eukaryotic cells the transcription is carried out by a complex enzyme
called RNA polymerase II (POL II). This polymerase moves along the DNA and
elongates the growing RNA chain one nucleotide at time in the 5’-to-3’ direction.
Since only a short segment of DNA is bound by the polymerase, multiple RNA
copies of the same gene can be made simultaneously. Right after the polymerase
has produced about 25 nucleotides of RNA, the capping of 5’ end occurs. In this
process a methylated guanine nucleotide is added to the 5’ end of the growing
mRNA. This cap structure indicates the 5’ end of eukaryotic mRNAs, and helps
the cell to distinguish mRNA from the other types of RNA present in the cell. The
5’ end cap is also essential for translation, splicing, polyadenylation, nuclear
export and protection from degradation (Cowling 2009). The addition of the 5’
end cap is an intricate process carried out by a specific protein complex, which
associates closely with POL II. The growing mRNA is then further processed in
an event called splicing. In splicing the non-coding sequences – termed introns –
are removed as lariat like structures while the protein coding sequences – termed
exons – are joined together. This task is done by a complicated protein apparatus
called spliceosome in close co-operation with POL II. Splicing is a typical feature
of eukaryote mRNA processing, since prokaryote mRNA doesn’t normally
contain introns. Diversity is also increased by splicing since many mRNAs can
have many different ways of getting spliced (Nilsen & Graveley 2010). The final
mRNA modification in eukaryotes is the addition of poly-A tail to the 3’ end of
RNA molecule. This action is yet again conducted in association with POL II.
First, certain proteins are attached to the 3’ end of the mRNA, and then the RNA
is cleaved. Next, a specific enzyme adds approximately 200 adenine nucleotides

21
to the 3’ end produced by the cleavage. After the poly-A tail is synthesized,
proteins called poly-A binding proteins attach onto it and determine the final
length of the tail. These proteins also take part in the trafficking and targeting of
the mRNA in the cytosol (Mangus et al. 2003). After these events the POL II
dissociates from the DNA and the transcription stops. The basics of mRNA
synthesis in a eukaryotic cell are illustrated in Figure 1.
2.1.2 mRNA trafficking and degradation
Following the transcription, the mRNA is exported from the nucleus. This process
is conserved from yeast to humans (Vinciguerra & Stutz 2004). However, the
mRNA export and trafficking has mainly been studied in mononucleated cells
(Weis et al. 2012). The export of the mRNA is tightly monitored, and only
successfully capped, spliced and polyadenylated mRNA is transported to the
cytoplasm. Thus the export of the mRNA is coupled to the transcription, securing
that only functional mRNA exits the nucleus (Jensen & Rosbash 2003). Prior to
the export the mRNA is coated with specific proteins – called RNA binding
proteins (RBP) – into a stable and exportable package termed messenger
ribonucleoprotein (mRNP) particles (Vinciguerra & Stutz 2004). Among these
proteins are mRNA export factors, which direct the export mRNA via the nuclear
pore complexes (NPC) to the cytoplasm. These export proteins are recruited by
certain adaptor proteins, which function often also as essential components of
other RNA processes, such as splicing or 3’ end processing (Kelly & Corbett
2009).
During the processing of mRNA a legion of proteins associates with it. Many
of them are then displaced following the completion of certain processes.
Eventually the mature mRNA is recognized by the export factors, which guide the
mRNA out of the nuclei through the NPC. In eukaryotes, mRNA is exported with
the 5’ end cap proceeding first through the NPC. In cytoplasm the export factors
are then displaced by other factors that further regulate the fate of the mRNA.
This kind of molecular wardrobe changes occur throughout the life cycle of an
mRNA and helps to co-ordinate mRNA biogenesis (Kelly & Corbett 2009).
In cytoplasm multiple fates await the exported mRNA; it can be localized to
discrete cellular destinations, can associate with ribosomes for translation or can
be degraded. Whatever the mRNA’s destiny is, the mRNA requires subtle and
accurate targeting. The mRNA localization therefore utilizes the nucleotide
sequence – usually in the 3’ untranslated region (UTR) of mRNA – and RBPs for

22
targeting the mRNAs to discrete cellular locations (St Johnston 2005). In spatially
polarized cells the mRNA trafficking to subcellular compartments is an
evolutionary conserved mechanism for the regulation of protein synthesis
(Donnelly et al. 2010). In neurons, for instance, the trafficking of mRNA is
considered to be more efficient than that of proteins, since a single mRNA can
produce dozens of copies of a protein. In the cytoplasm of eukaryote cells
mRNAs are transported as large RNP complexes. The formation of these
complexes is co-ordinated by nucleotide sequences and secondary RNA structures
frequently located in UTRs of the mRNAs (Jambhekar & Derisi 2007, Kiebler &
Bassell 2006). The active transport of mRNAs in cytoplasm occurs on the
cytoskeleton with the aid of the molecular motor proteins (Bullock 2011), for
example using microtubule-plus-end transport, utilizing kinesin motor proteins
(Kiebler & Bassell 2006). During transportation the mRNAs are commonly
translationally silenced by specific proteins (Holt & Bullock 2009).
Degradation of mRNA is also an important way of controlling gene
expression. Over the past several years, enzymatic regulation of mRNA decay has
been studied closely, and the cytoplasmic sites, where the mRNA turnover
happens (called P-bodies and stress granules), have been discovered (Balagopal &
Parker 2009, Garneau et al. 2007, Sheth & Parker 2003, Eulalio et al. 2007).
P-bodies are dynamic structures, where the mRNA degradation, surveillance,
repression and RNA-mediated gene silencing occurs (Eulalio et al. 2007). Stress
granules are considered to be consisting of mRNAs, which are stalled in process
of translation initiation (Balagopal & Parker 2009). In order to initiate mRNA
degradation, the exchange of RBPs must be done to inhibit translation and to
promote degradation (Balagopal & Parker 2009). After that either the 5’ end cap
or the 3’ poly-A-tail with their associated proteins must be compromised or the
mRNA must be cleaved internally by endonucleases. In eukaryotic cells, most of
the mRNA is degraded beginning with poly-A-tail shortening, followed by either
direct 3’ to 5’ direction degradation or removal of the 5’ end cap following 5’ to 3’
direction degradation. In these cases, degradation is conducted by exonucleases.
Another way is the internal cleavage of the mRNA followed by the degradation
by endonucleases. Other paths for mRNA decay also exist: Faulty mRNA is
degraded in the nucleus but translation dependent degradation, which is
connected to mRNA surveillance detecting aberrant mRNP stuctures, can also
occur in the cytoplasm. The RBPs and UTRs have also an essential role in the
mRNA stability thereby regulating mRNA turnover (Garneau et al. 2007).

23
Fig. 1. The production of a protein by a eukaryotic cell. First, the DNA unwinds to
enable the synthesis of an mRNA. The mRNA is then modified and, eventually,
exported from the nucleus, the defective mRNA being degraded. Subsequently, the
protein synthesis begins at the ribosomes. Finally, the protein product is further
modified to be fully functional, while the faulty proteins are degraded.
2.1.3 Protein synthesis
The main goal of the mRNA is to guide the protein synthesis (translation) in the
cytoplasm. In this event the nucleotide sequence of the mRNA codes and
produces – with the assistance of the ribosome – an amino acid chain i.e. a
polypeptide, which then, in turn, will fold into a functional protein. In eukaryotic
cells the translation occurs in the millions of ribosomes, which are complex
cellular structures consisting of several rRNAs and more than 50 proteins (Steitz
& Moore 2003). When a cytosolic protein is produced, the translation occurs in
the cytoplasm in the free ribosomes, whereas when a membrane protein or a
secreted protein is produced, the ribosome complex is translocated to the
endoplasmic reticulum (ER) during the translation. Usually multiple ribosomes

24
can be attached to a single mRNA thus making the translation more efficient.
These structures are called polysomes (Warner et al. 1963).
Translation includes four phases: iniation, elongation, translocation and
termination. When a ribosome meets an mRNA, it binds to the 5’ end cap of the
mRNA and starts to read the mRNA’s nucleotide sequence in triplets called
codons. For each of these possible triplets, only one particular amino acid of the
available twenty is accepted. The selected amino acids are brought to the
ribosome by tRNA, which recognizes the codons with its complementary
anticodon sequence. The initiation of the translation occurs, when the ribosome
finds a single specific codon. The initiation itself also requires specific initiation
factors, which are located both in the mRNA and in the ribosome. After initiation
the growing polypeptide chain is elongated by adding an amino acid to its
carboxyl terminus (C-terminus). The other end of the protein is called amino
terminus (N-terminus). The elongation phase is also driven forward and improved
by multiple proteins called elongation factors. In the translocation phase the
ribosome moves three nucleotides – one codon – at a time along the mRNA to the
5’ to 3’ direction. Then the polypeptide chain is again elongated by a single amino
acid, and the ribosome moves a codon forward. One of the specific three stop
codons marks the end of translation. These are not recognized by any tRNA and
tell the ribosome to cease the translation. Yet again auxiliary proteins called
release factors are required in this process. As a result, a water molecule is added
to the polypeptide chain freeing the completed protein to the cytoplasm.
Consequently the ribosome releases the mRNA and can now continue to translate
new protein (Ramakrishnan 2002).
The process of gene expression is not over when the translation is completed.
The newly synthesized polypeptide chain must fold up into its three-dimensional
conformation (Fig. 1), sometimes bind small-molecule cofactors, be modified by
protein kinases or other enzymes – meaning glycosylation, phosphorylation,
acetylation etc. – and occasionally assemble with other protein subunits with
which it functions. Consequently, the new protein can modified with hundreds of
different ways (Jha & Komar 2011). All the information needed for the above
mentioned steps is coded into the amino acid sequence of the protein. When a
protein folds into its final conformation, it reaches the state of lowest free energy
burying most of its hydrophopic residues in an interior core. Another important
point is the protein’s ability to fold rapidly. In some cases the folding begins
immediately when the protein emerges from the ribosome, but conventionally,
however, the folding is assisted by special proteins called molecular chaperones

25
such as heat shock proteins, which guide and correct the folding of proteins
(Young et al. 2004).
2.1.4 Protein trafficking and degradation
After the translation the fate of the new protein depends on its amino acid
sequence, which in some cases contains sorting signals that locate the new protein
outside the cytosol. Most of the proteins do not contain this signal, and therefore
remain in the cytosol. A plethora of proteins, however, are directed to the nucleus,
mitochondrion, peroxisome or ER by a specific sorting signal. These sorting
signals can also further guide the transport of proteins from the ER to other
destinations within the cell.
Fundamentally, protein trafficking includes three different ways in which
proteins can be transported. First, in gated transport, proteins move between the
cytosol and the nucleus through the NPCs, which form a selective gate allowing
active transport of macromolecules and free diffusion of smaller molecules.
Secondly, in transmembrane transport, proteins move from the cytosol into the
ER, mitochondria or peroxisome through a protein translocator, which is a special
membrane penetrating tunnel structure. Thirdly, in vesicular transport, proteins
move from the ER through the exocytic route to the outer regions of the cell or
even outside the utilizing vesicular membrane carriers. Of course, proteins can
also enter cells via the endocytic pathway. In general, each transport mode
demands a sorting signal in the protein itself as well as a complementary sorting
receptor in the corresponding final location. Sometimes even additional targeting
proteins are required (Schatz & Dobberstein 1996). Usually protein sorting
signals are located in the amino acid sequence of the protein and are 15–60
residues long. These signal sequences are typically found in the N-terminus and
are removed by specific enzymes once the sorting process is complete. The signal
sequences can also be internal stretches of amino acids, which are not removed
when the sorting is finished. Occasionally sorting signals can even be three-
dimensional structures on the protein’s surface, called signal patches.
The protein sorting into the nucleus, mitochondrions or peroxisomes occurs
through a post-translation mechanism (Agarraberes & Dice 2001). The nuclear
import, for instance, requires a nuclear localization signal in the transported
protein and specific nuclear import receptors that bind both to the transported
protein and to the NPC. Sorting to the mitochondrions – on the other hand –
requires also a specific sorting signal, but the unfolded state of the transported

26
protein is necessary as well. Peroxisomes instead only require a specific signal of
three amino acids for a protein to be imported.
The protein sorting into the ER is somewhat different when compared to
other organelles, since it usually happens co-translationally i.e. the ER captures
the selected proteins from cytosol while they are being synthesized (Rapoport et al. 1996). These proteins can be divided into two classes: transmembrane proteins
that only partly translocate across the ER and remain attached to it, and water-
soluble proteins that translocate across the ER into the subsequent lumen.
Transmembrane proteins can either function in the ER or be transported further to
the other membranes of the cell, whereas water-soluble proteins are destined
either for residence in the lumen of the ER or other organelle, or for secretion out
of the cell. The protein translocation to the ER begins when the ER signal
sequence arises from the ribosome. A specific protein, the signal recognition
particle, then binds to the nascent polypeptide and the ribosome and halts the
translation (Keenan et al. 2001). This complex is next directed to the respective
receptor and to the subsequent translocator protein on the ER that forms a tunnel
through which the polypeptide is translocated into the ER. The signal sequence is
cleaved, and the mature protein is released into the ER. The ribosome also
detaches from the translocator protein, which in turn closes the tunnel between
the cytosol and the ER (Agarraberes & Dice 2001). The translocated protein is
eventually folded and glycosylated in the ER. Indeed, most of the soluble and
membrane-bound proteins that are manufactured in the ER are glycosylated and
thus called glycoproteins. In contrast, very few of the cytosolic proteins are
glycosylated. The glycosylation is performed by an ER-bound enzyme called
oligosaccharyl transferase (OST) (Yan & Lennarz 2005), whereas the folding of
the proteins is assisted by ER chaperones such as binding protein and calnexin
(CLN) (Gething 1999).
The fate that awaits dysfunctional proteins is degradation. Usually, if a
protein has a large exposed region of hydrophopic amino acids on its surface, it is
considered abnormal; it has not folded correctly when leaving the ribosome,
suffered an injury partly unfolding it, or failed to find its normal partner subunit
in bigger protein complex. Since misfolded proteins can be potentially harmful to
the cells, cells have developed elaborate quality control mechanisms (Kostova &
Wolf 2003). The apparatus that destroys aberrant proteins is called proteasome,
and it is widely dispersed in cytosol and nucleus (Baumeister et al. 1998). The ER
associated dysfunctional proteins are also degraded by protesome, since they are
retrotranslocated from the ER back to the cytosol. Generally proteasomes

27
annihilate proteins that have been specifically marked for destruction by the
attachment of a recognition tag by a protein called ubiquitin. Commonly, tagging
with ubiquitin results in the degradation of the marked protein – however,
occasionally ubiquitylation has a completely different meaning, for instance,
endocytosis. Ultimately, it is the number of and the way in which ubiquitin
molecules are linked together which determines how the cell reads the ubiquitin
message (Hershko & Ciechanover 1998).
2.2 Exocytic trafficking
Thousands of membrane and secretary proteins exit the ER while trafficking to
various cellular destinations including the Golgi apparatus, lysosomes, peroximes
and the PM. In eukaryotic cells this exocytic trafficking is mediated by vesicles
that allow cells to transport proteins and lipids in a highly regulated manner and
yet preserve the membrane identity of different organelles. This vesicular
trafficking can be divided into four distinctive phases: budding from the donor
membrane, moving through the cell, tethering and docking to the target
membrane, and membrane fusion. The vesicle budding, as well as the cargo
selection, is mediated by protein coats, which are dynamic proteins cycling on
and off membranes (Bonifacino & Lippincott-Schwartz 2003, McMahon & Mills
2004). These coats bend flat membranes into round buds, which eventually lead
to the release of coated vesicles. The three most common coat proteins are coat
protein I (COPI), coat protein II (COPII) and clathrin (Barlowe et al. 1994,
Malhotra et al. 1989, Pearse 1975). COPI is involved in intra-Golgi trafficking
and in retrograde trafficking from the Golgi apparatus to the ER, while COPII
mediates anterograde trafficking from the ER to the Golgi apparatus. Clathrin, on
the other hand, is associated with the endocytic trafficking (Angers & Merz 2011).
After pinching off from the donor membrane, vesicles are transported to their
destination either by diffusion or by motor-mediated transport along cytoskeleton
track (Hehnly & Stamnes 2007). After the coat proteins are released, tethering and
docking begins. At the target membrane tethering factors make the first contact
between the membranes (Sztul & Lupashin 2006). This process is followed by
docking, during which soluble N-ethylmaleimide sensitive factor attachment
protein receptors (SNAREs) on both membranes fuse together forming a complex
that initiates membrane fusion (Jahn & Scheller 2006, Sollner et al. 1993).
Vesicle targeting to a specific membrane is thought to be mediated by the
tethering factors as well as by the SNAREs, since only certain cognate SNAREs

28
catalyze fusion. However, the coordination of vesicle trafficking also requires
small proteins called Rab GTPases, which have specific membrane localization in
the cell, thus regulating tethering and docking events. Rab GTPases are also
essential for vesicle budding, uncoating and motility (Stenmark 2009).
Additionally, vesicle coats are nowadays also thought to take part into the
targeting process (Angers & Merz 2011).
2.2.1 ER-to-Golgi trafficking
As already mentioned, anterograde trafficking from the ER to the Golgi apparatus
is mediated by COPII coated vesicles. COPII and the associated proteins are
responsible for cargo sorting and vesicle formation (Barlowe et al. 1994). The
COPII vesicles bud from special ER domains called transitional ER or ER exit
sites (ERES) (Bannykh et al. 1996). In most animal cells ERES are dispersed
throughout the entire ER network. The proteins exiting the ER are under strict
quality control and only the properly folded and assembled proteins can leave the
ER (Ellgaard & Helenius 2003). The selection of the cargo proteins to the COPII
vesicles is a regulated process, which is mediated by exit signals on the cargo
proteins themselves or on specific receptor proteins. The exit signals guiding
soluble proteins out of the ER for transport to the Golgi apparatus and beyond are
not, however, well understood.
After export vesicles have budded from the ER they lose their coat proteins,
and COPII complexes are recycled back to the ER for reuse (Forster et al. 2006).
Consequently, uncoated vesicles begin to fuse with each other. The new structures
formed between the ER and the Golgi apparatus are called vesicular tubular
clusters (VTC) or ER-Golgi intermediate complex. They are quite short-lived
structures since they move quickly along microtubules to the Golgi apparatus to
deliver their cargo (Appenzeller-Herzog & Hauri 2006). However, it is not exactly
known how vesicles from the ERES move to the VTC: both microtubule
dependent and independent routes are proposed to exist (Zanetti et al. 2011).
Immediately after VTC form, they begin to bud off vesicles of their own. Unlike
the ER-to-Golgi vesicles, these retrograde trafficking vesicles are coated with
COPI and carry back ER resident proteins and cargo receptors, which have
escaped the ER. This retrieval pathway to the ER uses specific sorting signals –
for instance, the KKXX or KDEL sequence – in the retrieved proteins or
specialized receptor proteins such as KDEL receptor, which binds to the proteins
facilitating the retrograde transport (Lee et al. 2004).

29
Eventually VTCs deliver their cargo – proteins and lipids – to the Golgi
apparatus, where further covalent modification occurs. Most commonly
oligosaccharides are removed or added, or glycosaminoglycan chains are added to
the proteins to form proteoglycans. Sulfation of certain amino acid on proteins
also occurs in the late Golgi compartment. Also lipid modification is done in the
Golgi apparatus (Munro 2011). In animal cells, a single Golgi apparatus is usually
located near the nucleus; however, in certain cells, such as skeletal muscle cells or
plant cells, the Golgi apparatus is dispersed into hundreds of individual stacks
found throughout the cytoplasm adjacent to the ERES (daSilva et al. 2004,
Rahkila et al. 1997). In fact, the Golgi apparatus is a polarized organelle
consisting of a series of flattened disc-like cisternae, which are further divided to
three functionally distinctive compartments termed cis-, medial- and trans-
cisternae. The cis- and trans-cisternae are both linked to specific sorting areas,
called cis-Golgi network and trans-Golgi network (TGN), respectively. The cis-
face of the Golgi apparatus is adjacent to the ER and the proteins and lipids pass
through the Golgi apparatus in cis-to-trans direction (Glick 2000). How this
movement occurs, is, nevertheless, disputed. One theory is that the transport
occurs by vesicles, whereas the other theory claims that the Golgi cisternae
themselves move and maturate continuously through the stack. Even the
combination model of these two theories has been proposed (Glick & Luini 2011,
Pelham 2001). Continual retrograde vesicular transport from the distal cisternae is
thought to keep the Golgi enzymes concentrated in the proper cisternae locations.
Eventually, the finished new proteins and lipids move to the TGN, which
packages them in transport vesicles and ships them to their specific destinations.
Figure 2 displays an overview of exocytic trafficking in mononucleated
mammalian cell.
2.2.2 Post-Golgi trafficking
The TGN is the final sorting station for newly synthesized proteins and lipids in
the exocytic pathway. From there proteins can be secreted in either a constitutive
or a regulated manner (Burgess & Kelly 1987), and this transport occurs via the
cytoskeleton of the cell, particularly through microtubules and their associated
motor proteins (Cole & Lippincott-Schwartz 1995). The regulated pathway
operates only in the specialized secretory cells and the secretory vesicles are
released only by an appropriate extracellular signal, whereas the constitutive
secretory pathway operates in all eukaryotic cells secreting proteins continuously

30
by vesicles from the TGN to the PM. In addition to plasmalemmal transport,
proteins can also be transported to the lysosomes or endosomes from the TGN,
and accordingly it is the place where the exocytic and endocytic pathways collide.
Generally, proteins are delivered to the PM by default from the TGN unless they
are diverted into other pathways or retained in the Golgi apparatus (Traub &
Kornfeld 1997). For instance, in the polarized cells, the transport from the TGN to
the PM is selective to ensure that different sets of membrane proteins, secreted
proteins and lipids are delivered to the specific domains of the PM (Schuck &
Simons 2004).
The exocytic and endocytic post-Golgi trafficking regulates the amount of
PM proteins, such as receptors, transporters and ion-channels, modulating
therefore the cell’s ability to function, communicate and adapt with its
environment. The exocytosis and endocytosis must be kept in balance to maintain
the same amount of different membranes. The small vesicles were once thought to
be the main way of transporting secreted proteins, but nowadays it is thought that
also larger more pleiomorphic and VTC-like transport vehicles exist also in the
post-Golgi trafficking (Stephens & Pepperkok 2001). In conclusion, the post-
Golgi trafficking is fairly well characterized even in live mononucleated cells
(Luini et al. 2008).
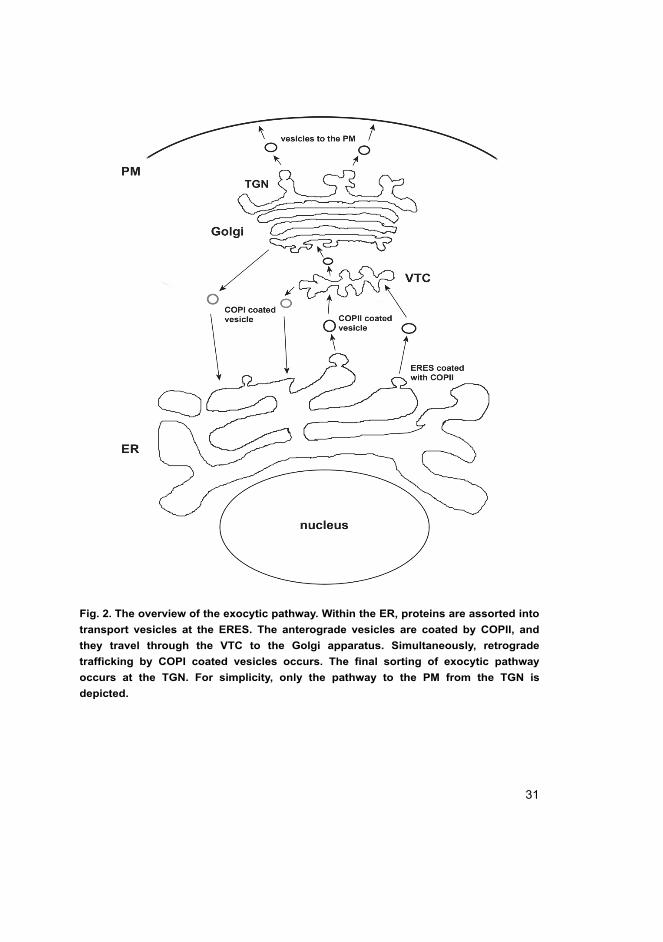
31
Fig. 2. The overview of the exocytic pathway. Within the ER, proteins are assorted into
transport vesicles at the ERES. The anterograde vesicles are coated by COPII, and
they travel through the VTC to the Golgi apparatus. Simultaneously, retrograde
trafficking by COPI coated vesicles occurs. The final sorting of exocytic pathway
occurs at the TGN. For simplicity, only the pathway to the PM from the TGN is
depicted.

32
2.3 Skeletal muscle cells
There are three types of muscle in vertebrates: smooth, cardiac and skeletal. The
latter two are collectively termed as cross striated muscle because of their cross
striation seen on light microscopy. These cross striations are formed by
sarcomeres, which are the basic force producing units in muscle cells.
Additionally, muscles consist of connective tissue layers, which surround an
individual muscle cell (endomysium), bundles of muscle cells (perimysium), and
the entire muscle (epimysium). The nerves and blood vessels associated the
muscle are embedded within the connective tissue.
Skeletal muscle is the most abundant tissue in human body and is responsible
for the movement of the joints and for posture maintenance. The adult skeletal
muscle cells (myofibers) can be divided into several types according to the
metabolic profile and myosin heavy chain (MHC) composition (Brooke & Kaiser
1970, Schiaffino et al. 1989). MHC is the main factor determining the contraction
velocity of myofibers. There are four types of mammalian myofibers: type I, IIa,
IIb and IId/x. The roman letter refers to the MHC isoform. Type I myofibers are
slow-twitch oxidative cells, type IIa myofibers are fast-twitch oxidative glycolytic
cells, while type IIb myofibers are fast-twitch non-oxidative glycolytic cells. Type
IId/x myofibers present an intermediate between types IIa and IIb, and is
expressed in humans instead of type IIb (Bottinelli & Reggiani 2000). However,
the classification of myofibers in the above mentioned types is not always
straightforward, since sometimes the phenotype defined by the metabolic profile
does not match that of the contractile properties especially when functional
demand of the myofibers is altered (Pette & Staron 2001).
2.3.1 Myogenesis
In vertebrates, all skeletal muscles – excluding some craniofacial muscles –
derive from the progenitors found in the somites. During somitogenesis the most
ventral part forms the mesenchymal sclerotome, which contains the precursors for
bone and cartilage. The most dorsal part of the somite, however, remains
epithelian and becomes dermamyotome, from which the skeletal muscle
originates (Bentzinger et al. 2012). Some of these myogenic precursor cells form
immediately terminally differentiated mononucleated muscle cells termed
myocytes. Only a fraction of myogenic progenitors terminally differentiate during
primary myotome formation, and conventionally stages involving different kinds

33
of myoblasts (embryonic and fetal myoblasts, and satellite cells) are required to
build adult skeletal muscle (Biressi et al. 2007).
Satellite cells, which reside under the basal lamina of myofibers, are essential
for secondary myogenesis during cellular damage or growth. In their
nonproliferative state they express regulatory genes, but when needed, the
satellite cells activate differentiation genes, undergo mitosis to fuse with the
damaged myofibers or form new ones, while some satellite cells return to
quiescence to maintain the progenitor pool (Bentzinger et al. 2012, Biressi et al. 2007).
Ultimately, skeletal muscle development is a highly regulated complex
process, where mesodermal precursor cells are selected to form myoblasts.
Myoblasts then fuse into myotubes, in which the assembly of myofilaments and
the production of muscle specific proteins begins. Myotubes are multinucleated
long cells, which then further differentiate into myofibers. During the
differentiation the architecture of the cellular organelles changes drastically, with
the consequent formation of a single huge functional unit. The cytoskeleton is
reorganized, and muscle specific organelles such as the sarcolemma, sarcoplasmic
reticulum (SR) and the tranverse tubules (T-tubules) are formed (Flucher 1992,
Tassin et al. 1985). Apparently, new intracellular pathways are needed also to
communicate with the new organelles and membrane domains, and consequently,
the Golgi apparatus is reorganized from a single polarized juxtanuclear to
multiple perinuclear and interfibrillar elements (Rahkila et al. 1996, Tassin et al. 1985). A part of the ER also becomes SR, and the localization of the ER becomes
more elaborate in the myofibers (Kaisto & Metsikko 2003). While ER mostly
functions as a site of protein synthesis, the SR is considered as a dynamic Ca2+
storage devoid of ribosomes.
2.3.2 Architecture of myofibers
Skeletal muscle consists of myofibers, which are enormous cylindrical cells
surrounded by a mosaic PM. The diameter of a vertebrate myofiber varies
between 10 and 100 µm, and the length can be up to several centimetres. The
characteristic structure of the myofiber is that the hundreds of nuclei are located
in the periphery of the cell right beneath the sarcolemma i.e. subsarcolemmally.
The cytoplasm of the myofiber is tightly packed with myofibrils, membrane
structures and the cytoskeleton, which anchors the contractile apparatus to the PM.
Myofibrils are composed of repeating units of thin actin and thick myosin

34
filaments. The thin actin filaments are attached at their barbed end into structures
called Z-discs (or Z-lines or Z-bands). The gap between two successive Z-discs
defines the sarcomere. The thick myosin filaments are located between the actin
filaments but do not reach the Z-discs when the myofiber is not fully contracted.
When observed under a light microscope, the center of the sarcomere consists of a
pale area, the H-zone, and a thin dense structure, the M-line. The M-line and H-
zone are parts of the A-band defined by a thick myosin filament, whereas the
region composed only of thin actin filaments and the Z-discs in the middle, is
termed the I-band. This actin-myosin filament system is further supported by two
large filament proteins, titin and nebulin. Figure 3 shows a summary of
myofiber’s structure.
According to the sliding filament theory, during muscle contraction, the thin
actin filaments slide along the thick myosin filaments towards the center of the
sarcomere. This is provoked by the attachment of the globular heads of the MHCs
to the actin filaments, which must be preceded by a conformational change in the
actin binding protein troponin to uncover a myosin binding site. This
conformational change is initiated by the binding of Ca2+ to one of the subunits in
the troponin complex. Once the myosin head is bound to the actin filament, the
head is bent as a result of ATP hydrolysis and release. The binding of a new ATP
molecule then releases myosin from the actin, and allows a new cycle of
contraction. The contraction continues as long as enough Ca2+ and ATP are
present. The Ca2+ is released from the SR in response to anaction potential and
pumped back to the SR by sarco/endoplasmic reticulum Ca2+ -ATPase (SERCA).
2.3.3 Plasma membrane and its specialized macrodomains
The PM of myofiber is a mosaic membrane consisting of various specialized
domains, which help to communicate with synaptic endplates of motor neurons,
transmit force to the tendon and conduct the incoming action potential to the deep
interior of the myofiber to stimulate Ca2+ release and contraction. In this thesis,
the term sarcolemma refers only to the lateral part of the myofiber’s PM thus
excluding the basement membrane and the T-tubules. Moreover, the sarcolemma
is divided into different structural and functional domains. Lipid rafts, which are
small heterogeneous, dynamic domains compartmentalizing cellular processes
(Simons & Ikonen 1997), and caveolae, which are flask-shaped invaginations in
the sarcolemma containing an abundance of caveolin 3 protein (CAV3) and which
are associated with various cellular functions (Parton & Simons 2007), are the

35
most common microdomains found at the sarcolemma (Parton & Simons 2007,
Simons & Ikonen 1997). Consequently, the whole sarcolemma can be seen to be
organized into caveolin associated raft and non-raft domains consisting of the
mosaic T-tubule domains, sarcolemmal caveolae and β-dystroglycan domains
(Kaakinen et al. 2012, Rahkila et al. 2001).
The T-tubules are tunnel-like structures continuous with the sarcolemma
running deep inside the myofiber where they are interconnected through
helicoidal structures (Peachey & Eisenberg 1978) or longitudinal tubules going
parallel to the long axis of the myofiber (Launikonis & Stephenson 2004).
Accordingly, this tubular part of the PM can be considered as a network. The
openings of the T-tubules are located in the sarcolemma at the A-I junction zone
(Rahkila et al. 2001). The overall orientation of the T-tubules is shown in the
Figure 3. Despite the continuum between the sarcolemma and the T-tubules they
share a different protein composition. For instance, dihydropyridine receptor
(DHPR) is only present in the T-tubules, not in the sarcolemma (Jorgensen et al. 1989), whereas dystrophin and dystoglycans – the components of the large
dystrophin glycoprotein complex – are found exclusively on the sarcolemma
(Ohlendieck et al. 1991). Inside the myofiber the T-tubules are connected with the
SR in the regions termed as triads, where the SR forms sac-like terminal cisternae
enriched with ryanodine receptor (RYR) Ca2+ channels. In the T-tubule, the
DHPR, which functions primarily as a voltage sensor, is located in the same
regions thus coupling the depolarization wave evoked at the sarcolemma to the
release of Ca2+ and, ultimately, to the muscle contraction.
The neuromuscular junction (NMJ) is an elaborate structure that serves to
communicate the electrical impulse from the motor neuron to the myofibers to
signal muscle contraction. This is the site at which acetylcholine released from
motor axon terminal binds to the corresponding receptor. Usually, only one NMJ
exists per myofiber. The folding of the sarcolemma and the abundance of
acetylcholine receptors are characteristic to the NMJ (Hughes et al. 2006).
Subsequently, the binding of the acetylcholine to its receptor evokes a
depolazation wave, which in turn is sensed by voltage sensitive sodium channels
at the NMJ. This results in an action potential, which spreads in all directions
along the PM and eventually leads to muscle contraction. Additionally, the NMJ
domain differs from normal sarcolemmal regions. Beneath the NMJ, the
microtubules form a dense network, the number of nuclei is increased and the
Golgi distribution is more random (Rahkila et al. 1997). Also, the high local

36
concentration of acetylcholine receptors and of several associated proteins and
their mRNA are hallmarks of the NMJ (Ralston et al. 1997).
The myotendinous junction (MTJ) is a functionally distinct compartment of
the sarcolemma, where the thin actin filaments terminate and the myofiber is
attached to a tendon. The MTJ is specialized in transmitting the sarcomere
generated force to the adjacent tendon and therefore requires exquisite structural
stability. The MTJ is enriched with components of the dystrophin glycoprotein
complex as well as the focal adhesion proteins (Tidball 1991). In the MTJ the
sarcolemma is extensively folded into finger-like extensions increasing the
surface of the lateral contacts between the myofilaments and the PM and thus also
reducing the surface tension (Berthier & Blaineau 1997).
2.3.4 SR, ER and the Golgi apparatus
In myofibers, the SR is considered as an equivalent of the smooth ER present in
other cell types. During primary myogenesis the SR develops as tubular
projections from the rough ER (RER) vesicles and then wraps around the
developing myofibrils (Ezerman & Ishikawa 1967). In adult myofibers, the SR
appears as a network of tubules and cisternae surrounding the myofibrils like a
net stocking and occupying the interfibrillar space (Sorrentino 2011). Primarily,
the SR is a dynamic Ca2+ storage organelle found in all muscle cells and it
maintains specialized associations with the myofibrils and the T-tubules (Rossi et al. 2008). The SR can be further divided into longitudinal SR (L-SR) and
junctional SR (jSR), which have distinct functions. The vast majority of SR is
formed by L-SR, which runs around the sarcomeres and is dedicated to Ca2+
uptake and contains the SERCAs, which exist in two isoforms in myofibers:
SERCA1 is expressed in fast-twitch fiber typer and SERCA2 in slow-twitch fiber
types (Periasamy & Kalyanasundaram 2007). The L-SR regularly merges into
larger membrane structures, termed terminal cisternae. In myofibers, two terminal
cisternae positioned on the opposite sides of one T-tubule form a triad. The SR
facing the terminal cisternae is called jSR. The jSR is the region, where SR-
specific proteins, like RYR, calsequestrin (CLQ), triadin and junctin reside
(Sorrentino 2011). The jSR specializes in Ca2+ release through RYRs as
mentioned earlier.
In adult myofibers RER proteins are located as clusters within the SR, and
similarly to mononucleated cells, the RER proteins show a clear staining also
around the myonuclei (Kaisto & Metsikko 2003, Rossi et al. 2008, Volpe et al.

37
1992). Interestingly, all SR/ER related proteins seem to be distributed in the SR
compartments located over the I-band and to a minor extent over the M-line in
middle of the A-band (Kaisto & Metsikko 2003, Rossi et al. 2008). The L-SR
covers most of the A- and I-band of the sarcomere, with differential density of
membranes, which is higher on the Z-discs in the middle of the I-band.
Consequently, immunostaining with SR protein antibodies and analysis with
fluorescence or confocal microscopes yields a stronger signal on the Z-discs and
much weaker signal on the vicinity of the M-line (Salanova et al. 2002). However,
it is suggested that absence of detectable quantity of proteins in the A-band
vicinity might be due to a lesser amount of SR. Therefore, the level of proteins is
below detection limit of light microscopy (Rossi et al. 2008). This observation is
further supported by immunoelectron microscopic studies that have found
SERCA greatly in the I-band, but scarcely also in the A-band (Jorgensen et al. 1982).
The Golgi apparatus is reorganized anew during myogenesis into hundreds of
dispersed elements which form a belt-like distribution around each myotube
nuclei and extend between the nuclei along microtubule tracts like strings of
pearls (Rahkila et al. 1997). In myofibers, each Golgi apparatus consists of
thousands of these small distributed elements. In electron microscopic studies
each of these elements is observed to be made of a small stack of cisternae, and
an average of these Golgi elements per myonuclei is one hundred (Ralston et al. 1999). The distribution of Golgi elements in the NMJ is constant and independent
of myofiber type, but otherwise the distribution of Golgi elements is dependent of
the myofiber type. In type I myofibers, approximately 75% of Golgi elements are
found within 1 µm beneath the sarcolemma and every nucleus is surrounded by a
belt of Golgi elements. In type IIb myofibers, on the contrary, most Golgi
elements are found in the core of the myofibers and most nuclei only possess
Golgi elements at their poles. The type IIa myofibers represent an intermediate
distribution pattern of Golgi elements. The microtubules, ERES and Golgi
elements are linked and affected by muscle activity. It is, however, unclear
whether one of them determines the localization of others, since very little is
known of the link between ERES and microtubules (Ralston et al. 1999, Ralston et al. 2001).

38
Fig. 3. The general architecture of myofiber. Myofiber is surrounded by the
sarcolemma, where the T-tubule openings reside at the borders of I-band and A-band.
The interior of a myofiber consists mostly of myofibrils, whose elaborate organization
is further depicted at the bottom of this figure. The interfibrillar space is filled by the
SR and the mitochondria. The myonuclei are found subsarcolemmally and are
surrounded by the SR/ER and by Golgi elements, which are also located within the
myofiber.
2.3.5 Ribosomes and mRNA
In mononucleated cells ribosomes attached to the RER clearly show its location.
This is not the case in myofibers and locating RER in myofibers merely by
morphology is difficult if not impossible. In ultrathin sections of a myofiber it is
hard to distinguish reliably single ribosomes from single glycogen particles,
although it is possible with careful analysis (Dolken et al. 1975). In myofibers
ribosomes have been found distributing evenly within the cells. In chicken
anterior latissimus dorsi muscle, ribosomes were even found to be located
between thick filaments, usually aligned in rows proposing that ribosomes are
located within the filament lattice to be available for local myosin protein
synthesis (Gauthier & Mason-Savas 1993). However, it was not shown which, if
any of the ribosomes were translationally active.
In myofibers specialized nuclei at the NMJ exist. They produce mRNA not
expressed in non-junctional nuclei. These mRNAs remain localized near their
nuclei of origin and their proteins products are transported to the junctional PM

39
above those nuclei (Jasmin et al. 1993). Studies with myotubes have proposed
that gene products of non-junctional nuclei are also restricted in the vicinity of
their nuclei of origin (Ralston & Hall 1992). Accordingly, the concept of nuclear
domain has been introduced. This concept describes that each myonucleus has its
own volume of influence where its gene products are targeted within the cell.
Nowadays it is thought that myonuclear domain size differs in different myofiber
types and can change during muscular adaption (Van der Meer et al. 2011).
Concerning myofibers, no data exist to answer how far the mRNAs reach from
the nuclei of origin. Instead, it is known that mRNAs encoding dystroglycan
(Mitsui et al. 1997) or Na+ channel (Awad et al. 2001) are located
subsarcolemmally following the localization of their protein products.
Intriguingly, mRNAs encoding MHC were also heavily concentrated
subsarcolemmally although the respective protein is a component of the
myofibrils that are located all over the myofibers (Dix & Eisenberg 1991).
Similarly, mRNAs encoding CLQ or DHPR, which typically distribute in the
interfibrillar SR, were also restricted to beneath the sarcolemma. Furthermore, the
mRNAs were expressed in every nucleus indicating no division of labor between
the nuclei (Nissinen et al. 2005). In conclusion, these findings imply that
translation in myofibers occurs subsarcolemmally, from where the protein
products further move into the interior of the cell.
2.3.6 Studying the trafficking of gene products
Myofibers form a very complex environment when it comes to studying the
trafficking of gene products. The overwhelming majority of protein trafficking
studies have been conducted with mononucleated cells and in some instances with
multinucleated osteoclasts (Coxon & Taylor 2008, Salo et al. 1996). Not many
studies concerning intracellular trafficking in adult myofibers have been carried
out, and the knowledge of intracellular trafficking in myofibers is still quite vague
compared to that of mononucleated cells. The methods allowing live imaging of
mRNA have been available only for a few years and thus experiments have been
mainly conducted with mononucleated cells (Bao et al. 2009). Since
multinucleated cells are methodologically more challenging than mononucleated
cells, no studies using live imaging of mRNA have been carried out.
Examining the trafficking of gene products in voluminous myofibers comes
with many issues. First of all is the dilemma of sufficient expression level of the
studied mRNA or protein. The only potent methods for transfection with

40
sufficient expression levels are viral carriers or in vivo electroporation (see
Methods for further details). Enveloped viruses, such as Semliki Forest virus
(SFV), are ingenious tools in studying protein transport. They enter the cell and
exploit the intracellular trafficking machinery for their replication. The viral
glycoproteins are subjected to the same posttranslational controls and processing
as endogenous proteins (Katz et al. 1977, Rothman & Lodish 1977), but are
expressed much more abundantly, which makes them easier to locate. Mutant
viruses have also been manufactured. A typical example is ts045, a temperature
sensitive mutant of vesicular stomatitis virus (VSV), whose transport can be
controlled by changing the incubation temperature. The expression of cloned
sequences using viral carriers have been used since 1980s (Olkkonen et al. 1994)
and it is a powerful tool for expressing modified or exogenous mRNA or proteins
in cells resistant to transfection methods, such as myofibers.
Also the detection of gene products in the voluminous myofibers can be
tricky due to low expression levels. Nowadays a conventional way of live protein
detection is the green fluorescent protein (GFP) and its derivatives. Originally
found in jellyfish and utilized by Chalfie and colleagues (1994), the GFP fusion
proteins have revolutionized the live cell imaging with many applications
(Chudakov et al. 2010). Various colour options are also derived from the
traditional green fluorescence, for instance yellow fluorescent protein (YFP).
When GFP is attached by genetic engineering techniques to the protein of interest,
the trafficking and whereabouts of the resulting fusion protein can be followed in
living cells. When a complementary DNA (cDNA) encoding such a fusion protein
is expressed in a cell, the protein is visible in a fluorescence microscope. The
examination of GFP fusion proteins can be combined with fluorescence recovery
after photobleaching (FRAP), and fluorescence loss in photobleaching, techniques,
where the GFP of selected regions of the cell is bleached using strong laser light
(Lippincott-Schwartz et al. 2000). GFP and its natural colour variants are thought
to be mostly neutral towards the physiology of the cell, but, nevertheless, fusing a
GFP to a protein of interest can impair the function of the latter and thus the
expression of this construct can have adverse effect on cellular function
(Wiedenmann et al. 2009). Therefore, some consideration must be used when
working with the GFP constructs.
The localization of the mRNA is usually observed in fixed cells through in
situ hybridization methods, in which specific nucleic acid sequences can be
detected within individual cells. The method is based on the complementary
binding of a labeled nucleic acid probe to complementary sequences in cells,

41
followed by visualization of target sequences within the cells (Bassell et al. 1994,
Mabruk 2004). This method, however, cannot be used with living cells, and
consequently alternative methods for live imaging of mRNA have been developed
(Dirks et al. 2001, Fusco et al. 2003), for review see (Bao et al. 2009). Since
there is no RNA analogy of GFP available for the creation of purely genetically
encoded fluorescent RNA molecules, the fluorescent molecules must be delivered
to an mRNA, normally via noncovalent binding to a cognate tag region of the
mRNA. Usually this is carried through an RBP labeled with GFP to allow
visualization (Armitage 2011, Bertrand et al. 1998). In the future these methods
will be applicable for the visualization of single mRNA of various kinds to
elucidate their precise localization and dynamics (Yamada et al. 2011).
2.4 LBs in mammalian cells
Lipid bodies (LBs) are cellular organelles consisting of a neutral lipid core
containing triglycerides, diacylglycerides and cholesterol esters (Thiele & Spandl
2008). They are coated with a monolayer of phospholipid and cholesterol that
includes the PAT proteins – perilipin, adipose differentiation related protein
(ADRP), tail-interacting protein of 47 kDa, S3-12 and OXPAT (Bickel et al. 2009)
– and an ample set of other proteins, such as CAV3. LBs are numerous and
humongous in size – up to 150 µm in diameter – in adipocytes, but they are also
present in all other cells types including skeletal muscle. In non-adipose cells
their size varies between 0.1–2.0 µm (Digel et al. 2010). In adipocytes LBs are
considered to be immobile, while in non-adipose cells LBs are found to be mobile
organelles interacting with various other cellular organelles (Murphy et al. 2009).
Formerly, LBs were regarded as inert lipid storage vessels, however owing to the
functional protein set in their coat, they are now considered dynamic organelles of
lipid metabolism and intracellular lipid trafficking. Furthermore, the coating
proteins regulate the storage and mobilization of the lipids (Bickel et al. 2009).
The formation of LBs is a much-debated issue. The prevailing model of LB
biogenesis suggests that the neutral lipids incorporate in the interspace between
the bilayer leaflets of the ER, followed by the budding out of cytoplasm oriented
membrane to form an LB (Beller et al. 2010). The origin of LB coat proteins
remains also partly unknown and it is thought that some of the coat proteins are
inserted at the ER, while some are delivered to the LB after the biogenesis and
budding (Londos et al. 1999). Another unsolved issue is whether LBs are

42
connected with the ER. The current FRAP data in mammals summarizes that the
larger LBs seem to be disconnected from the ER (Digel et al. 2010).
2.4.1 LBs in skeletal muscle and insulin resistance
In skeletal muscle, LBs are found in type I myofibers, but are practically absent in
type IIb myofibers while type IIa myofibers represent an intermediate expression
pattern (Malenfant et al. 2001, Shaw et al. 2009). Accordingly, the LB specific
coat proteins OXPAT and ADRP follow this distribution pattern (Minnaard et al. 2009). Previously it has been shown that the intramuscular lipid content is
decreased by physical exercise in exercising muscle, but increased in non-
exercising muscle, indicating that the amount of the LBs can be regulated
(Schrauwen-Hinderling et al. 2003). On the other hand, it has been demonstrated
that elevated fatty acid levels induce the formation of LBs in a plethora of
different kinds of mononucleated cells (Pol et al. 2004). Moreover, ER stress in
several cell types plays an essential role in inducing LB formation, which is
associated with metabolic diseases, mainly obesity and type II diabetes (Hapala et al. 2011).
Occasionally, the coating proteins regulating the lipids within the LBs are
subject to disturbances that result in lipid degradation intermediates, which act as
cell signaling molecules (Bickel et al. 2009). This is highly relevant in myofibers
and therefore the LBs in skeletal muscle cells have recently gained interest in the
research of obesity and type II diabetes. Furthermore, the amount of LBs in
myofibers is linked with insulin resistance (Goodpaster et al. 2000, Russell et al. 1998). This lipid-induced insulin resistance is mediated by insulin desensitizing
lipid intermediates such as diacylglycerol, ceramide and protein kinase C (Chavez et al. 2003, Itani et al. 2002, Yu et al. 2002). Nowadays, however, it is thought
that insulin resistance is a complex metabolic disorder that defies explanation by a
single etiological pathway. Results from recent studies suggest a strong
association between insulin resistance and the accumulation of ceramides and
diacylglycerols. Moreover, the presence of LBs in myofibers, as well as the ER
stress are suggested to be obviously linked to insulin resistance (Samuel &
Shulman 2012).

43
3 Aims of the study
Myofibers are humongous cylindrical cells, which are tightly packed with a
contractile apparatus, endomembrane systems and numerous nuclei. It is clear that
the intracellular organization and trafficking of these elaborate cells must differ
from that of mononucleated cells. However, today rather vague information exists
on the membrane dynamics of myofibers. The purpose of this study was to
examine the localization of mRNA in the myofibers and to characterize the
membrane dynamics of the myofibers. The specific aims were:
1. To localize the endogenous mRNA in myofibers and determine whether
translation occurs exclusively beneath the sarcolemma.
2. To characterize the mobility of cytoplasmic and especially membrane
proteins in myofibers and observe the range of protein movement from a
single nucleus.
3. To clarify the details of the exocytic pathway in skeletal muscle cells.
4. To analyze LB formation and dynamics in myofibers.

44

45
4 Materials and methods
4.1 Cell culture of L6 myoblasts, myotubes and BHK cells (II)
L6 myoblasts were purchased from ATCC and were grown on plastic or glass
bottom dishes (WillCo Wells, Netherlands) in DMEM (Sigma) containing a 7%
fetal calf serum. After a two-day growth period, the myoblasts were induced to
differentiate into myotubes as described earlier (Rahkila et al. 1998). The
myotubes were used for experiments four days after the induction of
differentiation. Primary myotubes were obtained from rat muscle satellite cells
via proliferation and cell-cell fusion as reported previously (Rahkila et al. 1998).
BHK cells were grown in Glasgow MEM supplemented with 5% fetal calf serum
and 10% tryptose phosphate broth.
4.1.1 Isolation of myofibers (I, II, III, IV)
Myofibers were isolated from the footpad FDB muscle of adult female Sprague-
Dawley rats. The FBD myofibers represent fast-twitch oxidative glycolytic type
IIa myofibers (Carlsen et al. 1985). The muscle was excised and treated with
collagenase II digestion followed by trituration by a pipette. The isolated
myofibers were cultivated on dishes coated with Matrigel (Becton-Dickinson
Labware) in Dulbecco’s MEM containing 5% horse serum, in an atmosphere of
5% CO2.
Additionally, bundles or individual myofibers from extensor digitorum longus
(EDL) muscle of adult female Sprague-Dawley rats were carefully prepared by
using fine forceps. Immediately following the preparation, the EDL myofibers
were fixed and used for immunofluorescence studies.
4.2 Recombinant mammalian expression plasmids (I, IV)
To produce an expression plasmid encoding the GFP-Dad1 fusion protein, the
cDNA of the GFP was amplified from a pEGFP-Actin vector (Clontech) and
Dad1 from isolated rat muscle mRNA by using RT PCR. Both cDNAs were
cloned into a pBluescript vector and then into a pEYFP-N1 expression vector
(Clontech) after excision of the EYFP insert. CAV3-YFP-N1 plasmid was
provided by Robert Parton (University of Queensland, Brisbane) and has been

46
described by Pol and colleagues (2004). All constructs were sequenced with an
ABI PRISMTM3130XL sequencer and a BigDye Terminator v1.1 cycle
sequencing kit (Applied Biosystems).
4.2.1 In vivo electroporation of recombinant plasmids (I, IV)
In vivo electroporation was performed on living rat FDB muscle. Isoflurane
anaesthesia was introduced to keep the rat asleep during the operation. An
appropriate plasmid was injected into the muscle through a small incision made
on the footpad. Thereafter, the foot was placed between custom made tweezer
electrodes and subjected to 10 pulses of a 200 V/cm electric field lasting for 20
ms each. A conductive gel was used to improve conduction. Eventually, the
incision wound was closed with tissue glue. The operated rat was sacrificed 2–24
h later, and the myofibers isolated as described above. All animal treatments were
conducted according to the national animal welfare laws that meet EU Directive
2010/63.
4.3 Preparation of recombinant SFV (I, II, III, IV)
Recombinant SFV (recSFV) were generally used to produce larger amounts of
recombinant proteins in myofibers. cDNA endocing VSV temperature sensitive
G-protein (tsG) tagged with GFP – tsG-GFP – in pCB6 vector has been described
by Scales and co-workers (1997). The insert was excised from the vector with
ClaI/BamHI digestion and was cloned to pSFV1 as depicted earlier (Kaisto &
Metsikko 2003). The cDNA encoding GFP-Dad1 was prepared as described by
Kaakinen and co-workers (2008) and cloned to pSFV. The cDNA encoding mouse
CAV3-YFP was excised from the plasmid CAV3-YFP-N1 that was provided by
Dr. Robert Parton. The excision was with KpNI/NotI digestion. The cDNA was
blunted and ligated to pSFV1 that was opened with Smal. The corresponding
recSFV particles were prepared according to Olkkonen et al. (1994). The
multiplicies of infections of the virus used were evaluated on BHK cell
monolayers and had titers approximately 108 infective particles/ml.
4.3.1 Other viruses used (II)
Influeza virus was of the WSN strain, and virus stocks were grown in MDCK
cells. The stocks were diluted with the culture medium, and viruses were

47
adsorbed to the myofibers for 1 h at 37 °C in an atmosphere of 5% CO2. The virus
was then removed, and the infection was allowed to proceed for various time
periods.
4.4 Immunofluorescence studies (I, II, III, IV)
Muscle cryosections or isolated myofibers on the culture dishes were fixed with
3% paraformaldehyde in PBS for 10 min, followed by a 5 min permeabilization
with 1% Triton X-100 in PBS. Blocking of non-specific antibody binding was
conducted using 1% BSA for 20 min. Primary antibody incubations (Table 1)
were carried out for 60 min at 37 °C, two hours at room temperature or overnight
at 4 °C. Alexa conjugated secondary antibodies were applied for the visualization
of bound primary antibodies. The samples were examined using a Zeiss LSM510
confocal microscope.
Table 1. List of primary antibodies and fluorophores used in the original publications.
Antibodies and fluorophores Source/reference Used in
Antibody against
ADRP pAb Progen IV
digoxigenin mAb Roche I, II
BrU mAb Sigma I
Calnexin pAb Stressgen IV
GM-130 mAb BD Biosciences III, IV
HA (WSN) pAb Rahkila et al. 1998 II
L26 Ribosomal protein pAb Abcam I
NP (WSN) pAb Martin and Helenius 1991 II
PDI mAb Koivu et al. 1987 IV
Puromycin mAb KeraFast I
SERCA mAb Sigma IV
SREBP mAb BD Pharmingen IV
VSV G protein pAb (VISA) Metsikkö et al. 1992 II, IV
Fluorophore
FITC-streptavidin Sigma I, II
LD540 Spandl et al. 2009 IV
Oil Red O Koopman et al. 2001 IV
TO-PRO-3 iodide Life Technologies I, IV

48
4.5 Live cell microscopy and FRAP (I, III, IV)
Imaging of living cells was used to observe the intracellular trafficking, as well as
in FRAP experiments. For live cell microscopy the cells were plated onto glass
bottomed dishes. To ensure proper pH, the culture medium was replaced by CO2
independent medium (Invitrogen) supplemented by glutamine and 0.5% horse
serum. A Zeiss LSM510 confocal microsope was utilized for imaging.
FRAP analyses were conducted using the 488 nm line of argon laser. A region
of interest was photobleached at 25% of maximum laser power with 100%
transmittance for 10–60 s. Recovery of fluorescence was monitored for 5–40 min
at timed scanning intervals of 5–60 s at minimum laser power and 5%
transmittance. Pixel intensities of regions of interest were measured and mobile
fractions calculated as described earlier (Feder et al. 1996). Plausible
photobleaching of the sample was corrected by multiplying the pixel intensity
values inside the region of interest with the fractional change of intensity in the
non-bleached area at each time.
4.6 In situ hybridization (I, II)
Transcripts encoding influenza virus HA were probed with a HPLC-purified
single strand DNA 40mer oligonucleotide labeled with digoxigenin (DIG) (Sigma
Aldrich) and complementary for the segment 1441–1480 of HA mRNA
(5´TATTGTGACTGGGTGTATATTCTGGAAAGGGAGATTGCTG-DIG). A
biotin labeled 40mer oligomer corresponding for the segment 1441–1480 of HA
mRNA vas used to detect HA vRNA in the infected nucleus. Samples were fixed
for 15 min with 4% paraformaldehyde in PBS and permeabilized with 0.5%
Triton X-100. Dual in situ hybridizations were performed for 2 h a t 37 °C at
probe concentrations of 2 µg/ml and the hydridization conditions were as
described (II). Biotin was visualized with FITC-streptavidin (Sigma) and DIG
was visualized with an antidigoxigenin antibody (Roche) using Alexa 568-
conjugated anti-mouse IgG as a secondary antibody. Poly- A tails of mRNAs were
detected with a 26mer poly-T oligonucleotide tagged with DIG. Hybridization
was performed at 25 °C for 2 h in hybridization conditions as described in (II) and
DIG was visualized with a specific monoclonal antibody and Alexa 488-
conjugated IgG. Samples were examined with a Zeiss LSM 510 laser scanning
confocal microscope.

49
4.7 Pulse labeling and SDS/PAGE (III)
Pulse labeling with [35S]methionine (100µCi/ml) (Perkin Elmer) was performed
on 30 mm diameter plastic dishes for 20 min at 39 °C. Cells were scraped with
PBS containing 1% Triton X-100 / 1% deoxycholate and 1 mM
phenylmethylsulfonyl fluoride (0.5 ml/dish). Insoluble material was removed by
centrifugation for 5 min at 10000g and the supernatants were subjected to
immunoprecipitation with anti-G protein antibodies and Protein A Sepharose (GE
Healthcare Life Sciences). SDS/PAGE was performed on 9% gels using a buffer
system (Laemmli 1970). Proteins of SFV that are abundant in infected cells were
used as standards. Briefly, SFV infected L6 myotubes were pulse labeled with
[35S]methionine and the cell lysates were loaded on the SDS/PAGE gels. Dried
gels were exposed to Kodak Biomax MR film.
4.8 Western blotting (I, III, IV)
Detergent extracted and TCA precipitated proteins were subjected to SDS/PAGE
in Laemmli buffer (Laemmli 1970) under reducing conditions. After
electrotransfer onto nitrocellulose, Ponceau S staining was performed to check
proper transfer. The blocking of unspecific binding was conducted with 5% non-
fat milk powder in PBS for two hours at room temperature or overnight at 4 °C.
The primary antibodies were applied in a solution containing 0.05% non-fat milk
powder in PBS for two hours at room temperature or overnight at 4 °C. Bound
primary antibodies were detected with horseradish peroxidase conjugated
secondary antibodies and enhanced chemiluminescence detection reagents (GE
Healthcare) using CL-XPosure film (Thermo Scientific).
4.9 Statistical testing (III)
Statistical testing was applied to verify the statistical significance of
measurements. Accordingly, analysis of variances was used. If unequal variances
were assumed between the groups, the Brown-Forsythe test was used to test
statistical significance. Additionally, between two groups Student’s t-test was
applied. P-values are given in parentheses when statistical testing was performed.
All the data are expressed as mean ± SD and n indicates the number of
determinations.

50

51
5 Results
5.1 The localization of mRNA and translation in myofibers (I)
To date, the precise localization of mRNA within the myofibers has been rather
obscure, and no convincing data exist whether all mRNA lie beneath the
sarcolemma or does interfibrillar space also contain mRNA and ribosomes.
Previous studies suggest that the mRNA localizes subsarcolemmally (Dix &
Eisenberg 1991, Mitsui et al. 1997, Nissinen et al. 2005), but some studies made
in the 1990s (Billeter et al. 1992, Gauthier & Mason-Savas 1993) propose that
interfibrillar translation may also exist. Here, our findings suggest that translation
in myofibers is concentrated around the myonuclei locating beneath the
sarcolemma, but also in interfibrillar spots locating within the cells.
5.1.1 Poly-A tails of endogenous mRNAs and ribosomes localize subsarcolemmally and perinuclearly
Since myofibers are major force generating cells, there are massive amounts of
mitochondria in the myofibers and therefore also mitochondrial mRNA. The
mitochondrial mRNA nevertheless lacks the poly-A tails that are present in the
nuclear mRNA products. Accordingly, to investigate the gross distribution of all
cellular mRNA in myofibers, we located the poly-A tails by using a DIG-labeled
poly-T oligonucleotide probe. Using in situ hybridization studies, it was found
that the probe was heavily concentrated around the nuclei, but it was also present
beneath the sarcolemma. Interestingly, a cross-striated distribution pattern
showing some intensity at the A-I junctional regions was also observed. In
addition to cultured myofibers, a similar subsarcolemmal pattern was also
observed in the EDL muscle cryosections. Nevertheless, the staining of the
interior parts of the myofibers could not be excluded with this method.
5.1.2 Bromouridine labeling also marks myonuclei and subsarcolemmal region
In situ hybridization labeling marks mRNA that has accumulated in the cytoplasm
over relatively long time periods. It has been shown that bromouridine (BrU)
enters the cells and is converted to bromouridinetriphostate that is incorporated

52
into the mRNA (Sierakowska et al. 1989). However, the splicing is kept intact
only if less than 50% of the uridines are replaced with BrU (Wansink et al. 1994).
We therefore labeled isolated myofibers with BrU at a relatively low
concentration to retain splicing and the nuclear export of the mRNA. At the BrU
concentration of 5 mM for 6 hours only nuclei were visible, and prolonging the
labeling time did not alter the distribution. Therefore we concluded that the
splicing and nuclear export of the mRNA was disturbed. With lower concentration
of BrU (varying between 1–2.5 mM) and long labeling time up to 16 hours, we
observed fluorescence within the nuclei, but also beneath the sarcolemma,
suggesting that the mRNA was efficiently exported from the nuclei. Importantly,
the subsarcolemmal and perinuclear distribution pattern was quite similar to that
which was previously noticed with poly-A tails.
5.1.3 Translation is concentrated in the vicinity of myonuclei and in interfibrillar spots
The mRNA localization experiments indicated that there are no detectable
amounts of the mRNA in the interior regions of the myofibers, suggesting that the
translation process was exclusively occurring beneath the sarcolemma. To
investigate where translation was occurring we utilized puromycylation of
nascent polypeptide chains and detection with specific antibodies. Puromycin
mimics tRNAtyr and terminates translation by covalently incorporating into the
nascent chain C-terminus (Pestka et al. 1972). David et al. (2012) recently
showed that chain elongation inhibitors, such as emetine, immobilize
puromycylated nascent polypeptide chains into ribosomes. We exploited this
finding to localize translating ribosomes with the anti-puromycin antibodies.
Accordingly, with 10 min puromycin labeling accompanying emetine we found
that translation was concentrated around the myonuclei. With longer labeling time
up to 60 min fluorescence was seen in the vicinity of the nuclei, but surprisingly
small dots also appeared within the myofibers. These interfibrillar spots seemed
to flank the Z-discs. When GM130 staining for Golgi elements was performed,
we noticed that Golgi elements and puromycin dots were located right next to
each other, although there was no full co-localization.
Finally, we studied whether ribosomes followed the subsarcolemmal
distribution pattern in the myofibers. For this purpose we used antibodies against
a 17 kDa eukaryotic ribosomal protein and accordingly, mitochondrial ribosomes
were not recognized. Immunofluorescence studies indicated that the ribosomal

53
distribution pattern was very heavily concentrated around the myonuclei but it
also followed A-I junctional regions. In EDL cryosections there was interfibrillar
staining present too, indicating that there were ribosomal subunits present in the
interfibrillar spaces. The anti-ribosomal antibodies used, however, recognized an
unrelated protein at 110 kDa region in addition to the relevant 17 kDa protein
when EDL homogenate was analyzed by Western blotting.
5.2 Dynamics of proteins in myofibers is versatile (I, II, III)
As voluminous multinucleated cells, the trafficking of proteins in myofibers must
be efficient, since only minor mRNA trafficking to interior parts of the cells
seems to occur. Here we found that rapid movement of both a cytosolic and a
membrane bound model protein occurs, but that also mobility restricted ER
subcompartments are present within the myofibers.
5.2.1 The Influenza A viral proteins travel a long distance from the nucleus of origin, whereas the viral mRNA does not
We infected myofibers at various Influenza A virus dilutions and used
immunofluorescence staining to localize viral proteins – nucleoprotein (NP) and
hemagglutinin (HA) / neuraminidase (NA) proteins – after a 24 h infection period.
With a virus concentration of 1.3 × 107 pfu/ml, we found that all myofibers
contained the viral proteins in question and that the proteins occupied the entire
length of the myofibers. A 1.3 × 106 pfu/ml virus concentration produced
myofibers with locally infected segments and myofibers infected from end to end,
while at a dose of 1.3 × 105 pfu/ml, there were only few locally infected
myofibers. The propagation of the infection was analyzed more closely using the
virus concentration of 1.3 × 106 pfu/ml. In these experiments NP and HA/NA
proteins were undetectable after a 12 h infection period, whereas after a 16 h
infection period they were detected. At this time point, NP staining was found in
several closely spaced nuclei within segments of the myofibers. The intensity of
the staining gradually diminished with growing distance from the nuclei of
highest intensity. The viral proteins were found to be expressed in less than half of
the myofibers, and the length of the segments, where NP containing nuclei were
observed, ranged from 100 µm to 400 µm. After a 24 h infection period, the viral
proteins in several myofibers were spread over the entire length of the cell.

54
Moreover, the fraction of myofibers exhibiting infection was significantly greater
than after a 16 h infection period.
Having shown that an influenza virus at a low dose is able to infect a single
nucleus in an isolated myofiber and that the infection does not propagate into the
other nuclei in that myofiber, we utilized the low-dose infection conditions and
marked the infected nuclei in the myofibers by in situ hybridization using a probe
specific for the viral RNA encoding the HA protein. An in situ hybridization
probe specifically detecting mRNA encoding HA was simultaneously applied. It
was observed that the mRNA was mainly located around the infected nucleus and
exhibited longitudinal stripes that did not reach far. These stripes most likely
represented mRNA associated with microtubules. Immunofluorescence staining
for the HA protein at that stage of infection instead showed a distribution pattern
extending about 200 µm into both longitudinal directions reflecting the fact that
proteins moved effectively within the myofibers.
5.2.2 Rapid transverse movement of cytoplasmic and membrane proteins occurs in myofibers
In mononucleated cells GFP-Dad1 has been reported to be almost fully mobile,
mobile fraction being 88% (Nikonov et al. 2002). In CHO cells, the plain GFP
within the ER was measured to be fully mobile, the fluorescence recovering
completely within 50 seconds (Dayel et al. 1999). Here, we examined the
mobility of plain GFP, which is a cytoplasmic protein, and GFP-Dad1, which is
an ER-bound protein, in the voluminous myofibers. The expression of the
constructs was carried out by using an SFV vector for over-expression. Previously,
the mobile fraction of plain GFP was reported to be 97.0 ± 1.4% (n = 4) in
myofibers (Papponen et al. 2009). Similarly, we confirmed that the plain GFP
displayed rapid mobility through the cytoplasm in these vast cells. FRAP
experiments showed totally free mobility of the plain GFP, the fluorescence
recovering completely within minutes, implying unhindered movement of
cytoplasmic proteins within tightly packed myofibers.
Next we analyzed the lateral mobility of GFP-Dad1 within the ER/SR system.
In over-expression situation, the GFP-Dad1 yields a typical ER pattern in
myofibers, the fluorescence seen at the Z-discs in the middle of the I-bands and
perinuclearly. Accordingly, photobleaching experiments were carried out to study
the lateral mobility of the GFP-Dad1. Fragments of single Z-discs were
photobleached, and the recovery of fluorescence was observed to be rapid.

55
Similarly, multiple adjacent Z-discs were photobleached and the recovery of
fluorescence was monitored. It turned out that the most lateral Z-discs recovered
fluorescence most rapidly, suggesting that movement from one Z-disc to another
occurred.
Lastly, we investigated whether there is a diffusion barrier between the
perinuclear ER and the interfibrillar ER/SR. During the over-expression situation,
the FRAP experiments indicated free mobility within the perinuclear ER and,
furthermore, between the perinuclear and the interfibrillar ER. These experiments
were carried out by photobleaching parts of or the entire perinuclear ER, and
observing the recovery of the fluorescence. We found out that no barrier seemed
to exist. When in vivo electroporation was performed to gain access to a more
physiological situation, where the expression level of GFP-Dad1 is considerably
lower, we noticed that the distribution of the GFP-Dad1 was more dot-like and
restricted to the vicinity of the myonuclei. Furthermore, the mobility of the
chimeric protein was observed to be considerably lower than in the viral
expression experiments. Accordingly, the most reasonable explanation is that the
low amount of the GFP-Dad1 was bound to the OST complex (Nikonov et al. 2002), and therefore the mobility of the protein was restricted within the ER/SR
system.
All in all, our results indicate that cytoplasmic and membrane associated
model proteins GFP and GFP-Dad1 show rapid movement in transverse direction
thus facilitating the trafficking of the proteins from the site of the synthesis to the
interior parts of the cylindrical vast myofibers.
5.2.3 Cellular subcompartments, where mobility is restricted, exist in myofibers
The tsG-GFP is retained in the ER at the non-permissive temperature due to a
folding defect (Hammond & Helenius 1994), and therefore it ought to be bound to
the chaperones. However, tsG-GFP has been shown to be fully mobile in
mononucleated cells (Nehls et al. 2000). The observation that in the myofibers the
protein does not fully co-localize with protein disulfide isomerise (PDI), suggests
that it resides in an ER subcompartment, where its mobility is restricted. In this
study, we investigated whether the mobility of the chimeric protein was restricted
at the restrictive temperature. FRAP experiments revealed that at 39 °C the tsG-
GFP protein exhibited a 24.9 ± 8.1% (n = 5) mobile fraction. This is significantly
lower than what we observed in mononucleated BHK cells – mobile fraction

56
being 62.0 ± 10.0% (n = 9) – with the same construct. To measure the mobility at
20 °C, the myofibers were incubated in the presence of Brefeldin A (10 µm/ml) to
keep the protein in the ER after the incubation at 39 °C. In these conditions the
tsG-GFP presented a 27.3 ± 6.9% (n = 5) mobile fraction. To find out whether this
was due to the high viscosity of the ER/SR environment, GFP-Dad1 was chosen
as a reference protein since it has been reported that over-expression of the GFP-
Dad1 results in a pool that is not associated with OST complex and is freely
mobile (Nikonov et al. 2002). Accordingly, we expressed GFP-Dad1 from the
recSFV that resulted in over-expression and observed that GFP-Dad1 displayed a
76.0 ± 8.8% (n = 5) mobile fraction. These findings are in accordance with the
idea suggested by the morphological data that tsG-GFP resides in an ER
subcompartment in myofibers.
5.3 Exocytic membrane trafficking in myofibers (III)
In differentiated myofibers the exocytic trafficking is totally reorganized when
compared to mononucleated cells. Owing to the fact that myofibers are colossal
cells with multiple nuclei, the membrane trafficking has to be arranged effectively.
In this study we found that ER-to-Golgi trafficking greatly differs from that of
most mammalian cells, while post-Golgi trafficking was observed to be similar to
that found in other mammalian cells.
5.3.1 Adult myofibers lack ER-to-Golgi transport carriers
It has been shown that a considerable fraction of tsG-GFP acquires
Endoglycosidase H resistance in myoblasts, myotubes and myofibers when
expressed from the recSFV vector (Kaisto & Metsikko 2003), indicating passage
through the Golgi apparatus. Here we examined the membrane trafficking of tsG-
GFP. After a 16 h (L6 cells) or a 24 h (myofibers) infection, the cells were
fluorescent, and the fluorescence intensity varied between cells in all
differentiation stages which were analyzed. Pulse-labeling of the cells with
[35S]methionine followed by immunoprecipitation, SDS/PAGE and fluorography
verified that a major band of approximately 93 kDa was synthesized at 39 °C,
corresponding to the assumed molecular weight of the myc-tagged tsG-GFP
chimera (Dalton & Rose 2001). Only minor additional bands were seen,
especially in myotubes. When tsG-GFP expressing cells were fixed after
incubation at 39 °C and subsequently immunostained for an ER marker protein –

57
PDI – considerable co-localization was observed in the L6 cells. In myofibers, the
GFP fluorescence was more restricted over the Z-discs than PDI (Kaisto &
Metsikko 2003), indicating that tsG-GFP resided in a subcompartment of the ER
in myofibers. Shifting the temperature from 39 °C to 20 °C, at which temperature
exported proteins should accumulate in the TGN (Matlin & Simons 1983),
resulted in intense fluorescent spots appearing in myofibers. Immunostaining with
the Golgi marker GM130 verified that these spots represented Golgi elements.
These structures were also seen when stained with antibodies against the G
protein, indicating that the fluorescence seen in the Golgi elements truly
represented the tsG-GFP.
Having shown that intact tsG-GFP was produced in the muscle cells and its
temperature dependence was as expected, we monitored the ER-to-Golgi
trafficking in live cells by time-lapse imaging after releasing the 39 ˚C
temperature block. In the L6 myoblasts and myotubes, many moving carriers
were seen when shifting the temperature to either 32 ˚C or 20 ˚C. The carriers
were usually tubular in shape. In the myoblasts, the carriers often moved towards
the Golgi apparatus. However, contrary to the situation in fibroblasts (Presley et al. 1997), we frequently observed randomly moving carriers. Especially in the
myotubes, the carrier movement was random, apparently since the Golgi
apparatus is already dispersed throughout the cytoplasm at this stage of
differentiation. In both myoblasts and myotubes, the carriers swiftly moved long
distances (>10 µm) along straight paths in a stop-and-go fashion, suggesting
microtubular involvement. The carriers commonly merged with the bright spots
shown to be Golgi elements. The average speed of the carriers in the L6
myoblasts was 429 ± 204 nm/s (n = 15) and in the L6 myotubes 279 ± 188 nm/s
(n = 15) (p = 0.046). Primary cultured rat myotubes also showed moving carriers
behaving in a similar way as those seen in the L6 myotubes.
Next we infected freshly isolated myofibers with the recSFV-tsG-GFP at
39˚C. When analyzing the fluorescent myofibers after releasing the 39˚C block,
we observed few moving fluorescent carriers. We did occasionally see small
slowly moving fluorescent spots, but they travelled very short distances (< 1 µm)
in irregular directions. Such spots were seen in only 20% of the 25 investigated
myofibers. Owing to the fact that spots did not collide with Golgi elements, we
concluded that these carriers were irrelevant in ER-to-Golgi trafficking. That the
ER-to-Golgi carriers were absent was demonstrated by photobleaching Golgi
elements soon after the temperature shift from 39˚C to 20˚C. The photobleached
Golgi elements were seen to recover fluorescence rapidly. Nevertheless, no

58
carriers merging with the Golgi elements were seen. Double staining with the
GM130 was used to verify that the spots represented Golgi elements. To exclude
the possibility that very rapid carrier movement occurred, we observed the cells at
500 ms intervals, but yet again, no carriers were seen. In these experiments the
confocal plane was adjusted just beneath the sarcolemma, since in the fast-twitch
FDB myofibers the majority of the Golgi elements – the expected targets of the
postulated carriers – resided at this plane (Ralston et al. 2001). Additionally, most
of the microtubules, serving as tracks for the vesicles in other cell types, have
been shown to locate just beneath the sarcolemma (Rahkila et al. 1997). Notably,
all the GM130 positive elements received tsG-GFP. Interestingly, during the
follow-up period, the Golgi elements showed no movement, thus being stationary
structures.
5.3.2 Post-Golgi vesicles are present in skeletal muscle cells
Post-Golgi trafficking was analyzed in recSFV-tsG-GFP infected myoblast,
myotubes and myofibers. Following the growth period at 39 ˚C, the cells were
incubated for 2 h at 20˚C and subsequently shifted to the temperature of 32˚C.
Accordingly, in the L6 myoblast and L6 myotubes as well as in primary myotubes,
vast amount of migrating tubular and vesicular carriers were observed. When
analyzing myofibers, we observed tubular and vesicular structures regularly
budding from the Golgi elements and moving around. Such carriers were,
nevertheless, considerably less frequent than in the myotubes and they travelled
clearly shorter distances (< 5 µm). Their average speed (164 ± 96 nm/s; n = 52)
was lower than in L6 myoblasts (266 ± 89 nm/s; n = 15) or in L6 myotubes
(468 ± 241 nm/s; n = 15) (p < 0.001). Interestingly, in COS cells, the speed of
post-Golgi carriers were up to 2.7 µm/s in the temperature of 32 °C (Hirschberg et al. 1998). Importantly, a remarkable fraction of the carriers were seen to bud from
the Golgi elements and, moreover, many of them were seen to disappear abruptly,
implying fusion with the PM. The fraction of fluorescence arriving to the
sarcolemma was quite low. However, since the intrinsic GFP fluorescence
intensity at the sarcolemma remained relative low and, furthermore, the intensity
of the fluorescence in the Golgi elements was not measurably reduced, we
concluded that tsG-GFP was not efficiently transported from the Golgi elements
to the PM.

59
5.4 LB formation in myofibers is inducible (IV)
Previously it has been shown that human type I myofibers contain considerable
more LBs than type II (Malenfant et al. 2001, Shaw et al. 2009). In addition to
human muscle, it has been shown that the rat soleus muscle type I myofibers
contain abundant LBs (Prats et al. 2006). In the present study we examined the
situation in the rat FDB myofibers, which are fast-twitch oxidative glycolytic type
IIa myofibers. When analyzing frozen sections from FDB muscle, we found that
only small fraction – below 1% of the three sections analyzed – of the myofibers
exhibited LBs when stained with the LD540 dye. To obtain a more
comprehensive view over the whole muscle cells, we analyzed isolated myofibers.
Surprisingly, it was found that the isolation procedure transiently induced massive
formation of LBs. Staining with the LD540 dye showed dots in a vast majority of
the myofibers at 4 h after the isolation while the rest of the myofibers did not
contain any dots. The amount of LD540 positive myofibers steadily decreased
upon time and leveled at approximately 2% after a 3 day cultivation period. At
this time point most of the myofibers showed no dots indicating LBs but just a
faint SR resembling staining pattern.
Next we observed the myofibers more specifically at 24 h from the isolation.
The size and number of the LBs varied remarkably between myofibers and also
within a single myofiber, their diameter being 753 ± 207 nm (n = 18).
Interestingly, the majority of the LB positive myofibers showed LBs within short
segments in the myofibers, apparently reflecting the fact that the high number of
LBs induced by the isolation procedure was gradually diminishing over the time
in the cell culture. However, the LBs were seen along the entire length of the
myofibers, too. Similarly, antibodies against the LB coat protein ADRP also
showed dots or ring-like structures in the LD540 positive myofibers. It is notable
that the LBs in FDB myofibers consistently flanked or overlaid the Z-discs. This
result is compatible with earlier EM findings (Meex et al. 2009), but differs from
the situation in the type I soleus muscle, where the LBs steadily distributed within
the myofibers (Prats et al. 2006).
5.4.1 LBs are immobile structures without a connection to the ER
In mononucleated cells LBs have been reported to exhibit microtubule dependent
movement (Pol et al. 2004). In this study we examined whether the LBs in
myofibers were equally mobile. Consequently, live FDB myofibers were stained

60
with the LD540 dye to visualize the LBs. Intriguingly, a time lapse series
indicated no movement whatsoever. Since the LBs were immobile and flanking
the Z-discs, it was possible that they were still connected to the ER/SR, where
they are postulated to be generated (Beller et al. 2010). To further analyze the
possible continuum between the LBs and the ER/SR, we performed FRAP
analyses. Because of LD540 dye’s possible diffusion from one LB to another, it is
not a suitable marker for photobleaching experiments. We therefore utilized
CAV3-YFP for this purpose since it was observed to accumulate in the LBs upon
expression from the recSFV vector. The FRAP experiments indicated that after
photobleaching the fluorescence did not return to the LBs during a 20 min follow-
up time period. Experiments with CAV3-YFP in live cells also verified the result
obtained with the LD540 dye, namely that the LBs were totally immobile
structures in the myofibers. However, the faint fluorescence over the Z-discs
returned within a few minutes reflecting the fact that within the ER/SR system
CAV3-YFP was clearly mobile. Accordingly, we concluded that the LB
membrane monolayer was not continuous with the ER/SR in the myofibers.
5.4.2 ER stress induces LBs accumulation in myofibers
After noticing that the isolation procedure induced LB formation in the FBD
myofibers, we examined the effect of other stress factors and found that elevated
temperature also induced LB formation. Consequently, myofibers were incubated
for 16 h at 41 °C, which resulted in the appearance of small LBs (diameter < 200
nm) in most of the myofibers. After a 24 h incubation at 41 °C the LBs were more
abundant and greater in size (mean diameter 400 nm) and they were found in
54.5 ± 10.8% (n = 4 dishes) of the myofibers. It is intriguing that the LBs steadily
distributed throughout the cells while the rest of the myofibers contained no LBs.
Next we examined whether viral infection could induce LB formation. Thus
myofibers were infected with SFV. It was discovered that after an 8 h infection
period there was no increase of LBs observed when compared to a non-infected
control dish. However, the infection induced considerable LB formation after a 20
h infection period. At this time point a 60.0 ± 17.4% (n = 2) fraction of the
myofibers exhibited abundant LBs that often distributed throughout the myofibers,
showing a mean diameter of 600 nm. The remaining 40% of the myofibers
contained no LBs although immunofluorescence staining with the virus specific
antibodies showed infection in the cells. Infection of myofibers with VSV also

61
increased the fraction of LB containing myofibers, which was 34.0 ± 9.9% (n = 2)
after a 20 h infection period.
Since SFV infection is known to cause unfolded protein response of the ER
(Barry et al. 2010), we next investigated the effect of tunicamycin that typically
induces ER stress via inhibition of glycosylation. In accordance, isolated
myofibers were subjected to tunicamycin treatment for 20 h. At 2 µg/ml
concentration the drug resulted in LBs being observed in a 25.9 ± 2.4% (n = 4)
fraction of the myofibers and in 48.6 ± 5.2% (n = 4) fraction at 10 µg/ml
concentration, implying that LBs were indeed synthesized under ER stress. The
heat induced and tunicamycin induced LB formation was reversible.
Consequently, after incubation at 41 °C for 24 h and then at 37 °C for additional
24 h, we found that the fraction of LB containing myofibers reduced to
15.3 ± 9.0% (n = 2). Similarly, after a 20 h treatment at 10 µg/ml concentration of
tunicamycin the fraction of LB containing myofiber was reduced to 9.3 ± 3.2
(n = 4) within 24 h after removal of the drug.
To verify that the tunicamycin concentrations used and the viral infection or
heat treatment really inflicted ER stress, we determined whether ER chaperone
levels were elevated. For this purpose we performed Western blotting with
antibodies against CLN and PDI. It was observed that CLN and PDI levels rose
only marginally at the 2 µg/ml concentration whereas the 10 µg/ml concentration
as well as the viral infection and the 41 °C temperature markedly elevated the
chaperone levels. Immunofluorescence staining after tunicamycin treatment (10
µg/ml) revealed no differences in CLN levels between myofibers containing LBs
and those devoid of them.
Interestingly, ER stress has been demonstrated to affect the biosynthesis of
cholesterol and triglycerides in the liver and Hela cell via activation of sterol
regulatory element binding protein (SREBP) (Colgan et al. 2007). This protein is
bound to the ER and its activation results in the release of the N-terminal portion,
which is a transcription factor and moves into the nucleus. We did not observe
significant activation-cleavage of SREBP1 upon Western blotting analysis, but
immunohistochemical staining with antibodies against the N-terminal portion of
SREBP consistently indicated shifting into the nuclei after treatment with
tunicamycin. This shift was seen in approximately 5–40% of the myofibers,
depending on the experiment, but was never seen in the control myofibers not
treated with the drug.
Since CAV3 has been shown to accumulate into LBs during overexpression
conditions (Pol et al. 2005), we expressed CAV3-YFP in isolated myofibers by

62
using the recSFV. It was observed that after a 24 h infection about half of the
myofibers exhibited YFP fluorescence indicating infection, and in about 80% of
the infected cells, CAV-YFP accumulated into ring-like structures typical of LBs.
Since the FDB muscle usually contains approximately 10% of LB positive
myofibers at this time point from the isolation, we conclude that like the parent
SFV also the recSFV infection induced LB formation. Compatible with these
findings, the CAV3-YFP ring structures showed a 1:1 double staining with the
LD540 dye. These findings propose that the FDB myofibers have the capability to
create LBs upon stress conditions and especially during ER stress.

63
6 Discussion
6.1 mRNA localization differs from the site of translation in
myofibers
Previous studies have shown that gene products of the nuclei beneath the NMJ do
not reach far (Jasmin et al. 1993, Moscoso et al. 1995). We studied here whether
the gene products of the non-junctional nuclei behaved similarly. We used an
influenza virus as a model in the FDB myofibers and were able to show that
mRNA products from a single infected nucleus did not reach far. Importantly, the
mRNA distribution occupied a region beneath the sarcolemma and did not
penetrate into the core regions of the myofibers. Contrary to the behavior of the
influenza virus mRNAs, the corresponding viral proteins reached hundreds of
micrometers away from the nucleus of origin.
Compatible with the behavior of the influenza virus gene products, several
previous studies have shown a subsarcolemmal distribution pattern for selected
mRNAs such as myosin, dystrophin, or CLQ. Nevertheless, some other studies
have reported that myosin mRNA (Billeter et al. 1992, Dix & Eisenberg 1991)
and also myoglobin mRNA (Mitsui et al. 1997) show an even distribution pattern
in cross-sections of myofibers. Therefore, there has not been a clear consensus on
the localization of the mRNAs encoding myofilament components. Here we
localized the entire mRNA pool in the myofibers by detecting their poly-A tails.
This probe located in the vicinity of the myonuclei and beneath the sarcolemma,
however, an interfibrillar signal could not be excluded. Similar results were
obtained when BrU incorporation into the synthesized mRNAs was studied.
Accordingly, BrU tagged mRNA was exported from the nuclei and occupied the
subsarcolemmal regions. Our results suggest that mRNAs spread into the
subsarcolemmal space and adopt a cross-striated distribution pattern over the A-I
junction regions. In conclusion, the in situ hybridization experiments could not
unambiguously resolve whether or not there was mRNA present in the interior
parts of the myofibers, leaving the question unanswered where translation
occurred in the myofibers.
To locate the site of translation in the myofibers we used puromycylation of
nascent polypeptide chains that were immobilized to ribosomes. Our results on
the location of translation show that, in addition to the perinuclear components,
there were mRNA species as well as ribosomes present in the interior regions of

64
the myofibers. Accordingly, the puromycylation experiments showed that
translation was not restricted to around the myonuclei and beneath the
sarcolemma to the extent as the total mRNA pool appeared to be. The ribosomal
antibody staining also supported this idea. Furthermore, our results argue that
there are small amounts of mRNAs present in the core regions and this fraction is
translating. A substantial amount of the individual transcripts that have been
localized subsarcolemmally encode ER targeted membrane proteins. The very
different distribution of puromycylated polypeptide chains when compared to that
of the total mRNA pool suggests that different mRNAs are localized differently in
the myofibers. Another possibility is that there is a subsarcolemmal pool of
mRNA that is translationally inactive. This pool may be comparable to the P-
bodies or stress granules that have been found in mononucleated cells and among
other things function as mRNA pools for later use (Balagopal & Parker 2009,
Eulalio et al. 2007). The discovery that after puromycylation fluorescent spots
appeared within the myofiber and that they were flanking the Golgi elements
elements suggests that they were sites of protein translocation into the ER. If so,
translocation in the myofibers is occurring in the vicinity of the Golgi elements
thus resembling the distribution of the ERES in the myofibers (Ralston et al. 2001). Concerning ER targeted proteins and their transcripts an essential feature
is that the SR is smooth-surfaced, which suggests a lack of ribosomes. Despite
this the SR contains chaperones, ribophorins, and translocons. Importantly, during
strenuous exercise myofibers are subject to severe ER stress (Kim et al. 2011)
leading to the unfolding of proteins implying that ER chaperones are needed to
compensate the stress and translocons may be needed to retrotranslocate proteins
that have not managed to refold.
In the light of our findings it remains possible that different mRNAs are
localized differently in FDB myofibers. The mRNAs in question encode proteins
that are involved in cell differentiation and therefore the translation must be
limited to a certain time and place in the cell (Shahbabian & Chartrand 2012). It
has been speculated that there is spatiofunctional association with the
sarcolemmal proteins and the subsarcolemmal transcripts (Luck et al. 1998,
Mitsui et al. 1997), but ultimately this seems unlikely. Muscle cells are terminally
differentiated, so there is little need for temporal or spatial regulation of
translation but owing to their large size some kind of mRNA targeting may be
required. It remains an enigma why mRNAs are mostly restricted beneath the
sarcolemma although it seems that proteins readily move into the core regions.
One reason might be the size of mRNAs possibly restricts their movement into

65
the interfibrillar space. Another possibility is that mRNA retention mechanisms
might exist in the subsarcolemmal space.
6.2 Protein mobility and its efficacy in vast myofibers
Proteins that are not attached to existing structures should freely move in the
cytoplasm and in the ER/SR system. Plain GFP and GFP-Dad1 represents such a
protein when over-expressed (Nikonov et al. 2002, Papponen et al. 2009). Having
shown that translation is concentrated around the myonuclei and in interfibrillar
dots, and that proteins readily move within the core regions of the myofiber, we
analyzed the mobility of two model proteins – one cytosolic, plain GFP, and one
membrane bound, GFP-Dad1 – within myofibers. Free mobility was enabled
using an overexpression situation through SFV infection. Then, FRAP
experiments were carried out, and as expected, mobility was found to be rapid in
cytosol, as well as within the ER/SR system. The GFP-Dad1 resides in a
compartment over the Z-discs, and therefore, an area of several adjacent Z-discs
comprising a confocal section was photobleached to examine whether the probe
moved back to the bleached section in longitudinal or transverse direction. We
found that the Z-discs in the middle of the bleached area did not recover
fluorescence as rapidly as those at the edges. Accordingly, slow movement in
longitudinal direction seemed to occur. Nonetheless, we concluded that new GFP-
Dad1 molecules arrived mainly to the bleached area in transverse direction. Next
we conducted FRAP experiments with perinuclear ER. Interestingly, no diffusion
barrier existed between the perinuclear ER and the interfibrillar ER, suggesting
free movement of membrane proteins within the whole ER/SR system in FDB
myofibers. In conclusion, these findings indicate that the tightly packed
architecture of the vast myofibers do not hinder the mobility of the marker
proteins.
The discovery that in myofibers the tsG-GFP exhibits a low mobile fraction
of 24.9% at 39 ºC indicates that the protein was not bound completely to the ER.
However, this value is substantially lower than the 62% mobile fraction we
observed in the BHK cells or the 100% mobile fraction reported in COS cells
(Nehls et al. 2000), although tsG-GFP was observed in higher temperature. In
myofibers – in contrast to mononucleated cells – the protein may be partially
aggregated or bound to immobile structures, but it may be segregated into an ER
subcompartment, too. The latter hypothesis is supported by the observation that
tsG-GFP at 39ºC is concentrated at the Z-discs despite the fact that chaperones

66
distribute over the entire I-bands areas (Kaakinen et al. 2008). That ER/SR
system is a continuous network enabling lateral movement of proteins is
supported by the finding that GFP-Dad1 exhibited a 76% mobile fraction in
myofibers. Compatible with this, an 88.8% mobile fraction was reported in
differentiated myotubes with GFP-tagged calcium pump of the SR (Cusimano et al. 2009). Also, when GFP-Dad1 was expressed through electroporation,
substantial decrease of mobility was observed. Plausibly, the chimeric protein was
bound to OST complex thus hindering the mobility.
6.3 Regression of Golgi associated trafficking in myofibers
In this study we observed that in L6 myoblasts and in partially differentiated
myotubes the ER-to-Golgi trafficking of tsG-GFP was similar to the trafficking
reported in several mononucleated cell types (Hirschberg et al. 1998, Presley et al. 1997, Scales et al. 1997). In these cells, ER-to-Golgi carriers were distinctively
detected and they presumably moved along microtubule tracks and merged with
Golgi elements. Interestingly, in the myotubes, there were carriers migrating back
and forth without a well-defined destination, reflecting the fact that there are no
microtubule organization centers in these multinucleated cells and hence, the
polarity of microtubules may be random. In mice myotubes, microtubules are
shown be arranged into longitudinal antiparallel arrays (Pizon et al. 2005), and
apparently this is the case in rat myotubes, too. In contrast, in mature FDB
myofibers, similar trafficking of ER-to-Golgi carriers was not seen although the
fluorescent probe shifted from the ER to the Golgi elements. Previously it has
been shown that a major fraction of the tsG-GFP – up to 61–82% – arrives to the
Golgi apparatus in all differentiation stages of muscle cells (Kaisto & Metsikko
2003). Since membrane carriers moving along microtubule tracks were not
observed in myofibers, although these cells possess a microtubule network
(Boudriau et al. 1993) and the Golgi elements are associated with the
microtubules (Rahkila et al. 1997), we infer that microtubules play no role in ER-
to-Golgi membrane trafficking in the myofibers.
The ERES in mononucleated cells are distributed evenly as punctuate
structures within the ER (Gorelick & Shugrue 2001), and the exported proteins
and lipids travel in vesicular containers along microtubules to the centrally
located Golgi apparatus. In myofibers, however, the Golgi apparatus is dispersed
into hundreds of single elements and the ERES are flanking the Golgi elements
both in myotubes (Lu et al. 2001) and in myofibers (Ralston et al. 2001). This

67
kind of Golgi organization is similar to that of nocodazole treated mononucleated
cells, where the Golgi apparatus is dispersed into elements flanking ERES (Ward et al. 2001). The situation in myofibers closely resembles that in Drosophila cells,
where the Golgi elements are usually dispersed in the cytoplasm and associated
with ERES (Kondylis & Rabouille 2009). Similar dispersion of Golgi elements
and ERES is also found in plant cells (daSilva et al. 2004, Hanton et al. 2009) or
in yeast, although in these cells the Golgi cisternae are not usually stacked (Levi et al. 2010). During the myogenic differentiation the organization of the Golgi
apparatus and the early exocytic pathway returns to an archetypal mode. In plant
cells the actin filament associated with Golgi elements are highly mobile, whereas
in muscle cells these structures are stationary, most likely due to microtubular
association (Rahkila et al. 1998). Therefore, the mechanism of the ER-to-Golgi
traffic differs from that of the plant cells. In the myotubes, vesicular trafficking
was observed although the ERES and the Golgi elements have been reported to
lie next to each other in these cells (Lu et al. 2001). The situation in myotubes
bears a resemblance to the trafficking in differentiated neurons in which highly
mobile carriers fuse to multiple Golgi outposts dispersed in the dendrites (Horton
& Ehlers 2003). One explanation is that ER-to-Golgi trafficking was only
partially rearranged in the myotubes and these cells partially retained the mode of
trafficking typical to mononucleated cells.
It makes sense that in the voluminous myofibers the ERES and the Golgi
elements localize right next to each other thus facilitating the membrane
trafficking. Moreover, it is reasonable that within these short distances no
vesicular or tubular carriers are needed. However, when it comes to the post-
Golgi trafficking of the muscle cells, we did not observe such profound difference
as with the ER-to-Golgi trafficking. Interestingly, the frequency of carrier
movement was quite low in the myofibers when compared to L6 cells. Our
finding here that no clear sarcolemmal fluorescence pattern was seen suggests
that tsG-GFP was not efficiently transported from the Golgi elements to the PM.
Prior studies with myotubes and myofibers show that the plain G-protein is
mostly retained in a GLUT4-containing post-Golgi compartment, while only a
small fraction of the protein arrives to the PM (Rahkila et al. 1998). Therefore a
plausible explanation for our findings here with tsG-GFP is that it behaved
similarly. We also found short-distance moving small post-Golgi vesicles, but it
remains indefinite whether we were following the GLUT4 route or another post-
Golgi route existing in the adult myofibers.

68
Interestingly, the regressed Golgi associated trafficking in myofibers may
have some significant implications. Just recently, a Japanese research group found
that disturbances within ER-to-Golgi trafficking in myofibers may lie behind the
myotoxicity of statins (HMG-CoA reductase inhibitors) (Sakamoto et al. 2011).
Sakamoto et al. (2011) found – with rat FBD myofibers – that fluvastatin
inhibited Rab1A, which is essential for ER-to-Golgi targeting, translocation from
cytosol to the ER. It was concluded that the suppression of Rab1 GTPase and the
following inhibition of ER-to-Golgi trafficking are involved in statin-induced
myotoxicity. The importance of this arises from the fact that previously no
consistent data existed on the myotoxicity of statins. Importantly, statins are used
world-wide by millions of people, and mild muscle pains affect 2–11% of the
users (Silva et al. 2007). Consequently, it is intriguing to speculate whether the
archetypal regression of ER-to-Golgi trafficking in myofibers plays an essential
role in explaining why skeletal muscle in particular is adversely affected by the
statin therapy.
6.4 ER stress, LBs and insulin resistance in myofibers
Formerly it has been shown that fast-twitch myofibers contain few LBs (Shaw et al. 2009). Here we have verified these results by showing that most of the FBD
myofibers do not contain any LBs. Interestingly, we found that the formation of
LBs is inducible in the FBD myofibers. Previously it has been reported that
exercise training causes an increase of intramyocellular lipid content in sedentary
muscles although it decreases it in exercising muscles (Goodpaster et al. 2001,
Schrauwen-Hinderling et al. 2003). This form of increase is possibly due to
elevated fatty acid levels in the serum, since muscle is also capable of storing
excessive lipids. Physical activity has been reported to decrease the LB size (He et al. 2004) and, moreover, in slow-twitch soleus myofibers the number of LBs
reduces upon electrical stimulation or epinephrine treatment (Prats et al. 2006).
One plausible explanation of this phenomenon could be ER stress, which has
been shown to promote lipolysis in adipocytes (Deng et al. 2012). Intriguingly, in
skeletal muscle ER stress can be stimulated by extreme endurance exercise (Kim et al. 2011). In this study we showed that the isolation procedure, heat shock,
viral infection, or tunicamycin treatment led to enormous accumulation of LBs in
cultured FBD myofibers. An ample amount of data shows that heat shock or
infection with certain enveloped viruses or treatment with drugs such as
tunicamycin induce ER stress – meaning accumulation of unfolded and misfolded

69
proteins in the ER lumen – resulting in unfolded protein response (Cao &
Kaufman 2012, Schroder & Kaufman 2005). Our data is compatible with those
obtained with yeast cells, in which tunicamycin or Brefeldin A which typically
induce ER stress, stimulated LB formation (Fei et al. 2009). Ultimately, it remains
somewhat obscure whether the isolation procedure caused ER stress that led to
LB formation. It is however notable that integrins mediate shear-stress activation
of SREBP1 in endothelial cells (Liu et al. 2002), and therefore the possibility of
integrin mediated SREBP activation exists in the LB formation during the
myofiber isolation procedure.
In mononucleated cells, ER stress affects the biosynthesis of fats in liver and
HeLa cells through activation of SREBP (Colgan et al. 2007, Werstuck et al. 2001). The SREBP1 can be found in skeletal muscle cells (Commerford et al. 2004), and especially the mRNA levels in the fast-twitch myofibers are
comparable to those of liver cells (Guillet-Deniau et al. 2002). Forced expression
of SREBP1 was also shown to stimulate the expression of lipogenic genes in
muscle cells. In this study we did not observe efficient cleavage of SREBP during
the formation of LBs. However, the protein was seen to shift into the nuclei under
ER stress. Although other mechanisms cannot be excluded, there is a possibility
of a minor activation level of SREBP in the voluminous ER/SR system of the
FDB myofibers leading to increased lipid synthesis and LB accumulation.
Interestingly, the activation of SREBP leads to increased synthesis of saturated
fatty acids that in turn are known for inducing ER stress in various cell types
(Guo et al. 2007, Samuel & Shulman 2012). Furthermore, using cultured cells
from obese mice, it has been shown that lipid accumulation induces ER stress
(Ozcan et al. 2004). An intriguing possibility is that lipid accumulation induces
ER stress leading to further lipid accumulation, thus creating a vicious circle in
the myofibers. Interestingly, we found that the conditions of ER stress induced
either enormous accumulation of LBs in the myofibers or none at all, hereby
being an on-off phenomenon. Differences of myofiber type do not explain this
observation, since over 90% of the FBD myofibers represent type IIa (Carlsen et al. 1985). Our findings here suggest a threshold level of ER stress which was
reached in some of the myofibers to trigger the circle.
It is a fascinating possibility that ER stress affiliated with LB accumulation in
muscle cells contributes to the development of type II diabetes. Even 15 years ago,
it was reported that the amount of LBs in muscle is linked with the insulin
resistance (Pan et al. 1997). However, nowadays the insulin resistance is
considered to be most likely a multietiological syndrome (Samuel & Shulman

70
2012), clearly tied closely together by disturbances in lipid metabolism in muscle
tissue. Therefore, it is reasonable to speculate whether the peculiarities of LB
dynamics in myofibers play some role in this complex process.

71
7 Conclusions
This study showed that a major fraction of various mRNAs remains in the vicinity
of their nuclei of origin subsarcolemmally, while protein products readily move
long distances within the myofibers in spite of the tightly packed contractile
apparatus. Surprisingly, the site of translation differed from the distribution of
mRNA, localizing in the vicinity of myonuclei but also to the core regions of the
myofibers. No diffusion barrier for proteins seemed to exist within the different
segments of the ER/SR system in myofibers. Exocytic trafficking from the ER to
Golgi in muscle cells totally differed from that found in mononucleated
mammalian cells. Accordingly, ERES in muscle cells lie next to the Golgi
elements and trafficking along microtubules typical of mononucleated cells does
not occur. Consequently, during myogenesis archetypal regression of the ER-to-
Golgi trafficking occurs. The trafficking from Golgi elements to sarcolemma
instead utilized microtubules. Interestingly, the ER was able to generate LBs
under stress conditions although fast-twitch myofibers normally do not contain
these structures. The LB formation seemed to be an on-off phenomenon,
apparently because LBs enhance ER stress and vice versa, creating a vicious
circle. Finally, LBs were found to be stationary structures in contrast to
observations in other cell types.
Ultimately, many basic clinical medical challenges are linked to myofibers.
For instance, type II diabetes affiliated with insulin resistance, and the
myotoxicity of the statins are dilemmas associated with the basic physiology and
cell biology of the myofibers. Since profound understanding of the very basics of
these colossal cells is still lacking, more cell biological research on myofibers is
needed in the future to resolve these problems.

72

73
References
Agarraberes FA & Dice JF (2001) Protein translocation across membranes. Biochim Biophys Acta 1513(1): 1–24.
Angers CG & Merz AJ (2011) New links between vesicle coats and Rab-mediated vesicle targeting. Semin Cell Dev Biol 22(1): 18–26.
Appenzeller-Herzog C & Hauri HP (2006) The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci 119(Pt 11): 2173–2183.
Armitage BA (2011) Imaging of RNA in live cells. Curr Opin Chem Biol 15(6): 806–812. Awad SS, Lightowlers RN, Young C, Chrzanowska-Lightowlers ZM, Lomo T & Slater
CR (2001) Sodium channel mRNAs at the neuromuscular junction: distinct patterns of accumulation and effects of muscle activity. J Neurosci 21(21): 8456–8463.
Balagopal V & Parker R (2009) Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Curr Opin Cell Biol 21(3): 403–408.
Bannykh SI, Rowe T & Balch WE (1996) The organization of endoplasmic reticulum export complexes. J Cell Biol 135(1): 19–35.
Bao G, Rhee WJ & Tsourkas A (2009) Fluorescent probes for live-cell RNA detection. Annu Rev Biomed Eng 11: 25–47.
Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M & Schekman R (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77(6): 895–907.
Barry G, Fragkoudis R, Ferguson MC, Lulla A, Merits A, Kohl A & Fazakerley JK (2010) Semliki forest virus-induced endoplasmic reticulum stress accelerates apoptotic death of mammalian cells. J Virol 84(14): 7369–7377.
Bassell GJ, Taneja KL, Kislauskis EH, Sundell CL, Powers CM, Ross A & Singer RH (1994) Actin filaments and the spatial positioning of mRNAS. Adv Exp Med Biol 358: 183–189.
Baumeister W, Walz J, Zuhl F & Seemuller E (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92(3): 367–380.
Beller M, Thiel K, Thul PJ & Jackle H (2010) Lipid droplets: a dynamic organelle moves into focus. FEBS Lett 584(11): 2176–2182.
Bentzinger CF, Wang YX & Rudnicki MA (2012) Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4(2): 10.1101/cshperspect.a008342.
Berthier C & Blaineau S (1997) Supramolecular organization of the subsarcolemmal cytoskeleton of adult skeletal muscle fibers. A review. Biol Cell 89(7): 413–434.
Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH & Long RM (1998) Localization of ASH1 mRNA particles in living yeast. Mol Cell 2(4): 437–445.
Bickel PE, Tansey JT & Welte MA (2009) PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta 1791(6): 419–440.
Billeter R, Messerli M, Wey E, Puntschart A, Jostarndt K, Eppenberger HM & Perriard JC (1992) Fast myosin light chain expression in chicken muscles studied by in situ hybridization. J Histochem Cytochem 40(10): 1547–1557.

74
Biressi S, Molinaro M & Cossu G (2007) Cellular heterogeneity during vertebrate skeletal muscle development. Dev Biol 308(2): 281–293.
Bonifacino JS & Lippincott-Schwartz J (2003) Coat proteins: shaping membrane transport. Nat Rev Mol Cell Biol 4(5): 409–414.
Bottinelli R & Reggiani C (2000) Human skeletal muscle fibres: molecular and functional diversity. Prog Biophys Mol Biol 73(2–4): 195–262.
Boudriau S, Vincent M, Cote CH & Rogers PA (1993) Cytoskeletal structure of skeletal muscle: identification of an intricate exosarcomeric microtubule lattice in slow- and fast-twitch muscle fibers. J Histochem Cytochem 41(7): 1013–1021.
Brooke MH & Kaiser KK (1970) Muscle fiber types: how many and what kind? Arch Neurol 23(4): 369–379.
Bullock SL (2011) Messengers, motors and mysteries: sorting of eukaryotic mRNAs by cytoskeletal transport. Biochem Soc Trans 39(5): 1161–1165.
Burgess TL & Kelly RB (1987) Constitutive and regulated secretion of proteins. Annu Rev Cell Biol 3: 243–293.
Cao SS & Kaufman RJ (2012) Unfolded protein response. Curr Biol 22(16): R622–6. Carlsen RC, Larson DB & Walsh DA (1985) A fast-twitch oxidative-glycolytic muscle
with a robust inward calcium current. Can J Physiol Pharmacol 63(8): 958–965. Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL & Summers SA
(2003) A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem 278(12): 10297–10303.
Chudakov DM, Matz MV, Lukyanov S & Lukyanov KA (2010) Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev 90(3): 1103–1163.
Cole NB & Lippincott-Schwartz J (1995) Organization of organelles and membrane traffic by microtubules. Curr Opin Cell Biol 7(1): 55–64.
Colgan SM, Tang D, Werstuck GH & Austin RC (2007) Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int J Biochem Cell Biol 39(10): 1843–1851.
Commerford SR, Peng L, Dube JJ & O'Doherty RM (2004) In vivo regulation of SREBP-1c in skeletal muscle: effects of nutritional status, glucose, insulin, and leptin. Am J Physiol Regul Integr Comp Physiol 287(1): R218–27.
Cowling VH (2009) Regulation of mRNA cap methylation. Biochem J 425(2): 295–302. Coxon FP & Taylor A (2008) Vesicular trafficking in osteoclasts. Semin Cell Dev Biol
19(5): 424–433. Cusimano V, Pampinella F, Giacomello E & Sorrentino V (2009) Assembly and dynamics
of proteins of the longitudinal and junctional sarcoplasmic reticulum in skeletal muscle cells. Proc Natl Acad Sci USA 106(12): 4695–4700.
Dalton KP & Rose JK (2001) Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279(2): 414–421.
daSilva LL, Snapp EL, Denecke J, Lippincott-Schwartz J, Hawes C & Brandizzi F (2004) Endoplasmic reticulum export sites and Golgi bodies behave as single mobile secretory units in plant cells. Plant Cell 16(7): 1753–1771.

75
Dayel MJ, Hom EF & Verkman AS (1999) Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys J 76(5): 2843–2851.
Deng J, Liu S, Zou L, Xu C, Geng B & Xu G (2012) Lipolysis response to endoplasmic reticulum stress in adipose cells. J Biol Chem 287(9): 6240–6249.
Digel M, Ehehalt R & Fullekrug J (2010) Lipid droplets lighting up: insights from live microscopy. FEBS Lett 584(11): 2168–2175.
Dirks RW, Molenaar C & Tanke HJ (2001) Methods for visualizing RNA processing and transport pathways in living cells. Histochem Cell Biol 115(1): 3–11.
Dix DJ & Eisenberg BR (1991) Distribution of myosin mRNA during development and regeneration of skeletal muscle fibers. Dev Biol 143(2): 422–426.
Dolken G, Leisner E & Pette D (1975) Immunofluorescent localization of glycogenolytic and glycolytic enzyme proteins and of malate dehydrogenase isozymes in cross-striated skeletal muscle and heart of the rabbit. Histochemistry 43(2): 113–121.
Donnelly CJ, Fainzilber M & Twiss JL (2010) Subcellular communication through RNA transport and localized protein synthesis. Traffic 11(12): 1498–1505.
Ellgaard L & Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4(3): 181–191.
Eulalio A, Behm-Ansmant I & Izaurralde E (2007) P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 8(1): 9–22.
Ezerman EB & Ishikawa H (1967) Differentiation of the sarcoplasmic reticulum and T system in developing chick skeletal muscle in vitro. J Cell Biol 35(2): 405–420.
Feder TJ, Brust-Mascher I, Slattery JP, Baird B & Webb WW (1996) Constrained diffusion or immobile fraction on cell surfaces: a new interpretation. Biophys J 70(6): 2767–2773.
Fei W, Wang H, Fu X, Bielby C & Yang H (2009) Conditions of endoplasmic reticulum stress stimulate lipid droplet formation in Saccharomyces cerevisiae. Biochem J 424(1): 61–67.
Flucher BE (1992) Structural analysis of muscle development: transverse tubules, sarcoplasmic reticulum, and the triad. Dev Biol 154(2): 245–260.
Forster R, Weiss M, Zimmermann T, Reynaud EG, Verissimo F, Stephens DJ & Pepperkok R (2006) Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr Biol 16(2): 173–179.
Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, Singer RH & Bertrand E (2003) Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr Biol 13(2): 161–167.
Garneau NL, Wilusz J & Wilusz CJ (2007) The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8(2): 113–126.
Gauthier GF & Mason-Savas A (1993) Ribosomes in the skeletal muscle filament lattice. Anat Rec 237(2): 149–156.
Gething MJ (1999) Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol 10(5): 465–472.
Glick BS (2000) Organization of the Golgi apparatus. Curr Opin Cell Biol 12(4): 450–456.

76
Glick BS & Luini A (2011) Models for Golgi traffic: a critical assessment. Cold Spring Harb Perspect Biol 3(11): a005215.
Goodpaster BH, He J, Watkins S & Kelley DE (2001) Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 86(12): 5755–5761.
Goodpaster BH, Theriault R, Watkins SC & Kelley DE (2000) Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism 49(4): 467–472.
Gorelick FS & Shugrue C (2001) Exiting the endoplasmic reticulum. Mol Cell Endocrinol 177(1–2): 13–18.
Guillet-Deniau I, Mieulet V, Le Lay S, Achouri Y, Carre D, Girard J, Foufelle F & Ferre P (2002) Sterol regulatory element binding protein-1c expression and action in rat muscles: insulin-like effects on the control of glycolytic and lipogenic enzymes and UCP3 gene expression. Diabetes 51(6): 1722–1728.
Guo W, Wong S, Xie W, Lei T & Luo Z (2007) Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am J Physiol Endocrinol Metab 293(2): E576–86.
Hammond C & Helenius A (1994) Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol 126(1): 41–52.
Hanton SL, Matheson LA, Chatre L & Brandizzi F (2009) Dynamic organization of COPII coat proteins at endoplasmic reticulum export sites in plant cells. Plant J 57(6): 963–974.
Hapala I, Marza E & Ferreira T (2011) Is fat so bad? Modulation of endoplasmic reticulum stress by lipid droplet formation. Biol Cell 103(6): 271–285.
He J, Goodpaster BH & Kelley DE (2004) Effects of weight loss and physical activity on muscle lipid content and droplet size. Obes Res 12(5): 761–769.
Hehnly H & Stamnes M (2007) Regulating cytoskeleton-based vesicle motility. FEBS Lett 581(11): 2112–2118.
Hershko A & Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67: 425–479.
Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD & Lippincott-Schwartz J (1998) Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J Cell Biol 143(6): 1485–1503.
Holt CE & Bullock SL (2009) Subcellular mRNA localization in animal cells and why it matters. Science 326(5957): 1212–1216.
Horton AC & Ehlers MD (2003) Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J Neurosci 23(15): 6188–6199.
Hughes BW, Kusner LL & Kaminski HJ (2006) Molecular architecture of the neuromuscular junction. Muscle Nerve 33(4): 445–461.
Itani SI, Ruderman NB, Schmieder F & Boden G (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 51(7): 2005–2011.

77
Jahn R & Scheller RH (2006) SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol 7(9): 631–643.
Jambhekar A & Derisi JL (2007) Cis-acting determinants of asymmetric, cytoplasmic RNA transport. RNA 13(5): 625–642.
Jasmin BJ, Lee RK & Rotundo RL (1993) Compartmentalization of acetylcholinesterase mRNA and enzyme at the vertebrate neuromuscular junction. Neuron 11(3): 467–477.
Jensen TH & Rosbash M (2003) Co-transcriptional monitoring of mRNP formation. Nat Struct Biol 10(1): 10–12.
Jha S & Komar AA (2011) Birth, life and death of nascent polypeptide chains. Biotechnol J 6(6): 623–640.
Jorgensen AO, Shen AC, Arnold W, Leung AT & Campbell KP (1989) Subcellular distribution of the 1,4-dihydropyridine receptor in rabbit skeletal muscle in situ: an immunofluorescence and immunocolloidal gold-labeling study. J Cell Biol 109(1): 135–147.
Jorgensen AO, Shen AC, MacLennan DH & Tokuyasu KT (1982) Ultrastructural localization of the Ca2+ + Mg2+-dependent ATPase of sarcoplasmic reticulum in rat skeletal muscle by immunoferritin labeling of ultrathin frozen sections. J Cell Biol 92(2): 409–416.
Kaakinen M, Kaisto T, Rahkila P & Metsikko K (2012) Caveolin 3, flotillin 1 and influenza virus hemagglutinin reside in distinct domains on the sarcolemma of skeletal myofibers. Biochem Res Int 2012: 497572.
Kaakinen M, Papponen H & Metsikko K (2008) Microdomains of endoplasmic reticulum within the sarcoplasmic reticulum of skeletal myofibers. Exp Cell Res 314(2): 237–245.
Kaisto T & Metsikko K (2003) Distribution of the endoplasmic reticulum and its relationship with the sarcoplasmic reticulum in skeletal myofibers. Exp Cell Res 289(1): 47–57.
Katz FN, Rothman JE, Knipe DM & Lodish HF (1977) Membrane assembly: synthesis and intracellular processing of the vesicular stomatitis viral glycoprotein. J Supramol Struct 7(3–4): 353–370.
Keenan RJ, Freymann DM, Stroud RM & Walter P (2001) The signal recognition particle. Annu Rev Biochem 70: 755–775.
Kelly SM & Corbett AH (2009) Messenger RNA export from the nucleus: a series of molecular wardrobe changes. Traffic 10(9): 1199–1208.
Kiebler MA & Bassell GJ (2006) Neuronal RNA granules: movers and makers. Neuron 51(6): 685–690.
Kim HJ, Jamart C, Deldicque L, An GL, Lee YH, Kim CK, Raymackers JM & Francaux M (2011) Endoplasmic reticulum stress markers and ubiquitin-proteasome pathway activity in response to a 200-km run. Med Sci Sports Exerc 43(1): 18–25.
Kondylis V & Rabouille C (2009) The Golgi apparatus: lessons from Drosophila. FEBS Lett 583(23): 3827–3838.
Konig J, Zarnack K, Luscombe NM & Ule J (2012) Protein-RNA interactions: new genomic technologies and perspectives. Nat Rev Genet 13(2): 77–83.

78
Kostova Z & Wolf DH (2003) For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J 22(10): 2309–2317.
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259): 680–685.
Launikonis BS & Stephenson DG (2004) Osmotic properties of the sealed tubular system of toad and rat skeletal muscle. J Gen Physiol 123(3): 231–247.
Lee MC, Miller EA, Goldberg J, Orci L & Schekman R (2004) Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol 20: 87–123.
Levi SK, Bhattacharyya D, Strack RL, Austin JR,2nd & Glick BS (2010) The yeast GRASP Grh1 colocalizes with COPII and is dispensable for organizing the secretory pathway. Traffic 11(9): 1168–1179.
Lippincott-Schwartz J, Roberts TH & Hirschberg K (2000) Secretory protein trafficking and organelle dynamics in living cells. Annu Rev Cell Dev Biol 16: 557–589.
Liu Y, Chen BP, Lu M, Zhu Y, Stemerman MB, Chien S & Shyy JY (2002) Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol 22(1): 76–81.
Londos C, Brasaemle DL, Schultz CJ, Segrest JP & Kimmel AR (1999) Perilipins, ADRP, and other proteins that associate with intracellular neutral lipid droplets in animal cells. Semin Cell Dev Biol 10(1): 51–58.
Lu Z, Joseph D, Bugnard E, Zaal KJ & Ralston E (2001) Golgi complex reorganization during muscle differentiation: visualization in living cells and mechanism. Mol Biol Cell 12(4): 795–808.
Luck G, Oberbaumer I & Blottner D (1998) In situ identification of neuronal nitric oxide synthase (NOS-I) mRNA in mouse and rat skeletal muscle. Neurosci Lett 246(2): 77–80.
Luini A, Mironov AA, Polishchuk EV & Polishchuk RS (2008) Morphogenesis of post-Golgi transport carriers. Histochem Cell Biol 129(2): 153–161.
Mabruk MJ (2004) In situ hybridization: detecting viral nucleic acid in formalin-fixed, paraffin-embedded tissue samples. Expert Rev Mol Diagn 4(5): 653–661.
Malenfant P, Joanisse DR, Theriault R, Goodpaster BH, Kelley DE & Simoneau JA (2001) Fat content in individual muscle fibers of lean and obese subjects. Int J Obes Relat Metab Disord 25(9): 1316–1321.
Malhotra V, Serafini T, Orci L, Shepherd JC & Rothman JE (1989) Purification of a novel class of coated vesicles mediating biosynthetic protein transport through the Golgi stack. Cell 58(2): 329–336.
Mangus DA, Evans MC & Jacobson A (2003) Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol 4(7): 223.
Matlin KS & Simons K (1983) Reduced temperature prevents transfer of a membrane glycoprotein to the cell surface but does not prevent terminal glycosylation. Cell 34(1): 233–243.

79
McMahon HT & Mills IG (2004) COP and clathrin-coated vesicle budding: different pathways, common approaches. Curr Opin Cell Biol 16(4): 379–391.
Meex RC, Schrauwen P & Hesselink MK (2009) Modulation of myocellular fat stores: lipid droplet dynamics in health and disease. Am J Physiol Regul Integr Comp Physiol 297(4): R913–24.
Minnaard R, Schrauwen P, Schaart G, Jorgensen JA, Lenaers E, Mensink M & Hesselink MK (2009) Adipocyte differentiation-related protein and OXPAT in rat and human skeletal muscle: involvement in lipid accumulation and type 2 diabetes mellitus. J Clin Endocrinol Metab 94(10): 4077–4085.
Mitsui T, Kawai H, Shono M, Kawajiri M, Kunishige M & Saito S (1997) Preferential subsarcolemmal localization of dystrophin and beta-dystroglycan mRNA in human skeletal muscles. J Neuropathol Exp Neurol 56(1): 94–101.
Moscoso LM, Merlie JP & Sanes JR (1995) N-CAM, 43K-rapsyn, and S-laminin mRNAs are concentrated at synaptic sites in muscle fibers. Mol Cell Neurosci 6(1): 80–89.
Munro S (2011) What is the Golgi apparatus, and why are we asking? BMC Biol 9: 63. Murphy S, Martin S & Parton RG (2009) Lipid droplet-organelle interactions; sharing the
fats. Biochim Biophys Acta 1791(6): 441–447. Nehls S, Snapp EL, Cole NB, Zaal KJ, Kenworthy AK, Roberts TH, Ellenberg J, Presley
JF, Siggia E & Lippincott-Schwartz J (2000) Dynamics and retention of misfolded proteins in native ER membranes. Nat Cell Biol 2(5): 288–295.
Nikonov AV, Snapp E, Lippincott-Schwartz J & Kreibich G (2002) Active translocon complexes labeled with GFP-Dad1 diffuse slowly as large polysome arrays in the endoplasmic reticulum. J Cell Biol 158(3): 497–506.
Nilsen TW & Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463(7280): 457–463.
Nissinen M, Kaisto T, Salmela P, Peltonen J & Metsikko K (2005) Restricted distribution of mRNAs encoding a sarcoplasmic reticulum or transverse tubule protein in skeletal myofibers. J Histochem Cytochem 53(2): 217–227.
Ohlendieck K, Ervasti JM, Snook JB & Campbell KP (1991) Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J Cell Biol 112(1): 135–148.
Olkkonen VM, Dupree P, Simons K, Liljestrom P & Garoff H (1994) Expression of exogenous proteins in mammalian cells with the Semliki Forest virus vector. Methods Cell Biol 43 Pt A: 43–53.
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH & Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306(5695): 457–461.
Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB & Storlien LH (1997) Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46(6): 983–988.
Papponen H, Kaisto T, Leinonen S, Kaakinen M & Metsikko K (2009) Evidence for gamma-actin as a Z disc component in skeletal myofibers. Exp Cell Res 315(2): 218–225.

80
Parton RG & Simons K (2007) The multiple faces of caveolae. Nat Rev Mol Cell Biol 8(3): 185–194.
Peachey LD & Eisenberg BR (1978) Helicoids in the T system and striations of frog skeletal muscle fibers seen by high voltage electron microscopy. Biophys J 22(2): 145–154.
Pearse BM (1975) Coated vesicles from pig brain: purification and biochemical characterization. J Mol Biol 97(1): 93–98.
Pelham HR (2001) Traffic through the Golgi apparatus. J Cell Biol 155(7): 1099–1101. Periasamy M & Kalyanasundaram A (2007) SERCA pump isoforms: their role in calcium
transport and disease. Muscle Nerve 35(4): 430–442. Pestka S, Rosenfeld H, Harris R & Hintikka H (1972) Studies on transfer ribonucleic acid-
ribosome complexes. XXI. Effect of antibiotics on peptidyl-puromycin synthesis by mammalian polyribosomes. J Biol Chem 247(21): 6895–6900.
Pette D & Staron RS (2001) Transitions of muscle fiber phenotypic profiles. Histochem Cell Biol 115(5): 359–372.
Pizon V, Gerbal F, Diaz CC & Karsenti E (2005) Microtubule-dependent transport and organization of sarcomeric myosin during skeletal muscle differentiation. EMBO J 24(21): 3781–3792.
Pol A, Martin S, Fernandez MA, Ferguson C, Carozzi A, Luetterforst R, Enrich C & Parton RG (2004) Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Mol Biol Cell 15(1): 99–110.
Pol A, Martin S, Fernandez MA, Ingelmo-Torres M, Ferguson C, Enrich C & Parton RG (2005) Cholesterol and fatty acids regulate dynamic caveolin trafficking through the Golgi complex and between the cell surface and lipid bodies. Mol Biol Cell 16(4): 2091–2105.
Prats C, Donsmark M, Qvortrup K, Londos C, Sztalryd C, Holm C, Galbo H & Ploug T (2006) Decrease in intramuscular lipid droplets and translocation of HSL in response to muscle contraction and epinephrine. J Lipid Res 47(11): 2392–2399.
Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ & Lippincott-Schwartz J (1997) ER-to-Golgi transport visualized in living cells. Nature 389(6646): 81–85.
Rahkila P, Alakangas A, Vaananen K & Metsikko K (1996) Transport pathway, maturation, and targetting of the vesicular stomatitis virus glycoprotein in skeletal muscle fibers. J Cell Sci 109 ( Pt 6)(Pt 6): 1585–1596.
Rahkila P, Luukela V, Vaananen K & Metsikko K (1998) Differential targeting of vesicular stomatitis virus G protein and influenza virus hemagglutinin appears during myogenesis of L6 muscle cells. J Cell Biol 140(5): 1101–1111.
Rahkila P, Takala TE, Parton RG & Metsikko K (2001) Protein targeting to the plasma membrane of adult skeletal muscle fiber: an organized mosaic of functional domains. Exp Cell Res 267(1): 61–72.
Rahkila P, Vaananen K, Saraste J & Metsikko K (1997) Endoplasmic reticulum to Golgi trafficking in multinucleated skeletal muscle fibers. Exp Cell Res 234(2): 452–464.

81
Ralston E & Hall ZW (1992) Restricted distribution of mRNA produced from a single nucleus in hybrid myotubes. J Cell Biol 119(5): 1063–1068.
Ralston E, Lu Z & Ploug T (1999) The organization of the Golgi complex and microtubules in skeletal muscle is fiber type-dependent. J Neurosci 19(24): 10694–10705.
Ralston E, McLaren RS & Horowitz JA (1997) Nuclear domains in skeletal myotubes: the localization of transferrin receptor mRNA is independent of its half-life and restricted by binding to ribosomes. Exp Cell Res 236(2): 453–462.
Ralston E, Ploug T, Kalhovde J & Lomo T (2001) Golgi complex, endoplasmic reticulum exit sites, and microtubules in skeletal muscle fibers are organized by patterned activity. J Neurosci 21(3): 875–883.
Ramakrishnan V (2002) Ribosome structure and the mechanism of translation. Cell 108(4): 557–572.
Rapoport TA, Jungnickel B & Kutay U (1996) Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu Rev Biochem 65: 271–303.
Rossi D, Barone V, Giacomello E, Cusimano V & Sorrentino V (2008) The sarcoplasmic reticulum: an organized patchwork of specialized domains. Traffic 9(7): 1044–1049.
Rothman JE & Lodish HF (1977) Synchronised transmembrane insertion and glycosylation of a nascent membrane protein. Nature 269(5631): 775–780.
Russell JC, Shillabeer G, Bar-Tana J, Lau DC, Richardson M, Wenzel LM, Graham SE & Dolphin PJ (1998) Development of insulin resistance in the JCR:LA-cp rat: role of triacylglycerols and effects of MEDICA 16. Diabetes 47(5): 770–778.
Sakamoto K, Wada I & Kimura J (2011) Inhibition of Rab1 GTPase and endoplasmic reticulum-to-Golgi trafficking underlies statin's toxicity in rat skeletal myofibers. J Pharmacol Exp Ther 338(1): 62–69.
Salanova M, Priori G, Barone V, Intravaia E, Flucher B, Ciruela F, McIlhinney RA, Parys JB, Mikoshiba K & Sorrentino V (2002) Homer proteins and InsP(3) receptors co-localise in the longitudinal sarcoplasmic reticulum of skeletal muscle fibres. Cell Calcium 32(4): 193–200.
Salo J, Metsikko K, Palokangas H, Lehenkari P & Vaananen HK (1996) Bone-resorbing osteoclasts reveal a dynamic division of basal plasma membrane into two different domains. J Cell Sci 109 ( Pt 2)(Pt 2): 301–307.
Samuel VT & Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148(5): 852–871.
Scales SJ, Pepperkok R & Kreis TE (1997) Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell 90(6): 1137–1148.
Schatz G & Dobberstein B (1996) Common principles of protein translocation across membranes. Science 271(5255): 1519–1526.
Schiaffino S, Gorza L, Sartore S, Saggin L, Ausoni S, Vianello M, Gundersen K & Lomo T (1989) Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J Muscle Res Cell Motil 10(3): 197–205.

82
Schrauwen-Hinderling VB, van Loon LJ, Koopman R, Nicolay K, Saris WH & Kooi ME (2003) Intramyocellular lipid content is increased after exercise in nonexercising human skeletal muscle. J Appl Physiol 95(6): 2328–2332.
Schroder M & Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74: 739–789.
Schuck S & Simons K (2004) Polarized sorting in epithelial cells: raft clustering and the biogenesis of the apical membrane. J Cell Sci 117(Pt 25): 5955–5964.
Shahbabian K & Chartrand P (2012) Control of cytoplasmic mRNA localization. Cell Mol Life Sci 69(4): 535–552.
Shaw CS, Sherlock M, Stewart PM & Wagenmakers AJ (2009) Adipophilin distribution and colocalization with lipid droplets in skeletal muscle. Histochem Cell Biol 131(5): 575–581.
Sheth U & Parker R (2003) Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300(5620): 805–808.
Sierakowska H, Shukla RR, Dominski Z & Kole R (1989) Inhibition of pre-mRNA splicing by 5-fluoro-, 5-chloro-, and 5-bromouridine. J Biol Chem 264(32): 19185–19191.
Silva M, Matthews ML, Jarvis C, Nolan NM, Belliveau P, Malloy M & Gandhi P (2007) Meta-analysis of drug-induced adverse events associated with intensive-dose statin therapy. Clin Ther 29(2): 253–260.
Simons K & Ikonen E (1997) Functional rafts in cell membranes. Nature 387(6633): 569–572.
Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P & Rothman JE (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362(6418): 318–324.
Sorrentino V (2011) Sarcoplasmic reticulum: structural determinants and protein dynamics. Int J Biochem Cell Biol 43(8): 1075–1078.
St Johnston D (2005) Moving messages: the intracellular localization of mRNAs. Nat Rev Mol Cell Biol 6(5): 363–375.
Steitz TA & Moore PB (2003) RNA, the first macromolecular catalyst: the ribosome is a ribozyme. Trends Biochem Sci 28(8): 411–418.
Stenmark H (2009) Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10(8): 513–525.
Stephens DJ & Pepperkok R (2001) Illuminating the secretory pathway: when do we need vesicles? J Cell Sci 114(Pt 6): 1053–1059.
Sztul E & Lupashin V (2006) Role of tethering factors in secretory membrane traffic. Am J Physiol Cell Physiol 290(1): C11–26.
Tassin AM, Paintrand M, Berger EG & Bornens M (1985) The Golgi apparatus remains associated with microtubule organizing centers during myogenesis. J Cell Biol 101(2): 630–638.
Thiele C & Spandl J (2008) Cell biology of lipid droplets. Curr Opin Cell Biol 20(4): 378–385.

83
Tidball JG (1991) Force transmission across muscle cell membranes. J Biomech 24 Suppl 1: 43–52.
Traub LM & Kornfeld S (1997) The trans-Golgi network: a late secretory sorting station. Curr Opin Cell Biol 9(4): 527–533.
Van der Meer SF, Jaspers RT & Degens H (2011) Is the myonuclear domain size fixed? J Musculoskelet Neuronal Interact 11(4): 286–297.
Vinciguerra P & Stutz F (2004) mRNA export: an assembly line from genes to nuclear pores. Curr Opin Cell Biol 16(3): 285–292.
Volpe P, Villa A, Podini P, Martini A, Nori A, Panzeri MC & Meldolesi J (1992) The endoplasmic reticulum-sarcoplasmic reticulum connection: distribution of endoplasmic reticulum markers in the sarcoplasmic reticulum of skeletal muscle fibers. Proc Natl Acad Sci USA 89(13): 6142–6146.
Wansink DG, Nelissen RL & de Jong L (1994) In vitro splicing of pre-mRNA containing bromouridine. Mol Biol Rep 19(2): 109–113.
Ward TH, Polishchuk RS, Caplan S, Hirschberg K & Lippincott-Schwartz J (2001) Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol 155(4): 557–570.
Warner JR, Knopf PM & Rich A (1963) A multiple ribosomal structure in protein synthesis. Proc Natl Acad Sci USA 49: 122–129.
Weis B, Schleiff E & Zerges W (2012) Protein targeting to subcellular organelles via mRNA localization.
Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK, Malinow MR & Austin RC (2001) Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest 107(10): 1263–1273.
Wiedenmann J, Oswald F & Nienhaus GU (2009) Fluorescent proteins for live cell imaging: opportunities, limitations, and challenges. IUBMB Life 61(11): 1029–1042.
Yamada T, Yoshimura H, Inaguma A & Ozawa T (2011) Visualization of nonengineered single mRNAs in living cells using genetically encoded fluorescent probes. Anal Chem 83(14): 5708–5714.
Yan A & Lennarz WJ (2005) Unraveling the mechanism of protein N-glycosylation. J Biol Chem 280(5): 3121–3124.
Young JC, Agashe VR, Siegers K & Hartl FU (2004) Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol 5(10): 781–791.
Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW & Shulman GI (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277(52): 50230–50236.
Zanetti G, Pahuja KB, Studer S, Shim S & Schekman R (2011) COPII and the regulation of protein sorting in mammals. Nat Cell Biol 14(1): 20–28.

84

85
Original publications
I Nevalainen M, Kaakinen M & Metsikkö K (2012) Localization of mRNA transcripts and translation activity in skeletal myofibers. Manuscript.
II Nevalainen M, Nissinen M, Kaakinen M & Metsikkö K (2010) Influenza virus infection in multinucleated skeletal myofibers. Exp Cell Res 316: 1784–1794.
III Nevalainen M, Kaisto T & Metsikkö K (2010) Mobile ER-to-Golgi but not post-Golgi membrane transport carriers disappear during the terminal myogenic differentiation. Cell Tissue Res 342: 107–116
IV Nevalainen M, Kaakinen M, Rahkila P & Metsikkö K (2012) Reversible stress-induced lipid body formation in fast twitch rat myofibers. Exp Cell Res 318: 2191–2199.
Reprinted with permission from Springer Verlag (III) and Elsevier (II, IV).
Original publications are not included in the electronic version of the dissertation.

86

A C T A U N I V E R S I T A T I S O U L U E N S I S
Book orders:Granum: Virtual book storehttp://granum.uta.fi/granum/
S E R I E S D M E D I C A
1179. Haapsamo, Helena (2012) A Follow-up study of children's communicativedevelopment : Associations to social-emotional and behavioural problems andcompetences and experienced maternal stress
1180. Norppa, Anna (2012) Association between periodontal and systemicinflammation : A study of pro- and anti-inflammatory mediators
1181. Karjalainen, Teemu (2012) Nitinol shape memory alloy in flexor tendon repair
1182. Saukkonen, Tuula (2012) Prediabetes and associated cardiovascular risk factors :A prospective cohort study among middle-aged and elderly Finns
1183. Hagman, Juha (2012) Resource utilization in the treatment of open angleglaucoma in Finland: an 11-year retrospective analysis
1184. Eskola, Pasi (2012) The search for susceptibility genes in lumbar discdegeneration : Focus on young individuals
1185. Kaivorinne, Anna-Lotta (2012) Frontotemporal lobar degeneration in Finland :Molecular genetics and clinical aspects
1186. Härkönen, Pirjo (2012) Elämäntyytyväisyys ja terveys : Voimavarasuuntautunutikääntyvien henkilöiden seurantatutkimus
1187. Laukkanen, Päivi (2012) Occurrence of high risk human papillomaviruses andcervical cancer among fertile-aged women in Finland
1188. Junttila, Eija (2012) Cardiovascular abnormalities after non-traumatic intracranialhemorrhage
1189. Hookana, Eeva (2012) Characteristics of victims of non-ischemic sudden cardiacdeath
1190. Murugan, Subramanian (2012) Control of nephrogenesis by Wnt4 signaling :Mechanisms of gene regulation and targeting of specific lineage cells by tissueengineering tools
1191. Nikkilä, Jenni (2013) PALB2 and RAP80 genes in hereditary breast cancerpredisposition
1192. Määttä, Mikko (2012) Assessment of osteoporosis and fracture risk : Axialtransmission ultrasound and lifestyle-related risk factors
1193. Kaakinen, Marika (2012) Genetic and life course determinants of cardiovascularrisk factors : Structural equation modelling of complex relations

ABCDEFG
UNIVERS ITY OF OULU P.O.B . 7500 F I -90014 UNIVERS ITY OF OULU F INLAND
A C T A U N I V E R S I T A T I S O U L U E N S I S
S E R I E S E D I T O R S
SCIENTIAE RERUM NATURALIUM
HUMANIORA
TECHNICA
MEDICA
SCIENTIAE RERUM SOCIALIUM
SCRIPTA ACADEMICA
OECONOMICA
EDITOR IN CHIEF
PUBLICATIONS EDITOR
Senior Assistant Jorma Arhippainen
University Lecturer Santeri Palviainen
Professor Hannu Heusala
Professor Olli Vuolteenaho
University Lecturer Hannu Heikkinen
Director Sinikka Eskelinen
Professor Jari Juga
Professor Olli Vuolteenaho
Publications Editor Kirsti Nurkkala
ISBN 978-952-62-0062-0 (Paperback)ISBN 978-952-62-0063-7 (PDF)ISSN 0355-3221 (Print)ISSN 1796-2234 (Online)
U N I V E R S I TAT I S O U L U E N S I S
MEDICA
ACTAD
D 1194
ACTA
Mika N
evalainen
OULU 2012
D 1194
Mika Nevalainen
GENE PRODUCT TARGETING INTO AND MEMBRANE TRAFFICKING FROMTHE ENDOPLASMIC/SARCOPLASMIC RETICULUM IN SKELETAL MYOFIBERS
UNIVERSITY OF OULU GRADUATE SCHOOL;UNIVERSITY OF OULU,FACULTY OF MEDICINE,INSTITUTE OF BIOMEDICINE,DEPARTMENT OF ANATOMY AND CELL BIOLOGY





























