Embed Size (px)
Citation preview

Molecular Genetics and Metabolism 80 (2003) 365–376
www.elsevier.com/locate/ymgme
Minireview
Gene expression in early ischemic renal injury: cluestowards pathogenesis, biomarker discovery, and novel therapeutics
Prasad Devarajan,a,b,c,* Jaya Mishra,a Suroj Supavekin,b Larry T. Patterson,a
and S. Steven Potterc
a Department of Nephrology, Cincinnati Children�s Hospital, Medical Center and Research Foundation, 3333 Burnet Avenue, MLC 7022,
Cincinnati, OH 45229-3039, USAb Department of Nephrology, Children�s Hospital at Montefiore, 111 E 210 Street, Bronx, NY 10467, USA
c Department of Developmental Biology, Cincinnati Children�s Hospital, Medical Center and Research Foundation, 3333 Burnet Avenue,
MLC 7022, Cincinnati, OH 45229-3039, USA
Received 8 July 2003; received in revised form 10 September 2003; accepted 10 September 2003
Abstract
Acute renal failure (ARF) represents a common and serious problem in clinical medicine. Renal ischemia–reperfusion injury (IRI) is
the major cause of ARF in the native and transplanted kidney. Several decades of research have provided successful therapeutic
approaches in animal models, but translational efforts in humans have yielded disappointing results. The major reasons for this
include a lack of early markers for ARF (and hence a delay in initiating therapy), and the multi-factorial nature of the disease. This
review focuses on the use of cDNA microarrays to elucidate the molecular genetic mechanisms underlying tubule cell apoptosis, and
to identify novel biomarkers for early renal IRI. Also presented is a comparative temporal analysis of cDNA microarray results
from mature kidneys following IRI and during normal nephrogenesis. Molecular genetic evidence for the notion that regeneration
recapitulates development in the kidney, and that injured tubule cells possess the capacity to de-differentiate to the earliest stages of
development, is presented. The implications of these findings to the ability of the kidney to repair itself and potential strategies for
accelerating recovery are briefly discussed.
� 2003 Elsevier Inc. All rights reserved.
Introduction
Acute renal failure (ARF) represents a significant andpersistent problem in clinical medicine with serious
consequences [1–12]. Renal ischemia–reperfusion injury
(IRI) is the major cause of ARF in the native [1,4], and
transplanted kidney [5]. Several decades of research have
provided successful therapeutic approaches in animal
models, but translational efforts in humans have yielded
disappointing results. An improved understanding of
the molecular mechanisms underlying early renal cellinjury will be critical for innovative and effective ther-
apy. The morphologic and biologic responses of renal
tubular cells to IRI include cell death, de-differentiation
* Corresponding author. Fax: +513-636-7407.
E-mail address: [email protected] (P. Devarajan).
1096-7192/$ - see front matter � 2003 Elsevier Inc. All rights reserved.
doi:10.1016/j.ymgme.2003.09.012
of viable cells, proliferation, re-differentiation, and res-
titution of a normal epithelium [1,19–22]. Apoptosis has
recently emerged as the major mechanism leading toearly tubule cell death following IRI [23–28] and down-
regulation of apoptosis may offer a novel therapeutic
approach to the amelioration of IRI in both the native
and the transplanted kidney. Attempts at unraveling the
molecular basis of the myriad apoptotic pathways in-
duced by IRI have been facilitated by recent advances in
functional genomics [60–67]. In this review, we will first
outline what is known about the apoptotic mechanismspertinent to renal IRI, and then present our genetic
analysis of this process using cDNA microarrays. We
will then review the current literature on molecular
mechanisms of tubule cell regeneration following IRI,
and present the pertinent cDNA microarray data with
special emphasis on translating this information to the
field of non-invasive biomarker discovery. In the final

366 P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376
part of this review, we will present a detailed compara-tive analysis of renal gene expression during develop-
ment and following IRI. We will provide molecular
genetic evidence for the hypothesis that injured tubule
cells possess the capacity to de-differentiate to the ear-
liest stages of development, and that the de-differenti-
ated tubule cells can persist in the injured mature kidney
for a relatively long period of time. The implications of
these findings to the ability of the kidney to repair itselfand potential strategies for accelerating recovery will be
briefly discussed.
Background
Acute renal failure
Acute renal failure (ARF) continues to represent a
very significant and potentially devastating problem in
clinical medicine [1–12]. The incidence of ARF is high
(about 200 cases per million adult population per year),
and varies from 5% of hospitalized patients to 30–50%
of patients in intensive care units. Despite significant
technical advances in therapeutics, the mortality and
morbidity associated with ARF remain dismally highand have not appreciably improved during the last four
decades [1–12]. For example, in a recent large study of
1095 patients with ARF [13], the survival rate was 60%
of cases; however, only half of the survivors attained full
recovery of renal function, with the other half displaying
either renal insufficiency or end-stage renal disease
(ESRD). Although this unacceptable prognosis may be
partly attributable to other co-morbid conditions, recentstudies have revealed that several outcome variables are
adversely affected by the presence of ARF, and that
ARF itself is a major risk factor for the development of
non-renal complications. In addition to its implications
on human life and its quality, the treatment of ARF also
represents an enormous financial burden to society [14],
and ARF-associated medical expenses are conserva-
tively estimated at over $8 billion per year in adultsalone. In addition, even sub-clinical episodes of ARF
significantly contribute to the development of ESRD, a
common clinical condition (nearly 400 new cases per
million adult population per year) with a mean survival
rate only slightly better than lung cancer and with
staggering economic implications.
Ischemia–reperfusion injury
Renal ischemia–reperfusion injury (IRI) is the major
cause of ARF in the native kidney [1,4], and an invari-
able occurrence in the transplanted kidney [15]. Com-
mon medical conditions leading to IRI in the native
kidney include cardiovascular disease, stroke, trauma,
dehydration, and surgical procedures. In addition to the
usual complications of ARF, IRI in the transplantedkidney results in delayed graft function [16], which sig-
nificantly increases the risk of graft loss and acute re-
jection [17,18]. Thus, even long-suffering ESRD patients
who finally get a kidney transplant are not spared from
the ravages of IRI. Pioneering work over several de-
cades has illuminated the roles of persistent vasocon-
striction, tubular obstruction, cellular metabolic
alterations, and the inflammatory response in the path-ogenesis of IRI [19–22]. While these studies have paved
the way for successful therapeutic approaches in animal
models, translational research efforts in humans have
yielded disappointing results. One of the major reasons
for this includes a paucity of early biomarkers for hu-
man ARF, which contributes to a delay in initiating
preventive and therapeutic measures that have been ef-
fective in ameliorating ARF in animals. The secondreason pertains to the multi-factorial nature of the dis-
ease. An improved understanding of the molecular
mechanisms underlying early renal cell injury will be
critical for innovative and effective therapy.
Molecular pathogenesis of renal IRI
It is now clear that the morphologic and biologicresponses of renal tubular cells to IRI are multifaceted,
and include cell death, de-differentiation of viable cells,
proliferation, re-differentiation, and restitution of a
normal epithelium [1,19–22], as illustrated in Fig. 1. The
molecular mechanisms underlying each of these re-
sponses are subjects of intense current investigations.
Although the term ‘‘acute tubular necrosis’’ has been
synonymous with ARF, apoptosis has recently emergedas an additional potential mechanism leading to early
tubule cell death following ischemic renal injury [23–28].
Several studies have documented that IRI in animal
models predictably leads to apoptosis of renal tubular
epithelial cells [29–42]. Importantly, this finding has now
been confirmed in biopsy samples from transplanted
cadaveric kidneys, which constitutes a human model for
renal IRI [43–45]. Furthermore, several renal tubule cellculture models of ischemic renal injury have now been
described, which reliably demonstrate the induction of
apoptosis following partial ATP depletion or hypoxia,
and necrotic cell death following more severe grades of
injury [46–52]. These findings have lead to the hypoth-
esis that down-regulation of apoptosis may offer a un-
ique and powerful therapeutic approach to the
amelioration of IRI in both the native and the trans-planted kidney. Considerable attention and efforts have
therefore been directed towards identification of the
molecular pathways involved in the tubule cell apoptosis
following IRI. In recent years, specific proteases be-
longing to the caspase family have surfaced as crucial
effectors of apoptosis [53,54]. Members of this family are
expressed as pro-enzymes and require activation by
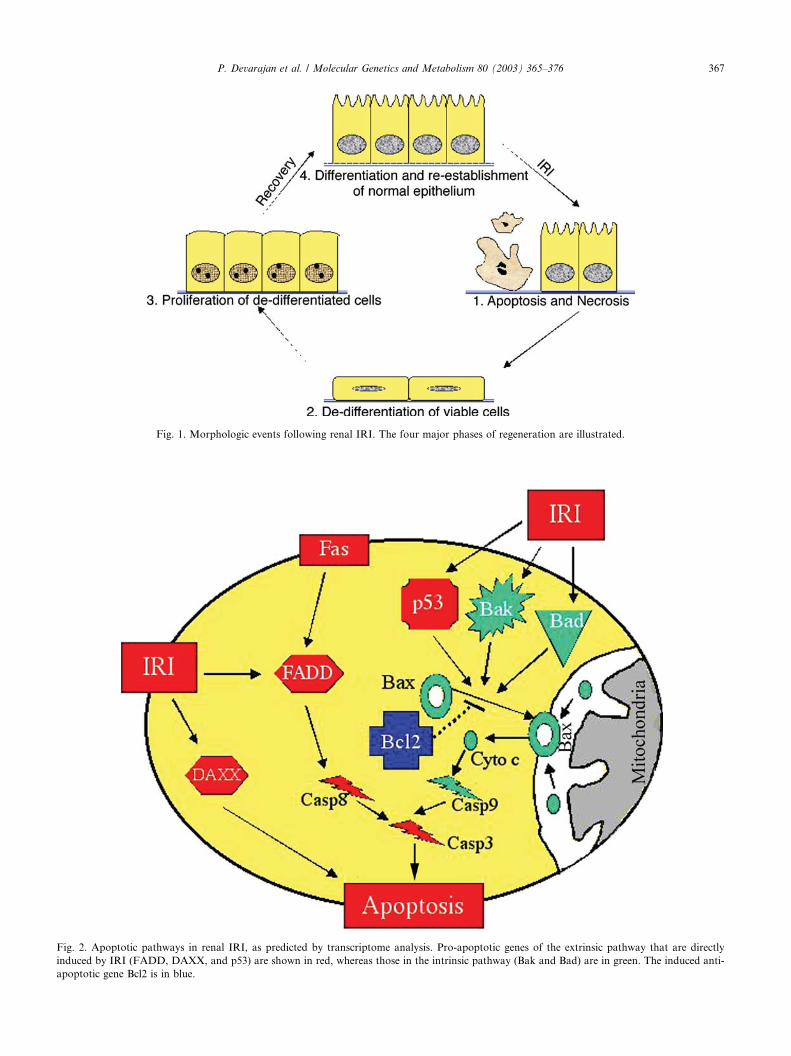
Fig. 1. Morphologic events following renal IRI. The four major phases of regeneration are illustrated.
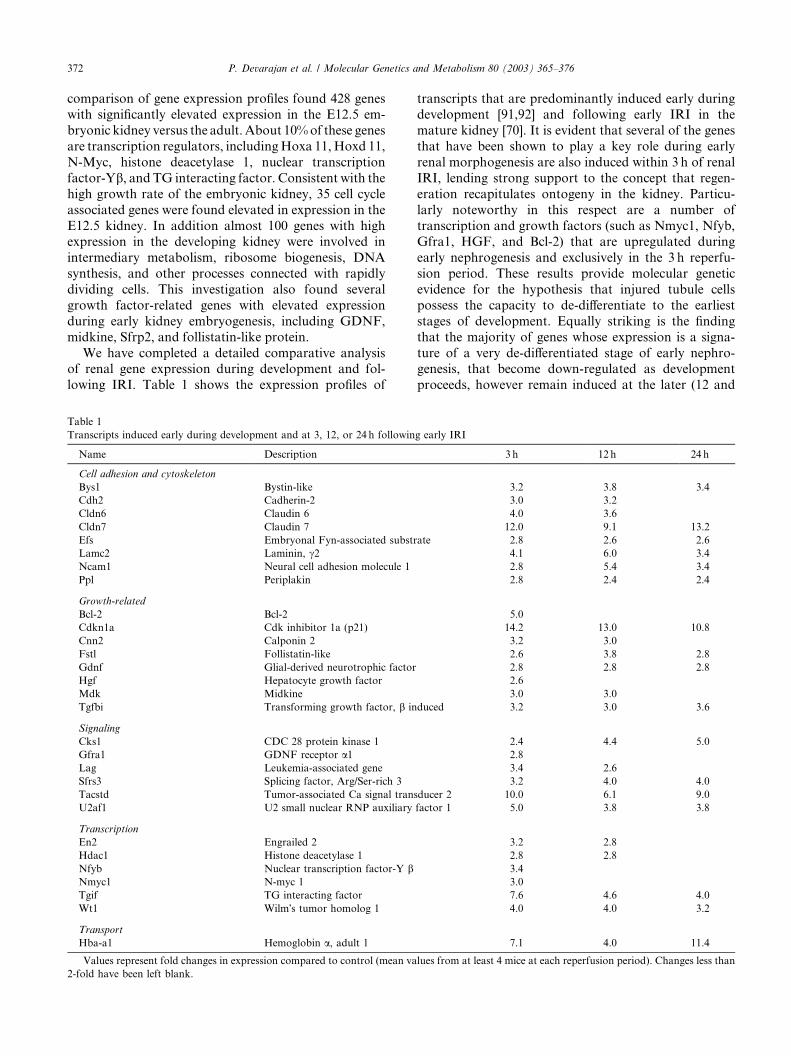
Fig. 2. Apoptotic pathways in renal IRI, as predicted by transcriptome analysis. Pro-apoptotic genes of the extrinsic pathway that are directly
induced by IRI (FADD, DAXX, and p53) are shown in red, whereas those in the intrinsic pathway (Bak and Bad) are in green. The induced anti-
apoptotic gene Bcl2 is in blue.
P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376 367

368 P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376
upstream stimuli in order to commit a cell into the ex-ecution phase of apoptosis. The major intracellular ap-
optotic pathways may be classified according to the type
of pro-caspase that is activated. Activation of the initi-
ator pro-caspase 8 results from signaling via cell surface
death receptors such as Fas (the ‘‘extrinsic’’ pathway)
and their ligands such as FADD and DAXX [55]. On
the other hand, activation of the initiator pro-caspase 9
is dependent primarily on ’’intrinsic’’ mitochondrialsignaling pathways regulated by members of the Bcl-2
family [56]. Activation of pro-apoptotic Bcl-2 family
members such as Bax and Bak can trigger a sequence of
events leading to release of mitochondrial cytochrome c
into the cytosol, and activation of pro-caspase 9 [57–59].
At least three potential levels of ‘‘cross-talk’’ exist be-
tween the extrinsic and intrinsic apoptotic pathways.
First, initial activation of caspase 8 via Fas can inducethe mitochondrial translocation of BID, a pro-apoptotic
member of the Bcl-2 family, with resultant cytochrome c
release and activation of caspase 9 [60]. Second, the p53
gene is a potent transcription factor that regulates ap-
optosis most notably by activating pro-apoptotic Bcl-2
family members as well as the Fas-dependent axis [61].
Third, both pathways culminate in the activation of
caspase 3, with subsequent entry into the ’’execution’’phase of apoptosis, resulting in DNA fragmentation and
cellular morphologic changes characteristic of apoptosis
[53]. The anti-apoptotic Bcl-2 family members such as
Bcl-2 itself play a pivotal protective role by preserving
mitochondrial structure and inhibiting cytochrome c
release [57–59].
Several of these apoptosis-related genes are expressed
and appear to be mechanistically operative in renal tu-bular cells following IRI. Attempts at unraveling the
molecular basis of these myriad apoptotic mechanisms
have been significantly facilitated by recent advances in
functional genomics that have yielded new tools for
genome-wide analysis of complex biologic processes
[60–67]. The DNA microarray methodologies provide
parallel and quantitative expression profiles of thou-
sands of genes, which when combined with bioinfor-matics can identify genes in a biologic pathway,
characterize the function of novel genes, analyze genetic
variation, detect disease subclasses, and identify drug
targets [60–67]. Others [68,69] and we [70] have recently
utilized these genome-wide approaches to analyze the
early renal response to ischemia both in vitro and in
animal models. Our results indicate that apoptosis plays
a major role and that a specific subset of apoptotic genesare induced in tubule cells following early IRI, as de-
tailed below.
Molecular mechanisms of recovery from renal IRI
Renal epithelial cells possess a remarkable ability to
regenerate and proliferate after IRI, a quality that is not
shared by the majority of other tissues. An improvedunderstanding of the molecular mechanisms of repair
may provide clues towards accelerating recovery fol-
lowing renal IRI. In the fully developed kidney, cell
division is minimal but can increase more than 10-fold
after acute injury [71]. Four major phases of this re-
generative process have been described [72–74], as il-
lustrated in Fig. 1. The first phase consists of cell
damage and cell death, during which tubule cells havebeen postulated to generate signals that initiate the re-
generative response. During this very early phase, renal
tubule cells up-regulate the expression of a number of
genes, including stress-activated protein kinases such as
MAP kinases [75,76] and transcription factors such as
c-Fos and Egr-1 [77]. The second phase is characterized
by the appearance of a large pool of de-differentiated
epithelial cells with flattened appearance and poorlydifferentiated brush borders [72–74]. These cells express
vimentin, a marker for multipotent embryonic mesen-
chymal cells [78]. Since the number of resident stem cells
in the native kidney and the number of bone marrow-
derived cells that appear in the post-injury kidney are
both very small, these cells must represent surviving or
sub-lethally injured tubule cells that have de-differenti-
ated [79]. De-differentiation of the tubular epithelial cellmay be a necessary pre-requisite to allow for spreading,
migration and re-population of cells over the denuded
basement membrane. The third phase is exemplified by a
marked increase in the number of proliferating tubule
epithelial cells [78–80] that express genes encoding for a
variety of growth factors such as IGF-1 [81,82], HGF
[83], and FGF [80]. The fourth phase is one of re-dif-
ferentiation, during which the normal tubular epithe-lium is restored with fully differentiated polarized cells.
Tubule epithelial cells in this stage display induction of a
variety of genes including NCAM [84], osteopontin [85],
CD44 [86], and TGF-b1 [87]. Others [68,69] and we [70]
have recently utilized genome-wide approaches to ana-
lyze the early global renal response to ischemia both in
vitro and in animal models. Our results have identified a
number of genes that are induced following early IRIand may play an important role in the recovery process,
as detailed below.
It is interesting to note that during recovery from
IRI, renal tubule cells recapitulate phases and processes
very similar to those during normal kidney develop-
ment. For instance, both the immature developing tu-
bule and the mature injured tubule cells display
apoptotic death as the mechanism for removing dam-aged or unwanted cells [23–27,88,89]. In both situa-
tions, de-differentiated mesenchymal cells undergo
proliferation, differentiation, and establishment of a
polarized epithelium [90]. However, many questions
remain unanswered. For example, it is unknown whe-
ther the inductive interactions between the metanephric
mesenchyme and ureteric bud that are characteristic of

P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376 369
and critical to the developing nephron also play a rolefollowing IRI. Also it is unclear which stage of devel-
opment the injured tubule cells de-differentiate to, and
how the patterns of gene expression resemble or differ
from each other during the two processes. The advent
of cDNA microarrays has, however, revolutionized the
global analysis of gene expression in the developing
kidney. Others [91] and we [92] have recently utilized
these approaches to catalogue gene expression duringkidney development. The results indicate that specific
subsets of genes are induced during various stages of
development. The results are detailed below, along with
an instructive and interesting comparison of the tem-
poral patterns of gene expression during renal devel-
opment and following IRI.
Analysis of gene expression during early recovery from
IRI
Maximally induced transcripts: clues for biomarker
discovery
We have utilized cDNA microarray technology and
stringent statistical analysis to define global changes inrenal gene expression during the early reperfusion peri-
ods following ischemic injury in an established mouse
model [70]. We have screened for changes in expression
of 9000 sequence-verified mouse genes at various early
points (3, 12, and 24 h) following renal IRI. We chose to
examine the immediate and early responses because (a) it
is well known that changes in gene expression occur soon
after an ischemic insult, (b) interventions aimed atameliorating injury and/or accelerating recovery may
need to target these early changes, (c) the temporal pat-
terns of these early morphologic and biologic changes
recapitulate those during renal development, and (d)
genes whose protein products are induced early after
injury may represent biomarkers that have hitherto elu-
ded discovery. We identified several transcripts that have
previously been shown to be over-expressed or repressedfollowing ischemic injury, thereby validating this tech-
nique. For example, our results have confirmed previous
reports documenting by alternative methods the upreg-
ulation of genes such as Bcl-2, Egr-1, c-Fos, HSP-70,
HGF, HB-EGF, IGF-1, TGFb1, p21, heme oxygenase 1,
and a-crystallin following renal IRI [70]. Similarly, pre-
viously described transcripts that are down-regulated
following ischemia that were also identified in our studyinclude NHE3, a-albumin, and members of the cyto-
chrome p450 family [70]. In addition, several genes not
previously associated with IRI were consistently shown
to be differentially expressed, and a detailed analysis of
these transcripts is beyond the scope of this minireview.
Surprisingly, several of the transcripts that were
maximally induced during early IRI were novel to the
field of acute renal failure. In a recent study [93], wehave further characterized one of these previously un-
recognized genes, namely neutrophil gelatinase-associ-
ated lipocalin (NGAL), because it encodes a small
secreted polypeptide that is protease resistant and con-
sequently might be readily detected in the urine. We
confirmed the marked upregulation of NGAL mRNA
by semi-quantitative RT-PCR and protein levels by
Western analysis in the early post-ischemic mouse kid-ney (both greater than 10-fold). NGAL protein expres-
sion was detected predominantly in PCNA-positive
proximal tubule cells that were undergoing proliferation
and regeneration. These findings strongly implicate a
role for this maximally induced gene and protein in the
repair process following IRI. Other recent studies have
also suggested that NGAL enhances the epithelial phe-
notype. During nephrogenesis, NGAL is expressed bythe penetrating ureteric bud, and triggers nephrogenesis
by stimulating the conversion of mesenchymal cells into
kidney epithelia [94]. Another lipocalin, glycodelin, has
been shown to induce an epithelial phenotype when
expressed in human breast carcinoma cells [95]. These
findings are especially pertinent to the mature kidney, in
which one of the well-documented responses to ischemic
injury is the remarkable appearance of de-differentiatedepithelial cells lining the proximal tubules [78]. An im-
portant aspect of renal regeneration and repair after
ischemic injury involves the reacquisition of the epithe-
lial phenotype, a process that recapitulates several as-
pects of normal development [96]. This suggests that
NGAL may be expressed by the damaged tubule in
order to induce re-epithelialization. Support for this
notion derives from the recent identification of NGALas an iron transporting protein that is complementary to
transferrin during nephrogenesis [94]. It is well known
that the delivery of iron into cells is crucial for cell
growth and development, and this is presumably critical
to post-ischemic renal regeneration just as it is during
ontogeny. Since NGAL appears to bind and transport
iron [94], it is also likely that NGAL may serve as a sink
for iron that is shed from damaged proximal tubuleepithelial cells. Because NGAL can be endocytosed by
the proximal tubule, the protein could potentially re-
cycle iron into viable cells. This might stimulate growth
and development, as well as remove iron, a reactive
molecule, from the site of tissue injury, thereby limiting
iron-mediated cytotoxicity.
Importantly, we have found that NGAL is easily
detected in the urine in the very first urine output fol-lowing ischemia in both mouse and rat models of ARF
[93]. The appearance of NGAL in the urine is related to
the dose and duration of renal ischemia, and precedes by
far the appearance of other known urinary markers such
as NAG and b2-microglobulin. The origin of NGAL
was confirmed in cultured human proximal tubule cells
subjected to in vitro ischemic injury, where NGAL

370 P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376
mRNA was rapidly induced in the cells and NGALprotein readily detectable in the culture medium within
one hour of mild ATP depletion. Our results indicate
that NGAL may represent an early, sensitive, non-in-
vasive urinary biomarker for ischemic renal injury that
compares very favorably with other biomarkers that
have been described. One of the best-studied examples is
kidney injury molecule-1 (KIM-1), a putative adhesion
molecule involved in renal regeneration that was alsofirst detected as a result of genomic analysis [103–105].
In a rat model of ischemia–reperfusion injury, KIM-1
was found to be upregulated 24–48 h after the initial
insult, rendering it a reliable but somewhat late marker
of tubular cell damage. Recent elegant studies have
shown that KIM-1 can be detected in the kidney biopsy
and urine of patients with ischemic acute tubular ne-
crosis [105]. However, this detection was documented inpatients with established ischemic renal damage, and the
utility of urinary KIM-1 measurement for the detection
of early subclinical injury has thus far not been vali-
dated. In another recent example, Cyr61 was found to
be a secreted cysteine-rich protein that is detectable in
the urine 3–6 h after ischemic renal injury [106]. How-
ever, this detection required a bioaffinity purification
step with heparin–Sepharose beads, and even after suchpurification several cross-reacting peptides were appar-
ent. In contrast, our study demonstrates that NGAL
was easily and rapidly detected as relatively clean im-
munoreactive peptides in Western blots with as little as
1 ll of the very first unprocessed urine output following
renal ischemia in both mice and rats. In addition, uri-
nary NGAL was evident even after very mild ‘‘subclin-
ical’’ renal ischemia, in spite of normal serum creatininelevels. Translational work in patients who are at risk for
developing sub-clinical forms of ischemic injury is in
progress. It is anticipated that early detection of sub-
clinical renal injury will enable clinicians to begin timely
interventional measures to prevent progression to overt
ARF. It is also anticipated that early detection will al-
low for institution of novel therapeutic approaches that
have thus far been met with limited success due to thelack of early biomarkers.
Induction of apoptosis-related genes: clues for pathogen-
esis and therapy
Our microarray analysis revealed for the first time a
marked upregulation of several pro-apoptotic genes in
the early post-ischemic period [70]. The most prominentchange was noted for FADD, which was upregulated
2.1-fold, 5.4-fold, and 6.1-fold at 3, 12, and 24 h of
reperfusion, respectively. Significant increases in ex-
pression were also noted at all reflow periods for DAXX
(1.9-fold, 2.5-fold, and 2.5-fold at 3, 12, and 24 h of
reperfusion, respectively), p53 (2.2-fold, 2.4-fold, and
2.2-fold at 3, 12, and 24 h of reperfusion, respectively),
and BAD (2.5-fold, 3-fold, and 2.5-fold at 3, 12, and24 h of reperfusion respectively). In contrast, the ex-
pression of BAK was significantly upregulated (3.2-fold)
only at 3 h of reflow. The immediate response to is-
chemic injury was also characterized by increased ex-
pression of the anti-apoptotic genes Bcl-2 and NF-jB.The finding of upregulated pro-apoptotic transcripts by
microarray analysis was confirmed by semi-quantitative
RT-PCR and immunohistochemistry [70]. In serial sec-tions, the majority of tubule epithelial cells that stained
positive for these pro-apoptotic factors also revealed
Tunel positive nuclei, implicating these pathways in the
cell death following IRI.
Products of the induced transcripts FADD and
DAXX are members of the ‘‘extrinsic’’ cell death path-
way, and are known to transduce apoptotic signals
emanating from cell surface receptors such as Fas, withresultant activation of caspase 8 [55]. Expression of Fas
has previously been documented in renal tubule cells
both in vivo [97] and in vitro [52], and upregulation of
Fas protein has been shown to occur in mouse kidney
after a 24 h reflow period following ischemia [97]. Sim-
ilarly, renal expression of FADD has been demonstrated
in vivo and in vitro [70]. Furthermore, we have previ-
ously demonstrated a rapid upregulation of Fas andFADD protein and activation of caspase 8 in cultured
tubule cells following an in vitro ischemic injury [52].
Our previous and present studies suggest that induction
of the Fas-FADD-caspase 8 pathway may play an im-
portant pathogenetic role in the initiation of tubule cell
apoptosis during the early reperfusion period. The pre-
cise role of DAXX in kidney cell apoptotic cascades is
unclear. However, it is worthwhile noting that DAXXmediates both Fas-dependent and TGF-b-induced ap-
optosis, and the marked renal induction of TGF-b fol-
lowing ischemia is well documented in our study [70].
Transcripts belonging to the intrinsic mitochondrial
apoptotic pathway, namely BAD and BAK, were also
consistently induced in our study [70]. These pro-apop-
totic molecules of the Bcl-2 family normally reside in the
cytosol, but can undergo activation and mitochondrialtranslocation following an apoptotic stimulus, leading
to release of cytochrome c and activation of caspase 9
[56–59]. Both molecules are expressed in kidney tubule
cells [70]. BAK was found to be transcriptionally up-
regulated exclusively at the 3 h reflow period, whereas
the induction of BAD was evident at all times examined.
It is interesting to note that the anti-apoptotic genes Bcl-
2 and NF-jB were also upregulated at earlier time pe-riods in our study. Bcl-2 plays a pivotal protective role
by inhibiting mitochondrial cytochrome c release, and
our findings confirm previous reports of enhanced tu-
bule cell Bcl-2 expression following renal ischemia–rep-
erfusion injury [40]. NF-jB comprises a family of
transcription factors that regulate the expression of
several genes involved in inflammation, proliferation,

P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376 371
and anti-apoptosis [98]. Our findings allow us to hy-pothesize that the simultaneous induction of pro- and
anti-apoptotic transcripts may account for the paucity
of apoptotic cells immediately following IRI [70].
However, the continued upregulation of extrinsic and
intrinsic pro-apoptotic pathway transcripts as observed
in the 12 and 24 h reperfusion periods, combined with a
diminished induction of survival factors such as Bcl-2
and NF-jB, may tilt the balance in favor of apoptosis.The p53 gene is a potent transcription factor that
regulates apoptosis most notably by activating pro-ap-
optotic Bcl-2 family members as well as the Fas-FADD
axis [61]. Both the extrinsic and intrinsic pathways cul-
minate in the activation of caspase 3, with subsequent
entry into the ‘‘execution’’ phase of apoptosis [53]. Our
overall findings suggest induction of multiple apoptotic
pathways, as illustrated in Fig. 2, and lend support tothe notion of caspase inhibition as a potential thera-
peutic tool in renal IRI. Indeed, recent animal studies
have revealed a promising role for caspase inhibitors in
ameliorating injury following renal ischemia [101,102].
From a translational perspective, this concept may be
especially pertinent to renal transplantation, wherein the
kidney is perfused with a defined solution and stored
until re-anastomosis. It will be important in futurestudies to explore the efficacy of adding cell-permeant
caspase inhibitors to the solutions used during cold
storage in ameliorating the IRI typical of kidney trans-
plantation.
It is important to recognize that one of the limitations
to using functional genomic approaches is the fact that
alterations in gene expression are not always predictive
of downstream functional and/or pathophysiologicpathways. Although our results have suggested activa-
tion of select pro-apoptotic pathways at the mRNA
level, additional post-transcriptional and post-transla-
tional events may be required to fully implicate these
factors in the programmed cell death following renal
IRI. For example, phosphorylation is a key event that
determines the activity of p53 and BAD, and the role
of FADD is dependent on complex protein–proteininteractions. The cDNA microarray results provide a
stepping stone, and it will be important in future studies
to fully characterize the biology of the identified apop-
totic factors in order to confirm their role in tubule
cell death.
Induction of regeneration-related genes: lessons from the
developing kidney
The advent of microarrays has revolutionized global
studies of gene expression in the developing kidney.
Using microarrays it is now possible to rapidly assay the
expression level of essentially every mouse or human
gene. New powerful target micro-amplification tech-
niques allow the application of this procedure to very
small samples. Valerius et al. [99] generated two clonalcell lines, mK3 and mK4, that by several criteria rep-
resented early metanephric mesenchyme and later in-
duced metanephric mesenchyme, respectively. For
example, the mK3 cells were able to induce branching
morphogenesis of ureteric bud in organ co-culture ex-
periments, and the mK4 cells showed a more cuboidal,
epithelial-like morphology. Microarray analysis of these
cells identified thousands of genes they expressed, pro-viding a detailed molecular fingerprint that confirmed
their developmental stages [99]. It was observed that the
mK3 cells expressed known markers of early meta-
nephric mesenchyme, including collagen I and vimentin,
while the mK4 cells expressed genes such as Pax-8, Pax-
2, Wnt-4, cadherin-6, collagen IV, and LFB3, diagnostic
of a somewhat later developmental stage during which
the metanephric mesenchyme undergoes epithelialtransformation. In addition, over 70 transcription fac-
tors were expressed in the mK3 cells. Six Hox genes were
expressed, Hoxc 9, Hoxa 11, Hoxc 8, Hoxa 9, Hoxa 10,
and Hoxa 5, in order of decreasing abundance. Their
expression in mK3 cells suggested they are expressed in
early, uninduced metanephric mesenchyme.
Stuart et al. [91] used Affymetrix gene chips to ex-
amine kidney development in the rat. Samples werecollected from E13, 15, 17, 19, newborn, one week, and
adult. K-means hierarchical cluster analysis identified
five groups of genes with distinct expression patterns.
The first group consisted of genes with highest expres-
sion in the earliest examined developing kidney and then
diminishing expression with time. The genes in this
group were largely associated with cell proliferation and
morphogenesis, including pleiotrophin, IGF-II, TGF-b2, ganglioside, frizzled, PDGF-R, Wilms tumor 1, and
Bcl2. The genes of group 2 showed highest expression in
the developing kidney at E15 to E19, with lower ex-
pression levels at earlier and later time points. Many of
these genes encoded proteins of the extracellular matrix
or cytoskeleton, including agrin, decorin, and osteo-
nectin. This group also included a number of morpho-
genetic effectors, including jagged, LIM1, HGF, FGF7,and BMP4. Group 3 contained genes that peaked late
during development, and consisted notably of retro-
transposons. The genes in groups 4 and 5 increased in a
linear fashion into adulthood. These included BMP7,
energy-related genes (such as aldolase, fructose bis-
phosphatase 2, hexokinase, and lactate dehydrogenase),
and transporters (such as aquaporin, H+ transporting
ATPase, and organic cation transporter).Affymetrix oligonucleotidemicroarrays have also been
used to study early kidney development in the mouse [92].
In this case the U74A GeneChips were used, with about
12,500 probe set arrays. Kidneys from E11.5, E12.5,
E13.5, E16.5, and adult were examined, as well as micro-
dissected E11.5 ureteric bud and metanephric mesen-
chyme and P1 total mouse as a reference. Stringent
372 P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376
comparison of gene expression profiles found 428 geneswith significantly elevated expression in the E12.5 em-
bryonic kidney versus the adult.About 10%of these genes
are transcription regulators, includingHoxa 11,Hoxd 11,
N-Myc, histone deacetylase 1, nuclear transcription
factor-Yb, and TG interacting factor. Consistent with the
high growth rate of the embryonic kidney, 35 cell cycle
associated genes were found elevated in expression in the
E12.5 kidney. In addition almost 100 genes with highexpression in the developing kidney were involved in
intermediary metabolism, ribosome biogenesis, DNA
synthesis, and other processes connected with rapidly
dividing cells. This investigation also found several
growth factor-related genes with elevated expression
during early kidney embryogenesis, including GDNF,
midkine, Sfrp2, and follistatin-like protein.
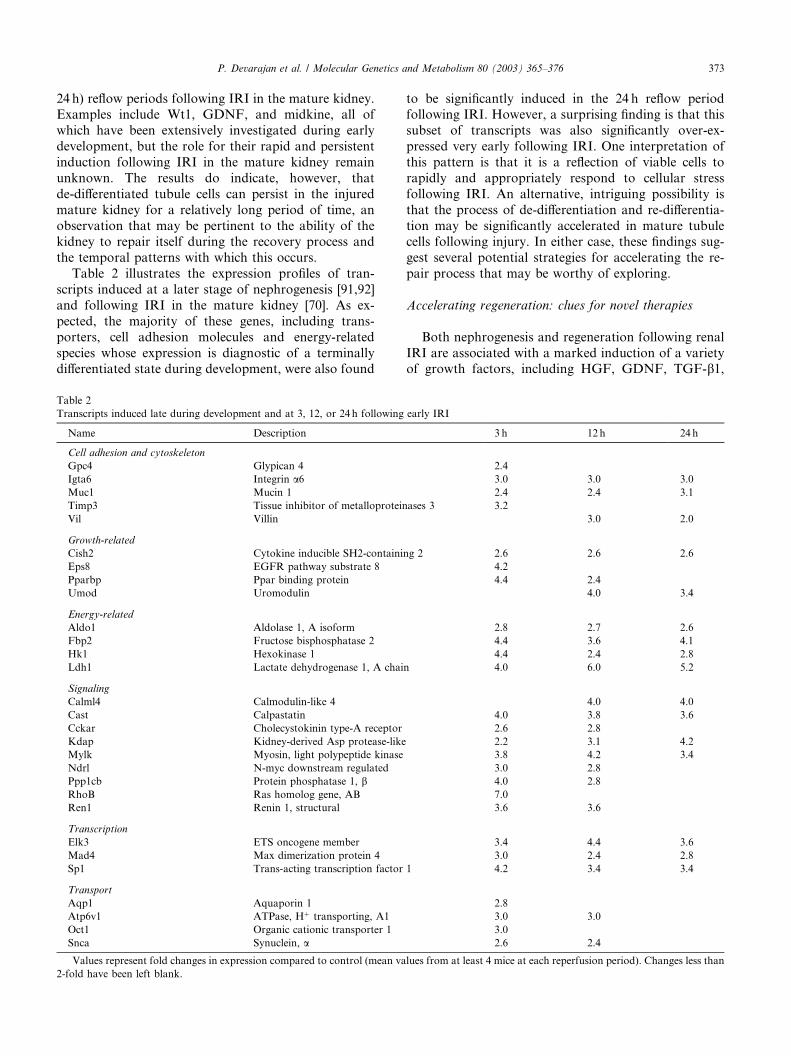
We have completed a detailed comparative analysisof renal gene expression during development and fol-
lowing IRI. Table 1 shows the expression profiles of
Table 1
Transcripts induced early during development and at 3, 12, or 24 h followin
Name Description
Cell adhesion and cytoskeleton
Bys1 Bystin-like
Cdh2 Cadherin-2
Cldn6 Claudin 6
Cldn7 Claudin 7
Efs Embryonal Fyn-associated substr
Lamc2 Laminin, c2Ncam1 Neural cell adhesion molecule 1
Ppl Periplakin
Growth-related
Bcl-2 Bcl-2
Cdkn1a Cdk inhibitor 1a (p21)
Cnn2 Calponin 2
Fstl Follistatin-like
Gdnf Glial-derived neurotrophic factor
Hgf Hepatocyte growth factor
Mdk Midkine
Tgfbi Transforming growth factor, b in
Signaling
Cks1 CDC 28 protein kinase 1
Gfra1 GDNF receptor a1Lag Leukemia-associated gene
Sfrs3 Splicing factor, Arg/Ser-rich 3
Tacstd Tumor-associated Ca signal tran
U2af1 U2 small nuclear RNP auxiliary
Transcription
En2 Engrailed 2
Hdac1 Histone deacetylase 1
Nfyb Nuclear transcription factor-Y bNmyc1 N-myc 1
Tgif TG interacting factor
Wt1 Wilm�s tumor homolog 1
Transport
Hba-a1 Hemoglobin a, adult 1
Values represent fold changes in expression compared to control (mean va
2-fold have been left blank.
transcripts that are predominantly induced early duringdevelopment [91,92] and following early IRI in the
mature kidney [70]. It is evident that several of the genes
that have been shown to play a key role during early
renal morphogenesis are also induced within 3 h of renal
IRI, lending strong support to the concept that regen-
eration recapitulates ontogeny in the kidney. Particu-
larly noteworthy in this respect are a number of
transcription and growth factors (such as Nmyc1, Nfyb,Gfra1, HGF, and Bcl-2) that are upregulated during
early nephrogenesis and exclusively in the 3 h reperfu-
sion period. These results provide molecular genetic
evidence for the hypothesis that injured tubule cells
possess the capacity to de-differentiate to the earliest
stages of development. Equally striking is the finding
that the majority of genes whose expression is a signa-
ture of a very de-differentiated stage of early nephro-genesis, that become down-regulated as development
proceeds, however remain induced at the later (12 and
g early IRI
3 h 12 h 24 h
3.2 3.8 3.4
3.0 3.2
4.0 3.6
12.0 9.1 13.2
ate 2.8 2.6 2.6
4.1 6.0 3.4
2.8 5.4 3.4
2.8 2.4 2.4
5.0
14.2 13.0 10.8
3.2 3.0
2.6 3.8 2.8
2.8 2.8 2.8
2.6
3.0 3.0
duced 3.2 3.0 3.6
2.4 4.4 5.0
2.8
3.4 2.6
3.2 4.0 4.0
sducer 2 10.0 6.1 9.0
factor 1 5.0 3.8 3.8
3.2 2.8
2.8 2.8
3.4
3.0
7.6 4.6 4.0
4.0 4.0 3.2
7.1 4.0 11.4
lues from at least 4 mice at each reperfusion period). Changes less than
P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376 373
24 h) reflow periods following IRI in the mature kidney.Examples include Wt1, GDNF, and midkine, all of
which have been extensively investigated during early
development, but the role for their rapid and persistent
induction following IRI in the mature kidney remain
unknown. The results do indicate, however, that
de-differentiated tubule cells can persist in the injured
mature kidney for a relatively long period of time, an
observation that may be pertinent to the ability of thekidney to repair itself during the recovery process and
the temporal patterns with which this occurs.
Table 2 illustrates the expression profiles of tran-
scripts induced at a later stage of nephrogenesis [91,92]
and following IRI in the mature kidney [70]. As ex-
pected, the majority of these genes, including trans-
porters, cell adhesion molecules and energy-related
species whose expression is diagnostic of a terminallydifferentiated state during development, were also found
Table 2
Transcripts induced late during development and at 3, 12, or 24 h following
Name Description
Cell adhesion and cytoskeleton
Gpc4 Glypican 4
Igta6 Integrin a6Muc1 Mucin 1
Timp3 Tissue inhibitor of metalloprotein
Vil Villin
Growth-related
Cish2 Cytokine inducible SH2-containi
Eps8 EGFR pathway substrate 8
Pparbp Ppar binding protein
Umod Uromodulin
Energy-related
Aldo1 Aldolase 1, A isoform
Fbp2 Fructose bisphosphatase 2
Hk1 Hexokinase 1
Ldh1 Lactate dehydrogenase 1, A chai
Signaling
Calml4 Calmodulin-like 4
Cast Calpastatin
Cckar Cholecystokinin type-A receptor
Kdap Kidney-derived Asp protease-like
Mylk Myosin, light polypeptide kinase
Ndrl N-myc downstream regulated
Ppp1cb Protein phosphatase 1, bRhoB Ras homolog gene, AB
Ren1 Renin 1, structural
Transcription
Elk3 ETS oncogene member
Mad4 Max dimerization protein 4
Sp1 Trans-acting transcription factor
Transport
Aqp1 Aquaporin 1
Atp6v1 ATPase, Hþ transporting, A1
Oct1 Organic cationic transporter 1
Snca Synuclein, a
Values represent fold changes in expression compared to control (mean va
2-fold have been left blank.
to be significantly induced in the 24 h reflow periodfollowing IRI. However, a surprising finding is that this
subset of transcripts was also significantly over-ex-
pressed very early following IRI. One interpretation of
this pattern is that it is a reflection of viable cells to
rapidly and appropriately respond to cellular stress
following IRI. An alternative, intriguing possibility is
that the process of de-differentiation and re-differentia-
tion may be significantly accelerated in mature tubulecells following injury. In either case, these findings sug-
gest several potential strategies for accelerating the re-
pair process that may be worthy of exploring.
Accelerating regeneration: clues for novel therapies
Both nephrogenesis and regeneration following renal
IRI are associated with a marked induction of a varietyof growth factors, including HGF, GDNF, TGF-b1,
early IRI
3 h 12 h 24 h
2.4
3.0 3.0 3.0
2.4 2.4 3.1
ases 3 3.2
3.0 2.0
ng 2 2.6 2.6 2.6
4.2
4.4 2.4
4.0 3.4
2.8 2.7 2.6
4.4 3.6 4.1
4.4 2.4 2.8
n 4.0 6.0 5.2
4.0 4.0
4.0 3.8 3.6
2.6 2.8
2.2 3.1 4.2
3.8 4.2 3.4
3.0 2.8
4.0 2.8
7.0
3.6 3.6
3.4 4.4 3.6
3.0 2.4 2.8
1 4.2 3.4 3.4
2.8
3.0 3.0
3.0
2.6 2.4
lues from at least 4 mice at each reperfusion period). Changes less than

374 P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376
IGF-1, IGF-2, and EGF. This has stimulated interro-gation of the effects of exogenously administered growth
factors for enhancing renal repair following IRI. Per-
haps the best studied example is that of IGF-1, which
clearly accelerates functional recovery and regeneration
of damaged tubular epithelium when administered prior
to, concomitant with, or even following renal IRI in
animal models [100]. However, in a prospective ran-
domized trial of IGF-1 in humans with establishedARF, no beneficial effects were discernible [96]. It has
been suggested that a delay in starting IGF-1 therapy
(due to lack of early biomarkers) might have contrib-
uted to the negative outcome [96]. Another explanation
suggested by the available microarray analysis is that the
molecular genetic response to IRI is multifaceted, with
upregulation of a number of growth factors that may act
in concert to accelerate regeneration, and that exoge-nous administration of a single growth factor may be
insufficient. It is likely that a combination of growth
factors (including factors previously not associated with
IRI such as PDGF-a, angiopoietin, and follistatin that
have emerged from our transcriptome analysis) admin-
istered early in the course of ARF (based on novel bi-
omarkers) will provide the effective therapy that has
eluded clinicians thus far.
Acknowledgments
Portions of the work described in this review weresupported by grants from the NIH to P.D. (DK53289,
DK52612), L.T.P. (DK02702), and S.S.P. (DK61916),
and a Fellowship from the Kidney and Urology Foun-
dation to S.S.
References
[1] R. Thadhani, M. Pascual, J.V. Bonventre, Acute renal failure, N.
Engl. J. Med. 334 (1996) 1448–1460.
[2] C.R. Nolan, R.J. Anderson, Hospital acquired acute renal
failure, J. Am. Soc. Nephrol. 9 (1998) 710–718.
[3] H. Brady, G. Singer, Acute renal failure, Lancet 346 (1995)
1533–1540.
[4] C.L. Edelstein, H. Ling, R. Schrier, The nature of renal cell
injury, Kid. Int. 51 (1997) 1341–1351.
[5] T.D. DuBose Jr., D.G. Warnock, R.L. Mehta, J.V. Bonventre,
M.R. Hammerman, B.A. Molitoris, M.S. Paller, N.J. Siegel, J.
Scherbenske, G.E. Striker, Acute renal failure in the 21st
century: recommendations for management and outcomes
assessment, Am. J. Kidney Dis. 29 (1997) 793–799.
[6] M.S. Paller, Acute renal failure: controversies, clinical trials, and
future directions, Semin. Nephrol. 18 (1998) 482–489.
[7] S. Klahr, S.B. Miller, Acute oliguria, N. Engl. J. Med. 338 (1998)
671–675.
[8] H. Schiffl, S.M. Lang, R. Fischer, Daily hemodialysis and the
outcome of acute renal failure, N. Engl. J. Med. 346 (2002) 305–
310.
[9] R.A. Star, Treatment of acute renal failure, Kidney Int. 54
(1998) 1817–1831.
[10] F. Liano, J. Pascual, Predictive factors and scoring, in: M.A.
Molitoris, W.F. Finn (Eds.), Acute renal failure, W.B. Saunders
Company, Philadelphia, 2001, pp. 507–518.
[11] P. Devarajan, Oliguria, in: E-medicine.com, Boston Medical
Publications, 2001.
[12] R. Woroniecki, P. Devarajan, Acute tubular necrosis, in:
E-medicine.com, Boston Medical Publications, 2001.
[13] S. Bhandari, J.H. Turney, Survivors of acute renal failure who
do not recover renal function, QJM 89 (1996) 415–421.
[14] M.B. Hamel, R.S. Phillips, R.B. Davis, N. Desbiens, A.F.
Connors, J.M. Teno, N. Wenger, J. Lynn, A.W. Wu, W.
Fulkerson, J. Tsevat, Outcomes and cost-effectiveness of
initiating dialysis and continuous aggressive care in seriously
ill hospitalized adults, Ann. Int. Med. 127 (1997) 195–202.
[15] A.A. Magee, J.J. Walshe, Acute renal failure in solid organ
transplantation, in: M.A. Molitoris, W.F. Finn (Eds.), Acute
renal failure, W.B. Saunders Company, Philadelphia, 2001, pp.
322–343.
[16] O.H. Koning, R.J. Ploeg, J.H. van Bockel, et al., Risk factors for
delayed graft function in cadaveric kidney transplantation.
European Multicenter Study Group, Transplantation 63 (1997)
1620–1628.
[17] A.O. Ojo, R.A. Wolfe, P.J. Held, et al., Delayed graft function:
risk factors and implications for renal allograft survival, Trans-
plantation 63 (1997) 968–974.
[18] C.Y. Lu, J.G. Penfield, M.L. Kielar, M.A. Vasquez, D.R.
Jeyarajah, Hypothesis: is renal allograft rejection initiated by the
response to injury during the transplant process?, Kid. Int. 55
(1999) 2157–2168.
[19] N.J. Siegel, S. Van Why, P. Devarajan, K.M. Gaudio, Patho-
genesis of acute renal failure, in: T.M. Barrett, E.D. Avner, W.E.
Harmon (Eds.), Pediatric nephrology, 4th ed., Williams and
Wilkins, Baltimore, 1999, pp. 1109–1118.
[20] N.J. Siegel, P. Devarajan, S.K. Van Why, Renal cell injury:
metabolic and structural alterations, Pediatric Res. 36 (1994)
129–136.
[21] T.A. Sutton, B.A. Molitoris, Mechanisms of cellular injury
in ischemic acute renal failure, Semin. Nephrol. 18 (1998) 490–
497.
[22] A.M. Sheridan, J.V. Bonventre, Cell biology and molecular
mechanisms of injury in ischemic acute renal failure, Curr. Opin.
Nephrol. Hypertens. 9 (2000) 327–434.
[23] J. Savill, Apoptosis and the kidney, J. Am. Soc. Nephrol. 5
(1994) 12–21.
[24] W. Lieberthal, J. Levine, Mechanisms of apoptosis and its
potential role in renal tubular epithelial cell injury, Am. J.
Physiol. 271 (1996) F477–F488.
[25] W. Lieberthal, J.S. Koh, J.S. Levine, Necrosis and apoptosis in
acute renal failure, Semin. Nephrol. 18 (1998) 505–518.
[26] N. Ueda, G.P. Kaushal, S.V. Shah, Apoptotic mechanisms in
acute renal failure, Am. J. Med. 108 (2000) 403–415.
[27] J.S. Levine, W. Lieberthal, Terminal pathways to cell death, in:
M.A. Molitoris, W.F. Finn (Eds.), Acute renal failure, W.B.
Saunders Company, Philadelphia, 2001, pp. 30–59.
[28] B.R. Bonegio, W. Lieberthal, Role of apoptosis in the patho-
genesis of acute renal failure, Curr. Op. Nephrol. Hypertens. 11
(2002) 301–308.
[29] M. Schumer, M.C. Colombel, I.S. Sawczuk, G. Gobe, J.
Connor, K.M. O�Toole, C.A. Olsson, G.J. Wise, R. Buttyan,
Morphologic, biochemical, and molecular evidence of apoptosis
during the reperfusion phase after brief periods of renal
ischemia, Am. J. Pathol. 140 (1992) 831–838.
[30] A. Shimizu, N. Yamanaka, Apoptosis and cell desquamation in
repair process of ischemic tubular necrosis, Virchows Archiv. B
Cell Pathol. 64 (1993) 171–180.
[31] R.A. Zager, S.M. Fuerstenberg, P.H. Baehr, D. Myerson, B.
Torok-Storb, An evaluation of antioxidant effects on recovery

P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376 375
from postischemic acute renal failure, J. Am. Soc. Nephrol. 4
(1994) 1588–1597.
[32] A. Yoshimura, T. Taira, T. Ideura, Expression of apoptosis-
related molecules in acute renal injury, Exp. Nephrol. 4 (1996)
15–18.
[33] T. Nakajima, T. Miyaji, A. Kato, N. Ikegaya, T. Yamamoto, A.
Hishida, Uninephrectomy reduces apoptotic cell death and
enhances renal tubular cell regeneration in ischemic ARF in
rats, Am. J. Physiol. 271 (1996) F846–F853.
[34] A.M. Raafat, M.T. Murray, T. McGuire, M. DeFrain, A.P.
Franko, R.S. Zafar, K. Palmer, L. Diebel, S.A. Dulchavsky,
Calcium blockade reduces renal apoptosis during ischemia
reperfusion, Shock 8 (1997) 186–192.
[35] R. Safirstein, Renal stress response and acute renal failure, Adv.
Ren. Replac. Ther. 4 (1997) 38–42.
[36] R.A. Zager, M. Iwata, D.S. Conrad, K.M. Burkhart, Y.
Igarashi, Altered ceramide and sphingosine expression during
the induction phase of ischemic acute renal failure, Kidney Int.
52 (1997) 60–70.
[37] S. Nogae,M.Miyazaki, N.Kobayashi, T. Saito, K. Abe,H. Saito,
P.K. Nakane, Y. Nakanishi, T. Koji, Induction of apoptosis in
ischemia-reperfusion model of mouse kidney: possible involve-
ment of Fas, J. Am. Soc. Nephrol. 9 (1998) 620–631.
[38] M. Daemen, M. Van de Ven, E. Heineman, W.A. Buurman,
Involvement of endogenous interleukin-10 and TNF alpha in
renal ischemia-reperfusion injury, Transplantation 67 (1999)
792–800.
[39] K.J. Kelly, Z. Plotkin, P.C. Dagher, Guanosine supplementation
reduces apoptosis and protects renal function in the setting of
ischemic injury, J. Clin. Invest. 108 (2001) 1291–1298.
[40] D.P. Basile, H. Liapis, M.R. Hammerman, Expression of bcl-2
and bax in regenerating rat remnant tubules following ischemic
injury, Am. J. Physiol. (Renal Physiol) 272 (1997) F640–F647.
[41] G. Gobe, X.-J. Zhang, D.A. Willgoss, E. Schoch, N.A. Hogg,
Z.H. Endre, Relationship between expression of Bcl-2 genes and
growth factors in ischemic acute renal failure in rat, J. Am. Soc.
Nephrol. 11 (2000) 454–467.
[42] C.-T. Chien, P.-H. Lee, C.-F. Chen, M.-C. Ma, M.-K. Lai, S.-M.
Hsu, De novo demonstration and co-localization of free-radical
production and apoptosis formation in rat kidney subjected to
ischemia/reperfusion, J. Am. Soc. Nephrol. 12 (2001) 973–982.
[43] A.T. Burns, D.R. Davies, A.J. McLaren, L. Cerundolo, P.J.
Morris, S.V. Fuggle, Apoptosis in ischemia/reperfusion injury of
human renal allografts, Transplantation 66 (1998) 872–876.
[44] R. Oberbauer, M. Rohrmoser, H. Regele, F. Muhlbacher, G.
Mayer, Apoptosis of tubular epithelial cells in donor kidney
biopsies predicts early renal allograft function, J. Am. Soc.
Nephrol. 10 (1999) 2006–2013.
[45] M.P. Castaneda, A. Swiatecka-Urban, M.M. Mitsnefes, D.
Feuerstein, F.J. Kaskel, V. Tellis, P. Devarajan, Activation of
mitochondrial apoptotic pathways in human renal allografts
following ischemia, Transplantation 76 (2003) 50–54.
[46] J. Allen, C. Winterford, R.A. Axelson, G.C. Gobe, Effects of
hypoxia on morphological and biochemical characteristics of
renal epithelial cells and tubule cultures, Renal Failure 14 (1992)
453–460.
[47] H. Hagar, N. Ueda, S.V. Shah, Role of reactive oxygen
metabolites in DNA damage and cell death in chemical hypoxic
injury to LLC-PK1 cells, Am. J. Physiol. 271 (1996) F209–215.
[48] H. Hagar, N. Ueda, S.V. Shah, Endonuclease induced DNA
damage and cell death in chemical hypoxic injury to LLC-PK1
cells, Kidney Int. 49 (1996) 355–361.
[49] H. Hagar, N. Ueda, S.V. Shah, Tyrosine phosphorylation in
DNA damage and cell death in hypoxic injury to LLC-PK1 cells,
Kidney Int. 51 (1997) 1747–1753.
[50] G.P. Kaushal, N. Ueda, S.V. Shah, Role of caspases (ICE/CED
3 proteases) in DNA damage and cell death in response to a
mitochondrial inhibitor, antimycin A, Kidney Int. 52 (1997) 438–
445.
[51] W. Lieberthal, S.A. Menza, J.S. Levine, Graded ATP depletion
can cause necrosis or apoptosis of cultured mouse proximal
tubular cells, Am. J. Physiol. 274 (Renal) (1998) F315–F327.
[52] R.L. Feldenberg, S. Thevananther, M. Del Rio, M. De Leon,
P. Devarajan, Partial ATP depletion induces Fas- and caspase-
mediated apoptosis in MDCK cells, Am. J. Physiol. 276 (Renal)
(1999) F837–F846.
[53] N.A. Thornberry, Y. Lazebnik, Caspases: enemies within,
Science 281 (1998) 1312–1316.
[54] G.M. Cohen, Caspases: the executioners of apoptosis, Biochem.
J. 326 (1997) 1–16.
[55] A. Ashkenazi, V.M. Dixit, Death receptors: signaling and
modulation, Science 281 (1998) 1305–1308.
[56] J.M. Adams, S. Cory, The Bcl-2 protein family: arbiters of cell
death, Science 281 (1998) 1322–1326.
[57] P. Saikumar, Z. Dong, J.M. Weinberg, M.A. Venkatachalam,
Mechanisms of cell death in hypoxia/reoxygenation injury,
Oncogene 17 (1998) 3341–3349.
[58] S.J. Korsmeyer, M.C. Wei, M. Saito, S. Meiler, K.J. Oh, P.H.
Schlesinger, Pro-apoptotic cascade activates BID, which oligo-
merizes BAK or BAX into pores that result in the release of
cytochrome c, Cell Death Differ. 7 (2000) 1166–1173.
[59] J.C. Goldstein, N.J. Waterhouse, P. Juin, G.I. Evan, D.R.
Green, The coordinate release of cytochrome c during apoptosis
is rapid, complete and kinetically invariant, Nat. Cell Biol. 2
(2000) 156–162.
[60] X. Luo, I. Budihardjo, H. Zou, C. Slaughter, X. Wang, Bid, a
Bcl-2-interacting protein, mediates cytochrome c release from
mitochondria in response to activation of cell surface death
receptors, Cell 94 (1998) 481–490.
[61] T.F. Burns, W.S. El-Deiry, The p53 pathway and apoptosis, J.
Cell Physiol. 181 (1999) 231–239.
[62] M. Kurella, L.-L. Hsiao, T. Yishida, J.D. Randall, G. Chow,
S.S. Sarang, R.V. Jensen, S.R. Gullans, DNA microarray
analysis of complex biologic processes, J. Am. Soc. Nephrol.
12 (2001) 1072–1078.
[63] H.C. King, A.A. Sinha, Gene expression profile analysis by
DNA microarrays, JAMA 286 (2001) 2280–2288.
[64] M. Schena, D. Shalon, R.W. Davis, P.O. Brown, Quantitative
monitoring of gene expression patterns with a complementary
DNA microarray, Science 270 (1995) 467–470.
[65] M.B. Eisen, P.T. Spellman, P.O. Brown, D. Botstein, Cluster
analysis and display of genome-wide expression patterns, Proc.
Natl. Acad. Sci. USA 95 (1998) 14863–14868.
[66] T.R. Golub, D.K. Slonim, P. Tamayo, C. Huard, M. Gaasen-
beek, J.P. Mesirov, H. Coller, M.L. Loh, J.R. Downing, M.A.
Caliguri, C.D. Bloomfield, E.S. Lander, Molecular classification
of cancer: class discovery and class prediction by gene expression
monitoring, Science 286 (1999) 531–537.
[67] D.J. Lockhart, E.A. Winzeler, Genomics, gene expression and
DNA arrays, Nature 405 (2000) 827–836.
[68] T. Yoshida, M. Kurelia, F. Beato, H. Min, J.R. Ingelfinger, R.L.
Stears, R.D. Swinford, S.R. Gullans, S.-S. Tang, Monitoring
changes in gene expression in renal ischemia-reperfusion in the
rat, Kidney Int. 61 (2002) 1646–1654.
[69] T. Yoshida, S.-.S. Tang, L.-L. Hsiao, R.V. Jensen, J.R.
ingelfinger, S.R. Gullans, Global analysis of gene expression in
renal ischemia-reperfusion in the mouse, Biochem. Biophys. Res.
Commun. 291 (2002) 787–794.
[70] S. Supavekin, W. Zhang, R. Kucherlapati, F.J. Kaskel, L.C.
Moore, P. Devarajan, Differential gene expression following
early renal ischemia-reperfusion, Kidney Int. 63 (2003) 1714–
1724.
[71] R.C. Harris, Growth factors and cytokines in acute renal failure,
Adv. Ren. Replace Ther. 4 (1997) 42–53.

376 P. Devarajan et al. / Molecular Genetics and Metabolism 80 (2003) 365–376
[72] R. Bacallao, L.G. Fine, Molecular events in the organization of
renal tubular epithelium: from nephrogenesis to regeneration,
Am. J. Physiol. (1989) F913–F924.
[73] F.C. Toback, Regeneration after acute tubular necrosis, Kidney
Int. 41 (1992) 226–246.
[74] T. Ichimura, J.V. Bonventre, Growth factors, signaling, and
renal injury and repair, in: M.A. Molitoris, W.F. Finn (Eds.),
Acute renal failure, W.B. Saunders Company, Philadelphia,
2001, pp. 101–118.
[75] J.F. Di Mari, R. Davis, R.L. Safirstein, MAPK activation
determines renal epithelial cell survival during oxidative injury,
Am. J. Physiol. 277 (1999) F195–F203.
[76] K.M. Park, A. Chen, J.V. Bonventre, Prevention of kidney
ischemia/reperfusion-induced functional injury and JNK, p38,
and MAPK kinase activation by remote ischemic pretreatment,
J. Biol. Chem. 276 (2001) 11870–11876.
[77] A.J. Ouellette, R.A. Malt, V.P. Sukhatme, J.V. Bonventre,
Expression of two immediate early genes, Egr-1 and c-fos, in
response to renal ischemia and during renal hypertrophy in mice,
J. Clin. Invest. 85 (1990) 766–777.
[78] R. Witzgall, D. Brown, C. Schwartz, J.V. Bonventre, Localiza-
tion of proliferating cell nuclear antigen, vimentin, c-fos, and
clusterin in the post-ischemic kidney. Evidence for a heteroge-
neous genetic response among nephron segments, and a large
pool of mitotically active and dedifferentiated cells, J. Clin.
Invest. 93 (1994) 2175–2188.
[79] J.V. Bonventre, Dedifferentiation and proliferation of surviving
epithelial cells in acute renal failure, J. Am. Soc. Nephrol. 14
(2003) S55–S61.
[80] T. Ichimura, J.A. Maier, T. Maciag, FGF-1 in normal and
regenerating kidney: expression in mononuclear, interstitial, and
regenerating epithelial cells, Am. J. Physiol. 269 (1995) F653–
F662.
[81] G. Andersson, E. Jennische, IGF-1 immunoreactivity is ex-
pressed by regenerating renal tubular cells after ischaemic injury
in the rat, Acta Physiol. Scand. 132 (1988) 453–457.
[82] S.A. Rogers, G. Ryan, M.R. Hammerman, Insulin-like growth
factors I and II are produced in the metanephros and are
required for growth and development in vitro, J. Cell Biol. 113
(1991) 1447–1453.
[83] M. Nagaike, S. Hirao, H. Tajima, S. Noji, S. Taniguchi, K.
Matsumoto, T. Nakamura, Renotropic function of hepatocyte
growth factor in renal regeneration after unilateral nephrectomy,
J. Biol. Chem. 266 (1991) 22781–22784.
[84] M. Abbate, D. Brown, J.V. Bonventre, Expression of NCAM
recapitulates tubulogenic development in kidneys recovering
from acute ischemia, Am. J. Physiol. 277 (1999) F454–F463.
[85] J.G. Kleinman, E.M. Worcester, A.M. Beshensky, A.M. Sher-
idan, J.V. Bonventre, D. Brown, Upregulation of osteopontin
expression by ischemia in rat kidney, Ann. N. Y. Acad. Sci. 760
(1995) 321–323.
[86] A.J. Lewington, B.J. Padanilam, D.R. Martin, M.R. Hammer-
man, Expression of CD44 in kidney after acute ischemic injury in
rats, Am. J. Physiol. 278 (2000) R247–R254.
[87] D.P. Basile, J.M. Rovak, D.R. Martin, M.R. Hammerman,
Increased TGF-b1 expression in regenerating rat renal tubules
following ischemic injury, Am. J. Physiol. 270 (1996) F500–F509.
[88] C. Koseki, D. Herzlinger, Q. al-Awqati, Apoptosis in meta-
nephric development, J. Cell Biol. 119 (1992) 1327–1333.
[89] H.S. Coles, J.F. Burne, M.C. Raff, Large-scale normal cell death
in the developing rat kidney and its reduction by epidermal
growth factor, Development 118 (1993) 777–784.
[90] J.A. Davies, C.E. Fisher, Genes and proteins in renal develop-
ment, Exp. Nephrol. 10 (2002) 102–113.
[91] R.O. Stuart, K.T. Bush, S.K. Nigam, Changes in global gene
expression patterns during development and maturation of the
rat kidney, Proc. Natl. Acad. Sci. USA 98 (2001) 5649–
5654.
[92] K. Schwab, L.T. Patterson, B.J. Aronow, R. Luckas, H.-C.
Liang, S.S. Potter, A catalogue of gene expression in the
developing kidney, Kidney Int. 64 (2003) 1588–1604.
[93] J. Mishra, Q. Ma, A. Prada, M. Mitsnefes, K. Zahedi, J. Yang, J.
Barasch, P. Devarajan, Identification of NGAL as a novel early
urinary biomarker for ischemic injury, J. Am. Soc. Nephrol. 14
(2003) 2534–2543.
[94] J. Yang, D. Goetz, J.-Y. Li, W. Wand, K. Mori, D. Setlik, T.
Du, H. Erdjument-Bromage, P. Tempst, R. Strong, J. Barasch,
An iron delivery pathway mediated by a lipocalin, Mol. Cell 10
(2002) 1045–1056.
[95] M. Kamarainen, M. Seppala, I. Virtanen, L.C. Andersson,
Expression of glycodelin in MCF-7 breast cancer cells induces
differentiation into organized acinar epithelium, Lab Invest. 77
(1997) 565–573.
[96] M.R. Hammerman, Recapitulation of phylogeny by ontogeny in
nephrology, Kidney Int. 57 (2000) 742–755.
[97] S. Nogae, M. Miyazaki, N. Kobayashi, Induction of apoptosis in
ischemia-reperfusion model of mouse kidney: possible involve-
ment of Fas, J. Am. Soc. Nephrol. 9 (1998) 620–631.
[98] C. Guijarro, J. Egido, Transcription factor-jB and renal disease,
Kidney Int. 59 (2001) 415–424.
[99] M.T. Valerius, L.T. Patterson, D.P. Witte, S.S. Potter, Micro-
array analysis of novel cell lines representing two stages of
metanephric mesenchyme differentiation, Mech. Dev. 112 (2002)
219–232.
[100] M.R. Hammerman, S.B. Miller, Therapeutic use of growth
factors in renal failure, J. Am. Soc. Nephrol. 5 (1994) 1–11.
[101] M.A.R.C. Daemen, C. Van�t Veer, G. Denecker, V.H. Heems-
kerk, T.G.A.M. Wolfs, M. Clauss, P. Vandenabeele, W.A.
Buurman, Inhibition of apoptosis induced by ischemia-reperfu-
sion prevents inflammation, J. Clin. Invest. 104 (1999) 541–
549.
[102] V.Y. Melnikov, S. Faubel, B. Siegmund, M.S. Lucia, D.
Ljubanovic, C.L. Edelstein, Neutrophil-independent mecha-
nisms of caspase-1- and IL-18-mediated ischemic acute
tubular necrosis in mice, J. Clin. Invest. 110 (2002) 1083–
1091.
[103] T. Ichimura, J.V. Bonventre, V. Bailly, H. Wei, C.A. Hession,
R.L. Cate, M. Sanicola, Kidney injury molecule-1 (KIM-1), a
putative epithelial cell adhesion molecule containing a novel
immunoglobulin domain, is up-regulated in renal cells after
injury, J. Biol. Chem. 271 (1998) 4135–4142.
[104] V. Bailly, Z. Zhang, W. Meier, R. Cate, M. Sanicola, J.V.
Bonventre, Shedding of kidney injury molecule-1, a putative
adhesion protein involved in renal regeneration, J. Biol. Chem.
277 (2002) 39739–39748.
[105] W.K. Han, W. Bailly, R. Abichandani, R. Thadani, J.V.
Bonventre, Kidney injury molecule-1 (KIM-1): a novel biomar-
ker for human renal proximal tubule injury, Kidney Int. 62
(2002) 237–244.
[106] Y. Muramatsu, M. Tsujie, Y. Kohda, B. Pham, A.O. Perantoni,
H. Zhao, S.-K. Jo, P.S.T. Yuen, L. Craig, X. Hu, R.A. Star,
Early detection of cysteine-rich protein 61 (CYR61, CCN1) in
urine following renal ischemia reperfusion injury, Kidney Int. 62
(2002) 1601–1610.





























