Embed Size (px)
Citation preview

Gaucher Disease
A Closer Look
Ray Molzon

Introduction
Lysosomal storage disease (Sphingolipidose)Deficiency of glucocerebrosidase causes
buildup of glucocerebrosideGaucher cells, store sphingolipid in spleen,
liver, bone marrow, alveolar spaces, brain tissue

Symptoms
AnemiaReduced platelet countBone demineralizationJaundiceHepatosplenomegalyNeurologic effects (ataxia, seizures, …)

Diagnosis
Three recognized types:Type I (Noncerebral juvenile)
Most common in Ashkenazi Jew lineage (1:450)
Type II (Infantile cerebral)1 in 100,000 live birthsDeath usually occurs w/in 1 year
Type III (Chronic neuropathic/Norbottnian)1 in 50,000 live births

The Gene
GBALocated on 1q21
11 exons, mRNA 1610bpPotential promoters: 2 TATA & 2 CAT
2 ATG start sites, both equally efficient
GBAPLocated ~16kb downstream of GBA

The Enzyme
Reaction catalyzed:D-glucosyl-N-acylsphingosine + H2O <=>
D-glucose + N-acylsphingosine
536 peptidesSignal peptide: 1-39aaGlu235 (acid/base catalyst) and Glu340
(nucleophile) predicted to be in active region5 potential glycosylation sites
Member of O-glycosyl hydrolase 30 family

Mutations
Over 150 GBA mutations identifiedNot all proven to cause disease
4 account for disease in 95% of Ashkenazi Jewish population, 50% of general pop.
Many found to be identical to mutations in pseudogene

L444P
Single bp substitution in exon 10Produces new cleavage site for NCiI
endonucleaseProtein has only 497 aa
Same mutation in GBAPHomozygosity associated w/ Type III
Found to be cause of Norbottnian subtypeRecombinant allele found w/ Type II

N370S
Single bp (A to G) in exon 9Found only in Type I patientsGene frequency of .035 in Ashkenazi
Much rarer in general population
Appears to prevent neuropathic symptoms in patients with L444P allele

84GG
Insertion mutation of G at cDNA 84Also very common in Ashkenazi populationN370S found to have .957 linkage
disequilibrium w/ neighboring PKLR A1 allele84GG has 1.00 linkage disequilibrium w/ A6 allele
of PKLRSupports hypothesis that both mutations originated
in individual founders
IVS2
Single bp substitution in splice donor site of intron 2Causes skipping of exon 2
Accounts for .034 of disease alleles in Ashkenzaki
84GG & IVS2 both result in no enzyme

Treatment
Splenectomy (rarely cures)Gene therapyEnzyme replacement
Placental glucorcerebrosidase (Ceredase)$382,200/year for 70kg patient!
Bone marrow transplantationTreatment of choice in advanced disease
Chemical chaperone treatmentFound to aid folding of N370S mutation
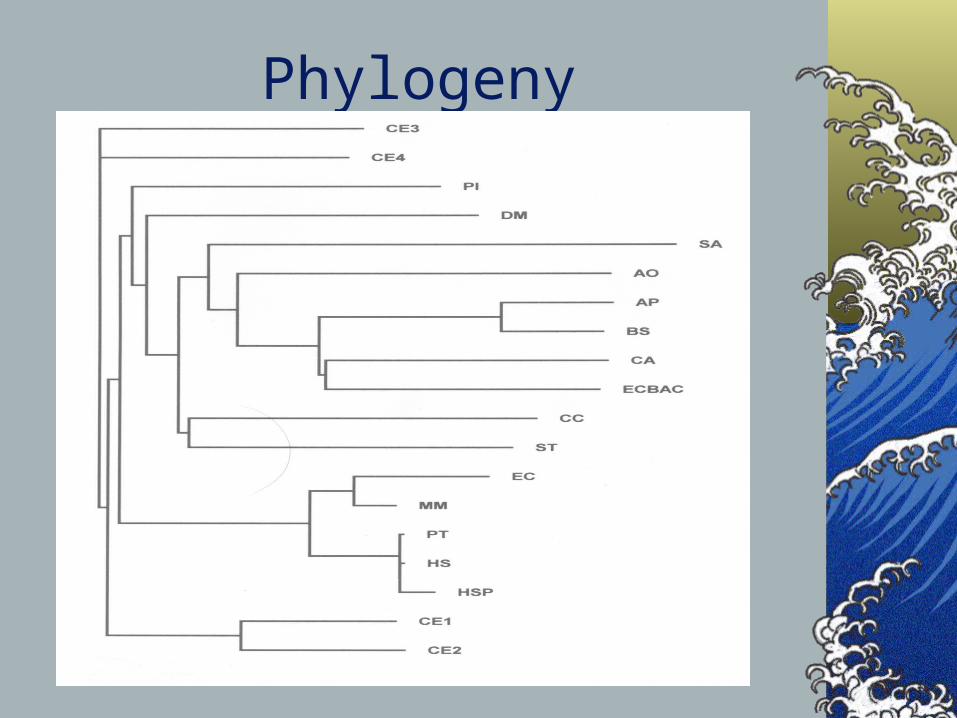
Phylogeny

Tidbits
Identity Matrix
Hs GBA Hs GBAP Pt GBAP Gg GBAP
Hs GBA 1.000 94.953 92.605 92.037Hs GBAP 1.000 92.605 92.037Pt GBAP 1.000 98.537Gg GBAP 1.000
Pseudogene is recent in evolutionary history

ReferencesObtained medical info from various sources:
Obtained genetic/molecular info from:
Used GCG for sequence comparisons



![Glucocerebrosidase and its relevance to Parkinson disease...Gaucher disease [16]. Patients with any neurologic involvement who do not fit into the category of type 2 Gaucher disease](https://img.pdfslide.us/doc/110x75/60b1554575e0fd564115a969/glucocerebrosidase-and-its-relevance-to-parkinson-disease-gaucher-disease-16.jpg)

























