Embed Size (px)
Citation preview

doi:10.1016/j.jmb.2004.10.091 J. Mol. Biol. (2005) 345, 1229–1241
Functional Solubilization of Aggregation-prone HIVEnvelope Proteins by Covalent Fusion with ChaperoneModules
Christian Scholz1*, Peter Schaarschmidt1, Alfred Michael Engel1
Herbert Andres1, Urban Schmitt1, Elke Faatz1, Jochen Balbach2 andFranz Xaver Schmid2
1Roche Diagnostics GmbHNonnenwald 2, D-82377Penzberg, Germany
2Laboratorium fur BiochemieUniversitat Bayreuth, D-95440Bayreuth, Germany
0022-2836/$ - see front matter q 2004 E
Abbreviations used: HIV-1, humavirus type 1; HIV-2, human immuno2; HTLV, human T-cell lymphotropimmunodeficiency virus; gp160, glypolypeptide precursor; gp41, gp21transmembrane glycoproteins fromHIV-2; gp41#, gp36#, ectodomain mgp36; MBP, maltose-binding proteinlysis D, cytosolic E. coli chaperone;protein A, periplasmic E. coli chaperprolyl cis/trans isomerase; SlyD*, Fvariants of SlyD and FkpA; GdmClchloride; Ni-NTA, nickel-nitrilotriacE-mail address of the correspond
The envelope proteins of human immunodeficiency virus (HIV) andhuman T-cell lymphotrophic virus (HTLV) mediate cell attachment andmembrane fusion. For HIV-1, the precursor protein gp160 is cleavedproteolytically into two fragments, the surface-associated receptor bindingsubunit gp120 and the membrane spanning subunit gp41, which isinvolved in membrane fusion during virus entry. Soluble and immuno-reactive variants of gp41 are essential for the reliable diagnosis of HIV-1infections. Hitherto, gp41 was solubilized by adding detergents, or in acidicor alkaline solvents. We find that covalent fusions with SlyD or FkpA, twohomodimeric Escherichia coli chaperones with peptidyl-prolyl isomeraseactivity, solubilize retroviral envelope proteins without compromising theirimmunological reactivity. gp41 from HIV-1, gp36 from HIV-2 and gp21from HTLV could be expressed in large amounts in the Escherichia colicytosol when fused with one or two subunits of SlyD or FkpA. The fusionproteins could be easily isolated and refolded, and showed high solubilityand immunoreactivity, thus providing sensitive and reliable tools fordiagnostic applications. Covalent fusions with SlyD or FkpA might bevaluable generic tools for the solubilization and activation of aggregation-prone proteins.
q 2004 Elsevier Ltd. All rights reserved.
Keywords: protein folding; SlyD; FkpA; retroviral ectodomain; PPIase-chaperones
*Corresponding authorIntroduction
Surface glycoproteins of enveloped viruses areessential for infection and host cell tropism. They
lsevier Ltd. All rights reserve
n immunodeficiencydeficiency virus typehic virus; SIV, simiancoprotein 160,and gp36,HIV-1, HTLV andutants of gp41 and; SlyD, sensitive toFkpA, FK506 bindingone; PPIase, peptidylkpA*, truncated, guanidiniumetate.ing author:
mediate both the attachment to the respective targetcells and the subsequent fusion of viral and cellularmembranes.1 The human immunodeficiency virustype 1 (HIV-1) envelope glycoprotein consists oftwo non-covalently associated subunits, gp120 andgp41, which originate from the proteolytic cleavageof the precursor protein gp160.2 gp120 initiatesinfection by its interaction with the receptor CD4and with several co-receptors of the chemokinereceptor family. The membrane-spanning subunitgp41 triggers membrane fusion. It consists of ahydrophobic N-terminal fusion peptide, followedby two heptad repeat sequences, which are con-nected by the disulfide-bridged apical loop. Thisextracellular part, the so-called ectodomain, isanchored in the virus envelope by a transmembraneregion. The N-heptad repeat of gp41 forms anintermolecular trimeric coiled coil to which thecorresponding C-helices pack in an antiparallelorientation.3–5 Probably, the ectodomain switches
d.

1230 In Vitro Refolding of Chaperone Fusion Proteins
from a six-helix bundle in the fusion-competent“tense form” to a “trimer of hairpin-intermediate”in the fusion-active extended form. This latterintermediate has been a prime target for attemptsto inhibit HIV-1 fusion and infection.3,6–10
The large structural rearrangements of gp41during membrane fusion probably require substan-tial conformational flexibility. Together with its highhydrophobicity, this dynamic character rendersgp41 a very difficult protein for recombinantproduction in a functional form. The proteinaggregates when diluted with physiological buffer,which interferes severely with diagnostic appli-cations. The solubility of the entire HIV-1 ecto-domain including the hydrophobic apical loopregion is very poor, even in the absence of thefusion peptide, and simple and reliable protocolsfor the refolding of gp41 after recombinantproduction are needed.
Moreover, for use in immunoassays, antigens areusually modified by the coupling of Lys orCys residues with N-hydroxy-succinimide ormaleimide derivatives. These reactions require anarrow pH range between 7.0 and 8.5 in which theprotein must be soluble. Thus, the strong tendencyof the gp41 ectodomain to aggregate betweenpH 4.0 and pH 9.0 has been a serious obstacle forroutine use in immunoassays. This highlights theurgent need to improve the solubility of gp41.
So far, most solubilizing attempts have focusedon small fragments of gp41 such as the immuno-dominant loop or parts of the heptad repeatregions.11–13 The structure of a protease-resistantfragment of gp41 from HIV-1, which forms thehexa-helix bundle, has been investigated3,4,5,14 aswell as the structure of gp41 from SIV.15–17
Furthermore, a soluble five-helix construct hasbeen designed in which the N and C-helices ofgp41 are connected by artificial linkers.6 Structuralinformation on the HIV-2 counterpart, i.e. the gp36ectodomain, is scarce, which might be due to thelower impact of HIV-2.
The ectodomains of gp41 from HIV-1 and of gp36from HIV-2 are immunodominant proteins.18 Sol-uble and native-like folded variants would thus beinvaluable tools not only for diagnostic
with grey boxes as well as the cysteine residues bridgingbetween the mutated ectodomains is 49%.
applications, but also to develop strategies forHIV vaccination. Here we report the use of twohomodimeric chaperones, FKP 506 binding proteinA (FkpA) and C-terminally truncated, cysteine-freesensitive to lysis D (SlyD*), as covalently linkedfolding helpers for the ectodomains of HIV-1, HIV-2or human T-cell lymphotrophic virus (HTLV).N-terminal fusions with one or two FkpA orSlyD* modules led to soluble and stable fusionproteins. Optimal results were obtained forfusion proteins in which one ectodomain waslinked with two peptidyl prolyl cis/transisomerase (PPIase)-chaperone modules.
Results
Fusion strategies
The HTLV envelope protein gp21 had beenexpressed successfully in fusion with maltose-binding protein (MBP) from Escherichia coli.19 Inanalogy, we fused MBP to the 536-681 fragment ofgp41 from HIV-1, but this fusion protein showed atendency to aggregate. In a second attempt, weintroduced four point mutations into the helicalheptad repeat 1 of the ectodomain (L555E, L566E,I573T, I580E, see Figure 1). We reasoned that thenegative charges of the three Glu residues at the aand d positions of the helical wheel projectionmight improve solubility while retaining the highhelical propensity. The I573T substitution wasintroduced to mimic gp36, which seemed to beslightly less aggregation-prone than gp41 whenrefolded in isolation (H.A., unpublished results). Infusion with MBP this mutant gp41 (gp41#) wasindeed more soluble, but it still aggregated slowlyover time (data not shown).
An important clue for an alternative fusionpartner came from the analysis of two host proteinsthat copurified with the MBP-gp41# fusionprotein. They were identified as SlyD and FkpAby N-terminal sequence analysis. These two E. coliproteins combine chaperone and PPIase functions.PPIases are ubiquitous enzymes that accelerateprolyl isomerizations during protein folding.20,21
Figure 1. Alignment of theectodomains gp41 (536-681) fromHIV-1 and gp36 (534-676) fromHIV-2. The two heptad repeatregions and the disulfide-bridgedapical loop (598-604) of gp41 arehighlighted with boxes. The fouramino acid residue motif (WNAS)in the gp41 ectodomain is markedby bold print. The four pointmutations in the gp41 and thethree point mutations in the gp36N-heptad repeat are underlaid
the immunodominant apical loop. The identity level of
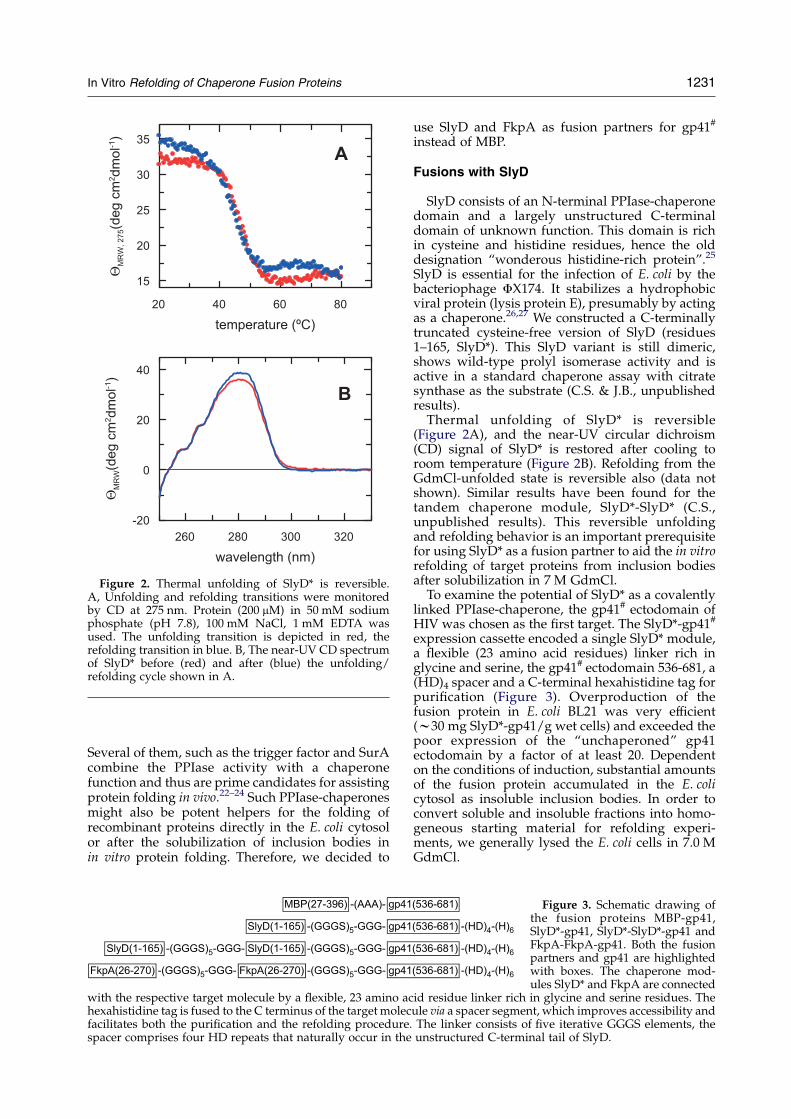
Figure 2. Thermal unfolding of SlyD* is reversible.A, Unfolding and refolding transitions were monitoredby CD at 275 nm. Protein (200 mM) in 50 mM sodiumphosphate (pH 7.8), 100 mM NaCl, 1 mM EDTA wasused. The unfolding transition is depicted in red, therefolding transition in blue. B, The near-UV CD spectrumof SlyD* before (red) and after (blue) the unfolding/refolding cycle shown in A.
In Vitro Refolding of Chaperone Fusion Proteins 1231
Several of them, such as the trigger factor and SurAcombine the PPIase activity with a chaperonefunction and thus are prime candidates for assistingprotein folding in vivo.22–24 Such PPIase-chaperonesmight also be potent helpers for the folding ofrecombinant proteins directly in the E. coli cytosolor after the solubilization of inclusion bodies inin vitro protein folding. Therefore, we decided to
with the respective target molecule by a flexible, 23 amino achexahistidine tag is fused to the C terminus of the target molecfacilitates both the purification and the refolding procedure.spacer comprises four HD repeats that naturally occur in the
use SlyD and FkpA as fusion partners for gp41#
instead of MBP.
Fusions with SlyD
SlyD consists of an N-terminal PPIase-chaperonedomain and a largely unstructured C-terminaldomain of unknown function. This domain is richin cysteine and histidine residues, hence the olddesignation “wonderous histidine-rich protein”.25
SlyD is essential for the infection of E. coli by thebacteriophage FX174. It stabilizes a hydrophobicviral protein (lysis protein E), presumably by actingas a chaperone.26,27 We constructed a C-terminallytruncated cysteine-free version of SlyD (residues1–165, SlyD*). This SlyD variant is still dimeric,shows wild-type prolyl isomerase activity and isactive in a standard chaperone assay with citratesynthase as the substrate (C.S. & J.B., unpublishedresults).Thermal unfolding of SlyD* is reversible
(Figure 2A), and the near-UV circular dichroism(CD) signal of SlyD* is restored after cooling toroom temperature (Figure 2B). Refolding from theGdmCl-unfolded state is reversible also (data notshown). Similar results have been found for thetandem chaperone module, SlyD*-SlyD* (C.S.,unpublished results). This reversible unfoldingand refolding behavior is an important prerequisitefor using SlyD* as a fusion partner to aid the in vitrorefolding of target proteins from inclusion bodiesafter solubilization in 7 M GdmCl.To examine the potential of SlyD* as a covalently
linked PPIase-chaperone, the gp41# ectodomain ofHIV was chosen as the first target. The SlyD*-gp41#
expression cassette encoded a single SlyD* module,a flexible (23 amino acid residues) linker rich inglycine and serine, the gp41# ectodomain 536-681, a(HD)4 spacer and a C-terminal hexahistidine tag forpurification (Figure 3). Overproduction of thefusion protein in E. coli BL21 was very efficient(w30 mg SlyD*-gp41/g wet cells) and exceeded thepoor expression of the “unchaperoned” gp41ectodomain by a factor of at least 20. Dependenton the conditions of induction, substantial amountsof the fusion protein accumulated in the E. colicytosol as insoluble inclusion bodies. In order toconvert soluble and insoluble fractions into homo-geneous starting material for refolding experi-ments, we generally lysed the E. coli cells in 7.0 MGdmCl.
Figure 3. Schematic drawing ofthe fusion proteins MBP-gp41,SlyD*-gp41, SlyD*-SlyD*-gp41 andFkpA-FkpA-gp41. Both the fusionpartners and gp41 are highlightedwith boxes. The chaperone mod-ules SlyD* and FkpA are connected
id residue linker rich in glycine and serine residues. Theule via a spacer segment, which improves accessibility andThe linker consists of five iterative GGGS elements, theunstructured C-terminal tail of SlyD.
1232 In Vitro Refolding of Chaperone Fusion Proteins
Matrix-assisted refolding and renaturing gel-filtration yield soluble SlyD*-gp41# fusionprotein
Soluble and immunoreactive fusion protein wasobtained by a coupled purification and refoldingprocedure. The unfolded protein in 7.0 M GdmClwas bound to a Ni-NTA resin, and renatured in situby passing 15 column volumes of refolding bufferover the column in an overnight reaction. Afterelution with imidazole, the fusion protein wassoluble in phosphate buffer (in the absence ofdetergents or other additives) and could beconcentrated to O40 mg/ml without detectableaggregation.
The SlyD*-gp41# fusion protein could also berenatured by size-exclusion chromatography. To
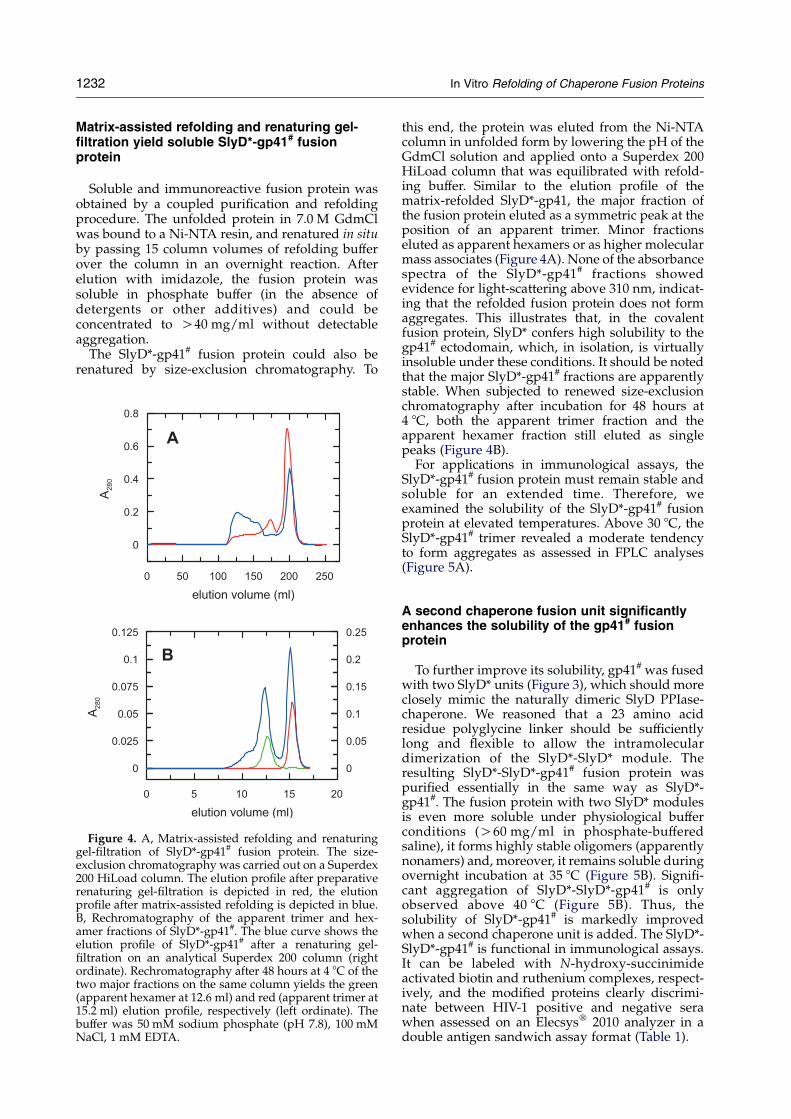
Figure 4. A, Matrix-assisted refolding and renaturinggel-filtration of SlyD*-gp41# fusion protein. The size-exclusion chromatography was carried out on a Superdex200 HiLoad column. The elution profile after preparativerenaturing gel-filtration is depicted in red, the elutionprofile after matrix-assisted refolding is depicted in blue.B, Rechromatography of the apparent trimer and hex-amer fractions of SlyD*-gp41#. The blue curve shows theelution profile of SlyD*-gp41# after a renaturing gel-filtration on an analytical Superdex 200 column (rightordinate). Rechromatography after 48 hours at 4 8C of thetwo major fractions on the same column yields the green(apparent hexamer at 12.6 ml) and red (apparent trimer at15.2 ml) elution profile, respectively (left ordinate). Thebuffer was 50 mM sodium phosphate (pH 7.8), 100 mMNaCl, 1 mM EDTA.
this end, the protein was eluted from the Ni-NTAcolumn in unfolded form by lowering the pH of theGdmCl solution and applied onto a Superdex 200HiLoad column that was equilibrated with refold-ing buffer. Similar to the elution profile of thematrix-refolded SlyD*-gp41, the major fraction ofthe fusion protein eluted as a symmetric peak at theposition of an apparent trimer. Minor fractionseluted as apparent hexamers or as higher molecularmass associates (Figure 4A). None of the absorbancespectra of the SlyD*-gp41# fractions showedevidence for light-scattering above 310 nm, indicat-ing that the refolded fusion protein does not formaggregates. This illustrates that, in the covalentfusion protein, SlyD* confers high solubility to thegp41# ectodomain, which, in isolation, is virtuallyinsoluble under these conditions. It should be notedthat the major SlyD*-gp41# fractions are apparentlystable. When subjected to renewed size-exclusionchromatography after incubation for 48 hours at4 8C, both the apparent trimer fraction and theapparent hexamer fraction still eluted as singlepeaks (Figure 4B).
For applications in immunological assays, theSlyD*-gp41# fusion protein must remain stable andsoluble for an extended time. Therefore, weexamined the solubility of the SlyD*-gp41# fusionprotein at elevated temperatures. Above 30 8C, theSlyD*-gp41# trimer revealed a moderate tendencyto form aggregates as assessed in FPLC analyses(Figure 5A).
A second chaperone fusion unit significantlyenhances the solubility of the gp41# fusionprotein
To further improve its solubility, gp41# was fusedwith two SlyD* units (Figure 3), which should moreclosely mimic the naturally dimeric SlyD PPIase-chaperone. We reasoned that a 23 amino acidresidue polyglycine linker should be sufficientlylong and flexible to allow the intramoleculardimerization of the SlyD*-SlyD* module. Theresulting SlyD*-SlyD*-gp41# fusion protein waspurified essentially in the same way as SlyD*-gp41#. The fusion protein with two SlyD* modulesis even more soluble under physiological bufferconditions (O60 mg/ml in phosphate-bufferedsaline), it forms highly stable oligomers (apparentlynonamers) and, moreover, it remains soluble duringovernight incubation at 35 8C (Figure 5B). Signifi-cant aggregation of SlyD*-SlyD*-gp41# is onlyobserved above 40 8C (Figure 5B). Thus, thesolubility of SlyD*-gp41# is markedly improvedwhen a second chaperone unit is added. The SlyD*-SlyD*-gp41# is functional in immunological assays.It can be labeled with N-hydroxy-succinimideactivated biotin and ruthenium complexes, respect-ively, and the modified proteins clearly discrimi-nate between HIV-1 positive and negative serawhen assessed on an Elecsysw 2010 analyzer in adouble antigen sandwich assay format (Table 1).
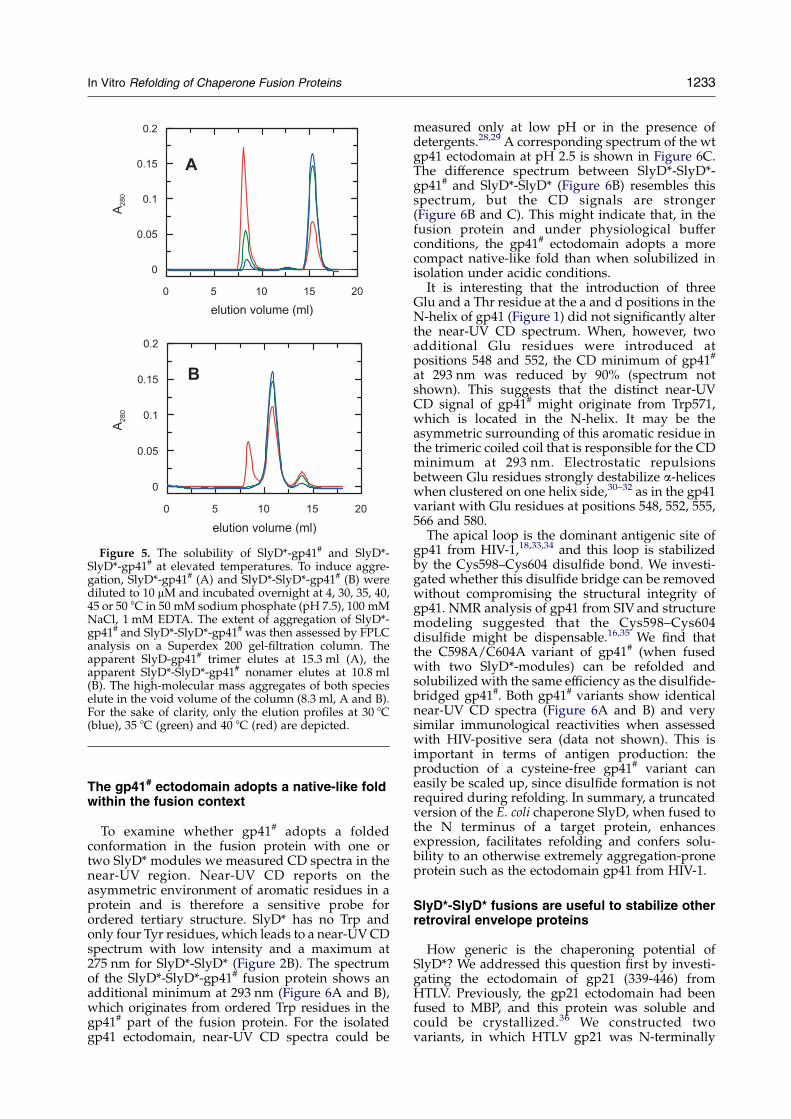
Figure 5. The solubility of SlyD*-gp41# and SlyD*-SlyD*-gp41# at elevated temperatures. To induce aggre-gation, SlyD*-gp41# (A) and SlyD*-SlyD*-gp41# (B) werediluted to 10 mM and incubated overnight at 4, 30, 35, 40,45 or 50 8C in 50 mM sodium phosphate (pH 7.5), 100 mMNaCl, 1 mM EDTA. The extent of aggregation of SlyD*-gp41# and SlyD*-SlyD*-gp41# was then assessed by FPLCanalysis on a Superdex 200 gel-filtration column. Theapparent SlyD-gp41# trimer elutes at 15.3 ml (A), theapparent SlyD*-SlyD*-gp41# nonamer elutes at 10.8 ml(B). The high-molecular mass aggregates of both specieselute in the void volume of the column (8.3 ml, A and B).For the sake of clarity, only the elution profiles at 30 8C(blue), 35 8C (green) and 40 8C (red) are depicted.
In Vitro Refolding of Chaperone Fusion Proteins 1233
The gp41# ectodomain adopts a native-like foldwithin the fusion context
To examine whether gp41# adopts a foldedconformation in the fusion protein with one ortwo SlyD* modules we measured CD spectra in thenear-UV region. Near-UV CD reports on theasymmetric environment of aromatic residues in aprotein and is therefore a sensitive probe forordered tertiary structure. SlyD* has no Trp andonly four Tyr residues, which leads to a near-UVCDspectrum with low intensity and a maximum at275 nm for SlyD*-SlyD* (Figure 2B). The spectrumof the SlyD*-SlyD*-gp41# fusion protein shows anadditional minimum at 293 nm (Figure 6A and B),which originates from ordered Trp residues in thegp41# part of the fusion protein. For the isolatedgp41 ectodomain, near-UV CD spectra could be
measured only at low pH or in the presence ofdetergents.28,29 A corresponding spectrum of the wtgp41 ectodomain at pH 2.5 is shown in Figure 6C.The difference spectrum between SlyD*-SlyD*-gp41# and SlyD*-SlyD* (Figure 6B) resembles thisspectrum, but the CD signals are stronger(Figure 6B and C). This might indicate that, in thefusion protein and under physiological bufferconditions, the gp41# ectodomain adopts a morecompact native-like fold than when solubilized inisolation under acidic conditions.It is interesting that the introduction of three
Glu and a Thr residue at the a and d positions in theN-helix of gp41 (Figure 1) did not significantly alterthe near-UV CD spectrum. When, however, twoadditional Glu residues were introduced atpositions 548 and 552, the CD minimum of gp41#
at 293 nm was reduced by 90% (spectrum notshown). This suggests that the distinct near-UVCD signal of gp41# might originate from Trp571,which is located in the N-helix. It may be theasymmetric surrounding of this aromatic residue inthe trimeric coiled coil that is responsible for the CDminimum at 293 nm. Electrostatic repulsionsbetween Glu residues strongly destabilize a-heliceswhen clustered on one helix side,30–32 as in the gp41variant with Glu residues at positions 548, 552, 555,566 and 580.The apical loop is the dominant antigenic site of
gp41 from HIV-1,18,33,34 and this loop is stabilizedby the Cys598–Cys604 disulfide bond. We investi-gated whether this disulfide bridge can be removedwithout compromising the structural integrity ofgp41. NMR analysis of gp41 from SIV and structuremodeling suggested that the Cys598–Cys604disulfide might be dispensable.16,35 We find thatthe C598A/C604A variant of gp41# (when fusedwith two SlyD*-modules) can be refolded andsolubilized with the same efficiency as the disulfide-bridged gp41#. Both gp41# variants show identicalnear-UV CD spectra (Figure 6A and B) and verysimilar immunological reactivities when assessedwith HIV-positive sera (data not shown). This isimportant in terms of antigen production: theproduction of a cysteine-free gp41# variant caneasily be scaled up, since disulfide formation is notrequired during refolding. In summary, a truncatedversion of the E. coli chaperone SlyD, when fused tothe N terminus of a target protein, enhancesexpression, facilitates refolding and confers solu-bility to an otherwise extremely aggregation-proneprotein such as the ectodomain gp41 from HIV-1.
SlyD*-SlyD* fusions are useful to stabilize otherretroviral envelope proteins
How generic is the chaperoning potential ofSlyD*? We addressed this question first by investi-gating the ectodomain of gp21 (339-446) fromHTLV. Previously, the gp21 ectodomain had beenfused to MBP, and this protein was soluble andcould be crystallized.36 We constructed twovariants, in which HTLV gp21 was N-terminally
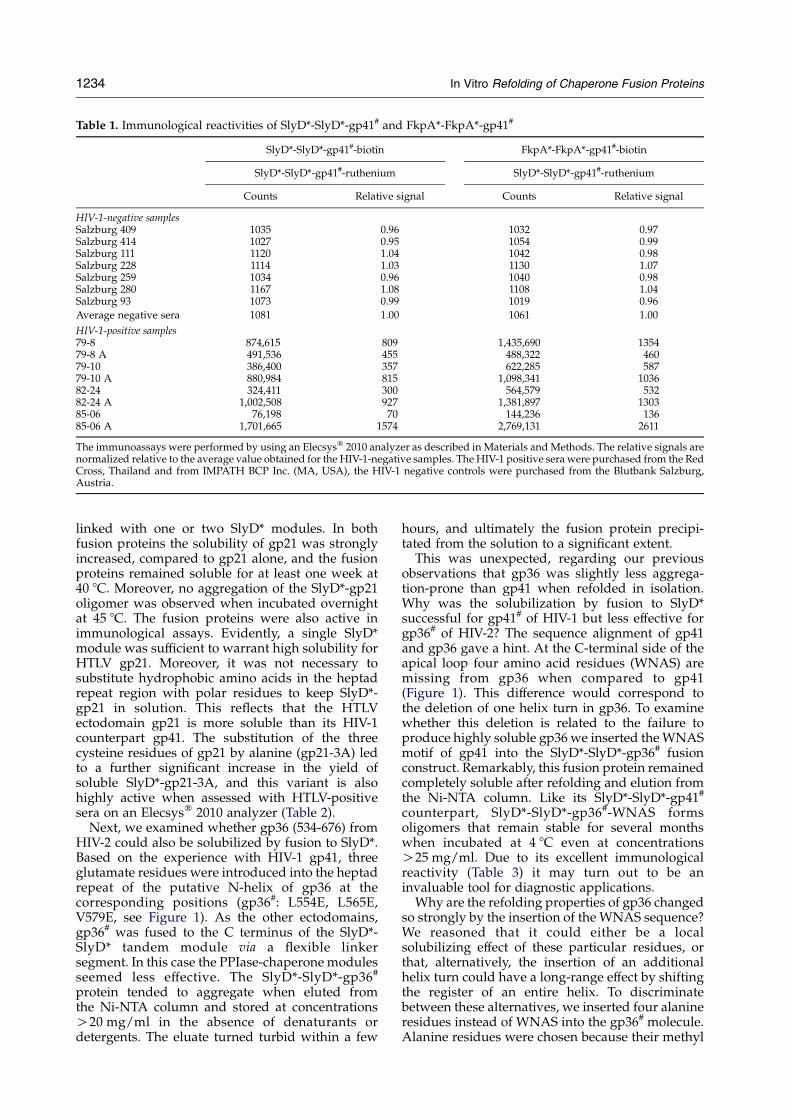
Table 1. Immunological reactivities of SlyD*-SlyD*-gp41# and FkpA*-FkpA*-gp41#
SlyD*-SlyD*-gp41#-biotin FkpA*-FkpA*-gp41#-biotin
SlyD*-SlyD*-gp41#-ruthenium SlyD*-SlyD*-gp41#-ruthenium
Counts Relative signal Counts Relative signal
HIV-1-negative samplesSalzburg 409 1035 0.96 1032 0.97Salzburg 414 1027 0.95 1054 0.99Salzburg 111 1120 1.04 1042 0.98Salzburg 228 1114 1.03 1130 1.07Salzburg 259 1034 0.96 1040 0.98Salzburg 280 1167 1.08 1108 1.04Salzburg 93 1073 0.99 1019 0.96
Average negative sera 1081 1.00 1061 1.00
HIV-1-positive samples79-8 874,615 809 1,435,690 135479-8 A 491,536 455 488,322 46079-10 386,400 357 622,285 58779-10 A 880,984 815 1,098,341 103682-24 324,411 300 564,579 53282-24 A 1,002,508 927 1,381,897 130385-06 76,198 70 144,236 13685-06 A 1,701,665 1574 2,769,131 2611
The immunoassays were performed by using an Elecsysw 2010 analyzer as described in Materials andMethods. The relative signals arenormalized relative to the average value obtained for the HIV-1-negative samples. The HIV-1 positive sera were purchased from the RedCross, Thailand and from IMPATH BCP Inc. (MA, USA), the HIV-1 negative controls were purchased from the Blutbank Salzburg,Austria.
1234 In Vitro Refolding of Chaperone Fusion Proteins
linked with one or two SlyD* modules. In bothfusion proteins the solubility of gp21 was stronglyincreased, compared to gp21 alone, and the fusionproteins remained soluble for at least one week at40 8C. Moreover, no aggregation of the SlyD*-gp21oligomer was observed when incubated overnightat 45 8C. The fusion proteins were also active inimmunological assays. Evidently, a single SlyD*module was sufficient to warrant high solubility forHTLV gp21. Moreover, it was not necessary tosubstitute hydrophobic amino acids in the heptadrepeat region with polar residues to keep SlyD*-gp21 in solution. This reflects that the HTLVectodomain gp21 is more soluble than its HIV-1counterpart gp41. The substitution of the threecysteine residues of gp21 by alanine (gp21-3A) ledto a further significant increase in the yield ofsoluble SlyD*-gp21-3A, and this variant is alsohighly active when assessed with HTLV-positivesera on an Elecsysw 2010 analyzer (Table 2).
Next, we examined whether gp36 (534-676) fromHIV-2 could also be solubilized by fusion to SlyD*.Based on the experience with HIV-1 gp41, threeglutamate residues were introduced into the heptadrepeat of the putative N-helix of gp36 at thecorresponding positions (gp36#: L554E, L565E,V579E, see Figure 1). As the other ectodomains,gp36# was fused to the C terminus of the SlyD*-SlyD* tandem module via a flexible linkersegment. In this case the PPIase-chaperonemodulesseemed less effective. The SlyD*-SlyD*-gp36#
protein tended to aggregate when eluted fromthe Ni-NTA column and stored at concentrationsO20 mg/ml in the absence of denaturants ordetergents. The eluate turned turbid within a few
hours, and ultimately the fusion protein precipi-tated from the solution to a significant extent.
This was unexpected, regarding our previousobservations that gp36 was slightly less aggrega-tion-prone than gp41 when refolded in isolation.Why was the solubilization by fusion to SlyD*successful for gp41# of HIV-1 but less effective forgp36# of HIV-2? The sequence alignment of gp41and gp36 gave a hint. At the C-terminal side of theapical loop four amino acid residues (WNAS) aremissing from gp36 when compared to gp41(Figure 1). This difference would correspond tothe deletion of one helix turn in gp36. To examinewhether this deletion is related to the failure toproduce highly soluble gp36 we inserted theWNASmotif of gp41 into the SlyD*-SlyD*-gp36# fusionconstruct. Remarkably, this fusion protein remainedcompletely soluble after refolding and elution fromthe Ni-NTA column. Like its SlyD*-SlyD*-gp41#
counterpart, SlyD*-SlyD*-gp36#-WNAS formsoligomers that remain stable for several monthswhen incubated at 4 8C even at concentrationsO25 mg/ml. Due to its excellent immunologicalreactivity (Table 3) it may turn out to be aninvaluable tool for diagnostic applications.
Why are the refolding properties of gp36 changedso strongly by the insertion of the WNAS sequence?We reasoned that it could either be a localsolubilizing effect of these particular residues, orthat, alternatively, the insertion of an additionalhelix turn could have a long-range effect by shiftingthe register of an entire helix. To discriminatebetween these alternatives, we inserted four alanineresidues instead of WNAS into the gp36# molecule.Alanine residues were chosen because their methyl
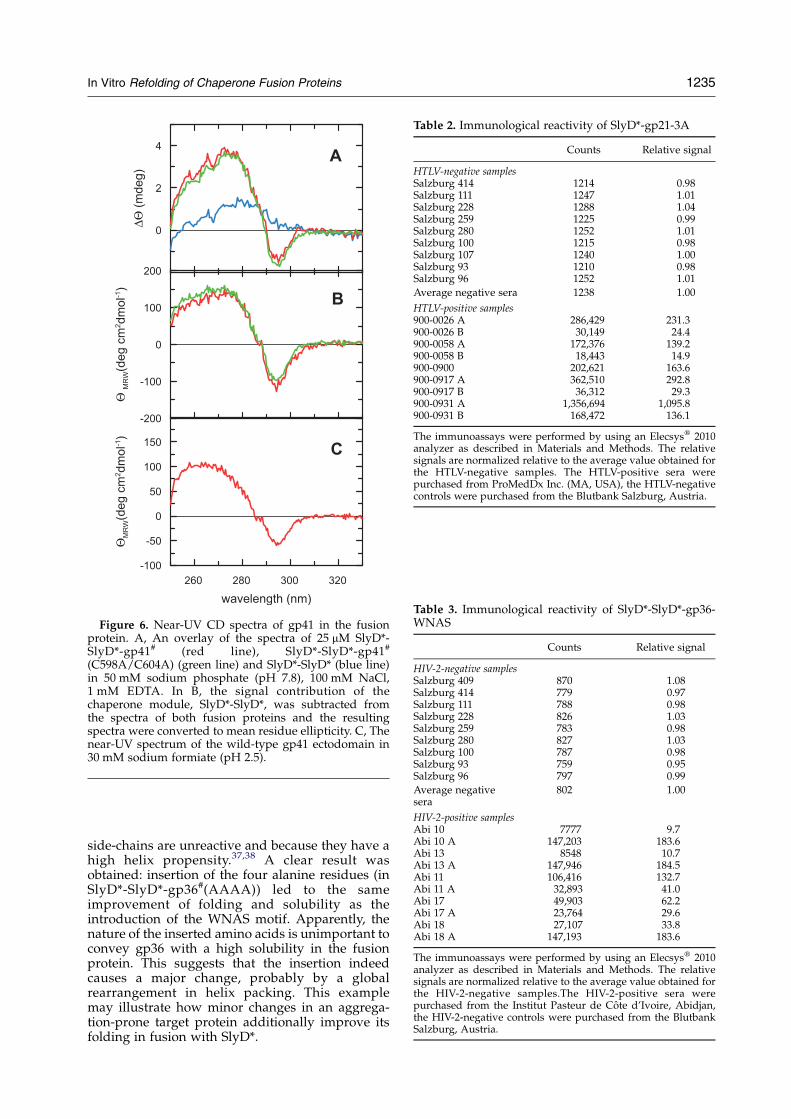
Table 2. Immunological reactivity of SlyD*-gp21-3A
Counts Relative signal
HTLV-negative samplesSalzburg 414 1214 0.98Salzburg 111 1247 1.01Salzburg 228 1288 1.04Salzburg 259 1225 0.99Salzburg 280 1252 1.01Salzburg 100 1215 0.98Salzburg 107 1240 1.00Salzburg 93 1210 0.98Salzburg 96 1252 1.01
Average negative sera 1238 1.00
HTLV-positive samples900-0026 A 286,429 231.3900-0026 B 30,149 24.4900-0058 A 172,376 139.2900-0058 B 18,443 14.9900-0900 202,621 163.6900-0917 A 362,510 292.8900-0917 B 36,312 29.3900-0931 A 1,356,694 1,095.8900-0931 B 168,472 136.1
The immunoassays were performed by using an Elecsysw 2010analyzer as described in Materials and Methods. The relativesignals are normalized relative to the average value obtained forthe HTLV-negative samples. The HTLV-positive sera werepurchased from ProMedDx Inc. (MA, USA), the HTLV-negativecontrols were purchased from the Blutbank Salzburg, Austria.
Table 3. Immunological reactivity of SlyD*-SlyD*-gp36-WNAS
Counts Relative signal
HIV-2-negative samplesSalzburg 409 870 1.08Salzburg 414 779 0.97Salzburg 111 788 0.98Salzburg 228 826 1.03Salzburg 259 783 0.98Salzburg 280 827 1.03Salzburg 100 787 0.98Salzburg 93 759 0.95Salzburg 96 797 0.99
Average negativesera
802 1.00
HIV-2-positive samplesAbi 10 7777 9.7Abi 10 A 147,203 183.6Abi 13 8548 10.7Abi 13 A 147,946 184.5Abi 11 106,416 132.7Abi 11 A 32,893 41.0Abi 17 49,903 62.2Abi 17 A 23,764 29.6Abi 18 27,107 33.8Abi 18 A 147,193 183.6
The immunoassays were performed by using an Elecsysw 2010analyzer as described in Materials and Methods. The relativesignals are normalized relative to the average value obtained forthe HIV-2-negative samples.The HIV-2-positive sera werepurchased from the Institut Pasteur de Cote d’Ivoire, Abidjan,the HIV-2-negative controls were purchased from the BlutbankSalzburg, Austria.
Figure 6. Near-UV CD spectra of gp41 in the fusionprotein. A, An overlay of the spectra of 25 mM SlyD*-SlyD*-gp41# (red line), SlyD*-SlyD*-gp41#
(C598A/C604A) (green line) and SlyD*-SlyD* (blue line)in 50 mM sodium phosphate (pH 7.8), 100 mM NaCl,1 mM EDTA. In B, the signal contribution of thechaperone module, SlyD*-SlyD*, was subtracted fromthe spectra of both fusion proteins and the resultingspectra were converted to mean residue ellipticity. C, Thenear-UV spectrum of the wild-type gp41 ectodomain in30 mM sodium formiate (pH 2.5).
In Vitro Refolding of Chaperone Fusion Proteins 1235
side-chains are unreactive and because they have ahigh helix propensity.37,38 A clear result wasobtained: insertion of the four alanine residues (inSlyD*-SlyD*-gp36#(AAAA)) led to the sameimprovement of folding and solubility as theintroduction of the WNAS motif. Apparently, thenature of the inserted amino acids is unimportant toconvey gp36 with a high solubility in the fusionprotein. This suggests that the insertion indeedcauses a major change, probably by a globalrearrangement in helix packing. This examplemay illustrate how minor changes in an aggrega-tion-prone target protein additionally improve itsfolding in fusion with SlyD*.

1236 In Vitro Refolding of Chaperone Fusion Proteins
Fusions with FkpA
Often it is desirable to have a choice of differentfusion partners for the protein to be folded andsolubilized. Particularly in immunological assaysthe fused chaperone might evoke cross-reactionsand thus lead to false-positive results. Therefore, wesearched for an alternative fusion module thatshares the chaperone properties but is phylogeneti-cally distinct from SlyD. As mentioned above, FkpAwas found as a persistent contaminant in earlypreparations of the MBP-gp41# fusion protein.FkpA is a homodimeric protein with prolyl iso-merase and chaperone functions39,40 and it issupposed to be involved in folding events in theE. coli periplasm.41,42 FkpA suppresses aggregationin vitro39,41,42 and facilitates the refolding ofaggregation-prone proteins in vivo.41–43
To examine its suitability as an alternative fusionpartner, the mature form of FkpA (FkpA 26-270)was linked to the N terminus of the target protein.The signal sequence of FkpA was omitted to retainthe fusion proteins in the host cytosol. As in theprevious experiments with SlyD*, the viral ecto-domains gp41# of HIV-1, gp21 of HTLV-1 andgp36#-WNAS of HIV-2 were fused to one or twoFkpA modules. All of these FkpA-X fusionproteins were overproduced very efficiently inE. coli BL21. They accumulated mainly as inclusionbodies in the cytosol, and all of them werecompletely soluble after refolding and elutionfrom the Ni-NTA column. Remarkably, the solu-bility of the tandem fusion proteins of the FkpA-FkpA-X or SlyD*-SlyD*-X type exceeded 50 mg/mlfor all retroviral ectodomains examined. Forinstance, FkpA-FkpA-gp36#-WNAS remainedsoluble in phosphate buffer at concentrations upto 85 mg/ml. Thus, SlyD* and FkpA modules areequally efficient as intramolecular PPIase-chaper-ones in the folding and solubilization of covalentlylinked target proteins.
The interaction between the chaperone modulesand the target proteins seems to be intramolecularrather than intermolecular. When the isolated gp41ectodomain and the FkpA-FkpA module were co-incubated (50 mM each) in 7.0 M GdmCl and thensubjected to renaturing gel-filtration, FkpA-FkpAwas eluted, but gp41 aggregated quantitatively onthe column. This suggests that gp41 and FkpA-FkpA do not form a stable intermolecular complexduring refolding. A similar result was obtainedwhen gp41 and FkpA-FkpA were co-incubated in30 mM sodium formate (pH 3.0) instead of 7.0 MGdmCl prior to the gel-filtration. In this case, gp41eluted as a high molecular mass aggregate. In bothexperiments, we do not observe significant associ-ation between gp41 and FkpA-FkpA. We concludethat the solubilizing interactions between chaper-one and target in the fusion proteins are largelyintramolecular in nature.
The prolyl isomerase activity of FkpA has beenlocalized to the C-terminal domain, whereas thechaperone function is confined to the N-terminal
domain.39,42,43 Therefore, we reasoned that theN-domain might be sufficient to warrant the highsolubility of FkpA-X fusion proteins. Indeed, wefind that a further truncated variant of FkpA(26-143, FkpA*), which completely lacks theC-terminal domain, retains its solubilizing featureswhen fused to the N terminus of the gp41#
ectodomain. The resulting fusion construct isindistinguishable from SlyD*-gp41# and FkpA-gp41# with respect to solubility and immunologicalreactivity (C.S., unpublished results). Thus, FkpA*(26-143) and FkpA (26-270) are comparable asfusion modules, and the size of the respective targetprotein might be the decisive factor in which one tochoose.
The extent of native structure that is reached bythe target proteins in fusion with the PPIase-chaperones is not known at present. It might infact be a native-like state that represents the globalminimum on the conformational energy landscape.This is indicated by the finding that gp41# in fusionwith SlyD* or FkpA folds to states that areindistinguishable with respect to solubility, CDspectra and immunological reactivity. Moreover,the renatured product seems to be independent ofthe refolding method. In addition to matrix-assistedrefolding, the gp41# fusion proteins were alsosuccessfully refolded by removing the denaturantby gel-filtration, rapid dilution and dialysis, albeitat lower yields. This independence of the refoldingmethod points to a well-defined native-like form ofthe renatured ectodomains, which might corre-spond to the post-fusion coiled coil trimer.
The most compelling evidence for a native-likefold within the SlyD*/FkpA fusion context arisesfrom immunological data. When SlyD*-SlyD*-gp41#
and FkpA-FkpA-gp41# were used pairwise in adouble antigen sandwich format, the modifiedproteins turned out to ensure a high sensitivitywhen assessed with HIV-positive sera (Table 1).Thus, the highly soluble gp41# fusion proteins seemto be well suited for the detection of anti-HIVantibodies, and it is conceivable that they may servea role in future vaccination attempts. Generally,retroviral ectodomains seem capable of elicitingstrong humoral immune response. Neutralizingantibodies that bind to a highly conserved regionin the C-terminal heptad repeat of gp41 have beenobserved in HIV-infected individuals.44,45 Also,neutralizing antibodies against the transmembraneenvelope protein p15E from the porcine endo-genous retrovirus were described recently.46
Discussion
The envelope proteins from human retrovirusesare notoriously prone to aggregation, not onlyin vitro, but also in vivo. Neurological lesions inbrain tissue of patients suffering from HIV-associ-ated dementia (HAD) are correlated with plaquesconsisting of gp41 aggregates.47 It is remarkablethat the neurological defects in HIVencephalopathy

In Vitro Refolding of Chaperone Fusion Proteins 1237
share pathological, radiological and clinical featureswith other encephalopathies such as Binswanger’sdisease.47 Apparently, the strong aggregation ten-dency of gp41, which is mediated especially by theapical loop region, cannot easily be coped with byHIV-infected individuals. This strong tendency toaggregate has hitherto rendered the retroviralectodomains gp41 and gp36 very hard targets forin vitro refolding.
Here, we refolded the ectodomains gp41, gp21and gp36 of HIV-1, HTLV and HIV-2, respectively,to highly soluble (O40 mg/ml in physiologicalbuffer), native-like and immunologically reactiveconformations by fusing them covalently withtruncated versions of the E. coli chaperones SlyDor FkpA. Both SlyD* and FkpA fold and unfoldreversibly, they are soluble at concentrations up to100 mg/ml, respectively, and the overproduction ofboth proteins is well tolerated in the E. coli cytosol.The flexible linkers apparently allow the chaperonemodules SlyD* and FkpA to access hydrophobicsurface regions of the target proteins.
In the E. coli cytosol all fusion proteins accumu-lated as inclusion bodies, indicating that thechaperone modules cannot prevent aggregationduring protein synthesis in vivo. After solubilizationand unfolding by a denaturant, however, they wereable to mediate the in vitro refolding of gp41, gp21and gp36 very efficiently. Matrix-assisted refold-ing48–50 turned out to be the most efficient way torenature the ectodomain fusion proteins. Refoldingby gel-filtration, by dialysis and by rapid dilutionyielded soluble, native-like folded retroviral ecto-domains as well, albeit with lower yields. Theconformational state of the ectodomains reached inthe refolded fusion proteins seems to be thermo-dynamically stable. Probably, it corresponds to thecoiled coil trimer that is attained after membranefusion in the infection cycle.
Successful expression of a complete gp41 variantin insect cells has been reported.51 Its solubility waspoor and could be increased from !0.4 mg/ml to15 mg/ml by adding gp41-specific Fab fragments.These Fab molecules probably bind to the mosthydrophobic region of the gp41 ectodomain, i.e. theapical loop region, and thereby mask these aggre-gation-prone patches. At first glance, the fusedchaperones FkpA and SlyD* might act in a similarfashion. However, the hydrophobic immuno-dominant epitopes of the gp41 ectodomain remainaccessible in the fusion proteins, which points to adynamic association/dissociation equilibriumrather than a tight binding. A rapid equilibriumbetween substrate binding and release has beenreported for the trigger factor, another chaperonefrom the FKBP family.52
In the fusion proteins, the PPIase-chaperonesSlyD*-SlyD* and FkpA-FkpA combine twoapparently opposite functions. On the one hand,the interactions with gp41# are strong enough toshield hydrophobic patches at the surface of gp41and to retain solubility. On the other hand, they aresufficiently weak to allow the immunoglobulins to
bind to the dominant antigenic epitopes on gp41,which are hydrophobic. Possibly, these two require-ments are met for SlyD*-SlyD*-gp41# because itexists in a dynamic equilibrium between a closedand an open form. In the closed form, hydrophobicregions are shielded and thus the fusion proteinremains soluble and does not aggregate. In the openform the antigenic sites are exposed, which allowsthe gp41# part to be functional in theimmunoassays.The affinity of the SlyD*-SlyD* fusion module for
the gp41# part is thus well balanced in the fusionprotein. If the affinity were too high, the proteinwould be perfectly soluble, but it would remain inthe closed conformation and thus inactive inimmunological assays. If the affinity were too low,gp41# would be active in the assays, but it wouldnot be sufficiently protected against aggregation.This latter scenario might reflect the situation ofgp41 when fused to MBP. As mentioned above, wehad experienced a significant aggregation tendencyof the MBP-gp41 and MBP-gp41# fusion proteins.Even though MBP has been attributed a highsolubilization potential and generic chaperoneproperties,53–55 it is obviously not as efficient asthe genuine chaperones SlyD* and FkpA.Seemingly, the embedding into a chaperone fusioncontext has a similarly solubilizing effect on gp41 asthe natural fusion to the gp120 moiety in theprecursor polypeptide gp160.The fused chaperone modules remain covalently
linked to the target protein, which limits ourrefolding technique to applications that tolerate aheterologous protein part. For immunologicaldiagnostics it might still provide invaluable anti-gens, which cannot be produced by othertechniques. It is promising that the covalentlylinked chaperone modules do not impair thespecific recognition of gp41 in immunologicalassays, even though the chaperones and theantibodies probably recognize the same hydro-phobic areas. They do not interfere with eachother because the affinity that is optimal for thechaperone function is probably several orders ofmagnitude lower than the affinity required in theantigen–antibody interaction.The fusion to either chaperone-PPIase, SlyD* or
FkpA, strongly improved the expression of all ourtarget proteins, which were retroviral ectodomains.It remains to be seen whether the strategy of fusinga protein with one or two SlyD or FkpA modules,overproduction as insoluble inclusion bodies andchaperone-mediated in vitro refolding can beapplied to other aggregation-prone proteins ofdiagnostic or therapeutic value.
Materials and Methods
Materials
Guanidinium chloride (A-grade) was purchased fromNIGU (Waldkraiburg, Germany). Completew EDTA-free

1238 In Vitro Refolding of Chaperone Fusion Proteins
protease inhibitor tablets, L-lysine, imidazole and EDTAwere from Roche Diagnostics GmbH (Mannheim,Germany), all other chemicals were analytical gradefrom Merck (Darmstadt, Germany). Ultrafiltration mem-branes (YM10, YM30) were purchased from Amicon(Danvers, MA, USA), microdialysis membranes(VS/0.025 mm) and ultrafiltration units (biomax ultrafreefilter devices) were from Millipore (Bedford, MA, USA).Cellulose nitrate and cellulose acetate membranes(1.2 mm/0.45 mm/0.2 mm) for the filtration of crudelysates were from Sartorius (Gottingen, Germany).
Cloning of expression cassettes
To indicate that the E. coli chaperones that we use asfusion partners are truncated, they are designated withasterisks (FkpA*, SlyD*). Also, gp41# and gp36# denotemutant variants of the HIV-1 and HIV-2 ectodomains,respectively. gp41# bears four point mutations in the N-heptad repeat (L555E, L566E, I573T, I580E), whereas threehydrophobic amino acid residues are substituted in gp36#
(L554E, L565E, V579E).The DNA fragments encoding SlyD* (residues 1 to 165,
Swiss Prot accession no. P30856), FkpA (residues 26 to270, Swiss Prot accession no. P45523) and FkpA* (residues26–143) were amplified by PCR from E. coli chromosomalDNA and inserted in the pET24a vector from Novagen(Madison, WI, USA) using NdeI and XhoI. To obtainSlyD*-SlyD*, DNA fragments encoding NdeI/BamHI-flanked SlyD*-(GGGS)2GG and BamHI/XhoI-flanked(GGGS)2GGG-SlyD* were amplified by PCR from thepET24a plasmid encoding SlyD*and inserted in pET24ausing NdeI and XhoI.The SlyD*-gp41# cassette was obtained by ligating the
DNA fragments encoding NdeI/BamHI-flanked SlyD*-(GGGS)2GG and BamHI/XhoI-flanked (GGGS)2GGG-gp41# into pET24a using NdeI and XhoI. The SlyD*fragment was PCR-generated from the SlyD*pET24aplasmid (see above). The gp41# fragment (residues 536to 681, Swiss Prot accession no. P03375) was amplifiedfrom a HIV-1 isolate by RT-PCR and found to match theoriginal BH10 clone described by Ratner et al.56 The pointmutations L555E, L566E, I573T and I580E were intro-duced into the expression cassette using the Quik-Changee Site-Directed Mutagenesis Kit from Stratagene(La Jolla, CA, USA). Construction of FkpA-gp41# wascarried out accordingly.SlyD*-gp21 (HTLV-1): The gp41# cassette of SlyD*-
gp41# was exchanged for a synthetic gene encoding(GGGS)2GGG-gp21(residues 339–446, Swiss Prot acces-sion number P14075) flanked by BamHI/XhoI restrictionsites. Construction of FkpA-gp21 was carried out accord-ingly. The expression cassette of the cysteine-free triplemutant SlyD*-gp21-3A (C393A, C400A, C401A) wasconstructed by using QuikChangee.To generate SlyD*-gp36#, the gp41# cassette of SlyD*-
gp41# was exchanged for a DNA fragment encoding(GGGS)2GGG-gp36# flanked by BamHI/XhoI restrictionsites. The gp36 fragment (residues 534 to 676, Swiss Protaccession no. P04577) was amplified from anHIV-2 isolateby RT-PCR and found to match the ROD isolate describedby Clavel et al.57 The point mutations L554E, L565E,V579E and the WNAS motif between residues 608 and609 were introduced into the expression cassette usingQuikChangee. Construction of FkpA-gp36# was carriedout accordingly. The SlyD*-SlyD*-gp41# cassette wasobtained by cleaving SlyD*-gp41# with BamHI andinserting a DNA fragment encoding BamHI/BamHIflanked (GGGS)2GGG-SlyD*-(GGGS)2GG which was
PCR-amplified from the SlyD*pET24a plasmid (seeabove). Construction of SlyD*-SlyD*-gp21, SlyD*-SlyD*-gp36# and SlyD*-SlyD*-gp36#-WNAS was carried outaccordingly. Introduction of point mutations C598A andC604A into SlyD*-SlyD*-gp41# and insertion of theAAAA motif between residues 608 and 609 in SlyD*-SlyD*-gp36# were carried out using QuikChangee.FkpA-FkpA-gp41#, FkpA-FkpA-gp21 and FkpA-FkpA-
gp36#-WNAS fusion constructs were obtained by cleav-ing the corresponding single FkpA plasmids with BamHIand inserting a DNA fragment encoding BamHI/BamHI-flanked (GGGS)2GGG-FkpA*-(GGGS)2GG which wasPCR-amplified from the FkpA pET24a plasmid (seeabove). The (HD)4 spacer segment was inserted betweenthe antigen and the C-terminal hexahistidine tag of thevarious expression constructs using QuikChangee.
Expression, purification and refolding of therecombinant fusion proteins
All SlyD*/FkpA fusion proteins were purified by usingthe same protocol. E. coli BL21(DE3) cells harboring theparticular pET24a expression plasmid were grown at37 8C in LB medium plus kanamycin (30 mg/ml) to anA600 nm of 1.0, and cytosolic overexpression was inducedby adding isopropyl-b-D-thiogalactoside to a final con-centration of 1 mM. Four hours after induction, cells wereharvested by centrifugation (20 minutes at 5000g), frozenand stored at K20 8C. For cell lysis, the frozen pellet wasresuspended in chilled 100 mM sodium phosphate (pH8.0), 7.0 M GdmCl, 10 mM imidazole and the suspensionwas stirred for two hours on ice to complete cell lysis.After centrifugation and filtration (cellulose nitratemembrane, 0.45 mm/0.2 mm), the lysate was loaded ontoa Ni-NTA column equilibrated with the lysis buffer. Thesubsequent washing step was varied from 10 mMimidazole for the FkpA-X- type fusion proteins to25 mM imidazole for the SlyD*-X type fusion proteins.At least 10–15 column volumes of washing buffer wereapplied. Then, the GdmCl solution was replaced by50 mM sodium phosphate (pH 7.8), 100 mM NaCl toinduce refolding of the matrix-bound protein. In order toavoid reactivation of copurifying proteases, a proteaseinhibitor cocktail (Completew, EDTA-free, Roche) wasincluded in the refolding buffer. A total of 15–20 columnvolumes of refolding buffer were applied in an overnightreaction. The native fusion protein was then eluted by250 mM imidazole in 50 mM sodium phosphate (pH 7.8),100 mMNaCl. Protein-containing fractions were assessedfor purity by SDS-PAGE and pooled. Finally, the proteinswere subjected to size-exclusion chromatography (Super-dex 200 HiLoad, Amersham Pharmacia) and the protein-containing fractions were pooled and concentrated in anAmicon cell (YM 10).Alternatively, the fusion proteins were eluted from the
Ni-NTA column by 7.0 M GdmCl in 50 mM sodiumphosphate (pH 2.5) after the washing step. Protein-containing fractions were pooled and subjected torenaturing gel-filtration on a Superdex 200 HiLoadcolumn pre-equilibrated with 50 mM sodium phosphate(pH 7.5), 100 mM NaCl, 1 mM EDTA. Aliquots of 5 ml ofthe protein (5–20 mg/ml) in 7.0 M GdmCl were appliedon the column and the elution of the renatured proteinwas monitored at 280 nm.
UV spectroscopy
Measurements were performed with a Uvikon XSdouble-beam spectrophotometer. The concentrations of

In Vitro Refolding of Chaperone Fusion Proteins 1239
the various ectodomain and chaperone variants weredetermined spectrophotometrically by using the follow-ing molar absorption coefficients (3280): 5120 MK1 cmK1
(SlyD*); 10,240 MK1 cmK1 (SlyD*-SlyD*); 14,650 MK1
cmK1 (FkpA); 3840 MK1 cmK1 (FkpA* (26-143));77,360 MK1 cmK1 (SlyD*-gp41#); 82,480 MK1 cmK1
(SlyD*-SlyD*-gp41#); 101,540 MK1 cmK1 (FkpA-FkpA-gp41#); 91,440 MK1 cmK1 (FkpA-FkpA-gp36#-WNAS);85,750 MK1 cmK1 (FkpA-FkpA-gp36#-AAAA); 29,160MK1 cmK1 (SlyD*-gp21-3A); 34,280 MK1 cmK1 (SlyD*-SlyD*-gp21-3A); 76,080 MK1 cmK1 (FkpA*(26-143)-gp41#). All values were calculated by using theprocedure described by Gill & von Hippel.58
CD spectroscopy
Near-UV CD spectra were recorded with a Jasco-720spectropolarimeter with a thermostated cell holder andconverted to mean residue ellipticity. The standard bufferwas 50 mM sodium phosphate (pH 7.5), 100 mM NaCl,1 mM EDTA. The pathlength was 0.5 cm or 1.0 cm, theprotein concentration was 20–200 mM. The band widthwas 1 nm, the scanning speed was 50 nm/minute at aresolution of 0.5 nm and the response was two seconds. Inorder to improve the signal-to-noise ratio, spectra weremeasured nine times and averaged.For the thermal unfolding transition of SlyD*, the
protein was diluted to a concentration of 200 mM in thestandard buffer. The unfolding transition was recorded at275 nm under gentle stirring, the pathlength of thecuvette was 1.0 cm. Heating and cooling rates were1 deg. C/minute, the response time was four seconds.To assess the reversibility of unfolding, near-UV CD
spectra of SlyD* and SlyD*-SlyD* were recorded beforeand after the thermally induced unfolding/refoldingcycle. The spectrum of the gp41 wild-type ectodomainwas recorded with a scan speed of 20 nm/minute, aresponse of one second and a band width of 1.0 nm. Theprotein concentration was 30 mM in 30 mM sodiumformate (pH 2.5). The pathlength of the cuvette was0.5 cm.For near-UV CD difference spectra, SlyD*-SlyD*-gp41#,
SlyD*-SlyD*-gp41# (C598A, C604A) and SlyD*-SlyD*were dialyzed against 50 mM sodium phosphate(pH 7.5), 100 mM NaCl, 1 mM EDTA and adjusted to aconcentration of 25 mM each. The spectrum of the SlyD*-SlyD* reference molecule was subtracted from the spectraof the gp41 fusion proteins.
Assessment of solubility after incubation at elevatedtemperatures
After gel-filtration, the apparent trimer fractions ofSlyD*-gp41# and SlyD*-gp21 and the apparent nonamerfraction of SlyD*-SlyD*-gp41# were collected and dilutedto 10 mM in 50 mM sodium phosphate (pH 7.5), 100 mMNaCl, 1 mM EDTA. Aliquots were incubated overnight at4 8C, 30 8C, 35 8C, 40 8C, 45 8C and 50 8C. The degree ofassociation/aggregation after different incubation timeswas assessed by rechromatography on an analytical gel-filtration column (Superdex 200 HR10/30). Elution of theproteins was monitored at 280 nm, the peak area wastaken as a direct measure of the fraction size.
Coupling of biotin and ruthenium moieties to thefusion proteins
The lysine 3-amino groups of the fusion ectodomainswere modified at protein concentrations of w10 mg/ml
with N-hydroxy-succinimide activated biotin and ruthe-nium labels, respectively. The label/protein molar ratiovaried from 2:1 to 5:1, depending on the respective fusionprotein. The reaction buffer was 150 mM sodium phos-phate (pH 8.0), 50 mM NaCl, 1 mM EDTA. The reactionwas carried out at room temperature for 15 minutes andstopped by adding buffered L-lysine to a final concen-tration of 10 mM. To avoid hydrolytic inactivation of thelabels, the respective stock solutions were prepared indried DMSO (seccosolv quality, Merck, Germany). DMSOconcentrations up to 15% in the reaction buffer were welltolerated by all fusion proteins studied. After thecoupling reaction, unreacted free label was removed bypassing the crude protein conjugate over a gel-filtrationcolumn (Superdex 200 HiLoad).
Immunological reactivity of the ectodomain fusionproteins
The immunological reactivity of the different fusionproteins was assessed in an automated Elecsysw 2010analyzer. Elecsysw is a registered trademark of the Rochegroup. Measurements were carried out in the doubleantigen sandwich format.Signal detection in Elecsysw 2010 is based on electro-
chemoluminescence. The biotin-conjugate (i.e. thecapture-antigen) is immobilized on the surface of astreptavidin-coated magnetic bead, whereas the detec-tion-antigen bears a complexed ruthenium cation (switch-ing between the redox states 2C and 3C) as the signalingmoiety. In the presence of a specific immunoglobulinanalyte, the chromogenic ruthenium complex is bridgedto the solid phase and emits light at 620 nm afterexcitation at a platinum electrode. The signal output isin arbitrary light units.SlyD*-gp41# was assessed for its reactivity against HIV-
1 positive sera at concentrations ofw750 ng/ml of SlyD*-gp41#-biotin andw750 ng/ml of SlyD*-gp41#-ruthenium.As for the other measurements, unlabeled SlyD*-SlyD*was implemented in the reaction buffer as an anti-interference substance to avoid immunological cross-reactions via the chaperone fusion unit.SlyD*-SlyD*-gp36#-WNASwas assessed for its reactivity
against HIV-2 positive sera at concentrations ofw150 ng/ml of SlyD*-SlyD*-gp36#-biotin andw100 ng/ml of SlyD*-SlyD*gp36#-ruthenium. SlyD*-gp21was assessed for its reactivity against HTLV-positive sera atconcentrations of w500 ng/ml of SlyD*-gp21-biotin andw500 ng/ml of SlyD*-gp21-ruthenium.For a pairwise assessment, biotinylated FkpA-FkpA-
gp41# and ruthenylated SlyD*-SlyD*-gp41# were usedeach at a concentration of 750 ng/ml and tested againstHIV-1 positive sera. At least five negative sera were usedas controls. All measurements were carried out induplicate.
Acknowledgements
Thisworkwas performed in the department LR-IRof Roche Diagnostics GmbH (Penzberg, Germany).We are indebted to our colleaguesDorothea Sizmann,Ariuna Bazarsuren and Zhixin Shao for fruitful andstimulating discussions. The excellent technicalassistance of Laurence Thirault, Franz Wagner,Susanne Burget, Sonja Freiburghaus, Sima

1240 In Vitro Refolding of Chaperone Fusion Proteins
Hassanzadeh-Makooi, Nicole Steinlein and MarkusEckl is gratefully acknowledged. We thank JoachimDenner (Robert Koch Institut, Berlin) and RalfBollhagen, Gabriele Pestlin, Helmut Lenz andToralf Zarnt (Roche Diagnostics GmbH, Penzberg)for critically reading the manuscript. J.B. wassupported by a grant from INTAS-2001-2347.
References
1. Eckert, D. M. & Kim, P. S. (2001). Mechanisms of viralmembrane fusion and its inhibition. Annu. Rev.Biochem. 70, 777–810.
2. Moulard, M. & Decroly, E. (2000). Maturation of HIVenvelope glycoprotein precursors by cellular endo-proteases. Biochim. Biophys. Acta Rev. Biomembr. 1469,121–132.
3. Chan, D. C., Fass, D., Berger, J. M. & Kim, P. S. (1997).Core structure of gp41 from the HIV envelopeglycoprotein. Cell, 89, 263–273.
4. Weissenhorn, W., Dessen, A., Harrison, S. C., Skehel,J. J. & Wiley, D. C. (1997). Atomic structure of theectodomain from HIV-1 gp41. Nature, 387, 426–430.
5. Lu, M., Blacklow, S. C. & Kim, P. S. (1995). A trimericstructural domain of the HIV-1 transmembraneglycoprotein. Nature Struct. Biol. 2, 1075–1082.
6. Root, M. J., Kay, M. S. & Kim, P. S. (2001). Proteindesign of anHIV-1 entry inhibitor. Science, 291, 884–888.
7. Doms, R. W. & Moore, J. P. (2000). HIV-1 membranefusion: targets of opportunity. J. Cell Biol. 151, F9–F13.
8. Chen, C. H., Matthews, T. J., Mcdanal, C. B.,Bolognesi, D. P. & Greenberg, M. L. (1995).A molecular clasp in the human-immunodeficiency-virus (HIV) type-1 Tm protein determines the anti-HIV activity of Gp41 derivatives–implication for viralfusion. J. Virol. 69, 3771–3777.
9. Chan, D. C., Chutkowski, C. T. & Kim, P. S. (1998).Evidence that a prominent cavity in the coiled coil ofHIV type 1 gp41 is an attractive drug target. Proc. NatlAcad. Sci. USA, 95, 15613–15617.
10. Eckert, D. M. & Kim, P. S. (2001). Design of potentinhibitors of HIV-1 entry from the gp41 N-peptideregion. Proc. Natl Acad. Sci. USA, 98, 11187–11192.
11. Gnann, J. W., Mccormick, J. B., Mitchell, S., Nelson,J. A. & Oldstone, M. B. A. (1987). Synthetic peptideimmunoassaydistinguishesHIV type-1 andHIV type-2infections. Science, 237, 1346–1349.
12. Gnann, J. W., Schwimmbeck, P. L., Nelson, J. A.,Truax, A. B. & Oldstone, M. B. A. (1987). Diagnosis ofaids by using a 12-amino acid peptide representing animmunodominant epitope of the human-immuno-deficiency-virus. J. Infect. Dis. 156, 261–267.
13. Dobeli, H., Andres, H., Breyer, N., Draeger, N.,Sizmann, D., Zuber, M. T., Weinert, B. & Wipf, B.(1998). Recombinant fusion proteins for the industrialproduction of disulfide bridge containing peptides:purification, oxidation without concatamer for-mation, and selective cleavage. Protein Expr. Purif.12, 404–414.
14. Lu, M. & Kim, P. S. (1997). A trimeric structuralsubdomain of the HIV-1 transmembrane glyco-protein. J. Biomol. Struct. Dynam. 15, 465–471.
15. Malashkevich, V. N., Chan, D. C., Chutkowski, C. T. &Kim, P. S. (1998). Crystal structure of the simianimmunodeficiency virus (SIV) gp41 core: conserved
helical interactions underlie the broad inhibitoryactivity of gp41 peptides. Proc. Natl Acad. Sci. USA,95, 9134–9139.
16. Caffrey, M., Cai, M. L., Kaufman, J., Stahl, S. J.,Wingfield, P. T., Covell, D. G. et al. (1998). Three-dimensional solution structure of the 44 kDa ecto-domain of SIV gp41. EMBO J. 17, 4572–4584.
17. Caffrey, M., Kaufman, J., Stahl, S., Wingfield, P.,Gronenborn, A. M. & Clore, G. M. (1999). Monomer-trimer equilibrium of the ectodomain of SIV gp41:insight into the mechanism of peptide inhibition ofHIV infection. Protein Sci. 8, 1904–1907.
18. Wang, J. J. G., Steel, S., Wisniewolski, R. & Wang, C. Y.(1986). Detection of antibodies to human T-lympho-tropic virus type-III by using a synthetic peptide of 21amino-acid-residues corresponding to a highly anti-genic segment of Gp41 envelope protein. Proc. NatlAcad. Sci. USA, 83, 6159–6163.
19. Kobe, B., Center, R. J., Kemp, B. E. & Poumbourios, P.(1999). Crystal structure of human T cell leukemiavirus type 1 gp21 ectodomain crystallized as amaltose-binding protein chimera reveals structuralevolution of retroviral transmembrane proteins. Proc.Natl Acad. Sci. USA, 96, 4319–4324.
20. Schmid, F. X., Mayr, L. M., Mucke, M. &Schonbrunner, E. R. (1993). Prolyl isomerases–role inprotein-folding. Advan. Protein Chem. 44, 25–66.
21. Schmid, F. X. (2002). Prolyl isomerases. Protein Fold.Cell, 59, 243–282.
22. Behrens, S., Maier, R., de Cock, H., Schmid, F. X. &Gross, C. A. (2001). The SurA periplasmic PPIaselacking its parvulin domains functions in vivo and haschaperone activity. EMBO J. 20, 285–294.
23. Scholz, C., Stoller, G., Zarnt, T., Fischer, G. & Schmid,F. X. (1997). Cooperation of enzymatic and chaperonefunctions of trigger factor in the catalysis of proteinfolding. EMBO J. 16, 54–58.
24. Zarnt, T., Tradler, T., Stoller, G., Scholz, C., Schmid,F. X. & Fischer, G. (1997). Modular structure of thetrigger factor required for high activity in proteinfolding. J. Mol. Biol. 271, 827–837.
25. Wulfing, C., Lombardero, J. & Pluckthun, A. (1994).An Escherichia coli protein consisting of a domainhomologous to Fk506-binding proteins (Fkbp) and anew metal-binding motif. J. Biol. Chem. 269, 2895–2901.
26. Bernhardt, T. G., Roof, W. D. & Young, R. (2000).Genetic evidence that the bacteriophage phi X174lysis protein inhibits cell wall synthesis. Proc. NatlAcad. Sci. USA, 97, 4297–4302.
27. Roof, W. D. & Young, R. (1995). Phi-X174 lysis requiresSlyd, a host gene which is related to the Fkbp familyof peptidyl-prolyl cis-trans isomerases. FEMSMicrobiol. Rev. 17, 213–218.
28. Caffrey, M., Cai, M. L., Kaufman, J., Stahl, S. J.,Wingfield, P. T., Gronenborn, A. M. & Clore, G. M.(1997). Determination of the secondary structure andglobal topology of the 44 kDa ectodomain of gp41 ofthe simian immunodeficiency virus by multi-dimensional nuclear magnetic resonancespectroscopy. J. Mol. Biol. 271, 819–826.
29. Wingfield, P. T., Stahl, S. J., Kaufman, J., Zlotnick, A.,Hyde, C. C., Gronenborn, A. M. & Clore, G. M. (1997).The extracellular domain of immunodeficiency virusgp41 protein: expression in Escherichia coli, purifi-cation, and crystallization. Protein Sci. 6, 1653–1660.
30. Scholtz, J. M. & Baldwin, R. L. (1992). The mechanismof alpha-helix formation by peptides. Annu. Rev.Biophys. Biomol. Struct. 21, 95–118.
31. Kohn, W. D., Kay, C. M. & Hodges, R. S. (1995).

In Vitro Refolding of Chaperone Fusion Proteins 1241
Protein destabilization by electrostatic repulsions inthe 2-stranded alpha-helical coiled-coil leucine-zipper. Protein Sci. 4, 237–250.
32. Kohn, W. D., Monera, O. D., Kay, C. M. & Hodges,R. S. (1995). The effects of interhelical electrostaticrepulsions between glutamic-acid residues in control-ling the dimerization and stability of 2-strandedalpha-helical coiled-coils. J.Biol.Chem.270, 25495–25506.
33. Oldstone, M. B. A., Tishon, A., Lewicki, H., Dyson,H. J., Feher, V. A., Assamunt, N. &Wright, P. E. (1991).Mapping the anatomy of the immunodominantdomain of the human immunodeficiency virus-Gp41transmembraneprotein–peptide conformationanalysisusing monoclonal-antibodies and proton nuclear-mag-netic-resonance spectroscopy. J. Virol. 65, 1727–1734.
34. Gnann, J. W., Nelson, J. A. &Oldstone, M. B. A. (1987).Fine mapping of an immunodominant domain in thetransmembrane glycoprotein of human-immuno-deficiency-virus. J. Virol. 61, 2639–2641.
35. Caffrey, M. (2001). Model for the structure of the HIVgp41 ectodomain: insight into the intermolecularinteractions of the gp41 loop. Biochim. Biophys. ActaMol. Basis Dis. 1536, 116–122.
36. Center, R. J., Kobe, B., Wilson, K. A., Teh, T., Howlett,G. J., Kemp, B. E. & Poumbourios, P. (1998). Crystal-lization of a trimeric human Tcell leukemia virus type1 gp21 ectodomain fragment as a chimera withmaltose-binding protein. Protein Sci. 7, 1612–1619.
37. Marqusee, S., Robbins, V. H. & Baldwin, R. L. (1989).Unusually stable helix formation in short alanine-basedpeptides.Proc.Natl Acad. Sci. USA, 86, 5286–5290.
38. Rohl, C. A., Fiori, W. & Baldwin, R. L. (1999). Alanineis helix-stabilizing in both template-nucleated andstandard peptide helices. Proc. Natl Acad. Sci. USA, 96,3682–3687.
39. Ramm, K. & Pluckthun, A. (2000). The periplasmicEscherichia coli peptidylprolyl cis,trans-isomeraseFkpA-II. Isomerase-independent chaperone activityin vitro. J. Biol. Chem. 275, 17106–17113.
40. Ramm, K. & Pluckthun, A. (2001). High enzymaticactivity and chaperone function are mechanisticallyrelated features of the dimeric E. coli peptidyl-prolyl-isomerase FkpA. J. Mol. Biol. 310, 485–498.
41. Bothmann, H. & Pluckthun, A. (2000). The peri-plasmic Escherichia coli peptidylprolyl cis,trans-isomerase FkpA-I. Increased functional expressionof antibody fragments with and without cis-prolines.J. Biol. Chem. 275, 17100–17105.
42. Arie, J. P., Sassoon, N. & Betton, J. M. (2001).Chaperone function of FkpA, a heat shock prolylisomerase, in the periplasm of Escherichia coli. Mol.Microbiol. 39, 199–210.
43. Saul, F. A., Arie, J. P., Normand, B. V. L., Kahn, R.,Betton, J. M. & Bentley, G. A. (2004). Structural andfunctional studies of FkpA from Escherichia coli, acis/trans peptidyl-prolyl isomerase with chaperoneactivity. J. Mol. Biol. 335, 595–608.
44. Muster, T., Steindl, F., Purtscher, M., Trkola, A., Klima,A., Himmler, G. et al. (1993). A conserved neutralizingepitope on Gp41 of human-immunodeficiency-virustype-1. J. Virol. 67, 6642–6647.
45. Zwick, M. B., Labrijn, A. F., Wang, M., Spenlehauer,
C., Saphire, E. O., Binley, J. M. et al. (2001). Broadlyneutralizing antibodies targeted to the membrane-proximal external region of human immuno-deficiency virus type 1 glycoprotein gp41. J. Virol.75, 10892–10905.
46. Fiebig, U., Stephan, O., Kurth, R. & Denner, J. (2003).Neutralizing antibodies against conserved domainsof p15E of porcine endogenous retroviruses: basis fora vaccine for xenotransplantation? Virology, 307,406–413.
47. Caffrey, M., Braddock, D. T., Louis, J. M., Abu-Asab,M. A., Kingma, D., Liotta, L. et al. (2000). Biophysicalcharacterization of gp41 aggregates suggests a modelfor the molecular mechanism of HIV-associatedneurological damage and dementia. J. Biol. Chem.275, 19877–19882.
48. Stempfer, G., HollNeugebauer, B. & Rudolph, R.(1996). Improved refolding of an immobilized fusionprotein. Nature Biotechnol. 14, 329–334.
49. Stempfer, G., HollNeugebauer, B., Kopetzki, E. &Rudolph, R. (1996). A fusion protein designed fornoncovalent immobilization: stability, enzymaticactivity, and use in an enzyme reactor. NatureBiotechnol. 14, 481–484.
50. Clark, E. D., Schwarz, E. & Rudolph, R. (1999).Inhibition of aggregation side reactions duringin vitro protein folding. Amyloid, Prions Other ProteinAggregates, 309, 217–236.
51. Weissenhorn, W., Wharton, S. A., Calder, L. J., Earl,P. L., Moss, B., Aliprandis, E. et al. (1996). Theectodomain of HIV-1 env subunit gp41 forms asoluble, alpha-helical, rod-like oligomer in theabsence of gp120 and the N-terminal fusion peptide.EMBO J. 15, 1507–1514.
52. Maier, R., Scholz, C. & Schmid, F. X. (2001). Dynamicassociation of trigger factor with protein substrates.J. Mol. Biol. 314, 1181–1190.
53. Bach, H., Mazor, Y., Shaky, S., Shoham-Lev, A.,Berdichevsky, Y., Gutnick, D. L. & Benhar, I. (2001).Escherichia colimaltose-binding protein as a molecularchaperone for recombinant intracellular cytoplasmicsingle-chain antibodies. J. Mol. Biol. 312, 79–93.
54. Kapust, R. B. & Waugh, D. S. (1999). Escherichia colimaltose-binding protein is uncommonly effective atpromoting the solubility of polypeptides to which it isfused. Protein Sci. 8, 1668–1674.
55. Waugh, D. S., Kapust, R. B. & Fox, J. D. (1999). E-colimaltose binding protein is uncommonly effective atinhibiting the aggregation of proteins to which it isfused. Abstr. Pap. Am. Chem. Soc. 217, U181–U181.
56. Ratner, L., Haseltine, W., Patarca, R., Livak, K. J.,Starcich, B., Josephs, S. F. et al. (1985). Completenucleotide-sequence of the Aids virus, HTLV-III.Nature, 313, 277–284.
57. Clavel, F., Guyader, M., Guetard, D., Salle, M.,Montagnier, L. & Alizon, M. (1986). Molecular-cloning and polymorphism of the human immune-deficiency virus type-2. Nature, 324, 691–695.
58. Gill, S. C. & von Hippel, P. H. (1989). Calculation ofprotein extinction coefficients from amino acidsequence data. Anal. Biochem. 182, 319–326.
Edited by C. R. Matthews
(Received 22 September 2004; received in revised form 28 October 2004; accepted 29 October 2004)





























