Embed Size (px)
Citation preview

Microporous and Mesoporous Materials 155 (2012) 75–81
Contents lists available at SciVerse ScienceDirect
Microporous and Mesoporous Materials
journal homepage: www.elsevier .com/locate /micromeso
Formation of a nanohybrid composite between mesostructured cellular silica foamand microporous copper trimesate
You-Kyong Seo a,b, Ji Woong Yoon a, U-Hwang Lee a, Young Kyu Hwang a,⇑, Chul-Ho Jun b,Jong-San Chang a,⇑a Research Group for Nanocatalyst, Biorefinery Research Center, Korea Research Institute of Chemical Technology (KRICT), P.O. Box 107, Yusung, Daejeon 305-600, Republic of Koreab Department of Chemistry, Center for Bioactive Molecular Hybrid, Yonsei University, Seodaemoonku, Seoul 120-749, Republic of Korea
a r t i c l e i n f o
Article history:Received 4 September 2011Received in revised form 26 December 2011Accepted 29 December 2011Available online 8 January 2012
Keywords:Metal-organic frameworkCu3(BTC)2
Mesostructured cellular silica foam (MCF)NanocompositeHydrophobicity
1387-1811/$ - see front matter � 2012 Elsevier Inc. Adoi:10.1016/j.micromeso.2011.12.053
⇑ Corresponding authors.E-mail addresses: [email protected] (Y.K. H
(J.-S. Chang).
a b s t r a c t
A metal-organic framework HKUST-1 or Cu3(BTC)2 was synthesized in mesopores of a COOH functional-ized mesostructured cellular silica foam (MCF(M)-COOH), resulting in a hydrophobic nanocomposite. TheCu3(BTC)2 formed in the MCF(M)-COOH has been prepared by a microwave method with sequentialincorporation of copper nitrate and 1,3,5-benzenetricarboxylic acid in a water/ethanol mixture contain-ing the MCF(M)-COOH. The nanocomposite was characterized by XRD, BET, FT-IR, TEM and solid state 13CNMR. The nanocomposite had a higher hydrophobic property than pure Cu3(BTC)2 and MCF(M)-COOH asa result of their hybridization. Moreover, the formation of the nanocomposite increased the sorption rateof cyclohexane vapor as compared with those of pure Cu3(BTC)2 and MCF(M)-COOH due to the increasedhydrophobicity.
� 2012 Elsevier Inc. All rights reserved.
1. Introduction
Metal-organic frameworks (MOFs), which are generally referredto as coordination polymers, are a new class of emerging nanopor-ous materials [1–3] because of their unique properties and versatileapplications [4–7]. In particular, the large surface area and porevolume are the best characteristics of MOFs for extending their abil-ity of sorption and separation of gases. For more applications it issometime necessary to modify the primary framework throughpre- or post-functionalization of ligands and/or metal sites [8,9].Another interesting attempt to improve the physico-chemical prop-erties of MOFs has been made to prepare MOF-based compositesthat are formed by physical or chemical integration of a basicMOF building block into inorganic or polymer substrates [10–14].There have been many reports on various methods to prepare com-posite materials: sequential formation of a core–shell MOF@MOFby the seeded growth technique [15], direct crystallization of aCu-MOF inside pores of silica beads by impregnation of a mixedsolution with metal ions and ligands and following evaporation ofthe solvent [16], one-pot hydrothermal synthesis of a MOFmesoporous alumina composite using triblock copolymers [17], amesoporous silica/MOFs composite for ammonia adsorption [18],surface modification or functionalization of nanoscale MOFs for
ll rights reserved.
wang), [email protected]
the coating of silica shells [19], etc. In addition to three-dimensionalhybridization, two-dimensional hybridization of membrane andthin film has also been investigated. For thin films of MOFs, it hasbeen demonstrated that the COOH-terminated surfaces of self-assembled monolayers (SAMs) induce the chelating coordination(as with BTC) of metal cations with surface COOH groups to createwell-defined orientations of MOF crystals [20]. These MOF compos-ites will be useful for direct use in gas separation and catalysts dueto the easy modification for a practical subject, i.e., membrane, pel-let or spherical type of solid [21]. However, a systematic method ofeffective hybridization between nanoscale MOFs and mesoporoussubstrates is still a challenging task because of the difficulty ofencapsulating microporous MOFs in the pores of mesoporousmaterials.
The COOH-functionalized mesoporous silica SBA-15 with lessthan 10 nm have been prepared by co-condensation and post-grafting using organosilane [22,23]. Recently, we proposed a meth-od to synthesize hierarchically ordered mesocellular mesoporoussilica called mesostructured cellular silica foam (MCF) with twodifferent large mesoporous of about 10 and 30 nm [24]. This mate-rial can be considered as a starting substrate that has sufficientlylarge pores to allow the accessibility of nanosized objects. In addi-tion to this, we reported microwave crystallization of a well-known cubic Cu-MOF, microporous copper trimesate (HKUST-1or Cu3(BTC)2), which is built up by paddle-wheel copper dimer mo-tifs connected by 1,3,5-benzenetricarboxylate (BTC) ligands [25].The Cu3(BTC)2 material is one of topical MOFs that have been

76 Y.-K. Seo et al. / Microporous and Mesoporous Materials 155 (2012) 75–81
widely studied for gas sorption, separation, and catalysis [26,27].The successful synthesis of two different types of porous materialsencourages us to study the preparation of a nanocomposite mate-rial through chemical integration of a nanosized MOF into a largemesoporous substrate.
Here, we report a new approach for successful hybridization toform a new nanocomposite in which a microporous Cu3(BTC)2 as anano-object is selectively crystallized within large pores of theMCF material through the surface COOH group.
2. Experimental
2.1. Preparation of a nanocomposite
Typically, for the cyano group functionalization, 1.62 g(0.279 mmol) of P123 was transferred into 100 ml volume polypro-pylene bottle and dissolved in a mixture of 33.33 g (1852 mmol) ofde-ionized water and 0.8 g (13.32 mmol) of acetic acid. Then, theresulting solution was heated to 60 �C and kept for 1 h in an oil bath.Another solution was prepared with 2.67 g (11 mmol) of sodiumsilicate and 0.23 g (1.0 mmol) of 4-(triethoxysilyl)butyronitrile(TESBN) as the silica source in 33.33 g of water. The molar composi-tion of the final solution was Na2SiO3:TESBN:P123:H2O:aceticacid = 1:0.091:0.025:336.4:1.21. The latter solution was droppedinto the former solution, aged at 60 �C for 1 h, transferred to a Teflonreactor, and hydrothermally reacted at 100 �C for 12 h. The reactorwas then cooled to room temperature. A white product was formedin the solution, filtered with de-ionized water and ethanol, and thendried at 100 �C for 12 h. In the next step, the COOH-functionalizedMCF (MCF-COOH) was prepared by oxidation of the nitrile groupin the MCF-CN with 48% H2SO4 at 95 �C. 1 g of MCF-CN was furthertreated in 120 ml of an aqueous 48% H2SO4 solution at 95 �C for1 day. To exclude the interference of surface silanol groups duringthe crystallization of the MOF, MCF-COOH was treated with meth-yltriethoxysilane (MTS). For example, 1 g MCF-COOH was reactedwith 0.89 g of MTS dissolved in 60 ml of toluene in a reflux conditionfor 12 h. This treatment led to the replacement of the surface silanolgroup by the methylsilane moiety, giving the white mesoporoussubstrate, MCF(M)-COOH. The polymer surfactant inside the poreswas removed during the oxidation [22]. The content of the surfaceCOOH groups measured by acid–base titration with 1 N NaOH was1.9 mmol/g-solid. The initial step for the hybridization processwas the selective coordination of copper cations with the terminalCOOH groups in the mesopores. The white MCF(M)-COOH solidwas immersed and stirred in the solution of 0.2 M copper nitratedissolved in a solvent mixture of ethanol and water (4:1 w/w). Then,the solvents of the resulting slurry solution were slowly evaporatedat 60 �C to produce the dry product with a pale blue color, Cu(II)/MCF(M)-COOH. After this process was repeated three times, the fi-nal loading of the copper was 2.46 mmol/g-solid. The final step wasthe crystallization to form Cu3(BTC)2 from the Cu(II)/MCF(M)-COOHby microwave-assisted heating at 140 �C for 30 min. The dried solidwas gently mixed in a solution of the BTC ligand (molar ratio of Cu/BTC = 1.5) dispersed in a solvent mixture of ethanol/water. Then,microwave irradiation was used to synthesize a crystalline coppertrimesate phase in the MCF(M)-COOH substrate, which is denotedas Cu-MOF/MCF(M)-COOH. For comparison, Cu3(BTC)2 was synthe-sized with microwave irradiation (microwave power, 400 W) at140 �C for 30 min as reported in the literature [24].
2.2. Characterization of materials
Powder X-ray diffraction patterns of the as-prepared and func-tionalized samples were obtained by a Rigaku diffractormeter (D/MAX IIIB, 2 kW) using Ni-filtered Cu Ka-radiation (40 kV, 30 mA,
k = 1.5406 Å) and a graphite crystal monochromator. The crystal sizeand morphology were examined using a scanning electron micro-scope (SEM:JEOL JSM-840A). For IR analyses, samples were pressed(10�7 Pa) into self-supported disk (2 cm2 area, 7–10 mg cm�2). Theywere placed in a quartz cell equipped with KBr windows. A mova-ble quartz sample holder permitted the adjustment of the pellet inthe infrared beam for spectra acquisition and to displace it into afurnace at the top of the cell for thermal treatments at 250 �C for12 h. IR spectra were recorded at room temperature. The BET sur-face area measurements were performed with N2 adsorption–desorption isotherms at liquid nitrogen temperature (77 K) afterdehydration in a vacuum at 423 K for 12 h using Micromeritics Tri-star 3000. The specific surface areas were evaluated using the Bru-nauer–Emmett–Teller (BET) method in a p/p0 range of 0.05–0.3.Pore size distribution curves were calculated using the adsorptionbranch of the isotherms and Non-Local-Density Functional (NLDFT)approaches and pore sizes were obtained from the peak positionsof the distribution curves. The pore volume was taken by a singlepoint method at p/p0 = 0.99. FE-TEM images were taken using aJEM-2100F instrument (JEOL) at the Korea Basic Science Institute(KBSI). 13C NMR spectra were recorded on a DSX 400 MHz spec-trometer equipped with a wideline and CP/MAS (15 kHz) probe-head. 13C NMR spectra were recorded by using contact times of3 ms.
2.3. Sorption kinetic experiments of cyclohexane
The vapor phase adsorption of cyclohexane was carried out at30 �C using a specially designed volumetric adsorption apparatusand a constant temperature oven. The adsorbed cyclohexane wasdetermined by analyzing the concentration of residual benzeneby FID–GC. The sorption kinetics of the cyclohexane of the dehy-drated samples were carried out at 30 �C by measuring continu-ously (1 s intervals) the vapor pressure of the cyclohexane fromabout 120 torr to an equilibrium pressure during adsorption. Thesamples (ca. 0.1 g) were dehydrated in a high vacuum (1 �10�5 torr) before adsorption, and the adsorbate (cyclohexane) waspurified by freeze–pump–thaw cycles. The sorption capacity wascalculated by ideal gas law at a constant temperature. The repro-ducibility of the sorption kinetics was confirmed at least threetimes, and the averaged results were derived from three indepen-dent experiments.
3. Results and discussion
Scheme 1 represents overall hybridization process to producesuch a nanocomposite from the crystallization of the Cu3(BTC)2
using the mesoporous MCF substrate. Before the preparation ofthe COOH-functionalized MCF (MCF-COOH), CN-functionalizedMCF (MCF-CN) was initially synthesized by co-condensation of so-dium silicate and 3-cyanopropyltriethoxysilane and a triblockcopolymer P123. Then, the MCF-COOH was prepared by oxidationof MCF-CN with 48% H2SO4 at 95 �C. The polymer surfactant insidethe pores is removed during the oxidation. In order to exclude theinterference of surface silanol groups during the crystallization ofMOF, MCF-COOH was treated with methyltriethoxysilane (MTS)in toluene at 100 �C for 12 h (see Section 2 for details). This treat-ment leads to the replacement of the surface silanol group by themethylsilane moiety, giving the white mesoporous substrate(MCF(M)-COOH). The initial step for the hybridization is theselective coordination of copper cations with the terminal COOHgroups in the mesopores (see Section 2 for details). The whiteMCF(M)-COOH solid was immersed and stirred in the solution of0.2 M copper nitrate dissolved in a solvent mixture of ethanol andwater (4:1 w/w). Then, solvents of the resulting slurry solution
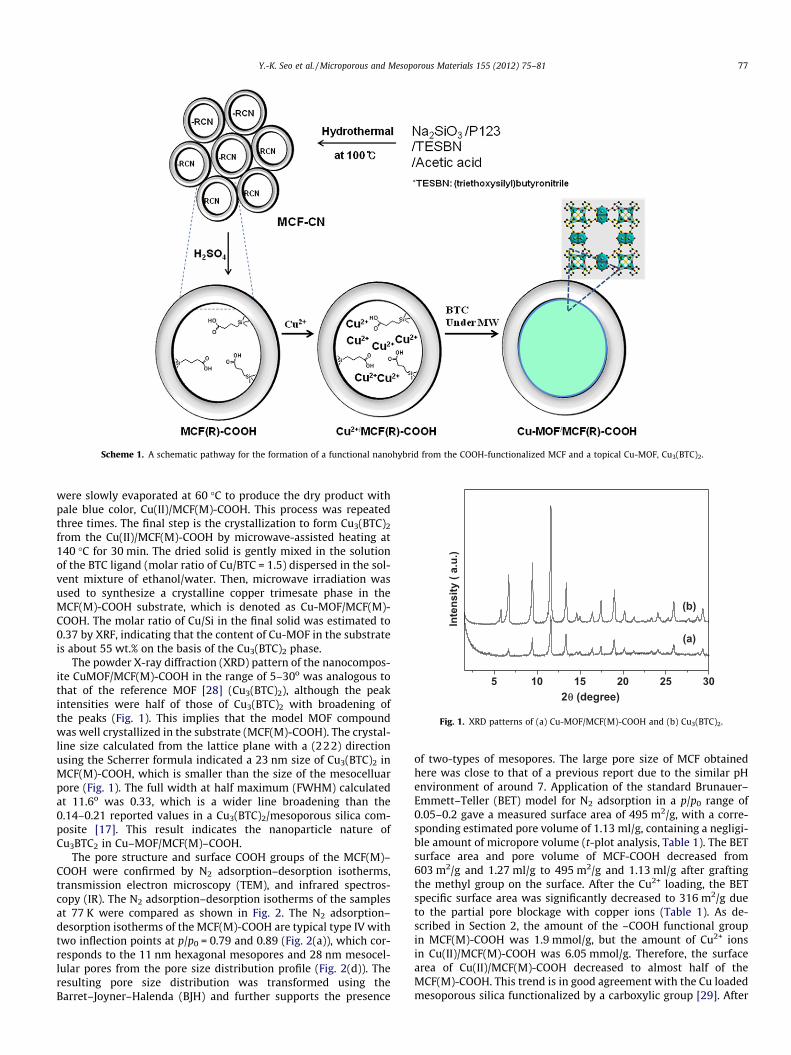
Scheme 1. A schematic pathway for the formation of a functional nanohybrid from the COOH-functionalized MCF and a topical Cu-MOF, Cu3(BTC)2.
5 10 15 20 25 30
(b)
Inte
nsity
( a.
u.)
2θ (degree)
(a)
Fig. 1. XRD patterns of (a) Cu-MOF/MCF(M)-COOH and (b) Cu3(BTC)2.
Y.-K. Seo et al. / Microporous and Mesoporous Materials 155 (2012) 75–81 77
were slowly evaporated at 60 �C to produce the dry product withpale blue color, Cu(II)/MCF(M)-COOH. This process was repeatedthree times. The final step is the crystallization to form Cu3(BTC)2
from the Cu(II)/MCF(M)-COOH by microwave-assisted heating at140 �C for 30 min. The dried solid is gently mixed in the solutionof the BTC ligand (molar ratio of Cu/BTC = 1.5) dispersed in the sol-vent mixture of ethanol/water. Then, microwave irradiation wasused to synthesize a crystalline copper trimesate phase in theMCF(M)-COOH substrate, which is denoted as Cu-MOF/MCF(M)-COOH. The molar ratio of Cu/Si in the final solid was estimated to0.37 by XRF, indicating that the content of Cu-MOF in the substrateis about 55 wt.% on the basis of the Cu3(BTC)2 phase.
The powder X-ray diffraction (XRD) pattern of the nanocompos-ite CuMOF/MCF(M)-COOH in the range of 5–30o was analogous tothat of the reference MOF [28] (Cu3(BTC)2), although the peakintensities were half of those of Cu3(BTC)2 with broadening ofthe peaks (Fig. 1). This implies that the model MOF compoundwas well crystallized in the substrate (MCF(M)-COOH). The crystal-line size calculated from the lattice plane with a (222) directionusing the Scherrer formula indicated a 23 nm size of Cu3(BTC)2 inMCF(M)-COOH, which is smaller than the size of the mesocelluarpore (Fig. 1). The full width at half maximum (FWHM) calculatedat 11.6o was 0.33, which is a wider line broadening than the0.14–0.21 reported values in a Cu3(BTC)2/mesoporous silica com-posite [17]. This result indicates the nanoparticle nature ofCu3BTC2 in Cu–MOF/MCF(M)–COOH.
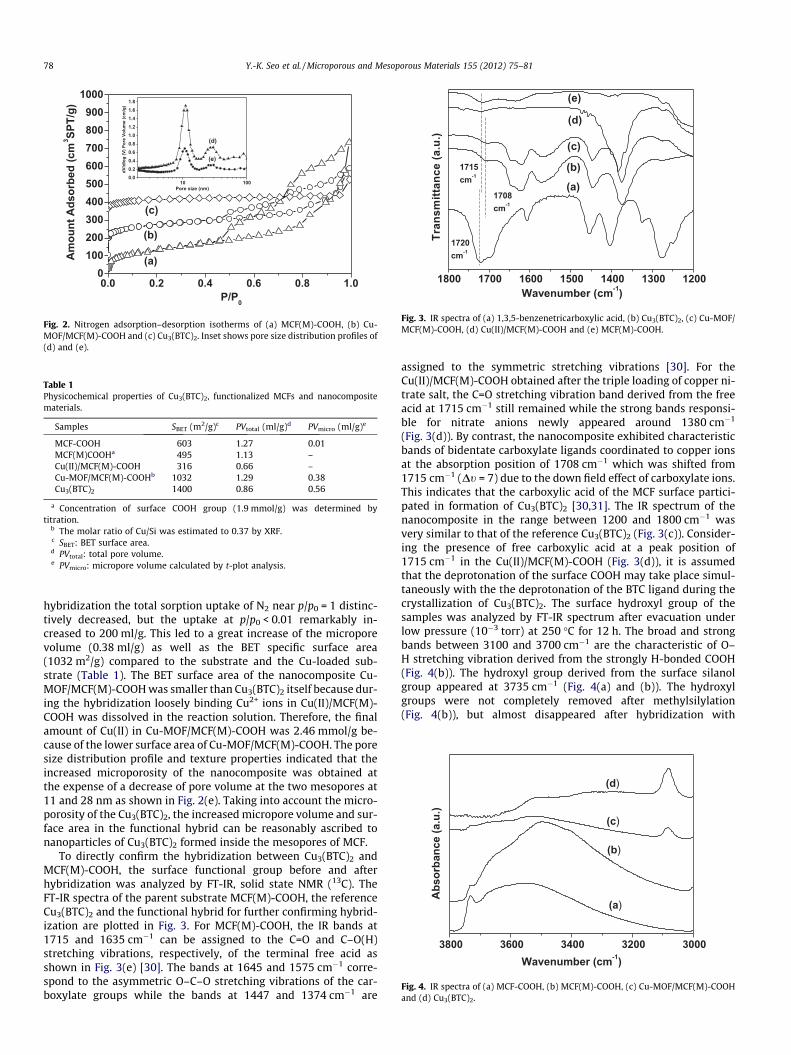
The pore structure and surface COOH groups of the MCF(M)–COOH were confirmed by N2 adsorption–desorption isotherms,transmission electron microscopy (TEM), and infrared spectros-copy (IR). The N2 adsorption–desorption isotherms of the samplesat 77 K were compared as shown in Fig. 2. The N2 adsorption–desorption isotherms of the MCF(M)-COOH are typical type IV withtwo inflection points at p/p0 = 0.79 and 0.89 (Fig. 2(a)), which cor-responds to the 11 nm hexagonal mesopores and 28 nm mesocel-lular pores from the pore size distribution profile (Fig. 2(d)). Theresulting pore size distribution was transformed using theBarret–Joyner–Halenda (BJH) and further supports the presence
of two-types of mesopores. The large pore size of MCF obtainedhere was close to that of a previous report due to the similar pHenvironment of around 7. Application of the standard Brunauer–Emmett–Teller (BET) model for N2 adsorption in a p/p0 range of0.05–0.2 gave a measured surface area of 495 m2/g, with a corre-sponding estimated pore volume of 1.13 ml/g, containing a negligi-ble amount of micropore volume (t-plot analysis, Table 1). The BETsurface area and pore volume of MCF-COOH decreased from603 m2/g and 1.27 ml/g to 495 m2/g and 1.13 ml/g after graftingthe methyl group on the surface. After the Cu2+ loading, the BETspecific surface area was significantly decreased to 316 m2/g dueto the partial pore blockage with copper ions (Table 1). As de-scribed in Section 2, the amount of the –COOH functional groupin MCF(M)-COOH was 1.9 mmol/g, but the amount of Cu2+ ionsin Cu(II)/MCF(M)-COOH was 6.05 mmol/g. Therefore, the surfacearea of Cu(II)/MCF(M)-COOH decreased to almost half of theMCF(M)-COOH. This trend is in good agreement with the Cu loadedmesoporous silica functionalized by a carboxylic group [29]. After
0.0 0.2 0.4 0.6 0.8 1.00
100200300400500600700800900
1000
(b)
(c)
Am
ount
Ads
orbe
d (c
m3 SP
T/g)
P/P0
(a)
10 1000.00.20.40.60.81.01.21.41.61.8
dV/d
log
(V) P
ore
Volu
me
(cm
/g)
Pore size (nm)
(e)
(d)
Fig. 2. Nitrogen adsorption–desorption isotherms of (a) MCF(M)-COOH, (b) Cu-MOF/MCF(M)-COOH and (c) Cu3(BTC)2. Inset shows pore size distribution profiles of(d) and (e).
Table 1Physicochemical properties of Cu3(BTC)2, functionalized MCFs and nanocompositematerials.
Samples SBET (m2/g)c PVtotal (ml/g)d PVmicro (ml/g)e
MCF-COOH 603 1.27 0.01MCF(M)COOHa 495 1.13 –Cu(II)/MCF(M)-COOH 316 0.66 –Cu-MOF/MCF(M)-COOHb 1032 1.29 0.38Cu3(BTC)2 1400 0.86 0.56
a Concentration of surface COOH group (1.9 mmol/g) was determined bytitration.
b The molar ratio of Cu/Si was estimated to 0.37 by XRF.c SBET: BET surface area.d PVtotal: total pore volume.e PVmicro: micropore volume calculated by t-plot analysis.
1800 1700 1600 1500 1400 1300 1200
1708cm-1
1715cm-1
(a)(b)
(d)
(e)
(c)
Tran
smitt
ance
(a.u
.)
Wavenumber (cm-1)
1720cm-1
Fig. 3. IR spectra of (a) 1,3,5-benzenetricarboxylic acid, (b) Cu3(BTC)2, (c) Cu-MOF/MCF(M)-COOH, (d) Cu(II)/MCF(M)-COOH and (e) MCF(M)-COOH.
3800 3600 3400 3200 3000
(d)
(c)
(b)
Abs
orba
nce
(a.u
.)
Wavenumber (cm-1)
(a)
Fig. 4. IR spectra of (a) MCF-COOH, (b) MCF(M)-COOH, (c) Cu-MOF/MCF(M)-COOHand (d) Cu3(BTC)2.
78 Y.-K. Seo et al. / Microporous and Mesoporous Materials 155 (2012) 75–81
hybridization the total sorption uptake of N2 near p/p0 = 1 distinc-tively decreased, but the uptake at p/p0 < 0.01 remarkably in-creased to 200 ml/g. This led to a great increase of the microporevolume (0.38 ml/g) as well as the BET specific surface area(1032 m2/g) compared to the substrate and the Cu-loaded sub-strate (Table 1). The BET surface area of the nanocomposite Cu-MOF/MCF(M)-COOH was smaller than Cu3(BTC)2 itself because dur-ing the hybridization loosely binding Cu2+ ions in Cu(II)/MCF(M)-COOH was dissolved in the reaction solution. Therefore, the finalamount of Cu(II) in Cu-MOF/MCF(M)-COOH was 2.46 mmol/g be-cause of the lower surface area of Cu-MOF/MCF(M)-COOH. The poresize distribution profile and texture properties indicated that theincreased microporosity of the nanocomposite was obtained atthe expense of a decrease of pore volume at the two mesopores at11 and 28 nm as shown in Fig. 2(e). Taking into account the micro-porosity of the Cu3(BTC)2, the increased micropore volume and sur-face area in the functional hybrid can be reasonably ascribed tonanoparticles of Cu3(BTC)2 formed inside the mesopores of MCF.
To directly confirm the hybridization between Cu3(BTC)2 andMCF(M)-COOH, the surface functional group before and afterhybridization was analyzed by FT-IR, solid state NMR (13C). TheFT-IR spectra of the parent substrate MCF(M)-COOH, the referenceCu3(BTC)2 and the functional hybrid for further confirming hybrid-ization are plotted in Fig. 3. For MCF(M)-COOH, the IR bands at1715 and 1635 cm�1 can be assigned to the C=O and C–O(H)stretching vibrations, respectively, of the terminal free acid asshown in Fig. 3(e) [30]. The bands at 1645 and 1575 cm�1 corre-spond to the asymmetric O–C–O stretching vibrations of the car-boxylate groups while the bands at 1447 and 1374 cm�1 are
assigned to the symmetric stretching vibrations [30]. For theCu(II)/MCF(M)-COOH obtained after the triple loading of copper ni-trate salt, the C=O stretching vibration band derived from the freeacid at 1715 cm�1 still remained while the strong bands responsi-ble for nitrate anions newly appeared around 1380 cm�1
(Fig. 3(d)). By contrast, the nanocomposite exhibited characteristicbands of bidentate carboxylate ligands coordinated to copper ionsat the absorption position of 1708 cm�1 which was shifted from1715 cm�1 (Dt = 7) due to the down field effect of carboxylate ions.This indicates that the carboxylic acid of the MCF surface partici-pated in formation of Cu3(BTC)2 [30,31]. The IR spectrum of thenanocomposite in the range between 1200 and 1800 cm�1 wasvery similar to that of the reference Cu3(BTC)2 (Fig. 3(c)). Consider-ing the presence of free carboxylic acid at a peak position of1715 cm�1 in the Cu(II)/MCF(M)-COOH (Fig. 3(d)), it is assumedthat the deprotonation of the surface COOH may take place simul-taneously with the the deprotonation of the BTC ligand during thecrystallization of Cu3(BTC)2. The surface hydroxyl group of thesamples was analyzed by FT-IR spectrum after evacuation underlow pressure (10�3 torr) at 250 �C for 12 h. The broad and strongbands between 3100 and 3700 cm�1 are the characteristic of O–H stretching vibration derived from the strongly H-bonded COOH(Fig. 4(b)). The hydroxyl group derived from the surface silanolgroup appeared at 3735 cm�1 (Fig. 4(a) and (b)). The hydroxylgroups were not completely removed after methylsilylation(Fig. 4(b)), but almost disappeared after hybridization with
200 150 100 50 0
200 150 100 50 0
200 150 100 50 0
(c)
*
C1,C1
C1,C1C2
C2
C3
Chemical Shift (ppm)
(b)
(a)
C3
C4
C4
*
'
'
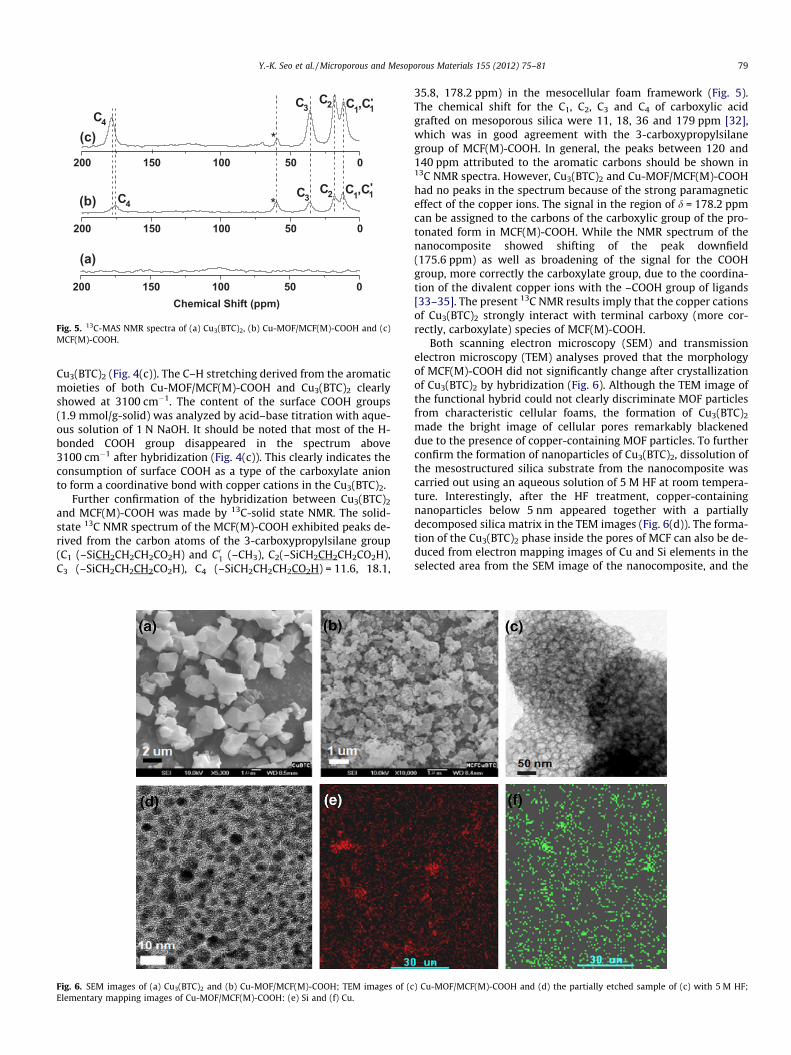
Fig. 5. 13C-MAS NMR spectra of (a) Cu3(BTC)2, (b) Cu-MOF/MCF(M)-COOH and (c)MCF(M)-COOH.
Y.-K. Seo et al. / Microporous and Mesoporous Materials 155 (2012) 75–81 79
Cu3(BTC)2 (Fig. 4(c)). The C–H stretching derived from the aromaticmoieties of both Cu-MOF/MCF(M)-COOH and Cu3(BTC)2 clearlyshowed at 3100 cm�1. The content of the surface COOH groups(1.9 mmol/g-solid) was analyzed by acid–base titration with aque-ous solution of 1 N NaOH. It should be noted that most of the H-bonded COOH group disappeared in the spectrum above3100 cm�1 after hybridization (Fig. 4(c)). This clearly indicates theconsumption of surface COOH as a type of the carboxylate anionto form a coordinative bond with copper cations in the Cu3(BTC)2.
Further confirmation of the hybridization between Cu3(BTC)2
and MCF(M)-COOH was made by 13C-solid state NMR. The solid-state 13C NMR spectrum of the MCF(M)-COOH exhibited peaks de-rived from the carbon atoms of the 3-carboxypropylsilane group(C1 (–SiCH2CH2CH2CO2H) and C01 (–CH3), C2(–SiCH2CH2CH2CO2H),C3 (–SiCH2CH2CH2CO2H), C4 (–SiCH2CH2CH2CO2H) = 11.6, 18.1,
Fig. 6. SEM images of (a) Cu3(BTC)2 and (b) Cu-MOF/MCF(M)-COOH; TEM images of (cElementary mapping images of Cu-MOF/MCF(M)-COOH: (e) Si and (f) Cu.
35.8, 178.2 ppm) in the mesocellular foam framework (Fig. 5).The chemical shift for the C1, C2, C3 and C4 of carboxylic acidgrafted on mesoporous silica were 11, 18, 36 and 179 ppm [32],which was in good agreement with the 3-carboxypropylsilanegroup of MCF(M)-COOH. In general, the peaks between 120 and140 ppm attributed to the aromatic carbons should be shown in13C NMR spectra. However, Cu3(BTC)2 and Cu-MOF/MCF(M)-COOHhad no peaks in the spectrum because of the strong paramagneticeffect of the copper ions. The signal in the region of d = 178.2 ppmcan be assigned to the carbons of the carboxylic group of the pro-tonated form in MCF(M)-COOH. While the NMR spectrum of thenanocomposite showed shifting of the peak downfield(175.6 ppm) as well as broadening of the signal for the COOHgroup, more correctly the carboxylate group, due to the coordina-tion of the divalent copper ions with the –COOH group of ligands[33–35]. The present 13C NMR results imply that the copper cationsof Cu3(BTC)2 strongly interact with terminal carboxy (more cor-rectly, carboxylate) species of MCF(M)-COOH.
Both scanning electron microscopy (SEM) and transmissionelectron microscopy (TEM) analyses proved that the morphologyof MCF(M)-COOH did not significantly change after crystallizationof Cu3(BTC)2 by hybridization (Fig. 6). Although the TEM image ofthe functional hybrid could not clearly discriminate MOF particlesfrom characteristic cellular foams, the formation of Cu3(BTC)2
made the bright image of cellular pores remarkably blackeneddue to the presence of copper-containing MOF particles. To furtherconfirm the formation of nanoparticles of Cu3(BTC)2, dissolution ofthe mesostructured silica substrate from the nanocomposite wascarried out using an aqueous solution of 5 M HF at room tempera-ture. Interestingly, after the HF treatment, copper-containingnanoparticles below 5 nm appeared together with a partiallydecomposed silica matrix in the TEM images (Fig. 6(d)). The forma-tion of the Cu3(BTC)2 phase inside the pores of MCF can also be de-duced from electron mapping images of Cu and Si elements in theselected area from the SEM image of the nanocomposite, and the
) Cu-MOF/MCF(M)-COOH and (d) the partially etched sample of (c) with 5 M HF;
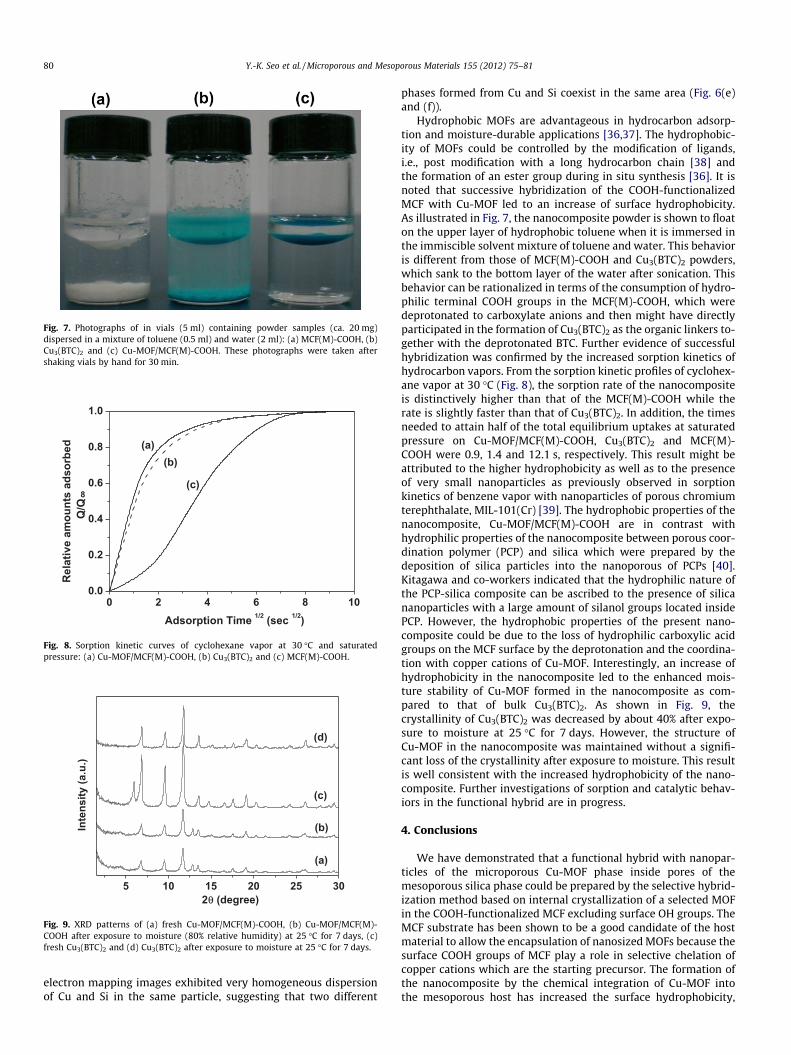
Fig. 7. Photographs of in vials (5 ml) containing powder samples (ca. 20 mg)dispersed in a mixture of toluene (0.5 ml) and water (2 ml): (a) MCF(M)-COOH, (b)Cu3(BTC)2 and (c) Cu-MOF/MCF(M)-COOH. These photographs were taken aftershaking vials by hand for 30 min.
0 2 4 6 8 100.0
0.2
0.4
0.6
0.8
1.0
(c)
(b)
Rel
ativ
e am
ount
s ad
sorb
edQ
/Q
Adsorption Time 1/2 (sec 1/2)
8
(a)
Fig. 8. Sorption kinetic curves of cyclohexane vapor at 30 �C and saturatedpressure: (a) Cu-MOF/MCF(M)-COOH, (b) Cu3(BTC)2 and (c) MCF(M)-COOH.
5 10 15 20 25 30
(d)
(c)
(b)Inte
nsity
(a.u
.)
2θ (degree)
(a)
Fig. 9. XRD patterns of (a) fresh Cu-MOF/MCF(M)-COOH, (b) Cu-MOF/MCF(M)-COOH after exposure to moisture (80% relative humidity) at 25 �C for 7 days, (c)fresh Cu3(BTC)2 and (d) Cu3(BTC)2 after exposure to moisture at 25 �C for 7 days.
80 Y.-K. Seo et al. / Microporous and Mesoporous Materials 155 (2012) 75–81
electron mapping images exhibited very homogeneous dispersionof Cu and Si in the same particle, suggesting that two different
phases formed from Cu and Si coexist in the same area (Fig. 6(e)and (f)).
Hydrophobic MOFs are advantageous in hydrocarbon adsorp-tion and moisture-durable applications [36,37]. The hydrophobic-ity of MOFs could be controlled by the modification of ligands,i.e., post modification with a long hydrocarbon chain [38] andthe formation of an ester group during in situ synthesis [36]. It isnoted that successive hybridization of the COOH-functionalizedMCF with Cu-MOF led to an increase of surface hydrophobicity.As illustrated in Fig. 7, the nanocomposite powder is shown to floaton the upper layer of hydrophobic toluene when it is immersed inthe immiscible solvent mixture of toluene and water. This behavioris different from those of MCF(M)-COOH and Cu3(BTC)2 powders,which sank to the bottom layer of the water after sonication. Thisbehavior can be rationalized in terms of the consumption of hydro-philic terminal COOH groups in the MCF(M)-COOH, which weredeprotonated to carboxylate anions and then might have directlyparticipated in the formation of Cu3(BTC)2 as the organic linkers to-gether with the deprotonated BTC. Further evidence of successfulhybridization was confirmed by the increased sorption kinetics ofhydrocarbon vapors. From the sorption kinetic profiles of cyclohex-ane vapor at 30 �C (Fig. 8), the sorption rate of the nanocompositeis distinctively higher than that of the MCF(M)-COOH while therate is slightly faster than that of Cu3(BTC)2. In addition, the timesneeded to attain half of the total equilibrium uptakes at saturatedpressure on Cu-MOF/MCF(M)-COOH, Cu3(BTC)2 and MCF(M)-COOH were 0.9, 1.4 and 12.1 s, respectively. This result might beattributed to the higher hydrophobicity as well as to the presenceof very small nanoparticles as previously observed in sorptionkinetics of benzene vapor with nanoparticles of porous chromiumterephthalate, MIL-101(Cr) [39]. The hydrophobic properties of thenanocomposite, Cu-MOF/MCF(M)-COOH are in contrast withhydrophilic properties of the nanocomposite between porous coor-dination polymer (PCP) and silica which were prepared by thedeposition of silica particles into the nanoporous of PCPs [40].Kitagawa and co-workers indicated that the hydrophilic nature ofthe PCP-silica composite can be ascribed to the presence of silicananoparticles with a large amount of silanol groups located insidePCP. However, the hydrophobic properties of the present nano-composite could be due to the loss of hydrophilic carboxylic acidgroups on the MCF surface by the deprotonation and the coordina-tion with copper cations of Cu-MOF. Interestingly, an increase ofhydrophobicity in the nanocomposite led to the enhanced mois-ture stability of Cu-MOF formed in the nanocomposite as com-pared to that of bulk Cu3(BTC)2. As shown in Fig. 9, thecrystallinity of Cu3(BTC)2 was decreased by about 40% after expo-sure to moisture at 25 �C for 7 days. However, the structure ofCu-MOF in the nanocomposite was maintained without a signifi-cant loss of the crystallinity after exposure to moisture. This resultis well consistent with the increased hydrophobicity of the nano-composite. Further investigations of sorption and catalytic behav-iors in the functional hybrid are in progress.
4. Conclusions
We have demonstrated that a functional hybrid with nanopar-ticles of the microporous Cu-MOF phase inside pores of themesoporous silica phase could be prepared by the selective hybrid-ization method based on internal crystallization of a selected MOFin the COOH-functionalized MCF excluding surface OH groups. TheMCF substrate has been shown to be a good candidate of the hostmaterial to allow the encapsulation of nanosized MOFs because thesurface COOH groups of MCF play a role in selective chelation ofcopper cations which are the starting precursor. The formation ofthe nanocomposite by the chemical integration of Cu-MOF intothe mesoporous host has increased the surface hydrophobicity,

Y.-K. Seo et al. / Microporous and Mesoporous Materials 155 (2012) 75–81 81
the water stability and the sorption rate of hydrophobic hydrocar-bon vapor. This work may provide a challengeable opportunity toexpand the application of the present approach to the selectivehybridization of other MOFs in the mesoporous MCF substrate.
Acknowledgements
This work was supported by the KCCS 2020 program funded bythe KCRC center (2011-0031982) and the Converging ResearchCenter Program (2011K000611) through the National ResearchFoundation of Korea funded by the Ministry of Education, Scienceand Technology.
References
[1] G. Férey, Chem. Soc. Rev. 37 (2008) 191–214.[2] D.J. Tranchemontagne, J.L. Mendoza-Cortés, M. O’Keeffe, O.M. Yaghi, Chem.
Soc. Rev. 38 (2009) 1257–1283.[3] T. Uemura, N. Yanai, S. Kitagawa, Chem. Soc. Rev. 38 (2009) 1228–1236.[4] Themed issue: metal-organic frameworks, Chem. Soc. Rev. 38 (2009) 1201–
1508.[5] L. Alaerts, C. Kirschhock, M. Maes, M. van der Veen, V. Finsy, A. Depla, J.
Martens, G. Baron, J. Denayer, D. De Vos, Angew. Chem., Int. Ed. 46 (2007)4293–4297.
[6] J.W. Yoon, Y.-K. Seo, Y.K. Hwang, J.-S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A.Vimont, M. Daturi, E. Bloch, P.L. Llewellyn, C. Serre, P. Horcajada, J.-M.Greneche, A.E. Rodrigues, G. Férey, Angew. Chem., Int. Ed. 49 (2010) 5949–5952.
[7] A.C. McKinlay, R.E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur, C. Serre,Angew. Chem., Int. Ed. 49 (2010) 6260–6266.
[8] Z. Wang, S. Cohen, Chem. Soc. Rev. 38 (2009) 1315–1329.[9] Y.K. Hwang, D.-Y. Hong, J.-S. Chang, S.H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M.
Daturi, C. Serre, G. Férey, Angew. Chem., Int. Ed. 47 (2008) 4144–4148.[10] C. Petit, B. Mendoza, T. Bandosz, Langmuir 26 (2010) 15302–15309.[11] C. Petit, T.J. Bandosz, Adv. Mater. 21 (2009) 4753–4757.[12] C.-Y. Sun, S.-X. Liu, D.-D. Liang, K.-Z. Shao, Y.-H. Ren, Z.-M. Su, J. Am. Chem. Soc.
131 (2009) 1883–1888.[13] M.G. Schwab, I. Senkovska, M. Rose, M. Koch, J. Pahnke, G. Jonschker, S. Kaskel,
Adv. Eng. Mater. 10 (2008) 1151–1155.[14] H.-L. Jiang, Q. Xu, Chem. Commun. 47 (2011) 3351–3370.
[15] K. Koh, A.G. Wong-Foy, A.J. Matzger, Chem. Commun. (2009) 6162–6164.[16] R. Ameloot, A. Liekens, L. Alaerts, M. Maes, A. Galarneau, B. Coq, G. Desmet, B.F.
Sels, J.F.M. Denayer, D.D. Vos, Eur. J. Inorg. Chem. (2010) 3735–3738.[17] J. Górka, P.F. Fulvio, S. Pikus, M. Jaroniec, Chem. Commun. 46 (2010) 6798–
6800.[18] Amanda M.B. Furtado, Jian. Liu, Yu. Wang, Douglas LeVan, J. Mater. Chem. 21
(2011) 698–6706.[19] W.J. Rieter, K.M.L. Taylor, W. Lin, J. Am. Chem. Soc. 129 (2007) 9852–9853.[20] E. Biemmi, C. Scherb, T. Bein, J. Am. Chem. Soc. 129 (2007) 8054–8055.[21] A.U. Czaja, N. Trukhan, U. Müller, Chem. Soc. Rev. 38 (2009) 1284–1293.[22] S. Shen, P.S. Chow, S. Kim, K. Zhu, R.B.H. Tan, J. Colloid. Inter. Sci. 321 (2008)
365–372.[23] H.H.P. Yiu, P.A. Wright, N.P. Botting, J. Mol. Catal. B: Enzym. 15 (2001)
81–92.[24] Y.-K. Seo, I. Suryanarayana, Y.K. Hwang, N. Shin, D.-C. Ahn, C.-H. Jun, J.-S.
Chang, J. Nanosci. Nanotechnol. 8 (2008) 3995–3998.[25] Y.-K. Seo, G. Hundal, I.T. Jang, Y.K. Hwang, C.-H. Jun, J.-S. Chang, Microporous
Mesoporous Mater. 119 (2009) 331–337.[26] S.S.Y. Chui, S.M.F. Lo, J.P.H. Charmant, A.G. Orpen, I.D. Williams, Science 283
(1999) 1148–1150.[27] K. Schlichte, T. Kratzke, S. Kaskel, Microporous Mesoporous Mater. 73 (2004)
81–88.[28] Q.M. Wang, D. Shen, M. B}ulow, M.L. Lau, S. Deng, F.R. Fitch, N.O. Lemcoff, J.
Semanscin, Microporous Mesoporous Mater. 55 (2002) 217–230.[29] C.-S. Chen, C.-C. Chen, C.-T. Chen, H.-M. Kao, Chem. Commun. 47 (2011) 2288–
2290.[30] C. Petit, J. Burress, T.J. Bandosz, Carbon 49 (2011) 563–572.[31] S. Vairam, S. Govindarajan, Thermochim. Acta 414 (2004) 263–270.[32] T.M. Suzuki, M. Mizutani, T. Nakamura, Y. Akimoto, K. Yano, Microporous
Mesoporous Mater. 116 (2008) 284–291.[33] R.J. Smernik, J.M. Oades, Geoderma 89 (1999) 219–248.[34] P. Horcajada, C. Serre, G. Maurin, N.A. Ramsahye, F. Balas, M. V-Regi, M.
Sebban, F. Taulelle, G. Férey, J. Am. Chem. Soc. 130 (2008) 6774–6780.[35] T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille, G.
Férey, Chem. Eur. J. 10 (2004) 1373–1382.[36] M.I.H. Mohideen, B. Xiao, P.S. Wheatley, A.C. McKinlay, Y. Li, A.M.Z. Slawin,
D.W. Aldous, N.F. Cessford, T. Düren, X. Zhao, R. Gill, K.M. Thomas, J.M. Griffin,S.E. Ashbrook, R.E. Morris, Nat. Chem. 3 (2011) 304–310.
[37] D. Ma, Y. Li, Z. Li, Chem. Commun. 47 (2011) 7377–7379.[38] J.G. Nguyen, S.M. Cohen, J. Am. Chem. Soc. 132 (2010) 4560–4561.[39] S.H. Jhung, J.-H. Lee, J.W. Yoon, C. Serre, G. Férey, J.-S. Chang, Adv. Mater. 19
(2007) 121–124.[40] T. Uemura, Y. Kadowaki, C.R. Kim, T. Fukushima, D. Hiramatsu, S. Kitagawa,
Chem. Mater. 23 (2011) 1736–1741.






![Fast and efficient synthesis of microporous polymer ......in organic electronics [8]. Among the microporous materials, conjugated microporous polymers (CMPs) [9,10] or porous aro-matic](https://img.pdfslide.us/doc/110x75/5ed931156714ca7f47695094/fast-and-efficient-synthesis-of-microporous-polymer-in-organic-electronics.jpg)






















