Embed Size (px)
Citation preview

This journal is c The Royal Society of Chemistry 2011 Chem. Commun., 2011, 47, 6957–6959 6957
Cite this: Chem. Commun., 2011, 47, 6957–6959
First enantioselective iron-porphyrin-catalyzed sulfide oxidation with
aqueous hydrogen peroxidewzPaul Le Maux and Gerard Simonneaux*
Received 23rd March 2011, Accepted 21st April 2011
DOI: 10.1039/c1cc11675d
The asymmetric oxidation of sulfides by H2O2 to give optically
active sulfoxides (ee up to 90%) was carried out in methanol and
water using chiral water-soluble iron porphyrins as catalysts.
The selective oxidation of sulfides to sulfoxides has attracted
much attention over the years after the pioneering work
of Kagan et al.1 and Modena et al.2 Sulfoxides constitute
chiral synthons in organic synthesis for the preparation of
biologically active compounds.3 They also serve as chiral
auxiliaries.4 Among all methods described so far,5 the
asymmetric oxidation of sulfides by metal catalysts is one of
the most attractive routes to optically active sulfoxides,3,4,6
and quite recently, even nontoxic and inexpensive iron
complexes have been developed successfully, using hydrogen
peroxide as an oxidant.7–10 Metalloporphyrins, widely studied
as models of hemes or cytochrome P-450, have been known to
exhibit the catalytic activity for monooxygenation, proceeding
via the formation of a high valency metal–oxygen complex
intermediate. However the asymmetric oxidation of sulfides
catalysed by chiral iron porphyrin is still unprecedented when
the oxidant is hydrogen peroxide. This is quite surprising
since the first metalloporphyrin-catalyzed oxygenation with
hydrogen peroxide reported the formation of sulfoxide from
sulfide11 and the enzymatic oxidation of sulfides to optically
active sulfoxides catalysed by peroxidases12–15 and other heme
proteins4 have been reported.
There are previously reported asymmetric homogeneous
iron-porphyrin-catalyzed sulfide oxidations in the literature
with iodosylbenzene as an oxidant.16–21 Many iron porphyrin–
H2O2 systems have been studied to get information on the
mechanism and nature of the active intermediates.22–25 The
two main obstacles when using hydrogen peroxide are the
high activity of many first-row transition metals in its
decomposition thereof, the so-called catalase reaction26 and
the catalyst destruction by hydroxyl radicals readily released
by homolytic H2O2 decomposition.
A chiral water-soluble iron Halterman porphyrin, due to the
presence of four sulfonate groups at the para-position,
was previously reported by our group and used for chiral
recognition of amino acids27 and catalytic carbene transfer in
water.28 Herein we report its use for catalytic asymmetric
sulfoxidation in polar solvents (water or methanol) by 35%
aqueous hydrogen peroxide (Scheme 1).
The starting point of the work described here was the
introduction of four sulfonate groups into an optically active
porphyrin with the aim of preparing water soluble porphyrins.
We choose a C2-symmetric group which contains two
norbornane groups fused to the central benzene ring, previously
reported by Halterman and Jan.29 The resulting metallo-
porphyrin28 after iron insertion (Fig. 1) is an electron-rich
iron(III) porphyrin which may tend to favour O–O homolysis
of hydroperoxide.22 Since it has been reported many times30,31
that solvent alcohols coordinate to the iron(III) porphyrin to
facilitate the heterolytic cleavage of the oxygen–oxygen bond,
we first use methanol as a solvent to favour the heterolytic
cleavage.
Oxidation of methyl phenyl sulfide was first examined with
this solvent at room temperature and lower temperatures.
The reaction is fast at room temperature though slight
over-oxidation to sulfones was observed (5%) and the
Fig. 1 Structure of the iron catalysts.
Scheme 1 Oxidation of sulfides by hydrogen peroxide.
Ingenierie Chimique et Molecules pour le Vivant, UMR CNRS 6226,Campus de Beaulieu, Universite de Rennes 1, 35042 Rennes, France.E-mail: [email protected];Fax: +33 (0)223235637; Tel: +33 (0)223236285w Electronic supplementary information (ESI) available: Experimentaldetails, chromatograms and visible spectra. See DOI: 10.1039/c1cc11675dz Dedicated to Professor Didier Astruc on the occasion of his 65thbirthday.
ChemComm Dynamic Article Links
www.rsc.org/chemcomm COMMUNICATION
Dow
nloa
ded
by U
nive
rsity
of
Hon
g K
ong
Lib
rari
es o
n 14
Mar
ch 2
013
Publ
ishe
d on
10
May
201
1 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CC
1167
5DView Article Online / Journal Homepage / Table of Contents for this issue
6958 Chem. Commun., 2011, 47, 6957–6959 This journal is c The Royal Society of Chemistry 2011
corresponding sulfoxide was obtained with 71% ee. At �20 1C,
the ee increases to 84% whereas the yield is still maintained
after 1 hour. We next examined the oxidation of other alkyl
aryl sulfides with 1 as the catalyst under identical conditions.
Similar levels of yields and enantioselectivities were obtained.
The enantioselectivities were high (between 78 and 90%) at
�20 1C, the best results being obtained with alkyl aryl sulfides
bearing electron-withdrawing groups onto the phenyl ring. It
is also interesting to note that ee is low when the phenyl group
is replaced by benzyl. The results are summarized in Table 1.
Our goal was not only to optimize this particular reaction,
but to use it as an indicator of the mechanism of oxygen atom
transfer. Because the sulfoxide yield depends on the efficiency
of the oxygen-atom transfer, and the degree of asymmetry
induction depends on the chirality of the catalyst, we can infer
the state of the catalyst from its catalytic activity in various
media. Furthermore, the enzymatic oxidation of sulfides to
sulfoxides in water by heme peroxidases is an important
process from both the mechanistic32–34 and synthetic point
of view allowing to get optically active sulfoxides with high
enantiomeric excess.12 Consequently, a systematic investigation
of the effect of the water content on the oxidation of methyl
phenyl sulfide at room temperature was first undertaken. The
results are summarized in Table 2, showing an interesting
phenomenon for the different combination of methanol/water.
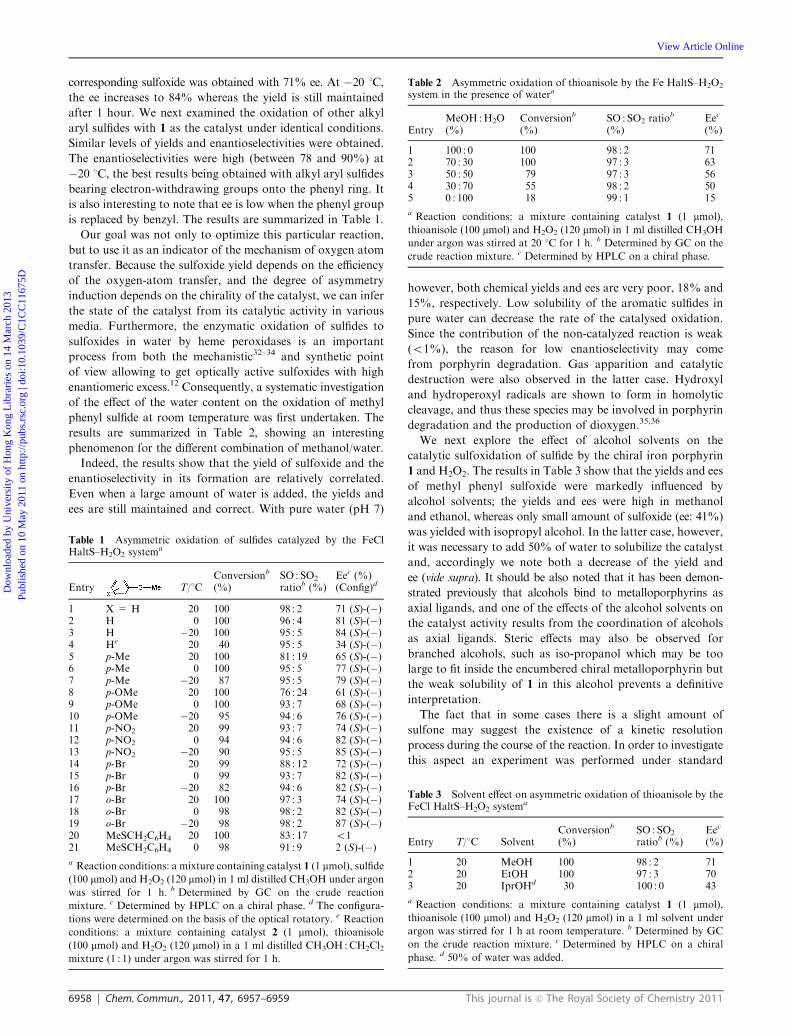
Indeed, the results show that the yield of sulfoxide and the
enantioselectivity in its formation are relatively correlated.
Even when a large amount of water is added, the yields and
ees are still maintained and correct. With pure water (pH 7)
however, both chemical yields and ees are very poor, 18% and
15%, respectively. Low solubility of the aromatic sulfides in
pure water can decrease the rate of the catalysed oxidation.
Since the contribution of the non-catalyzed reaction is weak
(o1%), the reason for low enantioselectivity may come
from porphyrin degradation. Gas apparition and catalytic
destruction were also observed in the latter case. Hydroxyl
and hydroperoxyl radicals are shown to form in homolytic
cleavage, and thus these species may be involved in porphyrin
degradation and the production of dioxygen.35,36
We next explore the effect of alcohol solvents on the
catalytic sulfoxidation of sulfide by the chiral iron porphyrin
1 and H2O2. The results in Table 3 show that the yields and ees
of methyl phenyl sulfoxide were markedly influenced by
alcohol solvents; the yields and ees were high in methanol
and ethanol, whereas only small amount of sulfoxide (ee: 41%)
was yielded with isopropyl alcohol. In the latter case, however,
it was necessary to add 50% of water to solubilize the catalyst
and, accordingly we note both a decrease of the yield and
ee (vide supra). It should be also noted that it has been demon-
strated previously that alcohols bind to metalloporphyrins as
axial ligands, and one of the effects of the alcohol solvents on
the catalyst activity results from the coordination of alcohols
as axial ligands. Steric effects may also be observed for
branched alcohols, such as iso-propanol which may be too
large to fit inside the encumbered chiral metalloporphyrin but
the weak solubility of 1 in this alcohol prevents a definitive
interpretation.
The fact that in some cases there is a slight amount of
sulfone may suggest the existence of a kinetic resolution
process during the course of the reaction. In order to investigate
this aspect an experiment was performed under standard
Table 1 Asymmetric oxidation of sulfides catalyzed by the FeClHaltS–H2O2 system
a
Entry T/1CConversionb
(%)SO : SO2
ratiob (%)Eec (%)(Config)d
1 X = H 20 100 98 : 2 71 (S)-(�)2 H 0 100 96 : 4 81 (S)-(�)3 H �20 100 95 : 5 84 (S)-(�)4 He 20 40 95 : 5 34 (S)-(�)5 p-Me 20 100 81 : 19 65 (S)-(�)6 p-Me 0 100 95 : 5 77 (S)-(�)7 p-Me �20 87 95 : 5 79 (S)-(�)8 p-OMe 20 100 76 : 24 61 (S)-(�)9 p-OMe 0 100 93 : 7 68 (S)-(�)10 p-OMe �20 95 94 : 6 76 (S)-(�)11 p-NO2 20 99 93 : 7 74 (S)-(�)12 p-NO2 0 94 94 : 6 82 (S)-(�)13 p-NO2 �20 90 95 : 5 85 (S)-(�)14 p-Br 20 99 88 : 12 72 (S)-(�)15 p-Br 0 99 93 : 7 82 (S)-(�)16 p-Br �20 82 94 : 6 82 (S)-(�)17 o-Br 20 100 97 : 3 74 (S)-(�)18 o-Br 0 98 98 : 2 82 (S)-(�)19 o-Br �20 98 98 : 2 87 (S)-(�)20 MeSCH2C6H4 20 100 83 : 17 o121 MeSCH2C6H4 0 98 91 : 9 2 (S)-(�)a Reaction conditions: a mixture containing catalyst 1 (1 mmol), sulfide
(100 mmol) and H2O2 (120 mmol) in 1 ml distilled CH3OH under argon
was stirred for 1 h. b Determined by GC on the crude reaction
mixture. c Determined by HPLC on a chiral phase. d The configura-
tions were determined on the basis of the optical rotatory. e Reaction
conditions: a mixture containing catalyst 2 (1 mmol), thioanisole
(100 mmol) and H2O2 (120 mmol) in a 1 ml distilled CH3OH :CH2Cl2mixture (1 : 1) under argon was stirred for 1 h.
Table 2 Asymmetric oxidation of thioanisole by the Fe HaltS–H2O2
system in the presence of watera
EntryMeOH :H2O(%)
Conversionb
(%)SO : SO2 ratio
b
(%)Eec
(%)
1 100 : 0 100 98 : 2 712 70 : 30 100 97 : 3 633 50 : 50 79 97 : 3 564 30 : 70 55 98 : 2 505 0 : 100 18 99 : 1 15
a Reaction conditions: a mixture containing catalyst 1 (1 mmol),
thioanisole (100 mmol) and H2O2 (120 mmol) in 1 ml distilled CH3OH
under argon was stirred at 20 1C for 1 h. b Determined by GC on the
crude reaction mixture. c Determined by HPLC on a chiral phase.
Table 3 Solvent effect on asymmetric oxidation of thioanisole by theFeCl HaltS–H2O2 system
a
Entry T/1C SolventConversionb
(%)SO : SO2
ratiob (%)Eec
(%)
1 20 MeOH 100 98 : 2 712 20 EtOH 100 97 : 3 703 20 IprOHd 30 100 : 0 43
a Reaction conditions: a mixture containing catalyst 1 (1 mmol),
thioanisole (100 mmol) and H2O2 (120 mmol) in a 1 ml solvent under
argon was stirred for 1 h at room temperature. b Determined by GC
on the crude reaction mixture. c Determined by HPLC on a chiral
phase. d 50% of water was added.
Dow
nloa
ded
by U
nive
rsity
of
Hon
g K
ong
Lib
rari
es o
n 14
Mar
ch 2
013
Publ
ishe
d on
10
May
201
1 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CC
1167
5D
View Article Online

This journal is c The Royal Society of Chemistry 2011 Chem. Commun., 2011, 47, 6957–6959 6959
asymmetric sulfide oxidation conditions using racemic sulf-
oxide as a substrate. After 1 hour reaction time, the conversion
of sulfoxide to sulfone was 30%. The ee of 10% for recovered
sulfoxide (in favor for the S-configured sulfoxide) revealed that
the sulfoxidation was indeed enantioselective. However, the
importance of the kinetic resolution under the experimental
conditions applied for the sulfide oxidation (very low amount
of sulfone) is very weak and consequently, the observed
enantioselectivity originates from the sulfide oxidation itself.
Even though the degradation of the catalyst is weak after 1
hour (o10% decrease of the Soret band, see ESIw), we decidedto study the time dependence of the asymmetric process in
more detail. Actually, most of the sulfoxidations are ended up
after 15 min.
Generally, in protic solvents which can act as a proton
donor, such as methanol and ethanol, the role of a cocatalyst
is not important, as shown by the unnecessary presence of a
cocatalyst with the iron system as reported by Traylor et al.37
Nevertheless, we noted a longer reaction time for complete
conversion of thioanisole after addition of 10 equivalents of
2-methylimidazole at room temperature with a slight increase
of the ee value: from 71% to 75%. All the results are
summarized in Table 4. As a possible explanation, imidazole
binds the iron center more strongly than methanol and may
decrease the rate of H2O2 consumption.
In conclusion, this investigation of H2O2 asymmetric oxidation
of sulfides in a protic solvent shows the practicability of the
process (absence of excess of oxidant and substrate, small
reaction time, room temperature reaction. . .) even though the
chiral catalyst does not bear a robust porphyrin ligand.
A protection of the oxoiron(IV) cation radical intermediate
from the two norbornane groups fused to the central benzene
ring is suggested since a similar reaction catalysed by
FeClTPPS yields to the destruction of the porphyrin ring.35
Ongoing work includes investigations of an extended range of
substrates, particularly those of pharmaceutical importance,
further optimization of the reaction medium and chiral
catalysts, and more experiments to precise the role of the
chirality in the mechanism.
Notes and references
1 P. Pitchen, E. Dunach, M. N. Deshmukh and H. B. Kagan, J. Am.Chem. Soc., 1984, 106, 8188.
2 F. Di Furia, G. Modena and R. Seraglia, Synthesis, 1984, 325.3 J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347,19.
4 I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651.5 K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetra-hedron, 2005, 61, 8315.
6 M. C. Carreno, G. H. Hernandez-Torres, M. Ribagorda andA. Urbano, Chem. Commun., 2009, 6129.
7 J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2004, 43, 4225.8 J. Legros and C. Bolm, Chem.–Eur. J., 2005, 11, 1086.9 H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940.10 H. Egami and T. Katsuki, Synlett, 2008, 1543.11 S. Oae, Y. Watanabe and K. Fujimori, Tetrahedron Lett., 1982, 23,
1189.12 S. Colonna, N. Gaggero, G. Carrea and P. Pasta, J. Chem. Soc.,
Chem. Commun., 1992, 357.13 R. Z. Harris, S. L. Newmyer and P. R. Ortiz de Montellano,
J. Biol. Chem., 1993, 268, 1637.14 M. P. J. van Deurzen, F. van Rantwijk and R. A. Sheldon,
Tetrahedron, 1997, 53, 13183.15 E. N. Kadnikova and N. M. Kostic, J. Org. Chem., 2003, 68, 2600.16 J. T. Groves and P. Viski, J. Org. Chem., 1990, 55, 3628.17 Y. Naruta, F. Tani and K. Maruyama, J. Chem. Soc., Chem.
Commun., 1990, 1378.18 Y. Naruta, F. Tani and K. Maruyama, Tetrahedron: Asymmetry,
1991, 2, 533.19 L. C. Chiang, K. Konishi, T. Aida and S. Inoue, J. Chem. Soc.,
Chem. Commun., 1992, 254.20 S. Inoue, T. Aida and K. Konoshi, J. Mol. Catal., 1992, 74, 121.21 Y. Ferrand, R. Daviaud, P. Le Maux and G. Simonneaux, Tetra-
hedron: Asymmetry, 2006, 17, 952.22 B. Meunier, Chem. Rev., 1992, 92, 1411.23 W. Nam, H. J. Han, S.-Y. Oh, Y. J. Lee, M.-H. Choi, S.-Y. Han,
C. Kim, S. K. Woo and W. Shin, J. Am. Chem. Soc., 2000, 122,8677.
24 E. Baciocchi, M. F. Gerini, O. Lanzalunga, A. Lapi and M. GraziaLo Piparo, Org. Biomol. Chem., 2003, 1, 422.
25 M. Wolak and R. van Eldik, Chem.–Eur. J., 2007, 13, 4873.26 I. W. C. E. Arends, Angew. Chem., Int. Ed., 2006, 45, 6250.27 I. Nicolas, S. Chevance, P. L. Maux and G. Simonneaux, Tetra-
hedron: Asymmetry, 2010, 21, 1788.28 I. Nicolas, P. Le Maux and G. Simonneaux, Tetrahedron Lett.,
2008, 49, 5793.29 R. L. Halterman and S. T. Jan, J. Org. Chem., 1991, 56, 5253.30 T. G. Traylor and F. Xu, J. Am. Chem. Soc., 1990, 112, 178.31 W. Nam, S.-Y. Oh, Y. J. Sun, J. Kim, W.-K. Kim, S. K. Woo and
W. Shin, J. Org. Chem., 2003, 68, 7903.32 E. Baciocchi, O. Lanzalunga, S. Malandrucco, M. Ioele and
S. Steenken, J. Am. Chem. Soc., 1996, 118, 8973.33 Y. Goto, T. Matsui, S.-i. Ozaki, Y. Watanabe and S. Fukuzumi,
J. Am. Chem. Soc., 1999, 121, 9497.34 Y. Watanabe, H. Nakajima and T. Ueno, Acc. Chem. Res., 2007,
40, 554.35 G. Lente and I. Fabian, Dalton Trans., 2007, 4268.36 N. A. Stephenson and A. T. Bell, J. Mol. Catal. A: Chem., 2007,
275, 54.37 T. G. Traylor, W. P. Fann and D. Bandyopadhyay, J. Am. Chem.
Soc., 1989, 111, 8009.
Table 4 Asymmetric oxidation of thioanisole by the FeCl HaltS–H2O2 system in the presence of 2-methylimidazolea
Entry T/1C2-Methylimidazole/mmol
Conversionb
(%)SO : SO2
ratiob (%)Eec
(%)
1 20 — 100 98 : 2 712 20 10 100 91 : 9 753 0 — 100 96 : 4 784 0 10 99 91 : 9 845 �20 — 100 95 : 5 826 �20 10 99 92 : 8 85.57d �20 10 61 92 : 8 90
a Reaction conditions: a mixture containing catalyst 1 (1 mmol),
2-methylimidazole (10 mmol) sulfide (100 mmol) and H2O2 (120 mmol)
in 1 ml distilled CH3OH under argon was stirred for 1 h. b Determined
by GC on the crude reaction mixture. c Determined by HPLC on a
chiral phase. d With 2-bromothioanisole.
Dow
nloa
ded
by U
nive
rsity
of
Hon
g K
ong
Lib
rari
es o
n 14
Mar
ch 2
013
Publ
ishe
d on
10
May
201
1 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CC
1167
5D
View Article Online





























