Embed Size (px)
Citation preview

Factors Contributing to NeuronalDegeneration in Retinas of ExperimentalGlaucomatous Rats
Xu Wang,1–3 Yee-Kong Ng,1 and Samuel Sam-Wah Tay1*1Department of Anatomy, Yong Loo Lin School of Medicine,National University of Singapore, Singapore2Retina and Optic nerve Research Laboratory, Dalhousie University, Halifax, Nova Scotia, Canada3Department of Physiology and Biophysics, Dalhousie University, Halifax, Nova Scotia, Canada
After our studies on ganglion cell degeneration in theglaucomatous retina, the current work further confirmedthe reduction of amacrine cells in the retina after theonset of glaucoma. Present study also tried to under-stand the possible mechanisms underlying neuronaldegeneration in the glaucomatous retina. Changes ofexpressions in immediate early genes (IEGs), glutamatereceptors (GluRs), calcium-binding proteins (CaBPs), 8-hydroxy-deoxyguanosine (8-OH-dG) and nitric oxidesynthase (NOS), as well as apoptotic-related factorsincluding caspase 3, bax, and bcl-2 were examined.IEGs such as c-fos and c-jun were induced in the retinaof the glaucomatous rat as early as 2 hr after the onsetof glaucoma and lasted up to 2 weeks. Expressions ofGluRs and CaBPs (i.e., parvalbumin and calbindin D-28k) were observed to be increased in the retinal gan-glion cell layer (GCL) and inner nuclear layer (INL) at 3days and 1 week after the onset of glaucoma. Theincrease occurred well before and during the phasewhere significant neuronal death was observed in theGCL and INL of the glaucomatous retinae. Induction of8-OH-dG was present in both the GCL and INL of theglaucomatous retina at 3 days after the onset of glau-coma before significant neuronal death was observed.Furthermore, confocal microscopy study showed thecomplete colocalization of immunohistochemical ex-pression of caspase 3 with glial fibrillary acidic protein(GFAP), but not with neuronal nuclei (NeuN). It indicatesthat astrocytes and Muller cells are involved in thepathological processes of neuronal death. The relation-ship between the linked factors and neuronal degener-ation is also discussed. VVC 2005 Wiley-Liss, Inc.
Key words: glaucoma; apoptotic factors; glutamatereceptors; calcium-binding proteins; retina
Glaucoma is defined best as an optic neuropathywith characteristic optic nerve head and associated visualfield changes (Thomas and Liesegang, 1996). Althoughstudies on the damage in visual function caused by glau-coma have been going on for quite a long time, thepathogenesis of optic neuropathy in glaucoma remains
very much debatable. Numerous works on glaucomatousretinopathy and optic nerve neuronopathy of clinicalpatients and experimental animals have been done in thepast few decades. Almost all the studies about glaucoma,however, have been focused on retinal ganglion cells(RGCs) death and optic nerve head damage (Glovinskyet al., 1991; Varma et al., 1993; Garcia-Valenzuela et al.,1995; Morrison et al., 1995; Johnson et al., 1996; Yanoffand Fine, 1996; Kerrigan et al., 1997; Laquis et al.,1998). There remain controversies and gaps regardingthe pathological processes of glaucoma and their under-lying mechanisms, which need to be investigated further.The main purpose of current studies is to use an animalmodel of glaucoma to establish the underlying mecha-nisms in neuronal degeneration or death in the retinaafter glaucoma, which include the aspects of apoptosis-related factors, glutamate receptors (GluRs), calcium-binding proteins (CaBPs), and immediate early genes(IEGs).
In our previous study, using the techniques of elec-tron microscopy (Wang et al., 2002b), we confirmedthat nuclei of dying ganglion cells in the rat during ele-vated intraocular pressure (IOP) undergo fragmentationof their chromatin as described in classical apoptosis,which had been reported by other laboratories (Garcia-Valenzuela et al., 1995; Kerrigan et al., 1997; Laquiset al., 1998). It is widely accepted that RGCs die viaapoptosis in glaucoma; however, little work has beendone on factors involved in the apoptotic pathway in
Xu Wang and Yee-Kong Ng contributed equally to this work.
Contract grant sponsor: National Medical Research Council (NMRC),
Singapore; Contract grant number: NMRC/0334/1999.
*Correspondence to: Samuel Sam-Wah Tay, Department of Anatomy,
Yong Loo Lin School of Medicine, National University of Singapore,
MD10, 4 Medical Drive, Singapore 117597.
E-mail: [email protected]
Received 6 July 2005; Revised 9 September 2005; Accepted 12 Septem-
ber 2005
Published online 4 November 2005 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/jnr.20679
Journal of Neuroscience Research 82:674–689 (2005)
' 2005 Wiley-Liss, Inc.

RGC death during glaucoma, and the mechanism ofganglion cell apoptosis is poorly understood (Nickells,1999). Research on apoptotic cell death in other areasshowed that the mechanism of apoptosis is controlled byspecific genes and their products that are activated in thedying cell (Nickells, 1999). Caspase 3 is a frequentlyactivated death protease. In mammals, caspases (princi-pally caspase 3) seem to be activated in a protease cas-cade that leads to inappropriate activation or rapid dis-ablement of key structural proteins and important signal-ing, homeostatic, and repair enzymes (reviewed byPorter and Janicke, 1999). Bax overexpression may causea decrease in permeability of the outer mitochondrialmembrane in some conditions, resulting in sensitivity tovoltage-dependent anion channel deficiency, reactiveoxygen species (ROS) production, and mitochondrialhyperpolarization. In other conditions, overexpression ofbax seems to form large pores in the outer mitochondrialmembrane that are capable of releasing cytochrome c(reviewed by Harris and Thompson, 2000). On theother hand, antiapoptotic bcl-2 family members, likebcl-2, have been shown to prevent the matrix swelling,ROS damage, release of cytochrome c, and loss ofmembrane potential associated with apoptosis. They maybind to bax and thus prevent bax-complex formation orchange the dynamics of the outer mitochondrial mem-brane in such a way as to disfavor the formation of baxcomplexes or insertion (Harris and Thompson, 2000).
Previous studies indicate that the neurotransmitterglutamate plays a key role in the visual system. Photore-ceptors, bipolar cells, and ganglion cells release glutamateto mediate the transfer of visual information from theretina to the brain (Brandstatter et al., 1998; Nakanishiet al., 1998). Glutamate neurotransmission has also beenimplicated to participate in neuronal plasticity and neu-rotoxicity (Nakanishi, 1992). Retinal neurons exhibit adiversity of GluRs, and overactivation of N-methyl-D-asparate (NMDA) and non-NMDA GluRs is believed toinduce excitotoxic cell death in these neurons (Yoshiokaet al., 1996; Thoreson and Witkosky, 1999). There aremany recent studies that show that elevated glutamatelevels exist in the vitreous humor of glaucoma patients,and the concentration of glutamate is suffice to killRGCs on its own. In addition, NMDA receptors(NMDARs) also have been shown to exist on ganglioncells and a subset of amacrine cells (Naskar et al., 1999;Osborne et al., 1999; Vorwerk et al., 1999; Haefligeret al., 2000). It is supposed that the increased IOP mayrepresent an initial insult that precipitates the productionof excessive glutamate and further cause the ganglion cellloss in the retina (Vorwerk et al., 1999). The majorcauses of cell death after activation of NMDARs are theinflux of calcium ions into cells and the generation offree radicals (Sucher et al., 1997; Osborne et al., 1999).
IEGs act as transcription factors affecting theexpression of other genes over longer periods of time.Rapid and transient expressions of these transcriptionfactors have been found after various neuronal injuriesand induction of transcription factors c-fos and c-jun
served as early markers of neuronal response to axonalinjury (Haas et al., 1993; Broude et al., 1997; Herdegenet al., 1997). In the visual system, it has been well estab-lished that IEG expression is induced by light stimulationand circadian regulation in the different components,including the retina, suprachiasmatic nucleus, and visualcortex (Caputto and Guido, 2000). The IEG expressionhave been observed in apoptotic neurons in the retinaduring development of neuronal degeneration as well asin the circadian visual system (Morin, 1994). Little stud-ies have been done, however, on the functions of IEGsin the retina after experimental glaucoma.
Based on the above reviews, the present workaimed to investigate the alteration of expression of c-fos,c-jun, caspase 3, bax, bcl-2, NMDAR1, GluR2, GluR2/3,and CaBPs in the glaucomatous retina using immunohis-tochemistry and reverse transcription-PCR (RT-PCR)techniques.
MATERIALS AND METHODS
Animals
Adult male Wistar rats (weighing 250 g) were used inthis study. In the handling and care of all animals, they werecarried out in accordance with the National Institute of HealthGuide for the Care and Use of Laboratory Animals (NIH Pub-lications No. 80-23) revised 1996. It is attested that all effortswere made to minimize the number of animals used and theirsuffering. All rats were bred and supplied by the LaboratoryAnimal Center, National University of Singapore, and kept inthe animal house before and after the experiments.
Surgical Procedure
After deep anesthesia, limbus-draining veins of the ratwere exposed and three of the four veins were cauterized usinga small vessel cauterizer (Fine Science Tools, Canada). Thedetailed surgical procedures have been reported previously(Wang et al., 2000a). Only the right eye of each rat was usedfor this operation. After operation, the rats were kept for 2 hrto 3 months before sacrificing them for various experiments.
Measurement of Intraocular Pressure
The IOPs of both eyes were measured by using a fac-tory-calibrated Tono-Pen XL tonometer (Mentor, Norwell,MA). Each IOP registered was an average of three consecu-tive measurements. IOP measurements were recorded beforeand a half-hour after cauterization, and then once every week,as well as immediately just before rats were sacrificed at differ-ent time intervals.
Immunohistochemistry
After deep anesthesia, rats were perfused first withRinger’s solution followed by 2% paraformaldehyde in 0.1 Mphosphate buffer (PB; pH 7.4). After perfusion, eyeballs wereremoved and postfixed in the same fixative for 2–4 hr beforethey were transferred into 0.1 M PB containing 15% sucroseand kept overnight at 48C. Frozen sections of the eye werecut at 20-lm thickness and mounted on chrome alum–gela-tin-coated slides. Mounted sections were washed for 20 min
Neuronal Degeneration in Glaucoma 675

in 0.1 M phosphate-buffered saline (PBS) at pH 7.4 contain-ing 0.1% Triton X-100, then blocked with 5% normal goatserum in PBS for 1 hr. Sections were then incubated over-night with primary antibodies respectively: polyclonal rabbitanti-caspase 3 (dilution 1:100; Santa Cruz Biotechnology,Santa Cruz, CA), bax (dilution 1:100; Pharmingen Chemicals,Singapore), calbindin D-28K (dilution 1:1,000; Swant, Swit-zerland), parvalbumin (dilution 1:1,000; Swant), NMDAR1(dilution 1:100; Chemicon International, Temecula, CA),GluR2 (dilution 1:100; Chemicon), GluR2/3 (dilution 1:100;Chemicon), polyclonal sheep anti-c-fos (dilution 1:1,000;Chemicon), and monoclonal mouse anti-choline acetyl trans-ferase (ChAT, dilution 1:500; Chemicon), 8-hydroxy-deoxy-guanosine (8-OH-dG, dilution 1:100; QED Bioscience Inc.),bcl-2 (dilution 1:100; Transduction Lab), c-jun (dilution 1:10;Transduction Lab), neuronal nitric oxide synthase (nNOS,dilution 1:100; Transduction Lab), and inducible NOS(iNOS, dilution 1:100; Transduction Lab). These antibodieswere diluted with PBS containing 0.1% Triton X-100. Afterincubation, sections were rinsed in PBS for 15 min andreacted with the Vectastain ABC Kit (PK4002; Vector Labo-ratories) against rabbit/mouse IgG (dilution 1:200) for 1 hr.Sections were then treated with 3,30-diaminobenzidine tetra-hydrochloride (Cat. No. 5637, Sigma Co.) as the peroxidasesubstrate. The sections were air-dried, counterstained with 1%methyl green, dehydrated, mounted in Permount, and cov-ered with coverslips. Negative controls for immunostainingconsisted of substituting normal serum in place of the primaryantibodies.
Immunofluorescence
For c-fos staining, after incubation overnight with pri-mary antibody, slides were washed in 0.1 M PBS-Triton X-100 (PBS-TX) three times for 15 min and placed in Alexa594-conjugated rabbit anti-sheep IgG (Molecular Probes) for1 hr. Slides were then rinsed at least three times with 0.1 MPBS before they were coverslipped with SlowFade AntifadeKits (Molecular Probes). Control sections were incubated asabove but without the primary antibodies.
Double Immunofluorescence Labeling of Caspase 3 andGlial Fibrillary Acidic Protein or Neuronal Nuclei
To investigate whether caspase 3 expression cells areneurons or astrocytes/Muller cells in the retina, double immu-nohistochemical labeling of caspase 3 with glial fibrillary acidicprotein (GFAP) and neuronal nuclei (NeuN) were carriedout. Retina sections on slides were washed in 0.1 M PBS-TXand incubated separately with monoclonal mouse antibodiesagainst GFAP and NeuN overnight at room temperature.After incubation, they were washed with 0.1 M PBS-TX for15 min and placed in Alexa 594-conjugated goat anti-mouseIgG for 1 hr before they were finally washed three times in0.1 M PBS. The sections were then incubated further withpolyclonal rabbit anti-caspase 3 antibody overnight at roomtemperature. After incubation, they were washed in 0.1 MPBS and placed in Oregon Green 488-conjugated goat anti-rabbit IgG for 1 hr, and finally washed three times in PBS.Slides were then air-dried, mounted with DAKO fluorescent
mounting medium, and covered with coverslips. Control sec-tions were incubated without primary antibodies.
Total RNA Extraction and Reverse Transcriptase-PCR
After deep anesthesia, rats were scarified and rat retinaewere removed rapidly and total RNA was isolated using theRNeasy Mini Kit (QIAGEN, Hilden, Germany) according tothe manufacturer’s protocol. The concentration and purity ofthe extracted RNA were evaluated by spectrophotometricabsorbance readings at 260 and 280 nm. A 50-ng amount oftotal RNA was submitted for reverse transcriptase (RT)-PCRby using the OneStep RT-PCR Kit (QIAGEN). The oligonu-cleotides used for PCR primers were: for GluR2, 50-CTA-TTTCCAAGGGGCGCTGAT-30 upstream and 50-CAG-TCCAGGAR RACACGCCG-30 downstream; for GluR3,50-TTCCGCTTTGCTGTGCAG-30 upstream and 50-AAT-GATGCGTCTGAATTC-30 downstream; and for NMDAR1,50-AACCTGCAGAACCGCAAG-30 upstream and 50-GCTT-GATGAGCAGGTCTATGC -30 downstream (Yoshioka et al.,1996). The amplification protocol involved denaturation at948C for 1 min, primer annealing at 558C for 1 min, andextension at 728C for 1 min. this cycle was repeated 30 times.Control amplifications were done either without RT or withoutRNA. After RT-PCR, amplification products (5 ll) wereresolved on 2% agarose gel stained with ethidium bromide andphotographed under a UV transilluminator.
RESULTS
Intraocular Pressure
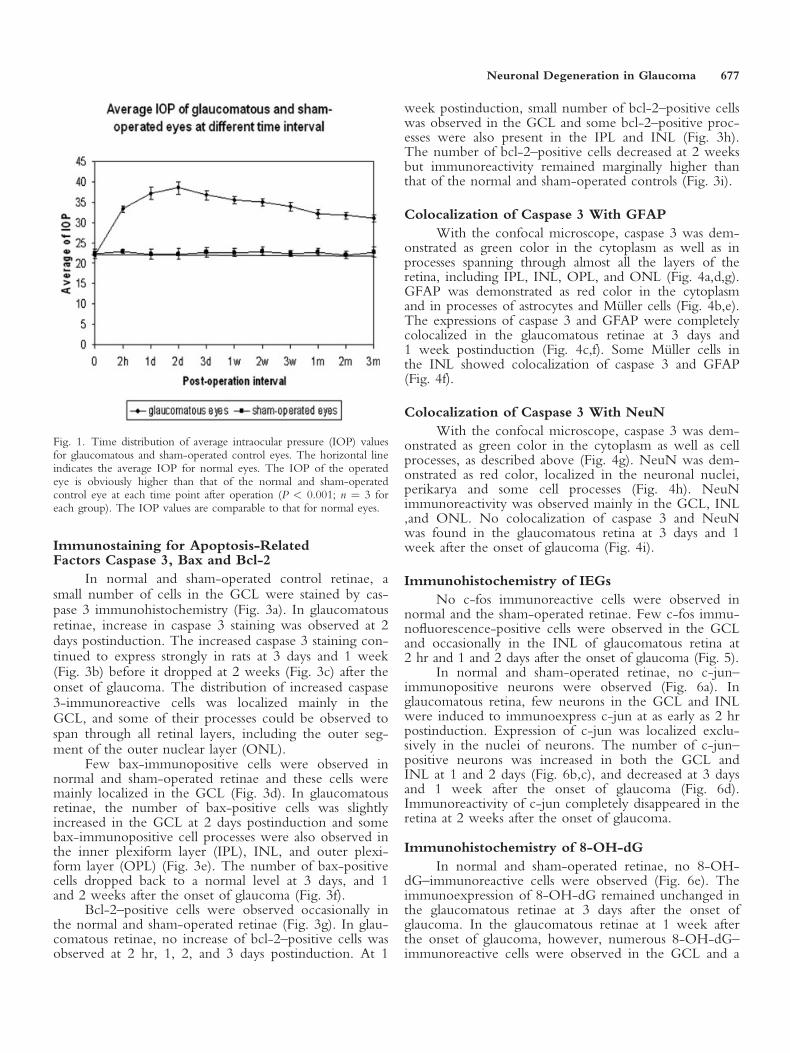
In the normal and sham-operated control eyes, theaverage IOP was 22.42 6 1.19 mm Hg. The mean IOPof the experimental eyes increased immediately afteroperation, and remained at a significantly elevated levelthroughout the entire length of the experiment (P <0.001; Fig. 1). The ocular tissues including cornea, lens,and sclera appeared normal throughout the experiment.
Alteration of Amacrine Cells
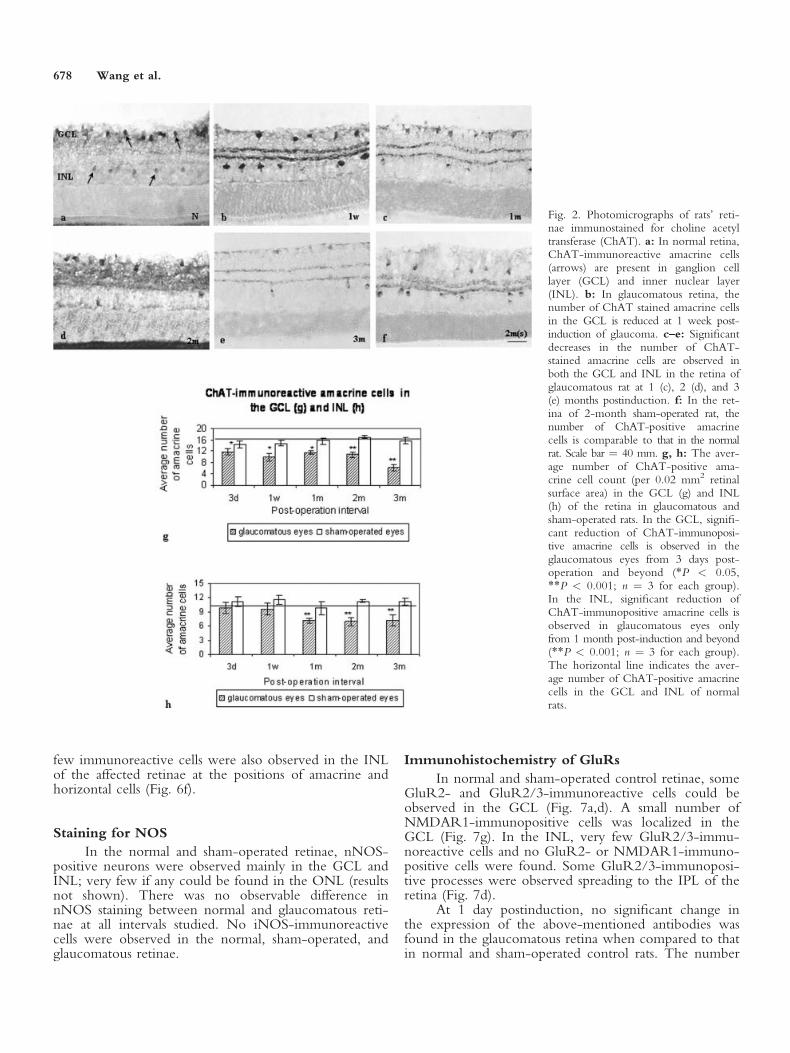
The monoclonal antibody ChAT was used as amarker to detect amacrine cells in the retina. In normalretina, ChAT-immunoreactive amacrine cells werepresent in both the ganglion cell layer (GCL) and innernuclear layer (INL; Fig. 2a). Cytoplasm and cell proc-esses of these amacrine cells were moderately labeled byChAT. The number of ChAT-stained amacrine cells inthe GCL dropped as early as 3 days after the onset ofglaucoma (P < 0.05; Fig. 2g). In the longer-survivingrats, the average number of ChAT-positive amacrinecells in the GCL showed further reduction and moresignificant decreases were observed at 2 and 3 monthspostinduction of glaucoma (P < 0.001; Fig. 2b–e,g). Asignificant drop in the number of ChAT-stained ama-crine cells in the INL of the glaucomatous retina, how-ever, was only observed from 1 month after the onset ofglaucoma and beyond (P < 0.001; Fig. 2c–e,h).
676 Wang et al.
Immunostaining for Apoptosis-RelatedFactors Caspase 3, Bax and Bcl-2
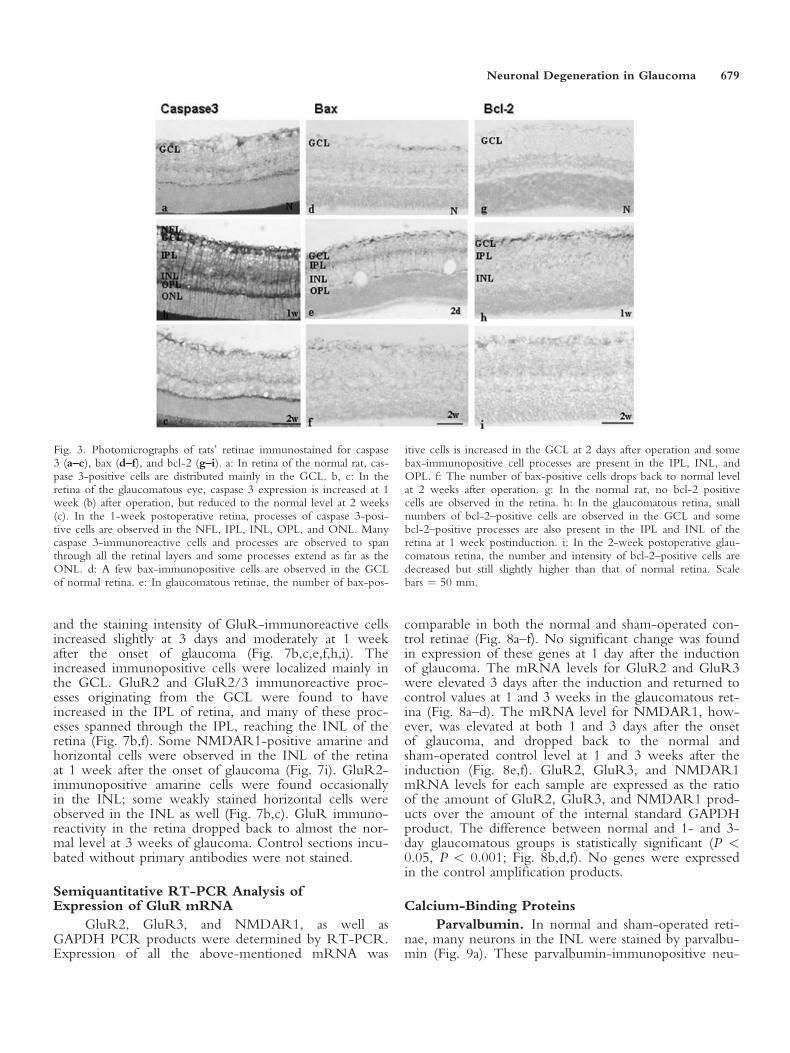
In normal and sham-operated control retinae, asmall number of cells in the GCL were stained by cas-pase 3 immunohistochemistry (Fig. 3a). In glaucomatousretinae, increase in caspase 3 staining was observed at 2days postinduction. The increased caspase 3 staining con-tinued to express strongly in rats at 3 days and 1 week(Fig. 3b) before it dropped at 2 weeks (Fig. 3c) after theonset of glaucoma. The distribution of increased caspase3-immunoreactive cells was localized mainly in theGCL, and some of their processes could be observed tospan through all retinal layers, including the outer seg-ment of the outer nuclear layer (ONL).
Few bax-immunopositive cells were observed innormal and sham-operated retinae and these cells weremainly localized in the GCL (Fig. 3d). In glaucomatousretinae, the number of bax-positive cells was slightlyincreased in the GCL at 2 days postinduction and somebax-immunopositive cell processes were also observed inthe inner plexiform layer (IPL), INL, and outer plexi-form layer (OPL) (Fig. 3e). The number of bax-positivecells dropped back to a normal level at 3 days, and 1and 2 weeks after the onset of glaucoma (Fig. 3f).
Bcl-2–positive cells were observed occasionally inthe normal and sham-operated retinae (Fig. 3g). In glau-comatous retinae, no increase of bcl-2–positive cells wasobserved at 2 hr, 1, 2, and 3 days postinduction. At 1
week postinduction, small number of bcl-2–positive cellswas observed in the GCL and some bcl-2–positive proc-esses were also present in the IPL and INL (Fig. 3h).The number of bcl-2–positive cells decreased at 2 weeksbut immunoreactivity remained marginally higher thanthat of the normal and sham-operated controls (Fig. 3i).
Colocalization of Caspase 3 With GFAP
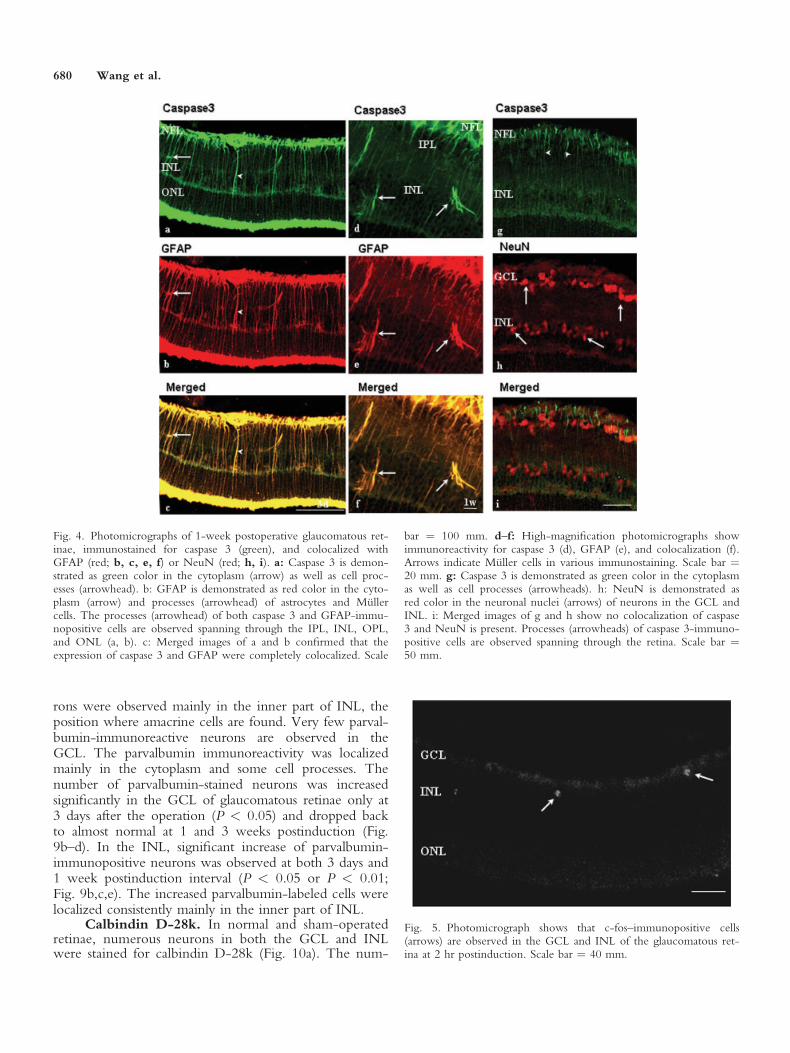
With the confocal microscope, caspase 3 was dem-onstrated as green color in the cytoplasm as well as inprocesses spanning through almost all the layers of theretina, including IPL, INL, OPL, and ONL (Fig. 4a,d,g).GFAP was demonstrated as red color in the cytoplasmand in processes of astrocytes and Muller cells (Fig. 4b,e).The expressions of caspase 3 and GFAP were completelycolocalized in the glaucomatous retinae at 3 days and1 week postinduction (Fig. 4c,f). Some Muller cells inthe INL showed colocalization of caspase 3 and GFAP(Fig. 4f).
Colocalization of Caspase 3 With NeuN
With the confocal microscope, caspase 3 was dem-onstrated as green color in the cytoplasm as well as cellprocesses, as described above (Fig. 4g). NeuN was dem-onstrated as red color, localized in the neuronal nuclei,perikarya and some cell processes (Fig. 4h). NeuNimmunoreactivity was observed mainly in the GCL, INL,and ONL. No colocalization of caspase 3 and NeuNwas found in the glaucomatous retina at 3 days and 1week after the onset of glaucoma (Fig. 4i).
Immunohistochemistry of IEGs
No c-fos immunoreactive cells were observed innormal and the sham-operated retinae. Few c-fos immu-nofluorescence-positive cells were observed in the GCLand occasionally in the INL of glaucomatous retina at2 hr and 1 and 2 days after the onset of glaucoma (Fig. 5).
In normal and sham-operated retinae, no c-jun–immunopositive neurons were observed (Fig. 6a). Inglaucomatous retina, few neurons in the GCL and INLwere induced to immunoexpress c-jun at as early as 2 hrpostinduction. Expression of c-jun was localized exclu-sively in the nuclei of neurons. The number of c-jun–positive neurons was increased in both the GCL andINL at 1 and 2 days (Fig. 6b,c), and decreased at 3 daysand 1 week after the onset of glaucoma (Fig. 6d).Immunoreactivity of c-jun completely disappeared in theretina at 2 weeks after the onset of glaucoma.
Immunohistochemistry of 8-OH-dG
In normal and sham-operated retinae, no 8-OH-dG–immunoreactive cells were observed (Fig. 6e). Theimmunoexpression of 8-OH-dG remained unchanged inthe glaucomatous retinae at 3 days after the onset ofglaucoma. In the glaucomatous retinae at 1 week afterthe onset of glaucoma, however, numerous 8-OH-dG–immunoreactive cells were observed in the GCL and a
Fig. 1. Time distribution of average intraocular pressure (IOP) valuesfor glaucomatous and sham-operated control eyes. The horizontal lineindicates the average IOP for normal eyes. The IOP of the operatedeye is obviously higher than that of the normal and sham-operatedcontrol eye at each time point after operation (P < 0.001; n ¼ 3 foreach group). The IOP values are comparable to that for normal eyes.
Neuronal Degeneration in Glaucoma 677
few immunoreactive cells were also observed in the INLof the affected retinae at the positions of amacrine andhorizontal cells (Fig. 6f).
Staining for NOS
In the normal and sham-operated retinae, nNOS-positive neurons were observed mainly in the GCL andINL; very few if any could be found in the ONL (resultsnot shown). There was no observable difference innNOS staining between normal and glaucomatous reti-nae at all intervals studied. No iNOS-immunoreactivecells were observed in the normal, sham-operated, andglaucomatous retinae.
Immunohistochemistry of GluRs
In normal and sham-operated control retinae, someGluR2- and GluR2/3-immunoreactive cells could beobserved in the GCL (Fig. 7a,d). A small number ofNMDAR1-immunopositive cells was localized in theGCL (Fig. 7g). In the INL, very few GluR2/3-immu-noreactive cells and no GluR2- or NMDAR1-immuno-positive cells were found. Some GluR2/3-immunoposi-tive processes were observed spreading to the IPL of theretina (Fig. 7d).
At 1 day postinduction, no significant change inthe expression of the above-mentioned antibodies wasfound in the glaucomatous retina when compared to thatin normal and sham-operated control rats. The number
Fig. 2. Photomicrographs of rats’ reti-nae immunostained for choline acetyltransferase (ChAT). a: In normal retina,ChAT-immunoreactive amacrine cells(arrows) are present in ganglion celllayer (GCL) and inner nuclear layer(INL). b: In glaucomatous retina, thenumber of ChAT stained amacrine cellsin the GCL is reduced at 1 week post-induction of glaucoma. c–e: Significantdecreases in the number of ChAT-stained amacrine cells are observed inboth the GCL and INL in the retina ofglaucomatous rat at 1 (c), 2 (d), and 3(e) months postinduction. f: In the ret-ina of 2-month sham-operated rat, thenumber of ChAT-positive amacrinecells is comparable to that in the normalrat. Scale bar ¼ 40 mm. g, h: The aver-age number of ChAT-positive ama-crine cell count (per 0.02 mm2 retinalsurface area) in the GCL (g) and INL(h) of the retina in glaucomatous andsham-operated rats. In the GCL, signifi-cant reduction of ChAT-immunoposi-tive amacrine cells is observed in theglaucomatous eyes from 3 days post-operation and beyond (*P < 0.05,**P < 0.001; n ¼ 3 for each group).In the INL, significant reduction ofChAT-immunopositive amacrine cells isobserved in glaucomatous eyes onlyfrom 1 month post-induction and beyond(**P < 0.001; n ¼ 3 for each group).The horizontal line indicates the aver-age number of ChAT-positive amacrinecells in the GCL and INL of normalrats.
678 Wang et al.
and the staining intensity of GluR-immunoreactive cellsincreased slightly at 3 days and moderately at 1 weekafter the onset of glaucoma (Fig. 7b,c,e,f,h,i). Theincreased immunopositive cells were localized mainly inthe GCL. GluR2 and GluR2/3 immunoreactive proc-esses originating from the GCL were found to haveincreased in the IPL of retina, and many of these proc-esses spanned through the IPL, reaching the INL of theretina (Fig. 7b,f). Some NMDAR1-positive amarine andhorizontal cells were observed in the INL of the retinaat 1 week after the onset of glaucoma (Fig. 7i). GluR2-immunopositive amarine cells were found occasionallyin the INL; some weakly stained horizontal cells wereobserved in the INL as well (Fig. 7b,c). GluR immuno-reactivity in the retina dropped back to almost the nor-mal level at 3 weeks of glaucoma. Control sections incu-bated without primary antibodies were not stained.
Semiquantitative RT-PCR Analysis ofExpression of GluR mRNA
GluR2, GluR3, and NMDAR1, as well asGAPDH PCR products were determined by RT-PCR.Expression of all the above-mentioned mRNA was
comparable in both the normal and sham-operated con-trol retinae (Fig. 8a–f). No significant change was foundin expression of these genes at 1 day after the inductionof glaucoma. The mRNA levels for GluR2 and GluR3were elevated 3 days after the induction and returned tocontrol values at 1 and 3 weeks in the glaucomatous ret-ina (Fig. 8a–d). The mRNA level for NMDAR1, how-ever, was elevated at both 1 and 3 days after the onsetof glaucoma, and dropped back to the normal andsham-operated control level at 1 and 3 weeks after theinduction (Fig. 8e,f). GluR2, GluR3, and NMDAR1mRNA levels for each sample are expressed as the ratioof the amount of GluR2, GluR3, and NMDAR1 prod-ucts over the amount of the internal standard GAPDHproduct. The difference between normal and 1- and 3-day glaucomatous groups is statistically significant (P <0.05, P < 0.001; Fig. 8b,d,f). No genes were expressedin the control amplification products.
Calcium-Binding Proteins
Parvalbumin. In normal and sham-operated reti-nae, many neurons in the INL were stained by parvalbu-min (Fig. 9a). These parvalbumin-immunopositive neu-
Fig. 3. Photomicrographs of rats’ retinae immunostained for caspase3 (a–c), bax (d–f), and bcl-2 (g–i). a: In retina of the normal rat, cas-pase 3-positive cells are distributed mainly in the GCL. b, c: In theretina of the glaucomatous eye, caspase 3 expression is increased at 1week (b) after operation, but reduced to the normal level at 2 weeks(c). In the 1-week postoperative retina, processes of caspase 3-posi-tive cells are observed in the NFL, IPL, INL, OPL, and ONL. Manycaspase 3-immunoreactive cells and processes are observed to spanthrough all the retinal layers and some processes extend as far as theONL. d: A few bax-immunopositive cells are observed in the GCLof normal retina. e: In glaucomatous retinae, the number of bax-pos-
itive cells is increased in the GCL at 2 days after operation and somebax-immunopositive cell processes are present in the IPL, INL, andOPL. f: The number of bax-positive cells drops back to normal levelat 2 weeks after operation. g: In the normal rat, no bcl-2 positivecells are observed in the retina. h: In the glaucomatous retina, smallnumbers of bcl-2–positive cells are observed in the GCL and somebcl-2–positive processes are also present in the IPL and INL of theretina at 1 week postinduction. i: In the 2-week postoperative glau-comatous retina, the number and intensity of bcl-2–positive cells aredecreased but still slightly higher than that of normal retina. Scalebars ¼ 50 mm.
Neuronal Degeneration in Glaucoma 679
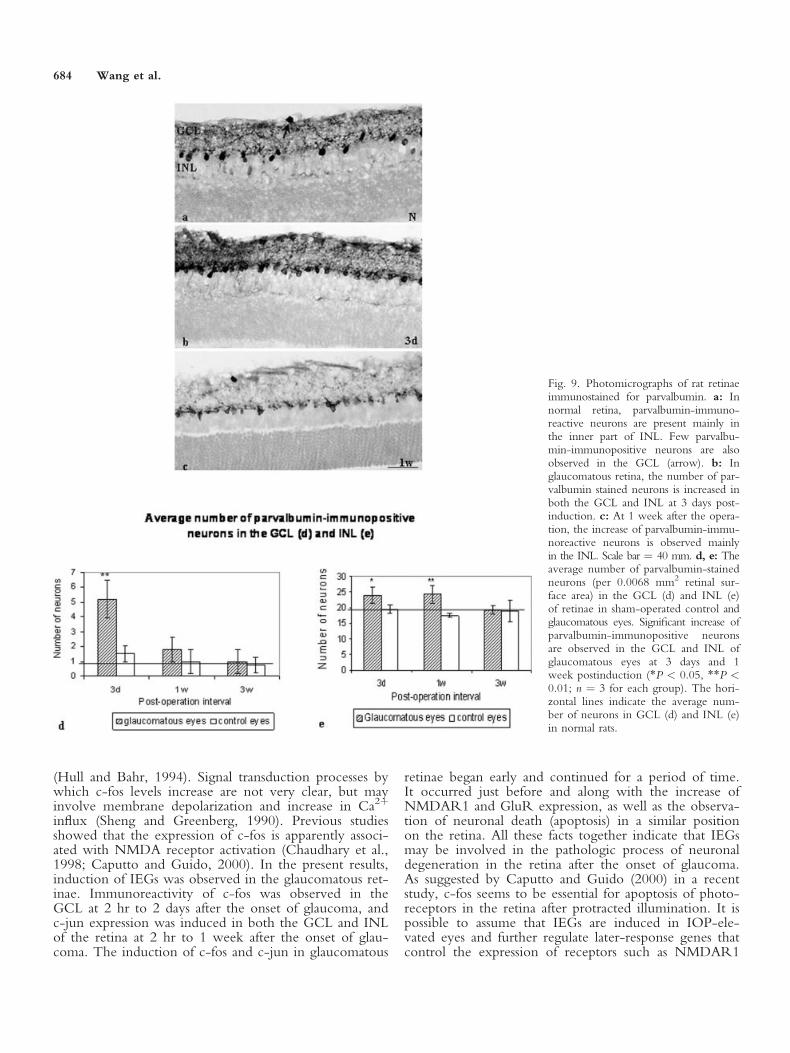
rons were observed mainly in the inner part of INL, theposition where amacrine cells are found. Very few parval-bumin-immunoreactive neurons are observed in theGCL. The parvalbumin immunoreactivity was localizedmainly in the cytoplasm and some cell processes. Thenumber of parvalbumin-stained neurons was increasedsignificantly in the GCL of glaucomatous retinae only at3 days after the operation (P < 0.05) and dropped backto almost normal at 1 and 3 weeks postinduction (Fig.9b–d). In the INL, significant increase of parvalbumin-immunopositive neurons was observed at both 3 days and1 week postinduction interval (P < 0.05 or P < 0.01;Fig. 9b,c,e). The increased parvalbumin-labeled cells werelocalized consistently mainly in the inner part of INL.
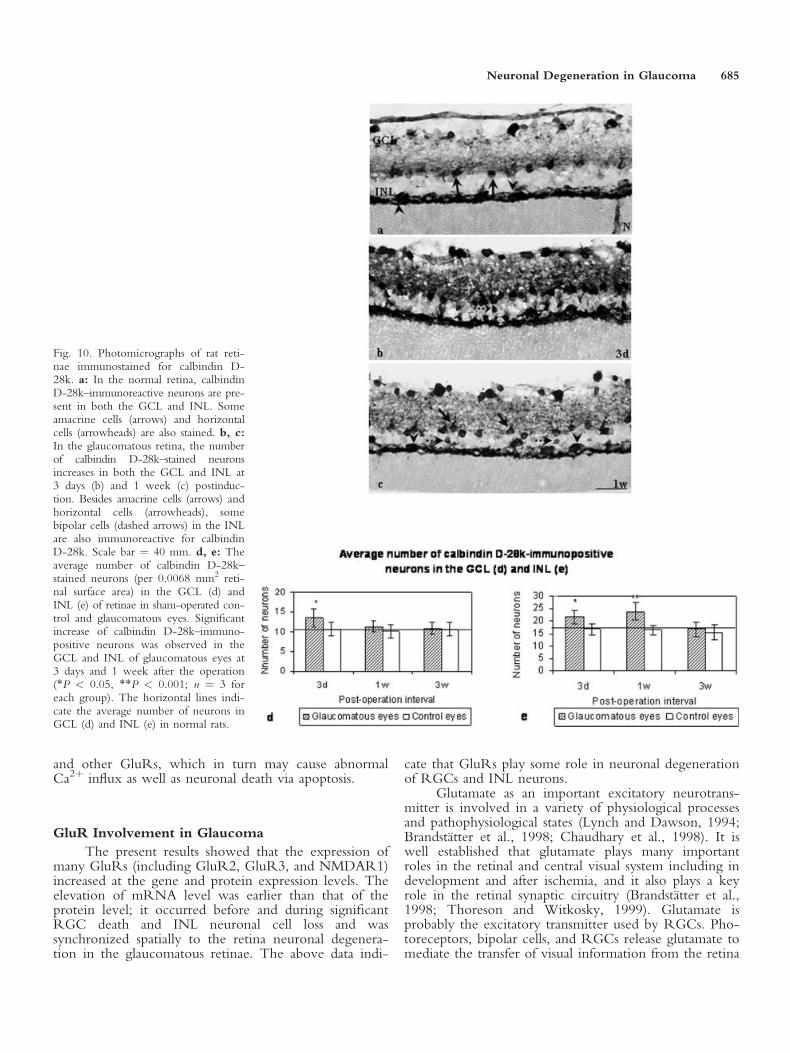
Calbindin D-28k. In normal and sham-operatedretinae, numerous neurons in both the GCL and INLwere stained for calbindin D-28k (Fig. 10a). The num-
Fig. 4. Photomicrographs of 1-week postoperative glaucomatous ret-inae, immunostained for caspase 3 (green), and colocalized withGFAP (red; b, c, e, f) or NeuN (red; h, i). a: Caspase 3 is demon-strated as green color in the cytoplasm (arrow) as well as cell proc-esses (arrowhead). b: GFAP is demonstrated as red color in the cyto-plasm (arrow) and processes (arrowhead) of astrocytes and Mullercells. The processes (arrowhead) of both caspase 3 and GFAP-immu-nopositive cells are observed spanning through the IPL, INL, OPL,and ONL (a, b). c: Merged images of a and b confirmed that theexpression of caspase 3 and GFAP were completely colocalized. Scale
bar ¼ 100 mm. d–f: High-magnification photomicrographs showimmunoreactivity for caspase 3 (d), GFAP (e), and colocalization (f).Arrows indicate Muller cells in various immunostaining. Scale bar ¼20 mm. g: Caspase 3 is demonstrated as green color in the cytoplasmas well as cell processes (arrowheads). h: NeuN is demonstrated asred color in the neuronal nuclei (arrows) of neurons in the GCL andINL. i: Merged images of g and h show no colocalization of caspase3 and NeuN is present. Processes (arrowheads) of caspase 3-immuno-positive cells are observed spanning through the retina. Scale bar ¼50 mm.
Fig. 5. Photomicrograph shows that c-fos–immunopositive cells(arrows) are observed in the GCL and INL of the glaucomatous ret-ina at 2 hr postinduction. Scale bar ¼ 40 mm.
680 Wang et al.
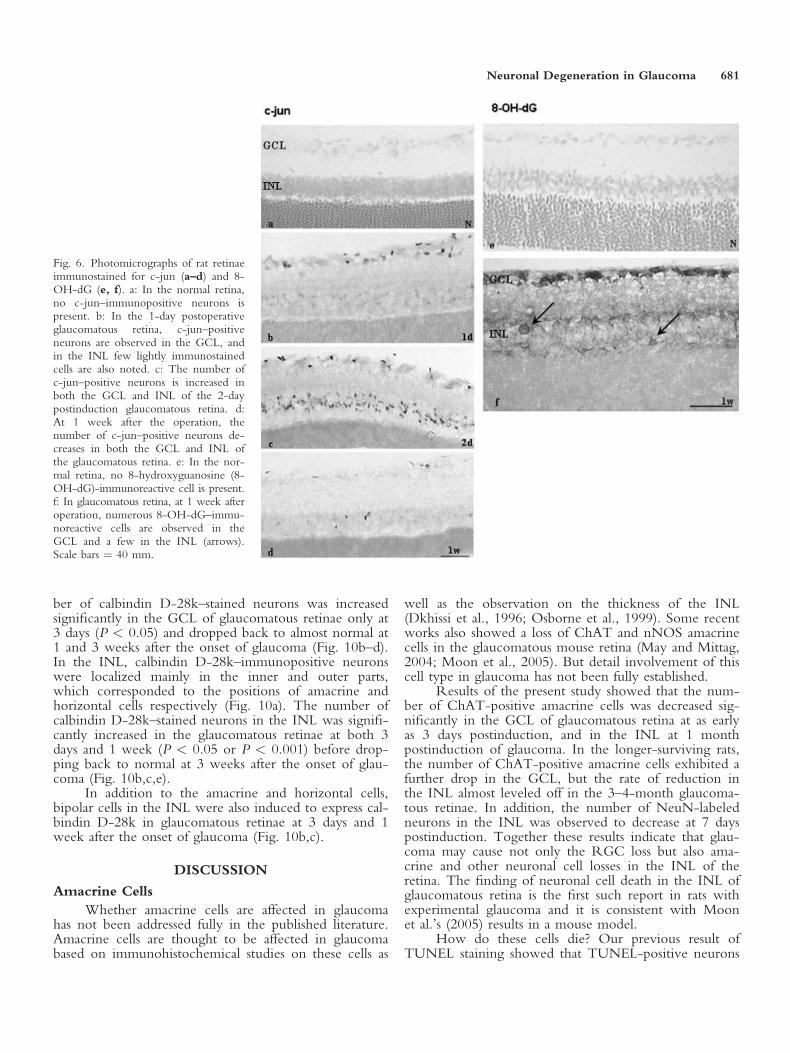
ber of calbindin D-28k–stained neurons was increasedsignificantly in the GCL of glaucomatous retinae only at3 days (P < 0.05) and dropped back to almost normal at1 and 3 weeks after the onset of glaucoma (Fig. 10b–d).In the INL, calbindin D-28k–immunopositive neuronswere localized mainly in the inner and outer parts,which corresponded to the positions of amacrine andhorizontal cells respectively (Fig. 10a). The number ofcalbindin D-28k–stained neurons in the INL was signifi-cantly increased in the glaucomatous retinae at both 3days and 1 week (P < 0.05 or P < 0.001) before drop-ping back to normal at 3 weeks after the onset of glau-coma (Fig. 10b,c,e).
In addition to the amacrine and horizontal cells,bipolar cells in the INL were also induced to express cal-bindin D-28k in glaucomatous retinae at 3 days and 1week after the onset of glaucoma (Fig. 10b,c).
DISCUSSION
Amacrine Cells
Whether amacrine cells are affected in glaucomahas not been addressed fully in the published literature.Amacrine cells are thought to be affected in glaucomabased on immunohistochemical studies on these cells as
well as the observation on the thickness of the INL(Dkhissi et al., 1996; Osborne et al., 1999). Some recentworks also showed a loss of ChAT and nNOS amacrinecells in the glaucomatous mouse retina (May and Mittag,2004; Moon et al., 2005). But detail involvement of thiscell type in glaucoma has not been fully established.
Results of the present study showed that the num-ber of ChAT-positive amacrine cells was decreased sig-nificantly in the GCL of glaucomatous retina at as earlyas 3 days postinduction, and in the INL at 1 monthpostinduction of glaucoma. In the longer-surviving rats,the number of ChAT-positive amacrine cells exhibited afurther drop in the GCL, but the rate of reduction inthe INL almost leveled off in the 3–4-month glaucoma-tous retinae. In addition, the number of NeuN-labeledneurons in the INL was observed to decrease at 7 dayspostinduction. Together these results indicate that glau-coma may cause not only the RGC loss but also ama-crine and other neuronal cell losses in the INL of theretina. The finding of neuronal cell death in the INL ofglaucomatous retina is the first such report in rats withexperimental glaucoma and it is consistent with Moonet al.’s (2005) results in a mouse model.
How do these cells die? Our previous result ofTUNEL staining showed that TUNEL-positive neurons
Fig. 6. Photomicrographs of rat retinaeimmunostained for c-jun (a–d) and 8-OH-dG (e, f). a: In the normal retina,no c-jun–immunopositive neurons ispresent. b: In the 1-day postoperativeglaucomatous retina, c-jun–positiveneurons are observed in the GCL, andin the INL few lightly immunostainedcells are also noted. c: The number ofc-jun–positive neurons is increased inboth the GCL and INL of the 2-daypostinduction glaucomatous retina. d:At 1 week after the operation, thenumber of c-jun–positive neurons de-creases in both the GCL and INL ofthe glaucomatous retina. e: In the nor-mal retina, no 8-hydroxyguanosine (8-OH-dG)-immunoreactive cell is present.f: In glaucomatous retina, at 1 week afteroperation, numerous 8-OH-dG–immu-noreactive cells are observed in theGCL and a few in the INL (arrows).Scale bars ¼ 40 mm.
Neuronal Degeneration in Glaucoma 681

were observed in the GCL and INL of glaucomatousretina at both 1 and 3 weeks post-glaucoma, and someof these cells were on the in the inner zone of the INLin the position of amacrine cells (Wang et al., 2002a).Furthermore, the previous results of transmission elec-tron microscopic (TEM) study also presented morpho-logic features of apoptotic neurons in the INL. Takentogether, these results indicate that neuronal deathincluding amacrine cells in the INL of glaucomatous ret-ina may also occur by apoptosis (Wang et al., 2002a)though we do not provide the colocalization of theapoptotic factors with ChAT-positive cells in the currentwork. Recent studies by Ullian et al. (2004), however,showed that amacrine cells were vulnerable to glutamateand NMDA excitotoxicity. This results suggests thatoverexpression of GluRs in the current study might bein response to the loss of ChAT-amacrine cells.
Apoptotic Pathway
In the present study, the changes of protein expres-sions of caspase 3, bax, and bcl-2 in the retina after theonset of glaucoma were documented and are consistentwith the general pathway of apoptosis in other areas. It
indicates that apoptosis of RGCs after glaucoma may beunder the same pathway or mechanism as that whichoccurs in other areas of the central nervous system(CNS). The results showed that the expressions of allthree proteins were increased from 2 days to 2 weeksafter the onset of glaucoma, and in a sequence from baxto caspase 3 to bcl-2. The bax- and bcl-2–positive cellsexhibited characteristics of neurons in the retina, espe-cially the RGCs. It is easy to understand that the over-expression of bax in the RGCs may cause neuronaldeath via apoptosis in these cells, and this is supportedby other studies using the retinal ischemia model(Kaneda et al., 1999). The results of the present study,however, showed that bcl-2 expression was increased.This is not consistent with some recent reports that bcl-2 expression decreased during RGC apoptosis after opticnerve crush or transection (Levin et al., 1997; Chaudh-ary et al., 1999). Overexpression of bcl-2 has beendemonstrated to be effective in preventing RGCs fromnatural and axotomy-induced cell death in neonataltransgenic mice (Bonfanti et al., 1996). Normally, down-regulation of bcl-2 and upregulation of bax are observedduring cell death via apoptosis. In the present results,however, bax and bcl-2 both showed upregulation. The
Fig. 7. Photomicrographs of rat retinae immunostained for GluRs:GluR2 (a–c), GluR2/3 (d–f) and NMDAR1 (g–i). a: In normal ret-ina, GluR2-immunoreactive cells are present in the GCL. b: Thenumber of GluR2-positive cells in the GCL is slightly increased at 3days after operation in glaucomatous retina. Arrows show GluR2-immunoreactive processes in the IPL. Immunopositive amacrine cells(arrowhead) are occasionally observed in the INL. c. Significantincrease in the number of GluR2-stained cells is observed in theGCL of the glaucomatous retina at 1 week after operation. Someweakly stained horizontal cells are present in the INL (arrowheads).d. In normal retina, GluR2/3-immunoreactive cells are present inthe GCL. Some immunopositive processes are spreading to the IPL(arrowheads). e. The number of GluR2/3-positive cells in the GCLincreased slightly at 3 days after operation in glaucomatous retina.
Arrowhead shows the process in the IPL emanating out from animmunoreactive cell in the GCL. f: Significant increase in the stain-ing intensity and number of GluR2/3 stained cells is observed in theGCL, 1 week postinduction of glaucoma. Many processes (arrows) ofGluR2/3-immunoreactive cells are observed spanning the IPL andreaching the INL of the retina. GluR2/3 immunoreactivity in theINL is also increased. g: In normal retina, some NMDAR1-immu-noreactive cells are present in the GCL (arrowheads). h: The numberof NMDAR1-positive cells increased marginally in the GCL at 3days postinduction in glaucomatous retina. i: Significant increase inthe intensity and number of NMDAR1-stained cells is observed inboth the GCL and INL, 1 week after the operation. Some amacrinecells (arrowheads) and horizontal cells (arrows) are also NMDAR1immunopositive. Scale bars ¼ 40 mm.
682 Wang et al.

possible reasons might be that the expression of bax andbcl-2 occurs in different neurons. RGCs that expressincreased bax will die via apoptosis; those that expressincreased bcl-2 will survive. It is possible that RGCsexpress both increased bax and bcl-2, however, andwhether the cell will die or survive is dependent onwhich expression is stronger (Cellerino et al., 2000).
Moreover, the present results showed that caspase3 and GFAP were well colocalized in the glaucomatousretina. It suggests that astrocytes and Muller cells overex-pressed caspase 3 after the onset of glaucoma. This find-ing reveals a novel hypothesis that astrocytes and Mullercells may be involved in the process of apoptosis in theglaucomatous retina. As it was suggested previously byTezel and Wax (2000) through their in vitro studies, theincreased production of death-promoting substancesincluding NO and tumor necrosis factor-a (TNF-a) thatwere produced by functional glial cells may result inRGC death after exposure to a stress condition.Although caspase 3 is essential for apoptotic processesassociated with the dismantling of the cell and the for-mation of apoptotic bodies, the activation of caspase 3 isnot by itself proof that it is required for apoptotic eventsand cell death (Porter and Janicke, 1999). Studies on theexpression of bax-like proteins imply that DNA frag-mentation requires functional caspase 3, but not all mor-phologic changes in dying cells require caspases (Xianget al., 1996; McCarthy et al., 1997; Gross et al., 1998).It may function before or at the stage when commit-ment to loss of cell viability is made. Exactly which cas-pase 3 dependent proteolytic events are required to causeapoptotic cell death remain largely unknown (Porter andJanicke, 1999). Unlike the report by McKinnon et al.(2002), our current work does not show any elevationof caspase 3 immunoactivation in the neurons in the ret-ina, possibly because our models were different fromtheirs. It also suggests that neuronal death in the retinaof our current model might not go through the apop-totic pathway via caspase 3. Astrocytes and Muller cellsthemselves survived, however, thereby suggesting thatnot all cells expressing caspase 3 will die.
Induction of Transcription Factors
In the visual system, c-fos and c-jun expressionshave been shown to be induced in RGCs and amacrinecells after light stimulation and optic nerve axotomy
Fig. 8.
Fig. 8. a, c, e: RT-PCR analysis demonstrating the expression ofGluR2 (a, upper panel; 539 base pairs [bp]), GluR3 (c, upper panel; 468bp), and NMDAR1 (e, upper panel; 333 bp). For comparison, expres-sion of GAPDH (528 bp) in gels is also shown (a, c, e, lower panels).b, d, f: Data represent mean optical density units 6 standard deviationof ethidium bromide-stained RT-PCR products for GluR2 (b), GluR3(d), and NMDAR1 (f). Marker, 100 bp DNA marker; N, normal; S1d,1 day sham-operated; S3d, 3 days sham-operated; G1d, 1 day glaucoma;G3d, 3 days glaucoma; G1w, 1 week glaucoma; G3w, 3 week glau-coma. *P < 0.05, **P < 0.001, significantly different from sham-oper-ated control values (n ¼ 3 for each group).
3
Neuronal Degeneration in Glaucoma 683
(Hull and Bahr, 1994). Signal transduction processes bywhich c-fos levels increase are not very clear, but mayinvolve membrane depolarization and increase in Ca2þ
influx (Sheng and Greenberg, 1990). Previous studiesshowed that the expression of c-fos is apparently associ-ated with NMDA receptor activation (Chaudhary et al.,1998; Caputto and Guido, 2000). In the present results,induction of IEGs was observed in the glaucomatous ret-inae. Immunoreactivity of c-fos was observed in theGCL at 2 hr to 2 days after the onset of glaucoma, andc-jun expression was induced in both the GCL and INLof the retina at 2 hr to 1 week after the onset of glau-coma. The induction of c-fos and c-jun in glaucomatous
retinae began early and continued for a period of time.It occurred just before and along with the increase ofNMDAR1 and GluR expression, as well as the observa-tion of neuronal death (apoptosis) in a similar positionon the retina. All these facts together indicate that IEGsmay be involved in the pathologic process of neuronaldegeneration in the retina after the onset of glaucoma.As suggested by Caputto and Guido (2000) in a recentstudy, c-fos seems to be essential for apoptosis of photo-receptors in the retina after protracted illumination. It ispossible to assume that IEGs are induced in IOP-ele-vated eyes and further regulate later-response genes thatcontrol the expression of receptors such as NMDAR1
Fig. 9. Photomicrographs of rat retinaeimmunostained for parvalbumin. a: Innormal retina, parvalbumin-immuno-reactive neurons are present mainly inthe inner part of INL. Few parvalbu-min-immunopositive neurons are alsoobserved in the GCL (arrow). b: Inglaucomatous retina, the number of par-valbumin stained neurons is increased inboth the GCL and INL at 3 days post-induction. c: At 1 week after the opera-tion, the increase of parvalbumin-immu-noreactive neurons is observed mainlyin the INL. Scale bar ¼ 40 mm. d, e: Theaverage number of parvalbumin-stainedneurons (per 0.0068 mm2 retinal sur-face area) in the GCL (d) and INL (e)of retinae in sham-operated control andglaucomatous eyes. Significant increase ofparvalbumin-immunopositive neuronsare observed in the GCL and INL ofglaucomatous eyes at 3 days and 1week postinduction (*P < 0.05, **P <0.01; n ¼ 3 for each group). The hori-zontal lines indicate the average num-ber of neurons in GCL (d) and INL (e)in normal rats.
684 Wang et al.
and other GluRs, which in turn may cause abnormalCa2þ influx as well as neuronal death via apoptosis.
GluR Involvement in Glaucoma
The present results showed that the expression ofmany GluRs (including GluR2, GluR3, and NMDAR1)increased at the gene and protein expression levels. Theelevation of mRNA level was earlier than that of theprotein level; it occurred before and during significantRGC death and INL neuronal cell loss and wassynchronized spatially to the retina neuronal degenera-tion in the glaucomatous retinae. The above data indi-
cate that GluRs play some role in neuronal degenerationof RGCs and INL neurons.
Glutamate as an important excitatory neurotrans-mitter is involved in a variety of physiological processesand pathophysiological states (Lynch and Dawson, 1994;Brandstatter et al., 1998; Chaudhary et al., 1998). It iswell established that glutamate plays many importantroles in the retinal and central visual system including indevelopment and after ischemia, and it also plays a keyrole in the retinal synaptic circuitry (Brandstatter et al.,1998; Thoreson and Witkosky, 1999). Glutamate isprobably the excitatory transmitter used by RGCs. Pho-toreceptors, bipolar cells, and RGCs release glutamate tomediate the transfer of visual information from the retina
Fig. 10. Photomicrographs of rat reti-nae immunostained for calbindin D-28k. a: In the normal retina, calbindinD-28k–immunoreactive neurons are pre-sent in both the GCL and INL. Someamacrine cells (arrows) and horizontalcells (arrowheads) are also stained. b, c:In the glaucomatous retina, the numberof calbindin D-28k–stained neuronsincreases in both the GCL and INL at3 days (b) and 1 week (c) postinduc-tion. Besides amacrine cells (arrows) andhorizontal cells (arrowheads), somebipolar cells (dashed arrows) in the INLare also immunoreactive for calbindinD-28k. Scale bar ¼ 40 mm. d, e: Theaverage number of calbindin D-28k–stained neurons (per 0.0068 mm2 reti-nal surface area) in the GCL (d) andINL (e) of retinae in sham-operated con-trol and glaucomatous eyes. Significantincrease of calbindin D-28k–immuno-positive neurons was observed in theGCL and INL of glaucomatous eyes at3 days and 1 week after the operation(*P < 0.05, **P < 0.001; n ¼ 3 foreach group). The horizontal lines indi-cate the average number of neurons inGCL (d) and INL (e) in normal rats.
Neuronal Degeneration in Glaucoma 685

to the brain (Sucher et al., 1997; Brandstatter et al.,1998). Glutamate can be toxic to neurons through anexcitotoxic pathway mediated primarily through theNMDA subtype of GluRs (Naskar et al., 2000). Eleva-tion of glutamate concentration in the vitreous body istoxic to RGCs in the rat, and elevated glutamate hasalso been detected in vitreous body of human and mon-keys with glaucoma (Chaudhary et al., 1998; Naskaret al., 1999). Pathological activation of glutamate recep-tors is thought to be a final common pathway leading toneuronal damage in the course of many neurological dis-eases through their involvement in excitotoxicity (Lynchand Dawson, 1994; Sucher et al., 1997).
In the present study, NMDAR1 mRNA wasincreased at 1 and 3 days and GluR3 mRNA wasincreased at 3 days after the onset of glaucoma beforeneuronal death was observed in the glaucomatous retinaat 1 week postinduction. The immunoreactivity ofNMDAR1 and GluR2/3 was increased at 3 days and 1week after glaucoma, and the increased NMDAR1-andGluR2/3-immunopositive cells were localized mainly inthe GCL of the retina where the RGCs were reducedsignificantly. Some NMDAR1-positive cells have beenobserved in the positions of amarine and horizontal cellsin the INL where significant neuronal changes were alsoobserved. Because retinal neurons exhibit a diversity ofGluRs and overactivation of NMDA and non-NMDAGluRs is believed to lead to excessive levels of intracel-lular calcium and induce excitotoxic cell death in theseneurons (Sucher et al., 1997; Chaudhary et al., 1998),the increase in NMDAR1 and GluR2/3 in the glau-comatous retina may account for the retinal neuronalchanges observed. It is possible that RGC and INL neu-ronal death in glaucoma is secondary to excitatory aminoacid-mediated toxicity, as well as the potent neurotoxiceffects of glutamate, kainate, and NMDA on these neu-rons. This notion is supported by previous studies usingvarious methods, including in vitro study, injectingNMDA into the vitreous body and detecting glutamatelevel in the vitreous body of glaucomatous patient(Sucher et al., 1991; Siliprandi et al., 1992; Dreyer et al.,1996; Vorwerk et al., 1996; Otori et al., 1998).
The role of GluR2, however, is thought to deter-mine the Ca2þ permeability in recombinant AMPAreceptors (Ozawa et al., 1998). Reduction in GluR2expression has been observed in vulnerable neurons inmany neurological insults including global ischemia andlimbic seizures. Downregulation of GluR2 gene expres-sion may serve as a molecular switch leading to the for-mation of Ca2þ-permeable AMPA receptors andenhanced toxicity of endogenous glutamate after a neu-rological insult (Pellegrini-Giampietro et al., 1997;Ozawa et al., 1998; Tanaka et al., 2000). In the presentstudy, the GluR2 mRNA level was increased at 3 daysafter the onset of glaucoma. The processes of GluR2-and GluR2/3-immunopositive cells have been found tobe increased in the IPL and further spanned to reach theINL of the retina at 3 days and 1 week after the onset ofglaucoma. This suggests that the enhanced expression of
GluR2 and GluR2/3 was localized mainly in the axonsand axon terminals of RGCs and possibly the processesof amacrine cells related to the bipolar cells in the IPL.Using the immunoelectron microscopy study, Ghoshet al. (2001) reported that amacrine cells expressedGluR2/3 at the output synapses of rod bipolar cell inmonkey retina. The upregulation of GluR2 in relationto increased protein and gene expression levels may be aresponse to prevent calcium conductance throughNMDAR1 and GluR3 and further protect these neuronsfrom degenerating in the retina after the onset of glau-coma.
Calcium-Binding Proteins
Ca2þ plays a key role in transmembrane signalingand the intracellular transmission of signals (Baimbridgeet al., 1992; Osborne et al., 1999). It is well establishedthat if the Ca2þ homeostasis of neurons is not well con-trolled, pathologic and toxicologic processes may occur(Osborne et al., 1999). The elevated cytosolic levels ofCa2þ may activate several destructive cascades that bearultimate responsibility for neuronal degeneration, such asdepolarization of mitochondrial membranes, productionof ROS, as well as activation of phospholipase A2,nucleases that act on DNA, and proteases that disruptribosomes, neurofilaments, and microtubules (Osborneet al., 1997, 1999).
Ca2þ, however, does not act alone. Many cellscontain a variety of cytosolic calcium-binding proteinsthat either modulate or mediate the actions of this ion(Baimbridge et al., 1992). Among the many CaBPs inthe nervous system, parvalbumin and calbindin D-28kare particularly striking in their abundance and in thespecificity of their distribution (Baimbridge et al., 1992).Their major roles are assumed to be Ca2þ buffering andtransport and regulation of various enzyme systems. Be-cause cellular degeneration is accompanied by impairedCa2þ homeostasis, CaBPs, in part, are thought as neuro-protective proteins (Heizmann and Braun, 1992).
The present immunohistochemical results for par-valbumin and calbindin D-28k showed increases in thenumbers of positive neurons containing these two CaBPs.In normal and sham-operated control rats, parvalbuminimmunoreactivity has been observed mainly in the loca-tion of amacrine cells in the innermost part of the INLbordering the IPL. Calbindin D-28k immunoreactivityhas been observed in neurons of the GCL and INL,including amacrine and horizontal cells, as well as fewbipolar cells. The staining pattern of these two CaBPswas observed to be in accord with previous reports inthe normal retina (Pasteels et al., 1990; Pochet et al.,1991; Uesugi et al., 1992).
There is a consensus that overactivation of gluta-matergic ionotropic receptors (NMDA and kainate/AMPA types) will lead to neuronal destruction becauseof the consequences of Ca2þ influx (Osborne et al.,1997; Chaudhary et al., 1998). Ca2þ that enters the cellthrough the NMDA receptor is capable of activating a
686 Wang et al.

variety of cellular enzymes, including proteases, kinases,phospholipases, and NOS (Lynch and Dawson, 1994).In the present study, the expressions of parvalbumin andcalbindin D-28k immunoreactivity increased only at 3days after the onset of glaucoma in the GCL and at 3days and 1 week after glaucoma in the INL. Theyoccurred only before significant neuronal death wasobserved in these two layers. In addition, the increase ofparvalbumin and calbindin D-28k immunoreactivityis well paralleled with the increased expression ofNMDAR1, GluR2, and GluR2/3. Upregulation ofNMDAR1 and GluR3 in ganglion, amacrine, and bipo-lar cells in the present study in the glaucomatous retinacould have resulted in the increase of intracellular Ca2þ
contents in these cells mediated through NMDAR1 andGluR3. The increased levels of CaBPs in amacrine andbipolar cells may function as a buffering reservoir tomaintain the Ca2þ homeostasis, and thus limit the num-ber of neurons dying from excessive calcium influx atthe early stage after the onset of glaucoma.
8-OH-dG and NO
8-OH-dG is the most commonly studied bio-marker for oxidative DNA damage (Floyd, 1990). Itsexpression has been found to be increased in many dis-eases associated with oxidative DNA damage (Hahmet al., 1998; Markesbery and Carney, 1999; Shen andOng, 2000). The present results showed that 8-OH-dGimmunoexpression was induced in RGCs and neuronsin the INL (most likely the amacrine and horizontalcells) of glaucomatous retinae at 3 days after the onset ofglaucoma. The induction of 8-OH-dG expression inglaucomatous retinae suggests that the oxidation ofDNA bases may be a causal factor in glaucoma-inducedneuronal cell death in the GCL and INL.
In recent years, NO has been one of the latest dis-coveries in the arena of messenger molecules in thebrain. In the visual system, it is produced in both pre-and postsynaptic structures and acts as a neurotransmitter(Cudeiro and Rivadulla, 1999). Previous studies havedemonstrated the apparent upregulation and induction ofcertain isoforms of NOS in the optic nerve head of theretina in many different diseases including glaucoma(Neufeld, 1999; Morgan, 2000). The results of thepresent study failed to document any observable differ-ence in the expression of NOS between normal andglaucomatous retinae, thereby indicating that NO maynot be involved in the pathological processes of neuronaldeath in the glaucomatous retina. Other unknown oxi-dative factors, however, may respond to the oxidativeDNA damage in the glaucomatous retinal neurons.Another possible reason for the failure to show NOSexpression could be due to the selection of specimensfor analysis. In the present study, sagittal sections throughthe whole retina have been used, as compared to someprevious studies, which used mainly retina around theoptic nerve head (Liu and Neufeld, 2001).
In summary, the present work and our earlier stud-ies (Wang et al., 2000a,b, 2002a,b) indicate thatincreased IOP in the glaucomatous eye could trigger aseries of biochemical and pathophysiological changes inthe microenvironment of the retina. It is hypothesizedthat IEGs are induced and they further regulate later-response genes that control the expression of numerousfactors, such as glutamate and Ca2þ, that are responsiblefor neuronal degeneration in the retina after the onset ofglaucoma. The imbalance of glutamate and Ca2þ levelsin the microenvironment of the retina may be one ofthe causal factors that could have promoted or aggra-vated apoptosis and oxidation of DNA bases, leadingeventually to the neuronal death in the glaucomatousretina via mainly apoptosis as was reported previously(Wang et al., 2002b). Our present study confirmed theinvolvement of some of the GluRs and CaBPs, and theirpossible interactions and roles in the neurodegenerationin glaucomatous retina were discussed.
REFERENCES
Baimbridge KG, Celio MR, Rogers JH. 1992. Calcium-binding proteins
in the nervous system. Trends Neurosci 15:303–308.
Bonfanti L, Strettoi E, Chierzi S, Cenni MC, Liu XH, Martinou JC,
Maffei L, Rabacchi SA. 1996. Protection of retinal ganglion cells from
natural and axotomy-induced cell death in neonatal transgenic mice
overexpressing bcl-2. J Neurosci 16:4186–4194.
Brandstatter JH, Koulen P, Wassle H. 1998. Diversity of glutamate
receptors in the mammalian retina. Vision Res 38:1385–1397.
Broude E, McAtee M, Kelley MS, Bregman BS. 1997. C-jun expression
in adult rat dorsal root ganglion neurons: Differential response after
central and peripheral axotomy. Exp Neurol 148:367–377.
Caputto BL, Guido ME. 2000. Immediate early gene expression within
the visual system: light and circadiam regulation in the retina and the
suprachiasmatic nucleus. Neurochem Res 25:153–162.
Cellerino A, Bahr M, Isenmann S. 2000. Apoptosis in the developing
visual system. Cell Tissue Res 301:53–69.
Chaudhary P, Ahmed F, Quebada P, Sharma SC. 1999 Caspase inhibitors
block the retinal ganglion cell death following optic nerve transection.
Brain Res Mol Brain Res 67:36–45.
Chaudhary P, Ahmed F, Sharma SC. 1998. MK801—a neuroprotectant
in rat hypertensive eyes. Brain Res 792:154–158.
Cudeiro J, Rivadulla C. 1999 Sight and insight—on the physiological
role of nitric oxide in the visual system. Trends Neurosci 22:109–116.
Dkhissi O, Chanut E, Versaux-Botteri C, Minvielle F, Trouvin JH,
Nguyen-Legros J. 1996 Changes in retinal dopaminergic cells and dop-
amine rhythmic metabolism during the development of a glaucoma-like
disorder in quails. Invest Ophthalmol Vis Sci 37:2335–2344.
Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA. 1996.
Elevated glutamate levels in the vitreous body of humans and monkeys
with glaucoma. Arch Ophthalmol 114:299–305.
Floyd RA. 1990. The role of 8-hydroguanine in carcinogenesis. Carcino-
genesis 11:1447–1450.
Garcia-Valenzuela E, Shareef S, Walsh J, Sharma SC. 1995. Programmed
cell death of retinal ganglion cells during experimental glaucoma. Exp
Eye Res 61:33–44.
Ghosh KK, Haverkamp S, Wassle H. 2001. Glutamate receptors in the
rod pathway of the mammalian retina. J Neurosci 21:8636–8647.
Glovinsky Y, Quigley HA, Dunkelberger RD. 1991. Retinal ganglion
cell loss is size dependent in experimental glaucoma. Invest Ophthalmol
Vis Sci 32:484–490.
Neuronal Degeneration in Glaucoma 687

Gross A, Jockel J, Wei MC, Korsmeyer SJ. 1998 Enforced dimerization
of BAX results in its translocation, mitochondrial dysfunction and apo-
ptosis. EMBO J 17:3878–3885.
Haas CA, Konath C, Kreutzberg GW. 1993. Differential expression
of immediate early genes after transection of the facial nerve. Neuro-
science 53:91–99.
Haefliger IO, Fleischhauer JC, Flammer J. 2000. In glaucoma, should en-
thusiasm about neuroprotection be tempered by the experience ob-
tained in other neurodegenerative disorders? Eye 14:464–472.
Hahm KB, Lee KJ, Kim JH, Cho SW, Chung MH. 1998 Helicobacter
pylori infection, oxidative DNA damage, gastric carcinogenesis, and re-
versibility by rebamipide. Dig Dis Sci 43(Suppl):72–77.
Harris MH, Thompson CB. 2000. The role of the Bcl-2 family in the
regulation of outer mitochondrial membrane permeability. Cell Death
Differ 7:1182–1191.
Heizmann CW, Braun K. 1992. Changes in Ca(2þ)-binding proteins in
human neurodegenerative disorders. Trends Neurosci 15:259–264.
Herdegen T, Skene P, Bahr M. 1997. The c-Jun transcription factor-
bipotential mediator of neuronal death, survival and regeneration. Trends
Neurosci 20:227–231.
Hull M, Bahr M. 1994. Differential regulation of c-jun expression in rat
retinal ganglion cells after proximal and distal optic nerve transection.
Neurosci Lett 178:39–42.
Johnson EC, Morrison JC, Farrell S, Deppmeier L, Moore CG, Mcginty
MR. 1996. The effect of chronically elevated intraocular pressure on
the rat optic nerve head extracellular matrix. Exp Eye Res 62:663–674.
Kaneda K, Kashii S, Kurosawa T, Kaneko S, Akaike A, Honda Y, Min-
ami M, Satoh M. 1999. Apoptotic DNA fragmentation and upregula-
tion of Bax induced by transient ischemia of the rat retina. Brain Res
815:11–20.
Kerrigan LA, Zack DJ, Quiglay HA, Smith SD, Pease ME. 1997.
TUNEL-positive ganglion cells in human primary open-angle glau-
coma. Arch Ophthalmol 115:1031–1035.
Laquis S, Chaudhary P, Sharma SC. 1998. The patterns of retinal gan-
glion cell death in hypertensive eyes. Brain Res 784:100–104.
Levin LA, Schlamp CL, Spieldoch RL, Geszvain KM, Nickells RW.
1997 Identification of the bcl-2 family of genes in the rat retina. Invest
Ophthalmol Vis Sci 38:2545–2553.
Liu B, Neufeld AH. 2001. Nitric oxide synthase-2 in human optic nerve
head astrocytes induced by elevated pressure in vitro. Arch Ophthalmol
119:240–245.
Lynch DR, Dawson TM. 1994. Secondary mechanisms in neuronal
trauma. Curr Opin Neurol 7:510–516.
Markesbery WR, Carney JM. 1999. Oxidative alterations in Alzheimer’s
disease. Brain Pathol 9:133–146.
May CA, Mittag T. 2004. Neuronal nitric oxide synthase (nNOS) posi-
tive retinal amacrine cells are altered in the DBA/2NNia mouse, a
murine model for angle-closure glaucoma. J Glaucoma 13:496–499.
McCarthy NJ, Whyte MK, Gilbert CS, Evan GI. 1997. Inhibition of
Ced-3/ICE-related proteases does not prevent cell death induced by
oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol
136:215–227.
McKinnon SJ, Lehman DM, Kerrigan-Baumrind LA, Merges CA, Pease
ME, Kerrigan DF, Ransom NL, Tahzib NG, Reitsamer HA, Levko-
vitch-Verbin H, Quigley HA, Zack DJ. 2002. Caspase activation and
amyloid precursor protein cleavage in rat ocular hypertension. Invest
Ophthalmol Vis Sci 43:1077–1087.
Moon JI, Kim IB, Gwon JS, Park MH, Kang TH, Lim EJ, Choi KR,
Chun MH. 2005. Changes in retinal neuronal populations in the
DBA/2J mouse. Cell Tissue Res 320:51–59.
Morgan JE. 2000. Optic nerve head structure in glaucoma: astrocytes as
mediators of axonal damage. Eye 14:437–444.
Morin LP. 1994. Full-length review: the circadian visual system. Brain
Res Brain Res Rev 67:102–127.
Morrison J, Farrell S, Johnson E, Deppmeier L, Moore CG, Frossmann
E. 1995. Structure and composition of the rodent lamina cribrosa. Exp
Eye Res 60:127–135.
Nakanishi S. 1992. Molecular diversity of glutamate receptors and impli-
cations for brain function. Science 258:597–603.
Nakanishi S, Nakajima Y, Masu M, Ueda Y, Nakahara K, Watanabe D,
Yamaguchi S, Kawabata S, Okada M. 1998. Glutamate receptors: brain
function and signal transduction. Brain Res Brain Res Rev 26:230–235.
Naskar R, Vorwerk CK, Dreyer EB. 1999. Saving the nerve from glau-
coma: memantine to caspaces. Semin Ophthalmol 14:152–158.
Naskar R, Vorwerk CK, Dreyer EB. 2000. Concurrent downregulation
of a glutamate transporter and receptor in glaucoma. Invest Ophthalmol
Vis Sci 41:1940–1944.
Neufeld AH. 1999. Nitric oxide: a potential mediator of retinal ganglion
cell damage in glaucoma. Surv Ophthalmol 43(Suppl):129–135.
Nickells RW. 1999. Apoptosis of retinal ganglion cells in glaucoma: an
update of the molecular pathways involved in cell death. Surv Ophthal-
mol 43(Suppl):151–161.
Osborne NN, Cazevieille C, Carvalho AL, Larsen AK, DeSantis L. 1997.
In vivo and in vitro experiments show that betaxolol is a retinal neuro-
protective agent. Brain Res 751:113–123.
Osborne NN, Ugarte M, Chao M, Chidlow G, Bae JH, Wood JP, Nash
MS. 1999. Neuroprotection in relation to retinal ischemia and rele-
vance to glaucoma. Surv Ophthalmol 43(Suppl):102–128.
Otori Y, Wei JY, Barnstable CJ. 1998. Neurotoxic effects of low doses
of glutamate on purified rat retinal ganglion cells. Invest Ophthalmol
Vis Sci 39:972–981.
Ozawa S, Kamiya H, Tsuzuki K. 1998. Glutamate receptors in the mam-
malian central nervous system. Prog Neurobiol 54:581–618.
Pasteels B, Rogers J, Blachier F, Pochet R. 1990. Calbindin and calreti-
nin localization in retina from different species. Vis Neurosci 5:1–16.
Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. 1997.
The GluR2 (GluR-B) hypothesis: Ca(2þ)-permeable AMPA receptors
in neurological disorders. Trends Neurosci 20:464–470.
Pochet R, Pasteels B, Seto-Ohshima A, Bastianelli E, Kitajima S, Van
Eldik LJ. 1991. Calmodulin and calbindin localization in retina from six
vertebrate species. J Comp Neurol 314:750–762.
Porter AG, Janicke RU. 1999. Emerging roles of caspase-3 in apoptosis.
Cell Death Differ 6:99–104.
Shen H, Ong C. 2000. Detection of oxidative DNA damage in human
sperm and its association with sperm function and male infertility. Free
Radic Biol Med 28:529–536.
Sheng M, Greenberg ME. 1990. The regulation and function of c-fos
and other immediate early genes in the nervous system. Neuron 4:477–
485.
Siliprandi R, Canella R, Carmignoto G, Schiavo N, Zanellato A, Zanoni
R, Vantini G. 1992. N-methyl-D-aspartate-induced neurotoxicity in
the adult rat retina. Vis Neurosci 8:567–573.
Sucher NJ, Aizenman E, Lipton SA. 1991. N-methyl-D-aspartate antago-
nists prevent kainate neurotoxicity in rat retinal ganglion cells in vitro.
J Neurosci 11:966–971.
Sucher NJ, Lipton SA, Dreyer EB. 1997. Molecular basis of glutamate
toxicity in retinal ganglion cells. Vision Res 37:3483–3493.
Tanaka H, Grooms SY, Bennett MV, Zukin RS. 2000. The AMPAR
subunit GluR2: still front and center-stage. Brain Res 886:190–207.
Tezel G, Wax MB. 2000. Increased production of tumor necrosis factor-
alpha by glial cells exposed to simulated ischemia or elevated hydrostatic
pressure induces apoptosis in cocultured retinal ganglion cells. J Neuro-
sci 20:8693–8700.
Thomas J, Liesegang MD. 1996. Concise review for primary-care physi-
cians. Glaucoma: changing concepts and future directions. Mayo Clin
Proc 71:689–694.
Thoreson WB, Witkovsky P. 1999. Glutamate receptors and circuits in
the vertebrate retina. Prog Retin Eye Res 18:765–810.
688 Wang et al.

Uesugi R, Yamada M, Mizuguchi M, Baimbridge KG, Kim SU. 1992.
Calbindin D-28k and parvalbumin immunohistochemistry in develop-
ing rat retina. Exp Eye Res 54:491–499.
Ullian EM, Barkis WB, Chen S, Diamond JS, Barres BA. 2004. Invul-
nerability of retinal ganglion cells to NMDA excitotoxicity. Mol Cell
Neurosci 26:544–557.
Varma R, Spaeth GL, Parker KW. 1993. The optic nerve in glaucoma.
Philadelphia: J.B. Lippincott Company. 354 p.
Vorwerk CK, Gorla MS, Dreyer EB. 1999. An experimental basis for
implicating excitotoxicity in glaucomatous optic neuropathy. Surv
Ophthalmol 43(Suppl):142–150.
Vorwerk CK, Lipton SA, Zurakowski D, Hyman BT, Sabel BA, Dreyer
EB. 1996. Chronic low-dose glutamate is toxic to retinal ganglion cells.
Toxicity blocked by memantine. Invest Ophthalmol Vis Sci 37:1618–
1624.
Wang X, Tay SS, Ng ,YK. 2000a. An immunohistochemical study of
neuronal and glial cell reactions in retinae of rats with experimental
glaucoma. Exp Brain Res 132:476–484.
Wang X, Tay SSW, Ng YK. 2000b. Nitric oxide, microglial activities
and neuronal cell death in the lateral geniculate nucleus of glaucoma-
tous rats. Brain Res 878:136–147.
Wang X, Tay SSW, Ng YK. 2002a. C-fos and c-jun expressions in nitric
oxide synthase immunoreactive neurons in the lateral geniculate nucleus
of experimental glaucomatous rats. Exp Brain Res 144:365–372.
Wang X, Tay SS, Ng YK. 2002b. An electron microscopic study of neu-
ronal degeneration and glial cell reaction in the retina of glaucomatous
rats. Histol Histopathol 17:1043–1052.
Xiang J, Chao DT, Korsmeyer SJ. 1996. BAX-induced cell death may
not require interleukin 1 beta-converting enzyme-like proteases. Proc
Natl Acad Sci USA 93:14559–14563.
Yanoff M, Fine BS. 1996. Ocular pathology. London: Mosby-Wolfe,
Times Mirror International Publishers Limited. 721 p.
Yoshioka A, Ikegaki N, Williams M, Pleasure D. 1996. Expression of N-
methyl-D-aspartate (NMDA) and non-NMDA glutamate receptor genes
in neuroblastoma, medulloblastoma, and other cell lines. J Neurosci Res
46:164–178.
Neuronal Degeneration in Glaucoma 689





























