Embed Size (px)
Citation preview

Calculation of total energies in multicomponent oxides
A. F. Kohan and G. Ceder
Department of Materials Science and Engineering
Massachusetts Institute of Technology
77 Massachusetts Ave. Room 13-4061
Cambridge, MA 02139
U.S.A.
phone #: (617)252-1507
Fax #: (617)258-6534
(October 4, 1996)
1

Abstract
The accuracy of di�erent total{energy methods to compute the properties
of multicomponent oxides is studied. These materials have typically large
unit cells and consequently, computer{running time considerations become
important. Many approximations are introduced in order to speed up calcu-
lations but at the expense of loosing accuracy. We show that while highly
sophisticated quantum{mechanics techniques such as pseudopotentials or the
full{potential linearized{augmented{plane{wave method can be used to ac-
curately compute materials properties, they may require prohibitively long
computer runs in oxides. On the other hand, simple potential models, or
even fast quantum{mechanics methods such as the spherical self{consistent
atomic deformation or the linear mu�n{tin orbital method (in the atomic
sphere approximation), are not always reliable to study oxides. Charge trans-
fer, breathing of the oxygen ions, and non{spherical charge relaxations are
some of the factors that can make any of these schemes fail. However, it is
not necessary to always use sophisticated techniques. We show that the self{
consistent semiempirical tight{binding formalism can be used as an interpo-
lation tool to extend the results of accurate calculations for a few compounds
in a system to the rest of them. This opens new possibilities for the use of ab
initio methods to study technologically{relevant materials properties, such as
the temperature behavior of oxides, since formation energies of many di�erent
compounds at 0 oK are a crucial input to these models.
I. INTRODUCTION
In the last decades, computer technology has been evolving at a very fast pace making
possible the use of powerful combinations of quantum{ and statistical{mechanics techniques
to study materials properties. The long sought goal of predicting materials properties before
2

they are even synthesized is �nally coming to a reality. Computational experiments, as
these calculations are usually called, have the advantage that they o�er full control of any
experimental condition and that they can be performed even under extreme values of such
conditions.
At the core of most of these methods lies a model that describes the energetics of the
system being studied. Nearly all physical properties are related to total energies (equations
of state) or their di�erences. By just computing total energies, valuable information such
as elastic properties, lattice constants, defect arrangements, and structural stability can
already be obtained. However, in most cases of interest for materials science, total{energy
calculations by themselves do not su�ce but need to be used in combination with statistical{
mechanics [1] or molecular{dynamics methods [2,3], as the temperature dependence and
time evolution of the system are usually essential to understand the properties of materials.
This adds a new dimension to the complexity of computing total energies since not only do
the methods have to be accurate but also fast. In all of the ab initio schemes developed
to study temperature{dependence behavior for real materials, total{energy methods are
usually the limiting step in the calculations. For example, temperature{composition phase
diagrams are one of the most important tools in designing and processing materials. Their
ab initio calculation usually requires the computation of formation energies ofmany di�erent
compounds at 0 oK with high accuracy [4{6] (of the order of a few hundredths of an eV).
Even for very simple systems, these calculations are very computer{time consuming if one
is looking for quantitative predictions.
Oxide materials present a particularly di�cult challenge to total{energy methods. They
usually have large unit cells, low symmetry, and a mixed ionic{covalent bonding. Under these
conditions, accurate quantum{mechanical techniques require extremely long computer runs.
Consequently, modeling in oxides has been mainly done using simple empirical potential
models [7]. These models have been successful in predicting defect energies, lattice con-
stants, and some elastic properties [8,9]. However, they are inadequate to make quantitative
predictions when computing phase diagrams [10]. It is an open question which of the more
3

elaborate, but more time consuming, quantum{mechanical models will improve this pic-
ture since some of them use approximations, such as spherically averaged electronic charge
densities or frozen electronic cores, that may not be su�ciently accurate in oxide systems.
The objective of this work is to evaluate the accuracy of di�erent total{energy tech-
niques for the calculation of formation energies. Several methods will be compared on two
problems: formation energies in the CaO{MgO system and polymorphic transition energies
between the phases of ZrO2 (zirconia). The former system presents a simple miscibility gap
in the temperature{composition phase diagram and consequently the di�erent CaO{MgO
compounds are expected to have positive formation energies. Zirconia is stable at room
temperature in a monoclinic structure (baddeleyite) and transforms into a tetragonal phase
(space group P42=nmc) at approximately 1180 oC [11]. Before melting, it transforms again,
but now into a cubic phase ( uorite{type structure) at approximately 2350 oC [12].
We will start in Section II by brie y introducing the di�erent techniques used in this
work. Then, in Section III, we apply these methods to compute formation energies and
cell parameters for compounds in the CaO{MgO system and to compute the di�erence in
energies between the observed ZrO2 phases. Finally, in Section IV, we discuss the e�ect
of the approximations used in the di�erent total{energy techniques and we show to what
degree they are acceptable in oxides.
II. TOTAL{ENERGY METHODS
The �rst total{energy calculations date back to the 1920s, when the main concern was
trying to understand cohesion in periodic solids [13,14]. Since then, many di�erent schemes,
from classical empirical models to highly sophisticated quantum{mechanical techniques have
been proposed and changed during the years. A major step was the introduction of density
functional theory [15] for the treatment of the many{body electronic problem. Currently,
most ab initio quantum{mechanical models are based on a local density approximation [16]
(LDA) to this theory or on improvements over the LDA by using gradient corrections [17,18].
4

Other methods, based on the Hartree{Fock approximation are also used [19].
Two di�erent approaches can be taken to compute total energies: We can assume a model
for the electronic density and solve for the energy, or we can directly solve Schr�odinger's
equation self consistently. Empirical potentials correspond to the �rst category and represent
one of the simplest energy models that can be used in oxides. In this case, the electronic
density and ionic charges are replaced by point charges centered at the ions. The energy
is computed as the sum of the electrostatic interaction and a short{range repulsive term
that represents the overlap of the electronic clouds. The value of the point charges can be
obtained from the ideal chemical valence or can be considered as a parameter of the potential.
On the other hand, in self{consistent quantum{mechanical methods no a priori assumption
is made about the electronic density (though in some cases spherical symmetry is assumed).
The main di�erence between the methods in the last category is in the basis they use to
expand the electronic density. A fast total{energy technique is fundamental for any attempt
of describing oxide's properties ab initio. Usually, fast methods imply using a large number
of approximations. Since accuracy is important to produce quantitative predictions, we will
evaluate di�erent total{energy techniques with di�erent degrees of approximations.
A two{body potential was used in this work to describe the short{range interaction
between ions with the following form:
V (rij) = Aije
��
rij
�ij
��
Cij
r6ij; (1)
where Aij, �ij, and Cij are parameters that depend on the identity of the ions i and j, and
rij is the distance between them. The polarizability of each ion was treated with a shell
model [20,21], where the total charge of the ion is split between a massless shell of charge
Qs and a core with charge Qc connected with an harmonic spring with constant ks. Thus,
the interaction between a core and a shell separated a distance d is given by,
V (ri) =1
2ksd
2: (2)
The ewald summation method [13] was used to compute the electrostatic energy of the point
charges. This is a completely classical approach where no contribution to the total energy
5

coming from the kinetic energy or exchange and correlation of the electrons is taken into
account explicitly. Point charges are �xed and do not depend on the environment.
Quantum{mechanical methods involve more complex, and consequently more time con-
suming calculations than potential models. One technique particularly fast in oxides is the
spherical self{consistent atomic deformation (SSCAD) model [22,23]. The SSCAD is a mod-
i�ed version of the Gordon and Kim electron gas theory [24]. The electronic charge density
is assumed to be localized around each ion, resulting in single{particle Schr�odinger's equa-
tion per ion, which is solved self consistently in the �eld of the other ions. The potential
energy term is spherically averaged. Consequently, its use is restricted in its present form, to
systems where the non-spherical components of the electronic charge density are negligible.
This condition is not necessarily satis�ed in all oxide materials and may lead to incorrect
predictions. In Ref. [25] it is demonstrated that a non{self{consistent version of the SSCAD
incorrectly predicts zirconia to be stable in the cubic phase (see Section III).
Other quantum{mechanical methods introduce less approximations, at the expense of
time performance. In this work we make use of two of these more accurate techniques: the
linear mu�n{tin orbital method in the atomic{sphere approximation [26,27] (LMTO{ASA)
and the pseudopotential (PP) method [28]. We will also compare all these techniques against
published results obtained with the linearized{augmented{plane{wave in its full potential
version [29,30] (FLAPW).
It is not our intention to review all these total{energy methods, instead we will brie y
mention the main approximations they use. In both LMTO and LAPW, space is divided
into spheres (generally centered around atoms) and a mu�n tin potential is used. Basis
functions are de�ned both in the interstitial region and within the spheres (MTO's in the
former method and APW's in the latter, but in both cases the energy dependence is linearized
around a given energy for the di�erent angular momenta). The basis sets used in these
methods are relatively small compared to the ones used in �xed basis methods such us
the pseudopotential plane wave technique. Usually, an order of magnitude less MTO's than
APW's are needed to accurately solve Schr�odinger's equation, making the LMTO a very fast
6

scheme. To make the LMTO less computational intensive it is frequently used in the atomic
sphere approximation [26,27] (ASA). In the ASA, all integrals over space are substituted by
integrals over space �lling spheres (Wigner{Seitz overlapping spheres) generally centered at
atoms, and the electronic density is spherically averaged within each sphere. The potential
is a superposition of spherical contributions from each sphere. In close{packed solids the
LMTO{ASA approximations are very accurate. Some corrections are sometimes used to
compensate for the sphere overlap (usually called \combined corrections" [27,31,32]) and
the symmetrization of the potential (usually called \mu�n tin corrections" [33]). In more
open structures the method is less accurate and empty spheres (not centered at atoms) are
usually used. A full{potential version of the LMTO, where no shape approximations are
made, has been developed [34] but we will not discuss it here. The full potential version
of the the LAPW also does not have shape approximations for the potential. In this case,
when the calculations are carefully performed, the only approximation within DFT is the
LDA.
Both LMTO and LAPW (or their full{potential versions) are used as all{electron meth-
ods with energy-linearized basis functions. On the other hand, in the pseudopotential
method only the wave functions of the valence electrons are computed (frozen core ap-
proximation) and they are usually expressed as a combination of plane waves. The frozen
core approximation is based on the fact that, in general, only valence electrons take part in
bonding while the core electrons, which are tightly bound to the nucleus, can be regarded
as frozen. The e�ect of the core electrons on the valence states is taken into account as a
potential that is added to the nuclear potential. This pseudopotential is chosen so that its
valence pseudo wave functions are equal to the actual all{electron wave functions beyond a
given core radius and so that they do not have nodes in the core region. Since the electrons
that participate in bonding usually reside beyond the core, using pseudopotentials is an
excellent approximation in most cases.
The use of the pseudopotential approximation not only allows for a reduction in the
number of plane{wave basis functions but also in the number of orbitals that need to be
7

computed. All{electron techniques need to accurately treat core energies that constitute a
large part of the total energy, while generally their e�ects are canceled out when energy dif-
ferences are taken. Consequently, the pseudopotential energy is much smaller and requires
a smaller relative accuracy than all-electron methods when computing formation energies.
The use of plane waves greatly simpli�es the calculations: Shape approximations are not
necessary and Pulay forces [35] are zero. The computational speed of the method depends
on the chemical identity of the elements that form the material. Sharply peaked valence
states, as in �rst row nonmetals such as oxygen, or in transition metals, require a large num-
ber of plane waves to be expanded. However, in the last years, new developments such as
Car{Parrinello molecular dynamics [3], conjugate{gradients [36], and optimized pseudopo-
tentials [37] have shown that pseudopotential calculations can be e�ciently performed for
any element in the periodic table [36].
In this work we will also use the semiempirical tight{binding method [38] in the self{
consistent form suggested in Ref. [39] and [40]. Contrary to empirical potential models, an
explicit treatment of the electronic structure is made and all the relevant terms for computing
total{energy di�erences in systems with charge transfer (such as oxides) are incorporated.
The diagonal terms of the Hamiltonian are not �xed but written as
"i;� = "oi� + uintrai� + uinter
i� + Vi� + gi�; (3)
where "oi� represents the kinetic energy and the interaction with its own ionic core (at site
i) of an electron at orbital �, and uintrai� and uinter
i� are the short{ and long{range interaction
of the electron in the orbital i� with the other electrons. Vi� represents the crystal �eld at
site i and gi� is a shift introduced by the orthogonalization of the basis functions [40,41] and
corresponds to
gi� = �X
i0�0 6=i�
si�;i0�0ti�;i0�0: (4)
si�;i0�0 are the overlap matrix elements of the original non{orthogonal basis functions and
ti�;i0�0 are o�{diagonal hopping terms. These terms are assumed to depend on the distances
between ions i and j.
8

The interaction between electrons, ui�, is written as
uintrai� =
X
�0
Ui�;i�0Qi�0 ; (5)
uinteri� =
X
i0 6=i;�0
Ui�;i0�0Qi0�0 ; (6)
where Ui�;i0�0 represents electron{electron two center integrals and Ui�;i�0 are intra{atomic
repulsion parameters [42]. Qi� is the occupation of the orbital i�
Qi� =occX
n;~k
j< 'i� j n~k >j2 : (7)
The sum over all occupied orbitals is associated with the number of electrons at site i.
Since Qi� depends on the solution eigenvectors, j n~k >, these eigenvectors need to be found
self consistently ('i� are the atomic{like basis functions). In this way, charge transfer is
incorporated into the formulation through the ui� terms, the e�ect of the crystal �eld (which
shifts the atomic eigenvalues when the solid is formed), and the hopping terms. The gi�,
on the other hand, is a repulsive term that represents an increase in the kinetic energy of
the electrons when they are brought together. Finally, the hopping integrals are essential
to determine the band structure of a solid and take into account the e�ect of lowering the
electronic energy when an electron is attracted not only by its own core but also by the
neighboring ones. Most of these terms are not explicitly accounted for in potential models,
and the charge on the ions is usually �xed, and independent of the environment of the
atom. The tight{binding model is not limited to a given kind of bonding and no shape
approximations for the ionic potentials need to be made.
This formulation of tight{binding is particularly fast since the time{consuming hopping
and overlap integrals are �tted to ab initio results. In this sense, this method represents a
much more sophisticated and accurate interpolation tool than potential models and is still
orders of magnitude faster than ab initio quantum{mechanical techniques.
We have reviewed the main approximations and we will now describe how they a�ect
the calculation of total{energy di�erences and cell parameters in oxide systems.
9

III. RESULTS
A. Ab initio Pseudopotential Results
In previous work [40] we reported formation energies obtained for 10 compounds in the
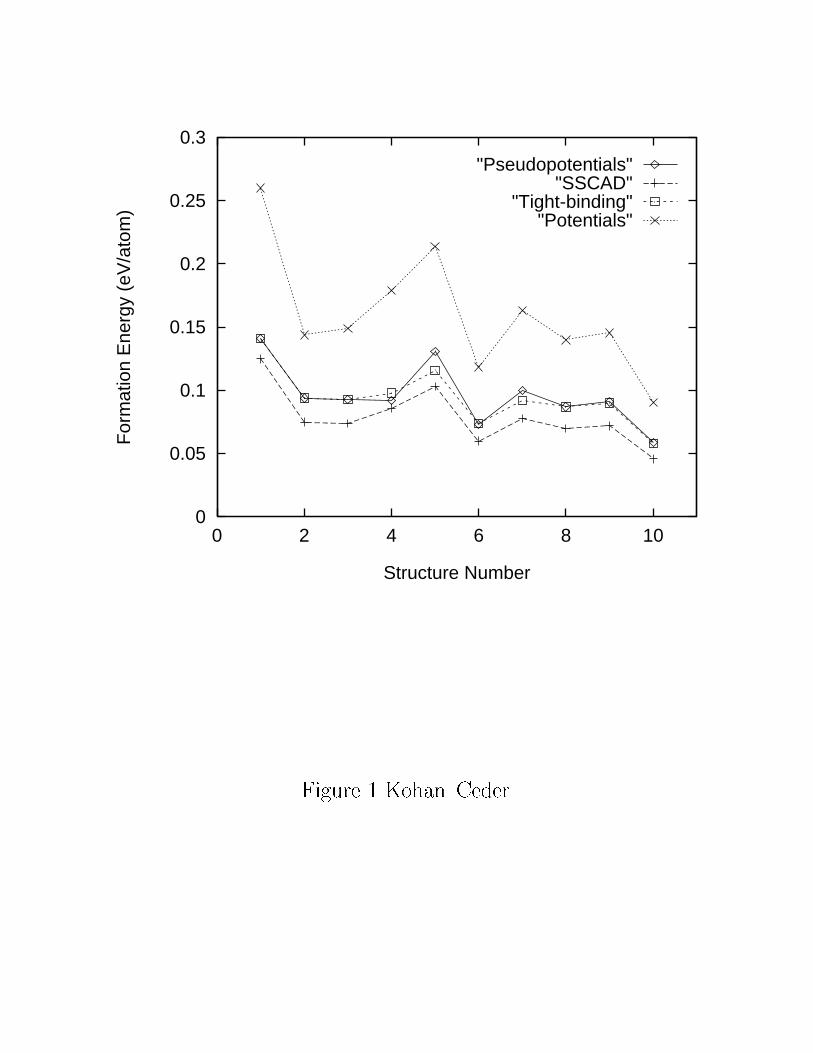
CaO{MgO system using the pseudopotential method (they are reproduced in Fig. 1). The
energy of some of those structures was also computed in Ref. [10] using the FLAPW. Table I
compares the FLAPW and PP results for the formation energies and lattice constants, the
largest absolute di�erence being 9 meV/atom. Since the FLAPW and pseudopotential cal-
culations were converged to within 3 meV, the results seem to agree well. These compounds
can not be observed experimentally since the formation energies are all positive. However
an indirect test of the accuracy of the formation values is obtained from the solubility limits
that can be predicted with these data. In the CaO{MgO phase diagram the computed sol-
ubility limits agree nicely with the experimental data as shown in Ref. [10]. (These values
were actually computed using the SSCAD but as we will show, they are close to the PP
results.)
We also computed the cubic to tetragonal transition in ZrO2. To our knowledge, this
is the �rst pseudopotential calculation of this sort. We did not compute the monoclinic
phase since it is very complex, with 12 atoms in the unit cell and 13 variables to optimize.
Performing this calculations with pseudopotentials requires a large number of supercomputer
hours. A single point calculation may take about 20 Cray90 cpu hours and the monoclinic
structure has 13 relaxation parameters. On the other hand the tetragonal phase takes about
40 minutes and has 3 relaxation parameters, and the cubic phase has just one relaxation
parameter and takes about 15 minutes.
We used non{local, optimized [37], Kleinman{Bylander [43] type pseudopotentials. We
considered the 4s, 4p and 4d orbitals in Zr as the valence orbitals, and we generated the
pseudopotential for a Zr+2 ionic con�guration. The Zr core radii used to generate the
pseudopotential were 1.3 a.u., 1.4 a.u., and 1.6 a.u. for the s, p, and d components. For
10

the oxygen pseudopotential the core radius was 0.8 a.u. for both the s and p components.
The energy functional was minimized employing the conjugate gradients technique and we
used the Perdew and Zunger [44] parameterization of the exchange{correlation energy. Ten
special Chadi{Cohen k points were chosen for the cubic phase and an equivalent set for the
tetragonal phase (12 k points). The energy cuto� was 700 eV.
The pseudopotential results are presented in Table II. The tetragonal phase is predicted
to have a lower energy than the cubic one, as one would expect from the experimental
information mentioned in Section I. The di�erence in energy between these two phases
agrees well with the estimated enthalpy of the transition measured at 2377 oC in Ref. [45].
The cell parameters for the cubic and tetragonal phases also agree well with experiment:
The reported numbers for the experiment are linear extrapolations to 0 oK using the data
from Ref. [12] for the tetragonal a and c, and the cubic a parameter (since the two phases
are very similar we assumed the same temperature dependence in both cases).
B. LMTO{ASA
The von Barth{Hedin [46] form for the exchange{correlation potential was used in all the
LMTO{ASA calculations in this work. Integrations in k{space where done in a 173 uniform
grid (considering crystal symmetry) and a s{p{d basis was used in the CaO{MgO system.
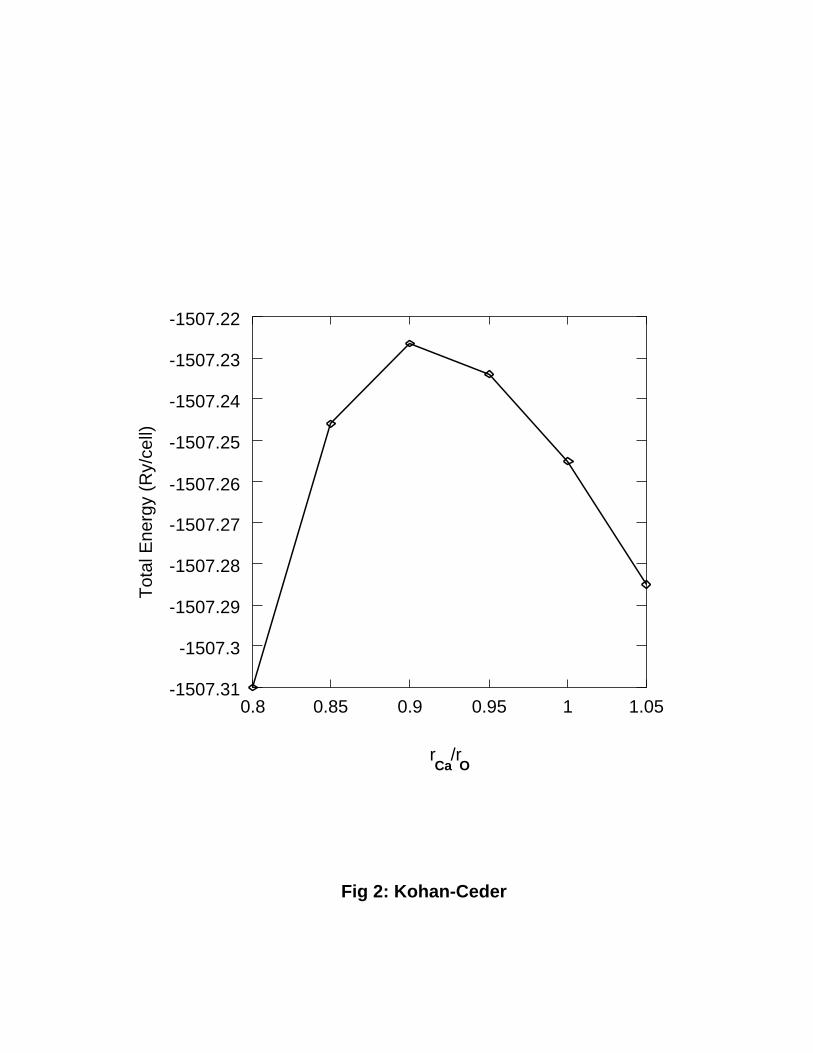
We found an important dependence of the total energies on the size of the atomic spheres
even when the \combined" and the \mu�n tin" corrections were used. For example, in
Fig. 2 we show the total energy versus sphere radius for the CaO structure. It is clear that
a criterion for choosing the relative sizes of the atomic spheres in the ASA is needed. By
choosing di�erent points in the curve, the formation energy of any compound in the system
can be switched from positive to negative.
For monoatomic solids the sphere radius is �xed (space{�lling condition). For all other
cases, where more than one atom is present in the unit cell, a criterion for choosing the
radii is needed. Four criteria are usually used. The �rst criterion is to just take equal{size
11

spheres. The second one is to make the atomic spheres charge neutral (in this way the
madelung energy error in the ASA vanishes). The third one is to minimize the formation
energy of the compound, though there is no variational principle that justi�es this procedure.
Finally, the last criterion, proposed by Andersen [31], gives an explicit formula for the sphere
radii in terms of Wigner{Seitz radii, bulk moduli, and elemental volumes. In metallic alloys
these four criteria usually agree and formation energy values are comparable to FLAPW
calculations [47].
In oxides, it does not make sense to make the spheres charge neutral. In general, we
found that the consistent use of any of these criteria along many compounds in the same
system does not provide accurate formation energies when compared to FLAPW or pseu-
dopotentials. For example, we found that both the Ca{ and Mg{rich L12 structures have
negative formation energies when using equal size spheres while all the other total{energy
methods predict them to be positive. Using a smaller radii for the cations than for the anions
makes the L12 structure formation energies positive but we could not �nd a consistent way
of setting the sphere sizes. We observed the same behavior in other oxide systems such us
ZrO2{CaO, Na2O, and Li{transition metal oxides.
C. SSCAD
In Ref. [10] we presented di�erent formation energies computed with the SSCAD for the
CaO{MgO system. Some of these values are reproduced in Fig. 1. The SSCAD values are
systematically lower than the pseudopotential and FLAPW results. The average error is of
the order of 17 %.
The phase transitions in ZrO2 were studied by Cohen, Mehl, and Boyer [25] using a
non{self{consistent version of the SSCAD (called the potential induced breathing (PIB)
method [48]). Their results are reproduced in Table II for comparison. Neither the tetragonal
phase nor the monoclinic phase were metastable at zero pressure. Cubic zirconia was found
to be the stable phase when compared to the rutile, the orthorhombic, and the cotunnite
12

structures.
D. Semiempirical tight-binding model
We showed in a previous work [40] that the semiempirical tight{binding method can
accurately reproduce the pseudopotential formation energies in the CaO{MgO system (see
Fig. 1). We will now apply the same scheme to study the phase transitions in zirconia.
The elements of the overlap matrix and the hopping integrals were �tted to the valence
bands of pseudopotential calculations for tetragonal ZrO2. Their distance dependence was
assumed to be the one proposed by Harrison [41,49]. The conduction bands were included
with a smaller weight (10 times) than the valence bands. Signi�cant departures from the
experimental lattice constants were penalized during the �t. In all the calculations, a s{
p{d and a s{p basis was used for zirconium and oxygen respectively. The results of this
calculations are shown in Table II. The di�erence in energy between the tetragonal and the
cubic phases are the same as in the pseudopotential method. This is a prediction of the
tight{binding scheme since only the tetragonal phase was included in the �t.
The monoclinic phase is predicted to have a lower energy than the tetragonal in agree-
ment with the fact that this is the observed phase at 0 oK. Not all the 13 crystallographic
parameters were relaxed for the monoclinic phase. The angle not �xed by the symmetry was
set to 90o and consequently, 12 (instead of 13) parameters were minimized. We found the
monoclinic phase to be unstable under variations of this angle. We think this is due to the
fact that we are evaluating the tight{binding parameters beyond the range in which they
were �tted. We do not discard also the possibility that more complex distance dependencies
of the overlap and hopping terms need to be considered. We are currently testing these
hypothesis. We expect that this will bring the energy di�erence between the tetragonal and
the monoclinic phase closer to the experimental values shown in Table II. Note that the
experimental monoclinic cell parameters are already well reproduced by the tight{binding
parameters we used.
13

E. Potential models
The potential results for the CaO{MgO system were computed in Ref. [10] and are
shown for comparison reasons in Fig. 1. Published potentials for this system overestimate
the formation energies by as much as 100%.
A Buckingham potential combined with a shell model were �tted to reproduce structural
parameters and dielectric constants of the tetragonal zirconia phase [50] (the O{O parame-
ters were taken from Ref. [21]). The predictions for the zirconia phase transitions are shown
in Table II and were taken from Ref. [50] and [51]. The monoclinic phase is the stable one
in this model.
IV. DISCUSSION
In Section III we presented total{energy studies of the CaO-MgO system and the poly-
morphic transitions in zirconia using di�erent total{energy techniques. They not only pro-
vide information regarding fundamental properties of materials but also new insights to
understand and improve them. We basically reported three approaches to perform these
calculations: highly accurate methods (in which the major approximation is the LDA), em-
pirical classical models, and a full range of techniques in between, from semiempirical to
fully ab initio schemes (that introduce di�erent approximations with the �nal objective of
improving the computational speed).
Full{potential methods, such as FLAPW or pseudopotentials, are clearly in the �rst
category. Their results usually compare well with experiments (see Table II). In our case,
the use of the frozen core and pseudopotential approximations simpli�ed considerably the
calculations without a large loss in accuracy. The di�erences between an all-electron method,
such as FLAPW, and the pseudopotential results were very small in the CaO{MgO system
as shown in Table I. On the other hand, for the tetragonal to cubic transition energy,
the agreement is not good (see Table II). However, we do not think this di�erence comes
14

from the pseudopotential approximations. The pseudopotential predictions are not only
much closer to the experimental values but also the FLAPW calculations [52] might have
not been fully converged.(In Ref. [52], a limited number of basis functions was used, which
introduced relative errors of the order of 1mRy/cell, compared to 0.1mRy/cell in our case,
and the relaxation of the tetragonal phase was not thoroughly performed.)
When there is an appreciable overlap between the valence and core orbitals the frozen{
core approximation can become an important source for error. In these cases some of the core
orbitals should be taken as valence orbitals reducing the size of the core. If a pseudopotential
is being used, this will also result in an deeper potential, considerably increasing the number
of plane waves and consequently, the computational demands of the method.
It is not uncommon for oxides to have structures more complex than the monoclinic
zirconia one. As we already mentioned, this structure requires prohibitively long PP runs.
Consequently, only the properties of simple oxides can be compute with highly accurate
techniques.
The LMTO{ASA method on the other hand, is orders of magnitude faster than the above
techniques but at the expense of loosing accuracy. The errors introduced by an incorrect
treatment of the overlap region and the spherical approximation can be very large in oxides.
As we showed in Section III B the computed formation energies can shift from being positive
to being negative according to how the atomic sphere sizes were chosen. We found that none
of the criteria usually used to set the sphere radii in metallic alloys gave consistently correct
formation energies in the CaO{MgO system. We could not �nd an equivalent criterion that
worked well in oxides. In metals, the error introduced by the ASA is less critical and it is
usually very small when using neutral spheres.It is expected that a full{potential non{ASA
LMTO method will overcome these di�culties in oxides and work is currently being done
in our group to test this.
The SSCAD is another fast quantum{mechanical technique. In Section III C we showed
that it underestimates the formation energies in the CaO{MgO, the larger di�erences being
of the order of 20%. The approximations introduced in the method clearly break down for the
15

zirconia phase transitions. The non{self consistent SSCAD predicts the cubic structure to be
the stable phase in contradiction with the experimental �ndings. Cohen et al. [25] suggested
that these discrepancies were related to non{spherical charge relaxations not included in the
model. Since many oxides have covalent bonding, signi�cant errors can be expected due to
this approximation. It is possible to avoid the spherical approximation in the SSCAD but
at the expense of an increase in the computational burden.
We already discussed in detail the accuracy of potential models in Ref. [10]. Although
they have been successfully used to study many oxide properties [8,9] they just render
qualitative agreement for phase diagrams. Most published potentials have been �tted to
energies on the scale of eV, while temperature e�ects are determined in the scale of several
meV. This is not the only source of error. Many body e�ects, charge transfer, oxygen
breathing, and other quantum{mechanical e�ects are di�cult to take into account within
the framework of potentials. However, the use of potentials is widespread since they are
simple and very fast. In the case of the CaO{MgO system, the ionic charge does not change
considerably from one structure to the other and a potential could be found that reproduces
the formation energies relatively well, but poorly predicts other properties such as elastic
constants [10]. We do not expect the same to happen in doped zirconias since the charge will
change when dopants and vacancies are introduced in the system [51]. This will seriously
limit the use of potentials in these materials.
A natural way of overcoming the shortfalls of potential models without going into the
complexities of full{potential ab initiomethods is the semiempirical tight{binding technique.
As we showed in Section IIID, this method provided, both in CaO-MgO and in ZrO2,
accurate energy di�erences comparable to the best quantum{mechanics techniques. Even
for monoclinic zirconia, the tight{binding cell parameters are in very good agreement with
the experimental values.
Although the need to use a self{consistent formalism (due to charge transfer e�ects)
slows down the tight{binding method compared to potentials, it is still orders of magnitude
faster than fully ab initio techniques.
16

V. CONCLUSIONS
We showed that full{potential quantum{mechanical methods are reliable tools to com-
pute total{energy di�erences on a scale relevant to the study of phase transitions. When
taken to their full capabilities their predictions compare well with experiments. Unfortu-
nately, these techniques are also the most computer{time demanding of all.
Oxides present a particularly di�cult case for any total{energy method due to the large
unit cells and low symmetry. Consequently, there are signi�cant bene�ts to using sim-
pler schemes. Approximations such as spherical averages of potentials, mapping space into
overlapping spheres, or replacing complex ion-ion interactions by simple pair potentials can
dramatically speed up total{energy calculations. However, we showed that even for the
relatively simple CaO{MgO system, most of these approximations break down.
Consequently, it is very important to accurately incorporate all the relevant e�ects in
any total{energy model. Our results in CaO{MgO and ZrO2 seem to indicate that a self{
consistent tight{binding model is capable of capturing the relevant physics of the problem
while retaining a low computational cost. The results are promising and more tests are
underway.
The scaling of the methods with the number of ions is another factor to be taken into
account. As computers are becoming more powerful and larger systems are being studied,
those methods that can be formulated to scale linearly in N will eventually succeed. Oxide
materials present a good case to test this new developments, since many di�erent mechanisms
are crucial to determine total{energy di�erences.
ACKNOWLEDGMENTS
This work was sponsored in part by the National Science Foundation under contract No.
DMR9501856, the National Institute for Health under contract No. 2-P30-ESO2109-16 and
the Petroleum Research Fund under contract No. ***********. Professor Joannopoulos
17

is gratefully acknowledged for providing us with the pseudopotential codes. We thank the
Pittsburgh Supercomputer Center for the possibility of using their C90 computer. We thank
Mark van Schilfgaarde for his permission to use the LMTO-ASA code.
18

FIGURES
FIG. 1. Formation energies for di�erent ordered structures in the CaO{MgO system obtained
with di�erent total{energy techniques. The number that identi�es each structure corresponds to
the one used in Ref. [40]. The tight{binding parameters were �tted only to structure number 1 in
the plot (and both CaO and MgO). The potential parameters were obtained from Ref. [21].
FIG. 2. Total energy for pure CaO as a function of the ratio between the Ca and the O sphere
radius in the LMTO{ASA. The volume of the cell was kept constant and corresponds to the
experimental value. Empty spheres, and mu�n tin and combined corrections were used to reduce
the errors introduced by the ASA. The total energy changes in a scale much larger than the scale
of the temperature e�ects in the system and a criterion is needed to choose the sphere size.
19

TABLES
TABLE I. Formation energies and cell parameters for the L12 structure in the CaO-MgO system
computed with the pseudopotential (PP) and the full{potential linearized{augmented{plane{wave
(FLAPW) methods. The CaO-MgO system orders on two interpenetrating fcc lattices. The L12
nomenclature identi�es the distribution of the Ca and Mg ions on the fcc cation lattice (that
corresponds to the minority species at the corners of the conventional fcc cell) while the oxygen fcc
sublattice remains fully occupied. Energy values are expressed in eV/ion and the cell parameters
are in �A. The values correspond to fully relaxed structures.
TABLE II. Cell parameters and structural energy di�erences for the experimentally observed
phases in zirconia at zero pressure. The energies are in eV per ZrO2 formula unit and the lattice
constants are in �A. All cell parameters were fully relaxed, including the internal positions, xi, yi,
and zi, that are shown here according to the Wycko� notation in reference [53]. The experimental
z value for the tetragonal structure was measured at 1295 oC. The tight{binding � angle was not
relaxed. PIB calculations for the tetragonal and monoclinic phases of zirconia are not reported in
the table since these structures are not predicted to be stable by this method.
20
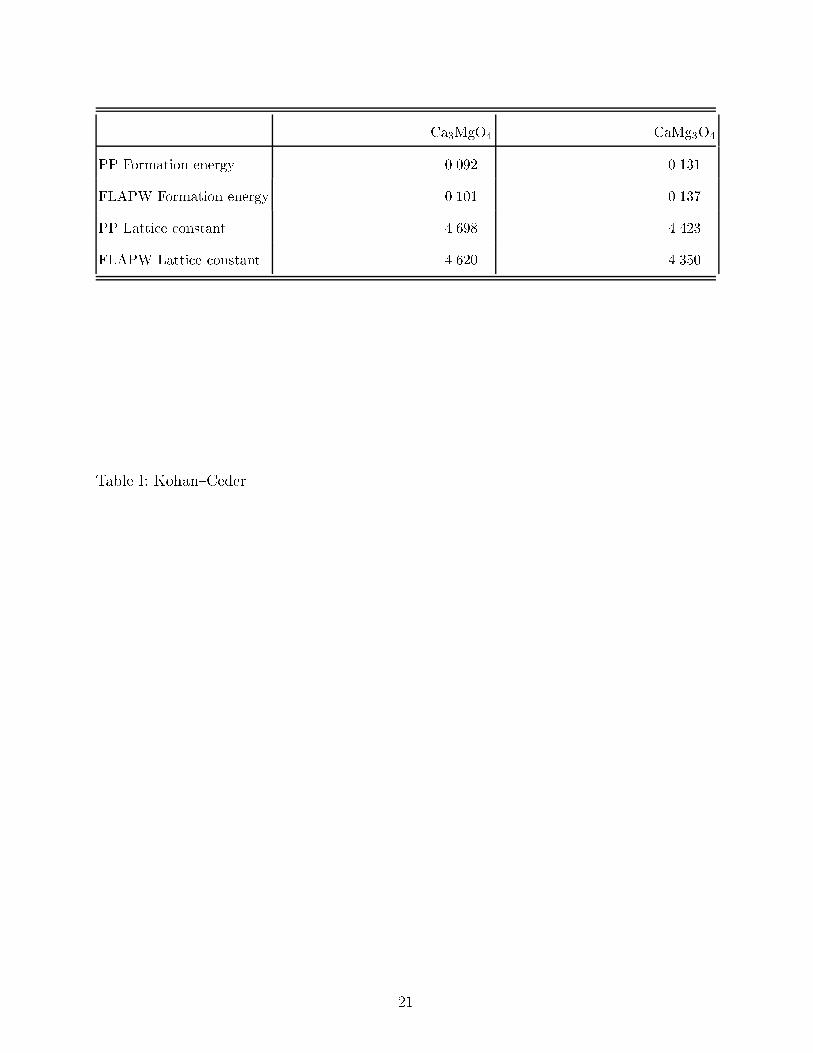
Ca3MgO4 CaMg3O4
PP Formation energy 0.092 0.131
FLAPW Formation energy 0.101 0.137
PP Lattice constant 4.698 4.423
FLAPW Lattice constant 4.620 4.350
Table I: Kohan{Ceder
21
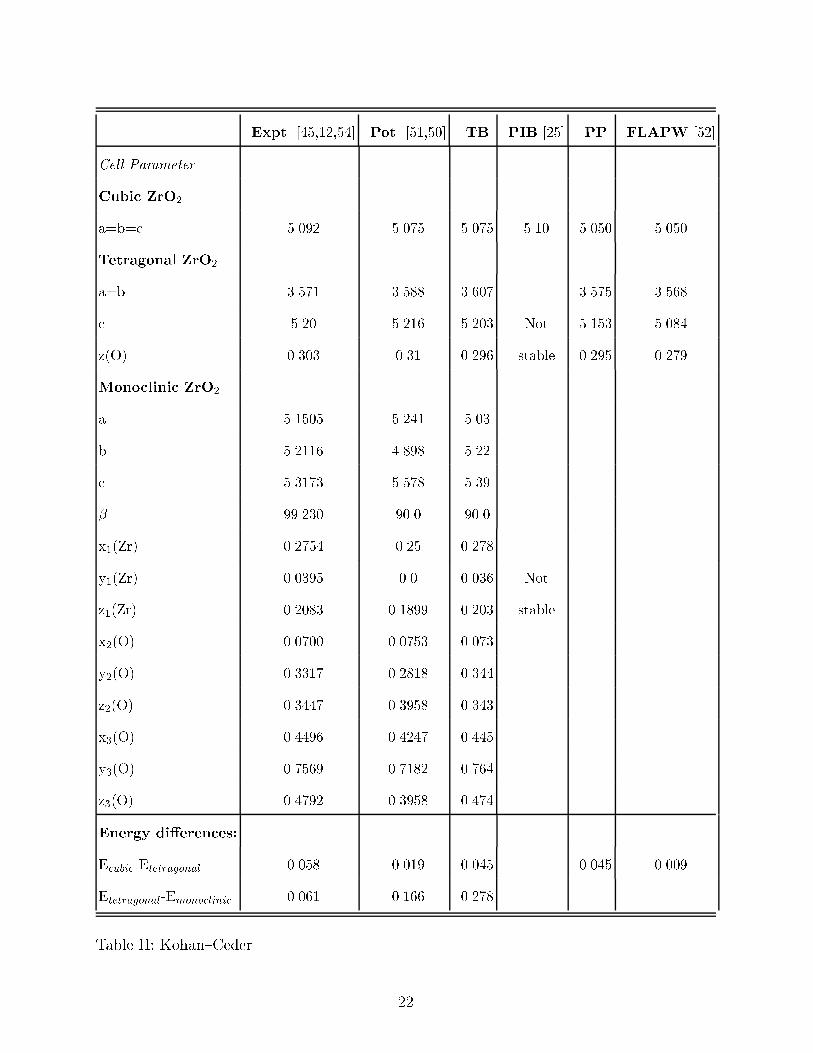
Expt. [45,12,54] Pot. [51,50] TB PIB [25] PP FLAPW [52]
Cell Parameter
Cubic ZrO2
a=b=c 5.092 5.075 5.075 5.10 5.050 5.050
Tetragonal ZrO2
a=b 3.571 3.588 3.607 3.575 3.568
c 5.20 5.216 5.203 Not 5.153 5.084
z(O) 0.303 0.31 0.296 stable 0.295 0.279
Monoclinic ZrO2
a 5.1505 5.241 5.03
b 5.2116 4.898 5.22
c 5.3173 5.578 5.39
� 99.230 90.0 90.0
x1(Zr) 0.2754 0.25 0.278
y1(Zr) 0.0395 0.0 0.036 Not
z1(Zr) 0.2083 0.1899 0.203 stable
x2(O) 0.0700 0.0753 0.073
y2(O) 0.3317 0.2818 0.344
z2(O) 0.3447 0.3958 0.343
x3(O) 0.4496 0.4247 0.445
y3(O) 0.7569 0.7182 0.764
z3(O) 0.4792 0.3958 0.474
Energy di�erences:
Ecubic-Etetragonal 0.058 0.019 0.045 0.045 0.009
Etetragonal-Emonoclinic 0.061 0.166 0.278
Table II: Kohan{Ceder
22

REFERENCES
[1] K. Binder and D. W. Heermann, Monte Carlo simulation in statistical physics (Springer{
Verlag, Berlin, 1988).
[2] M. P. Allen and D. J. Tildesley, Computer simulation of liquids (Oxford University
Press, New York, 1987).
[3] R. Car and M. Parrinello, Physical Review Letters 55 (1985) 2471.
[4] G. Ceder, Computational Materials Science 1 (1993) 144.
[5] F. Ducastelle, Order and Phase Stability in Alloys (North{Holland, Amsterdam, 1991).
[6] A. Zunger, in Statics and Dynamics of Alloy Phase Transformations (Plenum Press,
New York, 1994).
[7] I. M. Torrens, Interatomic Potentials (Academic Press, New York, 1972).
[8] R. E. Watson, S. C. Parker, and A. Wall, J. Phys. Condens. Matter. 4 (1992) 2097.
[9] S. C. Parker and G. D. Price, Adv. Solid{State Chem 1 (1989) 295.
[10] P. D. Tepesch et al., Journal of the American Ceramic Society 79 (1996) 2033.
[11] D. K. Smith and H. W. Newkirk, Acta Cryst. 18 (1965) 983.
[12] P. Aldebert and J.-P. Traverse, Journal of the American Ceramic Society 68 (1985) 34.
[13] P. P. Ewald, Ann. Phys. 64 (1921) 253.
[14] E. Wigner and F. Seitz, Physical Review B 43 (1933) 804.
[15] P. Hohenberg and W. Kohn, Physical Review 136 (1964) 864.
[16] W. Kohn and L. Sham, Physical Review 140 (1965) 1133.
[17] D. C. Langreth and M. J. Mehl, Physical Review B 28 (1983) 1809.
[18] A. D. Becke, Physical Review A 38 (1988) 3098.
23

[19] C. Pisani, R. Dovesi, and C. Roetti, Hartree{Fock Ab Initio Treatment of Crystalline
Systems (Springer{Verlag, Berlin, 1988).
[20] J. B. G. Dick and A. W. Overhauser, Physical Review 112 (1958) 90.
[21] G. V. Lewis and C. R. A. Catlow, Journal of Physics C: Solid State Physics 18 (1985)
1149.
[22] L. L. Boyer and M. J. Mehl, Ferroelectrics 150 (1993) 13.
[23] H. T. Stokes, L. L. Boyer, and M. J. Mehl, Physical Review B (1996) .
[24] R. G. Gordon and Y. S. Kim, The Journal of Chemical Physics 56 (1972) 3122.
[25] R. E. Cohen, M. J. Mehl, and L. L. Boyer, Physica B 150 (1988) 1.
[26] H. L. Skriver, The LMTO Method (Springer, Berlin, 1984).
[27] O. K. Andersen, Physical Review B 12 (1975) 3060.
[28] M. Payne et al., Reviews of Modern Physics 64 (1992) 1045.
[29] E. Wimmer, H. Krakauer, M. Weinert, and A. J. Freeman, Physical Review B 24 (1981)
864.
[30] H. J. F. Jansen and A. J. Freeman, Physical Review B 30 (1984) 561.
[31] O. K. Andersen, O. Jepsen, and M. Sob, in Statics and Dynamics of Alloy Phase Trans-
formations (Springer Lecture Notes, New York, 1987).
[32] A. M. Bratkovsky and S. Y. Savrasov, Journal of Computational Physics 88 (1990) 243.
[33] N. E. Christensen and S. Satpathy, Physical Review Letters 55 (1985) 600.
[34] K. H. Weyrich, Physical Review B 37 (1988) 10269.
[35] P. Pulay, Molecular Physics 17 (1969) 197.
[36] M. C. Payne et al., Reviews of Modern Physics 64 (1992) 1045.
24

[37] A. M. Rappe, K. M. Rabe, E. Kaxiras, and J. D. Joannopoulos, Physical Review B 41
(1990) 1227.
[38] J. C. Slater and G. F. Koster, Physical Review 94 (1954) 1498.
[39] J. A. Majewski and P. Vogl, Physical Review Letters 57 (1986) 1366.
[40] A. F. Kohan and G. Ceder, Physical Review B 54 (1996) 805.
[41] W. A. Harrison, Electronic Structure and the Properties of Solids (Dover Publications,
New York, 1989).
[42] W. A. Harrison, Physical Review B 31 (1985) 2121.
[43] L. Kleinman and D. Bylander, Physical Review Letters 48 (1982) 1425.
[44] J. P. Perdew and A. Zunger, Physical Review B 23 (1981) 5048.
[45] R. J. Ackermann, E. G. Rauh, and C. A. Alexander, High Temperature Science 7 (1975)
304.
[46] U. von Barth and L. Hedin, Journal of Physics C 5 (1972) 1629.
[47] C. Wolverton and A. Zunger, Physical Review B 50 (1994) 10548.
[48] L. L. Boyer et al., Physical Review Letters 54 (1985) 1940.
[49] S. Froyen and W. A. Harrison, Physical Review B 20 (1979) 2420.
[50] A. Dwivedi and A. N. Cormak, Philosophical Magazine A 61 (1990) 1.
[51] E. V. Stefanovich, A. L. Shluger, and C. R. A. Catlow, Physical Review B 49 (1994)
11560.
[52] H. J. F. Jensen, Physical Review B 43 (1991) 7267.
[53] T. I. U. of Crystallography, International Tables for Crystallography (Kluwer Academic
Publishers, Dordrecht, 1989).
25

[54] C. J. Howard, R. J. Hill, and B. E. Reichert, Acta Cryst. 44 (1988) 116.
26
0
0.05
0.1
0.15
0.2
0.25
0.3
0 2 4 6 8 10
For
mat
ion
Ene
rgy
(eV
/ato
m)
Structure Number
"Pseudopotentials""SSCAD"
"Tight-binding""Potentials"
Figure 1 Kohan{Ceder
Tot
al E
nerg
y (R
y/ce
ll)
r /r Ca O
-1507.31
-1507.3
-1507.29
-1507.28
-1507.27
-1507.26
-1507.25
-1507.24
-1507.23
-1507.22
0.8 0.85 0.9 0.95 1 1.05
Fig 2: Kohan-Ceder