Embed Size (px)
Citation preview

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Feb. 1989, p. 446-453 Vol. 55, No. 20099-2240/89/020446-08$02.00/0
Expression of a Cloned Bacillus thuringiensis Delta-EndotoxinGene in Bacillus subtilis
S. CALOGERO,1 A. M. ALBERTINI,1t C. FOGHER, R. MARZARI,3 AND A. GALIZZI1*Dipartimento di Genetica e Microbiologia "A.Buzzati-Traverso," Universitei di Pavia, 27100 Pavia,' Istituto di Genetica,
Facolta di Agraria, Universita Cattolica Sacro Cuore, 29100 Piacenza,2 and Dipartimento di Biologia,Universitai di Trieste, 34100 Triestej, Italy
Received 11 July 1988/Accepted 21 November 1988
The entire coding region of the Bacillus thuringiensis HD73 crystal protein gene was subclohed from plasmidpJWK20 into the integration vector pUG2-15. This plasmid expresses chloramphenicol resistance whenintegrated into the Bacillus subtilis chromosome in the outH locus near the recE region. The correct molecularorganization of the integrated plasmid was verified by hybridization to Southern blots of chromosomal DNAdigests. Production of the toxic crystal protein was monitored at different time points during the life cycle ofB. subtilis. Toxicity assays against Anagasta (Ephestia) larvae, direct electron microscopy crystal detection, andimmunoblotting assays proved that the expression of the gene in B. subtilis is time regulated and restrictedmainly to the sporulation stage. RNase protection experiments defined the transcription initiation start pointand the transcription timing. All tests were made in a strain containing one to three copies of the integratedplasmid and in a strain subjected to an amplification regimen.
The gram-positive sporeformer Bacillus thuringiensis iswell known for its ability to produce, during sporulation,proteinaceous crystals highly toxic to insect larvae. A vastnumber of subspecies of B. thuringiensis have been de-scribed which differ in plasmid patterns and specificity ofaction of the protein, also called delta-endotoxin. Several ofthe genes encoding the toxin have been cloned, sequenced,and expressed in various hosts (4, 22). B. thuringiensisstrains may contain more than one gene coding for delta-endotoxin; therefore, cloning of the toxin gene in a differenthost is a necessary step in the study of the organization andregulation of expression of the genes and in the identificationof the gene products and their modes of action. In addition,molecular cloning may be useful for applicative purposes,since it allows the production of a single entomocidal poly-peptide without the possibilities of genetic rearrangementsand subsequent modifications of the commercial product.Among the possible hosts for the delta-endotoxin gene,Bacillus subtilis should represent a bacterium of choicebecause of its phylogenetic relationship to B. thuringiensis.
It can be expected that the transcriptional and transla-tional mechanisms of B. subtilis are very similar to those ofB. thuringiensis, and therefore it should be possible to obtainfaithful expression of the cloned genes. That this is indeedthe case has been shown by Shivakumar et al. (20), who firstreported the correct expression of the B. thuringiensisdelta-endotoxin gene in B. subtilis, using a replicative plas-mid. In B. subtilis, segregational and structural instability ofplasmids has often been observed (6, 14). One way ofcircumventing both problems is to use integrational vectors,which cannot replicate autonomously in B. subtilis but whichcan integrate into the chromosome if they carry a DNAfragment homologous to a chromosomal sequence. PlasmidpJH101 (10) is well suited to the purpose because it is stableonce inserted into the chromosome and has the additionaladvantage of allowing gene amplification through growth of
* Corresponding author.t Present address: Facolta di Agraria, Universita di Udine, 33100
Udine, Italy.
the bacteria in increasing concentrations of chloramphenicol(3).We have recloned the crystal protein gene of B. thurin-
giensis subsp. kurstaki HD73 into B. subtilis, making use ofan integrational plasmid derived from pJH101. In the newhost, the toxin gene was expressed during sporulation andthe gene product was toxic to lepidopteran larvae. Afteramplification of the plasmid inserted into the chromosome,relatively high yields of crystal protein were obtained, andbipyramidal crystals similar to those in B. thuringiensis werefound in B. subtilis. The transcriptional start sites in B.subtilis were found to correspond to the start sites observedin B. thuringiensis.
MATERIALS AND METHODSBacterial strains and growth. The B. subtilis strain used
was PB1424 (hisB2 trpC2 metD4). Transformation of com-petent cells was performed by the method of Hoch et al. (13);selection was for chloramphenicol resistance (5 ,ug/ml) onnutrient agar (Difco Laboratories, Detroit, Mich.). Forgrowth and sporulation, nutrient broth (Difco) supplementedwith MnCl2 (10-5 M) was used. B. thuringiensis subsp.kurstaki HD73 (strain 4D4 of the Bacillus Genetic StockCenter, Ohio State University, Columbus) was grown at30°C in Tris-G medium (5).
Escherichia coli HB101 (F- hsdS20 recA13 ara-14 proA2lacYJ galK2 rpsL20 xyl-5 mtl-l supE44) and JM103 [supE44thi rpsL20 endA hspR4 A(lac-proAB) F' traD36 proAB lacIqZAM15] were used for transformation by the calcium shockprocedure (9) and plasmid propagation. The medium usedwas LB medium (tryptone [Difco], 10 g; yeast extract, 5 g;NaCl, 10 g; water to 1 liter; pH 7.1).Plasmid construction. The source of the delta-endotoxin
gene was plasmid pJWK20 (ATCC 31997), described byKronstad and Whiteley (15). The integrative vector waspUG2-15, a derivative of pJH101 (10) containing 1.7 kilo-bases of B. subtilis chromosomal DNA (10). p5, a derivativeof pSGMU2 (11) containing 400 base pairs (bp) derived fromthe B. subtilis ot-amylase gene, was used as an intermediatefor the final construction.
446
on May 15, 2018 by guest
http://aem.asm
.org/D
ownloaded from
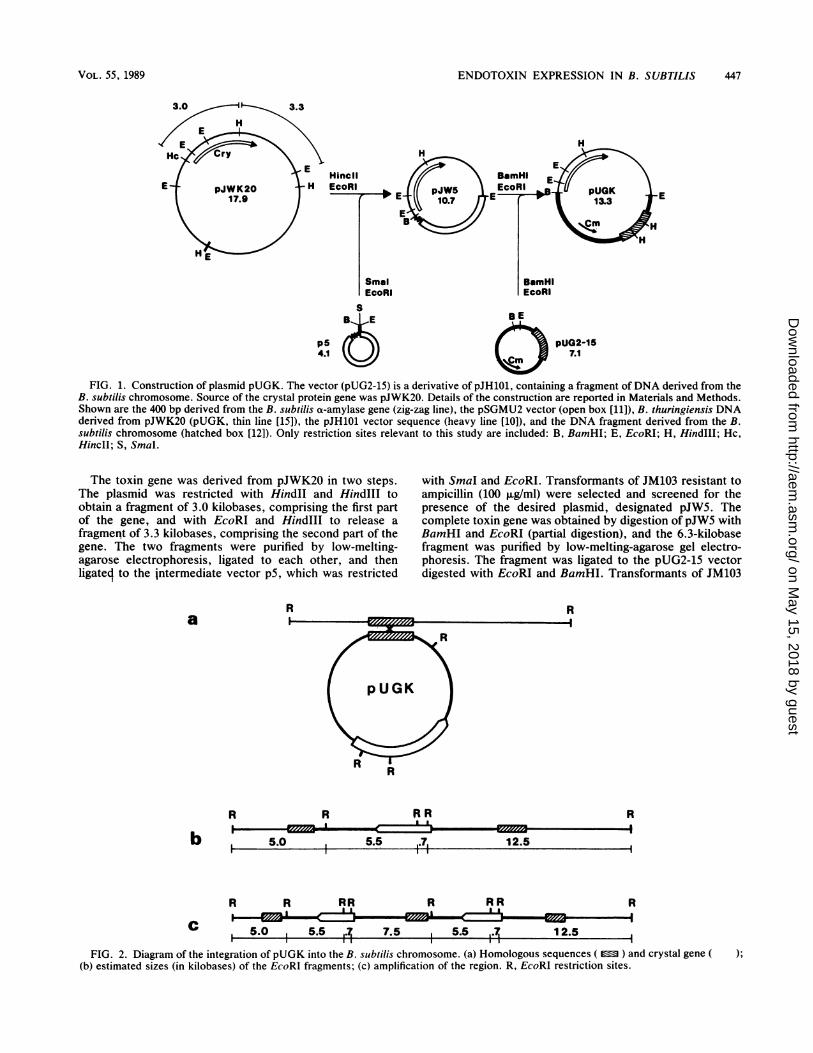
ENDOTOXIN EXPRESSION IN B. SUBTILIS 447
3.0 3.3
H
E pJWK20 H17.9
HE
HincilEcoRI
rTt
SmalEcoRI
B4..E BE
ps4 pUG2-15
FIG. 1. Construction of plasmid pUGK. The vector (pUG2-15) is a derivative of pJH101, containing a fragment ofDNA derived from theB. subtilis chromosome. Source of the crystal protein gene was pJWK20. Details of the construction are reported in Materials and Methods.Shown are the 400 bp derived from the B. subtilis a-amylase gene (zig-zag line), the pSGMU2 vector (open box [11]), B. thuringiensis DNAderived from pJWK20 (pUGK, thin line [15]), the pJH101 vector sequence (heavy line [10]), and the DNA fragment derived from the B.subtilis chromosome (hatched box [12]). Only restriction sites relevant to this study are included: B, BamHI; E, EcoRI; H, HindIlI; Hc,HincII; S, SmaI.
The toxin gene was derived from pJWK20 in two steps.The plasmid was restricted with HindII and HindlIl toobtain a fragment of 3.0 kilobases, comprising the first partof the gene, and with EcoRI and HindIII to release afragment of 3.3 kilobases, comprising the second part of thegene. The two fragments were purified by low-melting-agarose electrophoresis, ligated to each other, and thenligateq to the jntermediate vector p5, which was restricted
Ra i
with SmaI and EcoRI. Transformants of JM103 resistant toampicillin (100 ,ug/ml) were selected and screened for thepresence of the desired plasmid, designated pJW5. Thecomplete toxin gene was obtained by digestion of pJW5 withBamHI and EcoRI (partial digestion), and the 6.3-kilobasefragment was purified by low-melting-agarose gel electro-phoresis. The fragment was ligated to the pUG2-15 vectordigested with EcoRI and BamHI. Transformants of JM103
R
R
R R R
b 5.0 5.5 7 12.5I I.II
R R RR R RR R
5.0 5.5 .7 7.5 5.5 .7 12.5
FIG. 2. Diagram of the integration of pUGK into the B. subtilis chromosome. (a) Homologous sequences ( ) and crystal gene(b) estimated sizes (in kilobases) of the EcoRI fragments; (c) amplification of the region. R, EcoRI restriction sites.
VOL. 55, 1989
on May 15, 2018 by guest
http://aem.asm
.org/D
ownloaded from
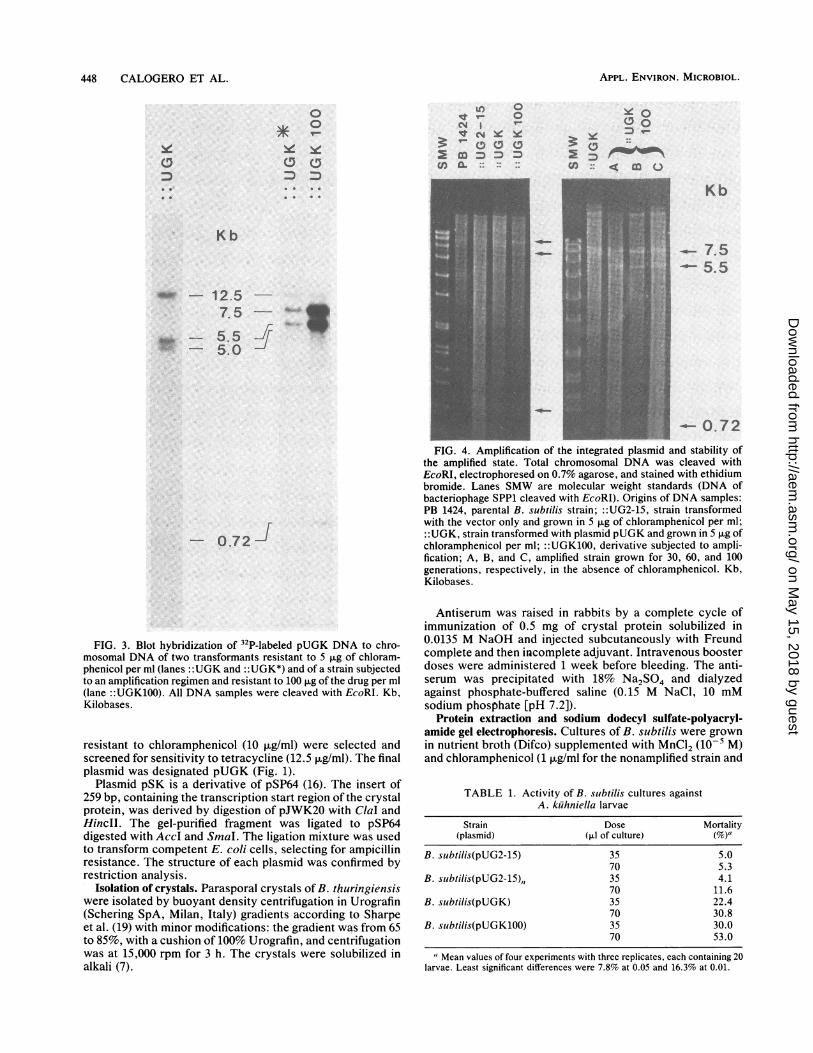
448 CALOGERO ET AL.
00,
FIG. 3. Blot hybridization of "P-labeled pUGK DNA to chro-
mosomal DNA of two transformants resistant to 5 pg of chloram-
phenicol per ml (lanes ::UGK and ::UGK*) and of a strain subjectedto an amplification regimen and resistant to 100 p.g of the drug per ml
(lane ::UGK100). All DNA samples were cleaved with EcoRI. Kb,
Kilobases.
resistant to chloramphenicol (10 p.g/ml) were selected and
screened for sensitivity to tetracycline (12.5 p.g/ml). The final
plasmid was designated pUGK (Fig. 1).
Plasmid pSK is a derivative of pSP64 (16). The insert of
259 bp, containing the transcription start region of the crystal
protein, was derived by digestion of pJWK2O with Clal and
HincII. The gel-purified fragment was ligated to pSP64
digested with Accl and Smal. The ligation mixture wa's used
to transform competent E. coli cells, selecting for ampicillinresistance. The structure of each plasmid was confirmed by
restriction analysis.
Isolation of crystals. Parasporal crystals of B. thuringiensiswere isolated by buoyant density centrifugation in Urografin
(Schering SpA, Milan, Italy) gradients according to Sharpeet al. (19) with minor modifications: the gradient was fr'om 65
to 85%, with a cushion of 100% Urografin, and centrifugationwas at 15,000 rpm for 3 h. The crystals were solubilized in
alkali (7).
to 0 ~
We v- 0
Kb_ ~~~~~~~7.:5-5.5
0.72
FIG. 4. Amplification of the integrated plasmid and stability of
the amplified state. Total chromosomal DNA was cleaved with
EcoRI, electrophoresed on 0.7% agarose, and stained with ethidium
bromide. Lanes SMW are molecular weight standards (DNA of
bacteriophage SPP1 cleaved with EcoRI). Origins of DNA samples:
PB 1424, parental B. subtilis strain; ::UG2-15, strain transformed
with the vector only and grown in p.g of chloramphenicol per ml;
::UGK, strain transformed with plasmid pUGK and grown in 5 p.g of
chloramphenicol per ml; ::UGK100, derivative subjected to ampli-
fication; A, B, and C, amplified strain grown for 30, 60, and 100
generations, respectively, in the absence of chloramphenicol. Kb,
Kilobases.
Antiserum was raised in rabbits by a complete cycle ofimmunization of 0.5 mg of crystal protein solubilized in0.0135 M NaOH and injected subcutaneously with Freundcomplete and then incomplete adjuvant. Intravenous boosterdoses were administered 1 week before bleeding. The anti-serum was precipitated with 18% Na2SO4 and dialyzedagainst phosphate-buffered saline (0.15 M NaCl, 10 mMsodium phosphate [pH 7.2]).
Protein extraction and sodium dodecyl sulfate-polyacryl-amide gel electrophoresis. Cultures of B. subtilis were grownin nutrient broth (Difco) supplemented with MnCl2 (10-5 M)and chloramphenicol (1 p.g/ml for the nonamplified strain and
TABLE 1. Activity of B. subtilis cultures againstA. kuhniella larvae
Strain Dose Mortality(plasmid) (,u1 of culture) (%)a
B. subtilis(pUG2-15) 35 5.070 5.3
B. subtilis(pUG2-15),, 35 4.170 11.6
B. subtilis(pUGK) 35 22.470 30.8
B. subtilis(pUGK100) 35 30.070 53.0
"Mean values of four experiments with three replicates, each containing 20larvae. Least significant differences were 7.8% at 0.05 and 16.3% at 0.01.
APPL. ENVIRON. MICROBIOL.
on May 15, 2018 by guest
http://aem.asm
.org/D
ownloaded from
ENDOTOXIN EXPRESSION IN B. SUBTILIS 449
200-
97~
68- AA8*2 0:000E f s _; f , j i S~
0
AJ : ::'g:I1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
FIG. 5. Enzyme immunoassay of crystal proteins produced in B. subtilis. Extracts of B. subtilis cells were resolved by sodium dodecylsulfate-polyacrylamide gel electrophoresis, and protein bands reacting with anticrystal antibody were analyzed. Lanes: 1 through 3,Coomassie blue staining of crystal proteins purified from B. thuringiensis subsp. kurstaki HD73, of an extract from log-phase cells of B.subtilis(pUGK100), and of an extract from a sporulating culture (T23) of B. subtilis(pUGK100), respectively; 4 through 8, immunoblots of cellextracts from B. subtilis(pUGK) (not amplified) in log phase and at To, T4, T7, and T18, respectively; 9 through 13, immunoblots of cell extractsfrom an amplified strain in log phase and at To, T4, T7, and T23, respectively; 14, immunoblot of purified crystal protein; 15 and 16,immunoblots of cell extracts of B. subtilis transformed with the vector only, taken at log phase and T18.
10 pLg/ml for the amplified strain). Samples of 15 ml werecollected at log phase (1.0 optical density unit at 560 nm) andat transition (To) between log and stationary phases. Latersamples (at 4, 7, 18, or 23 h after the end of exponentialgrowth [T4, T7, T18, or T23]) were of 10 ml. Further growthwas stopped by rapid chilling, and the cells were collected bycentrifugation. The cell pellets were frozen at -20°C andthen thawed in 1/20 volume of sonication buffer (10 mM Trishydrochloride [pH 7.4], 5 mM MgCl2, 1 mM phenylmethyl-sulfonate fluoride, 10 mM sodium metabisulfite) containingRNase 1 (50 pLg/ml). The cells were ruptured by sonication inthe presence of glass beads (0.1-mm diameter). The sampleswere kept in ice during treatment, and sonication was inpulses for a total of 1 min. The glass beads were allowed tosediment, and the supernatant was divided into portions of200 ,ul each and lyophilized.The method of Towbin et al. (21) was used to detect the
crystal protein immunologically. Proteins resolved by so-dium dodecyl sulfate-polyacrylamide gel electrophoresiswere transferred electrophoretically to nitrocellulose sheetswashed with 50 mM Tris hydrochloride-200 mM NaCl
r-> r*
containing 0.1% Nonidet P-40 and then incubated with theantiserum. After a wash with the same buffer, the nitrocel-lulose sheets were incubated with peroxidase-conjugatedsheep anti-rabbit immunoglobulin G antiserum (UnitedStates Biochemical Co., Cleveland, Ohio). The immunocom-plexes were then visualized in the presence of hydrogenperoxide and 4-chloro-1-naphthol as substrates (GIBCOLaboratories, Grand Island, N.Y.; Bethesda Research Lab-oratories, Inc., Gaithersburg, Md.).
Insect toxicity tests. B. subtilis cultures were tested fortoxicity by feedings to neonatal larvae of Anagasta (Ephes-tia) kuhniella (8). A 50-ml sample of bacterial culture [at T23for strain PB1424(pUGK100) and at T18 for all other strainstested] were mixed with 20 g of wheat flour and lyophilized.Portions (50 and 100 mg, corresponding to 35 and 70 JJl,respectively, of the original culture) of the mixture wereadded to wheat flour up to 500 mg per sample. Each samplecontained 20 larvae. The dead larvae were scored after 48 hof free feeding at 25°C.
Electron microscopy. B. subtilis PB1424(pUGK100) and B.thuringiensis subsp. kurstaki HD73 were grown on Tris-G
Hinc 11 Bt II Bt I AUG Cf :a I
259 bp
145 (±5) nt
180(±5) nt
RNA PROBE294 nt
FIG. 6. Promoter region of the crystal protein gene. BtI and BtII indicate the two start sites of transcription; AUG is the start codon. Theanti-RNA probe used for RNase protection experiments is shown by the leftward line, the heavy portion of which represents the sequencecomplementary to the B. thuringiensis antisense DNA strand. An interpretation of the origin of the transcripts detected is shown by therightward arrows. Estimated sizes are given in nucleotides (nt) at the right.
.§ mfflnMm.mWFmjjjmmp-
VOL. 55, 1989
14 -
on May 15, 2018 by guest
http://aem.asm
.org/D
ownloaded from
450 CALOGERO ET AL.
agar plates. An agar block with a single colony was removedfrom the plates. Fixation was with 1% (vol/vol) glutaralde-hyde in 0.15 M sodium cacodylate buffer, dehydration was inan ethanol series, and drying was by the critical-pointmethod.Other methods. Chromosomal DNA extraction, agarose
gel electrophoresis, labeling of DNA probes, and blot hy-bridization were carried out as described previously (3).RNA extraction and RNase mapping were also performed asdescribed elsewhere (2). Briefly, plasmid pSK linearizedwith EcoRI was the template for synthesis of the antisenseprobe. The labeled probe was hybridized to in vivo RNA;after RNase digestion, the RNA-RNA duplexes were recov-ered by ethanol precipitation. Samples were denatured for 5min at 90°C and analyzed on 5% acrylamide gels containing8 M urea. The gels were exposed to X-ray film for 48 h.
RESULTS
Construction of an integrative plasmid carrying the toxingene. The source of the gene coding for the crystal proteinwas plasmid pJWK20, carrying a 13.6-kbp fragment derivedfrom B. thuringiensis subsp. kurstaki HD73 (15). A segmentof 6.3 kbp comprising the coding portion of the toxin wassubcloned in two steps in a derivative of pSGMU2 (11)containing an insert obtained from the B. subtilis a-amylasegene (Fig. 1). Since we encountered some difficulties withpJW5, the toxin gene was moved from it into pUG2-15, aderivative of pJH101 (10) carrying a PstI fragment of 1.7 kbporiginating from the chromosome of B. subtilis and locatedbetween dnaA and recE (12). The final plasmid, calledpUGK (Fig. 1), was thus a derivative of pJH101 carrying theentire coding sequence for the crystal protein and 1.7 kbphomologous to a chromosomal sequence of B. subtilis.The plasmid is unable to replicate in B. subtilis and can
transform competent cells to chloramphenicol resistance byhomologous recombination between the plasmid and thechromosome (10). Transformants resistant to 5 p,g of chlor-amphenicol per ml were obtained in high yield after 24 h ofincubation at 37°C. Southern analysis of the DNA extractedfrom the transformants showed that the plasmid was cor-rectly inserted into the chromosome by a Campbell-likemechanism (Fig. 2). The DNA of some of the transformantsexamined gave a pattern consistent with the presence of asingle plasmid integrated into the chromosome. After diges-tion with EcoRI and hybridization to pUGK labeled by nicktranslation, four bands, corresponding to 12.5, 5.5, 5.0, and0.72 kbp, appeared in the autoradiographs (Fig. 3, lane::UGK). With the pUG2-15 vector used as a probe only twobands could be seen, one of 12.5 and the other of 5.0 kbp(data not shown). The results obtained with other transform-ants showed the presence of an additional band of 7.5 kbpwhen probed with both plasmids, which suggested amplifi-cation of the inserted plasmid (Fig. 3, lane ::UGK*). Thisfinding is in accordance with previous results regardingduplication of inserted sequences at concentrations of chlor-amphenicol as low as 5 pug/ml (3). Four transformants weresubjected to an amplification regimen by exposure to in-creasing concentrations of antibiotic. DNA extracted fromstrains resistant to 100 ,ug of chloramphenicol per ml,digested with EcoRI, and stained with ethidium bromideshowed the presence of three prominent bands of 7.5, 5.5,and 0.72 kb, respectively, as expected for samples in whichthe integrated plasmid has been amplified (Fig. 4, lane::UGK100). Hybridization analysis confirmed that the bandswere indeed derived from pUGK (Fig. 3, lane ::UGK100).
No attempt has been made to measure the copy number ofthe amplified plasmids; however, from our previous experi-ence (3) and data from Piggot and Curtis (17), we presumethat the unit is repeated between 10 and 20 times.
It was desirable to know whether the amplified state couldbe maintained in the absence of selective pressure. To thisend, one amplified strain was grown for approximately 30,60, and 100 generations in the absence of chloramphenicol.DNA was extracted, restricted with EcoRI, and analyzed bygel electrophoresis and ethidium bromide staining. No ap-parent loss of copies was observed for any sample (Fig. 4,lanes A, B, and C).
Antigenic analysis of recombinant B. subtilis strains. Cellextracts were prepared from B. subtilis PB1424(pUGK), anda derivative [PB1424(pUGK100)] subjected to amplification.The extracts were obtained from different stages of the cellcycle; i.e., in exponential growth, at the beginning of thestationary phase (TO) and at T4, T7, T18, and T23. Westernblot (immunoblot) analysis of the cell extracts was per-formed with polyclonal antibodies raised against purifiedcrystal proteins of the B. thuringiensis donor strain. The B.subtilis extracts contained a polypeptide antigen with elec-trophoretic mobility the same as (or very similar to) that ofthe dissolved B. thuringiensis crystal protein (Fig. 5). Theantigen was missing from a B. subtilis strain transformedwith the vector plasmid pUG2-15 and amplified. In strainPB1424(pUGK), bearing one or few copies of the plasmid,the antigen was first detected at T4, whereas in the amplified
ua. 0E 0 _
:;0tWi W 0 t-444
-276
FIG. 7. mRNA mapping of the crystal protein gene. TheseRNase protection experiments used as a probe RNA synthesized invitro with plasmid pSK. RNA was derived from cells of B.subtilis(pUGK100) in log phase and at To, T2, T'4 and T'6. Numbersrefer to length (in bases) of RNA standards obtained from the pSP64template cut at various positions before in vitro transcription.Arrows indicate positions of the two longer transcripts observed.
APPL. ENVIRON. MICROBIOL.
on May 15, 2018 by guest
http://aem.asm
.org/D
ownloaded from
ENDOTOXIN EXPRESSION IN B. SUBTILIS 451
strain the antigen was present at a low level even duringexponential growth. In these latter strains, samples obtainedlate during sporulation showed, in addition to the 133-kilodalton polypeptide, a number of discrete bands of lowermolecular size (in the range of 72 to 53 kilodaltons), possiblyas a result of degradation of the crystal protein. The totalamount of antigen present at T23 represented about 5% oftotal protein.
Insect toxicity bioassay. The B. subtilis strains expressingthe crystal protein antigen were assayed for lethality toneonate A. kuhniella (European flour moth) larvae by incor-poration of samples of total cultures into an artificial diet.The results indicated that the recombinant B. subtilis strainswere toxic to the lepidopteran larvae (Table 1). With culturesof strains containing few copies of pUGK, we observed 30%mortality at a dose corresponding to 70 ,ul of total culture(see Materials and Methods for the conditions of bioassay).Mortality was higher (up to 53%) at the same dose forcultures obtained from strains subjected to an amplificationregimen. Controls, which were cultures of the strain con-taining the vector pUG2-15, both amplified and nonampli-fied, had no toxic effect in A. kuhniella larvae. The differencein toxicity between amplified and nonamplified strains bear-ing pUGK was less than expected but could be explained bythe partial degradation of the delta-endotoxin in the ampli-fied cultures (Fig. 5) and by the presence of multiple copiesof the integrated plasmid in the strain maintained in 5 ,ug ofchloramphenicol per ml, which we consider nonamplified(see, for instance, Fig. 3, lane ::UGK*). Under the same
assay conditions, cultures of B. thuringiensis HD73 werefive times more effective than pUGK100. It should bestressed that the bioassay results obtained from this type ofexperiment, in which the entire bacterial culture is added tothe feeding diet, can be considered only qualitative. For thisreason, any comparison with the activity observed in the B.thuringiensis preparation for the purpose of estimating thepotency of the B. subtilis preparation is unrealistic.
Transcriptional start sites in B. subtilis. It has been re-ported that in B. thuringiensis subsp. kurstaki HDI-Dipel,transcription of the crystal protein gene is initiated from twostart sites (22). It was of interest to know whether the B.subtilis transcriptional mechanism was capable of initiatingtranscription at the same sites as are utilized by B. thurin-giensis. We used RNase protection experiments to study thein vivo start sites of transcription in B. subtilis. In vivo RNAwas hybridized to labeled anti-mRNA synthesized in vitroby phage SP6 RNA polymerase on a template consisting of259 bp of the toxin gene. The DNA extended from 153 bpupstream to 106 bp downstream of the AUG codon (Fig. 6).After hybridization to the RNA extracted from vegetativecells and cells at various stages of sporulation, the sampleswere treated with RNase and the protected fragments wereresolved on denaturing urea-acrylamide gels (Fig. 7). B.subtilis cells harboring plasmid pUGK showed protection ofthe RNA probe. The two longer protected fragments hadlengths of 180 + 5 and 145 + 5 nucleotides, respectively. Thelength of the transcripts was estimated by comparison withsingle-stranded RNA molecules synthesized from the pSP64
FIG. 8. Scanning electron micrographs of crystals from B. thuringiensis (a) and B. subtilis(pUGK-100) (b). Bars indicate 1 ,um.
VOL. 55, 1989
on May 15, 2018 by guest
http://aem.asm
.org/D
ownloaded from

452 CALOGERO ET AL.
template, cut at specific sites before the in vitro transcrip-tion. Since the nucleotide sequence of the toxin gene ofstrain HD73 was identical to the sequence of HDI-Dipel, 175nucleotides preceding and 444 nucleotides following theATG codon (1), the deduced start sites roughly (±5 bases)corresponded to the BtI and BtII start sites determined byWong et al. (23) for the HDI-Dipel strain. We cannot excludethat in B. subtilis the shorter protected fragments representadditional initiation sites. For the two main transcripts, therewere differences in time of appearance during the cell cycleof B. subtilis, at least in the amplified strains. The shortertranscript was already evident during vegetative growth, wasbarely detectable at the beginning of the stationary phase,and, starting from T4, became more abundant. The longertranscript was detected only starting from T2. The presenceof the delta-endotoxin transcript during vegetative growthwas in accordance with the immunoblotting data reportedabove.
Production of bipyramidal crystals from B. subtilis. Sporu-lating colonies of a B. subtilis clone bearing an amplifiedcrystal protein gene were shown by scanning electron mi-croscopy to contain bipyramidal crystals (Fig. 8b). Thecrystals were well-shaped bipyramids, very similar to thoseobserved in B. thuringiensis under the same growth condi-tions (Fig. 8a). The only difference found was a higherheterogeneity in size in B. subtilis than in B. thuringiensis.Well-formed crystals in B. subtilis recombinant strains havebeen observed by Shivakumar et al. (20).
DISCUSSIONUse of pJH101 as an integrational vector for cloning and
expressing the toxin gene of B. thuringiensis in B. subtiliswas successful. The recombinant clones produced a 133-kilodalton polypeptide antigenically indistinguishable fromthe solubilized B. thuringiensis crystal protein. The productwas toxic to lepidopteran larvae. In strains with a limitednumber of copies of the integrated plasmid, expression of thedelta-endotoxin was restricted to the sporulation stage.Therefore, it appears that expression in B. subtilis is regu-lated by the same type of mechanisms as act in B. thurin-giensis. After amplification of the toxin gene, we observedan increase in delta-endotoxin accumulation, paralleled byan increase in toxicity to larvae and by bipyramidal crystalproduction. The production of well-defined bipyramidalcrystals in B. subtilis has been reported by Shivakumar et al.(20) for a strain carrying a recombinant multicopy plasmid.The amplified state achieved by growing the transformants inincreasing concentrations of chloramphenicol appeared to bestable for at least 100 generations, even in the absence ofselective pressure (Fig. 4).
Synthesis of the delta-endotoxin in strains with the ampli-fied plasmid was not concomitant with sporulation; theantigen was already detectable during vegetative growth. Asimilar dissociation of expression of the toxin gene fromsporulation has been observed in B. subtilis strains withmultiple copies of the gene on a plasmid (20). Such an effectcould be due to a high copy number of the promoter region,with subsequent escape from regulation, or to the absence ofregulatory regions in the cloned DNA. We deliberatelyexcluded from our construction a large portion of DNAsequence upstream of the promoter region in order to avoidthe negative controlling elements reported by Wong et al.(23) and Schnepf et al. (18).Our experiments aimed at defining the in vivo start sites of
transcription show that the toxin gene is transcribed by B.subtilis RNA polymerase correctly, giving rise to transcripts
of the same size as those found in B. thuringiensis. Thetiming of utilization of the two tandem promoters is alsoreminiscent of the situation found in B. thuringiensis (23).The regulatory elements of the toxin genes should be ame-nable to physiological and genetic studies in B. subtilis.
ACKNOWLEDGMENTS
We thank A. Bianchi for the electron micrographs and F. Scoffonefor expert technical assistance.
This work was supported in part by grants from the Ministerodella Pubblica Istruzione (Rome).
LITERATURE CITED1. Adang, M. J., M. J. Staver, T. A. Rocheleau, J. Leighton, R. F.
Barker, and D. V. Thompson. 1985. Characterized full-lengthand truncated plasmid clones of the crystal protein of Bacillusthuringiensis subsp. kurstaki HD-73 and their toxicity to Mand-uca sexta. Gene 36:289-300.
2. Albertini, A. M., T. Caramori, D. J. Henner, E. Ferrari, and A.Galizzi. 1987. Nucleotide sequence of the outB locus of Bacillussubtilis and regulation of its expression. J. Bacteriol. 169:1480-1484.
3. Albertini, A. M., and A. Galizzi. 1985. Amplification of achromosomal region in Bacillus subtilis. J. Bacteriol. 162:1203-1211.
4. Andrews, R. E., R. M. Faust, H. Wabiko, K. C. Raymond, andL. A. Bulla, Jr. 1987. The biotechnology of Bacillus thuringien-sis. Crit. Rev. Biotechnol. 6:163-232.
5. Aronson, A. I., N. Angelo, and S. C. Holt. 1971. Regulation ofextracellular protease production in Bacillus cereus T: charac-terization of mutants producing altered amounts of protease. J.Bacteriol. 106:1016-1025.
6. Bron, S., and E. Luxen. 1985. Segregational instability ofpUB110-derived recombinant plasmids in Bacillus subtilis. Plas-mid 14:235-244.
7. Bulla, L. A., Jr., L. I. Davidson, K. J. Kramer, and B. L. Jones.1979. Purification of the insecticidal toxin from the parasporalcrystal of Bacillus thuringiensis subsp. kurstaki. Biochem.Biophys. Res. Commun. 91:1123-1126.
8. Burgerjon, A., and C. Yamvrias. 1959. Titrage biologique despreparation a base de Bacillus thuringiensis Berliner vis-a-vis deAnagasta (Ephestia) kuhniella Zell. C. R. Acad. Sci. Paris249:2871-2872.
9. Dagert, M., and S. D. Ehrlich. 1979. Prolonged incubation incalcium chloride improves the competence of Escherichia colicells. Gene 6:23-28.
10. Ferrari, F. A., A. Nguyen, D. Lang, and J. A. Hoch. 1983.Construction and properties of an integrable plasmid for Bacil-lus subtilis. J. Bacteriol. 154:1513-1515.
11. Fort, P., and J. Errington. 1985. Nucleotide sequence andcomplementation analysis of a polycistronic sporulation operon,spoVA, in Bacillus subtilis. J. Gen. Microbiol. 131:1091-1105.
12. Gianni, M., and A. Galizzi. 1986. Isolation of genes preferen-tially expressed during Bacillus subtilis spore outgrowth. J.Bacteriol. 165:123-132.
13. Hoch, J. A., M. Barat, and C. Anagnostopoulos. 1967. Transfor-mation and transduction in recombination-defective mutants ofBacillus subtilis. J. Bacteriol. 93:1925-1937.
14. Janniere, L., and S. D. Ehrlich. 1987. Recombination betweenshort repeated sequences is more frequent in plasmids than inthe chromosome of Bacillus subtilis. Mol. Gen. Genet. 210:116-121.
15. Kronstad, J. W., and H. R. Whiteley. 1984. Inverted repeatsequences flank a Bacillus thuringiensis crystal protein gene. J.Bacteriol. 160:95-102.
16. Melton, D. A., P. A. Krieg, M. R. Rebagliati, T. Maniatis, K.Linn, and M. R. Green. 1984. Efficient in vitro synthesis ofbiologically active RNA and RNA hybridization probes fromplasmids containing a bacteriophage SP6 promoter. NucleicAcids Res. 12:7035-7056.
17. Piggot, P. J., and C. A. M. Curtis. 1987. Analysis of the
APPL. ENVIRON. MICROBIOL.
on May 15, 2018 by guest
http://aem.asm
.org/D
ownloaded from

ENDOTOXIN EXPRESSION IN B. SUBTILIS
regulation of gene expression during Bacillus subtilis sporula-tion by manipulation of the copy number of spo-lacZ fusions. J.Bacteriol. 169:1260-1266.
18. Schnepf, H. E., H. C. Wong, and H. R. Whiteley. 1987. Expres-sion of a cloned Bacillus thuringiensis crystal protein gene inEscherichia coli. J. Bacteriol. 169:4110-4118.
19. Sharpe, E. S., K. W. Nickerson, J. N. Aronson, and L. A. Bulla,Jr. 1975. Separation of spores and parasporal crystals of Bacil-lus thuringiensis in gradients of certain X-ray contrastingagents. Appl. Microbiol. 30:1052-1055.
20. Shivakumar, A. G., G. J. Gundling, T. A. Benson, D. Casuto,M. F. Miller, and B. P. Spear. 1986. Vegetative expression of
the 5-endotoxin genes of Bacillus thuringiensis subsp. kurstakiin Bacillus subtilis. J. Bacteriol. 166:194-204.
21. Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretictransfer of proteins from polyacrylamide gels to nitrocellulosesheets: procedure and some applications. Proc. Natl. Acad. Sci.USA 76:4350-4354.
22. Whiteley, H. R., and H. E. Schnepf. 1986. The molecular biologyof parasporal crystal body formation in Bacillus thuringiensis.Annu. Rev. Microbiol. 40:549-576.
23. Wong, H. C., H. E. Schnepf, and H. R. Whiteley. 1983. Tran-scriptional and translational start sites for the Bacillus thurin-giensis crystal protein gene. J. Biol. Chem. 258:1960-1967.
VOL. 55, 1989 453
on May 15, 2018 by guest
http://aem.asm
.org/D
ownloaded from





























