Embed Size (px)
Citation preview

Exploring the structure of oligo- and polysaccharides Synthesis and NMR spectroscopy studies
Hanna Jonsson

©Hanna Jonsson, Stockholm 2010 Cover picture “Bacteria runs NMR” ISBN 978-91-7447-041-3 Printed in Sweden by US-AB, Stockholm 2010 Distributor: Department of Organic Chemistry, Stockholm University

All I ever want is sugar. -Andy Warhol
Mina mångåriga akademiska studier har lett mig fram till, den för hela den vetenskapliga forskningen
epokgörande slutsatsen, att jag är bäst. -Ur Grallimatik
Tage Danielsson


Abstract
A deeper understanding of the diversity of carbohydrates and the many ap-plications of oligo- and polysaccharides found in nature are of high interest. Many of the processes involving carbohydrates affect our everyday life. This thesis is based on six papers all contributing to an extended perspective of carbohydrate property and functionality. An introduction to carbohydrate chemistry together with a presentation of selected carbohydrate synthesis and analysis methods introduces the reader to the research field. The first paper is an NMR spectroscopy reinvestigation of the structures of the O-antigens from the lipopolysaccharides (LPS) of Escherichia coli O124 and Shigella dysenteriae type 3. The repeating units were concluded to be built of identical branched pentasaccharides now with the correct anomeric con-figurations. Paper II is a structural investigation of the O-antigen from the LPS of E. coli O74 which is built of branched tetrasaccharide repeating units including the uncommon monosaccharide D-Fuc3NAc. Paper III is a con-formational study of a rhamnose derivative, using NMR spectroscopy and X-ray crystallography. The benzoyl ester group positioned at C4 prefers an “eclipsed” conformation in the crystal as well as in solution. The use of site-specifically 13C-labeled compounds in conformational studies is discussed in Papers IV and V. The disaccharide α-L-Rhap-(1→2)-α-L-Rhap-OMe was synthesized together with two 13C-isotopologues and studied with NMR spectroscopy to give five J-couplings related to torsion angles φ and ψ. The trisaccharide α-L-Rhap-(1→2)[α-L-Rhap-(1→3)]-α-L-Rhap-OMe was syn-thesized with 13C-labeling at two positions which presented a solution to a problem of overlapping signals in the 1H NMR spectrum. The site-specific labeling also facilitated the measurement of two 3JCC and two 2JCH coupling constants. Finally, chapter 6 gives a short introduction to glycosynthase chemistry and discusses the synthesis of α-glycosyl fluorides. A novel cyclic heptasaccharide was synthesized from α-laminariheptaosyl fluoride using a mutant of the enzyme laminarase 16A and subsequently analyzed by NMR spectroscopy.

List of publications
This thesis is based on the following papers referred to as their Roman nu-merals I-VI. I Structural studies of the O-antigenic polysaccharides from
Shigella dysenteriae type 3 and Escherichia coli O124, a rein-vestigation K. Hanna M. Jonsson, Andrej Weintraub and Göran Widmalm Carbohydrate Research 2006, 341, 2986–2989.
II Structural determination of the O-antigenic polysaccharide from Escherichia coli O74 K. Hanna M. Jonsson, Andrej Weintraub and Göran Widmalm Carbohydrate Research 2009, 344, 1592–1595.
III Methyl 4-O-benzoyl-2,3-O-isopropylidene-α-L-rhamno-pyranoside K. Hanna M. Jonsson, Lars Eriksson and Göran Widmalm Acta Crystallographica C62 2006, o447-o449.
IV Studies on the conformational flexibility of α-L-Rhap-(1→2)-α-L-Rhap-OMe using molecular simulation and 13C-site-specific labeling: a model for a commonly occurring disac-charide in bacterial polysaccharides K. Hanna M. Jonsson, Elin Säwén and Göran Widmalm In manuscript

V NMR analysis of conformationally dependent nJC,H and nJC,C in the trisaccharide α-L-Rhap-(1→2)[α-L-Rhap-(1→3)]-α-L-Rhap-OMe and a site-specifically labeled isotopologue thereof K. Hanna M. Jonsson, Göran Widmalm In manuscript
VI Synthesis of cyclic β-glucan Using Laminarinase 16A Glyco-synthase Mutant from the Basidiomycete Phanerochaete chrysosporium Jonas Vasur, Rie Kawai, K. Hanna M. Jonsson, Göran Widmalm, Åke Engström, Martin Frank, Evalena Andersson, Henrik Hans-son, Zarah Forsberg, Kiyohiko Igarashi, Masahiro Samejima, Mats Sandgren, Jerry Ståhlberg Journal of the American Chemical Society 2010, 132, 1724-1730.
Paper I and II were reprinted with kind permission from Elsevier, paper III from IUCr and Paper VI from ACS.

Contents
Chapter 1: Introduction to carbohydrate chemistry..........................11 1.1 Carbohydrates in bacteria ................................................................................13 1.2 Short summary of carbohydrate nomenclature...........................................14
Chapter 2: Selected methods of carbohydrate synthesis.................17 2.1 Protecting group chemistry ..............................................................................17 2.2 O-Glycosylation methods..................................................................................18
Chapter 3: Methods of carbohydrate analysis ....................................21 3.1 Sugar analysis .....................................................................................................21 3.2 NMR spectroscopy analysis ..............................................................................22
3.2.1 Methods of structural investigation .......................................................23 3.2.2 Methods of conformational analysis ......................................................25 3.2.3 Selected NMR techniques for J-coupling measurements ..................26
Chapter 4: Structural studies of bacterial polysaccharides (Papers I & II)..............................................................................................................29
4.1 Introduction .........................................................................................................29 4.2 Structural investigation of the O-antigenic polysaccharides from E. coli
O124 and S. dysenteriae type 3 (Paper I)...........................................................30 4.3 Structural investigation of the O-antigen of E. coli O74 (Paper II) ........33
Chapter 5: Conformational analysis of rhamnose-containing saccharides (Paper III, IV & V) ..............................................................37
5.1 Synthesis of 13C-labeled rhamnopyranoside oligosaccharides .................38 5.2 Conformational study of a rhamnose derivative (Paper III).....................40 5.3 Studies on the conformational flexibility of α-L-Rhap(1→2)-α-L-Rhap-
OMe (Paper IV)...........................................................................................................41 5.4 NMR studies on of the trisaccharide α-L-Rhap-(1→2)[α-L-Rhap-(1→3)]-
α-L-Rhap-OMe (Paper V) ..........................................................................................44
Chapter 6: Enzymatic synthesis using glycosynthases (Paper VI).47 6.1 A short introduction to glycosynthase chemistry ........................................47 6.2 Synthesis of α-glycosyl fluoride donors.........................................................49 6.3 Investigating a cyclic heptaglucan synthesized by mutant laminarase
16A (Paper VI) ...........................................................................................................51

Chapter 7: Concluding remarks and future prospects ......................54
Acknowledgements ...................................................................................56
References ..................................................................................................58

Abbreviations
1DLR One dimensional long-range CPS Capsular polysaccharide DAST Diethylaminosulfur trifluoride DOSY Diffusion-ordered spectroscopy DQF-COSY Double-quantum filtered correlated spectroscopy EIEC Enteroinvasive Escherichia coli Fuc Fucose GalNAc N-acetyl galactosamine GLC Gas-liquid chromatography GlcNAc N-acetyl glucosamine HECADE (Heteronuclear Couplings from ASSCI-domain experiments
with E.COSY-type crosspeaks) HETCOR Heteronuclear correlation HMBC Heteronuclear multiple bond correlation HSQC Heteronuclear single quantum correlation LPS Lipopolysaccharide MD Molecular dynamics Me Methyl NIS N-iodosuccinimide NOESY Nuclear Overhauser effect spectroscopy OAc O-acetyl OBz O-benzoyl p pyranose Rha Rhamnose TOCSY Total correlation spectroscopy TSP Sodium 3-methylsilyl-(2,2,3,3-2H4)-propanoate

11
Chapter 1: Introduction to carbohydrate chemistry
Today when carbohydrates are mentioned one immediately thinks of the main source of energy obtained from our food. People in general also distin-guish between sugar and carbohydrates, the former being the dangerous white food-sweetener and the latter being healthy slow-digesting “good” energy. For a carbohydrate chemist, the difference only lies in the use of the words; sugar is the common word and carbohydrate is the scientifically cor-rect term. Of course, carbohydrates are not only found in the food we eat, they are also the major constituent of wood and the shells of insects and shellfish, and they are present in almost all cell walls of living tissue. They contribute as signaling substances recognized by many proteins and define our blood group antigens. Carbohydrates are the most abundant of the bio-logical substances, produced in thousands of tons per year by photosynthesis in plants and microorganisms.
Carbohydrate chemistry was founded by the German researcher Hermann Emil Fischer (1852-1919)1 who already in 1890 established the stereochemi-cal nature and isomerism of the common sugars. The same year he also managed to synthesize glucose, fructose and mannose starting from glycerol. In the 1890s, Fischer determined the stereochemical configuration of all the known monosaccharides and exactly foretold the possible isomers, not proven again until 1951. The work of Fischer was very extensive and it is admirable to have achieved all this knowledge without the assistance of the techniques carbohydrate chemists have to today, including NMR spectros-copy, mass spectrometry and the possibility to purchase all kinds of syn-thetic chemicals commercially. Fischer became a Nobel Prize laureate in 1902 for his work on carbohydrates and purine synthesis.
The vast variety of applications we find for carbohydrates are mainly due to the big diversity of the molecules. Up to today a few hundred different monosaccharides have been found in nature (only ~10 expressed in human)2 and since they can occur in different ring sizes and configurations, the total collection of monosaccharide types is very large. Added to these properties are the multiple hydroxyl groups which lead to an almost endless number of

12
combinations possible when the monosaccharides are linked together as oligo- and/or polysaccharides. As an example, when three different natural hexoses are linked together there is a possibility to form 6144 different linear trisaccharides and even more if branches are allowed.3 When compared to natural amino acid oligo- or polymers, for which the corresponding number of combinations is 6, an understanding of the multiformity of carbohydrates is generated. The hydroxyl functionalities also contribute to the many chal-lenges experienced by synthetic (carbohydrate) chemists trying to produce oligo- and polysaccharides in the laboratory.
When realizing how many areas carbohydrates are involved in and how they might affect our lives, it is not difficult to justify the intense research in the field of carbohydrate chemistry. The applications of the results are many and new areas of use are constantly evolving. Chapter 4 (Paper I & II) in this thesis describes the structural elucidation of the lipopolysaccharides (LPS) of two Escherichia coli serotypes (and one Shigella dysenteriae type), which is important for the delineation of such pathogenic bacteria and can indirectly contribute to the development of vaccines against the diseases caused by these bacteria.4,5,6 Chapter 5 (Paper IV & V) describes the conformational studies of some rhamnose-containing di- and trisaccharides which eventually should lead to a better understanding of the structure−activity relationships these oligosaccharides and their related polysaccharides (PS) are involved in. One should also not underestimate the importance of the basic research per-formed at university institutions all over the world leading to development of methodology and education of new researchers. Finally, Chapter 6 (Paper VI) shows how directed enzymatic synthesis can be used to simplify the time-consuming work of oligo- and polysaccharide synthesis.

13
1.1 Carbohydrates in bacteria
As mentioned above, carbohydrates have many different functions in nature, and the domain Bacteria use them e.g. as stabilizers and signaling molecules in their membrane. Bacteria have a dual effect on human beings; they can serve both as non-pathogenic members of the human colonic flora, as do some species of E. coli, or be a severe risk to our health, as do other types of E. coli. No matter the function, the study of bacteria is a well-examined re-search field.4, 7
One way to classify bacteria is to differentiate between Gram-positive and Gram-negative bacteria. The classification comes from the Gram staining method,8 where the bacteria are subjected to ink coloring. Gram positive bacteria lack an extra outer membrane, which leads to greater absorption of the ink, hence a positive response to the staining. It is in this extra outer membrane, possessed by Gram-negative bacteria, that the LPS is embedded; the carbohydrate containing biomolecule that is of high interest in our stud-ies. Both the LPS and the flagellin of Gram-negative bacteria bind to recep-tors of the immune system causing signaling cascades and thereby serving as identification to the host, are the bacteria hostile or cooperating. The LPS is an endotoxin,7 a toxic molecule that, when released from the membrane, is dangerous. It consists of the lipid A, the harmful part, an inner and an outer core and the O-antigen (Figure 1.1). The O-antigen in turn is composed of up to 60 repeating units of 2-8 monosaccharide residues. Amino acids and phosphodiesters can also be a part of the O-antigen, further expanding the diversity of the LPS. The LPS covers ca. 40 % of the cell surface and is of great importance in all bacterial immunology studies.4
n
O-antigen outer core lipid Ainner core Figure 1.1: Schematic picture of the LPS embedded in the outer cell wall of Gram-negative bacteria.
Bacterial polysaccharides can also exist in the form of capsules made out of either monosaccharides making a homopolymer or from repeating units like the O-antigen of the LPS. Capsular polysaccharides (CPS) can be found in the membranes of both Gram-positive and Gram-negative bacteria and are

14
also an interesting target for investigations of immunological properties.9,10 They will not be discussed further in this thesis.
1.2 Short summary of carbohydrate nomenclature
The word carbohydrate originates from the time when it was thought that carbon-containing compounds, possessing hydrogen and oxygen in the molar ratio of 2:1, must be built out of carbon and water (hydro), hence having the empirical formula Cn(H2O)n. Later they were concluded to be polyhydroxy-lated organic substances rather than hydrates of carbon and terms as saccha-rides, sugars and glycans refer to the same class of compounds. The word saccharide comes from the word for sugar. In Latin it means saccharum, revealing the sweet taste of the initially described carbohydrates. The term glyco is often used in combinations with other biomolecules such as in gly-coproteins; proteins with carbohydrate chains attached to them. Discussed here are some basic features of carbohydrate nomenclature. For a detailed description, the IUPAC/IUBMB websites provide substantial informa-tion.11,12
Carbohydrates exist as aldoses or ketoses, which are polyhydroxyaldehydes and polyhydroxyketones, respectively. The identity of the sugar is deter-mined by the chain length, i.e. the number of carbons, and the chiral configu-ration of the carbons. The absolute configuration of a sugar unit is deter-mined from the highest numbered chiral carbon in the chain and is denoted D (latin: dexter = right) or L (latin laevus = left) from the direction of the hy-droxyl group on the parent carbon in the Fischer projection, carbon 5 in Fig-ure 1.2.
C1
C2** OHH
C3* HHO
C4* OHH
C5* OHH
C6
H O
OHH
H
Figure 1.2: Fischer projection of D-glucose.

15
Being a polyhydroxyaldehyde, the monosaccharide can react with itself to form a cyclic hemiacetal. The two forms (cyclic and acyclic) are in equilib-rium with each other in aqueous solution and this equilibrium is shifted to-wards the more energetically favourable ring form, demonstrated on D-glucose in Figure 1.3. The process is called mutarotation and is catalyzed by either dilute acid or base. Thermodynamic equilibrium is established and for D-glucopyranose the molar ratio of α:β is 4:6. The open form is only present in trace amounts.
OHO
HO
OH
HOHO
OHOHO
HO
HOHO
+
α-configuration
β-configuration
OHH
HHO
OHH
OHH
CH2OH
CHO
Figure 1.3: Schematic picture of the equilibrium between the open aldehyde form (drawn in Fischer projection) and the pyranose 4C1 forms (drawn in chair conforma-tions) of D-glucose.
The ring closure occurs via nucleophilic attack by the oxygen positioned at C5 (or C4) on the carbonyl carbon and a 6-membered (5-membered) ring, a pyranose (furanose), is formed. The ring closure generates a new stereogenic centre at C1, the anomeric centre, with two possible configurations, α or β. The resulting two compounds are diastereomers. For D-glucose in pyranose form (Figure. 1.3), the α-anomer has the hydroxyl group located axial to the ring and the β-anomeric hydroxyl group is located equatorial, when the molecule is in its most stable conformation (4C1).
In general, a cyclohexane derivative is most stable (i.e., has the lowest en-ergy) in the chair conformation when the larger substituent is in equatorial position. For pyranosides, this rule is also true, but because of the anomeric effect,13 the form in which an electronegative substituent at C1 is in axial position is surprisingly stable. The sugar ring is not, as cyclohexane, built of only carbons but contains an oxygen atom with two lone pairs of electrons. When one of these electron pairs is overlapping with the anti-bonding orbital of the substituent bond on C1 the whole molecule is stabilized. This in-creases the amount of axial configuration of an electronegative substituent at the anomeric centre, pictured as in Figure 1.4.

16
Stabilisation No stabilisation
O O
nπ
σ*
Figure 1.4: The anomeric effect explains why axial substituents occur at the ano-meric position in the sugar ring to a surprisingly large extent.
For glycopyranosides preferred conformations have also been found for exo-cyclic glycosidic bonds, summarized by Lemieux as the exo-anomeric ef-fect.14,15 It has a strong influence on the φ torsion angle and can be important in conformational studies. The effect is mainly explained by favorable or-bital overlap in certain conformations and is stronger for β-glycosides.

17
Chapter 2: Selected methods of carbohydrate synthesis
The research field of carbohydrate synthesis is constantly expanding as new methods and applications are evolving. Due to the complexity and diversity of the carbohydrate molecules, the synthesis pathways towards oligosaccha-rides can differ vastly even if they contain the same monosaccharide units. Oligosaccharide synthesis is also time-consuming; project duration is meas-ured in months or years even if executed in a specialist laboratory.16 Carbo-hydrate synthesis can be divided into two main fields; protecting group chemistry and glycosylation methods. Both fields occupy many research groups around the world and there is a strong need to automate the process of synthesis by e.g. programmable one-pot synthesis and/or enzymatic syn-thesis (discussed in chapter 6).17 In this thesis, the methods of carbohydrate synthesis related to paper II-VI and a few other common synthesis paths are presented.
2.1 Protecting group chemistry
The diversity of oligo- and polysaccharides is mainly explained by the many hydroxyl groups pointing in different directions on the monosaccharide units and thereby generating many linkage possibilities. The major part of the synthesis work involves protecting and deprotecting these nucleophilic func-tionalities to prevent unwanted side-reactions. Depending on the application and stability requirements of the building blocks, there are several different protecting groups to choose from.18
Protection of the anomeric position is usually performed before the other OH-functionalities due to its special reactivity, but also to “lock” the mono-saccharide in an α- or β- configuration if it will be at the reducing end of an oligosaccharide. The most common method is the Fischer glycosylation with methanol and an acid.18 During a Fischer-glycosylation, the hemiacetal is readily reacted with an alcohol (often methanol) to form the acetal, called a glycoside. Although the reaction is acid catalyzed and reversible, a relatively

18
stable glycoside is formed. Harsh acidic conditions, like sulfuric acid and/or acetic acid and heat, are usually required for cleavage.19
A common method for hydroxyl group protection is to use acyl groups such as acetyl or benzoyl derivatives. They are easily introduced to the sugar us-ing pyridine as a catalyst and removed under basic conditions. Acyl groups are relatively stable under acidic conditions and can withstand catalytic hy-drogenation conditions. They can also be very helpful in the glycosylation process (see section 2.2). A disadvantage of this type of protecting groups is the risk of acyl migration, a phenomenon first reported by Fischer in 192020 but still challenging for many researchers.21 Using hydrazine acetate in DMF it is possible to deprotect the anomeric position selectively, opening up for e.g. fluorination to create a glycosyl donor (see section 6.2).
Ether protecting groups, like benzyl or allyl ethers, are also very common. They are resistant to strong base as well as to milder acidic conditions. Ethers are formed employing a base, e.g. sodium hydride, in a polar aprotic solvent together with the respective alkyl halide.22 The benzyl groups are commonly removed by catalytic hydrogenation and this procedure is usually a quantitative reaction. The allyl groups are more complicated to remove since they have to be isomerized by a metal catalyst forming a labile enol ether intermediate which can be removed under mild acidic conditions.18
The simultaneous protection of two hydroxyl groups in a monosaccharide can be carried out by the use of acetals. This is an acid catalyzed reaction using e.g. acetone or benzaldehyde derivatives. The isopropylidene group normally attaches to vicinal hydroxyl groups positioned cis on the ring to form five-membered derivatives, 1,3-dioxolanes. They are cleaved by treat-ment in aqueous acid. Benzylidene acetals, on the other hand, are more sta-ble as six-membered rings, with the phenyl ring in the equatorial position. When removed there is a choice of removing the whole group or to open it selectively.23 All acetals are stable against bases and nucleophilic attacks.
2.2 O-Glycosylation methods
Glycosylation reactions or “couplings” as they are called in common speech are the reactions conjoining two (or more) monosaccharide derivatives to form an oligosaccharide. In a typical glycosylation procedure, a glycosyl donor (electrophile) is reacted with an acceptor (a nucleophile) in a dry sol-vent. Often a promoter is needed to start up the reaction. The main challenge regarding the glycosylation reactions is the stereochemical outcome of the reaction, i.e. whether the α- or β-anomer is formed. By neighbouring group

19
participation, e.g. by an ester group at C2, the 1,2-trans glycoside is strongly favored. An acyloxonium ion intermediate is formed from the oxocarbenium ion produced initially from the donor. The ring is then cleaved at C1 by the acceptor, the ester group is blocking attack from cis-direction yielding the 1,2-trans oriented O-glycosidic linkage (Figure 2.1). In the case of D-mannose the α-configuration is extra favored due to the anomeric effect and the β-configuration is often completely excluded as a product.
OAcOAcO
OAcAcO
L
oxocarbenium ion
acyloxycarbenium ion
OAcOAcO
OAcAcO
OR
RO
H
Promotor
- L
OAcOAcO
OAc
O O
-H
OAcOAcO
OAcAcO
OAcOAcO
OAcAcO
Figure 2.1: Neighboring group participation exemplified by the formation of a 1,2-trans glycoside of D-mannose.
The corresponding 1,2-cis glycosides are more difficult to obtain. In such cases, the protecting group at C2 has to be a non-participating group like an O-alkyl ether group. A chiral auxiliary positioned at C2 is also an option.24
The classical donor used is a glycosyl bromide or chloride, as in the Koenigs-Knorr method,25 which can be activated by silver salts such as sil-ver triflate or silver carbonate. The electrophilic intermediate formed (the oxycarbenium ion) reacts with a suitable alcohol to form the product. An advantage of the Koenigs-Knorr method is the easily synthesized donor molecules; glycosyl bromides are obtained in one step from the acetylated sugars using HBr in acetic acid. A disadvantage is the lability of the glycosyl halides; they are sensitive to moisture and might decompose when stored too long.
It is also possible to use fluoride as the halide donor functionality.26 Glycosyl fluorides are more stable than the corresponding chlorides or bromides and can even be stored in aqueous solution for a limited time period. The fluo-ride can be introduced at the anomeric position with several different re-agents e.g. Selectfluor27, diethylaminosulfur trifluoride (DAST) or hydrogen fluoride (HF)28 depending on the already existing anomeric substituent. Se-lectfluor is used for conversion of thioglycosides into α-glycoside fluorides, however it is also known to oxidize the thio group creating glycosyl sulfox-ides which may reduce the yield and complicate the synthesis. DAST con-

20
verts free hydroxyl groups to fluorides and when applied to the anomeric position, a mixture of α/β-fluorides is obtained. Since the α-glycosyl fluoride is more stable, and often the desired anomeric configuration, additional treatment with HF (in pyridine) will give solely α-anomer. HF in pyridine can also be applied directly on O-acetyl derivatives which have β-configuration.
Apart from the Koenigs-Knorr method described above, the use of thiogly-cosides as donors is also very effective and widely examined.29 Thioglyco-sides can be generated from glycosyl halides or directly from acetylated hexopyranoses in the presence of Lewis acids such as BF3
.Et2O. They are more stable than the corresponding halide donors, thus making the sugar unit open to further modifications if needed. To form an O-glycoside, the thioglycosides have to be activated by a thiophilic promoter, such as N-iodosuccinimide (NIS).

21
Chapter 3: Methods of carbohydrate analysis
As the title of this thesis suggests, the main focus should be set on the struc-tural features and conformational preferences of carbohydrates. The analysis of oligo- and polysaccharides can be a difficult task due to the variety and many degrees of freedom of carbohydrate structures. Since new methods are constantly being developed, and the computer programs to analyze the data are becoming more advanced, more and more information is revealed. In carbohydrate chemistry, the most common analysis techniques are nuclear magnetic resonance (NMR) spectroscopy,30 mass spectrometry (MS)31 and high performance liquid chromatography (HPLC).32 They can both deter-mine structural features and serve as evidence of synthetic products. When examining stereochemistry and conformations, X-ray crystallography can also be the method of choice. Information about the molecule in the solid state is then achieved, and this might not be optimal since the natural envi-ronment of these biomolecules is aqueous media. In recent years, the devel-opments of computational methods such as molecular dynamics (MD) simu-lations have gained momentum in the carbohydrate research field. This en-ables the possibility to study three-dimensional structures as well as interac-tions between carbohydrates and, for example, proteins on a theoretical level.
Described in more detail here are the methods used in this thesis to deter-mine the structures of bacterial polysaccharides and conformational prefer-ences of rhamnose derivatives.
3.1 Sugar analysis
The structural studies of polysaccharides or LPS usually start with a wet chemistry sugar analysis to elucidate the monosaccharide identities and their respective absolute configuration (D/L). The polysaccharide is hydrolyzed with a strong acid, e.g. trifluoroacetic acid (TFA) at 100-120 °C. The hy-drolysis procedure is usually straightforward, but must sometimes be opti-mized depending on the tendency of the released monosaccharide units to degrade or not even hydrolyze from the polysaccharides in the first place. A

22
novel approach is to use microwave dielectric heating.33 This method some-times gives cleaner reactions. The hydrolysis is followed by reduction of the anomeric centres using sodium borohydride and subsequent acetylation re-sulting in alditol acetates ready for gas liquid chromatography (GLC) analy-sis.34 The retention times of the alditol acetates are compared to those of naturally occuring or synthetic standards giving the identity of the monosac-charide units. However, if the polysaccharide contains uronic acid(s) it will be the subject of methanolysis after the hydrolysis. An O-methyl glycoside is formed, the acid function is transformed into an ester group and the sugar derivative can then be acetylated and analyzed by GLC.
The absolute configuration of the unique monosaccharide units can be de-termined by butanolysis of the hydrolysed PS followed by acetylation and GLC analysis.35 When using (+)-2-butanol, an optically active alcohol, the enantiomeric sugars react with the chiral alcohol and form diastereomeric glycosides that have different physical properties, and can therefore be sepa-rated by GLC. Also here standards are used as retention time references.
3.2 NMR spectroscopy analysis
A very powerful method for analysis, and the method of choice in this thesis, is NMR spectroscopy.30 It is a non-destructive method providing a lot of information in a reasonable amount of time. Its major drawbacks are the low sensitivity (when compared to e.g. MS) and the somewhat expensive equip-ment. NMR spectroscopy theory is based on the principle of nuclear spin, an intrinsic angular momentum every magnetic nucleus possesses. When a molecule is placed in a magnetic field, this momentum is aligned in either the same direction as or the opposite direction to the field, resulting in two states separated by an energy difference, ΔE. The energy gap ΔE, and there-fore the resonance frequency, also depends on the chemical environment of the nucleus in a molecule, an effect known as the chemical shift. As the resonance frequency of a certain nucleus is dependent on the magnetic field strength, a recalculation to the chemical shift (in parts per million, ppm) is performed. Most elements have at least one naturally occurring NMR active isotope, unfortunately it is not always the most common one. The natural abundance of 1H is close to 100% and it is therefore easily detected by NMR spectroscopy. The detectable 13C and 15N isotopes are less abundant, only 1.1% and 0.4%, respectively, leading to longer experiment times.
Magnetic nuclei are affected by the surroundings and especially by the clos-est other magnetic nuclei resulting in spin-spin or J couplings. This feature leads to split resonances that can reveal how many neighboring protons a

23
certain proton has, and also whether another NMR active nucleus, like 19F, is attached to the molecule. The coupling pattern can complicate the spectrum and therefore decoupling is often applied. The magnitude of the J-couplings are not dependent on the magnetic field strength but can be more easily ex-tracted at higher field strength due to better resolution.
The use of spin-simulation36 can be very helpful when assigning chemical shifts and coupling constants in a molecule. The problem with “hard cou-pling” and overlapping peaks is overcome and chemical shifts can be as-signed with higher accuracy. In the process an experimental spectrum is fitted iteratively to a calculated spectrum using an appropriate spin simula-tion software, such as PERCH.37,38 When the two spectra appear exactly the same, the chemicals shifts and coupling constants are obtained.
The samples of oligo- and polysaccharides are often prepared in D2O or a mixture of H2O and D2O if exchangeable protons are to be observed. As an internal chemical shift reference TSP (δH = 0.0 ppm) is often used for 1H-NMR. The 13C chemical shifts are commonly referenced to external 1,4-dioxane in D2O (δC = 67.40 ppm).39
3.2.1 Methods of structural investigation
A structural investigation of a polysaccharide often starts with sugar analy-sis. When the identities of the monosaccharide units have been revealed, it is time to determine their relative amounts, how many sugars are present in the repeating unit, their connectivities, if the chain is branched etc. A 1H NMR spectrum of the PS gives information about the number of monosaccharide units in the repeating unit by counting the resonances in the anomeric region (4.4-5.5 ppm). The common hexoses show up here as well as in 13C NMR spectra (95-110 ppm). If there are resonances just downfield 1 ppm in the 1H NMR spectrum it is a sign of CH3-groups of e.g. a fucose or a rhamnose residue. Resonances close to 2 ppm reveal N-acetyl and/or O-acetyl func-tionalities. From the splitting of the anomeric peaks in 1H spectra (JH1,H2) the anomeric configuration can be established; a J-coupling of ~4 Hz indicates the α-configuration and a value of ~8 Hz indicates the β-form for common monosaccharides like D-glucopyranose and D-galactopyranose. The corre-sponding values of JC1,H1 are ~170-175 Hz for the α-form and ~160-165 Hz for β-form obtained from a coupled 13C NMR experiment.40
Since most polysaccharide NMR spectra suffer from peak overlap in the ring region (δH 3.1-4.4), 2D NMR techniques are often applied. The proton chemical shifts are linked to their respective carbon by a 1H,13C-HSQC NMR experiment41 or, when the resonances in the 13C dimension overlap too

24
much, a 13C,1H-HETCOR NMR experiment42. To distinguish methylene protons (CH2) from methyl (CH3) and methine (CH), a multiplicity-edited 1H,13C-HSQC experiment43 can be recorded. Methylene protons then have opposite phase and are often plotted in a different color (see Figure 6.2). The protons in each spin system can be assigned using 1H,1H-TOCSY and/or 1H,1H-DQF-COSY experiments, both allowing the magnetization to travel over bonds with the help of J-couplings and thereby connecting the pro-tons.44 The longer the mixing time in a 1H,1H-TOCSY experiment, the fur-ther away the magnetization can travel, if the J-couplings are large enough that is. Polysaccharides contain not only protons and carbons but sometimes also nitrogen and even phosphorous.45 Their respective chemicals shifts can be assigned and correlated to 1H using e.g. 1H,15N-HSQC and 1H,31P-TOCSY46 experiments. The exchangeable amide protons, measured in a H2O-D2O mixture, have J-couplings to the ring protons and can be appointed correctly in the ring by a 1H,1H-TOCSY with water suppression.
Once the separate monosaccharide units have been assigned, the substitution pattern within the repeating unit is established using 1H,1H-NOESY NMR experiments,47 connecting protons close in space, and a 1H,13C-HMBC NMR experiment,48 showing long-range couplings over the glycosidic linkage (Figure 3.1). By comparing the chemical shifts of the carbons in the PS with the respective carbons in the unique monosaccharides the glycosylation shifts (ΔδC) are revealed. They are usually in the range of 2-10 ppm and indicate connection points.
OHOHO
HO
OH
OHO OH
OH
OH
C1'
H1'
O4 C4
H4
Figure 3.1: Correlations over the glycosidic linkage, here demonstrated on cello-biose, can be measured with 1H,1H-NOESY (dashed line) and/or 1H,13C-HMBC (solid line) NMR experiments.

25
3.2.2 Methods of conformational analysis
Not only is the identity of the carbohydrate components in a polysaccharide important for its interaction with for example a protein but also its three-dimensional structure. The polysaccharide fits into the active site of a protein like a “key in a lock” and it must be folded, bended or rotated in the correct way for the protein to recognize and modify the carbohydrate. To determine the three-dimensional structure of a polysaccharide, or a part of it, the rela-tive conformations around the glycosidic linkages between the monosaccha-ridee units are analyzed to determine the most populated torsion angles. The torsion angles φ (phi) and ψ (psi) are originally defined by IUPAC49 with heavy atoms but for carbohydrate NMR spectroscopists it is more conven-ient to use nuclei detectable by NMR.50 Hence, the torsional angles around the glycosidic linkage, φH and ψH, are defined as (Figure 3.2):
φH = H1'-C1'-O4-C4
ψH = C1'-O4-C4-H4
φφH ψH
OHOHO
HO
OH
OHO OH
OH
OH
C1'
H1'
O4 C4
H4
Figure 3.2: Schematic picture of the torsion angles φH and ψH demonstrated in cel-lobiose.
The value of φ is influenced by steric effects and electronic effects, the main one being the exo-anomeric effect.14 The ψ torsion angle is mainly influ-enced by steric effects.
The torsional angles can not be measured directly with any NMR experi-ments, but since they are related to coupling constants through Karplus rela-tionships, NMR spectroscopy is a very valuable technique for conforma-tional studies. In 1959, Karplus formulated an equation (Equation 1) that relates coupling constants to torsion angles51 and since then, many variants of this equation have been presented.52,53
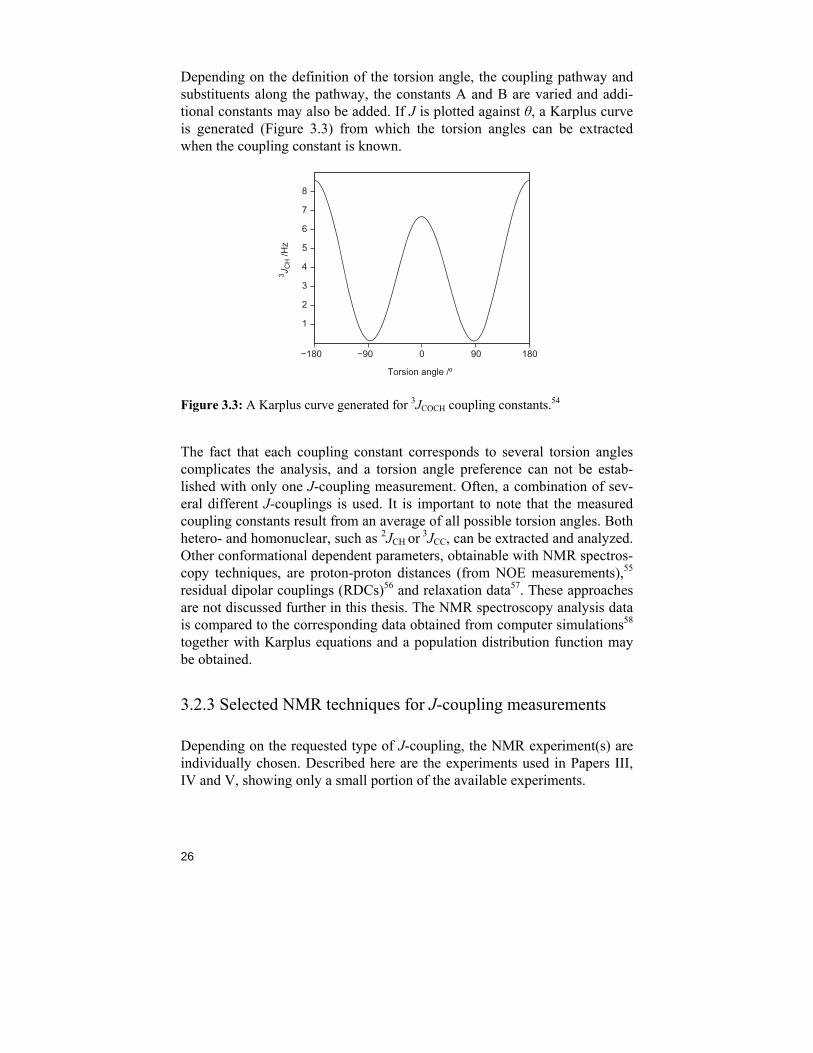
J = A cos2 θ − B (Equation 1)
26
Depending on the definition of the torsion angle, the coupling pathway and substituents along the pathway, the constants A and B are varied and addi-tional constants may also be added. If J is plotted against θ, a Karplus curve is generated (Figure 3.3) from which the torsion angles can be extracted when the coupling constant is known.
−180 −90 0 90 180
1
2
3
4
5
6
7
8
Torsion angle /º
3 J CH /H
z
Figure 3.3: A Karplus curve generated for 3JCOCH coupling constants.54
The fact that each coupling constant corresponds to several torsion angles complicates the analysis, and a torsion angle preference can not be estab-lished with only one J-coupling measurement. Often, a combination of sev-eral different J-couplings is used. It is important to note that the measured coupling constants result from an average of all possible torsion angles. Both hetero- and homonuclear, such as 2JCH or 3JCC, can be extracted and analyzed. Other conformational dependent parameters, obtainable with NMR spectros-copy techniques, are proton-proton distances (from NOE measurements),55 residual dipolar couplings (RDCs)56 and relaxation data57. These approaches are not discussed further in this thesis. The NMR spectroscopy analysis data is compared to the corresponding data obtained from computer simulations58 together with Karplus equations and a population distribution function may be obtained.
3.2.3 Selected NMR techniques for J-coupling measurements
Depending on the requested type of J-coupling, the NMR experiment(s) are individually chosen. Described here are the experiments used in Papers III, IV and V, showing only a small portion of the available experiments.

27
For measuring 3JCH coupling constants, a proton-detected long-range ex-periment with one or several site-selective 13C-pulse(s) followed by Ha-damard transformation (1DLR) is suitable.59 The resulting spectrum gives doublets with the 3JHH-couplings in phase and 3JCH-couplings in anti-phase (Figure 5.3). If needed, a band-selective homo-decoupling during the acqui-sition can be applied to simplify the coupling pattern. To extract the long-range coupling constants from the spectrum in a more accurate fashion, a J-doubling procedure can be used.60
Another long-range experiment is the 2D 1H,13C-J-HMBC where a low-pass J-filter is used to suppress the 1JCH couplings.61 Due to low resolution in the indirect (F1) dimension a scale factor (κ) is used to scale the coupling(s). Both 2JCH and 3JCH coupling constants can be obtained.
Heteronuclear nJCH coupling constants can also be measured by a heteronu-clear 2D HSQC-TOCSY-type experiment named 1H,13C-HSQC-HECADE, reported by Kósminskí and co-workers in 1997.62 From the direct dimension (1H), which has better resolution, the long-range JCH coupling constants are extracted. In the indirect dimension (X-nucleus), the cross-peaks are split by 1JCH multiplied by a factor (S), giving the opportunity to alter the separation to prevent e.g. resonance overlap (Figure 3.4).
3.533.553.57
91.5
92.5
93.5
94.5
1H /ppm
13C
/ppm
1 JC
H x
S
2JCH
Figure 3.4: Selected part of a 1H,13C-HSQC-HECADE NMR experiment of [1-13C]-D-glucose demonstrating the cross-peak H2-C1 in α-D-Glcp. In the indirect dimen-sion (13C) the 1JCH coupling constant multiplied by a factor (S) can be measured; here S = 1. The cross-peak is tilted to the left (dashed line) and the 2JCH coupling constant can be extracted in the 1H dimension.

28
The sign of a coupling constant is generally thought to depend on the num-ber of bonds involved in the coupling pathway. Geminal (two-bond) cou-plings are said to be negative and vicinal (three-bond) couplings are posi-tive.63 In reality, however, there are studies that have proven that this is not always the case. In particular, 2JC,H in sugars can have both negative and positive values and the sign might change for different 2J-couplings even within a monosaccharide residue.64,65 The HSQC-HECADE experiment en-ables the determination of the relative signs of the J-couplings through a tilting of the cross-peaks in the direct dimension. The tilting of the peaks is compared to the tilting of the cross-peaks in a reference compound, and thus the sign is found.66
Not only heteronuclear coupling constants are used for conformational stud-ies but also homonuclear JCC couplings.67 They can be extracted from 13C detected experiments like ordinary 1D NMR experiments with proton de-coupling. The measurements are facilitated by 13C labeling at key positions.

29
Chapter 4: Structural studies of bacterial polysaccharides (Papers I & II)
4.1 Introduction
Escherichia coli is a Gram-negative bacterial species commonly found in the colonial flora of both animal and man. In the intestinal environment, the bacteria exist in symbiosis with their host and rarely cause diseases. There are however strains that have acquired different virulence genes and there-fore become pathogenic. These strains have evolved specific characteristics that allow them to invade and colonize sites they do not normally inhabit, which causes different forms of abnormal behavior of the host. Three gen-eral syndromes are resulted from an infection of pathogenic E. coli: enteric diarrhoeal disease, urinary tract infections and sepsis/meningitis. Among the intestinal pathogens there are different categories (pathotypes), e.g. EPEC (enteropathogenic E. coli) and EIEC (enteroinvasive E. coli) differentiated on the mechanism by which they cause the infection.68 The various patho-types are designated as O:K:H serotypes, where O is the O-antigen (from the LPS), K is the CPS and H is the flagella antigen. Up to this date around 180 different O-antigens and over 100 capsular polysaccharides have been identi-fied.69 EIEC are both genetically and pathogenically related to Shigella spp. and several studies have shown cross-reactivity between E. coli serotypes and Shigella serotypes. 70,71 There is even a study suggesting Shigella should be classified within the E. coli species.72 Considering this it is not a big sur-prise that Paper I suggests the identical O-antigen structures for E. coli O124, an EIEC, and S. dysenteriae type 3. Shigella bacteria cause diseases like diarrhea and dysentery and like E. coli continue to threaten public health mainly in the developing world due to poor sanitation. Shigella strains are grouped into four subgroups, S. dysenteriae, S. flexneri, S. boydii and S. son-nei, and serogrouped based only on O-antigen since they lack the H and K antigens.73

30
4.2 Structural investigation of the O-antigenic polysaccharides from E. coli O124 and S. dysenteriae type 3 (Paper I)
The reported cross-reactivities between E. coli O164, E. coli O124 and S. dysenteriae type 3 indicate structural similarities in their respective O-antigen polysaccharides, both regarding their carbohydrate content and ap-parent three-dimensional structure.70,71 In studies from 1976 and 1977, Dmitriev and co-workers74,75 presented structural investigations of the O-antigens of E. coli O124 and S. dysenteriae type 3 containing structural in-consistencies, and compared to E. coli O16476 they seem to lack one key feature for the same 3-dimensional structure; the same anomeric configura-tion of the monosaccharide units. Therefore, a reinvestigation of the O-antigens of E. coli O124 and S. dysenteriae type 3 was performed.
From the investigation of E. coli O164, it was concluded that structural elu-cidation by NMR spectroscopy can be performed directly on the LPS. This is due to the fact that the main part of the LPS is the O-antigen; the contribu-tion from these resonances will be much bigger than from the resonances of the core and lipid A parts. There was no reason to doubt the identity of the reported monosaccharide contents of E. coli O124 and S. dysenteriae type 3 and consequently the O-antigens should consist of pentasaccharide units having D-Glc, 4-O-[(R)-1-carboxyethyl]-D-Glc, D-Gal and D-GalNAc as components.
A 1H NMR spectrum with five resonances in the anomeric region confirmed the presence of five monosaccharide residues. From a HSQC spectrum (Fig-ure 4.1) together with TOCSY spectra (different mixing times), the reso-nances of 1H and 13C were assigned to the residues and presented in Table 1, Paper I. Both LPSs were compared to each other with HSQC spectra and the resonances therein were, in principle, superimposable.

31
13C
/ppm
1H /ppm3.54.04.55.0
6065707580859095
100105110
55
Figure 4.1: Selected part of a 1H,13C-HSQC NMR spectrum of the LPS from S. dysenteriae type 3 in D2O at 45 °C.
The monosaccharide residues were denoted A-E in order of decreasing chemical shift of the anomeric proton resonances. The JH1,H2 and JH1,C1 cou-pling constants gave the anomeric configurations of the monosaccharide units. The spin system of A could be assigned to a →6)-β-D-Galf-(1→ resi-due due to the high chemical shift of C1 and low JH1,H2 value as well as a large glycosylation shift of C6. Residue B had a JH1,H2 coupling constant of 3.7 Hz, indicating an α-configuration, which was confirmed by the JH1,C1 coupling constant of 173 Hz. The spin system was assigned to an α-D-Glcp residue substituted at C6 due to the larger glycosylation shift, ΔδC 7.0. The third monosaccharide unit, C, revealed a galacto-configuration since the magnetization transfer in TOCSY NMR spectra stopped at H4 due to a small JH4,H5 coupling constant (<1 Hz). The low chemical shift of C2, 52.3 ppm, indicates an N-acetyl substitution and a JH1,H2 of 8.0 Hz leads the interpreta-tion to β-D-GalpNAc. The residue is glycosylated at C3, ΔδC3 7.3. Residue D is also a β-galacto-sugar, JH1,H2 7.7 Hz. It is disubstituted, ΔδC3 7.7 and ΔδC4 6.5 which means the repeating unit is branched. The monosaccharide is as-signed to →3,4)-β-D-Galp-(1→. Finally, residue E is the unusual 4-O-[(R)-1-carboxyethyl]-β-D-Glcp-(1→, identified and synthesized by Kochetkov and co-workers in 1977.77 In the HMBC spectrum, a cross-peak was ob-served between C1’ and H4 of residue E, thereby confirming the presence and location of the 1-carboxyethyl substituent in residue E. The chemical shift differences of the monosaccharide unit showed no further divergence, placing this residue at the end of the side-chain.

32
1H /ppm
13C
/ppm
4.44.54.64.74.84.95.05.1
707274767880828486
68
3.43.63.84.04.2
10199 D4 B
A
C3
B C
D3
A6
B6
D E
Figure 4.2: Selected sections of the 1H,13C-HMBC NMR spectrum of the LPS from E. coli O124 in D2O at 45 °C. Key correlations over the glycosidic linkages are denoted.
Table 4.1: Inter-residue correlations observed in the 1H,13C-HMBC NMR spectrum from the O-antigen part of the LPS from S. dysenteriae type 3. Residue Anomeric
atom Residue Atom
An H-1 C C-3 An C-1 C H-3 Ba H-1 D C-4 Bn C-1 D H-4 Cn H-1 D C-3 Ca C-1 D H-3 Da H-1 A C-6 En H-1 B C-6
a Additional correlations found in the 1H,13C-HMBC NMR spectrum from the O-antigen part of the LPS from E. coli O124.
The sequence of the monosaccharide units was determined from a HMBC experiment (Table 4.1 and Figure 4.2). Starting from the terminal residue of the side-chain, there is a cross-peak from E to C6 in residue B which in turn is connected to C4 in saccharide D. Also, residue C has a glycosidic linkage to D, but to C3, that is, the β-D-Galp confirming this residue to be the branching point of the chain. Residue D is linked to C6 in A, and residue A is connected to C through position C3 which leads to the following repeating unit: D A C →3)-β-D-Galp-(1→6)-β-D-Galf-(1→3)-β-D-GalpNAc-(1→ ||4-O-(R)-1-carboxyethyl-β-D-Glcp-(1→6)-α-D-Glcp-(1→4) E B

33
In a review from 2006,78 it is suggested that when there is only one β-D-GalpNAc residue in the repeating unit, it should be found at the reducing end of the biological repeating unit, as depicted above. In conclusion, the O-antigens from E. coli O124 and S. dysenteriae type 3 are identical to that from E. coli O164, but for the 4-O-[(R)-1-carboxyethyl]-substituent of the terminal sugar residue of the side-chain in the polymer, thereby making the reported cross-reactivity understandable. The reinvestigation resulted in new assignments of the anomeric configurations in the monosaccharide units.
4.3 Structural investigation of the O-antigen of E. coli O74 (Paper II)
E. coli O74:K-:H39 is a strain that belongs to the Sa-like toxin producing E. coli (STEC), most often found in cattle but pathogenic to human.79 Cross-reactivity has been reported between E. coli O74 and E. coli O2 as well as E. coli K45,80 suggesting structural similarities in their respective O-antigens. In a study from 1967, it was suggested that they all contain the uncommon sugar N-acetyl-3-amino-3,6-D-dideoxygalactose (D-Fuc3NAc) only found in a few other species.81 The main challenges in this structural study were to elaborate the O-antigen structure and the identity of this sugar residue on a small amount of material. Only 2 mg of the polysaccharide was available.
The primary structure of the polysaccharide O-antigen was analyzed with NMR spectroscopy (both 1D and 2D experiments) and sugar analysis. Sugar analysis revealed 2-deoxy-2-aminoglucose and 2-amino-3,6-deoxy-galactose. The galacturonic acid residue was found using methanolysis under acidic conditions followed by GLC analysis as an acetylated methyl ester derivative. Determination of the absolute configuration of the main compo-nents showed they all have the D-configuration.
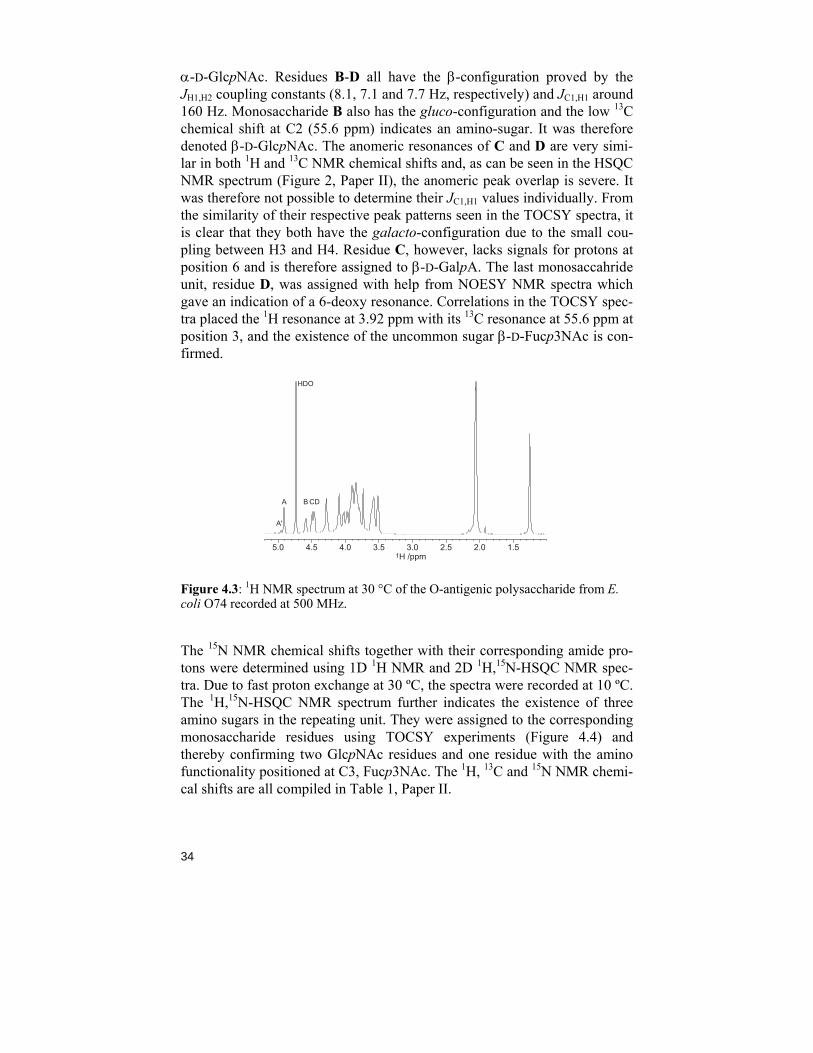
A 1H NMR spectrum (Figure 4.3) of the polysaccharide O-antigen revealed four resonances, 4.91, 4.59, 4.49 and 4.46 ppm, in the anomeric region, de-noted A-D with decreasing chemical shifts. The resonance at 1.25 ppm (3H) indicated a 6-deoxy carbohydrate and the three resonances just downfield 2 ppm (9H) serve as evidence for three N-acetylated carbohydrates. The HSQC NMR spectrum of the polysaccharide (Figure 2, paper II) shows four resonances in the anomeric region pointing towards as many hexopyranosyl residues. The H1 resonance in residue A has a JH1,H2 coupling constant of 3.8 Hz and a JC1,H1 coupling constant of 173 Hz and the α-configuration is con-cluded. From the analysis of TOCSY spectra, it is clear that residue A has the gluco-configuration and the 1H/13C resonances at 4.03/53.1 ppm indicate
34
α-D-GlcpNAc. Residues B-D all have the β-configuration proved by the JH1,H2 coupling constants (8.1, 7.1 and 7.7 Hz, respectively) and JC1,H1 around 160 Hz. Monosaccharide B also has the gluco-configuration and the low 13C chemical shift at C2 (55.6 ppm) indicates an amino-sugar. It was therefore denoted β-D-GlcpNAc. The anomeric resonances of C and D are very simi-lar in both 1H and 13C NMR chemical shifts and, as can be seen in the HSQC NMR spectrum (Figure 2, Paper II), the anomeric peak overlap is severe. It was therefore not possible to determine their JC1,H1 values individually. From the similarity of their respective peak patterns seen in the TOCSY spectra, it is clear that they both have the galacto-configuration due to the small cou-pling between H3 and H4. Residue C, however, lacks signals for protons at position 6 and is therefore assigned to β-D-GalpA. The last monosaccahride unit, residue D, was assigned with help from NOESY NMR spectra which gave an indication of a 6-deoxy resonance. Correlations in the TOCSY spec-tra placed the 1H resonance at 3.92 ppm with its 13C resonance at 55.6 ppm at position 3, and the existence of the uncommon sugar β-D-Fucp3NAc is con-firmed.
1H /ppm
HDO
1.52.02.53.03.54.04.55.0
A B CD
A'
Figure 4.3: 1H NMR spectrum at 30 °C of the O-antigenic polysaccharide from E. coli O74 recorded at 500 MHz.
The 15N NMR chemical shifts together with their corresponding amide pro-tons were determined using 1D 1H NMR and 2D 1H,15N-HSQC NMR spec-tra. Due to fast proton exchange at 30 ºC, the spectra were recorded at 10 ºC. The 1H,15N-HSQC NMR spectrum further indicates the existence of three amino sugars in the repeating unit. They were assigned to the corresponding monosaccharide residues using TOCSY experiments (Figure 4.4) and thereby confirming two GlcpNAc residues and one residue with the amino functionality positioned at C3, Fucp3NAc. The 1H, 13C and 15N NMR chemi-cal shifts are all compiled in Table 1, Paper II.

35
3.5
4.0
8.08.18.28.38.48.5
4.5
1H /ppm
1 H /p
pm
B D A A'
H1
H2 H3
H1
H2
Figure 4.4: Selected part from a 1H,1H-TOCSY spectrum (τmix 60 ms) of the O-antigenic polysaccharide from E. coli O74 recorded in H2O/D2O at 10 °C. Correla-tions from the amide protons into the ring protons are denoted.
A band-selective constant-time 1H,13C-HMBC spectrum was recorded with a selective pulse centered around the carbonyl signals. This spectrum lends further evidence of the two GlcpNAc residues with correlations from a car-bonyl resonance at 175.5 ppm to 4.03 (A2) and 3.90 (B2) ppm, respectively. The resonance at 175.2 ppm has a correlation to a peak at 3.92 ppm, con-firming the N-acetyl group placement at D3. Finally, the existence of a uronic acid was proven by a correlation from 174.0 ppm, which is also a broad peak in the 13C NMR spectrum, to 4.09 ppm, H5 in residue C.
Due to the rareness of D-Fucp3NAc and the fact that it had not yet been as-signed with NMR chemical shifts, it was synthesized and assigned in the same way as the polysaccharide. Starting from ethyl 3-amino-2,4-di-O-benzyl-3-deoxy-1-thio-β-D-fucopyranoside, the amine functionality was first acetylated with acetic anhydride. The thioethyl group at the anomeric posi-tion was then removed using NIS as promoter and H2O as nucleophile, yield-ing the product in two anomeric configurations, α and β. Finally, the benzyl groups were removed by catalytic hydrogenation using Pd(OH)2/C and H2. The final product was purified first on a P2 size-exclusion gel plug followed by a 20 mm C-18 Sep-Pak® column eluted with H2O resulting in N-acetyl 3-deoxy-3-amino-D-fucopyranoside (D-Fucp3NAc).
The sequence of the residues in the repeating unit was determined by analyz-ing NOESY and HMBC experiments; a summary of the connectivities is displayed in Table 1, Paper II. The anomeric resonances, both 1H and 13C, of residue A shows a three-bond heteronuclear correlation to C4 in both dimen-sions and in turn has glycosylation shifts at C3 of ΔδC 9.6 ppm and at C6 of ΔδC 7.3 ppm. Its substitution by residue D at position C3 is verified by corre-lations in both NOESY and HMBC spectra. Linked to A6 is residue B,

36
proven by a cross-peak in HMBC at 4.59/69.1 ppm. Together with the analy-sis of the individual sugar units, the repeating unit of the O-antigen of O74 is determined to be:
A C B →6)-α-D-GlcpNAc-(1→4)-β-D-GalpA-(1→3)-β-D-GlcpNAc-(1→ || β-D-Fucp3NAc-(1→3) D
Present in the 1H NMR spectrum is a resonance at 4.95 ppm, denoted A' in figure 4.3. This resonance, together with its spin system, was analyzed with HSQC and TOCSY spectra to give →3)-α-D-GlcpNAc-(1→, the second last residue of the polysaccharide chain. From this it is concluded that the above depicted repeating unit is also the biological repeating unit, having →3)-β-D-GlcpNAc-(1→ at its reducing end.78 The repeating unit is identical to that found in the capsular antigen of E. coli serotype K4582 and their cross-reactivity is explained. The not so common monosaccharide N-acetyl-3-amino-3,6-dideoxy-D-galactopyranoside (D-Fucp3NAc) had also been found e.g. in the O-antigen of E. coli O2 but in the α-configuration.83 As a β-anomer it has been found, according to the Bacterial Carbohydrate Structure DataBase,84 only in the O-antigen of Proteus vulgaris O45 as a 4-O-acetylated derivative.85

37
Chapter 5: Conformational analysis of rhamnose-containing saccharides (Paper III, IV & V)
The major constituent of the O-antigen polysaccharide from e.g. Sella flex-neri86 and some plant-associated bacteria54,87 is L-rhamnose, typically found as α-(1→2)- or α-(1→3)-linked fragments joined to other L-rhamnose units. The LPS of the bacteria, in which the O-antigen is found, has a key function in the interaction between bacteria and plant, either in symbiosis or pathogenesis. Due to multiple glycosidic linkages, conformational studies on polysaccharides involve many degrees of freedom. It is therefore more con-venient to use a smaller fragment as a model system. The disaccharide α-L-Rhap-(1→2)-α-L-Rhap-OMe (1) was chosen as a model for these polysac-charides and its conformational preferences around the glycosidic linkage was studied with respect to the torsional angles φ and ψ (Figure 5.1).
O
O
O
Me
OH
OMe
OHO
HO
HOMe
φφ2 ψ2
OOH
OHHO
Me
ψ3
φ3
O
O
O
Me
OH
OMe
OHHO
HO
HOMe
φ ψ
(1) (2)
Figure 5.1: Schematic picture of α-L-Rhap-(1→2)-α-L-Rhap-OMe (1) and α-L-Rhap-(1→2)[α-L-Rhap-(1→3)]-α-L-Rhap-OMe (2) with torsion angles indicated.
The trisaccharide α-L-Rhap-(1→2)[α-L-Rhap-(1→3)]-α-L-Rhap-OMe (2) (Figure 5.1) is not found in the above mentioned polysaccharide but since it has both α-(1→2)- and α-(1→3)-linkages it is a suitable model for a larger oligosaccharide. It can also be seen as a branched carbohydrate chain inter-

38
esting for studies of possible simultaneous movements of the rhamnose resi-dues.
Traditionally, conformational analysis with NMR spectroscopy is based on measurements of nuclear Overhauser effects (NOE) and transglycosidic het-eronuclear coupling constants.50 The development of new Karplus relation-ships for additional coupling constants88 led us to extend the experimental methods used for conformational studies. In the study of 1, the measure-ments of 3JCH, 2JCH and 3JCC coupling constants were combined with molecu-lar dynamics (MD) simulations to generate a picture of the conformational preferences. The conformational preferences for 2 were investigated by measurements of similar coupling constants. For both molecules, 13C site-specific labeling facilitated the measurements of JCC coupling constants, and for 2 it also resolved a problem of spectral overlap in 1H NMR spectra.
X-ray crystallography is also a method of choice for conformational studies. However, it requires crystals of adequate quality, which can be a challenge to produce. Usually, it is easier to crystallize molecules soluble in organic media, as the case of 4, and a comparison of the conformation in crystal to the confirmation in solution was obtained (Section 5.2).
5.1 Synthesis of 13C-labeled rhamnopyranoside oligosaccharides
The synthetic strategies for both α-L-Rhap-(1→2)-α-L-Rhap-OMe (1) and α-L-Rhap-(1→2)[α-L-Rhap-(1→3)]-α-L-Rhap-OMe (2) were planned to minimize the number of steps on all levels due to the limited amount of 13C-labeled L-rhamnose. In 1998, Söderman et al. presented a procedure for the synthesis of 1.89 This pathway was adopted with one exception; only benzoyl esters were used as protecting groups to reduce the deprotection to one sin-gle step. The synthesis of 2 was presented in a paper by Eklund et al., but was also modified with respect to the protecting group strategy; solely ben-zoyl esters were used here as well. Described and discussed here are the synthesis procedures for the non-labeled oligosaccharides, but the same rou-tines were used and similar yields were obtained for the isotopologues [1'-13C]-α-L-Rhap-(1→2)-α-L-Rhap-OMe (1-c1'), [2'-13C]-α-L-Rhap-(1→2)-α-L-Rhap-OMe (1-c2') and [2'-13C]-α-L-Rhap-(1→2)[[2''-13C]-α-L-Rhap-(1→3)]-α-L-Rhap-OMe (2-c2).
Starting from compound 3,90 the acceptors were synthesized essentially as described by Norberg et. al. (Scheme 5.1).91 First, a benzoyl group was in-

39
troduced at C4 to protect this hydroxyl group. The product, 4, was isolated by crystallization from ethanol/pentane. The crystals were analyzed further by X-ray crystallography, see section 5.3 (Paper III). From compound 4, acceptor 5 was obtained by removing the isopropylidene group with aqueous acid under slight heating. It was used without further purification in the gly-cosylation with 7 to form trisaccharide 9. Selective protection of OH3 as a benzoyl ester was performed at low temperature, –40 °C, to prevent the axial OH2 from reacting. It is known that axial groups react slower than equatorial groups in the pyranose ring; a carbohydrate can therefore be regioselectively protected.18 A minor amount of by-product was formed but the main prod-uct, 6, was collected in 69 % yield over two steps to be used in the glycosy-lation with 7 to form disaccharide 8.
OBzOHO
OH
OMe
OHO
OMe
OO
OBzO
OMe
OO
OBzOBzO
OH
OMe
3 4 5 6
a b c
Scheme 5.1: Synthesis of methyl 3,4-di-O-benzoyl-α-L-rhamnopyranoside (6). a) BzCl, pyridine, 0 °C, 78% b) acetic acid (80%, aq.), 80 °C c) BzCl, pyri-dine/CH2Cl2, −40 °C, 69%.
The donor, 7, was synthesized according to a method developed by Ness and co-workers in 1951.92 Full protection of L-rhamnose with benzoyl esters was followed by introduction of a bromide at C1 (α-anomer, Scheme 5.2). The glycosylation reaction of 7 with acceptor 6 was performed with a two-fold excess of acceptor using silver (I) triflate as promoter. Standard glycosyla-tion procedure suggests an excess of donor because of its tendency to de-compose, especially if the conditions are not dry enough. Due to the limited amount of 13C-labeled L-rhamnose, an excess of acceptor was used instead to maximize the donor consumption. This argument was however not applica-ble to the glycosylation of 7 with acceptor 5 to form trisaccharide 9; a 3-fold excess of donor was needed to glycosylate both hydroxyl groups. Only the α-anomeric glycosides were formed in both reactions due to neighbouring group participation from the benzoyl ester at C2. The oligosaccharides were deprotected with sodium methoxide and purified by gel permeation chroma-tography with water as the eluent. The low yield of compound 2 is explained by a remarkable amount of isolated methyl α-L-Rhap-(1→2)[-α-L-Rhap-(1→3)]-4-O-benzoyl α-L-Rhap. Repeated treatment under basic conditions can increase the obtained amount of 2.

40
OHOHO
OH
OMeO
BzOBzO
OBz
Br OBzO
ROO
OMe
OBzO
BzOOBz
OHO
ROO
OMe
OHO
HOOH
7 8 R=Bz9 R=2,3,4-tri-O-benzoyl α-L-Rhap
1 R=H2 R=α-L-Rhap
a, b c or d e
Scheme 5.2: Synthetic pathway of compounds 1 and 2 starting from L-rhamnose. a) BzCl, pyridine, 60 °C b) HBr/ HOAc, r.t. 84% c) 6, AgTOf, CH2Cl2 (dry), −40 °C, 82% d) 5, AgTOf, CH2Cl2 (dry), −40 °C, 43% e) NaOMe/MeOH, r.t., 1 79%, 2 23%.
5.2 Conformational study of a rhamnose derivative (Paper III)
The intermediate compound 4, methyl 4-O-benzoyl-2,3-O-isopropylidene-α-L-Rhap (Figure 5.2), in the total synthesis of 1 and 2 was studied in order to examine its conformational preferences around the ester group at C4. In a study from González-Outeiriño and co-workers in 2004,93 it was concluded that acetyl ester groups of cyclic alcohols prefer a staggered conformation when no flanking groups are present. However, if the acetyl group is flanked by equatorial substituents, as in a sugar, the preferred conformation is nearly “eclipsed”. This means that the torsion angle θ1, corresponding to H4-C4-O4-C10 in 4 (Figure 5.2), is usually observed in a syn conformation (0-20°) and θ2 (C4-O4-C10-O10 in 4) has a preference for the cis conformation (0-20°). A Karplus-type relationship derived by González-Outeiriño was used (Equation 2).
3JCH = 3.1 cos2θ – 1.25 cos θ + 2.35 (Equation 2)
C4O
C10 O4O
O
OMe
O H4θθ2 θ1
Figure 5.2: Schematic picture of methyl 4-O-benzoyl-2,3-O-isopropylidene-α-L-Rhap 4 with θ1 and θ2 marked.
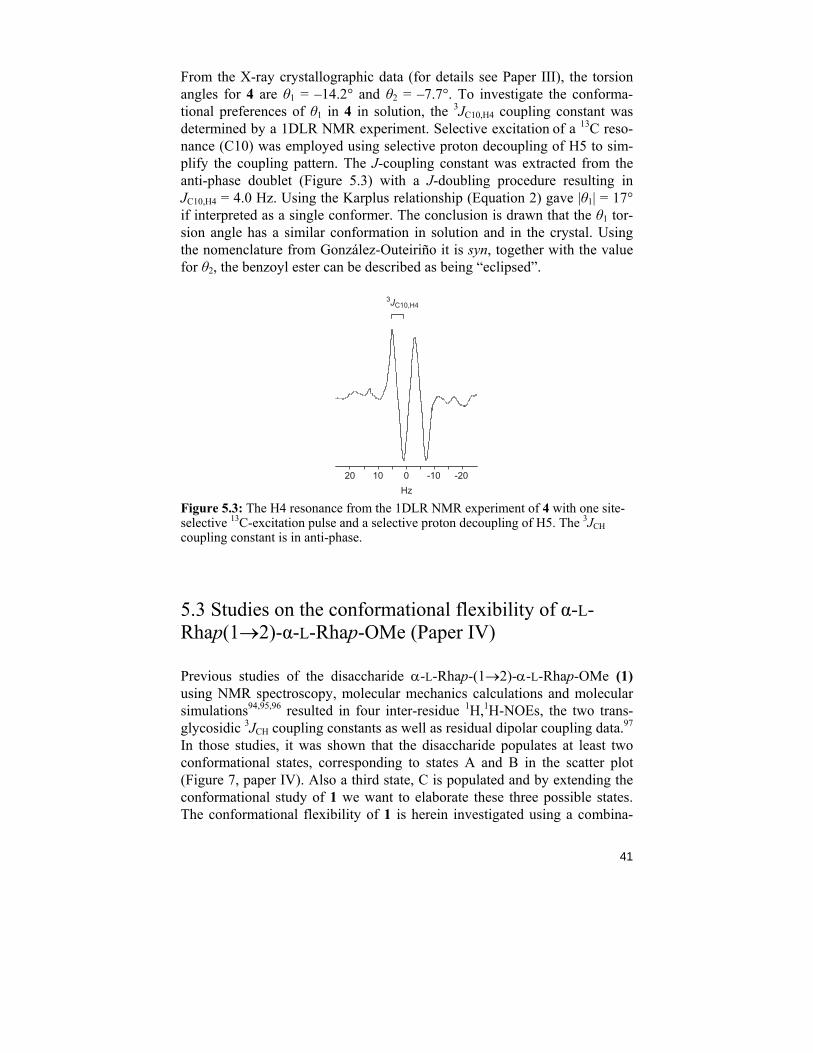
41
From the X-ray crystallographic data (for details see Paper III), the torsion angles for 4 are θ1 = –14.2° and θ2 = –7.7°. To investigate the conforma-tional preferences of θ1 in 4 in solution, the 3JC10,H4 coupling constant was determined by a 1DLR NMR experiment. Selective excitation of a 13C reso-nance (C10) was employed using selective proton decoupling of H5 to sim-plify the coupling pattern. The J-coupling constant was extracted from the anti-phase doublet (Figure 5.3) with a J-doubling procedure resulting in JC10,H4 = 4.0 Hz. Using the Karplus relationship (Equation 2) gave |θ1| = 17° if interpreted as a single conformer. The conclusion is drawn that the θ1 tor-sion angle has a similar conformation in solution and in the crystal. Using the nomenclature from González-Outeiriño it is syn, together with the value for θ2, the benzoyl ester can be described as being “eclipsed”.
Hz
3JC10,H4
01020 -20-10
Figure 5.3: The H4 resonance from the 1DLR NMR experiment of 4 with one site-selective 13C-excitation pulse and a selective proton decoupling of H5. The 3JCH coupling constant is in anti-phase.
5.3 Studies on the conformational flexibility of α-L-Rhap(1→2)-α-L-Rhap-OMe (Paper IV)
Previous studies of the disaccharide α-L-Rhap-(1→2)-α-L-Rhap-OMe (1) using NMR spectroscopy, molecular mechanics calculations and molecular simulations94,95,96 resulted in four inter-residue 1H,1H-NOEs, the two trans-glycosidic 3JCH coupling constants as well as residual dipolar coupling data.97 In those studies, it was shown that the disaccharide populates at least two conformational states, corresponding to states A and B in the scatter plot (Figure 7, paper IV). Also a third state, C is populated and by extending the conformational study of 1 we want to elaborate these three possible states. The conformational flexibility of 1 is herein investigated using a combina-

42
tion of nuclear magnetic resonance (NMR) spectroscopy techniques and computer simulations. In 1999, Serianni and co-workers presented new Kar-plus-type relationships for evaluating conformationally dependent 3JCC cou-pling constants over the glycosidic linkage related to both φ and ψ.54 To facilitate the measurements of JCC and nJCH, site-specific 13C-labeling was used. Two isotopologues to 1 were synthesized; [1'-13C]-α-L-Rhap-(1→2)-α-L-Rhap-OMe (1-c1') and [2'-13C]-α-L-Rhap-(1→2)-α-L-Rhap-OMe (1-c2').
Homonuclear carbon-carbon coupling constants were readily measured from 1D 13C NMR experiments with proton decoupling recorded for 1-c1' and 1-c2'. Sufficient resolution enhancement was applied for better interpretation. The coupling constants are compiled in Table 5.1. The carbon-carbon J-couplings are affected not only by the torsion angle but also by the disposi-tion of terminal electronegative substituents such as hydroxylgroups.54, 67 When the torsion angle is 180°, the atoms defining it create a plane. If one of the terminal carbons in this plane has a hydroxyl group as a substituent that lies in the plane, it is said to have an in-plane effect on the 3JCC related to this torsion angle. The coupling constant will be enhanced by approximately 0.6 Hz. For this reason, two different Karplus equations are used to interpret the measured 3JCC-values, one when in-plane effect occurs (Equation 3) and one where it does not (Equation 4).54
3JCC = 6.17 cos2 θ – 0.51 cos θ + 0.30 (Equation 3) [in-plane]
JC1',C3 = 4.96 cos2 ψ + 0.63 cos ψ – 0.01 (Equation 4)
The φC2-torsion angle is defined by C2'-C1'-O2-C2, and OH2' has an in-plane effect on the corresponding coupling constant. Because of this, Equa-tion 3 is used (Figure 5.4, solid line) to evaluate JC2',C2. The ψC-torsion angle is defined by two pathways, C1'-O2-C2-C1 and C1'-O2-C2-C3, where the latter does not experience an in-plane effect by any hydroxyl groups. For this reason Equation 4 is used to evaluate JC1',C3 (Figure 5.4, dotted line). The former torsion experiences an in-plane effect from OH1 and Equation 3 is used for interpretation of JC1',C1 (Figure 5.4, solid line).
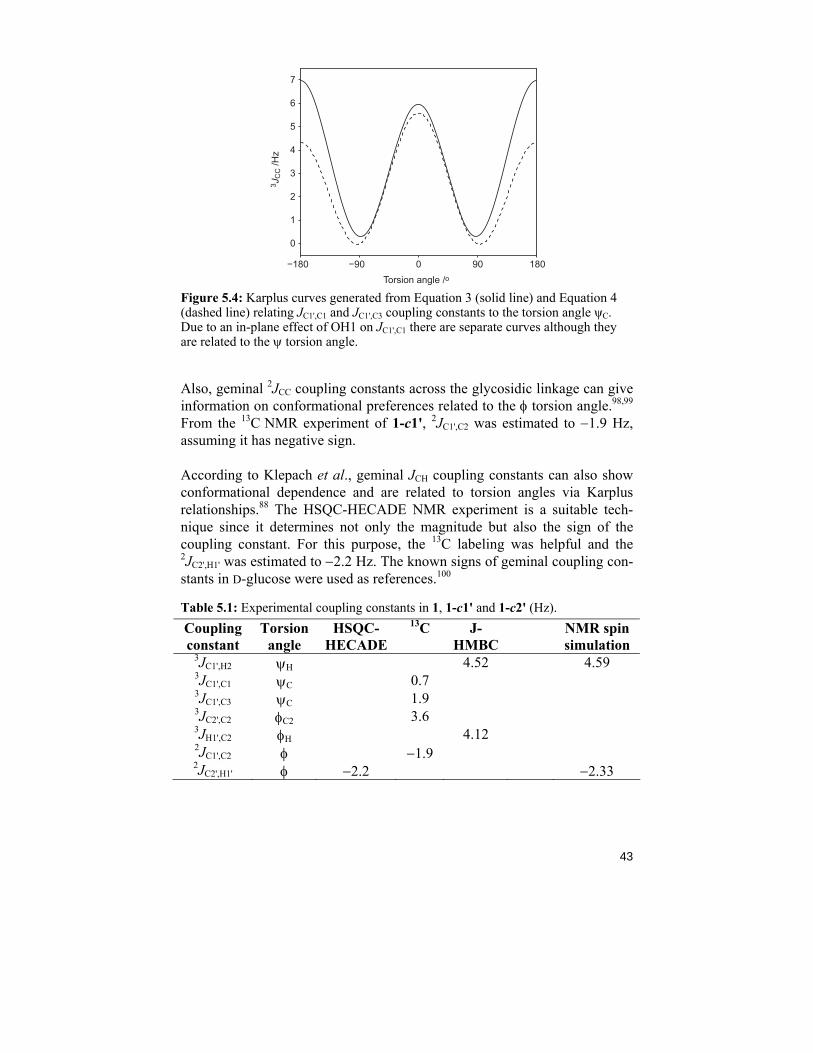
43
Torsion angle /o3 J
CC /H
z−180 −90 0 90 180
0
1
2
3
4
5
6
7
Figure 5.4: Karplus curves generated from Equation 3 (solid line) and Equation 4 (dashed line) relating JC1',C1 and JC1',C3 coupling constants to the torsion angle ψC. Due to an in-plane effect of OH1 on JC1',C1 there are separate curves although they are related to the ψ torsion angle.
Also, geminal 2JCC coupling constants across the glycosidic linkage can give information on conformational preferences related to the φ torsion angle.98,99 From the 13C NMR experiment of 1-c1', 2JC1',C2 was estimated to −1.9 Hz, assuming it has negative sign.
According to Klepach et al., geminal JCH coupling constants can also show conformational dependence and are related to torsion angles via Karplus relationships.88 The HSQC-HECADE NMR experiment is a suitable tech-nique since it determines not only the magnitude but also the sign of the coupling constant. For this purpose, the 13C labeling was helpful and the 2JC2',H1' was estimated to −2.2 Hz. The known signs of geminal coupling con-stants in D-glucose were used as references.100
Table 5.1: Experimental coupling constants in 1, 1-c1' and 1-c2' (Hz). Coupling constant
Torsion angle
HSQC-HECADE
13C J-HMBC
NMR spin simulation
3JC1',H2 ψH 4.52 4.59 3JC1',C1 ψC 0.7 3JC1',C3 ψC 1.9 3JC2',C2 φC2 3.6 3JH1',C2
φH 4.12 2JC1',C2 φ −1.9 2JC2',H1' φ −2.2 −2.33

44
In addition to the experimental values obtained directly from NMR spectra, the 1D spectra were also subjected to NMR spin simulation. 1H and 13C NMR chemical shifts and coupling constants were refined using the PERCH software, and a full NMR assignment at 37 °C was obtained (Table 2, Paper IV). By iterating a 1H NMR spectrum together with the corresponding cou-pled 13C spectrum of 1-c1' and 1-c2', respectively, the heteronuclear 3JC1',H2 and 2JC2',H1' coupling constants could be determined. The agreement between the different NMR interpretation methods is good and the result is seven coupling constants related to torsion angles φ and ψ that can be used for conformational analysis of 1.
MD-simulations of 1 resulted in trajectories from which the relative popula-tion of the three states (A, B and C) was calculated (Figure 7, Paper IV). Via the above-mentioned Karplus relationships, average spin-spin coupling con-stants were calculated and compared to those experimentally determined. The agreement is good but a refinement of the force field used in the MD simulations is needed to make a perfect match. The average conformation given by the glycosidic torsion angles in 1 was φ = 39° and ψ = –38°, shown in Figure 8, Paper IV.
5.4 NMR studies on of the trisaccharide α-L-Rhap-(1→2)[α-L-Rhap-(1→3)]-α-L-Rhap-OMe (Paper V)
The trisaccharide 2 can, because of vicinal substitution, be regarded as a branched oligosaccharide, with the main degrees of freedom around the two glycosidic linkages, Figure 5.1. In a study from 2005,101 it was concluded to be a highly flexible system with the conformational equilibrium mainly in the exo-anomeric state. A non-exo conformation was also populated to a minor extent and the two rhamnose branches (denoted with ' and '') move in a somewhat correlated fashion. The study was carried out with a combina-tion of computer simulations and NMR spectroscopy measurements like 1H,1H T-ROESY experiments, 13C relaxation measurements and heteronu-clear 3JCH coupling constants. However, due to spectral overlap in the 1H NMR spectrum, the 3JC1',H2 coupling constant could not be determined and a gap remained in the table of measured coupling constants.
The heteronuclear transglycosidic coupling constants were measured with the 1DLR NMR experiment described in section 3.2.3. Due to the H2' and H2 resonances appearing at almost the same chemical shift (ΔδH 0.004), there is a risk of disturbance from the 2JC1',H2' coupling constant when 3JC1',H2
45
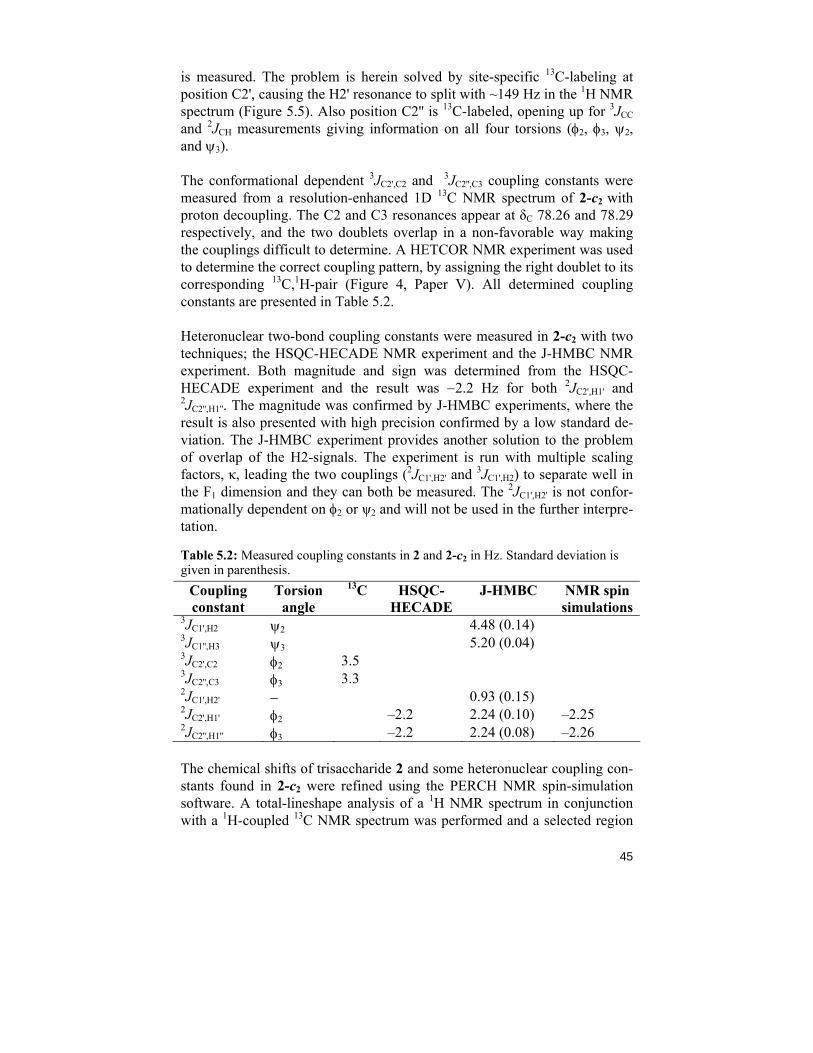
is measured. The problem is herein solved by site-specific 13C-labeling at position C2', causing the H2' resonance to split with ~149 Hz in the 1H NMR spectrum (Figure 5.5). Also position C2'' is 13C-labeled, opening up for 3JCC and 2JCH measurements giving information on all four torsions (φ2, φ3, ψ2, and ψ3).
The conformational dependent 3JC2',C2 and 3JC2'',C3 coupling constants were measured from a resolution-enhanced 1D 13C NMR spectrum of 2-c2 with proton decoupling. The C2 and C3 resonances appear at δC 78.26 and 78.29 respectively, and the two doublets overlap in a non-favorable way making the couplings difficult to determine. A HETCOR NMR experiment was used to determine the correct coupling pattern, by assigning the right doublet to its corresponding 13C,1H-pair (Figure 4, Paper V). All determined coupling constants are presented in Table 5.2.
Heteronuclear two-bond coupling constants were measured in 2-c2 with two techniques; the HSQC-HECADE NMR experiment and the J-HMBC NMR experiment. Both magnitude and sign was determined from the HSQC-HECADE experiment and the result was −2.2 Hz for both 2JC2',H1' and 2JC2'',H1''. The magnitude was confirmed by J-HMBC experiments, where the result is also presented with high precision confirmed by a low standard de-viation. The J-HMBC experiment provides another solution to the problem of overlap of the H2-signals. The experiment is run with multiple scaling factors, κ, leading the two couplings (2JC1',H2' and 3JC1',H2) to separate well in the F1 dimension and they can both be measured. The 2JC1',H2' is not confor-mationally dependent on φ2 or ψ2 and will not be used in the further interpre-tation.
Table 5.2: Measured coupling constants in 2 and 2-c2 in Hz. Standard deviation is given in parenthesis.
Coupling constant
Torsion angle
13C HSQC-HECADE
J-HMBC NMR spin simulations
3JC1',H2 ψ2 4.48 (0.14) 3JC1'',H3 ψ3 5.20 (0.04) 3JC2',C2 φ2 3.5 3JC2'',C3 φ3 3.3 2JC1',H2' − 0.93 (0.15) 2JC2',H1' φ2 –2.2 2.24 (0.10) –2.25 2JC2'',H1'' φ3 –2.2 2.24 (0.08) –2.26
The chemical shifts of trisaccharide 2 and some heteronuclear coupling con-stants found in 2-c2 were refined using the PERCH NMR spin-simulation software. A total-lineshape analysis of a 1H NMR spectrum in conjunction with a 1H-coupled 13C NMR spectrum was performed and a selected region

46
is presented in Figure 5.5. Shown in the picture are also the resonances of C2' and C2'' in 2-c2 giving a nice presentation of 13C resonances with dddd coupling patterns.
a
b
1H /ppm
4.2 4.1 4.0 3.9
H2
H2'H2''
H2''H3
H2'
71.7 70.571.5 70.313C /ppm
c C2' C2''
Figure 5.5: Selected regions from NMR spectra of the site-specifically 13C-labeled 2-c2, where the H2 resonances are denoted. Spin-simulation with the PERCH soft-ware resulted in a) iterated spectrum, b) experimental spectrum. c) Selected region from a 1H-coupled 13C NMR spectrum showing the two C2 resonances as dddd.
In conclusion, we have shown that by using site-specific 13C-labeling, four new spin-spin coupling constants can be obtained for trisaccharide 2 and a problem of spectral overlap can be resolved. By using additional NMR spec-troscopy techniques (J-HMBC), supplementary conformational dependent J-couplings were added to the list of parameters.

47
Chapter 6: Enzymatic synthesis using glycosynthases (Paper VI)
6.1 A short introduction to glycosynthase chemistry
In nature, carbohydrates are often found as polymers, like in starch and cel-lulose. These big molecules are synthesized and degraded by enzymes spe-cialized in forming and breaking glycosidic linkages. Among the groups of enzymes, one can find glycoside hydrolases (or glycosidases), a family of enzymes that hydrolyse glycosidic linkages via inverting or double-displacement retaining mechanisms. In the latter, the stereochemical out-come of the product is the same as for the starting sugar (Scheme 6.1). Some of the glycosidases also exhibit transglycosylation, a process that first hydro-lyses a glycosidic linkage and then attaches another carbohydrate residue to the substrate. The mechanism was first presented in 1953102 but refinements have recently been published.103,104
OHOHO O
OH
HOR
OO
H
O
O
OHOHO O
OH
HO R'
OO
H
O
O
OHOHO
OH
HO
OO
O
O
H
OR'-HOR
Scheme 6.1: Schematic overview of the double-displacement retaining mechanism, by which some glycoside hydrolases are operating. Scheme is adapted from Hult et al. (2003).104
First an enzyme-substrate complex is formed and the glycosidic linkage of the substrate (drawn in Scheme 6.1 in the β-anomeric configuration) is cleaved via protonation from the acid/base functionality, often a glutamic acid. Simultaneously the carbohydrate residue experiences an attack from a catalytic nucleophile and a covalent bond to the enzyme is formed (step 1 in Scheme 6.1). An attack from water on the glycosyl-enzyme intermediate, assisted by the acid/base functionality, releases a hydrolyzed sugar. How-ever, if the water molecule is replaced by another nucleophile, e.g. another

48
carbohydrate residue, transglycosylation occurs and the result is retention of the β-anomeric configuration (step 2 in Scheme 6.1).
Glycosidases are of current interest for many reasons, one being the possibil-ity to use them in the chemoenzymatic synthesis of oligo- or polysaccha-rides.105,106 The yields for the transglycosylation reactions are low, only 5-20%,103 since the product can be further hydrolysed by the wild-type en-zyme. The transglycosylation pathway leads to the kinetically most favour-able product, but due to the high concentration of water present, the thermo-dynamically stable product, the hemiacetal, will dominate. Two general strategies have been worked out to overcome this problem:107
• The use of activated donors, e.g. glycosyl fluorides, together with the wild-type enzyme under controlled reaction conditions will fa-vor the “kinetic” transglycosylation products.
• The use of mutant enzymes, glycosynthases, with the active site modified to prevent hydrolysis.
The most powerful tool is a combination of these two methods. The glyco-synthases do not have the catalytic nucleophile in the active site; it has been replaced by a small non-nucleophilic residue, e.g. serine (Scheme 6.2) hence making the mutant enzymes unable to hydrolyze glycosidic linkages. The many advantages of glycosyl fluorides, especially in the α-anomeric form, have led to their extensive use in glycosynthase research.108 They are, despite the good leaving-group properties of the fluoride ion, relatively stable in water solution. The size of the fluorine is comparable to that of a hydroxyl group which leads to little steric hindrance in the active site. Glycosyl fluo-rides also have the advantage over e.g. aryl glycosides that they seem to be universally processed by all glycosidases.108 For analysis of these kinds of processes, it is worth noting that the naturally occuring isotope of fluorine, 19F, has a spin number of ½ and is detectable with NMR spectroscopy.
OHOHO
OH
HO
OHO
CH3
O R'-FOHO
HO
OH
HO
OO
CH3
F
H
OR'
Scheme 6.2: Schematic picture over the active site of a mutant glycosidase, where the catalytic nucleophile has been replaced by an alanine residue enabling the trans-glycosylation of a glycosyl fluoride donor.

49
6.2 Synthesis of α-glycosyl fluoride donors
To serve as substrates for mutant laminarase 16A glycosynthases, α-laminarobiosyl fluoride (αL2F, 10) and α-laminariheptaosyl fluoride (αL7F) were chosen. The heptasaccharide αL7F, was utilized in Paper VI and de-scribed in section 6.3. Compound 10 will be used in further studies of these particular mutants. Described and discussed here is the synthesis of 10; αL7F was assembled the same way (Scheme 6.3).
A peracetylated carbohydrate in the β-D-configuration is readily fluorinated with pyridinium poly(hydrogen fluoride) (HF/pyridine) at room temperature leading to the thermodynamically stable α-pyranosyl fluoride.109 Since per-acetylation using acetic anhydride (Ac2O) and pyridine leads to an α/β-anomeric mixture, and the α-anomer is not successfully converted, a direct fluorination with HF/pyridine could not be accomplished. The obvious solu-tion to the problem would be to exclusively synthesize the β-anomer using Ac2O and NaOAc110 but the precipitation of the product is difficult to obtain when working with small amounts of material, as is the case in this particu-lar project. For this reason, additional steps were included in the synthetic pathway. Starting from 50 mg laminaribiose it was first peracetylated using Ac2O and pyridine. The anomeric position at the reducing end was depro-tected with hydrazine acetate followed by fluorination using diethylamino-sulfur trifluoride (DAST), known to give an α/β-anomeric mixture in di-chloromethane.111 This was proven by the homonuclear coupling constants of the anomeric resonances in the 1H NMR spectrum. Due to the high stabil-ity of the α-D-glucopyranosyl fluoride it remained intact when treated with HF/pyridine while the β-D-glucopyranosyl fluoride was successfully trans-formed. The peracetylated α-laminaribiosyl fluoride was readily deprotected under basic conditions and 10 was obtained in 50% overall yield.

50
a b
c d
e
10
OHOHO
HO
OH
OHOOHO
OH
OH
OAcOAcO
AcO
OAc
OAcOO
AcO
OAc
OAc
OAcOAcO
AcO
OAc
OAcOO
AcO
OAc
OH
OAcOAcO
AcO
OAc
OAcOO
AcO
OAc
F
OAcOAcO
AcO
OAc
OAcOO
AcO
OAc
F
OHOHO
HO
OH
OHOOHO
OH
F
Scheme 6.3: Synthetic pathway to α-laminaribiosyl fluoride. a) Ac2O, pyridine, r.t. b) Hydrazine acetate, 50 °C c) DAST, −20 °C d) HF/pyridine, −23 °C, 78% over 4 steps e) NaOMe/MeOH, r.t., 64%.
A HSQC NMR spectrum was recorded of 10 in D2O for NMR chemical shifts assignments,112 depicted in Figure 6.1. In the anomeric region, two resonances are visible: one at δH/δC 4.35/103.7, corresponding to the ano-meric position of the terminal β-D-glucose residue, and one at δH/δC 5.70/108.3 corresponding to the anomeric position at the reducing end. This latter peak has two coupling constants characteristic for a fluorine-containing carbohydrate, 2JF,H1 53.2 Hz and 1JF,C1 224.1 Hz, both indications of an α-D-pyranosyl fluoride.113
51
3.54.55.5
70
80
90
100
110
60
1H /ppm
13C
/ppm
2JFH
1JFC
Figure 6.1: The 1H,13C-HSQC NMR spectrum of 10. The characteristic heteronu-clear coupling constants, JF,X, in the anomeric region provides evidence of an α-D-glucopyranosyl fluoride.
6.3 Investigating a cyclic heptaglucan synthesized by mutant laminarase 16A (Paper VI)
Laminarase 16A (Lam16A) is a glucosidase that can be found in e.g. the wood-decaying fungus Phanerochaete chrysosporium. It is an enzyme with broad substrate specificity. It randomly hydrolyses linear β-1,3 glucans, branched β-1,3/1,6 glucans, and also β-1,3-1,4 glucans (e.g. lichenan). Its exact role in the fungus is not known. Lam16A has 298 amino acids and a molecular weight of 36 kDa.114 The wild-type enzyme operates, both when hydrolyzing and transglycosylating, via the double-displacement retaining mechanism (Scheme 6.1). A crystal structure of Lam16A was presented in 2006, the first crystallographic representation of a non-specific 1,3(4)-β-D-glucanase from glycosidase hydrolase family 16.115
Lam16A was mutated to give Lam16A E115S, where the proposed nucleo-phile glutamic acid (E) 115 was exchanged for a serine (S) amino acid resi-due. The mutant enzyme was crystallized together with both laminarihep-taose (L7) and αL7F. Interestingly, both substrates fit very well into the cata-lytic site and bind in an arch, occupying both the donor and acceptor sites of
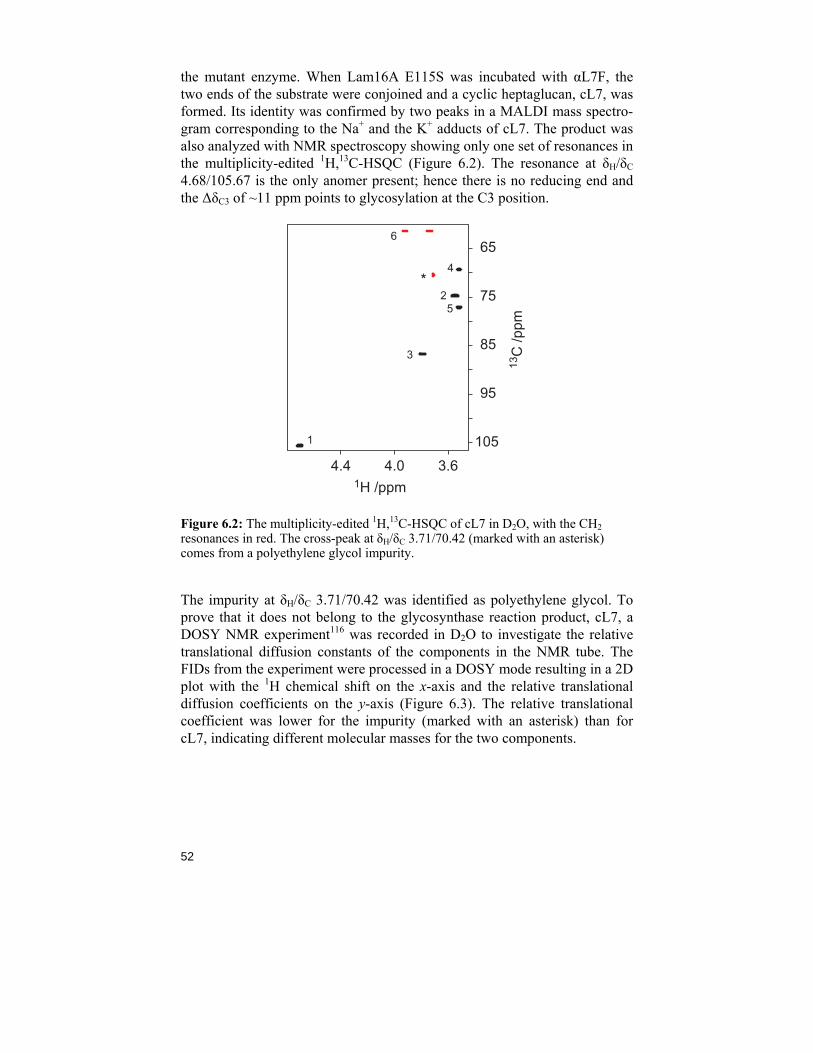
52
the mutant enzyme. When Lam16A E115S was incubated with αL7F, the two ends of the substrate were conjoined and a cyclic heptaglucan, cL7, was formed. Its identity was confirmed by two peaks in a MALDI mass spectro-gram corresponding to the Na+ and the K+ adducts of cL7. The product was also analyzed with NMR spectroscopy showing only one set of resonances in the multiplicity-edited 1H,13C-HSQC (Figure 6.2). The resonance at δH/δC 4.68/105.67 is the only anomer present; hence there is no reducing end and the ΔδC3 of ~11 ppm points to glycosylation at the C3 position.
3.64.04.4
65
75
85
95
105
1H /ppm
13C
/ppm
6
4*
25
3
1
Figure 6.2: The multiplicity-edited 1H,13C-HSQC of cL7 in D2O, with the CH2 resonances in red. The cross-peak at δH/δC 3.71/70.42 (marked with an asterisk) comes from a polyethylene glycol impurity.
The impurity at δH/δC 3.71/70.42 was identified as polyethylene glycol. To prove that it does not belong to the glycosynthase reaction product, cL7, a DOSY NMR experiment116 was recorded in D2O to investigate the relative translational diffusion constants of the components in the NMR tube. The FIDs from the experiment were processed in a DOSY mode resulting in a 2D plot with the 1H chemical shift on the x-axis and the relative translational diffusion coefficients on the y-axis (Figure 6.3). The relative translational coefficient was lower for the impurity (marked with an asterisk) than for cL7, indicating different molecular masses for the two components.
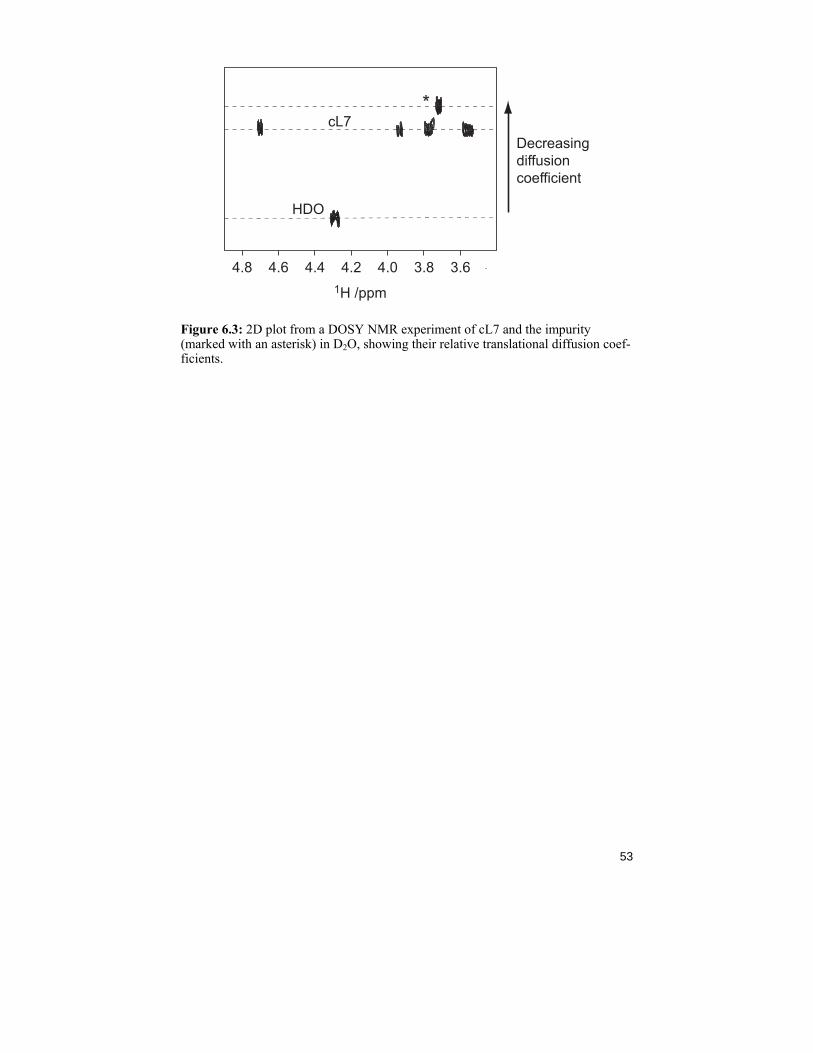
53
3.63.84.04.24.44.64.81H /ppm
Decreasingdiffusioncoefficient
HDO
cL7*
Figure 6.3: 2D plot from a DOSY NMR experiment of cL7 and the impurity (marked with an asterisk) in D2O, showing their relative translational diffusion coef-ficients.

54
Chapter 7: Concluding remarks and future prospects
During the years it has taken me to write and finish this thesis I have come to understand how complex and wide the world of carbohydrates is. Just as life is dependent on the ability to reproduce itself, using DNA, it is also depend-ent on communicating with other living species and/or defending itself against them. Carbohydrates play many roles to human life and cells, not only as fuel or blood group antigens but also as lubricant for the intestines and the insulating clothes we wear.
Mapping of bacteria, or more specifically the O-antigen of bacteria, is a critical part of the research regarding one of the biggest mortality problems the developing world is facing; the outbreaks of (water-borne) bacterial dis-eases causing diarrhea and other life-threatening infections. The structural elucidation of the O-antigens of E. coli O124 and O74 and S. dysenteriae. type 3 can provide basic information to the development for vaccines against these lethal infections.
The conformational studies of rhamnose-containing oligosaccharides have shown that site-specific 13C-labeling successfully contributes to the estima-tion of homonuclear JCC coupling constants as well as nJCH by facilitating the measurements and resolving a problem with spectral overlap in 1H NMR spectra. A comparison of the results from the NMR spectroscopy measure-ments of disaccharide 1 with the results from the MD simulations, shows that they are in agreement. For an even better result, a refinement of the force-field used is necessary and in progress. A better understanding of the three-dimensional structure of α-(1→2)- and α-(1→3)-linked rhamno-oligosaccharides has been obtained, and the knowledge can now be applied to e.g. the polysaccharides of S. flexneri. In the collection of J-couplings in trisaccharide 2, a gap still remains; the 3JC1',H2 is still to be determined with the 1DLR NMR experiment.
The incubation of αL7F with mutant Lam16A E115S resulted a cyclic hepta-saccharide (cL7) identified by MS and NMR spectroscopy. This is the first reported synthesis of a cyclic β-glucan from one substrate molecule and a

55
sensational discovery. The capacity of glycosynthases is surprising and the next step is to see what the incubation of Lam16A E115S with αL2F will result in; how many steps can the oligosaccharide chain be elongated, can longer chains than heptasaccharides be cyclized, can the reaction be sped up etc? Several other mutant variants have been developed and they will be investigated regarding their kinetics towards the synthesized αL2F and αL7F. It is demonstrated that glycosynthases can be utilized in synthesis of oligosaccharides, and even though every single enzyme might have a spe-cific substrate preference, the principle of enzymatic synthesis is applicable in carbohydrate synthesis.

56
Acknowledgements
During my time as a chemistry student I have met many people who have inspired, motivated and supported me to achieve my doctoral de-gree in organic chemistry. My warmest gratitude goes out to:
Professor Göran Widmalm, for accepting me as a graduate student, for al-ways (always) having time for bigger and smaller discussions, for pushing me when needed and listening when needed; for being the best supervisor ever! Professor Jan-Erling Bäckvall, for showing interest in my research. Docent Ian Cumstey, for linguistic improvements of this thesis. Technologie Doctor Jens Landström, Philosophie Licentiate Elin Säwén, Philosophie Licentiate Magnus Lundborg and Mona Svensson for helping me to develop my writing skills. Past and present members of the GW-group for inspiring alumni-evenings and interesting projects to inherit. Ulrika for motivation and friendship, Tara for a nice trip to Canada, Jennie for all the NMR knowledge and fun, Jens for letting me try to teach you German, Elin for Winnerbäck-support, Mona for fashion in the lab, MagnusL for patiently providing computer assistance, Jerk for music inspiration, Robert for veggie-food ideas, Christoffer Go Naturvetarna and Carolina for extending my geography knowledge. Kristina Romare, Pia Seffers, Joakim Staffas for keeping our equipment in good shape and Ola Andersson for computer assistance. Britt Eriksson for keeping the department running. Torbjörn Astlind for always taking time helping me with the NMR spectrometers and sharing all your tricks. Professor Pino Pilotti for fun during chemistry courses and Docent Björn Lüning for being a great inspirer when it comes to teaching and living. The nice people at the Department of Organic Chemistry for making this such a wonderful working place, especially Jesper for friendship also outside

57
work, Catta for being so sweet, JennyAM for all family and literature advice, Linnéa S for the nice trip to Oslo and Linnéa B for the baby-guidance. Professor Jerry Ståhlberg, Philosophie Doctor Jonas Vasur and the group at SLU for fruitful collaboration. Professor Andrej Weintraub, Docent Lars Eriksson and Philosophie Licentiate Elin Säwén at SU for nice collabora-tions. Professor Helena Grennberg, Professor Olle Matsson and Professor Adolf Gogoll for excellent guidance throughout my undergraduate studies in Upp-sala. All my chemistry-friends from UU; Amazing when you think about it, we all made it to a doctoral degree! Astra Zenecas resestipendium till minne av Nils Löfgren, Resestipendie ur Jubileumsdonationen Knut och Alice Wallenbergs Stiftelse, Kemistsamfundet samt förlagsföreningen Acta Chemica Scandinavica för att ni generöst bidragit till konferensresor.
♥ ♥ ♥ ♥ ♥ ♥ ♥ ♥ ♥ ♥ ♪ ♪ ♪ ♪ ♪ ♪ ♪ ♥ ♥ ♥ ♥ ♥ ♥ ♥ ♥ ♥ ♥ Mina underbara vänner utanför universitetets värld som ger mig kraft och mod att utvecklas som människa. Ni skänker värme och glädje när jag behöver det som mest! Mina härliga, stora familj: mamma & pappa för att ni alltid tror att jag kan, Emma, Per och Frida för att ni finns bara ett telefonsamtal bort samt Ellinor och bonusbrorsan Niklas för att ni gör familjen just stor och härlig. Min underbara lilla familj: Per ♥ Tack för att du alltid lyssnar och stöttar, du gör mig varm i hjärtat. Amanda, fantastiska lilla björn som får mig att skratta varje dag. Ziggy, för att du verkligen tycker att jag är bäst.

58
References
1 http://nobelprize.org/, Accessed in March 2010. 2 Lindberg, B. in Polysaccharides 1998, ed. Dumitriu, S., Marcel Dekker, New York, USA, p. 237-273. 3 Laine, R. A. Glycobiology 1994, 4, 759-767. 4 Weintraub, A. Carbohydr. Res. 2003, 338, 2539-2547. 5 Ekelöf, K.; Garegg, P. J.; Olsson, L.; Oscarson, S. Pure & Appl. Chem. 1997, 69, 1847-1852. 6 Ernst, B.; Magnani, J. L. Nat. Rev. 2009, 8, 661-677. 7 Salyers, A. A.; Whitt, D. D. Bacterial Pathogenesis, A molecular Approach 1994, ASM Press, Washington D.C., USA. 8 Gram, H. C. “Über die isolierte Färbung der Schizomyceten in Schnitt- und Trockenpräparaten“, Fortschritte der Medizin, 1884, 2, 185-189. 9 Whitfield, C. Annu. Rev. Biochem. 2006, 75, 39-68. 10 Ovodov, Y. S. Biochemistry (Moscow) 2006, 71, 937-954. 11 http://www.chem.qmul.ac.uk/iupac/, Accessed in March 2010. 12 http://www.chem.qmul.ac.uk/iubmb/, Accessed in March 2010. 13 Kamerling, J. P. ”Basic Concepts and Nomenclature Recommendations in Carbo-hydrate Chemistry”, Comprehensive Glycoscience, 2007, edited by Kamerling, J P., Elsevier Ltd., p. 19-22. 14 Lemieux, R. U.; Koto, S. Tetrahedron 1974, 30, 1933-1944. 15 Thøgersen, H.; Lemieux, R. U.; Bock, K.; Meyer, B. Can. J. Chem. 1982, 60, 44-57. 16 Stallforth, P.; Lepenies, B.; Adibekian, A.; Seeberger, P. H. J. Med. Chem. 2009, 52, 5561-5577. 17 Sears, P.; Wong, C.-H. Science 2001, 291, 2344-2350. 18 Lindhorst, T. K. Essentials of Carbohydrate Chemistry, 2nd revised and updated version, 2003, Wiley-VCH Verlag GmbH & Co. KGa, Weinheim. 19 Schmidt, R. R.; Stumpp, M. Liebigs Ann. Chem. 1983, 7, 1249-1256. 20 Fischer, E. Berichten der Deutschen Chemischen Gesellschaft zu Berlin 1920, 53, 1621-1633. 21 Roslund, M. U.; Aitio, O.; Wärnå, J.; Maaheimo, H.; Murzin, D. Y.; Leino, R. J. Am. Chem. Soc. 2008, 130, 8769-8772. 22 Smith, M. B.; March, J. March`s Advanced Organic Chemistry, 5th ed., 2001, John Wiley & Sons, Inc., USA, p. 677. 23 Garegg, P. J. ”Regioselective Cleavage of O-Benzylidene Acetals Benzyl Ethers”, Preparative carbohydrate chemistry, 1997, edited by Stephen Hanessian, Marcel Dekker Inc., New York, p. 53-68. 24 Kim, J.-H.; Yang, H.; Khot, V.; Whitfield, D.; Boons, G.-J. Eur. J. Org. Chem. 2006, 22, 5007-5028.

59
25 Koenigs, W.; Knorr, E. Berichten der Deutschen Chemischen Gesellschaft zu Berlin, 1901, 34, 957-981. 26 Cumpstey, I.; Fairbanks, A. J.; Redgrave, A. J. Org. Lett. 2001, 3, 2371-2374. 27 Nyffeler, P. T.; Durón, S. G.; Burkart, M. D.; Vincent, S. P.; Wong, C.-H. Angew. Chem. Int. Ed. 2005, 44, 192-212. 28 Yokoyama, M. Carbohydr. Res. 2000, 327, 5-14. 29 Garegg, P. J., Adv. Carbohydr. Chem. Biochem., 2004, 59, 69-134. 30Hore, P. J. Nuclear Magnetic Resonance 1995, Oxford chemistry primers, Oxford Science publications, Antony Rowe Ltd, Chippenham, Wiltshire, UK. 31 Lundström S. L.; Li, J.; Deadman, M. E.; Hood, D. W.; Moxon, E. R.; Schweda, E. K. H. Biochemistry 2008, 47, 6028-6038. 32 Rakotomanga, S.; Baillet, A.; Pellerin, F.; Baylocq-Ferrier, D. J. Pharm. Biomed. Anal.1992, 10, 587-591. 33Ali, T.; Urbina, F.; Weitraub, A.; Widmalm, G. Carbohydr. Res. 2005, 340, 2010-2014. 34Sawardeker, J. S.; Sloneker, J. H.; Jeanes, A. Analytical Chem. 1965, 37, 1602-1604. 35Leontein, K.; Lönngren, J. Meth. Carbohydr. Chem. 1993, 9, 87–89. 36 Budzelaar, P. H. M. Simulation of one-dimensional NMR spectra – a companion to the gNMR User Manual 1995-1999, Cherwell Scientific Ltd, Oxford, UK. 37 Laatikainen R.; Niemitz M.; Weber U.; Sundelin J.; Hassinen T.; Vepsäläinen, J. J. Magn. Reson., Ser. A, 1996, 120, 1-10. 38 http://www.perchsolutions.com/, Accessed March 2010. 39Claridge, T.D.W., High-resolution NMR Techniques in Organic Chemistry 1999, Elsevier Ltd, Oxford, UK. 40 Bundle, D. R.; Lemieux, R. U. Meth. Carbohydr. Chem. 1976, 7, 79-86. 41 Kay, L.E.; Keifer, P.; Saarinen, T. J. Am. Chem. Soc. 1992, 114, 10663-10665. 42 Bax, A. J. Magn. Reson. 1983, 53, 517. 43 Palmer, A. G., III; Cavanagh, J.; Wright, P. E.; Rance, M. J. Magn. Reson. 1991, 93, 151-170. 44 Bax, A.; Freeman, R. J. Magn. Reson. 1981, 44, 542-561. 45 Vilchez, S.; Lundborg, M.; Urbina, F.; Weintraub, A.; Widmalm, G. Carbohydr. Res. 2009, 344, 2528-2532. 46 Kellog, G. W. J. Magn. Reson. 1992, 98, 176-182. 47 Kumar, A.; Ernst, R. R.; Wüthrich K. Biochem. Biophys. Res. Comm., 1980, 95, 1-6. 48 Bax, A.; Summers, M. F. J. Am. Chem. Soc. 1986, 108, 2093-2094. 49 http://www.chem.qmul.ac.uk/iupac/misc/biop.html, Accessed in March 2010 50 Widmalm, G., “General NMR Spectroscopy of Carbohydrates and Conforma-tional Analysis in Solution”, Comprehensive Glycoscience, 2007, edited by Kamer-ling, J P., Elsevier Ltd, p. 110. 51 Karplus, M. J. Chem. Phys. 1959, 30, 11-15. 52 Karplus, M. J. Am. Chem. Soc. 1963, 85, 2870-2871. 53 Coxon, B. Adv. Carbohydr. Chem. Biochem. 2009, 62, 17-82. 54 Cloran, F.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 1999, 121, 9843-9851. 55 Neuhaus, D.; Williamson, M. The nuclear Overhauser effect in structural and conformational analysis 1989, VCH Publishers, Inc., New York, USA.

60
56 Vliegenthart, J. F. G.; Woods, R. J. NMR Spectroscopy and Computer Modeling of Carbohydrates 2006, ACS Symposium series 930, Oxford University Press, USA. 57 Ravindranathan, S; Feng, X.; Karlsson, T.; Widmalm, G.; Levitt, M. H. J. Am. Chem. Soc. 2000, 122, 1102-1115. 58 French, A. D.; Brady, J. W. Computer Modeling of Carbohydrate Molecules 1990, ACS Symposium Series 430, American Chemical Society, USA. 59 Nishida, T.; Widmalm, G.; Sándor, P. Magn. Reson. Chem. 1996, 34, 377-382. 60 Garza-Garcia, A.; Ponzanelli-Velazquez, G.; del Rio-Portilla, F. J. Magn. Reson. 2001, 148, 214-219. 61 Meissner, A.; Sörensen Winneche, O. Magn. Reson. Chem. 2001, 39, 49-52. 62 Kósminskí, W.; Nanz, D. J. Magn Reson. 1997, 124, 383-392. 63 Bock, K.; Pedersen, C. Acta Chem. Scand. B 1977, 31, 354-358. 64 Oikawa, M., Adachi, S., Kusumoto, S., Org. Lett. 2005, 7, 661-664. 65 Podlasek, C. A.; Wu, J.; Stripe, W. A.; Bondo, P. B.; Serianni, A. S. J. Am. Chem. Soc. 1995, 117, 8635-8644. 66 Nolis, P.; Parella, T. J. Magn. Reson. 2005, 176, 15-26. 67 Bose, B.; Zhao, S.; Stenutz, R.; Cloran, F.; Bondo, P. B.; Bondo, G.; Hertz, B.; Carmichael, I.; Serianni, A. S.; J. Am. Chem. Soc. 1998, 120, 11158-11173. 68 Kaper, J. B.; Nataro, J. P.; Mobley, H. L. T. Nat. Rev. Microbiol. 2004, 2, 123-140. 69 Ørskov, F.; Ørskov, I. Can. J. Microbiol. 1992, 38, 699-704. 70 Rowe, B.; Gross, R. J.; Woodroof, D. P. Int. J. Syst. Bacteriol. 1977, 27, 15-18. 71 Cheasty, T.; Rowe, B. J. Clin. Microbiol. 1983, 17, 681-684. 72 Pupo, G. M.; Lan, R.; Reeves, P. R. Proc. Natl. Acad. Sci. USA, 2000, 97, 10567-10572. 73 Liu, B.; Knirel, Y. A.; Perepelov, A. V.; Sechenkova, S. N.; Wang, Q.; Reeves, P. R.; Wang, L. FEMS Microbiol. Rev. 2008, 32, 627-653. 74 Dmitriev, B. A.; Lvov, V. L.; Kochetkov, N. K.; Jann, B.; Jann, K. Eur. J. Biochem. 1976, 64, 491-498. 75 Dmitriev, B. A.; Lvov, V. L.; Kochetkov, N. K. Carbohydr. Res. 1977, 56, 207-209. 76 Linnerborg, M.; Weintraub, A.; Widmalm, G. Eur. J. Biochem. 1999, 266, 460-466. 77 Kochetkov, N. K.; Dmitriev, B. A.; Lvov, V. L. Carbohydr. Res. 1977, 54, 253-259. 78 Stenutz, R.; Weintraub, A.; Widmalm, G. FEMS Microbiol. Rev. 2006, 30, 382-403. 79 Blanco, M.; Padola, N. L.; Kruger, A.; Sanz, M. E.; Blanco, J. E.; Gonzalez, E. A.; Dahbi, G.; Mora, A.; Bernardez, M. I.; Etcheverria, A. I.; Arroyo, G. H.; Luc-chesi, P. M.; Parma, A. E.; Blanco, J. Int. Microbiol. 2004, 7, 269-76. 80 Ørskov, F.; Ørskov, I. Meth. Microbiol. 1984, 14, 43-112. 81 Molinaro, A.; Silipo, A.; Lanzetta, R.; Newman, M.-A.; Dow, J. M.; Parrilli, M. Carbohydr. Res. 2003, 338, 277-281. 82 Grue, M. R.; Parolis, H.; Parolis, L. A. S. Carbohydr. Res. 1993, 248, 191-198. 83 Jansson, P.-E.; Lennholm, H.; Lindberg, B.; Lindquist, U. Carbohydr. Res. 1987, 161, 273-279. 84 http://www.glyco.ac.ru/bcsdb/start.shtml, Accessed in March 2010.

61
85 Perepelov, A. V.; Bartodziesjka, B.; Senchenkova, S. N.; Shashkov, A. S.; Rozalski, A.; Knirel, Y. A. Carbohydr. Res. 2003, 338, 327-331. 86 Wehler, T.; Carlin, N. I. A. Eur. J. Biochem. 1988, 176, 471-476. 87 Molinaro, A.; Newman, M.-A.; Lanzetta, R.; Parilli, M. Eur. J. Org. Chem. 2009, 34, 5887-5896. 88 Klepach, T. E.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 2005, 127, 9781-9793. 89 Söderman, P.; Oscarson, S.; Widmalm G. Carbohydr. Res. 1998, 312, 233-237. 90 Rainer, H.; Scharf, H.-D.; Runsink, J. Liebigs Ann. Chem. 1992, 103-107. 91 Norberg, T.; Oscarson, S.; Szöni, M. Carbohydr. Res. 1986, 152, 301-304. 92 Ness, R. K.; Hewitt, G. F. Jr.; Hudson, C. S. J. Am. Chem. Soc. 1951, 73, 296-300. 93 González-Outeiriño, J.; Nasser, R.; Anderson, J.E. J. Org. Chem. 2005, 70, 2486-2493. 94 Hardy, B. J.; Egan, W.; Widmalm, G. Int. J. Biol. Macromol. 1995, 17, 149-160. 95 Hardy, B. J.; Bystricky, S.; Kovac, P.; Widmalm, G. Biopolymers 1997, 41, 83-96. 96 Widmalm, G.; Byrd, R. A.; Egan, W. Carbohydr. Res. 1992, 229, 195-211. 97 Landersjö, C.; Stevensson, B.; Eklund, R.; Östervall, J.; Söderman, P.; Widmalm, G.; Maliniak, A. J. Biomol. NMR 2006, 35, 89-101. 98 Cloran, F.; Carmichael, I.; Serianni, A. S. J. Am. Chem. Soc. 2000, 122, 396-397. 99 Carmichael, I.; Serianni, A. S. Carbohydr. Res. 1996, 280, 177-186. 100 Oikawa, M.; Adachi, S.; Kusumoto, S. Org. Lett. 2005, 7, 661-664. 101 Eklund, R.; Lycknert, K.; Söderman, P.; Widmalm, G. J. Phys. Chem. B 2005, 109, 19936-19945. 102 Koshland, D. E. Biol. Rev. 1953, 28, 416-436. 103 Jahn, M.; Withers, S. G. Biocatal. and Biotransform., 2003, 21, 159-166. 104 Hult, K.; Berglund, P. Curr. Opin. Biotechnol. 2003, 14, 395-400. 105 Shaikh, F. A.; Withers, S. G. Biochem. Cell. Biol. 2008, 86, 169-177. 106 Boltje, T. J.; Buskas, T.; Boons, G.-J. Nat. Chem. 2009, 1 (8), 611-622. 107 Faijes, M.; Planas. A.; Carbohydr. Res. 2007, 342, 1581-1594. 108 Williams, S. J.; Withers, S. G. Carbohydr. Res. 2000, 327, 27-46. 109 Hayashi, M.; Hashimoto, S.; Noyori, R. Chem. Lett. 1984, 1747-1750 110 Lindhorst, T. K. ”Essentials of Carbohydrate Chemistry” 2nd ed., 2003, Wiley-VCH Verlag, GmbH & Co. KGaA, Weinheim, p 47 111 Fauré, R.; Saura-Valls, M.; Brumer, H. III; Planas, A.; Cottaz, S.; Driguez, H. J. Org. Chem. 2006, 71, 5151-5161. 112 Unpublished material, manuscript in preparation. 113 Michalik, M.; Mein, M.; Frank, M. Carbohydr. Res. 2000, 327, 185-218. 114 Kawai, R.; Igarashi, K.; Yoshida, M.; Kitaoka, M.; Samejima, M. Appl. Micro-biol. Biotechnol. 2006, 71, 898-906. 115 Vasur, J.; Kawai, R.; Larsson, A. M.; Igarashi, K.; Sandgren, M.; Samejima, M.; Ståhlberg, J. Acta Cryst. 2006, D62, 1422-1429. 116 Morris, K. F.; Johnson, C. S. J. Am. Chem. Soc. 1992, 114, 3139-3141.





























