Embed Size (px)
Citation preview

Chemical Physics Letters 382 (2003) 217–225
www.elsevier.com/locate/cplett
Exploring the electronic structure of 2,6-stelladionefrom momentum space I: the p-dominant molecular
orbitals in the outer valence shell
Feng Wang a,*, Michael J. Brunger b, Ian E. McCarthy b, Dave A. Winkler c
a Centre for Molecular Simulation, Swinburne University of Technology, Hawthorn, Melbourne, Vic. 3122, Australiab School of Chemistry, Physics and Earth Sciences, Flinders University, GPO Box 2100, Adelaide, SA 5001, Australia
c Division of Molecular Sciences, CSIRO, Private Bag 10, Clayton South MDC, Vic. 3169, Australia
Received 17 June 2003; in final form 25 September 2003
Abstract
The p-electron dominant contributions to the outer valence shell of 2,6-stelladione (C8H8O2) are analyzed using
binding energy spectra and the orbital momentum distributions obtained by experimental and theoretical electron
momentum spectroscopy. The binding energy spectra are given for azimuthal angles / ¼ 0� and 10�, respectively, inorder to reveal information of the s- and p-electron dominant characteristics in these molecular orbitals. The wave-
functions in configuration space are directly mapped into momentum space using the plane wave impulse approxi-
mation. This work focuses on the interpretation of the electronic structural information and bonding mechanism of the
molecule in momentum space. In particular, p-electron dominant contributions of the strained organic compound are
used to support our findings.
� 2003 Elsevier B.V. All rights reserved.
1. Introduction
Tricyclo[3.3.0.03:7]octane-2,6-dione (C8H8O2),
also known as 2,6-stelladione, is a highly sym-
metric compound. It has been a key compound inthe synthesis of model structures for studying a
stepwise Cope rearrangement and also in the study
of long range interactions [1]. Additionally, 2,6-
* Corresponding author. Fax: +61392145075.
E-mail address: [email protected] (F. Wang).
0009-2614/$ - see front matter � 2003 Elsevier B.V. All rights reserv
doi:10.1016/j.cplett.2003.09.151
stelladione has served as a reference structure to
investigate energy transfer reactions as a function
of the strain energy of the r-frame between donor
and receptor substitutions [2]. However, as this
compound has been synthesized relatively recently[1], only limited X-ray crystallographic data [2]
and a photoelectron spectrum [1] are available in
the literature at this time to provide information
on its geometry and electronic structure. This sit-
uation is actually worse than it might otherwise
appear as the photoelectron spectrum provides us
with an incomplete range of the binding energies
in the outer valence shell of the molecule, only
ed.

218 F. Wang et al. / Chemical Physics Letters 382 (2003) 217–225
the first three molecular orbitals having been tab-
ulated [1]. Our most recent electron momentum
spectroscopy (EMS) study, the first EMS mea-
surement on this compound, attempted to address
(at least in part) some of this paucity in informa-
tion for this molecule [3]. For example, with thehelp of the EMS binding energy spectra, we were
able to reanalyze the photoelectron spectrum [1] to
give the complete binding energies of 2,6-stelladi-
one in the outer valence shell [3].
Theoretical studies of 2,6-stelladione have been
also limited, in this case by the size of the com-
pound, with only a few ab initio calculations using
Hartree–Fock (HF) method HF/3-21G [2], re-stricted HF (RHF) method RHF/6-31G* and a
single point calculation using the second order
M€ooller–Plesset perturbation theory (MP2) MP2/
6-31G*//RHF/6-31G* [1] being performed. These
rather low level calculations were able to provide
some reliable geometric information of the com-
pound, as confirmed by the X-ray crystallographic
data [2]. However electronic structural informationof this compound, depending on the specific mo-
lecular properties under study, needs the inclusion
of electron correlation energy. As a result, density
functional theory (DFT), together with the DGauss
DFT triple zeta with valence polarized (TZVP)
basis set [7], calculations were employed in the
present work to simulate the orbital momentum
distributions (MDs). In this work, we will focus onthe p-dominant atomic orbital (AO) contributions
to the molecular orbitals (MOs) of 2,6-stelladione
in the outer valence shell of its ground electronic
state (X1A1). Most of our analysis is conducted in
momentum space, which is the Fourier transform
analogue of the more familiar configuration space.
2. Molecule orientation
The ground electronic state of 2,6-stelladione
(X1A1) is highly symmetric, with a point group
symmetry of D2d. In this work, the tricyclo octane–
carbon skeleton of 2,6-stelladione is orientated in a
Cartesian coordinate system with the oxygen at-
oms located along the vertical z-axis. This is shownin Fig. 1, with the x-axis in the plane and the y-axisout-of-plane. Fig. 1 also gives the atom numbering
order of the eight carbon, two oxygen and eight
hydrogen atoms. The origin of the Cartesian co-
ordinate system coincides at the symmetry centre
of the molecule�s D2d point group.
3. The binding energy spectra of 2,6-stelladione
In molecular orbital theory anMO of a molecule
is considered as a linear combination of the com-
ponentAOs (LCAO),where this linear combination
does not change the nature of the AOs. As a result,
together with the MO coefficients in the wavefunc-
tion, the AOs essentially contain and reflect theelectronic structural information of the bonding
mechanism for the molecule under study (the con-
cept of �atoms-in-molecules� [5]). The �shape� ornature (s, p, d,. . .) of the AOs is determined by the
azimuthal quantum number l with l ¼ 0; 1; 2; . . . ;respectively, which relates to the EMS binding en-
ergy spectra and MDs through the azimuthal angle
/ (through momentum conservation).The EMS experiment is also called (e, 2e)
scattering in which an in-coming electron interacts
with the target (the sample molecule) leading to
ionization so that two electrons are scattered out
from the interaction region [6]. In the experiment,
the energies of the two outgoing electrons A and B
are equal and the polar angle is h ¼ 45� with re-
spect to the direction of the incident electronbeam. The total energy (sum of the energies of A
and B) is 1500 eV. The binding energy range of
interest (�f ¼ 7� 16 eV) is stepped through se-
quentially at each of a chosen set of azimuthal
angle / using a binning mode [4] through the
range of / ¼ 0�–30�. This is equivalent [3] to
sampling different target electron momenta P,
where
P ¼ ð2PA cos h
��P0Þ2 þ 4P2
A sin2 h sin2 /
2
� ��1=2:
ð1Þ
Note that for closed shell molecules the total
momentum is conserved so that both linear and
angular momenta should conserve in the (e, 2e)
scattering process. Therefore the angular momen-
tum of the system is correlated indirectly via

Fig. 1. The molecule orientation of 2,6-stelladione (C8H8O2, X1A1) in space.
F. Wang et al. / Chemical Physics Letters 382 (2003) 217–225 219
Eq. (1). For example when the azimuthal angle is
/ ¼ 0�, the corresponding momentum is P � 0
a.u., and the resulting binding energy spectrum of
the molecule indicates the MOs which are domi-
nated by the s-AOs (l ¼ 0) of the component at-
oms. Such electronic information thus provides
direct details for the bonding mechanism of themolecule.
In its electronic ground state, 2,6-stelladione
has component atoms with occupied s-AOs and
p-AOs. It is clear from our discussion above that
when the azimuthal angle in the EMS technique
is tuned to zero degree, the binding energy spec-
trum of 2,6-stelladione at this angle (/ ¼ 0�) in-
dicates s-electron domination status in the outervalence shell of the molecule. Alternatively, as
there are no l > 1 orbitals involved in the bond-
ing of this molecule, the binding energy spectrum
at / 6¼ 0� provides information of the p-electron
domination in the outer valence shell of the
molecule. Therefore, from the binding energy
spectra at zero and non-zero azimuthal angles,
bonding information for the p-dominant MOs in
the outer valence shell can be recognized. The
bonding mechanism information provided by theBES can be further identified in our later orbital
momentum distribution analysis using the EMS
experiment and simulation.
Fig. 2 shows the experimental BES for the
ground electronic state of 2,6-stelladione at a
total energy of 1500 eV [3]. The BES for / ¼ 0�and / ¼ 10� are shown in (a) and (b), respec-
tively. The vertical bars with numbers in Fig. 2agive the positions of the orbital energy and the
orbital number (the highest occupied MO is
numbered as 1), which also apply to Fig. 2b BES.
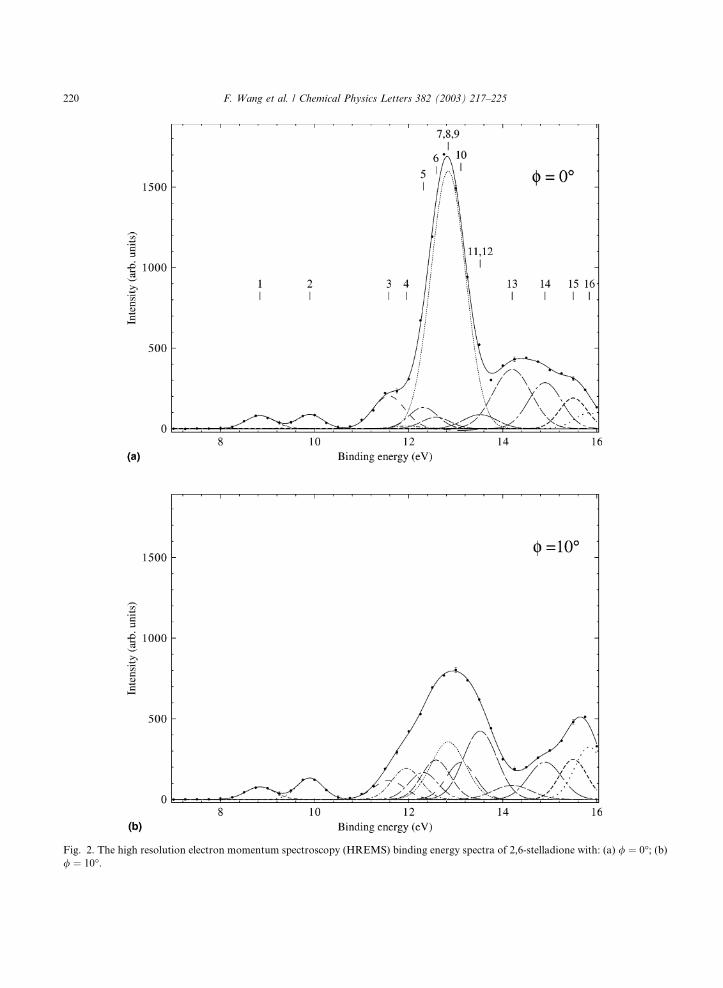
Fig. 2. The high resolution electron momentum spectroscopy (HREMS) binding energy spectra of 2,6-stelladione with: (a) / ¼ 0�; (b)/ ¼ 10�.
220 F. Wang et al. / Chemical Physics Letters 382 (2003) 217–225

F. Wang et al. / Chemical Physics Letters 382 (2003) 217–225 221
The MOs, such as 6b2 and 5b1 (their corre-
sponding positions are marked 4 and 10), with
peaks only apparent in the / 6¼ 0� spectrum are
almost certainly dominated by the p-electron
AOs. In addition, it has been observed that the
MOs are likely to be dominated by p-electrons, iftheir peak intensities in the binding energy spec-
tra experience large changes with respect to / 6¼0� and / ¼ 0� in the experiment. In the present
study the 4b3 & 3b2 and 5a MOs are thus also
likely to be p-dominant MOs.
4. Momentum distributions of the p-dominantmolecular orbitals
The p-electron dominant MOs, namely, 6b2,
5b1, 4b3 & 3b2 and 5a, of 2,6-stelladione can be
uniquely identified by their MDs, obtained from
both experimental and quantum mechanical sim-
ulation techniques. Under the Born–Oppenheimer
approximation for the target and ion wavefunc-tion, the EMS cross section for randomly-orien-
tated molecules is given in the plane-wave impulse
approximation by [4],
r ¼ KZ
dXjhPWN�1f jWN
i ij2; ð2Þ
where the terms in this equation are defined in our
previous study [3]. The overlap of the initial andfinal electronic wavefunctions is a one-electron
quantity known as the Dyson orbital. It may be
approximated by an MO, wjðPÞ, using the same
independent-particle model for the target and ion.
Eq. (2) reduces to,
r ¼ KSðf Þj
ZdXjwjðPÞj2; ð3Þ
where the spectroscopic factor Sðf Þj is the proba-
bility of one-hole configuration j being in the ion
wavefunction WN�1f [6].
5. Analysis for the p-dominant molecular orbitals
Details of the quantum mechanical calculationsand the transform of the wavefunction for the 2,6-
stelladione molecule have been reported elsewhere
[3]. To further extract information for the p-elec-
tron dominant MOs in the outer valence shell,
RHF/6-31G** calculations are performed using
GAMESS [8] and the molecular point group
symmetry information has been imposed in the
calculations. Table 1 gives the Mulliken atomicpopulation analysis on each of the p-dominated
MOs, based on the RHF/6-31G** calculations. As
these MOs are p-dominated and in the outer va-
lence shell of the molecule, it is clear from Table 1
that the oxygen and carbon atoms, possibly their
2p-AOs, are dominant. Hydrogen atoms make
important secondary contributions to some of the
MOs, such as 5b1 and 4b3, but are virtually elim-inated in 6b2. Indeed compared to the other MOs
in Table 1, 6b2 is the only MO where the hydrogen
1s-AO contributions are apparently negligible. As
a result, the bonding in 6b2 can be considered as
being due to �pure� p-contributions and its MD in
Fig. 3 supports this finding. Furthermore, as can
also be seen from this table, b2 is dominated by the
two ketone oxygen atoms, Oð1Þ and Oð2Þ (0.34), andthe two methylene carbon atoms (CH2), i.e., Cð9Þand Cð10Þ (0.23), with the central four equivalent
carbon atoms, i.e., Cð5Þ, Cð6Þ, Cð7Þ and Cð8Þ (0.17),
making some secondary contributions. This is be-
cause Oð1Þ, Oð2Þ, Cð3Þ, Cð4Þ, Cð9Þ and Cð10Þ are con-
fined in the same plane by symmetry, pz- (of Oð1Þand Oð2Þ) and px-AO (Cð9Þ and Cð10Þ) contributions
of the appropriate atoms are therefore expected(the distance between, say, Oð1Þ to Cð9Þ is 3.19 �AA).
Further analysis of the 6b2 orbital wavefunction
shows that the py-AOs of the methylene carbons
Cð9Þ and Cð10Þ overlap with px-AOs of Cð6Þ, Cð6Þ and
Cð7Þ and Cð8Þ, where Cð7Þ and Cð8Þ locate in the back
side (negative y) of the xz-plane and Cð7Þ and Cð8Þlocate in the front side (positive y) of the xz-plane,contributing to the strained six-ring r-bond of thecarbon skeleton.
The contributions from the s-AOs to the rest of
the p-dominant MOs can not be neglected, indi-
cating the possible effects of hybridization. As
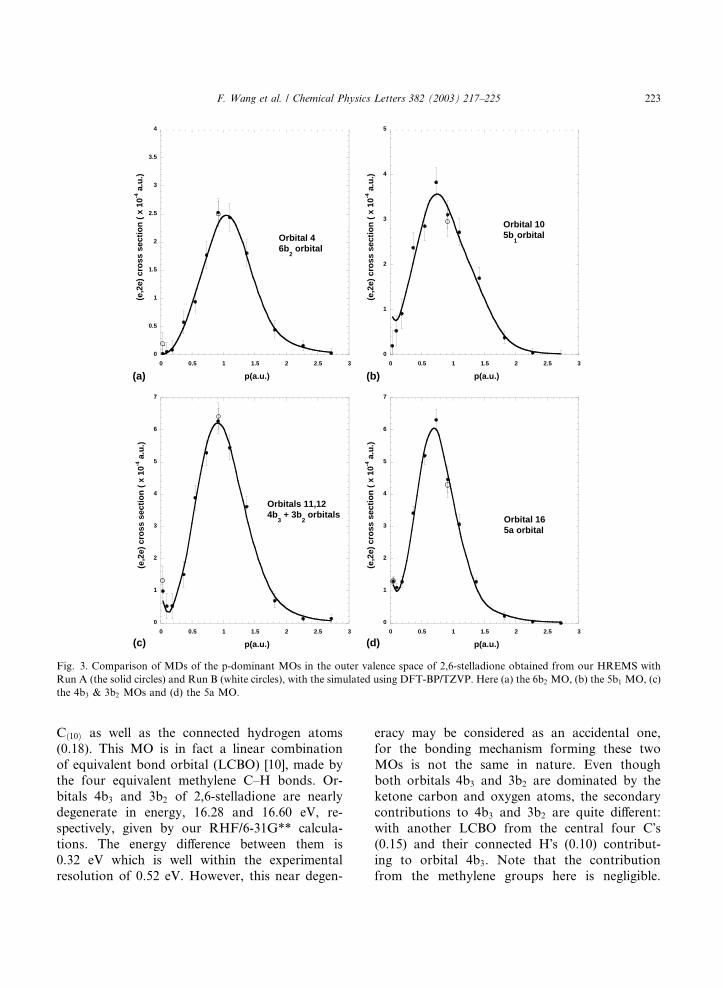
Fig. 3 shows, all the orbital MDs of the p-domi-
nant MOs exhibit a local minimum in the MD at
small P before they start to climb (that is, the sign
of its second derivative changes), except for the 6b2
MO. This indicates that those MOs except for 6b2
are dominated by the carbon p-AOs. Fig. 3 also
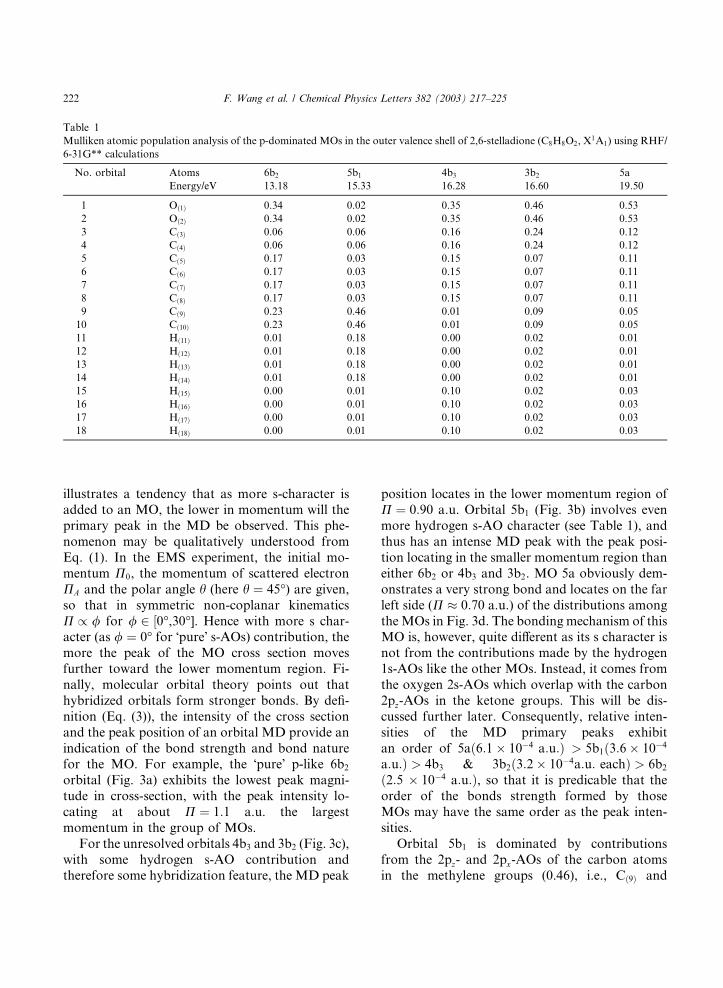
Table 1
Mulliken atomic population analysis of the p-dominated MOs in the outer valence shell of 2,6-stelladione (C8H8O2, X1A1) using RHF/
6-31G** calculations
No. orbital Atoms 6b2 5b1 4b3 3b2 5a
Energy/eV 13.18 15.33 16.28 16.60 19.50
1 Oð1Þ 0.34 0.02 0.35 0.46 0.53
2 Oð2Þ 0.34 0.02 0.35 0.46 0.53
3 Cð3Þ 0.06 0.06 0.16 0.24 0.12
4 Cð4Þ 0.06 0.06 0.16 0.24 0.12
5 Cð5Þ 0.17 0.03 0.15 0.07 0.11
6 Cð6Þ 0.17 0.03 0.15 0.07 0.11
7 Cð7Þ 0.17 0.03 0.15 0.07 0.11
8 Cð8Þ 0.17 0.03 0.15 0.07 0.11
9 Cð9Þ 0.23 0.46 0.01 0.09 0.05
10 Cð10Þ 0.23 0.46 0.01 0.09 0.05
11 Hð11Þ 0.01 0.18 0.00 0.02 0.01
12 Hð12Þ 0.01 0.18 0.00 0.02 0.01
13 Hð13Þ 0.01 0.18 0.00 0.02 0.01
14 Hð14Þ 0.01 0.18 0.00 0.02 0.01
15 Hð15Þ 0.00 0.01 0.10 0.02 0.03
16 Hð16Þ 0.00 0.01 0.10 0.02 0.03
17 Hð17Þ 0.00 0.01 0.10 0.02 0.03
18 Hð18Þ 0.00 0.01 0.10 0.02 0.03
222 F. Wang et al. / Chemical Physics Letters 382 (2003) 217–225
illustrates a tendency that as more s-character isadded to an MO, the lower in momentum will the
primary peak in the MD be observed. This phe-
nomenon may be qualitatively understood from
Eq. (1). In the EMS experiment, the initial mo-
mentum P0, the momentum of scattered electron
PA and the polar angle h (here h ¼ 45�) are given,
so that in symmetric non-coplanar kinematics
P / / for / 2 ½0�,30�]. Hence with more s char-acter (as / ¼ 0� for �pure� s-AOs) contribution, the
more the peak of the MO cross section moves
further toward the lower momentum region. Fi-
nally, molecular orbital theory points out that
hybridized orbitals form stronger bonds. By defi-
nition (Eq. (3)), the intensity of the cross section
and the peak position of an orbital MD provide an
indication of the bond strength and bond naturefor the MO. For example, the �pure� p-like 6b2
orbital (Fig. 3a) exhibits the lowest peak magni-
tude in cross-section, with the peak intensity lo-
cating at about P ¼ 1:1 a.u. the largest
momentum in the group of MOs.
For the unresolved orbitals 4b3 and 3b2 (Fig. 3c),
with some hydrogen s-AO contribution and
therefore some hybridization feature, the MD peak
position locates in the lower momentum region ofP ¼ 0:90 a.u. Orbital 5b1 (Fig. 3b) involves even
more hydrogen s-AO character (see Table 1), and
thus has an intense MD peak with the peak posi-
tion locating in the smaller momentum region than
either 6b2 or 4b3 and 3b2. MO 5a obviously dem-
onstrates a very strong bond and locates on the far
left side (P � 0:70 a.u.) of the distributions among
theMOs in Fig. 3d. The bonding mechanism of thisMO is, however, quite different as its s character is
not from the contributions made by the hydrogen
1s-AOs like the other MOs. Instead, it comes from
the oxygen 2s-AOs which overlap with the carbon
2pz-AOs in the ketone groups. This will be dis-
cussed further later. Consequently, relative inten-
sities of the MD primary peaks exhibit
an order of 5að6:1� 10�4 a:u:Þ > 5b1ð3:6� 10�4
a:u:Þ > 4b3 & 3b2ð3:2� 10�4a:u: eachÞ > 6b2
ð2:5 � 10�4 a:u:Þ, so that it is predicable that the
order of the bonds strength formed by those
MOs may have the same order as the peak inten-
sities.
Orbital 5b1 is dominated by contributions
from the 2pz- and 2px-AOs of the carbon atoms
in the methylene groups (0.46), i.e., Cð9Þ and
0
0.5
1
1.5
2
2.5
3
3.5
4
0 0.5 1 1.5 2 2.5 3
(e,2
e) c
ross
sec
tio
n (
x 1
0-4 a
.u.)
p(a.u.)
Orbital 46b
2 orbital
0
1
2
3
4
5
0 0.5 1 1.5 2 2.5 3(e
,2e)
cro
ss s
ecti
on
( x
10-4
a.u
.)p(a.u.)
Orbital 105b
1orbital
0
1
2
3
4
5
6
7
0 0.5 1 1.5 2 2.5 3
(e,2
e) c
ross
sec
tio
n (
x 1
0-4 a
.u.)
p(a.u.)
Orbitals 11,124b
3 + 3b
2 orbitals
0
1
2
3
4
5
6
7
0 0.5 1 1.5 2 2.5 3
(e,2
e) c
ross
sec
tio
n (
x 1
0-4 a
.u.)
p(a.u.)
Orbital 165a orbital
(a) (b)
(d)(c)
Fig. 3. Comparison of MDs of the p-dominant MOs in the outer valence space of 2,6-stelladione obtained from our HREMS with
Run A (the solid circles) and Run B (white circles), with the simulated using DFT-BP/TZVP. Here (a) the 6b2 MO, (b) the 5b1 MO, (c)
the 4b3 & 3b2 MOs and (d) the 5a MO.
F. Wang et al. / Chemical Physics Letters 382 (2003) 217–225 223
Cð10Þ as well as the connected hydrogen atoms
(0.18). This MO is in fact a linear combination
of equivalent bond orbital (LCBO) [10], made bythe four equivalent methylene C–H bonds. Or-
bitals 4b3 and 3b2 of 2,6-stelladione are nearly
degenerate in energy, 16.28 and 16.60 eV, re-
spectively, given by our RHF/6-31G** calcula-
tions. The energy difference between them is
0.32 eV which is well within the experimental
resolution of 0.52 eV. However, this near degen-
eracy may be considered as an accidental one,
for the bonding mechanism forming these two
MOs is not the same in nature. Even thoughboth orbitals 4b3 and 3b2 are dominated by the
ketone carbon and oxygen atoms, the secondary
contributions to 4b3 and 3b2 are quite different:
with another LCBO from the central four C�s(0.15) and their connected H�s (0.10) contribut-
ing to orbital 4b3. Note that the contribution
from the methylene groups here is negligible.

224 F. Wang et al. / Chemical Physics Letters 382 (2003) 217–225
Hence, the bonding mechanism for orbital 4b3
appears to be from through-bond interactions
[9]. On the other hand, the secondary contribu-
tions to orbital 3b2 are from the six-ring carbon
frame with the central four carbons of Mulliken
population 0.07 and the methylene carbons of0.09. The role of the two hydrogen groups, that
is, the methylene hydrogens Hð11Þ, Hð12Þ, Hð13Þ &
Hð14Þ and the central hydrogens Hð15Þ, Hð16Þ, Hð17Þ& Hð18Þ, is nearly equivalent, yielding the Mul-
liken population of approximately 0.02, respec-
tively. As a result, in 3b2, the LCBO of the
methylene carbons seems to interact with the
ketone groups by through-space interactions [9].The MD of the 5a orbital is quite different
from the other MOs in Fig. 3. The intensity of
its MD peak is apparently higher than those of
the other p-dominant MOs, almost twice as
much as the rest of the MOs in this Figure.
Additionally its MD peak position is at the
lowest momentum (see Fig. 3). Although a sec-
ondary contribution from groups of LCBO ofthe central carbons is observed in Table 1, the
primary contribution forming 5a is from the
overlap between 2s-AOs and 2pz-AOs of both
oxygen and carbon in the ketone groups, so that
this MO is dominated by the 2pz-AOs and 2s-
AOs �head-on� r-bond. As a result, the 5a orbital
exhibits a strong bond nature (high intensity)
and s-character (low momentum location).
6. Conclusions
The p-electron contribution is dominant in
several molecular orbitals in the outer valence
shell, namely, orbitals 6b2, 5b1, 4b3 & 3b2 and
5a of the 2,6-stelladione molecule. Our workindicated that the binding energy spectra at zero
and non-zero azimuthal angles / provided pre-
liminary information on the s- and p-contribu-
tions to the valence MOs of the compound,
which could be confirmed by an orbital based
momentum distribution analysis using both ex-
periment and simulation. Detailed MO analysis
of the p-dominant MOs of 2,6-stelladione hasbeen given using experimental binding energy
spectra, experimental and simulated momentum
distributions of the molecular orbitals, and has
been also assisted by the Mulliken atomic orbital
population analysis. In conclusion, it was evident
that EMS observes the behavior of the molecular
orbitals through the azimuthal angle, and there-
fore, reflects the momentum based informationof the molecular orbitals, which is not always
obvious in configuration space. As the p-domi-
nated molecular orbitals such as 6b2 and 5a can
be r bonds, depending on the direction of the
bonding, the orbital momentum distributions
obtained from EMS can describe the atomic
orbital nature from which the molecular orbitals
are determined. This work also has attempted tointerpret the EMS binding energy spectra and
momentum distributions using standard molecu-
lar spectral analysis, that is, indicating that the
electronic structure and the bonding mechanism
of the molecule link to the peak position in
momentum and intensity of the orbital momen-
tum distributions.
Acknowledgements
One of the authors (FW) would like to ac-knowledge the Australian Partnership for Ad-
vanced Computing (APAC) for using the Compaq
SC AlphaServer Cluster National Facilities. FW
also acknowledges the HPCCC group of CSIRO
for computer resources.
References
[1] R. Gleiter, B. Gaa, C. Sigwart, H. Lange, O. Borzyk, F.
Rominger, H. Irngartinger, T. Oeser, Eur. J. Org. Chem.
(1998) 171.
[2] R. Gleiter, H. Lange, O. Borzyk, J. Am. Chem. Soc. 118
(1996) 4889.
[3] K.L. Nixon, F. Wang, L. Campbell, T. Maddern, D.A.
Winkler, R. Gleiter, P. Loeb, E. Weigold, M.J. Brunger,
J. Phys. B: At. Mol. & Opt. Phys 36 (2003) 3155.
[4] I.E. McCarthy, E. Weigold, Rep. Prog. Phys. 54 (1991)
789.
[5] Moffitt, W. Proc. Roy. Soc. (London) A210 (1951)
224.
[6] E. Weigold, I.E. McCarthy, Electron Momentum Spec-
troscopy, Kluwer Academic/Plenum Publishers, New
York, 1999.

F. Wang et al. / Chemical Physics Letters 382 (2003) 217–225 225
[7] N. Godbout, D.R. Salahub, J. Andzelm, E. Wimmer, Can.
J. Chem. 70 (1992) 560.
[8] M.W. Schmidt et al., J. Comput. Chem. 14 (1993)
1347.
[9] R. Hoffmann, Acc. Chem. Res. 4 (1971) 1.
[10] C.W.N. Cumper, Wave Mechanics For Chemists,
Heinemann Educational Book Ltd., London,
1966.





























