Embed Size (px)
Citation preview

No.154
2
No.154
1. Introduction
As symbolized by awarding of the 2001 Nobel Prize in chemistry to K. B. Sharpless, R. Noyori, and W. S. Knowles, the catalytic synthesis of chiral molecules is of crucial importance in fine chemistry in order to ensure the supply of useful and valuable organic compounds. The importance of this research topic has stimulated chemists worldwide to develop numerous methods of asymmetric catalysis, although the process of developing new catalysts involves tedious effort even when modern methods are used. One way to overcome the time-consuming aspect of the process may be to use the technology of combinatorial chemistry for the discovery and optimization of catalysts. For efficient exploration of novel asymmetric catalysts using the combinatorial approach, a convenient technology of high-throughput screening (HTS) is required for analyzing both catalytic activity (chemical yield) and enantiomeric excess. Although recent remarkable work has resulted in some useful HTS systems, a more powerful analytical system is required in pursuit of wide application, easy handling, and practical use in broad asymmetric catalysis.
We have succeeded in the development of new HTS system by coupling circular dichroism (CD) detection and solid phase reaction. Here we wish to demonstrate the use of “Solid-phase Catalysis/CD-HTS” for the combinatorial development of a novel chiral imidazoline-aminophenol (IAP) ligand in asymmetric metal catalysis. Furthermore, the chemistry of imidazoline-consisting ligand was extended to the imidazolidine-consisting ligand, which promoted us a design and development of Bis(imidazolidine)-pyridine (PyBidine) ligand. Using IAP and PyBidine as the new type of chiral ligands, unique metal-catalyzed asymmetric reactions are conducted to provide novel highly functionalized chiral compounds having multiple stereogenic centers.
2. Solid-phase Catalysis/CD-HTS
A new HTS system has been developed by coupling circular dichroism (CD) detection and solid phase reaction (Figure 1). In this system, the origin of chirality is restricted
Figure 1. Solid-phase catalysis/CD HTS.
Research ArticleResearch Article
Exploration and Design of Novel Asymmetric Catalysts:
Development of Imidazoline-aminophenol (IAP) and
Bis(imidazolidine)pyridine (PyBidine)
Prof. Dr. Takayoshi Arai
Department of Chemistry, Graduate School of Science, Chiba University

No.154
3
by the solid support. When the catalytic asymmetric reaction is examined using an achiral substrate in solution, no asymmetric induction occurs; therefore, when the solution is analyzed using CD, no significant signal should be detected. Because any two enantiomers have exactly opposite CD values at each wavelength, the CD detector could analyze a single appropriate wavelength and record a positive or negative signal corresponding to the amount of excess enantiomer. Regarding the use of CD in the HTS, Mikami et al. reported pioneering works on the screening of solution-phase asymmetric catalysts.2) In their system, the chiral catalyst and ligand are separated using achiral column, and the efficiency was examined using “g-factor”. Although our system (Figure 1) isn’t possible to use the g-factor due to the UV adsorption of co-existing substrate and/or undesired products, the introduction of reaction mixture to the CD detector without purification provides a “direct monitoring system of reaction mixture”.
3. A L i b r a r y o f C h i r a l I m i d a z o l i n e -Aminophenols: Discovering an Efficient Reaction Sphere
The reason why we have interested in the imidazoline-consisting chiral ligand is summarized in Figure 2. A representative example of the chiral ligand chemistry can be seen in the famous bis(oxazoline) ligands, in which the nitrogen atom of a N,O-containing five-membered ring of the oxazoline coordinates to the metal cation to form the complex. The bis(oxazoline)-metal complexes have been utilized in numerous asymmetric reactions. However, even in the beautiful success of bis(oxazoline) ligands in asymmetric catalysis, regulating the electron density of the oxazoline ring has been remained an unsolved problem. One way to overcome the limitation of electronic variation on the oxazoline ring would be by a rational design of a chemical analog (e.g. imidazoline) of oxazoline. Imidazoline, the N,N-analog of N,O-consisting oxazoline,
L1-L8
CuClC1-C8
L9-L16
CuClC17-C25
Cu(OAc)2C9-C16 C26-C32
Cu(OAc)2
SClO
O
1
2
N N
Ph Ph
Cl
N N
Cl
S
SO
O
O
O
3
4
3
N N
Ph Ph
NH
SO
O
N N
Ph Ph
NH
SO
O
N N
NH
SO
O
N N
NH
SO
O
Ph
Ph
Ph
Ph
L1
L2
L3
L4
L5
L6
L7
L8
L9
L10
L11
L12
L13
L14
L15
5
6
7
8
L16
5: 6: 7: 8:
HN N
Ph Ph
Cl
HN N
Cl
H2N Ph
H2N Ph
O
H
HO
O
H
HO
O
H
HO
Br
O
H
HOBr
Br
a
b
b
c
N N
Ph Ph
N
SO
O
Ph
OH
R
N N
Ph Ph
N
SO
O
Ph
OH
R
N N
N
SO
O
Ph
OH
R
N N
N
SO
O
Ph
OH
R
R=H, 5-t-Bu, 5-Br, 3,5-diBr
H2N Ph
4H2N Ph
5
6
7
8
c
5
6
7
8
c
5
6
7
8
c
Catalyst Preparation
Ligand Synthesis
O N
N NR
i) the comparison of ligand ability of imidazoline with that of oxazoline
ii) the introduction of a linker (R) for immobilization
N N
Easinessof
SynthesisCoordination
Manner
N NR
Tuning of electron density
similar similar
difficult
easy
Scheme 1. Library of imidazoline-aminophenol ligands (L1-L16) and the Cu catalysts (C1-C32): a) chloromethylated imidazoline, triethylamine, CH2Cl2; b) corresponding amine, KI, MeCN; c) corresponding salicylaldehyde; then NaBH3CN, MeOH.
Figure 2. Imidazoline as a ligand.
No.154
4
No.154
would be reasonably considered to sufficiently meet such an electronically tunable demand (Figure 2-i). Since the oxazoline oxygen atom is replaced by a NR group in the imidazoline, the basicity of the ligating nitrogen could be accurately adjusted with a selection of either electron-withdrawing or -donating substituent (R). Moreover, in a planning of the library synthesis of imidazoline-consisting ligands on a solid support, the substituent (R) at the nitrogen atom in the imidazoline would be fascinating for the introduction of the linker for the immobilization (Figure 2-ii). Using an imidazoline, an amine, and a salicylaldehyde as the building blocks, the synthesis of the imidazoline-aminophenol library was formulated as shown in Scheme 1. After immobilization of the two types of chiral chloromethylated imidazolines (1, 2) onto the polystyryl-sulfonyl chloride, a nucleophilic substitution using (R)- or (S)-phenylethylamine (3, 4) provided the four combinations of polymer-supported imidazoline-amines. Further reductive alkylation using salicylaldehydes (5-8) provided a series of imidazoline-aminophenol ligands (L1-L16). After classification of the library by the types of chiral imidazoline, the metal catalysts (C1-C32) were prepared by reaction with CuCl or Cu(OAc)2. In all cases, the polystyrene beads turned to green, which suggested the formation of Cu complexes. With the total of 32 polymer supported chiral catalysts (C1-C32) in hand, the Cu-catalyzed Henry reaction was examined to evaluate the efficiency of the reaction sphere.3,4) After carrying out the asymmetric Henry reaction of nitromethane and o-nitrobenzaldehyde for 48 h, each reaction mixture was
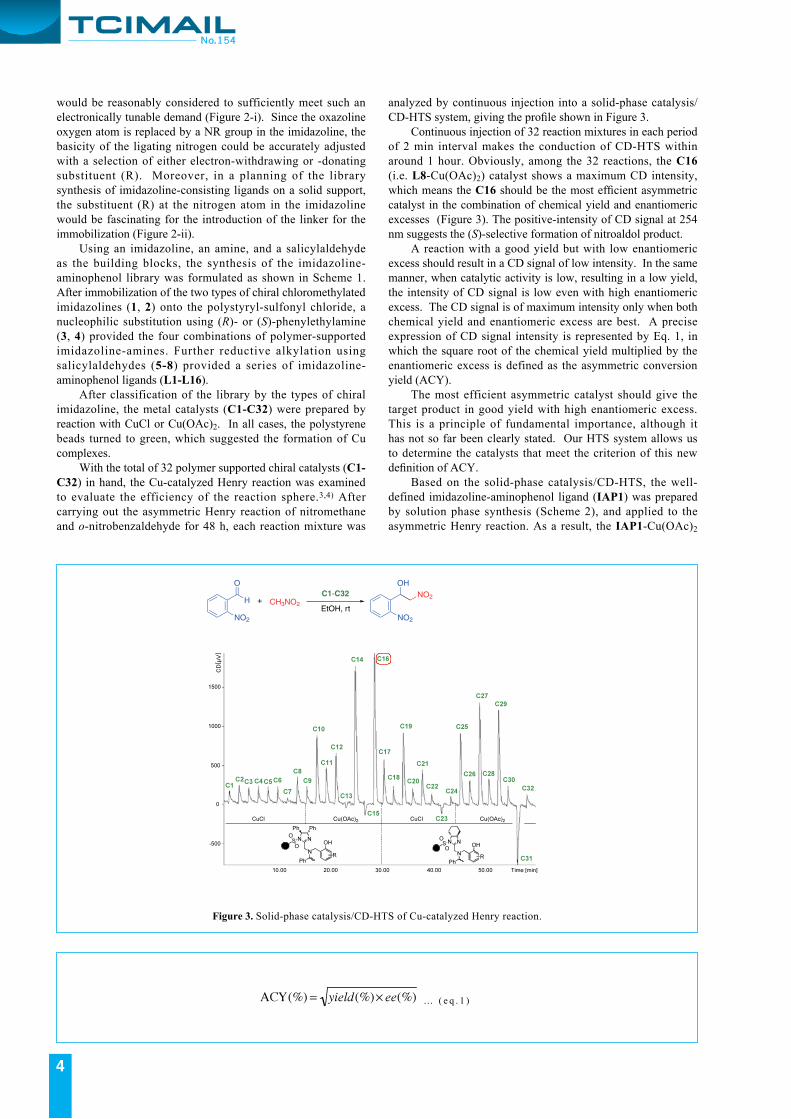
analyzed by continuous injection into a solid-phase catalysis/CD-HTS system, giving the profile shown in Figure 3. Continuous injection of 32 reaction mixtures in each period of 2 min interval makes the conduction of CD-HTS within around 1 hour. Obviously, among the 32 reactions, the C16 (i.e. L8-Cu(OAc)2) catalyst shows a maximum CD intensity, which means the C16 should be the most efficient asymmetric catalyst in the combination of chemical yield and enantiomeric excesses (Figure 3). The positive-intensity of CD signal at 254 nm suggests the (S)-selective formation of nitroaldol product. A reaction with a good yield but with low enantiomeric excess should result in a CD signal of low intensity. In the same manner, when catalytic activity is low, resulting in a low yield, the intensity of CD signal is low even with high enantiomeric excess. The CD signal is of maximum intensity only when both chemical yield and enantiomeric excess are best. A precise expression of CD signal intensity is represented by Eq. 1, in which the square root of the chemical yield multiplied by the enantiomeric excess is defined as the asymmetric conversion yield (ACY). The most efficient asymmetric catalyst should give the target product in good yield with high enantiomeric excess. This is a principle of fundamental importance, although it has not so far been clearly stated. Our HTS system allows us to determine the catalysts that meet the criterion of this new definition of ACY. Based on the solid-phase catalysis/CD-HTS, the well-defined imidazoline-aminophenol ligand (IAP1) was prepared by solution phase synthesis (Scheme 2), and applied to the asymmetric Henry reaction. As a result, the IAP1-Cu(OAc)2
H
O
NO2
+ CH3NO2
OHNO2
NO2
C1-C32
EtOH, rt
Figure 3. Solid-phase catalysis/CD-HTS of Cu-catalyzed Henry reaction.
… ( e q . 1 )

No.154
5
N N
Cl
Ph Ph
Ts N N
NH
Ph Ph
Ts
Ph
N N
N
Ph Ph
Ts
Ph
OHBr
X
a b
IAP1: X = BrIAP2: X = NO2
N N
Ph Ph
N
SO
O
Ph
OHBr
Br
C16
Cu(OAc)2
R H
OCH3NO2
NH
+
+ R'NO2
IAP 1-Cu(OAc)2
IAP 1-CuOTf
R
OHNO2
NH
R'
NO2
NH
+ R'NO2
IAP 2-CuOTfNH R'
NO2
NO2
OHNO2
EtOH, rt, 40-48 h
98%, 94% ee
OHNO2
76%, 95% ee
OCH3
OHNO2
90%, 90% ee
OHNO2
81%, 91% ee
OHNO2
99%, 90% ee
NH
NO2
O2N
orIAP 2-CuOTf
IAP 1 (20 h): 99%, 80% ee IAP 2 (12 h): 99%, 81% ee
toluene, rtHFIP (2 eq.)
(10 mol %)
NH
NO2
Ph
IAP 1 (28 h): 97%, 83% ee IAP 2 (15 h): 99%, 81% ee
NH
NO2
IAP 1 (98 h): 50%, 85% ee IAP 2 (72 h): 80%, 80% ee
NH
NO2
IAP 1 (47 h): 99%, 75% ee IAP 2 (8 h): 98%, 72% ee
NH
NO2
Cl
(IAP 1 (38 h): 64%, 89% ee) IAP 2 (24 h): 97%, 92% ee
toluene, 0 °C
NH
NO2
(IAP 1 (21 h): 10%, 55% ee) IAP 2 (24 h): 61%, 75% ee
NH
NO2
CH3
(IAP 1 (72 h): 34%, 83% ee) IAP 2 (24 h): 78%, 83% ee
NH
NO2
(IAP 1 (43 h): 52%, 52% ee) IAP 2 (24 h): 66%, 80% ee
S
a) Henry Reaction
b) Friedel-Crafts Reaction using Indole
c) Friedel-Crafts Reaction using Pyrrole
Scheme 2. Synthesis of imidazoline-aminophenols (IAP1 and IAP2): a) (S)-phenylethylamine, KI, DMF; b) 3,5-dibromosalicyl aldehyde (for IAP1) or 3-bromo-5-nitrosalicyl aldehyde (for IAP2), NaBH3CN, MeOH.
Scheme 3. IAP-Cu catalyzed asymmetric reactions.
catalyst provide the Henry adduct in good yield with up to 95% ee (Scheme3-a).5) When a new and unique chiral ligand is successfully explored, we can examine the utility in various asymmetric reactions. For the example, a catalytic asymmetric Friedel-Crafts alkylation5) of indole with nitroalkene was catalyzed by IAP1-CuOTf complex to give the product with highly enantioselective manner (Scheme 3-b).6) This catalyst system was also extensively applied to the catalytic asymmetric Friedel-Crafts reaction of nitroalkenes with pyrrole, which is less reactive substrate than the indole. For the less reactive pyrrole, newly synthesized nitro-substituted imidazoline-aminophenol (IAP2) is effective to increase the Lewis acidity of Cu catalyst to furnishing the smooth catalytic asymmetric Friedel-Crafts reaction with up to 92% ee (Scheme 3-c).7) In some reactions, the addition of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) is effective for improving both chemical yields and enantiomeric excesses.
4. IAP-Cu Catalyzed Asymmetric Friedel-Crafts/Henry(FCH) Reaction and Friedel-Crafts/Protonation(FCP) Reaction
After the success on the exploration of novel IAP-metal catalyst, the next research project is the development of new asymmetric reactions. We have interested in tandem multi component coupling reaction for providing complex molecules. The obtained compounds having multiple stereogenic centers would provide a fascinating scaffold for facilitating diversity-oriented synthesis, and for reducing the number of chemical steps in target-oriented synthesis. When the IAP-Cu(OTf)2 complex acts as a Lewis-acid catalyst in the Friedel-Crafts reaction of indole with nitroalkene, the nucreophilic attack of indole to the Cu-activated nitroalkene will generate the Cu-nitronate as the coupled intermediate (Scheme 4). Typically the Cu-nitronate is protonated to give the Friedel-Crafts adduct. However, this coupled intermediate is an

No.154
6
No.154
equivalent of the Cu-nitronate for the Henry reaction. According to the reaction mechanism proposed by Evans, the Cu complex bearing moderately basic charged counter anion (e.g. OAc-) would work for generation of a nitronate species.4)
Because the Friedel-Crafts reaction of indole with aldehyde was relatively slow, we planed the tandem Friedel-Crafts/Henry (FCH) reaction of indole, nitroalkene, and aldehyde using the IAP-Cu catalyst. When 1 eq. of benzaldehyde was added to the conventional Friedel-Crafts reaction mixture of indole with nitrostyrene (1:1), the desired three-component coupling FCH product was obtained. The addition of HFIP was effective in enhancing the catalytic reaction, and to perform the reaction with highly
diastereoselective manner. Using the optimized conditions, the first catalytic asymmetric FCH reaction was established as shown in Scheme 5.8)
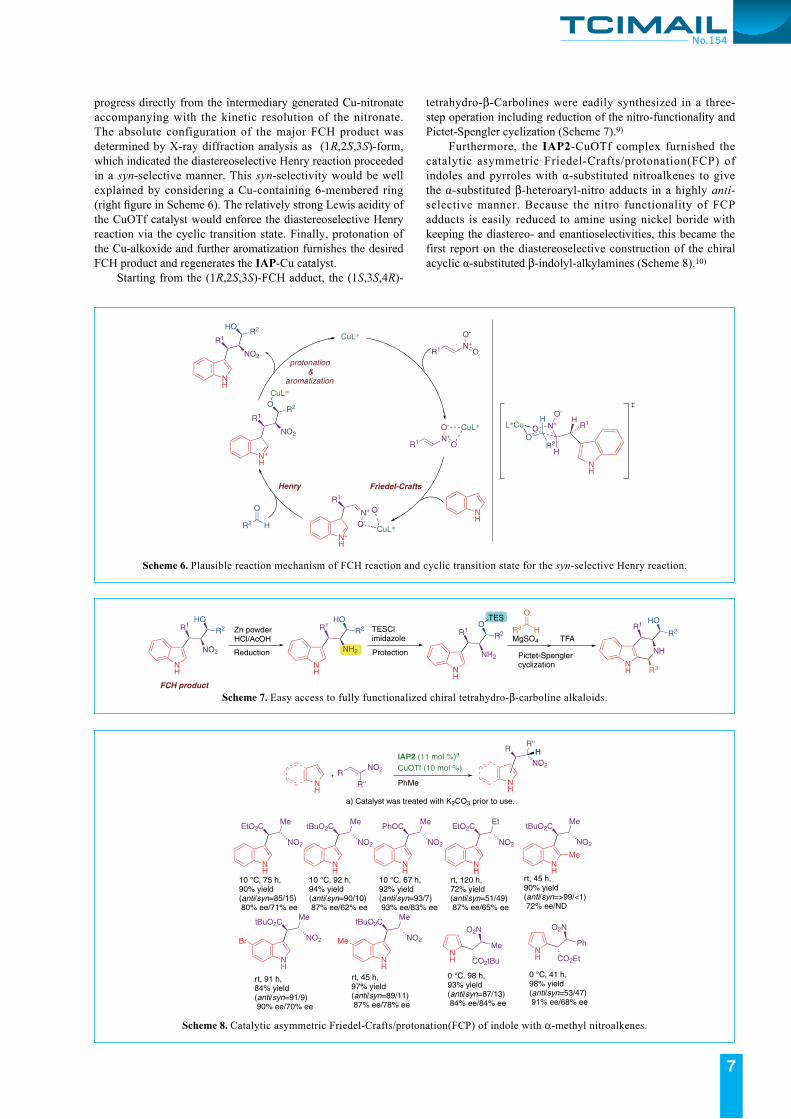
Scheme 6 shows a plausible reaction mechanism of the FCH reaction. At the first step, the nitroalkene is activated by the Lewis acid IAP-Cu catalyst to start the enantioselective Friedel-Crafts reaction. Then, the diastereoselective Henry reaction would be promoted through the Cu nitronate as planned. We confirmed that the reaction of the isolated Friedel-Crafts product and the aldehyde did not occur under the reaction conditions. The enantiomeric excess of FCH product was greatly improved to 99% from ca. 80% ee of the Friedel-Crafts adduct (Scheme 3b). Thus, the Henry reaction is considered to
R1 NO2NH
+R2 H
O+
NH
R1
NO2
R2HO
IAP1-CuOTf (10 mol %)HFIP (2 eq.)
PhMe
NH
Ph
NO2
HO
rt, 29%, ds= 1:9:0, 87% ee0 °C, 84%, ds= 1:16:0, 90% ee (99)a
Br
NH
Ph
NO2
HO
0 °C, 82%, ds= 1:10:0, 90% ee (99)a
Cl
NH
Ph
NO2
HO
0 °C, 90%, ds= 1:10:0, 90% ee (99)a
NO2
NH
Ph
NO2
HO
rt, 82%, ds= 1:3:0, 99% ee
NH
Ph
NO2
HO
rt, 79%, ds= 1:7:0, 99% ee
NH
NO2
HO
rt, 76%, ds= 1:2:0major: 98% eeminor: 97% ee
NH
NO2
HO
0 °C, 66%, ds= 3:4:0major: 99% eeminor: 99% ee
Br
NH
NO2
HO
rt, 76%, ds= 1:2:0major: 90% eeminor: not determined
Ph
N
Ph
NO2
HO
Me0 °C, 84%, ds= 2:3:0major: 99% eeminor: 99% ee
a Enantiomeric excess after single recrystalization
CH3NO2
ligand-Cu(OAc)2
N+O
O-
Culigand
OAc
O
HR N
O
CuO
R
HOAc
ligand
O-
Cu nitronate 6-membered tansition state
NO2R
OH
N+
R
N+ O-
OCu-IAP
RNO2
NH
+IAP-CuOTf R' H
O
NH
** R
NO2
R'HO
Cu-nitronate
Mechanism of Cu-catalyzed Henry reaction
Design of Friedel-Crafts/Henry reaction
H
Scheme 5. Catalytic asymmetric Friedel-Crafts/Henry(FCH) reaction.
Scheme 4. Design of new tandem Friedel-Crafts/Henry reaction.
No.154
7
progress directly from the intermediary generated Cu-nitronate accompanying with the kinetic resolution of the nitronate. The absolute configuration of the major FCH product was determined by X-ray diffraction analysis as (1R,2S,3S)-form, which indicated the diastereoselective Henry reaction proceeded in a syn-selective manner. This syn-selectivity would be well explained by considering a Cu-containing 6-membered ring (right figure in Scheme 6). The relatively strong Lewis acidity of the CuOTf catalyst would enforce the diastereoselective Henry reaction via the cyclic transition state. Finally, protonation of the Cu-alkoxide and further aromatization furnishes the desired FCH product and regenerates the IAP-Cu catalyst. Starting from the (1R,2S,3S)-FCH adduct, the (1S,3S,4R)-
tetrahydro-β-Carbolines were eadily synthesized in a three-step operation including reduction of the nitro-functionality and Pictet-Spengler cyclization (Scheme 7).9)
Furthermore, the IAP2-CuOTf complex furnished the catalytic asymmetric Friedel-Crafts/protonation(FCP) of indoles and pyrroles with α-substituted nitroalkenes to give the α-substituted β-heteroaryl-nitro adducts in a highly anti-selective manner. Because the nitro functionality of FCP adducts is easily reduced to amine using nickel boride with keeping the diastereo- and enantioselectivities, this became the first report on the diastereoselective construction of the chiral acyclic α-substituted β-indolyl-alkylamines (Scheme 8).10)
NH
RNO2
R'' NH
R
NO2
HR''
IAP2 (11 mol %)a
CuOTf (10 mol %)
a) Catalyst was treated with K2CO3 prior to use.
PhMe
NH
Me
NO2
tBuO2C
10 °C, 92 h, 94% yield(anti/syn=90/10) 87% ee/62% ee
NH
Me
NO2
PhOC
10 °C, 67 h, 92% yield(anti/syn=93/7) 93% ee/83% ee
NH
Et
NO2
EtO2C
rt, 120 h, 72% yield(anti/syn=51/49) 87% ee/65% ee
NH
Me
NO2
EtO2C
10 °C, 75 h, 90% yield(anti/syn=85/15) 80% ee/71% ee
NH
Me
NO2
tBuO2C
rt, 45 h, 90% yield(anti/syn=>99/<1) 72% ee/ND
Me
NH
Me
NO2
tBuO2C
rt, 91 h, 84% yield(anti/syn=91/9) 90% ee/70% ee
Br
NH
Me
NO2
tBuO2C
rt, 45 h, 97% yield(anti/syn=89/11) 87% ee/78% ee
Me
NH CO2tBu
Me
O2N
0 °C, 98 h, 93% yield(anti/syn=87/13) 84% ee/84% ee
NH CO2Et
Ph
O2N
0 °C, 41 h, 98% yield(anti/syn=53/47) 91% ee/68% ee
NH
R1
NO2
HOR2
NH
R1
NH2
HOR2
NH
R1
NH2
O
R2
TES
NH
NH
R1 HO
R2
R3
O
HR3
Reduction Protection Pictet-Spenglercyclization
Zn powderHCl/AcOH
TESClimidazole MgSO4 TFA
FCH product
NH
R1 N+
O
O-CuL*
R1 N+
O
O- CuL*
NH
NO2
R1R2HO
NH
N+
R1
O-
O-CuL*R2 H
O
NH
NO2
R1R2O
CuL*
Friedel-CraftsHenry
protonation&
aromatization
N+
O
L*Cu O
H
H
R2
O-
HR1
NH
+
+
Scheme 8. Catalytic asymmetric Friedel-Crafts/protonation(FCP) of indole with α-methyl nitroalkenes.
Scheme 7. Easy access to fully functionalized chiral tetrahydro-β-carboline alkaloids.
Scheme 6. Plausible reaction mechanism of FCH reaction and cyclic transition state for the syn-selective Henry reaction.

No.154
8
No.154
5. [3+2] Cycloaddition of Iminoesters with Nitroalkenes for Constructing Novel Chiral pyrrolidines
After the success of the control of three contiguous strereogenic centers using FCH reaction, the project moved on the control of four contiguous strereogenic centers on the cyclic compounds. Pyrrolidines, 5-membered N-heterocycles, are widely observed in biologically significant compounds. Due to the importance of pyrrolidines in pharmaceutical research, catalytic asymmetric 1,3-dipolar cyclization has been successfully utilized for the stereoselective construction of the pyrrolidine ring. The reaction of the azomethine ylide generated from an iminoester and a nitroalkene is a fascinating reaction system, in which an additional nitro functionality can be introduced on the pyrrolidine ring. In this particular reaction, the combination of four stereogenic centers can provide up to eight diastereomers. When a trans-nitroalkene is utilized, the stereoconjunction between the 3- and 4-positions is fixed, and four diastereomers are possible, classified as endo, exo, endo’,
and exo’ isomers (Scheme 9).11)
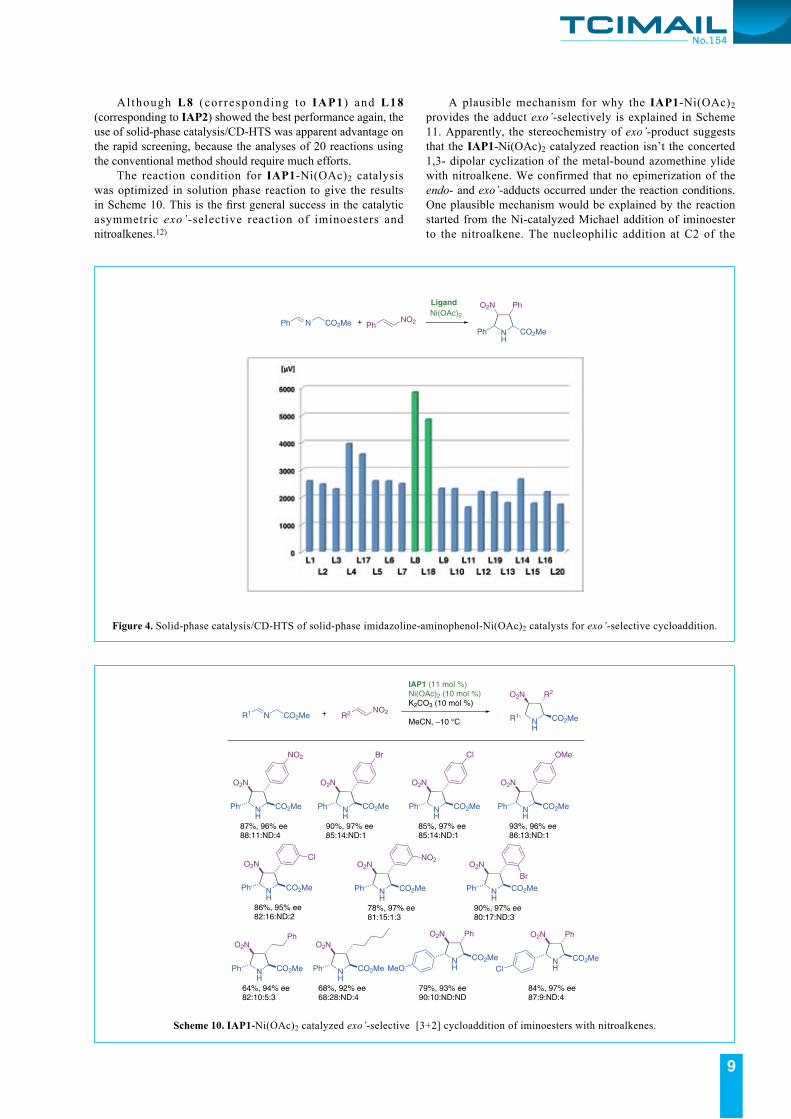
There are many outstanding works on the catalytic asymmetric endo- and exo-selective pyrrolidine ring construction. However, before starting our research, there are no reports on exo’-product formation even in the racemic form. The study was stated from the screening of the appropriate metal salt for giving the exo’-adduct selectively, and we found that Ni(OAc)2 facilitated the production of the exo’-product. The isolated exo’-product showed small, but unique CD signal at 280 nm. Based on these preliminary information, the “Solid-phase catalysis/CD-HTS” using the solid-phase imidazoline-aminophenol-Ni(OAc)2 catalysts was re-examined for exploring the appropriate catalysts for the asymmetric exo’-selective 1,3-dipolar cyclization. With including the p-nitro phenol derivatives, 20 solid-phase catalysts were prepared as coded in Table 1, and the CD-HTS were conducted in Figure 4. By monitoring the CD signals at 280 nm, the solid-phase L8-Ni(OAc)2 and L18-Ni(OAc)2 catalysts recorded superior CD intensities among the 20 solid-phase catalysts.
R1 N CO2R2 + R3 NO2
NH
R3O2N
R1 CO2R2
endo
NH
R3O2N
R1 CO2R2
exo
NH
R3O2N
R1 CO2R2
endo'
NH
R3O2N
R1 CO2R2
exo'
Scheme 9. Possible diastereomers generated in the pyrrolidine synthesis using an iminoester and a trans-nitroalkene.
L R1,R1 R2 R3 R4 L R1,R1 R2 R3 R4
L1 A C H H L9 B C H H
L2 A C H tBu L10 B C H tBu
L3 A C H Br L11 B C H Br
L4 A C Br Br L12 B C Br Br
L17 A C Br NO2 L19 B C Br NO2
L5 A D H H L13 B D H H
L6 A D H tBu L14 B D H tBu
L7 A D H Br L15 B D H Br
L8 A D Br Br L16 B D Br Br
L18 A D Br NO2 L20 B D Br NO2
Table 1. Ligand (L) code.
N N
NR2
OHSO
O
R1 R1
R3
R4
A = (S,S)-diphenylB = (R,R)-cyclohexylC = (R)-phenetylD = (S)-phenetyl
R1, R1:
R2:
No.154
9
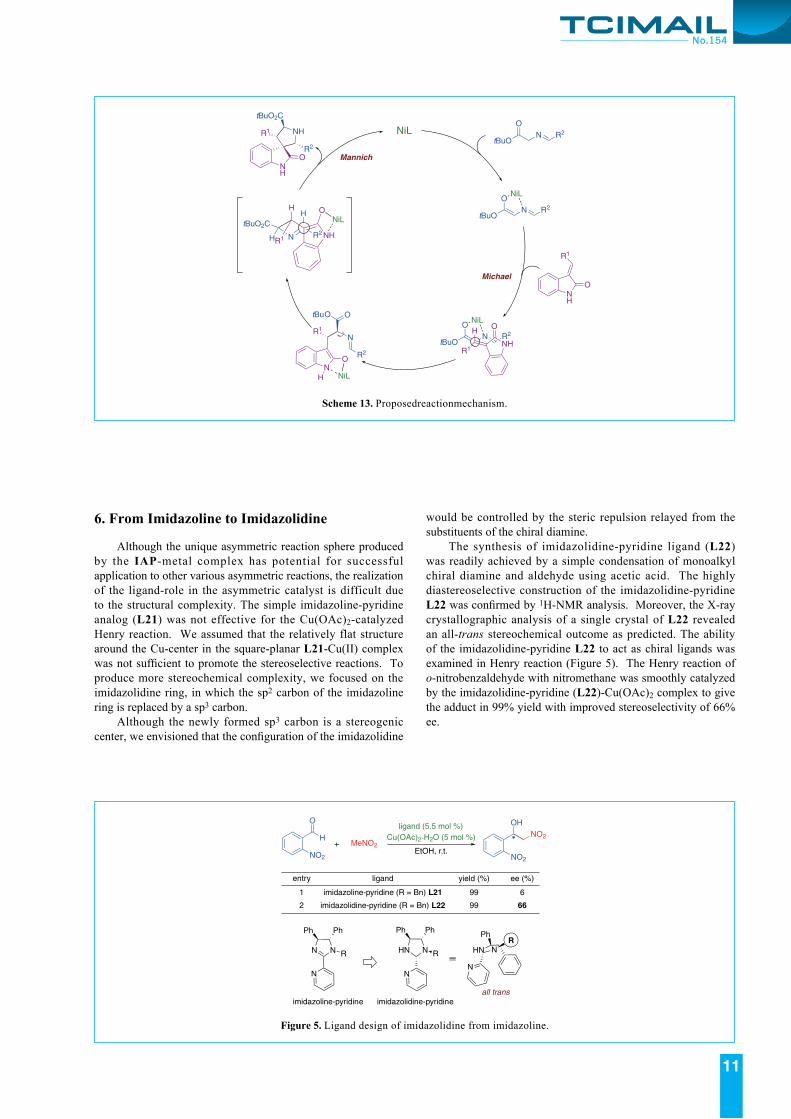
Al though L8 (corresponding to IAP1 ) and L18 (corresponding to IAP2) showed the best performance again, the use of solid-phase catalysis/CD-HTS was apparent advantage on the rapid screening, because the analyses of 20 reactions using the conventional method should require much efforts. The reaction condition for IAP1-Ni(OAc)2 catalysis was optimized in solution phase reaction to give the results in Scheme 10. This is the first general success in the catalytic asymmetric exo’-selective reaction of iminoesters and nitroalkenes.12)
A plausible mechanism for why the IAP1-Ni(OAc)2 provides the adduct exo’-selectively is explained in Scheme 11. Apparently, the stereochemistry of exo’-product suggests that the IAP1-Ni(OAc)2 catalyzed reaction isn’t the concerted 1,3- dipolar cyclization of the metal-bound azomethine ylide with nitroalkene. We confirmed that no epimerization of the endo- and exo’-adducts occurred under the reaction conditions. One plausible mechanism would be explained by the reaction started from the Ni-catalyzed Michael addition of iminoester to the nitroalkene. The nucleophilic addition at C2 of the
R2 NO2
IAP1 (11 mol %)Ni(OAc)2 (10 mol %)K2CO3 (10 mol %)
MeCN, –10 °C+
NH
CO2Me
O2N R2
R1R1 N CO2Me
NH
CO2Me
O2N Ph
NH
CO2Me
O2N Ph
MeO Cl
NH
CO2Me
O2N
Ph NH
CO2Me
O2N
Ph NH
CO2Me
O2N
Ph
NO2 Br Cl
NH
CO2Me
O2N
Ph
OMe
NH
CO2Me
O2N
Ph
NO2
NH
CO2Me
O2N
Ph
Br
NH
CO2Me
O2N
Ph
Ph
NH
CO2Me
O2N
Ph
NH
CO2Me
O2N
Ph
Cl
87%, 96% ee88:11:ND:4
90%, 97% ee85:14:ND:1
85%, 97% ee85:14:ND:1
93%, 96% ee86:13:ND:1
86%, 95% ee82:16:ND:2
78%, 97% ee81:15:1:3
90%, 97% ee80:17:ND:3
64%, 94% ee82:10:5:3
68%, 92% ee68:28:ND:4
79%, 93% ee90:10:ND:ND
84%, 97% ee87:9:ND:4
Scheme 10. IAP1-Ni(OAc)2 catalyzed exo’-selective [3+2] cycloaddition of iminoesters with nitroalkenes.
Figure 4. Solid-phase catalysis/CD-HTS of solid-phase imidazoline-aminophenol-Ni(OAc)2 catalysts for exo’-selective cycloaddition.
LigandNi(OAc)2
Ph N CO2Me + PhNO2
NH
PhO2N
Ph CO2Me

No.154
10
No.154
nitroalkene would be managed by an interaction between the nitro functionality and Ni center for controlling the nucleophilic reaction in anti-selective manner. However, after the anti-selective Michael addition, because the neutral Ni center cannot coordinate to both the nitro functionality and the iminoester, the Ni atom would spontaneously flip to the nitronate for opening the cyclic intermediate. Before the subsequent Mannich reaction, the C-N single bond must rotate to give the exo’-isomer via the most stable transition state. DFT calculations at the level of 6-31G/B3LYP also suggest that the exo’-adduct is the most stable among the four possible isomers depicted in Scheme 9.
Recently, the IAP1-Ni(OAc)2 enabled the catalytic asymmetric exo'-selective [3+2] cycloaddition for constructing stereochemically diversified spiro[pyrrolidin-3,3'-oxindole]s (Scheme 12).13)
Using the IAP1-Ni(OAc)2, the reaction proceeds in a similar stepwise Michael-Mannich reaction to provide the exo'-adducts as the stable isomers. The reaction would proceed by replacing the counter-substrate of nitroalkene in the previous study13) with the alkenyl amide part of methyleneindolinone to give (3R,4’S)-product as shown in Scheme 13.
NiX2
R2 NO2
Michael
Mannich
NH
O2N R2
R1
O
OMe
*N
MeO
OR1
NMeO
OR1
Ni
*
R1NMeO
R2
O
N+O- O
Ni
*X
NMeO
O
R1
Ni
*
R2
N+
-OO
C3
exo'
NR1
H
H
N+
O
O
Ni
R2
H
H
CO2Me*
X
X
X
NH
O
NH
R1
tBuO2C
R2IAP1 (11 mol %)Ni(OAc)2 4H2O (10 mol %)
NEt3 (10 mol %), MeOH, 0 °C
exo'NH
O
R2
NH
O
NH
CO2tBu
98% yielddr=>99/<196% ee
NH
O
NH
CO2tBu
96% yielddr=97/396% ee
Me
NH
O
NH
CO2tBu
89% yielddr=98/297% ee
Cl
NH
O
NH
CO2tBu
90% yielddr=>99/<199% ee
Br
NH
O
NH
CO2tBu
MeO2C
99% yielddr=80/18/297% ee
NH
O
NH
CO2tBu
90% yielddr=98/297% ee
BrNH
O
NH
CO2tBu
87% yielddr=99/195% ee
NH
O
NH
CO2tBu
84% yielddr=97/399% ee
Br
R1 N CO2Me
Scheme 11. Plausible reaction mechanism on exo’-selective [3+2]-cycloaddition.
Scheme 12. Catalytic asymmetric synthesis of exo'- spiro[pyrrolidin-3,3'-oxindole]s.
No.154
11
6. From Imidazoline to Imidazolidine
Although the unique asymmetric reaction sphere produced by the IAP-metal complex has potential for successful application to other various asymmetric reactions, the realization of the ligand-role in the asymmetric catalyst is difficult due to the structural complexity. The simple imidazoline-pyridine analog (L21) was not effective for the Cu(OAc)2-catalyzed Henry reaction. We assumed that the relatively flat structure around the Cu-center in the square-planar L21-Cu(II) complex was not sufficient to promote the stereoselective reactions. To produce more stereochemical complexity, we focused on the imidazolidine ring, in which the sp2 carbon of the imidazoline ring is replaced by a sp3 carbon. Although the newly formed sp3 carbon is a stereogenic center, we envisioned that the configuration of the imidazolidine
would be controlled by the steric repulsion relayed from the substituents of the chiral diamine. The synthesis of imidazolidine-pyridine ligand (L22) was readily achieved by a simple condensation of monoalkyl chiral diamine and aldehyde using acetic acid. The highly diastereoselective construction of the imidazolidine-pyridine L22 was confirmed by 1H-NMR analysis. Moreover, the X-ray crystallographic analysis of a single crystal of L22 revealed an all-trans stereochemical outcome as predicted. The ability of the imidazolidine-pyridine L22 to act as chiral ligands was examined in Henry reaction (Figure 5). The Henry reaction of o-nitrobenzaldehyde with nitromethane was smoothly catalyzed by the imidazolidine-pyridine (L22)-Cu(OAc)2 complex to give the adduct in 99% yield with improved stereoselectivity of 66% ee.
NiL
NH
O
R1
NH
O
NH
R2
tBuO2C
R1
Michael
Mannich
N
R2
OO
R1
NO
NiLH
tBu
tBuO2C
NH
H
R2 NH
H O
R1
NiL
NO
tBuOR2
NtBuO
R2O
NiL
NtBuO
R2O
NiLH
R1NH
O
Scheme 13. Proposedreactionmechanism.
NO2
H
O
MeNO2
NO2
∗∗ NO2
OH
EtOH, r.t.
Cu(OAc)2-H2O (5 mol %)+
ligand (5.5 mol %)
entry
1
2
ligand yield (%) ee (%)
imidazolidine-pyridine (R = Bn) L22
imidazoline-pyridine (R = Bn) L21 99 6
99 66
N
HN N
PhPh
R
N
N N
PhPh
imidazoline-pyridine imidazolidine-pyridine
R HN N
Ph
N
R
all trans
Figure 5. Ligand design of imidazolidine from imidazoline.

No.154
12
No.154
7. Development of Bis(imidazolidine)pyridine (PyBidine)
With employing imidazolidine, the new C2-symmetric bis(imidazolidine)pyridine ligand (PyBidine) was designed and easily synthesized in a single condensation of 2,6-pyridyl aldehyde and optically active (S,S)-diphenylethylene diamine (Scheme 14). The newly developed PyBidine ligand readily provided the metal complex with various metal salts. The structure of PyBidine-Cu(OTf)2 complex was determined by X-ray crystallographic analysis of a single crystal as an aqua complex (Figure 6). Two triflate anions occupy the apical positions, and the imidazolidines of PyBidine coordinate to the Cu with keeping the proton at the nitrogen atoms. The terdentate-coordination feature of PyBidine w a s c o m p a r e d w i t h t h e f a m o u s C 2 - s y m m e t r i c a l bis(oxazolinylpyridine) ligand (i.e., pybox) in Figure 7.15, 16) In the pybox-metal complex, pyridine and oxazoline rings lie sprawled on the equatorial plane in coordination with the metal center, and two alkyl groups at the stereogenic center of the oxazoline are arranged as “chiral fences.” In the current PyBidine-Cu(OTf)2 complex, two imidazolidine rings stand
perpendicular to the equatorial terdentate coordination plane. Thus, the imidazolidine rings themselves act as the “chiral fences” in the C2-symmetric PyBidine-Cu(OTf)2 to shield the first and third quadrants (Figure 7). The catalytic activity was demonstrated in the [3+2] cycloaddition of iminoesters with nitroalkenes. Using PyBidine-Cu(OTf)2 catalyst, the highly endo-selective reaction of iminoesters and nitroalkenes was achieved to give the adducts in up to 99% ee (Scheme 15).17)
Based on the selective formation of (2S,3R,4S,5S)-pyrrolidine using (S,S,S,S)-PyBidine-Cu(OTf)2 catalyst, the enantioface selection in the [3+2] cycloaddition is explained as follows. The Cu-catalyst would form the Cu-bound iminoester (or Cu-azomethine ylide). When the iminoester coordinates to the Cu using the equatorial and the upper apical site, the nitroalkene approaches from the second quadrant (Figure 7). In this reaction sphere, for giving the (2S,3R,4S,5S)-pyrrolidine, the nitrogen atom of the iminoester should stand at the upper apical site of Cu to react with nitroalkene using re-face of imine. The following access of the nitroalkene to the activated iminoester with avoiding the steric interaction is appropriate to give the product in endo-selective manner. (Concerted mechanism is also possible to be considered.)
O
NO
N N
O
R1R1
M
NN
N N
N
R1R1
M
R1R1
R2R2
pybox-Metal complex PyBidine-Metal complex
N M N N MN
R1
R1
N
N
R1
R1
NO
NR1
R1
H H
H
H
Figure 7. Structural comparison of PyBidine-metal complex to pybox-metal complex.
Figure 6. X-ray structure of PyBidine-Cu(OTf)2-H2O complex. Counteranions and protons are omitted for clarity.
117.5°
N
N
CuN
N
N
O
93.2°
A
B
PyBidine
NOHC CHO
AcOH (2 eq), CH2Cl2
H2N
Ph Ph
NH2
+ BnClBnHN
Ph Ph
NH2DMF, 35 °C
NNH
N N
HNPh
Ph
Bn
Ph
Bn
Ph
46% yield
99% yield
CsOH•H2O
Scheme 14. Synthesis of bis(imidazolidine)pyridine ligand (PyBidine).
No.154
13
Cu(OTf)2*N
MeO
OR1
NMeO
OR1
Cu
*
R2 NO2
A
NMeO
O
R1
Cu
*
R2
N+
-OO
C3
NH
R2O2N
R1 CO2Me
endo
Ph N COOMe PhNO2
NH
NH
O2N
Ph COOMe Ph COOMe
PhPh O2NligandCu(OTf)2
NN
HNNH
NBn
Ph
Ph
Bn
Ph
Ph
PyBidine
endo exo
entry ligand yield (%) endo:exo ee (%) (endo)
1
2
3
pybox
pybim
PyBidine
trace
trace
96 99:1 99
NN
NN
NBn
Ph
Ph
Bn
Ph
Ph
pybim
NO
NN
O
pybox
Nishiyama, H. et al.15) Beller, M. et al.16)
+ +
NH
O2N
COOMe
Ph
22 h, 96% yieldendo/exo=99/1ee of endo=99% ee
NH
O2N
COOMe
Ph
21 h, 87% yieldendo/exo=95/5ee of endo=98% ee
NH
O2N
COOMe
Ph
22 h, 78% yieldendo/exo=97/3ee of endo=99% ee
NH
O2N
COOMe
Ph
26 h, 75% yieldendo/exo=96/4ee of endo=94% ee
NH
O2N
COOMe
Ph
24 h, 63% yieldendo/exo=95/5ee of endo=93% ee
NH
O2N
COOMe
Ph
22 h, 78% yieldendo/exo=97/3ee of endo=94% ee
MeO MeO
OMe Cl
NH
O2N
COOMePh
21 h, 82% yieldendo/exo=97/3ee of endo=94% ee
NH
O2N
COOMePh
22 h, 84% yieldendo/exo>99/1ee of endo=94% ee
NH
O2N
COOMePh
21 h, 80% yieldendo/exo=95/5ee of endo=98% ee
OMe Cl
a) The catalyst was prepared using Cs2CO3, and Et3N was used in the catalysis. b) Using 10 mol % catalyst.
NH
O2N
COOMePh
Me
21 h, 68% yieldendo/exo=98/2ee of endo=99% ee
a
a, b
R1 N COOMe + R2 NO2
NH
O2N
COOMeR1NH
O2N R2
COOMeR1
R2
endo exo
+Cu(OTf)2 (5 mol %)
(S,S)-PyBidine (5.5 mol %)
r.t., time
dioxane (0.2 M)Cs2CO3 (0.1 eq)R3
R3 R3
Scheme 16. Plausible reaction mechanism for endo-selective [3+2]-cycloaddition.
Figure 8. Remarkable ligand-acceleration effect of PyBidine.
Scheme 15. PyBidine-Cu(OTf)2 catalyzed endo-selective [3+2] cycloaddition of iminoesters with nitroalkenes.
Noteworthy, the pybox-Cu(OTf)2 complex15 and the pybim-Cu(OTf)2 complex16 gave a trace amount of the adduct under the similar conditions (Figure 8). Although the highly selective reaction is reasonably explained by using the well-
organized reaction sphere having PyBidine-Cu complex, it is hard to explain the high catalyst activity. The NH functionality in the PyBidine would play a significant role for activating the substrates in catalysis.

No.154
14
No.154
8. Conclusion
A library-based study using imidazoline-aminophenol ligands succeeded in the creation of a unique reaction sphere. Clearly, predicting the actual catalyst activity of IAP1 and IAP2 simply from its molecular structure is still quite hard even in modern chemistry, and the result reported herein is difficult to achieve without the use of combinatorial technology. Using IAP-Cu catalyst, a novel Friedel-Crafts/Henry (FCH) reaction was successfully conducted for generating three contiguous stereogenic centers. The first exo’-[3+2] cycloaddition of iminoesters with nitroalkenes was achieved by IAP-Ni catalyst. As a designer chiral ligand, new bis(imidazolidine)pyridine (PyBidine) was also introduced for the Cu-catalyzed highly endo-selective [3+2]-cycloaddition of iminoesters with nitroalkenes. The newly provided highly functionalized molecules in this research will be fascinating toward the development of biologically significant compounds for promoting pharmaceutical and medicinal science.
Acknowledgements
This work has been conducted with my students in the laboratory of organic synthetic chemistry, Chiba University. I show my sincere thanks to their hard work. Especially, main players of the current paper are Dr. Naota Yokoyama, Ms. Atsuko Awata, Ms. Makiko Wasai, Mr. Masakiko Watanabe, Ms. Asami Mishiro, and Ms. Kuniko Suzuki. This work was supported by the Industrial Technology Research Grant Program in 2006 from the New Energy and Industrial Technology Development Organization (NEDO) of Japan, a series KAKENHI of Grant-in-Aid for Young Scientists (A), Scientific Research (B), Scientific Research on Priority Areas “Chemistry of Concerto Catalysis”, and for Exploratory Research, and the Workshop on Chirality in Chiba University (WCCU).
References
1) Arai, T.; Watanabe, M.; Fujiwara, A.; Yokoyama, N.; Yanagisawa, A. Angew. Chem. Int. Ed. 2006, 45, 5978-5981.
2) (a) Ding, K.; Ishii, A.; Mikami, K. Angew. Chem. Int. Ed. 1999, 38, 497-501. (b) Mikami, K.; Angelaud, R.; Ding, K.; Ishii, A.; Tanaka, A.; Sawada, N.; Kudo, K.; Senda, M. Chem. Eur. J. 2001, 7, 730-737. (c) Reetz, M. T.; Kühling, K. M.; Hinrichs, H.; Deege, A. Chirality 2000, 12, 479-482. (d) Long, J.; Hu, J.; Shen, X.; Ji, B.; Ding, K. J. Am. Chem. Soc. 2002, 124, 10-11.
3) Shibasaki, M.; Gröger, H. In Comprehensive Asymmetric Catalysis I-III; Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H. Eds.; Springer-Verlag: Berlin-Heidelberg, 1999, Chapter 29.3.
4) Evans, D. A.; Seidel, D.; Rueping, M.; Lam, H. W.; Shaw, J. T.; Downey, C. W. J. Am. Chem. Soc. 2003, 125, 12692-12693.
5) Poulsen, T. B.; Jφrgensen, K. A. Chem. Rev. 2008, 108, 2903-2915.
6) Arai, T.; Yokoyama, N.; Yanagisawa, A. Chem. Eur. J. 2008, 14, 2052-2059.
7) Yokoyama, N.; Arai, T. Chem. Commun. 2009, 3285-3287.8) Arai, T.; Yokoyama, N. Angew. Chem. Int. Ed. 2008, 47,
4989-4992.9) Arai, T.; Wasai, M.; Yokoyama, N. J. Org. Chem. 2011,
76, 2909-2912.10) Arai, T.; Awata, A.; Wasai, M.; Yokoyama, N.; Masu, H. J.
Org. Chem. 2011, 76, 5450-5456.11) (a) Gothelf, K. V.; Jφrgensen, K. A. Chem. Rev. 1998, 98,
863-909. (b) Pellissier, H. Tetrahedron, 2007, 63, 3235-3285.
12) Arai, T.; Yokoyama, N.; Mishiro, A.; Sato, H. Angew. Chem. Int. Ed. 2010, 49, 7895-7898.
13) Awata, A.; Arai, T. Chem. Eur. J. 2012, 18, 8278-8282.14) Arai, T.; Suzuki, K. Synlett 2009, 3167-3170.15) Nishiyama, H.; Sakaguchi, H.; Nakamura, T.; Horihata, M.;
Kondo, M.; Itoh, K. Organometallics 1989, 8, 846.16) Bhor, S.; Anilkumar, G.; Tse, M. K.; Klawonn, M.; Döbler,
C.; Bitterlich, B.; Grotevendt, A.; Beller, M. Org. Lett. 2005, 7, 3393-3396.
17) Arai, T.; Mishiro, A.; Yokoyama, N.; Suzuki, K.; Sato, H. J. Am. Chem. Soc. 2010, 132, 5338-5339.
Introduction of the author :
Takayoshi Arai Professor of Department of Chemistry, Graduate School of Science, Chiba University
Prof. Dr. Takayoshi Arai accomplished his master degree from the University of Tokyo in 1994, and went on to the doctoral course. In 1995, he was appointed as an assistant professor of Prof. Shibasaki’s group at the University of Tokyo, and received his Ph.D. degree in 1998 from the University of Tokyo. In 1997, he moved to the Institute of Scientific and Industrial Research, Osaka University. After carrying out a postdoc in Prof. S. L. Schreiber's group at the Harvard University in USA, he moved to Chiba University as an associate professor in 2003, and became a full professor in 2010. He received the Incentive Award in Synthetic Organic Chemistry, Japan in 2008.His current research interests focus on the exploration of new asymmetric catalysts and development of new synthetic methodology for creating the biologically significant compounds.



![Review Iridium catalysts for the asymmetric …lac.dicp.ac.cn/pdf1/zs/9.pdffrom the industrial viewpoint [31], and of green chemistry from the perspective of pharmaceutical manufacturers](https://img.pdfslide.us/doc/110x75/5b31cdf27f8b9a2c328cd3a5/review-iridium-catalysts-for-the-asymmetric-lacdicpaccnpdf1zs9pdffrom-the.jpg)

























