Embed Size (px)
Citation preview

Evolutionary approach to predicting the binding siteresidues of a protein from its primary sequenceYan Yuan Tsenga and Wen-Hsiung Lia,b,1
aDepartment of Ecology and Evolution, University of Chicago, Chicago, IL 60637; and bBiodiversity Research Center, Academia Sinica, Tapei 115, Taiwan
Contributed by Wen-Hsiung Li, February 10, 2011 (sent for review January 29, 2011)
Protein binding site residues, especially catalytic residues, play acentral role in protein function. Because more than 99% of the ∼12million protein sequences in the nonredundant protein databasehave no structural information, it is desirable to develop methodsto predict the binding site residues of a protein from its primarysequence. This task is highly challenging, because the binding siteresidues constitute only a small portion of a protein. However, thebinding site residues of a protein are clustered in its functionalpocket(s), and their spatial patterns tend to be conserved in evo-lution. To take advantage of these evolutionary and structuralprinciples, we constructed a database of ∼50,000 templates (calledthe pocket-containing segment database), each of which includesnot only a sequence segment that contains a functional pocket butalso the structural attributes of the pocket. To use this database,we designed a template-matching technique, termed residue-matching profiling, and established a criterion for selecting tem-plates for a query sequence. Finally, we developed a probabilisticmodel for assigning spatial scores to matched residues betweenthe template and query sequence in local alignments using a set ofselected scoring matrices and for computing the binding likelihoodof each matched residue in the query sequence. From the likeli-hoods, one can predict the binding site residues in the query se-quence. An automated computational pipeline was developed forour method. A performance evaluation shows that our methodachieves a 70% precision in predicting binding site residues at60% sensitivity.
functional surface | residue matching technique | spatial template | splitpocket
The binding site residues of a protein are essential for thefunction of the protein. Thus, computational methods have
been developed to predict and characterize protein binding sites(1–5). Because the structure of a protein provides much insightinto its function, more than 60,000 protein structures have beenexperimentally determined [in the Protein Data Bank (PDB)](6). Among them, ∼25,000 are bound structures (7, 8), each ofwhich includes its ligand(s) or interactions with other proteins.Computational methods have been developed to identify thebinding site residues in this simplest case (3). For unboundstructures, which do not include any ligand, it is more difficult topredict their binding site residues. Currently, the precision ofmethods for predicting the binding site residues in an unboundstructure ranges from 40% to 60% (4). An even more difficultcase is for proteins that have only primary sequences and nostructural information and no structurally similar homologs.More than 99.5% of the ∼12 million protein sequences in thenonredundant (NR) protein database have no 3D structures.Therefore, it is strongly desirable to be able to predict the bind-ing site residues of a protein from its primary sequence alone.However, difficulties arise from the fact that the binding siteresidues constitute only a small portion of a protein sequence.Three strategies have been applied to this challenging task.
First, support vector machine (SVM) methods and neural net-work methods (9, 10) have been used to extract useful in-formation from sequences and structures. These machine-learning methods require training data derived from sequence or
structural alignments, sequence profiles, and evolutionary anal-ysis. So far, the prediction precision is only about 50% at 20%sensitivity (11). Second, from experimentally derived structuralcoordinates of proteins, one may predict the coordinates ofa new protein to model the native protein folding and structurefrom its primary sequence by homology modeling (12). After thetheoretical structural coordinates are obtained, predictive toolssuch as CASTp (13) and SplitPocket (8) can be applied toidentify putative binding site residues. For example, Adamianet al. (14) identified the determinants of ρ1 GABAc receptorassembly and channel gating by detecting the binding surfaces ofthe ligand-gated ion channel located on the transmembrane.Third, a promising strategy is to assemble a library of pre-computed structural templates (15–19). Then, for an uncharac-terized protein, one can search for putative templates in thelibrary in the hope of matching attributes of the query with thoseof characterized templates. This approach has proved useful toidentify binding site residues of proteins that have structuralcoordinates. However, the focus was on the applicability oftemplates to structures but not sequences. For example, one ofthe active site templates curated by Meng et al. (17) has beenused to detect divergent members in the enolase superfamily.These knowledge-based templates require manual extractionfrom structures and therefore, are difficult to collect in a large-scale manner. One major difficulty is that a large-scale collectionrequires an automated pipeline to establish a diversified tem-plate database for function prediction and characterization in ahigh-throughput manner. A recent study (5) developed an au-tomated computational method, called signature of local activeregions (SOLAR), to construct a basic set of consensus tem-plates of binding surfaces and used the templates to characterizemetalloendopeptidase and nicotinamide adenine dinucleotidebinding proteins.In this study, we have developed a method that uses only
sequences to identify their binding site residues. Note that thebinding site residues of a protein include all substrate bindingresidues and nonbinding residues in the binding sites of theprotein; these include catalytic residues and binding residues.Our approach is based on evolutionary and structural principlesand is different from the approaches described above. We notethat the binding site residues of a protein are usually clustered inthe functional pocket(s) of the protein and possess geometriccharacteristics (Fig. 1A) and that their spatial patterns tend tobe well-conserved in evolution. A key step to use these twoprinciples is to build an extensive database of templates, each ofwhich is a sequence segment that contains a functional pocket(surface). This database is called pocket-containing segmentdatabase (PSD) (Fig. 1B). For each template in PSD, we alsoinclude geometric, biological, and evolutionary information of
Author contributions: Y.Y.T. and W.-H.L. designed research; Y.Y.T. performed research;Y.Y.T. analyzed data; and Y.Y.T. and W.-H.L. wrote the paper.
The authors declare no conflict of interest.1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1102210108/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1102210108 PNAS | March 29, 2011 | vol. 108 | no. 13 | 5313–5318
EVOLU
TION

the binding site residues. This database takes advantage of thefact that functionally relevant residues are much better con-served than the rest of the protein. To use this database, wedesign a template-matching technique, termed residue matchingprofiling (RMP) (Fig. 1C). Because binding surfaces are usuallyhighly conserved in evolution (20, 21), we use their spatial pat-terns to infer the binding site residues in the query sequence. Itworks by selecting a good template from PSD and using thebinding site residues in the template to match those in a querysequence. Thus, another key step is to establish a criterion forselecting templates for a query sequence; we call this criterionthe efficacy of a template. The third important step is to developa probabilistic model for assessing the binding propensity of eachpotential binding site residue in the query sequence. Using thismodel, we assign a spatial score to each pair of matched residuesbetween the template and the query sequence in local alignmentsusing a set of selected scoring matrices, and then, we computethe binding likelihoods of these residues in the query sequence(Fig. 1C). From the likelihoods, we select the putative bindingsite residues in the query sequence. As will be shown, our ap-proach can effectively identify the binding site residues ofa protein from its sequence alone, even when the sequenceidentity between the query and the selected template is below30%. This high performance attests to the evolutionary principlethat the spatial patterns of functionally important amino acidresidues in a protein, such as binding site residues, tend to bewell-conserved in evolution. To carry out the above tasks, wedevelop an automated pipeline.
ResultsDatabase of Spatial Templates. As noted above, the binding siteresidues are usually clustered in functional pockets. Thus, thedatabase of functional pockets that we established previously(i.e., SplitPocket) (8) can be used as a library of binding siteresidues. The number of binding site residues in a functionalpocket in SplitPocket ranges from 5 to 200, with a mean of 30(Fig. S1A). However, because the binding site residues of a pro-tein are usually dispersed over the primary sequence, it is diffi-cult to align them to any query sequence. Therefore, wetransformed SplitPocket into the PSD in which each sequencesegment is the shortest subsequence of the primary sequencethat contains the functional pocket of interest, starting from the
first residue to the last residue of the pocket (Fig. 1B). Thefunctional pocket of a ligand-bound structure is called a splitpocket, because it is split by the ligand (3). PSD also contains thespatial attributes of binding site residues of each pocket stored inSplitPocket, including residue composition, relative distancesbetween pocket residues, residue solvent accessible area, andphysicochemical features (SI Materials and Methods, Filtering Outthe Predicted Residues Located in a Protein Core). Moreover, forthe residues with biological annotations, we mapped the anno-tated coordinates from the feature tables in UniprotKB/Swiss-Prot (22) to the corresponding positions in PSD templates. PSDcurrently holds 48,289 entries from 24,882 structures. Fig. S1Bshows the length distributions of the spatial patterns and tem-plates in PSD; the template length ranges from 7 to 500 residues,with a mean of 211.
Predicting the Binding Site Residues of a Structural Genomics Target.Proteins targeted by structural genomics projects are proteins ofunknown functions. They usually have no known homolog, evenwhen the threshold of sequence identity is set as low as 25%. Asan example of the application of our method, we considerEscherichia coli biotin synthesis protein (BIOH) (256 aa; Swiss-Prot P13001), a protein targeted by structural genomics. Aftermatching this query against the spatial templates in PSD, weobtain 65 potential template hits. We arbitrarily select PDB1wprchain A as our template, because its sequence identity withP13001 is only 22.7%. Although the selected template has a lowsequence identity with the query, it is a good template, because ithas an efficacy of 2.43, which is considerably higher than the cut-off of 1.25 (Materials and Methods has the definition of efficacy).After determining a local alignment, the structural and evolu-tionary information of the binding site residues of the template(PDB1wpr.A) are transferred to the matched or similar residuesin the query (P13001). For each aligned pair of residues, weassign a score to the predicted residue in the query according toa spatial scoring system for computing a residue matching pro-file. For example, W22 of the query (P130001) is aligned to F27 ofthe template (PDB1wpr.A), and the pair W22-F27, a nonperfectmatch, has a spatial score of 1.32; however, the pair S82-S96,a perfect match, has the highest spatial score of 6. Because we donot know the date of divergence between the query and thetemplate, we sample various scoring matrices to conduct evolu-
L273G274Q275G276C277
F278G279V281A293K295
L297M314V323L325I336
T338Y340M341G344S345
L347D348D386R388A390
N391L393A403D404F405
L407A408I411Y416K423
P425
E7 E9 K11 F35 D37
Y39 W53 R55 R57 K65
H76 Y79 R125D145D147
V155E157A192K193L194
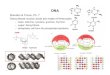
Ψ(T) 1.25≥
20x20
converting
query
RMPcollecting PSD templates
scoring matrices
selectingPSD templates
repeat alignmentswith different matrices
PSD
pocket residuesPSD template
pocket residues
SplitPocket
A B C
Fig. 1. Outline of constructing PSD and conducting RMP. (A) The database SplitPocket (in cyan) contains 48,289 functional surfaces (in green) with theirspatial patterns that consist of site-specific binding site residues. (B) Each pocket in SplitPocket is transformed into a PSD template. A template sequence is theshortest subsequence of the primary sequence of interest that starts from the first residue and goes to the last residue of the pocket. The pocket residues inthe template sequence are indicated in different colors to signify that the spatial pattern of each pocket residue is transformed to the template sequence byfilling in nonpocket residues (in gray). (C) A selected template is repeatedly aligned with the query sequence each time with a scoring matrix in the set ofselected scoring matrices. RMP then predicts the binding site residues for the query by generating a binding profile of likelihoods.
5314 | www.pnas.org/cgi/doi/10.1073/pnas.1102210108 Tseng and Li

tionary matching (Materials and Methods). In addition, by for-mulating a probabilistic model (Fig. 2) we obtain the 12 predictedresidues that have a binding likelihood higher than the cut-offvalue (Table S1). Table S2 shows a comparison of our RMPmethod with psi-BLAST (23) and hidden Markov models(HMMER) (24). RMP predicts 12 site-specific residues inP13001, whereas both psi-BLAST and HMMER give continuoussubsequences that do not include all of the actual binding siteresidues (Table S2). In this example, RMP achieves a precisionas high as 0.83 with a specificity of 99.2% and sensitivity (recall)of 83%, whereas psi-BLAST has a precision of 0.06 witha specificity of 81.0% and a sensitivity of 25%.The BIOH (P13001) structure was solved by the Midwest
Center for Structural Genomics, and its function was identifiedand validated by a panel of enzymatic assays. Fig. 3 shows thepredicted binding region with the 12 predicted residues mappedonto the 3D structure of P13001. Among them, 10 of the 12predicted residues are correct (Fig. 3 A and B). Moreover, thecatalytic triad (S82, D207, and H235) is perfectly matched with thethree residues on the spatial template of PDB1wpr.A (Fig. 3C).Only two predicted residues (V165 and L166) are false positives,and they are also located on the surface.
Importance of Selecting a Good Template. We use a protein fromBacillus halodurans (Q9K901; 192 residues) as a query to showthe importance of having a good template.Using Q9K901 as the query to psi-BLAST PDB, we find
PDB3cng (a nudix hydrolase from Nitrosomonas europaea) as thebest hit. The aligned segment is from positions 57–142 of thePDB3cng.A sequence. The records for PDB3cng.A in PDB,which specify the residues comprising a functional or ligandbinding site of the protein, indicate that the residues in the activesite (C4, C7, C26, C29, N36, I40, T77, E85, E98, L99, Q110, Y112, F148,R149, and L171) of PDB3cng.A have been experimentally tested.We select the six residues (T77, E85, E98, L99, Q110, and Y112)within the aligned segment to match the binding site residues ofQ9K901, but none of them is correctly matched in the psi-BLASTalignment. Thus, PDB3cng.A is not an effective template.For comparison, from PSD, the template derived from
PDB3bhd.A, a human thiamine triphosphatase, is selected, be-cause it has the highest efficacy (3.4) among all potential PSDtemplates. With this template, we extract the spatial pattern ofthe 20 binding site residues from its binding site (Fig. S2).
Among them, the five annotated residues (E9, K11, Y39, H76, andY79) are clustered in the active site of the human thiamine tri-phosphatase. Applying our RMP method (Fig. 4A), we predict20 putative binding site residues in the query sequence (192residues); each predicted residue is assigned an RMP score anda likelihood from our probabilistic model (Materials and Meth-ods). We are able to identify 13 well-aligned residues, which arefunctionally important (true positives in Fig. S2A). In particular,these 13 residues include four of five annotated residues: E8-E9,K10-K11, Y39-Y39, and E77-H76 (the first residue of each pair isfrom the query). Note that the query and the template have afull-length sequence identity <30%, but their aligned binding siteresidues show highly similar spatial patterns (Fig. S2B). Fig. 4Bshows that the binding site residues of the two proteins arealigned along the diagonal, suggesting evolutionary conservationof the specific patterns of binding site residues.The query (Q9K901) protein was targeted by Joint Center for
Structural Genomics. Although its structure coordinates havebeen determined (PDB2gfg), its biochemical function has notbeen characterized, and its class, architecture, topology, homol-ogous (CATH) superfamily (25) fold signature is not yetassigned. Applying our SplitPocket algorithm for shape analysis(Fig. 5), we identified the actual binding surface in PDB2gfg.Athat consists of 20 residues with a similar spatial pattern to that of
A B C
S82−S96
H235−H247
D207−D219
L166
S82
D207H235
V165
Fig. 3. The binding site residues of E. coli BIOH identified by the techniqueof RMP. (A) The 12 predicted binding site residues are clustered tightly intoa cavity (in green). Among them, S82, D207, and H235 are the catalytic residues(colored in red, yellow, and blue, respectively). (B) V165 and L166 (in brown)are false positives. (C) The side chains of the catalytic residues of BIOH(P13001:PDB1m33; chain A) and those of the hit template (PDB1wpr; chainA) are superimposed.
9K22
W24
L81
W82
S83
L11
1F14
3F14
9X15
0G15
4A15
5R16
1L16
2K16
5V16
6L17
0X17
5V17
6L18
0L18
1E18
2I18
3L18
4K18
5T18
6V19
2L20
3Y20
4G20
5Y20
7D20
8G20
9L21
0V21
9D22
1L22
8Y23
4A23
5H23
8F25
8S
0.00
0.02
0.04
0.06
0.08
0.10
0.12
Sequence Position
Lik
elih
oo
d
82S
207D
235H
111F 83
L22
W16
5V20
9L16
6L14
3F81
W24
L17
6L16
1L18
2I18
6V23
4A23
8F14
9X15
4A16
2K18
1E18
3L19
2L18
0L22
1L15
0G17
0X17
5V20
3Y21
9D25
8S20
4G20
5Y21
0V22
8Y15
5R18
4K18
5T20
8G 9K
0.00
0.02
0.04
0.06
0.08
0.10
0.12
Sequence Position
A B
Fig. 2. Residue-matching profile (RMP). The query protein is P13001 [Escherichia coli biotin synthesis protein (BIOH)], and the template is PDB1wpr.A. (A) Thenormalized likelihoods of the predicted residues in the query P13001. (The likelihoods are normalized by the sum of the likelihoods.) The x axis shows thesequence coordinate and the amino acid at the position. (B) The same binding profile as in A, but the residues are ranked in the decreasing order of like-lihoods. The catalytic triad (S82, D207, and H235) of P13001 is composed of the three residues with the highest likelihoods: 0.123, 0.097, and 0.091.
Tseng and Li PNAS | March 29, 2011 | vol. 108 | no. 13 | 5315
EVOLU
TION

PDB3bhd.A. Thus, PDB2gfg from B. halodurans and PDB3bhdfrom humans are homologous but have undergone deep di-vergence. Our shape analysis indicates that their binding surfacesare highly similar, because 13 of the 20 binding site residues arealigned with a sequence identity of 50% and a root mean squaredeviation (RMSD) of 2.6 Å at a P value of 10−7 (as opposed tothe cut-off P value of 10−4) (1, 3). On the basis of spatial patterns,the significant P value implies that these two binding sites poten-tially perform similar molecular functions. Thus, their biochemicalfunctions (thiamine triphosphatase; i.e., EC 3.6.1.28 and geneontology annotations) of PDB3bhd and Q9K901 are likely similar.
Performance of the Spatial Template Approach. We evaluated ourmethod on a diverse set of 145 tested sequences using precisionrecall (PR) curves as described in SI Materials and Methods,Method for Performance Evaluation. The performances at theresidue level are obtained for selected spatial templates witha range of sequence identities with the query defined as (i) α(sequence identity ≤ 80%, (ii) β (sequence identity ≤ 60%), and(iii) γ (sequence identity ≤ 35%). The areas under the obtainedcurves were used to analyze the influence of the spatial templateselection criterion (α, β, and γ) on the precision of discoveringbinding site residues. The curves were obtained by increasing thethreshold likelihood value of a binding residue from 0 to 0.25 witha 0.0025 increment. (The likelihoods were normalized by the sumof the likelihoods.) Fig. 6A shows that the method achieves 83%,80%, and 72% precision at 50% recall (sensitivity) for the α, β,and γ templates, respectively. Overall, the PR-areas under curve(AUC) for α, β, and γ are 0.80, 0.77, and 0.70, respectively. Thepair-wise comparisons indicate that the shapes of spatial tem-plates have been highly conserved, because the performancedecreases slowly. This is potentially useful when a template hasa high efficacy, even if its sequence identity with the query is below35%. Note that the curve slopes are seen at a steady state. At 60%recall, the spatial template approach achieves a precision >70%.In Fig. 6B, we assessed the accuracy across the range of possiblevalues of threshold for template γ and obtained an optimal like-lihood threshold (0.0175) of a binding residue.Predicting the catalytic residues of a protein from its primary
sequence is an even more challenging task, because the un-balance between the number of true positives and the number oftrue negatives is even more extreme than in the case of bindingsite residues. However, as described in SI Materials and Methods,Predictions of Enzyme Catalytic Residues by Selecting SpatialTemplates, our RMP method achieves a precision of 57% ata sensitivity of 50% (Fig. S3).
DiscussionAssessing the Spatial Scores for a Residue Matching Profile. Based ona large-scale analysis of protein binding surfaces (Fig. S1A), thenumber of binding site residues comprises only ∼10% of a pro-tein. Besides binding site residues, other residues may also bewell-conserved in evolution, including the residues that are sub-ject to the constraints for structural stability and protein folding(26, 27). How to separate these two types of residues requiresa good strategy when only the primary sequence of the protein isavailable. Our strategy hinges on establishing a large database ofspatial templates and a spatial scoring system. With the scoringsystem, we are able to generate an RMP to distinguish the resi-dues involved in the function from those involved in stability ina large-scale computation. Moreover, we filter out the residues ina protein core, because they are unlikely involved in any bindingreactivity (SI Materials and Methods, Filtering Out the PredictedResidues Located in a Protein Core). In this study, we focused oncollecting the binding site residues of proteins to construct anextensive database of spatial templates and developing a criterionfor selecting templates and an RMP technique. We found thata template with a high-efficacy value achieves good performance,even when its sequence identity with the query is low. Our studysuggests that the likelihood of a binding site residue has an op-timal threshold of 0.0175 in the iterative alignments of the evo-lutionary matching schema (3, 19, 26).
Assessing the Evolutionary Relationships and Structural Folds of PSDTemplates. A large-scale sequence comparison of PSD templatesreveals conservation in local regions, even when the overall iden-tity between two sequences is low. Interestingly, the selectedtemplates consist of ∼21% of the CATH fold domains that areindeed associated with functions. Note that not all classified folddomains are directly involved in biochemical reactions. Thesespatial templates yield clues of conserved domains that are oftenadopted by proteins as modules to fulfill diverse biological func-tions. For example, by sequence comparison, a total of 430 folddomains in PSD is identified and mapped into a subset of theCATH (2,178 categories in version 3.2) homologous superfamilies.Essentially, they are functional substructures across the fold space.The PSD templates with structure coordinates are proteins thatcan be used for substructural predictions and designing a bindingregion. Especially for a query protein with structural coordinates,the prediction of its binding site residues becomes much easier ifan appropriate template can be found in PSD. For example, thequery protein Q9K901 has structural coordinates (PDB2gfg).From PSD, we find the template PDB3bhd.A, and using foot-printing Pockets Of Proteins (fPOP) (7) to conduct a surface
0 0.02 0.04 0.06 0.08 0.1 0.12 0.14 0.16 0.18 0.2
50
100
150
200 1
50 100
150 200
0
0.05
0.1
0.15
0.2
Likelihood
Q9K901(query) PDB3bhd
(template)
E8
K10
Y39 E6
R55
R57
K68
E77
N37
R12
5A
53D
138
D14
7E
151
E15
7L8
2N
79I1
50C
136
F18
5
0.00
0.05
0.10
0.15
0.20
Sequence Position
Lik
elih
oo
d
A B
Fig. 4. Identifying the putative binding site residues of the query sequence (Q9K901) from B. halodurans. (A) The binding likelihoods of the 20 predictedbinding site residues. (B) The diagonalized alignment of the binding site residues between Q9K901 and PBD3bhd.A indicates the high similarity between thespatial patterns of the binding site residues in the two proteins. Functionally important residues (in solid black) have higher binding likelihoods.
5316 | www.pnas.org/cgi/doi/10.1073/pnas.1102210108 Tseng and Li

comparison (alignment), we identify all of the 20 binding siteresidues of Q9K901 (Fig. 5).
Inferring Function by PSD Templates. Each of the PSD templatescontains the spatial pattern of a functional surface, which includesstructural, functional, and evolutionary information about theprotein, such as the spatial distances between binding site resi-dues. The conservation of a spatial pattern is largely caused byfunctional constraints and allows us to infer the binding siteresidues of a protein from those of a remote homolog (Fig. 4B).Moreover, comparing the spatial patterns of two proteins allowsone to perform functional inference (19) in a reliable manner.
Materials and MethodsThe materials used are described in SI Materials and Methods, Data Used forPerformance Evaluation. Some methods are also described in SI Materialsand Methods.
Defining Spatial Matching Scores and Evaluating Templates. Let m be thenumber of residues in template T and denote tj ∈ T ; j ∈ f1; ::;mg by t�j if tj isa binding site residue. Let n be the number of residues in query sequence Q.
Let B be a 20 × 20 scoring matrix (see below), and for two amino acids μ andb, let θμb ∈ B be their log-odd score. In the alignment between query se-quence Q and template T, assume that ri ∈ Q is aligned with tj ∈ T. Then, fortj = μ, residues ri ∈ Q are classified into four categories Ωμ
k ; k ∈ f1; 2; 3; 4g,with the following spatial matching scores fμri (Eq. 1):
fμri ≡ λσtj
þ
8>>><>>>:
τρμ1; if ri ¼ t�j ðit matches a binding site residueÞ:τρμ2; if ri ≈ t�j ðit is similar to a binding site residueÞ:τρμ3; if ri ≠ t�j ðit is not matched or similar to a binding site residueÞ:τρμ4 ¼ 0; if ri is aligned to tj ða non-binding site residueÞ:
;
[1]
where σtj ∈ f0; 1g is a biological annotation of the amino acid residue tj intemplate T from UniprotKB/Swiss-Prot and PDB, λ; τ ∈ ℜþare two scalingparameters to be estimated, and ρμk ∈ ½0; 1� is defined as follows (Eq. 2):
ρμk ≡1
kΩμkk
Xb∈Ωμ
k
θμb − θμmin
θμmax − θμmin
; [2]
where k k means the number of elements in the set, θμmax ¼ maxðθμbÞ, andθμmin ¼ minðθμbÞ for θμb ∈ B.
The value of ρμk is directly computed from the scoring matrix B, whereasparameters λ and τ can be estimated from the input of tested sequences (seebelow). Note that we set ρμ4 ¼ 0; that is, when a residue in Q is aligned to anonbinding residue, its score is 0, because we are only interested in bindingsite residues. In this approach, we can evaluate the quality of a templateusing spatial matching scores. Thus, we define the efficacy of template T as(Eq. 3)
ΨðTÞ ≡
Prifri
kAðri ; t�j Þk; [3]
where Aðri ; t�j Þ is the set of aligned residue pairs (i.e., the set of residues inthe query sequence that are matched to binding site residues in T). A highefficacy value means that a high percentage of the binding site residues inthe template are matched to the residues in the query sequence.
Parameter Estimation and the Criterion for Selecting Templates. The param-eters λ and τ are related to biological annotation and evolutionary conser-vation. Here, we empirically obtain λ ≈ 2 and τ ≈ 4 (Fig. S4) by evaluating theperformance of the predicted residues of 100 sequences randomly selectedfrom PSD, with geometric measurements and experimental annotations ofbinding site residues from UniprotKB/Swiss-Prot. The method of the PR curvedescribed in SI Materials and Methods, Method for Performance Evaluationis used to assess the final prediction. We repeat the process until the pre-diction performance reaches a steady state.
The selection of templates from PSD for producing RMPs can have a sig-nificant effect on the prediction. Spatial scores are used to determine thequality of a template by how well the binding site residues on a template arealigned with those on a query sequence. The majority of the templates havethe number of binding site residues ranging from 10 to 50 (3) (Fig. S1A).Therefore, an alignment length of a template T that has ≥50 residues and anassociated efficacy of ≥1.25 means that most of the aligned pairs are similarresidues. In particular, the efficacy for a template usually has a higher valuewhen the first and last residues are similar or perfect matches to the querysequence. Therefore, these two residues can be used to screen templates inPSD. If the efficacy of a template is ≥1.25, it is qualified as a template. Atemplate with an efficacy ≥2.5 is considered efficacious. In this way, wesample all potential templates in PSD. We apply the above criterion toscreen potential templates in PSD.
Matching a Query Sequence Against PSD to Select Templates. We match thequery sequence Q against the templates in the PSD by screening sequencealignments with the Smith–Waterman algorithm (28). We gather the hitswith their Smith–Waterman scores and rank the selected hits with their ef-ficacy values. By evaluating the efficacy of each hit template, we can rapidlyselect useful templates. Within an optimal local alignment between a queryand a hit template, the hit template is used to transfer its spatial patterns tothe corresponding positions in the primary sequence of the query protein.Iteratively, we collect such hit templates to create an RMP based on thefollowing probabilistic model.
K 53 R55 R57 K65
H76 Y79 R125D145D147V155E157A192K193L194E4 E6 E8 K10 N37 Y39 A53 R55 R57 68
L75 E77 R125C136E149E151E153N180K181I182E7 E9 K11 F35 D37 Y39 W
template (PDB3bhd.A)
3bhd.A
backbond
2gfg.A
surface
3bhd.A
surface
rotation matrix (4x4)
Q9k901(PDB2gfg.A)
surface alignment
backbond
2gfg.A
A B
C D
F
E
Fig. 5. Shape analysis of the query structure (PDB2gfg.A) and the template(PDB3bhd.A). (A) The actual binding surface of the query. (B) The splitpocket (actual binding surface) of the template. (C) The structural alignmentis performed based on the superimposition of the two binding surfaces asshown in D by the specific rotation matrix in E, which was computed by themethod of fPOP (7). The spatial matching has a RMSD of 2.6 Å at a significantP value of 6.93 × 10−7. (F) In terms of the binding site residues, the sequenceidentity of the two spatial patterns is as high as 50%, although the full-lengthsequences of the two proteins have a sequence identity of only ∼34%.
Tseng and Li PNAS | March 29, 2011 | vol. 108 | no. 13 | 5317
EVOLU
TION

Computing the Binding Likelihood of a Site-Specific Residue. To compute thebinding propensity of a residue ri ∈ Q, i = {1, . . . , n}, we formulate a prob-abilistic model for computing the binding likelihood of ri. In the context ofprotein sequence, the method looks for matched or similar binding siteresidues and attempts to enumerate all possible combinatory compositionsof a binding shape similar to the template. Under the assumption that eachunit evolutionary time interval is equally likely, the probability pμ
ri for residueri to be a specific binding residue μ is calculated as (Eq. 4)
pμri ≡
PTz ;Bw ;Ch
fμriP
Tz ;Bw ;Ch
Pnk¼1 f
μrk
; [4]
where Bw ∈ S are the similarity scoring matrices, Ch ∈ G are the gap penaltiesin the Smith–Waterman algorithm, and Tz ∈ H are the selected templates.
Because it isdifficult toknowhowlongagothequery sequenceseparatedfromthe selected template, we use a set of scoring matrices (S) that represent various
degrees of sequence divergence (3, 19, 26). The degree of divergence, however, ismeasured by the rate of residue substitution in a 20 × 20 matrix. Specifically, weselect BLOSUM45, BLOSUM50, BLOSUM62, BLOSUM80, and BLOSUM90 (29) asthe set of scoringmatrices (S). Using these scoringmatrices, we perform templateevaluation and the matching between a targeted sequence and a selected tem-plate. Then, we use each of them to locate putative binding resides in a querysequence by matching their spatial pattern to that of the binding site residuesin the template. For all selected templates, we conduct iterative alignments toassess the potential binding site residues of the query sequence using the abovescoring matrices. We thereby obtain the binding likelihoods of site-specific posi-tions of functionally important residues for the query sequence. This approach,called residue matching profiling (RMP), is carried out in a fully automated pipe-line, and themethodcanbeused topredict thebinding regionsofanovelprotein.
ACKNOWLEDGMENTS. We thank Dr. Herbert Edelsbrunner for helpfuldiscussions and Dr. Jie Liang for the Volbl package. This study was supportedby National Institutes of Health Grant GM30998.
1. Binkowski TA, Adamian L, Liang J (2003) Inferring functional relationships of proteinsfrom local sequence and spatial surface patterns. J Mol Biol 332:505–526.
2. Xie L, Bourne PE (2007) A robust and efficient algorithm for the shape description ofprotein structures and its application in predicting ligand binding sites. BMCBioinformatics 8(Suppl 4):S9.
3. Tseng YY, Li WH (2009) Identification of protein functional surfaces by the concept ofa split pocket. Proteins 76:959–976.
4. Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA (2009) Predictingprotein ligand binding sites by combining evolutionary sequence conservation and 3Dstructure. PLOS Comput Biol 5:e1000585.
5. Dundas J, Adamian L, Liang J (2011) Structural signatures of enzyme binding pocketsfrom order-independent surface alignment: A study of metalloendopeptidase andNAD binding proteins. J Mol Biol 406:713–729.
6. Berman HM, et al. (2000) The protein data bank. Nucleic Acids Res 28:235–242.7. Tseng YY, Chen ZJ, Li WH (2010) fPOP: Footprinting functional pockets of proteins by
comparative spatial patterns. Nucleic Acids Res 38:D288–D295.8. Tseng YY, Dupree C, Chen ZJ, Li WH (2009) SplitPocket: Identification of protein
functional surfaces and characterization of their spatial patterns. Nucleic Acids Res 37:W384–W389.
9. Gutteridge A, Bartlett GJ, Thornton JM (2003) Using a neural network and spatialclustering to predict the location of active sites in enzymes. J Mol Biol 330:719–734.
10. Petrova NV, Wu CH (2006) Prediction of catalytic residues using Support VectorMachine with selected protein sequence and structural properties. BMCBioinformatics 7:312.
11. Fischer JD, Mayer CE, Söding J (2008) Prediction of protein functional residues fromsequence by probability density estimation. Bioinformatics 24:613–620.
12. Eswar N, et al. (2007) Comparative protein structure modeling using MODELLER. CurrProtoc Protein Sci 50:2.9.1–2.9.31.
13. Dundas J, et al. (2006) CASTp: Computed atlas of surface topography of proteins withstructural and topographical mapping of functionally annotated residues. NucleicAcids Res 34:W116–W118.
14. Adamian L, et al. (2009) Structural model of rho1 GABA(C) receptor based onevolutionary analysis: Testing of predicted protein-protein interactions involved inreceptor assembly and function. Protein Sci 18:2371–2383.
15. Binkowski TA, Freeman P, Liang J (2004) pvSOAR: Detecting similar surface patternsof pocket and void surfaces of amino acid residues on proteins. Nucleic Acids Res 32:W555–W558.
16. Boeckmann B, et al. (2003) The SWISS-PROT protein knowledgebase and itssupplement TrEMBL in 2003. Nucleic Acids Res 31:365–370.
17. Meng EC, Polacco BJ, Babbitt PC (2004) Superfamily active site templates. Proteins 55:962–976.
18. Torrance JW, Bartlett GJ, Porter CT, Thornton JM (2005) Using a library of structuraltemplates to recognise catalytic sites and explore their evolution in homologousfamilies. J Mol Biol 347:565–581.
19. Tseng YY, Dundas J, Liang J (2009) Predicting protein function and binding profile viamatching of local evolutionary and geometric surface patterns. J Mol Biol 387:451–464.
20. Glaser F, et al. (2003) ConSurf: Identification of functional regions in proteins bysurface-mapping of phylogenetic information. Bioinformatics 19:163–164.
21. Xie L, Bourne PE (2008) Detecting evolutionary relationships across existing foldspace, using sequence order-independent profile-profile alignments. Proc Natl AcadSci USA 105:5441–5446.
22. Wu CH, et al. (2006) The Universal Protein Resource (UniProt): An expanding universeof protein information. Nucleic Acids Res 34:D187–D191.
23. Altschul SF, et al. (1997) Gapped BLAST and PSI-BLAST: A new generation of proteindatabase search programs. Nucleic Acids Res 25:3389–3402.
24. Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14:755–763.25. Orengo CA, et al. (1997) CATH—a hierarchic classification of protein domain
structures. Structure 5:1093–1108.26. Tseng YY, Liang J (2006) Estimation of amino acid residue substitution rates at local
spatial regions and application in protein function inference: A Bayesian Monte Carloapproach. Mol Biol Evol 23:421–436.
27. Tourasse NJ, Li WH (2000) Selective constraints, amino acid composition, and the rateof protein evolution. Mol Biol Evol 17:656–664.
28. Smith TF, Waterman MS (1981) Identification of common molecular subsequences.J Mol Biol 147:195–197.
29. Henikoff S, Henikoff JG (1992) Amino acid substitution matrices from protein blocks.Proc Natl Acad Sci USA 89:10915–10919.
0.2 0.4 0.6 0.8 1.0
0.2
0.4
0.6
0.8
1.0
Recall (Sensitivity)
Pre
cisi
on
: 0.80: 0.77: 0.70
0.0 0.2 0.4 0.6 0.8 1.0
0.2
0.4
0.6
0.8
1.0
0.01
0.16
0.31
0.46
0.61
Recall (Sensitivity)
0.01 0.03 0.050.45
0.55
0.65
Threshold
Acc
ura
cy
A B
Fig. 6. Performances of selected spatial templates evaluated by PR curves. (A) The AUC for three sets of templates (α, β, and γ). The AUC decreases only 7%when the AUC of the γ-templates is compared with that of the β-templates. (B) The PR curve for the γ-templates is colored according to the spectrum bar ofthreshold on the right. In Inset, the accuracy of 65% is estimated when the optimal threshold of likelihood is set to 0.0175.
5318 | www.pnas.org/cgi/doi/10.1073/pnas.1102210108 Tseng and Li