Embed Size (px)
Citation preview

EVOLUTION, SPECIATION, AND GENOMICS
OF THE ANDEAN HUMMINGBIRDS
Coeligena bonapartei AND Coeligena helianthea
Catalina Palacios
Laboratorio de Biología Evolutiva de Vertebrados
Departamento de Ciencias Biológicas
Universidad de los Andes
Advisor
Carlos Daniel Cadena PhD
Dissertation committee
Sangeet Lamichhaney PhD
Juan Armando Sánchez PhD
Dissertation submitted in partial fulfillment of the requirements for obtaining the title of
Doctor of Philosophy in Biological Sciences
Bogotá, Colombia
2020

To Tim Minchin and Trevor Noah
To Martina, Emilia, Zoe, Lucia, Antonia, Eva, Gia and Maya
Little bits of light of a better future
“Emilia aprendió a no despreciar nada.
Menos que nada el lenguaje de los hechos,
el que mira en cada peripecia algo único,
aunque derive su conocimiento
de doctrinas generales”
Ángeles Mastretta. Mal de Amores. 1996

Continuidad
Las hojas caen en el oscuro silencio
Desde lo alto de los árboles del bosque
Sobre las aguas plateadas de los ríos corrientes
Hasta el latir profundo de las sombras
Al suelo húmedo donde aguardan los hongos.
Mientras tanto,
Los corazones rojos y húmedos laten desbocados
Al paso de las alas que se agitan
Las lenguas se pronuncian desde el techo de la cabeza
Viajan hacia las corolas
El azúcar a raudales entra a sus sistemas
Atraviesa como hilos los intestinos
Se deslíe y en segundos es sangre
Viaja entre balsas cargadas de oxígeno
Se atropella contra las paredes oscuras
Atraviesa las membranas
Entra al ciclo, uno, dos, tres ATPs son casi 30
Se hace energía, se va en el aire
Retumban los cuerpos a cada impulso
Alas adelante, alas atrás, van al vuelo
Un milisegundo quietos, vuelven al vuelo
Prisioneros de su historia buscan continuarse
Una generación, va otra
Las lenguas desde el cráneo
Los insectos, el néctar, los nidos, los huevos
La humedad
Al interior del bosque
Al borde del bosque
Va otra generación y sin anuncio no son los mismos
Las envolturas de unas plumas que se rompen
La luz que le da paso a los colores se refleja
Uno dorado, el otro rosa
No son los mismos, aunque estén cerca
En el laberinto de sus genes algo ha cambiado
¿Qué los diferencia?
Mientras tanto,
Al húmedo suelo donde los hongos aguardan
Hasta el latir oscuro de las sombras
Sobre las corrientes plateadas de las aguas del río
Desde los árboles altos de los bosques
En el profundo silencio
Caen las hojas.

Index
Acknowledgments ................................................................................................................. 5
Abstract ................................................................................................................................. 7
Resumen .............................................................................................................................. 8
Introduction ........................................................................................................................... 9
“We’re one but we are not the same”1 – Shallow genetic divergence and distinct
phenotypic differences between two Andean hummingbirds: Speciation with gene flow? –
Chapter 1 ............................................................................................................................ 14
How much is too little? – Complete mitochondrial genomes do not distinguish
phenotypically distinct lineages of Andean Coeligena hummingbirds – Chapter 2 ............ 48
Seeking gold and roses – Genomic differentiation and evolution of lineages of Andean
Coeligena Hummingbirds – Chapter 3 ................................................................................ 76
Conclusions ...................................................................................................................... 108

Acknowledgments
I am very grateful to all institutions and people who contributed to this work. I thank the
Universidad de los Andes, the Vicerrectoría de Investigación, and the Facultad de
Ciencias for funding through research grants and the teaching assistantship program. I
thank Colciencias for funding through a PhD scholarship and to Colfuturo for managing it. I
thank the Fundación para la promoción de la investigación y la tecnología from the Banco
de la República for funding. I thank the Moore Laboratory of Zoology at the Occidental
College, especially John McCormack, Eugenia Zarza, and Whitney Tsai. I thank the
Lovette Lab at the Cornell Lab of Ornithology, especially Irby Lovette, Leonardo
Campagna, and Bronwyn Butcher. I thank Christopher Witt at the University of New
Mexico. I thank the Jarvis Lab and the Vertebrate Genome Lab at Rockefeller University,
especially Erich Jarvis and Sadye Paez. I thank the Museo de Historia Natural at the
Universidad de los Andes, the Instituto de Ciencias Naturales at the Universidad Nacional
de Colombia, and the Instituto de Investigación de Recursos Biológicos Alexander Von
Humboldt.
I am deeply grateful to my advisor Daniel Cadena; you supported my ideas no matter how
unusual they were, from games to genomes, you helped me find the resources, the
people, and the way to achieve our work with the best possible quality. In our discussions
and lab meetings your recurrent come-back to theory and to the scientific method helped
me think not only about a singular problem but about the broad picture of questions and
hypotheses; having you as my advisor was a fortunate privilege. I thank all my coauthors;
collaborating with you was a pleasure and your contributions to this work were essential. I
am very grateful to Leonardo Campagna; I had a great time working with you, I learned a
lot, and I enjoyed our conversations; It was a pleasure to find a peer to talk about genes,
genomes, lab work, and so many other varied topics. I am extremely grateful to the
members of my advisory committee, my qualifying exam, and my candidacy exam, Silvia
Restrepo, Andrew Crawford, and Reto Burri; during each occasion I found in you the
knowledge and the encouragement to continue. I thank Gary Stiles whom I owe all my love
for hummingbirds. I thank the Good Will Runners with whom I ran so many kilometers. I
thank my D&D fellows with whom I lived so many adventures. I thank Pedro Andrés
Oróstegui, who knows how to look beyond, and whose views helped me find the way back

countless times. I thank my mother, my father, my sister, and my brother, always by my
side and in my heart.
With all my brain and all my heart, I owe my gratitude to my Friends. Life and discussions,
scientific or not, are always more lovely and interesting with you. To Tania Pérez, Manuel
Franco, María José Contreras, Camila Gómez, Nick Bayly, Andrés Quiñones, Paola
Montoya, María Alejandra Meneses, Laura Céspedes, Mateo Dávila, Melanie Ramírez,
Natalia Gutiérrez, Valentina Gómez, Ghislaine Cárdenas, Laura Mahecha, Santiago
Herrera, Lina Gutiérrez, Luisa Dueñas, Mileidy Betancourth, Jorge Avendaño, Paulo
Pulgarín, Oscar Laverde, David Ocampo, Lucas Barrientos, Ana Aldana, Vicky Flechas,
Andrew Crawford, Leonardo Campagna, Erich Jarvis, Andrés Cuervo, Carlos Guarnizo,
Juan Luis Parra, Catalina Cruz, Iván Páez, Fabian Salgado, Andrea Paz, Gerriet Janssen,
Will Vargas, Sauk Naranjo, Alfa, Milo, and Logan Luminus Veroandi. Without your
constant support and love none of this work would have ever been possible.
I thank you, for reading this document!

Abstract
The evolutionary mechanisms driving lineage differentiation in a system and the genetic
basis of phenotypic differentiation remain central issues in evolutionary biology. In the
Colombian Andes, two hummingbird species, Coeligena bonapartei and C. helianthea,
show striking phenotypic differences in coloration but low genetic differentiation in a
phylogeny of the Coeligena genus. These species comprise five allopatric and sympatric
subspecies. In this thesis I studied the evolution of the lineages in C. bonapartei and C.
helianthea to identify the evolutionary mechanisms driving their divergence and the genetic
basis of their phenotypic differences. I sequenced for the first time the genomes of a
population sample of hummingbirds and I used tools from population genetics,
comparative genomics, niche modeling, and comparative morphology to clarify the
evolutionary relationships among lineages of Coeligena; to test the hypothesis that they
diverged in the presence of gene flow; and to identify candidate genes related to their
phenotypic differences. I found that C. bonapartei and C. helianthea comprise at least four
evolutionary lineages. The evolution of the lineages C. b. eos and C. b. consita was likely
driven by genetic drift in the absence of gene flow, whereas C. b. bonapartei and C.
helianthea diverged in the presence of gene flow driven by natural selection, sexual
selection, or both. The low genetic differentiation among the mitochondrial genomes of
these hummingbirds supports their recent divergence and the occurrence of introgression.
The landscape of genomic differentiation among these lineages shows many peaks
containing genes possibly related to their phenotypic differences. These genes support the
idea that structural coloration in hummingbirds is a polygenic trait closely linked to feather
development. My work contributes to understanding of comparative genomics in
hummingbirds and how species are formed in megadiverse mountain systems in the
tropics.

Resumen
El efecto de los mecanismos evolutivos en la divergencia de un sistema dado, y las bases
genéticas de las diferencias fenotípicas son aún temas centrales en evolución. En la
cordillera Oriental de los Andes colombianos, habitan las especies de colibríes Coeligena
bonapartei y C. helianthea, que son muy diferentes en coloración, pero mostraron baja
divergencia genética en una filogenia del género. Estas especies tienen 5 subespecies
descritas entre alopátricas y simpátricas. En esta tesis estudié la evolución de C.
bonapartei y C. helianthea para identificar los mecanismos evolutivos asociados a su
divergencia y las bases genéticas de sus diferencias fenotípicas. Secuencié por primera
vez los genomas de una muestra poblacional de colibríes, y utilicé herramientas de
genómica comparativa, modelos de nicho, y comparaciones morfológicas, para aclarar las
relaciones filogenéticas entre estos linajes, evaluar la hipótesis de que divergieron en
presencia de flujo génico, e identificar genes candidatos asociados a sus diferencias
fenotípicas. Encontré que C. bonapartei y C. helianthea comprenden al menos 4 linajes
evolutivos. Los linajes C. b. eos y C. b. consita evolucionaron dirigidos principalmente por
deriva genética y sin flujo génico, mientras que C. b. bonapartei y C. helianthea
divergieron en presencia de flujo génico dirigidos por selección natural, sexual o ambas.
La baja diferenciación genética entre los genomas mitocondriales de estos linajes soporta
su reciente divergencia y eventos de introgresión. El paisaje de diferenciación genómica
entre estos linajes muestra varios picos con genes posiblemente asociados a sus
diferencias fenotípicas, que apoyan la idea de que la coloración estructural en colibríes es
un rasgo poligénico estrechamente ligado al desarrollo de las plumas. Con esta tesis
contribuí al desarrollo de la genómica comparativa en colibríes y al entendimiento de
cómo se forman las especies en los sistemas montañosos megadiversos de los trópicos.

Introduction
Biodiversity is a result of the ongoing process of evolution, whereby various mechanisms
playing out in lineage-specific contexts determines which lineages persist, go extinct, or
form new lineages through the process of speciation. Mutation and recombination
ultimately underlie the origin of differentiation, whereas drift, gene flow, and selection are
evolutionary mechanisms proximately governing the persistence or loss of differentiation
among lineages (Coyne & Orr, 2004; Price, 2007). Although lack of gene flow typically
drives speciation when lineages are allopatric (Mayr, 1963) whereas selection is
responsible for differentiation between sympatric lineages (Nosil, 2012), in any particular
case the question of what evolutionary mechanisms drove lineage differentiation is a
central issue in speciation research. Did recently diverged, sympatric lineages differentiate
in the present of gene flow? If so, and selection is strong enough to overcome the
homogenizing effect of gene flow, on which traits is selection acting on? How do different
evolutionary forces shape patterns of genomic differentiation between lineages? Genomic
approaches have furthered our ability to answer these kinds of questions and to
understand the role that the interplay among evolutionary mechanisms has played in the
differentiation and persistence of species (Lamichhaney et al., 2017; Ottenburghs et al.,
2017; Toews et al., 2016a).
The genomic landscape of divergence among lineages may consist of large and few
regions of high differentiation relative to background levels of differentiation (genomic
continents of differentiation, Michel et al., 2010), or may involve multiple, relatively small
regions of differentiation (peaks or islands of differentiation, Feder & Nosil, 2010).
Genomic regions of high differentiation may have contributed to speciation (Turner, Hahn,
& Nuzhdin, 2005), or may reflect processes such as genetic conflict, genetic drift, variable
mutation rates, chromosomal structure and selective sweeps (Kelley et al., 2012).
However, peaks of differentiation are expected to be associated with phenotypic
differentiation and speciation, especially if they exist between recently diverged species
having had limited time to accumulate neutral divergence (Nosil & Feder, 2012). Linking
patterns seen in comparisons of the genomes of species to evolutionary processes is
central to studies on the genomics of speciation. Among animals, studies in birds have led
the field of genomics, since the pioneering sequencing of the first avian genome (ICGSC
International Chicken Genome Sequencing Consortium, 2004) up to the consolidation of

one of the largest and better-quality genomic data sets currently available (Koepfli, Paten,
& Brien, 2015; Rhie et al., 2020). In particular, avian genomics has provided data relevant
for understanding different scenarios of speciation (Edwards et al., 2005; Toews et al.,
2016b), and new studies in closely related taxa contribute to identify genes and genomic
regions related to phenotypic differentiation and speciation. However, studies on
comparative genomics of neotropical birds are still required to understand how the
evolutionary mechanisms drive phenotypic differentiation and speciation in this region.
In this thesis, I worked on the speciation genomics of hummingbirds, a lineage that has
radiated in the Andes of tropical South America where up to 200 species exist (from 363 in
the family Trochilidae, Restall, Rodner, & Lentino, 2007).Out of the nine crown-clades of
the hummingbird family, two clades are mainly restricted to the high Andes: the Coquettes
and the Brilliants (McGuire, Witt, Remsen, Dudley, & Altshuler, 2008). The genus
Coeligena is a member of the Brilliants clade, formed by medium-sized hummingbirds with
straight and long bills with a striking variation in coloration phenotypes among species
(Hilty & Brown, 1986). Some species exhibit dull or dark colors (e.g. brown and black in C.
coeligena, C. wilsoni, and C. prunellei), whereas others exhibit brilliant and lighter colors
(e.g. green, golden and white in C. orina, C. bonapartei, and C. phalerata; Parra, 2010).
Coeligena hummingbirds inhabit mostly humid montane forests in the Andes from Bolivia
to Venezuela where they feed on flowers in the interior or the edge of the forest. A
multilocus phylogeny of Coeligena resolved the evolutionary relationships among species
of the genus (Parra, Remsen, Alvarez-Rebolledo, & McGuire, 2009). In this phylogeny the
species C. bonapartei and C. helianthea stood out as unusual because their sequences
revealed shallow divergence and lack of reciprocal monophyly although they show striking
phenotypic differences.
C. bonapartei and C. helianthea differ strongly in plumage coloration. Both species have
bright green crowns and violet gorgets, but males of C. bonapartei are largely golden
green with fiery golden underparts and rumps, whereas those of C. helianthea are largely
blackish with rose bellies and aquamarine rumps (Hilty & Brown, 1986). Although females
are paler than males, they are also distinctly different. C. bonapartei and C. helianthea are
similar in other traits such as ecology, behavior and morphology; both species feed on
flowering plants in the lower levels of the forests (Hilty & Brown, 1986) and inhabit cloud
forests in elevations between 2000 and 3500m in the Cordillera Oriental of the Andes in
Colombia and Venezuela (Ayerbe-Quiñones, 2015). In this group, five subspecies are

recognized based on differences in plumage coloration and distribution. Three subspecies
are recognized in C. bonapartei: C. b. bonapartei is distributed from Cundinamarca
(Sabana de Bogotá), Boyacá, and Santander departments in the western slope of the
Cordillera Oriental of Colombia; C. b. consita is endemic to the Serranía de Perijá
(northeastern Colombia and northwestern Venezuela); and C. b. eos is from the Cordillera
de Mérida in Venezuela. The latter taxon has been treated as a different species for some
authors but currently is recognized as a subspecies (Remsen et al., 2018). Two
subspecies are recognized in C. helianthea: C. h. helianthea is from Cundinamarca
(Sabana de Bogotá), Boyacá, Santander, and Norte de Santander departments in the
eastern slope of the Cordillera Oriental; and C. h. tamai is from the Tamá Massif in the
border between Colombia (south of Norte de Santander) and Venezuela. In sum, C. b.
consita and C. b. eos are allopatric to all other subspecies, C. b. bonapartei and C. h.
helianthea are parapatric with a zone of sympatry in the south of their ranges, and C. h.
tamai is possibly parapatric to C. b. bonapartei and C. h. helianthea. The recent origin,
diversity of phenotypes, and geographic ranges of these hummingbirds make them
appropriate subjects for studies on the forces leading to speciation in a complex landscape
and on the genetic basis of species differences.
The evolutionary history of C. bonapartei and C. helianthea is unclear due to their similarity
in genetics and overall ecology, their plumage differentiation, and the combination of
allopatric and parapatric geographical distributions of different lineages. In this thesis I
studied the evolution of C. bonapartei and C. helianthea, with a focus on assessing the
evolutionary mechanisms underlying their divergence and speciation. I employed genetic
and genomic resources, niche modeling, morphological comparisons, phylogenetic
reconstructions, and population genetics analyses to answer three main questions: (1)
Does the previously reported lack of genetic divergence between C. bonapartei and C.
helianthea hold with a larger sampling of individuals and genetic markers, and if it does,
could such a pattern be consistent with the hypothesis of divergence with gene flow? (2)
What are the patterns of population structure and the degree of differentiation in the
mitochondrial genomes of the lineages of these hummingbirds? And (3) How is the
genomic landscape of differentiation between C. bonapartei and C. helianthea, and what
candidate genes for phenotypic differentiation can be identified in these species? My work
contributes to understanding how the interaction among evolutionary mechanisms drives
the processes of phenotypic differentiation and speciation in the complex and biodiverse
landscape of the tropical Andes in which these awesome hummingbirds evolved.

References
Ayerbe-Quiñones, F. (2015). Colibríes de Colombia (First). Wildlife Conservation Society.
Coyne, J. A., & Orr, H. A. (2004). Speciation. Sunderland, MA: Sinauer Associates.
Edwards, S. V., Kingan, S. B., Calkins, J. D., Balakrishnan, C. N., Jennings, W. B.,
Swanson, W. J., & Sorenson, M. D. (2005). Speciation in birds: Genes, geography,
and sexual selection. Proceedings of the National Academy of Sciences,
102(Supplement 1), 6550–6557. doi: 10.1073/pnas.0501846102
Feder, J. L., & Nosil, P. (2010). The Efficacy of Divergence Hitchhiking in Generating
Genomic Islands During Ecological Speciation. Evolution, 64(6), 1729–1747. doi:
10.1111/j.1558-5646.2009.00943.x
Hilty, S. L., & Brown, W. L. (1986). A guide to birds of Colombia. Princeton, New Jersey:
Princeton University Press.
ICGSC International Chicken Genome Sequencing Consortium. (2004). Sequence and
comparative analysis of the chicken genome provide unique perspectives on
vertebrate evolution. Nature, 432(7018), 695–716. doi: 10.1038/nature03154
Kelley, J. L., Passow, C. N., Plath, M., Rodriguez, L. A., Yee, M.-C., & Tobler, M. (2012).
Genomic resources for a model in adaptation and speciation research:
characterization of the Poecilia mexicana transcriptome. BMC Genomics, 13(1), 652.
doi: 10.1186/1471-2164-13-652
Koepfli, K., Paten, B., & Brien, S. J. O. (2015). The Genome 10K Project : A Way Forward.
doi: 10.1146/annurev-animal-090414-014900
Lamichhaney, S., Han, F., Webster, M. T., Andersson, L., Grant, B. R., & Grant, P. R.
(2017). Rapid hybrid speciation in Darwin’s finches. Science. doi:
10.1126/science.aao4593
Mayr, E. (1963). Animal Species and Evolution. Cambridge, Massachusetts: Harvard
University Press.
McGuire, J. A., Witt, C. C., Remsen, J. V., Dudley, R., & Altshuler, D. L. (2008). A higher-
level taxonomy for hummingbirds. Journal of Ornithology, 150(1), 155–165. doi:
10.1007/s10336-008-0330-x
Michel, A. P., Sim, S., Powell, T. H. Q., Taylor, M. S., Nosil, P., & Feder, J. L. (2010).
Widespread genomic divergence during sympatric speciation. doi:
10.1073/pnas.1000939107/-
/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1000939107
Nosil, P. (2012). Ecological Speciation (1st ed.). Retrieved from

http://ukcatalogue.oup.com/product/9780199587117.do#.UWMcL5PU-Ak
Nosil, P., & Feder, J. L. (2012). Genomic divergence during speciation: causes and
consequences. Philosophical Transactions of the Royal Society of London. Series B,
Biological Sciences, 367(1587), 332–342. doi: 10.1098/rstb.2011.0263
Ottenburghs, J., Kraus, R. H. S., van Hooft, P., van Wieren, S. E., Ydenberg, R. C., &
Prins, H. H. T. (2017). Avian introgression in the genomic era. Avian Research, 8(1),
30. doi: 10.1186/s40657-017-0088-z
Parra, J. L. (2010). Color evolution in the hummingbird genus Coeligena. Evolution, 64(2),
324–335. doi: 10.1111/j.1558-5646.2009.00827.x
Parra, J. L., Remsen, J. V., Alvarez-Rebolledo, M., & McGuire, J. A. (2009). Molecular
phylogenetics of the hummingbird genus Coeligena. Molecular Phylogenetics and
Evolution, 53(2), 425–434. doi: 10.1016/j.ympev.2009.07.006
Price, T. (2007). Especiation in Birds. Boulder, Colorado: Roberts & Company.
Remsen, J. V., Areta, J. I., Cadena, C. D., Claramunt, S., Jaramillo, A., Pacheco, J. F., …
Zimmer, K. J. (2018). Version 1 December 2018. A classification of the bird species of
South America. Retrieved from
http://www.museum.lsu.edu/~Remsen/SACCBaseline.htm
Restall, R., Rodner, C., & Lentino, M. (2007). Birds of Northern South America. Yale
University Press.
Rhie, A., Mccarthy, S. A., Fedrigo, O., Damas, J., Formenti, G., London, S. E., …
Friedrich, S. R. (2020). Towards complete and error-free genome assemblies of all
vertebrate species. 1–56.
Toews, D. P. L., Campagna, L., Taylor, S. A., Balakrishnan, C. N., Baldassarre, D. T.,
Deane-Coe, P. E., … Winger, B. M. (2016a). Genomic approaches to understanding
population divergence and speciation in birds. The Auk, 133(1), 13–30. doi:
10.1642/auk-15-51.1
Toews, D. P. L., Campagna, L., Taylor, S. A., Balakrishnan, C. N., Baldassarre, D. T.,
Deane-Coe, P. E., … Winger, B. M. (2016b). Genomic approaches to understanding
population divergence and speciation in birds. The Auk, 133(1), 13–30. doi:
10.1642/AUK-15-51.1
Turner, T. L., Hahn, M. W., & Nuzhdin, S. V. (2005). Genomic islands of speciation in
Anopheles gambiae. PLoS Biology, 3(9), e285. doi: 10.1371/journal.pbio.0030285

“We’re one but we are not the same”1 – Shallow genetic divergence and
distinct phenotypic differences between two Andean hummingbirds:
Speciation with gene flow? – Chapter 1
Developed in collaboration with Silvana Garcia1, Juan Luis Parra2, Andrés Cuervo3, Gary
Stiles4, John McCormack5, and Carlos Daniel Cadena1
1 Laboratorio de Biología Evolutiva de Vertebrados, Departamento de Ciencias Biológicas,
Universidad de Los Andes, Carrera 1 No. 18 A 10, Bogotá, Colombia
2 Grupo de Ecología y Evolución de Vertebrados, Instituto de Biología, Universidad de
Antioquia, Calle 67 No. 53-108, Medellín, Colombia
3 Instituto de Investigación de Recursos Biológicos Alexander von Humboldt, Villa de
Leyva, Boyacá, Colombia
4 Instituto de Ciencias Naturales, Universidad Nacional de Colombia, Bogotá, Colombia
5 Moore Laboratory of Zoology, Occidental College, Los Angeles, California, USA
This chapter was published as a research paper in The Auk
https://academic.oup.com/auk/article-
abstract/136/4/ukz046/5556799?redirectedFrom=fulltext
Abstract
Ecological speciation can proceed despite genetic interchange when selection counteracts
the homogenizing effects of migration. We tested predictions of this divergence-with-gene-
flow model in Coeligena helianthea and C. bonapartei, 2 parapatric Andean hummingbirds
with marked plumage divergence. We sequenced putatively neutral markers
(mitochondrial DNA [mtDNA] and nuclear ultraconserved elements [UCEs]) to examine
genetic structure and gene flow, and a candidate gene (MC1R) to assess its role
underlying divergence in coloration. We also tested the prediction of Gloger’s rule that
darker forms occur in more humid environments, and examined morphological variation to
assess adaptive mechanisms potentially promoting divergence. Genetic differentiation
between species was low in both ND2 and UCEs. Coalescent estimates of migration were
consistent with divergence with gene flow, but we cannot reject incomplete lineage sorting
reflecting recent speciation as an explanation for patterns of genetic variation. MC1R

variation was unrelated to phenotypic differences. Species did not differ in macroclimatic
niches but were distinct in morphology. Although we reject adaptation to variation in
macroclimatic conditions as a cause of divergence, speciation may have occurred in the
face of gene flow driven by other ecological pressures or by sexual selection. Marked
phenotypic divergence with no neutral genetic differentiation is remarkable for Neotropical
birds, and makes C. helianthea and C. bonapartei an appropriate system in which to
search for the genetic basis of species differences employing genomics.
Resumen
La especiación ecológica puede ocurrir en presencia de flujo génico si la selección
contrarresta el efecto homogeneizador de la migración. Evaluamos predicciones del
modelo de divergencia con flujo génico en Coeligena helianthea y C. bonapartei, dos
especies parapátricas de colibríes altoandinos con diferencias marcadas en su coloración.
Secuenciamos marcadores putativamente neutrales (ADN mitocondrial [mtADN] y
elementos ultraconservados nucleares [UCEs]) para evaluar estructura genética y flujo
génico, y secuenciamos un gen candidato (MC1R) para evaluar su papel en la divergencia
en coloración. También evaluamos la regla de Gloger, que señala que organismos de
coloraciones más oscuras habitan ambientes más húmedos, y examinamos variación
morfológica para analizar posibles mecanismos adaptativos que podrían haber promovido
la divergencia entre estas especies de colibríes. Encontramos baja diferenciación genética
entre las especies tanto en ND2 como en UCEs. Los estimadores de migración fueron
consistentes con el modelo de divergencia con flujo génico, pero no podemos rechazar la
posibilidad de que los patrones de variación genética reflejen separación incompleta de
linajes debida a especiación reciente. La variación en MC1R fue también muy baja y no
estuvo asociada con las diferencias en coloración. Las dos especies no se diferencian en
el nicho macroclimático que ocupan, aunque son distintas morfológicamente. Aunque
rechazamos la variación en condiciones macroclimáticas como una causa de la
divergencia en estos colibríes, estas especies podrían haberse diferenciado en presencia
de flujo génico debido a otras presiones de selección ecológicas o por selección sexual.
La marcada diferenciación fenotípica sin diferenciación genética en marcadores neutrales
que documentamos es excepcional en aves neotropicales y hace de C. helianthea y C.
bonapartei un sistema ideal para buscar las bases genéticas de la divergencia entre
especies utilizando genómica.

Introduction
New species often arise when geographic isolation of populations allows for divergence
via genetic drift or selection (Mayr 1963, Coyne and Orr 2004). Central to this speciation
model are the ideas that geographic isolation restricts gene flow, thus allowing for
differentiation, and that speciation without geographic isolation is unlikely because gene
flow homogenizes populations (Coyne and Orr 2004). Alternatively, the divergence-with-
gene-flow model proposes that speciation is possible without geographic isolation if natural
selection is sufficiently strong to counteract the homogenizing effect of gene flow (Gavrilets
1999, Nosil 2008, Pinho and Hey 2010, Martin et al. 2013, Morales et al. 2017). Under this
model, phenotypic differentiation may develop in the face of gene flow owing to divergent
selection acting on traits directly associated with reproduction or on traits associated with
those involved in reproduction through pleiotropic effects (Schluter 2001, Servedio 2016).
Assortative mating or selection against hybrids may further facilitate the completion of
reproductive isolation (Coyne and Orr 2004, Schluter 2009, Fitzpatrick et al. 2009).
Several studies provide evidence that natural selection can promote phenotypic
divergence among populations despite gene flow (e.g. Smith 1997; Morgans et al. 2014;
Fitzpatrick et al. 2015) and this may lead to speciation (Hey 2006, Nosil 2008). However,
documenting speciation with gene flow is complicated because of the difficulty of
determining whether shared genetic variation between species is a consequence of
divergence in the presence of migration, hybridization ocurring after speciation, or
incomplete lineage sorting due to recent or rapid divergence (Hey 2006, Pinho and Hey
2010). This difficulty has been partly overcome thanks to the development of tools to
estimate migration between pairs of populations under alternative demographic scenarios
(Hey and Nielsen 2004, 2007; Beerli 2006, Kuhner 2006, Durand et al. 2011). Some
studies using such tools have found incomplete lineage sorting as the cause for lack of
genetic differentiation (Nosil et al. 2009, Wall et al. 2013, Suh et al. 2015), whereas others
support population divergence despite gene flow (Green et al. 2010, Rheindt et al. 2014,
Supple et al. 2015, Kumar et al. 2017). However, compelling evidence that population
divergence has scaled up to the formation of different species in the face of gene flow
remains limited. Nonetheless, the finding that the evolutionary histories of various
organisms are characterized by substantial cross-species genetic exchange (e.g.
Novikova et al. 2016; Zhang et al. 2016; Kumar et al. 2017) implies that attention should

be devoted to understanding the selective mechanisms maintaining species as distinct
entities in the face of gene flow.
In birds, plumage traits are often targets of natural selection. This results in adaptations for
foraging and flight efficiency (Zink and Remsen 1986), camouflage (Zink and Remsen
1986) or conspicuousness (Endler 1993), thermoregulation (Walsberg 1983), and
protection against pathogens (Burtt and Ichida 2004, Goldstein et al. 2004, Shawkey et al.
2007), among others. Because plumage traits are also critical in mate selection and
species recognition, plumage divergence may drive lineage diversification (Price 2007,
Servedio et al. 2011, Hugall and Stuart-Fox 2012, Maia et al. 2013). A frequently observed
pattern in presumably adaptive plumage variation is Gloger’s rule, which states that birds
with darker plumage coloration occur in more humid environments than lighter-colored
conspecifics (Delhey 2019). This pattern is often attributed to adaptation to reduce
bacterial degradation of plumage in humid conditions where bacteria are most abundant
because melanin (the pigment responsible for black plumage color) confers resistance
against these microbes (Goldstein et al. 2004, Peele et al. 2009, Amar et al. 2014).
Because differences in melanic pigmentation can serve as cues for mate choice and
species recognition (Uy et al. 2009), adaptive differentiation in plumage coloration might
thus drive the origin of reproductive isolation. However, we are unaware of studies
explicitly relating the evolution of melanic plumage coloration by natural selection to
population divergence or speciation in the presence of gene flow (but see Cooper and Uy
2017; see also Rosenblum et al. 2017; Pfeifer et al. 2018 for examples involving skin
pigmentation in other animals).
Here, we test the divergence-with-gene-flow model of speciation as an explanation for the
evolution of two Andean hummingbird species, Coeligena helianthea (Blue-throated
Starfrontlet) and Coeligena bonapartei (Golden-bellied Starfrontlet). We studied these
species because: (1) they have largely parapatric ranges in a topographically complex
area of the Andes over which environmental conditions, hence selective pressures, may
differ (Figure 1); (2) They seemingly lack genetic differentiation in neutral markers (Parra
et al. 2009, McGuire et al. 2014) as expected under divergence with gene flow; (3) They
exhibit distinct phenotypic differences (plumage in C. helianthea is considerably darker
than in C. bonapartei) and no hybrids have been reported even where they coexist locally
(except perhaps for a few old specimens; Fjeldså & Krabbe, 1990); and (4), because
variation in melanic pigmentation may reflect adaptation to different environments,

divergence in plumage traits between these hummingbird species might have been driven
by natural selection.
The apparent lack of genetic differentiation between C. helianthea and C. bonapartei
(Parra et al. 2009, McGuire et al. 2014) despite their distinct differences in traits potentially
under selection may reflect divergence with gene flow, contemporary hybridization, or
incomplete lineage sorting (Hey 2006, Suh et al. 2015, Sonsthagen et al. 2016). We here
evaluate predictions of the divergence-with-gene-flow model of speciation and consider
evolutionary mechanisms driving divergence between these species by first addressing
the following questions: (1) does the lack of genetic differentiation between C. helianthea
and C. bonapartei persist with a much larger and geographically extensive sampling and
additional molecular markers relative to earlier work (Parra et al. 2009)?, and (2) are
patterns of genetic variation consistent with a model of divergence in the face of gene
flow? We next asked (3) is color divergence associated with genetic variation in the MC1R
gene, a candidate underlying melanic coloration in various bird species and other
vertebrates? To examine possible mechanisms through which natural selection might have
driven population differentiation we examined whether phenotypic divergence may be
attributable to adaptation to contrasting macro-environmental conditions by asking (4) is C.
helianthea with darker plumage distributed in more humid environments as predicted by
Gloger’s rule? and (5) is there morphometric variation between species which may
suggest adaptation to alternative microhabitats or resources?
Materials and Methods
Study system
Coeligena helianthea inhabits mostly the eastern slope of the Cordillera Oriental of the
Northern Andes from western Meta in Colombia to the Táchira Depression in Venezuela,
and comprises two subspecies: C. h. helianthea occupies most of the range, whereas C.
h. tamai occurs in the Tamá Massif in the border between Colombia and Venezuela
(Figure 1). The distribution of C. bonapartei is not continuous and three subspecies are
recognized: (1) C. b. bonapartei ranges along the western slope of the Cordillera Oriental
in Cundinamarca, Boyacá, and western Santander in Colombia, (2) C. b. consita is
restricted to the Serranía del Perijá, and (3) C. b. eos is endemic to the Cordillera de
Mérida in the Venezuelan Andes (Hilty and Brown 1986; Hilty 2003. Figure 1). Some
authors consider eos a distinct species (Donegan et al. 2015, del Hoyo et al. 2018), but it

is currently treated as a subspecies of C. bonapartei (Remsen et al. 2018). Although the
distributions of C. helianthea and C. bonapartei are not sympatric for the most part, the
nominate subspecies co-occur regionally in Cundinamarca and Boyacá (Gutiérrez-Zamora
2008).
Coeligena helianthea and C. bonapartei differ strikingly in plumage coloration. Although
both species have bright green crowns and violet gorgets, males of C. helianthea are
considerably darker, with a largely greenish back with a rose belly and aquamarine rump;
males of C. bonapartei are largely golden green with fiery gold underparts and rump.
Females are paler than males, but also differ distinctly in plumage, especially in their lower
underparts (Hilty and Brown 1986, Parra 2010). The differences in coloration between
species may reflect variation in the melanin content of feathers (D’Alba et al. 2014),
differences in the nanostructure of feather barbules (Greenewalt et al. 1960), or both.
Tissue samples and DNA sequencing protocols
We collected specimens in Colombia and Venezuela, and obtained tissue samples from
the collections of the Instituto Alexander von Humboldt (IAvH), the Museo de Historia
Natural de la Universidad de los Andes (ANDES), and the Colección Ornitológica Phelps
(Table S1). Our sampling included a total of 62 individuals: 38 specimens of C. bonapartei
(12 C. b. bonapartei, 5 C. b. consita, and 21 C. b. eos) and 24 specimens of C. helianthea
(7 C. h. helianthea, 17 C. h. tamai). Subspecies were assigned based on taxonomic
determination of museum specimens or by geography. We extracted DNA from tissue
samples using either a QIAGEN DNeasy Tissue Kit (Qiagen, Valencia, CA, USA) following
the manufacturer’s instructions or a standard phenol/chloroform extraction protocol. For 60
specimens we amplified by PCR (Appendix A) and sequenced 1,041 base pairs (bp) of the
mitochondrial ND2 gene, and used the data for range-wide phylogeographic and
population genetic analyses (Genbank accession numbers: MG874354-874409). We used
published sequences of C. lutetiae (McGuire et al. 2007, Parra et al. 2009) and C. orina
(McGuire et al. 2014) as outgroups in phylogenetic analyses.
We used a subset of 36 individuals to assess whether color differentiation between
species is associated with nucleotide substitutions in the coding region of the
melanocortin-1 receptor gene (MC1R), a locus responsible for melanic pigmentation in
several birds and other vertebrates (Mundy 2005, Roulin and Ducrest 2013). We amplified
by PCR (Appendix A) and sequenced 788 bp of the 945 bp of the MC1R locus for 6

individuals of C. h. helianthea, 10 C. h. tamai, 8 C. b. bonapartei, 1 C. b. consita and 11 C.
b. eos. All PCR products were cleaned and sequenced in both directions by Macrogen Inc.
or at the sequencing facilities of the Universidad de los Andes (Genbank accession
numbers: MG880079-880114). We assembled, edited, and aligned sequences of the ND2
and MC1R genes using BioEdit 7.2.5 (Hall 1999) and Geneious 9.1.5
(http://www.geneious.com/; Kearse et al., 2012), employing the MUSCLE algorithm (Edgar
2004) and manual editing.
We also employed a sequence capture approach to acquire data from regions flanking
ultraconserved elements (UCEs; Faircloth et al. 2012) for a small sample of 1 individual of
C. h. helianthea, 4 C. h. tamai, 1 C. b. bonapartei, and 1 C. b. consita to obtain a
preliminary overview of genetic divergence between these taxa at a genomic level. We
used a standard library preparation protocol (http://ultraconserved.org/; Faircloth & Glenn,
2012) and enriched the pool of samples for 5,060 UCE loci using the MYbaits_Tetrapods-
UCE-5K probes. We sequenced the pool after quantification using 250 bp paired-end
Illumina MiSeq. Following the PHYLUCE pipelines (Faircloth 2015), we used
Illuminoprocessor (Faircloth 2013) and Trimmomatic (Bolger et al. 2014) to trim reads,
discarded adapter contamination and low-quality bases, and assembled the reads into
contigs using a kmer = 50 and ABySS (Simpson et al. 2009). We aligned the contigs
against the original UCE probes to identify contigs matching UCE loci using LASTZ (Harris
2007). Among the individuals, we aligned unphased sequences of UCE loci using the
default MAFFT v7.13 algorithm (Katoh and Standley 2013). Finally, we pulled out UCE loci
from the Anna’s Hummingbird (Calypte anna) genome (Gilbert et al. 2014, Zhang et al.
2014) to use them as outgroup.
For phylogenetic analyses we used a concatenated alignment of 2,313 UCE loci shared at
least among 3 individuals including the outgroup. Of these, 1,465 loci were present in all
the individuals (mean locus length = 615.1 bp, mean number of individuals per locus in the
incomplete matrix = 7.3). We generated a second concatenated alignment of 1,604 loci
shared among all Coeligena specimens (i.e. without the outgroup). Of these, 389 loci
showed no variation, 75 had only indels (informative or not), 615 had singletons and
indels, and 525 (32.8%) had informative sites (polymorphic sites with each variant
represented in at least two individuals). We used the latter 525 loci or a subset of these for
population genetic analyses (Appendix A), but because our sample size was low we
treated the results from these data as preliminary and interpreted them with caution.

Phylogenetic and Population Genetic Analysis
We used maximum-likelihood and Bayesian inference methods to reconstruct phylogenies
from the CIPRES Portal (http://www.phylo.org/) or locally. We selected a single partition
and the TNR substition model as the best-fit for our ND2 data according to the corrected
Akaike Information Criterion (cAIC) in PartionFider 2.1.1 (Guindon et al. 2010, Lanfear et
al. 2017). To analyze UCE data we used a concatenated alignment of all 2,313 loci for
which we specified 16 partitions and nucleotide substitution models for each partition
following CloudForest analysis (Crawford and Faircloth 2011). We conducted maximum-
likelihood analyses in RAxML (Stamatakis 2014) using the GTR+GAMMA model and non-
parametric bootstrapping under the autoMRE stopping criterion for ND2 and UCE data.
We conducted Bayesian analyses in Mr.Bayes v3.2 (Ronquist et al. 2012). The MCMC
parameters consisted of two runs with four chains ran for 15 million generations sampling
every 100 generations for the ND2 data, and ran for 25 million generations sampling every
500 generations for the UCE data. We discarded the first 10% generations as burn-in
before estimating the consensus tree and posterior probabilities. To account for
heterogeneity among gene trees, we also conducted a species-tree analysis of our data
set of 525 UCE loci using the program ASTRAL (Zhang et al. 2018; details in Appendix A).
We also conducted Bayesian analyses in BEAST 2 (Bouckaert et al. 2014) using the ND2
data to estimate divergence times using a strict clock model of evolution, assuming a Yule
model prior or a coalescent constant-size population prior. We used a substitution rate of
2.5% divergence per million year for ND2 (Smith and Klicka 2010). MCMC runs consisted
of 50 million generations, sampling trees every 1,000 generations, and discarding the first
15,000 trees (30%) as burn-in. Convergence and effective sample sizes of parameter
estimates for Mr.Bayes and Beast 2 results were examined using Tracer 1.7.1 (Rambaut
et al. 2018).
To further examine relationships among ND2 haplotypes, we used an alignment of 885 bp
for which complete data were available for all individuals to construct a median-joining
haplotype network in Network 5.0.0.1 (http://www.fluxus-engineering.com/; Bandelt,
Forster, & Röhl, 1999). To examine genetic structure between species, we calculated Fst
with R package hierfstat (Goudet and Jombart 2015, R Core Team 2017) and AMOVAs
with R package ade4 (Dray and Dufour 2007, R Core Team 2017) assessing significance
using 10,000 permutations (Script S1). Also, we used Structure 2.3.4 (Pritchard et al.
2000) to assess population structure using our 293 SNP data set from UCE loci; because

our sample size was limited, we consider population genetic analyses using UCEs
preliminary and thus do not report on them in detail in the main text (Appendix A).
Testing for Divergence with Gene Flow
To examine whether there has been geen flow between C. helianthea and C. bonapartei
we used Migrate 3.2.1 (Beerli 2009) to estimate the following demographic parameters:
effective population size scaled by mutation rate (), time scaled by the mutation rate (T),
and migration scaled by mutation rate (M = (m / µ)). If there has been gene flow between
species after speciation, then the posterior distributions of M should exclude values of
zero. We also tried to distinguish scenarios of divergence with gene flow and hybridization
following secondary contact using the option of parameter estimation for different moments
through time in Migrate, but because results were unreliable we chose not to report on
these analyses (Appendix A).
For Migrate analyses we employed a ND2 alignment including 23 individuals of C.
helianthea and 17 individuals of C. b. bonapartei/consita (i.e. excluding C. b. eos, which
we found to be genetically distinct; see below). We also used 293 SNPs from our UCE
data for Migrate analyses (Appendix A). Because inference of gene flow requires neutrally
evolving markers, we first confirmed that our data sets met this assumption by calculating
Tajima’s D using DNAsp 5.1 (Librado and Rozas 2009). We determined prior maximum
values for the parameters and M for each species based on several test runs. In final
analyses aimed to estimate gene flow we set prior values to 0.15 for for both species,
and to 1,000 for M in both directions. We ran Migrate in the CIPRES Portal
(http://www.phylo.org/) using a long chain of 3,000 million steps (sampling 1,000,000 steps
recorded every 3,000 steps) with a burn-in of 1,000,000 steps.
MC1R gene analyses
We compared variable sites in MC1R sequences between our study species and
translated sequences to aminoacids to check for synonymous and non-synonymous
substitutions. As reference for comparisons we used sequences of Anna’s Hummingbird
(Calypte anna) and Chimney Swift (Chaetura pelagica) predicted from genome
annotations (Zhang et al. 2014). Because these comparisons revealed no variation
potentially implied in phenotypic variation (see results), we did not conduct any additional
analysis.

Examining the selective regime: niches and morphological differentiation
We tested the hypothesis that natural selection underlies phenotypic divergence in color
between C. heliathea and C. bonapartei through macroclimatic differences in the regions
occupied by these species. Specifically, we tested the prediction of Gloger’s rule that C.
helianthea (with darker plumage) occurs in environments with more humid conditions than
C. bonapartei, and examined whether other macroclimatic conditions that may promote
adaptation differ between environments occupied by these hummingbirds. We examined
ecological differentiation among C. helianthea, C. b. bonapartei/consita and C. b. eos
(which we found to be genetically distinct; see below) using occurrence data,
environmental variables, and measurements of niche overlap (Broennimann et al. 2012).
In addition to the locality data associated with specimens included in molecular analyses,
we obtained occurrence data from eBird (http://ebird.org/content/ebird/), Vertnet
(http://vertnet.org/), GBIF (http://www.gbif.org/), Xeno-canto (http://www.xeno-canto.org/),
and the ornithological collection of the Instituto de Ciencias Naturales of the Universidad
Nacional de Colombia (http://www.biovirtual.unal.edu.co/en/), for a total of 242 records.
After eliminating duplicates and excluding non-reliable locations we retained 196 records
for analysis: 85 of C. helianthea, 75 of C. b. bonapartei/consita, and 36 of C. b. eos (Table
S2).
To delimit the accessible areas for each species we used ecoregions as defined by
Dinerstein et al. (2017). We used all the ecoregions with occurrence records as the
environmental background available for the analysis of niche overlap. We obtained climatic
data from WorldClim (http://www.worldclim.org/ Hijmans et al. 2005), CliMond
(https://www.climond.org/ Kriticos et al., 2012), and EarthEnv
(http://www.earthenv.org/cloud Wilson and Jetz 2016). We clipped layers for ecoregions
and climatic variables to our study area (i.e. longitude -76 to -70 degrees, latitude 3 to 12
degrees) and excluded variables highly correlated to others (r > 0.70) within this area
using the package usdm in R (Naimi 2015, R Core Team 2017). We conducted niche
overlap analyses using 11 variables: three related to temperature, three related to
precipitation, four related to cloudiness, and one related to air moisture (Table S3).
We extracted climatic data from 10,000 points from the background environment and from
the 196 occurrence records and performed a principal component analysis (PCA) to
summarize climatic variation using the package ade4 in R (Dray and Dufour 2007, R Core
Team 2017). With the two first PCA axes, we plotted the densities of each taxon in climatic

space relative to the background using the package ecospat in R (Broennimann et al.
2016, R Core Team 2017). We also used this package to estimate the D statistic (Warren
et al. 2008) to quantify niche overlap (D = 0 indicates different niches, and D = 1 indicates
identical niches), and we performed similarity tests (1,000 iterations) to assess whether
niches are less similar (niche divergence) than expected by chance given background
climatic variation (Script S2). Significant niche divergence with the darker C. helianthea
occupying more humid areas would be consistent with adaptive divergence following
Gloger’s rule, whereas no significant differences in niches would suggest that adaptation
to distinct climatic conditions cannot account for phenotypic differentiation between
species.
We also assessed whether there is morphometric differentiation between species which
may reflect adaptation to different microhabitats or food resources (Stiles 2008) by
measuring 17 traits related to beak, wing, tail and leg morphology (Table S4). We
measured morphological variables from 35 live individuals (17 females and 18 males) of C.
h. helianthea and 46 individuals (23 females and 23 males) of C. b. bonapartei. Using
these data we asked whether individuals of different species and sexes are distinguishable
in multivariate space employing linear discriminant analysis (LDA) using the package
MASS in R (Venables and Ripley 2002, R Core Team 2017). We built ANOVA models to
test for mean differences in all individual variables among species and sexes
simultaneously. Because a few of the variables were not normally distributed according to
Shapiro-Wilk tests, we used Kruskal-Wallis tests for comparisons involving such variables.
Shapiro-Wilk tests, Kruskal-Wallis tests and ANOVAs were performed using basic
functions in R (R Core Team 2017).
Results
Does lack of genetic differentiation between C. helianthea and C. bonapartei persist
with greater sampling and additional markers?
We found low genetic differentiation between C. b. bonapartei and C. b. consita, but both
taxa were markedly differentiated from C. b. eos. Therefore, hereafter we treat C. b.
bonapartei and C. b. consita as a single group, which we refer to as C. b.
bonapartei/consita. Divergence in ND2 of both C. helianthea and C. b. bonapartei/consita
relative to C. b. eos was high, with siginificant Fst values of 0.56 and 0.52, respectively (p
< 0.001 in both cases), and relatively high fractions of genetic variance (59.4% and 52.0%,

respectively) existing between groups in AMOVA. In contrast, ND2 data showed little to no
differentiation between C. helianthea and C. b. bonapartei/consita. Although differentiation
as measured by Fst was significant (p = 0.03), the Fst value was very low (0.07) and only
1.7% of the variance was partitioned between these two taxa in AMOVA, with 98.3% of the
variance existing among individuals within taxa.
Phylogenetic analyses using ND2 data showed that C. b. eos forms a strongly supported
clade (posterior probability PP = 1.0, maximum-likelihood bootstrap MLbs = 87%), which is
sister to a clade lacking strong support (PP = 0.84, MLbs = 69%) formed by C. helianthea
and C. b. bonapartei/consita (Figure 2A). Within the latter clade, relationships among
populations appeared to be determined more by geography than by current species-level
taxonomy: most sequences of the northern subspecies C. h. tamai and C. b. consita
formed a strongly supported clade (PP = 1.0, MLbs = 85%), whereas the majority of
sequences of southern subspecies C. h. helianthea and C. b. bonapartei formed another
moderately supported clade (PP = 0.95, MLbs = 61%).
Haplotype networks confirmed the above findings (Figure 2B): (1) C. helianthea and C. b.
bonapartei/consita shared haplotypes, whereas C. b. eos did not share any haplotypes
with the other taxa; and (2) haplotype groups were more consistent with geography than
with taxonomy. However, networks showed that the latter pattern is not perfect because
two individuals of C. b. bonapartei (from the south) had the haplotype most common in the
north, one C. h. tamai (from the north) had the haplotype most common in the south, and
one C. b. bonapartei had an intermediate haplotype.
Likewise, UCE nuclear markers for 7 individuals did not reveal genetic differentiation
between C. helianthea and C. bonapartei (no data were available for C. b. eos). The
phylogeny estimated using 2,313 concatenated UCE loci showed a well-supported clade
including all sequences of C. helianthea nested within a clade in which the earliest
diverging branches were the samples of (1) C. b. consita and (2) C. b. bonapartei (Figure
2C). The same topology was obtained with the species-tree analysis which considers
gene-tree heterogeneity (Appendix A). Because our data set of 293 SNPs obtained form
UCEs met the assumption of neutrality (Tajima’s D = 1.5; p > 0.1), we were able to use
them for analyses of population genetic structure. Differentiation between species in these
markers was not significant (Fst = 0.2, p = 0.5), and the most likely number of genetic
clusters estimated in Structure was one (K = 1, prob(k =1) = 0.99), although we caution our
sample was small.

We estimated that divergence between the clade formed by C. helianthea and C.
bonapartei lineage and its sister group formed by C. lutetiae and C. orina occurred ca.
0.40 million years ago (95% credibility interval 0.27 to 0.58 million years ago) using a Yule
model prior. Using a coalescent constant-size population prior, the time of divergence was
0.74 million years ago (95% credibility interval 0.48 to 1.01 million years ago, Figure 5 in
Appendix A). Because the latter divergence time is more consistent with published
estimates based on broader phylogenetic frameworks (Parra et al. 2009, McGuire et al.
2014), we focus on estimates using the coalescent constant-size population prior. Given
this model, the time of divergence between the C. helianthea + C. b. bonapartei/consita
clade and C. b. eos was estimated at 0.31 million years ago (95% credibility interval 0.17
to 0.46 million years ago). Because C. helianthea and C. b. bonapartei/consita wre not
reciprocally monophyletic in the ND2 gene tree, we were unable to date their divergence,
but it must be more recent than the time of their divergence from C. b. eos.
Are patterns of genetic variation consistent with divergence in the face of gene
flow?
Our ND2 data set fit the assumption of neutrality (Tajima’s D = -0.68 p > 0.1), which
allowed us to use it for gene flow inference. The analyses suggested that there has been
gene flow from C. helianthea to C. b. bonapartei/consita, whereas gene flow in the other
direction could not be estimated reliably. Mean estimates of migration (M = m / µ) were in
all cases different from zero: M = 725.1 from C. helianthea to C. b. bonapartei/consita and
446.1 from C. b. bonapartei/consita to C. helianthea. However, the estimated posterior
probability distributions of M were wide: 95% credibility intervals ranged from 284.7 to
1,000 from C. helianthea to C. b. bonapartei/consita, and from 0.0 to 628 from C.
helianthea to C. b. bonapartei/consita (Figure 3). Analyses of UCE markers with our limited
sample, however, were inconclusive as to whether patterns of variation are best explained
by gene flow between C. helianthea and C. b. bonapartei/consita, or by incomplete lineage
sorting (Appendix A).
Is color divergence associated with genetic variation in MC1R?
Of the 36 Coeligena individuals sampled for MC1R, 32 shared a haplotype (excluding
ambiguous positions). Genetic variation at MC1R was limited to three individuals of C.
helianthea and one individual of C. bonapartei, and involved changes in four sites. Only
one change was non-synonymous (Ser275 [AGC] → Arg275 [AGG] at nucleotide site

825), but it was present in a single C. helianthea (Andes-BT 1126) with typical plumage
coloration. These results reveal no association between MC1R genotype and species-
specific color phenotypes in C. helianthea and C. bonapartei.
Is C. helianthea with darker plumage distributed in more humid environments as
predicted by Gloger’s rule?
We found no support for the prediction that the more darkly colored C. helianthea occurs in
more humid environments than C. b. bonapartei/consita: the climatic niches of these taxa
overlap considerably (D = 0.65, Figure 4A) and we found no evidence for significant niche
divergence relative to background climate (p = 0.99). Niche overlap between C. b. eos and
C. b. bonapartei/consita and C. helianthea was considerably lower (D = 0.07 and 0.10,
respectively, Figure 4B), but relative to the background niche differences were not
significant (p = 0.70 and 0.76, respectively).
Is there morphometric variation between species that may suggest adaptations to
alternative microhabitats or resources?
Morphometric data showed differences between C. h. helianthea and C. b. bonapartei and
between females and males of each taxon: LDA analysis distinguished species/sex with a
low classification error of 1.2%. The two most relevant variables in the LD function were
wing loading (coefficients: LD1 = 240.2, LD2 = 287.8, and LD3 = 120.0), and wing taper
(coefficients: LD1 = -39.7, LD2 = -42.6, and LD3 = -23.4). ANOVA or Kruskal-Wallis tests
showed significant differences in 11 morphological variables between species, and in 14
variables between sexes (Figure 8 in Appendix A). The three variables that differed the
most between species and sexes were length of extended wing (ANOVA coefficients: -3.4
species and 5.9 sex), total culmen (ANOVA coefficients: 2.3 species and 2.2 sex), and
length of tail (ANOVA coefficients: 1.0 species and 3.7 sex). Coeligena b. bonapartei has
longer wings, shorter bills and shorter tails than C. h. helianthea (p = < 0.001 in all cases),
and females have shorter wings, longer bills and shorter tails than males in both species (p
= < 0.001 in all cases). Our analyses further revealed that the magnitude of morphometric
differences between sexes varied by species. For example, females of C. helianthea are
the smallest of the four groups (i.e. combinations of species and sexes), but males of C.
helianthea are the largest.

Discussion
Coeligena helianthea and C. bonapartei are closely related species of hummingbirds from
the Northern Andes differing distinctly in plumage coloration, but we found a striking lack of
genetic differentiation between them in a mitochondrial gene (ND2) and in 2,313 UCE
markers broadly scattered across the genome (Figure 2). Considering that low genomic
divergence is typically associated with low differentiation in coloration in other Andean
birds (Winger 2017), the strong phenotypic differences between C. helianthea and C.
bonapartei in the absence of neutral genetic differentiation are remarkable, and make
these species an appropriate system in which to search for the genetic basis and adaptive
significance of phenotypic differences involved in speciation (see Campagna et al. 2017).
However, we found no evidence that MC1R (a candidate gene associated with melanic
pigmentation in a variety of vertebrates) underlies phenotypic variation, and found no
support for the hypothesis that Gloger’s rule (adaptation to geographic variation in
humidity) or other macroclimatic niche differences (Figure 4) are associated with
phenotypic divergence between these species. Nonetheless, our finding that C. h.
helianthea and C. b. bonapartei differ in morphometric traits (Figure 8) potentially related
to habitat and resource use is consistent with the hypothesis that natural selection may
have played a role in their divergence. In addition, as we discuss below, phenotypic
divergence in coloration with little genetic differentiation may reflect sexual selection.
Because coalescent estimates of migration based on ND2 and UCE data suggested that
C. helianthea and C. b. bonapartei/consita may have experienced migration (Figures 3 and
7), their phenotypic divergence might have arisen or been maintained by selection in the
face of gene flow. However, preliminary analyses with UCE data did not allow us to rule
out incomplete lineage sorting as an explanation for the low genetic divergence between
these species. However, we note that divergence with gene flow and incomplete lineage
sorting are not mutually exclusive explanations of shallow genetic differentiation
(Kutschera et al. 2014), especially when speciation occurs rapidly due to selection (Suh et
al. 2015, McLean et al. 2016). Because our sample size for UCEs markers was admittedly
small, more extensive data and analyses of genome-wide variation are required to reach
more definitive conclusions. Complete genome analyses may help clarify the influence of
introgression and incomplete lineage sorting on patterns of genetic variation (Suh et al.
2015), may allow reducing uncertainty in the estimation of population genetic parameters

(Hey and Nielsen 2004), and may allow identifying the genetic basis of phenotypic
differences (Bourgeois et al. 2016, Toews et al. 2016, Campagna et al. 2017).
We found no variation between species in the coding region of MC1R, a gene associated
with variation in plumage coloration in several other birds (Theron et al. 2001, Mundy
2004, Doucet et al. 2004, Baião et al. 2007, Gangoso et al. 2011). Nevertheless, other
studies have shown no association between plumage coloration differences and variation
in the coding region of MC1R (MacDougall-Shackleton et al. 2003, Cheviron et al. 2006,
Haas et al. 2009). As in these latter studies, our work suggests that differences in
coloration between C. helianthea and C. bonapartei are controlled by other genetic
mechanisms which may include genes agonists or antagonists of MC1R in the melanin
metabolic pathway, regions regulating the expression of MC1R or other genes (Theron et
al. 2001), and genes controlling traits of feather structure influencing the production of
structural colors (Shawkey et al. 2003).
We found no support for Gloger’s rule because the darker C. helianthea does not occur in
more humid environments than the more lightly colored C. bonapartei (Figure 4).
Nevertheless, adaptation to different environmental conditions may occur at a finer scale,
where habitat differences might select for plumage traits that, for instance, stand out from
the background augmenting signal efficacy (Endler 1993, Brumfield and Braun 2001).
Indeed, we found that the species differ in morphometric traits (e.g. C. bonapartei has
longer wings and shorter tails than C. helianthea, Figure 8) typically associated with use of
different microhabitats or foraging behaviors. Variation in such traits can affect flight speed
or the relative ability to maneuver in open vs. closed environments (Altshuler et al. 2010,
Ortega-Jimenez et al. 2014). To the extent that morphological differences may reflect
adaptations to different resources between species (Altshuler and Dudley 2002) and
between sexes within species of hummingbirds (Temeles and Kress 2010), our data are
consistent with a role for selection driving morphological divergence, but the adaptive
value of phenotypic variation, if any, remains to be discovered. Considering that C.
bonapartei often occurs along forest edges whereas C. helianthea is more frequently
found in forest interior (Hilty and Brown 1986), studies of the functional consequences of
phenotypic differences would be especially useful to assess any potential role of natural
selection in driving and maintaining divergence.
Knowledge of the timing of speciation also might allow one to make inferences about
historical processes that could have promoted divergence between C. helianthea and C.

bonapartei. We estimated that the most recent common ancestor of these species
diverged from C. b. eos between 0.17 and 0.46 million years ago (Figure 5). Therefore,
divergence between C. helianthea and C. bonapartei must be more recent, potentially
coinciding with some of the last glaciations of the Pleistocene when high-altitude
environments were uninhabitable and forests likely retreated, resulting in the isolation and
divergence of populations (Vuilleumier 1969, Ramírez-Barahona and Eguiarte 2013).
Under this scenario, C. helianthea and C. bonapartei may have diverged in allopatry and
their lack of genetic differentiation could be a result of hybridization after secondary
contact, with the existence of two groups of haplotypes reflecting geography (north and
south) more than taxonomy (i.e. plumage phenotype, Figure 2) reflecting divergence in
allopatry followed by range expansions by both species and subsequent hibridization in
both areas. It thus remains possible that different selective regimes promoted speciation in
these hummingbirds if their divergence occurred across environments with contrasting
climatic conditions in the Pleistocene even if they occupy similar environments at present.
Although such a hypothesis might be partly testable by modeling historical climates and
potential distributions, one would still be faced with the question of what evolutionary
forces might maintain C. helianthea and C. bonapartei as distinct given that they occur in
regional sympatry in the same macroenvironments in the present.
Aside from natural selection, another plausible explanation for the origin and maintenance
of phenotypic distinctiveness in plumage, given the strong sexual dichromatism in C.
helianthea and C. bonapartei, is that their differentiation was promoted by sexual selection
(Price 1998, 2007). Sexual selection is thought to be a powerful force driving speciation in
birds and other organisms (Campagna et al. 2012, 2017; Harrison et al. 2015), and some
examples exist of speciation due to sexual selection with gene flow (Servedio 2016). Of
direct relevance to our system, a study comparing sexually selected (i.e. gorget and crown
coloration) and non-sexually selected traits among Coeligena species found that sexual
selection may be an important driver of phenotypic differentiation, but that it is probably
insufficient for speciation to be completed unless it acts in concert with natural selection
(Parra 2010; see also Servedio and Boughman 2017). To assess the plausibility of the
hypothesis that sexual selection is involved in the divergence and speciation of C.
helianthea and C. bonapartei, one should test for associations among components of
males’ fitness, signaling traits (i.e. coloration, songs), and female preferences. Genomic
analyses examining whether there are genetic and signatures of selection acting on
regions associated with sexual traits (Charlesworth 2009, Huang and Rabosky 2015,

Kirkpatrick 2017) would further help to test the hypothesis of divergence driven by sexual
selection.
Another explanation for our results showing no genetic differentiation despite marked
phenotypic differences and patterns of population genetic structure better reflecting
geography than plumage phenotype is that C. helianthea and C. bonapartei are not
different species but rather morphs within a single species which have become
differentially sorted in different areas. While this possibility is intriguing and parapatric
populations lacking neutral genetic differences yet exhibiting distinct plumages are treated
as conspecific in other birds (e.g. Joseph et al., 2006; Poelstra et al., 2014; but see
Aguillon et al., 2018), we stress that with the exception of a handful of old specimens there
is no current evidence of hybridization that would suggest that C. helianthea and C.
bonapartei are conspecific. Furthermore, because plumage differences between them are
quite striking in the context of differences among undisputed species in the genus and
other hummingbirds (Remsen et al. 2018), we believe they are best treated as distinct
species given existing evidence.
In conclusion, our study provides evidence that the formation of two species of Andean
hummingbirds likely occurred recently, rapidly and possibly in the face of gene flow,
suggesting some form of selection played a role maintaining phenotypic differences and
driving speciation. However, because the main selective mechanism we examined (i.e.
adaptation to contrasting macroclimatic conditions) appears not to operate in C. helianthea
and C. bonapartei, we conclude that ecological pressures that we did not consider directly
or sexual selection may have been involved in their divergence. Future studies should thus
aim to test predictions of hypotheses of natural and sexual selection acting on this system.
Regardless of the selective processes involved, in line with previous research, our study
suggests that selection may play an important role in maintaining phenotypic differences
that could lead to speciation in tropical montane birds (Cadena et al. 2011, Winger and
Bates 2015). Finally, the shallow genetic divergence that we observed between these
species suggest that their genomes are unlikely to have been substantially affected by
processes occurring after speciation (e.g. post-speciation divergence by drift), which
makes this system especially promising for work on the genomics of speciation. Studies
aiming to understand the genetic underpinnings of species differences employing genomic
approaches (e.g. Campagna et al. 2017; Stryjewski and Sorenson 2017) will be an

important complement to increasing knowledge of the geographic and ecological context
of speciation in tropical montane birds.
Acknowledgments
We thank the Facultad de Ciencias at Universidad de Los Andes for financial support
through the Proyecto Semilla program. For providing tissue samples and supporting
fieldwork, we thank the Museo de Historia Natural de la Universidad de los Andes
(ANDES), the Instituto Alexander von Humboldt (IAvH) and the Colección Ornitológica
Phelps (Jorge Perez-Emán and Miguel Lentino). We also thank Whitney Tsai, Paola
Montoya, Camila Gómez and Laura Céspedes for their help with laboratory work and
analyses. The manuscript was improved thanks to comments by L. Campagna, E. Jarvis
and anonymous reviewers, and discussion with Irby Lovette’s and Daniel Cadena’s lab
groups. All the authors confirm that we do not have any conflicts of interest to declare.
Contributions: Conceived the idea S.G.R., C.D.C. and J.L.P. Performed the experiments
and analyzed the data: C.P. and S.G.R. Contributed substantial materials, resources and
funding A.M.C., F.G.S., J.E.M and C.D.C. All authors contributed to write the paper lead by
C.P. and C.D.C.

Figures
Figure 1. Geographical distribution and sampled localities of C. helianthea and C.
bonapartei. Black dots correspond to localities of specimens sampled for genetic markers.
Colored dots correspond to occurrence data obtained from public databases. All localities
were used for niche overlap analyses. Polygons correspond to the likely distributions of the
subspecies according to elevational limits (Ayerbe-Quiñones 2015) and occurrence data.
The tealed polygon in the south corresponds to the region where species are sympatric.
Illustrations courtesy of Lynx Edicions (del Hoyo et al. 2018).

Figure 2. ND2 phylogenetic reconstructions and haplotype network show lack of
divergence between C. helianthea and C. b. bonapartei/consita. The ND2 (1,041bp) gene
tree (A) and ND2 (885bp) haplotype network (B) show C. helianthea and C. b.
bonapartei/consita in a single group separate from C. b. eos. Most specimens of the
northern subspecies C. h. tamai and C. b. consita cluster together, whereas southern
subspecies C. h. helianthea and C. b. bonapartei form another group, suggesting that
population structure more strongly reflects geography (i.e. north-south differentiation) than
taxonomy based on plumage phenotype. The phylogenetic reconstruction based on UCEs
(2,313 loci shared by at least 3 individuals including the outgroup, C) shows C. helianthea
nested within C. b. bonapartei/consita. Numbers at the right of the individuals in the tips of
the trees correspond to the sampling locality (Table S1). Illustrations courtesy of Lynx
Edicions (del Hoyo et al. 2018).

Figure 3. Posterior probability distributions of the migration parameter M = m / µ based on
ND2 data (1,041bp) suggest gene flow from C. helianthea to C. b. bonapartei (right), but
gene flow could not be estimated reliably from C. b. bonapartei to C. helianthea (left).
Colors and lines correspond to the limits of the intervals accumulating 50% (darker colors),
and 95% (medium colors) of the probability density.
Figure 4. Because C. helianthea and C. b. bonapartei/consita do not differ in climatic
niches, their phenotypic differences are not attributable to Gloger’s rule. The climatic
niches of C. helianthea and C. b. bonapartei/consita overlap considerably (D = 0.65) (A).

The climatic niche of C. b. eos overlaps very little with C. helianthea (D = 0.10) and C. b.
bonapartei/consita (D = 0.07) climatic niches (B). Nevertheless, relative to the background
the differences between the niches are not significant in any case (p>0.1).
References
Aguillon, S. M., L. Campagna, R. G. Harrison, and I. J. Lovette (2018). A flicker of hope:
Genomic data distinguish Northern Flicker taxa despite low levels of divergence. The
Auk 135:748–766.
Altshuler, D. L., and R. Dudley (2002). The ecological and evolutionary interface of
hummingbird flight physiology. Journal of Experimental Biology 205:2325–2336.
Altshuler, D. L., M. Princevac, H. Pan, and J. Lozano (2010). Wake patterns of the wings
and tail of hovering hummingbirds. Animal Locomotion:273–284.
Amar, A., A. Koeslag, G. Malan, M. Brown, and E. Wreford (2014). Clinal variation in the
morph ratio of Black Sparrowhawks Accipiter melanoleucus in South Africa and its
correlation with environmental variables. Ibis 156:627–638.
Ayerbe-Quiñones, F. (2015). Colibríes de Colombia. First. Wildlife Conservation Society.
Baião, P. C., E. Schreiber, and P. G. Parker (2007). The genetic basis of the plumage
polymorphism in Red-Footed Boobies (Sula sula): a Melanocortin-1 receptor (MC1R)
analysis. Journal of Heredity 98:287–92.
Bandelt, H.-J., P. Forster, and A. Röhl (1999). Median-joining networks for inferring
intraspecific phylogenies. Molecular Biology and Evolution 16:37–48.
Beerli, P. (2006). Comparison of Bayesian and maximum-likelihood inference of population
genetic parameters. Bioinformatics 22:341–345.
Beerli, P. (2009). How to use MIGRATE or why are Markov Chain Monte Carlo programs
difficult to use? Population Genetics for Animal Conservation.
Bolger, A. M., M. Lohse, and B. Usadel (2014). Trimmomatic: A flexible trimmer for
Illumina sequence data. Bioinformatics 30:2114–2120.
Bouckaert, R., J. Heled, D. Kühnert, T. Vaughan, C. H. Wu, D. Xie, M. A. Suchard, A.
Rambaut, and A. J. Drummond (2014). BEAST 2: A Software Platform for Bayesian
Evolutionary Analysis. PLoS Computational Biology 10:1–6.
Bourgeois, Y. X. C., J. A. M. Bertrand, B. Delahaie, J. Cornuault, T. Duval, B. Milá, and C.
Thébaud (2016). Candidate Gene Analysis Suggests Untapped Genetic Complexity
in Melanin-Based Pigmentation in Birds. Journal of Heredity 107:327–335.
Broennimann, O., V. Di Cola, and A. Guisan (2016). ecospat: Spatial Ecology

Miscellaneous Methods. [Online.] Available at https://cran.r-
project.org/package=ecospat.
Broennimann, O., M. C. Fitzpatrick, P. B. Pearman, B. Petitpierre, L. Pellissier, N. G.
Yoccoz, W. Thuiller, M.-J. Fortin, C. Randin, N. E. Zimmermann, C. H. Graham, and
A. Guisan (2012). Measuring ecological niche overlap from occurrence and spatial
environmental data. Global Ecology and Biogeography 21:481–497.
Brumfield, R. T., and M. J. Braun (2001). Phylogenetic relationships in bearded manakins
(Pipridae: Manacus) indicate that male plumage color is a misleading taxonomic
marker. The Condor 103:248–258.
Burtt, E. H., and J. M. Ichida (2004). Gloger’s Rule, feather-degrading bacteria, and color
variation among Song Sparrows. The Condor 106:681–686.
Cadena, C. D., Z. A. Cheviron, and W. C. Funk (2011). Testing the molecular and
evolutionary causes of a “leapfrog” pattern of geographical variation in coloration.
Journal of Evolutionary Biology 24:402–414.
Campagna, L., P. Benites, S. C. Lougheed, D. A. Lijtmaer, A. S. Di Giacomo, M. D. Eaton,
and P. L. Tubaro (2012). Rapid phenotypic evolution during incipient speciation in a
continental avian radiation. Proceedings of the Royal Society B: Biological Sciences
279:1847–1856.
Campagna, L., M. Repenning, L. F. Silveira, C. Suertegaray Fontana, L. Tubaro, Pablo,
and I. J. Lovette (2017). Repeated divergent selection on pigmentation genes in a
rapid finch radiation. Science Advances 3:e1602404.
Charlesworth, B. (2009). Fundamental concepts in genetics: Effective population size and
patterns of molecular evolution and variation. Nature Reviews Genetics 10:195–205.
Cheviron, Z. A., S. J. Hackett, and R. T. Brumfield (2006). Sequence variation in the
coding region of the melanocortin-1 receptor gene (MC1R) is not associated with
plumage variation in the blue-crowned manakin (Lepidothrix coronata). Proceedings
of the Royal Society B: Biological Sciences 273:1613–8.
Cooper, E. A., and J. A. C. Uy (2017). Genomic evidence for convergent evolution of a key
trait underlying divergence in island birds. Molecular Ecology 26:3760–3774.
Coyne, J. A., and H. A. Orr (2004). Speciation. Sinauer Associates, Sunderland, MA.
Crawford, N. G., and B. C. Faircloth (2011). CloudForest. [Online.] Available at
https://github.com/ngcrawford/CloudForest.
D’Alba, L., C. Van Hemert, K. A. Spencer, B. J. Heidinger, L. Gill, N. P. Evans, P.
Monaghan, C. M. Handel, and M. D. Shawkey (2014). Melanin-based color of

plumage: role of condition and of feathers’ microstructure. Integrative and
Comparative Biology 54:633–644.
Delhey, K. (2019). A review of Gloger’s rule, an ecogeographical rule of colour: definitions,
interpretations and evidence. Biological Reviews. https://doi.org/10.1111/brv.12503
Dinerstein, E., D. Olson, A. Joshi, C. Vynne, N. D. Burgess, E. Wikramanayake, N. Hahn,
S. Palminteri, P. Hedao, R. Noss, M. Hansen, et al. (2017). An ecoregion-based
approach to protecting half the terrestrial realm. BioScience 67:534–545.
Donegan, T., A. Quevedo, J. C. Verhelst, O. Cortés-Herrera, T. Ellery, and P. Salaman
(2015). Revision of the status of bird species occurring or reported in Colombia 2015,
with discussion of BirdLife International’s new taxonomy. Conservación Colombiana
23:3–48.
Doucet, S. M., M. D. Shawkey, M. K. Rathburn, H. L. Mays, and R. Montgomerie (2004).
Concordant evolution of plumage colour, feather microstructure and a melanocortin
receptor gene between mainland and island populations of a fairy-wren. Proceedings
of the Royal Society of London. Series B: Biological Sciences 271:1663–70.
Dray, S., and A. B. Dufour (2007). The ade4 package: implementing the duality diagram
for ecologists. Journal of Statistical Software 22:1–20.
Durand, E. Y., N. Patterson, D. Reich, and M. Slatkin (2011). Testing for ancient admixture
between closely related populations. Molecular Biology and Evolution 28:2239–2252.
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high
throughput. Nucleic Acids Research 32:1792–1797.
Endler, J. A. (1993). Some general comments on the evolution and design of animal
communication systems. Philosophical transactions of the Royal Society of London.
Series B, Biological sciences 340:215–25.
Faircloth, B. C. (2013). Illumiprocessor: a Trimmomatic wrapper for parallel adapter and
quality trimming.
Faircloth, B. C. (2015). PHYLUCE is a software package for the analysis of conserved
genomic loci. Bioinformatics 32:786–788.
Faircloth, B. C., and T. C. Glenn (2012). Not all sequence tags are created equal:
Designing and validating sequence identification tags robust to indels. PLoS ONE
7:e42543.
Faircloth, B. C., J. E. McCormack, N. G. Crawford, M. G. Harvey, R. T. Brumfield, and T.
C. Glenn (2012). Ultraconserved elements anchor thousands of genetic markers
spanning multiple evolutionary timescales. Systematic Biology 61:717–26.

Fitzpatrick, B. M., J. A. Fordyce, and S. Gavrilets (2009). Pattern, process and geographic
modes of speciation. Journal of Evolutionary Biology 22:2342–2347.
Fitzpatrick, S. W., J. C. Gerberich, J. A. Kronenberger, L. M. Angeloni, and W. C. Funk
(2015). Locally adapted traits maintained in the face of high gene flow. Ecology
Letters 18:37–47.
Fjeldså, J., and N. Krabbe (1990). Birds of the High Andes. Apollo Books, Svendborg,
Denmark.
Gangoso, L., J. M. Grande, A. L. Ducrest, J. Figuerola, G. R. Bortolotti, J. A. Andrés, and
A. Roulin (2011). MC1R-dependent, melanin-based colour polymorphism is
associated with cell-mediated response in the Eleonora’s falcon. Journal of
Evolutionary Biology 24:2055–2063.
Gavrilets, S. (1999). A dynamical theory of speciation on holey adaptive landscapes. The
American Naturalist 154:1–22.
Gilbert, M. P., E. D. Jarvis, B. LI, C. LI, C. V. Mello, The_Avian_Genome_Consortium, J.
Wang, and G. Zhang (2014). Genomic data of the Anna’s Hummingbird (Calypte
anna). [Online.] Available at http://dx.doi.org/10.5524/101004.
Goldstein, G., K. R. Flory, B. A. Browne, S. Majid, J. M. Ichida, and E. H. Burtt (2004).
Bacterial degradation of black and white feathers. The Auk 121:656–659.
Goudet, J., and T. Jombart (2015). hierfstat: Estimation and Tests of Hierarchical F-
Statistics. [Online.] Available at https://cran.r-project.org/package=hierfstat.
Green, R. E., J. Krause, A. W. Briggs, T. Maricic, U. Stenzel, M. Kircher, N. Patterson, H.
Li, W. Zhai, M. H. Y. Fritz, N. F. Hansen, et al. (2010). A draft sequence of the
Neandertal genome. Science 328:710–722.
Greenewalt, C. H., W. Brandt, and D. D. Friel (1960). The iridescent colors of hummingbird
feathers. Proceedings of the American Philosophical Society 104:249–253.
Guindon, S., J. F. Dufayard, V. Lefort, M. Anisimova, W. Hordijk, and O. Gascuel (2010).
New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing
the performance of PhyML 3.0. Systematic Biology 59:307–321.
Gutiérrez-Zamora, A. (2008). Las interacciones ecológicas y estructura de una comunidad
altoandina de colibríes y flores en la cordillera oriental de Colombia. Ornitología
Colombiana 7:17–42.
Haas, F., M. A. Pointer, N. Saino, A. Brodin, N. I. Mundy, and B. Hansson (2009). An
analysis of population genetic differentiation and genotype-phenotype association
across the hybrid zone of carrion and hooded crows using microsatellites and MC1R.

Molecular Ecology 18:294–305.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and
analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41:95–98.
Harris, R. S. (2007). Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, The
Pennsylvania State University.
Harrison, P. W., A. E. Wright, F. Zimmer, R. Dean, S. H. Montgomery, M. A. Pointer, and
J. E. Mank (2015). Sexual selection drives evolution and rapid turnover of male gene
expression. Proceedings of the National Academy of Sciences of the USA 112:4393–
4398.
Hey, J. (2006). Recent advances in assessing gene flow between diverging populations
and species. Current Opinion in Genetics & Development 16:592–596.
Hey, J., and R. Nielsen (2004). Multilocus methods for estimating population sizes,
migration rates and divergence time, with applications to the divergence of Drosophila
pseudoobscura and D. persimilis. Genetics 167:747–60.
Hey, J., and R. Nielsen (2007). Integration within the Felsenstein equation for improved
Markov chain Monte Carlo methods in population genetics. Proceedings of the
National Academy of Sciences of the USA 104:2785–2790.
Hijmans, R. J., S. E. Cameron, J. L. Parra, P. G. Jones, and A. Jarvis (2005). Very high
resolution interpolated climate surfaces for global land areas. International Journal of
Climatology 25:1965–1978.
Hilty, S. L. (2003). Birds of Venezuela. 2nd edition. Princeton University Press, Princeton,
New Jersey.
Hilty, S. L., and W. L. Brown (1986). A guide to birds of Colombia. Princeton University
Press, Princeton, New Jersey.
del Hoyo, J., A. Elliott, J. Sargatal, D. A. Christie, and E. de Juana (2018). Handbook of
the Birds of the World Alive. [Online.] Available at http://www.hbw.com/.
Huang, H., and D. L. Rabosky (2015). Sex-linked genomic variation and its relationship to
avian plumage dichromatism and sexual selection. BMC Evolutionary Biology 15:199.
Hugall, A. F., and D. Stuart-Fox (2012). Accelerated speciation in colour-polymorphic
birds. Nature 485:631–634.
Joseph, L., T. Wilke, J. Ten Have, and R. Terry Chesser (2006). Implications of
mitochondrial DNA polyphyly in two ecologically undifferentiated but morphologically
distinct migratory birds, the masked and white-browed woodswallows Artamus spp. of
inland Australia. Journal of Avian Biology 37:625–636.

Katoh, K., and D. M. Standley (2013). MAFFT multiple sequence alignment software
version 7: Improvements in performance and usability. Molecular Biology and
Evolution 30:772–780.
Kearse, M., R. Moir, A. Wilson, S. Stones-Havas, M. Cheung, S. Sturrock, S. Buxton, A.
Cooper, S. Markowitz, C. Duran, T. Thierer, et al. (2012). Geneious Basic: An
integrated and extendable desktop software platform for the organization and analysis
of sequence data. Bioinformatics 28:1647–1649.
Kirkpatrick, M. (2017). The evolution of genome structure by natural and sexual selection.
Journal of Heredity 108:3–11.
Korneliussen, T. S., A. Albrechtsen, and R. Nielsen (2014). ANGSD: analysis of next
generation sequencing data. BMC Bioinformatics 15:356.
Kriticos, D. J., B. L. Webber, A. Leriche, N. Ota, I. Macadam, J. Bathols, and J. K. Scott
(2012). CliMond: Global high-resolution historical and future scenario climate surfaces
for bioclimatic modelling. Methods in Ecology and Evolution 3:53–64.
Kuhner, M. K. (2006). LAMARC 2.0: Maximum likelihood and Bayesian estimation of
population parameters. Bioinformatics 22:768–770.
Kumar, V., F. Lammers, T. Bidon, M. Pfenninger, L. Kolter, M. A. Nilsson, and A. Janke
(2017). The evolutionary history of bears is characterized by gene flow across
species. Scientific Reports 7:46487.
Kutschera, V. E., T. Bidon, F. Hailer, J. L. Rodi, S. R. Fain, and A. Janke (2014). Bears in
a forest of gene trees: Phylogenetic inference is complicated by incomplete lineage
sorting and gene flow. Molecular Biology and Evolution 31:2004–2017.
Lanfear, R., P. B. Frandsen, A. M. Wright, T. Senfeld, and B. Calcott (2017). Partitionfinder
2: New methods for selecting partitioned models of evolution for molecular and
morphological phylogenetic analyses. Molecular Biology and Evolution 34:772–773.
Librado, P., and J. Rozas (2009). DnaSP v5: a software for comprehensive analysis of
DNA polymorphism data. Bioinformatics 25:1451–1452.
MacDougall-Shackleton, E. a, L. Blanchard, S. a Igdoura, and H. L. Gibbs (2003).
Unmelanized plumage patterns in Old World leaf warblers do not correspond to
sequence variation at the melanocortin-1 receptor locus (MC1R). Molecular Biology
and Evolution 20:1675–81.
Maia, R., D. R. Rubenstein, and M. D. Shawkey (2013). Key ornamental innovations
facilitate diversification in an avian radiation. Proceedings of the National Academy of
Sciences of the United States of America 110:10687–92.

Martin, S. H., K. K. Dasmahapatra, N. J. Nadeau, C. Slazar, J. R. Walters, F. Simpson, M.
Blaxter, A. Manica, J. Mallet, and C. D. Jiggins (2013). Genome-wide evidence for
speciation with gene flow in Heliconius butterflies. Genome Research 23:1817–1828.
Mayr, E. (1963). Animal Species and Evolution. Harvard University Press, Cambridge,
Massachusetts.
McGuire, J. A., C. C. Witt, D. L. Altshuler, and J. V. Remsen (2007). Phylogenetic
systematics and biogeography of hummingbirds: Bayesian and maximum likelihood
analyses of partitioned data and selection of an appropriate partitioning strategy.
Systematic Biology 56:837–856.
McGuire, J. A., C. C. Witt, J. V. Remsen, A. Corl, D. L. L. Rabosky, D. L. L. Altshuler, R.
Dudley, A. Corl, D. L. L. Rabosky, D. L. L. Altshuler, and R. Dudley (2014). Molecular
phylogenetics and the diversification of hummingbirds. Current Biology 24:910–916.
McLean, B. S., D. J. Jackson, and J. A. Cook (2016). Rapid divergence and gene flow at
high latitudes shape the history of Holarctic ground squirrels (Urocitellus). Molecular
Phylogenetics and Evolution 102:174–188.
Morales, A. E., N. D. Jackson, T. A. Dewey, B. C. O’Meara, and B. C. Carstens (2017).
Speciation with Gene Flow in North American Myotis Bats. Systematic Biology
66:440–452.
Morgans, C. L., G. M. Cooke, and T. J. Ord (2014). How populations differentiate despite
gene flow: sexual and natural selection drive phenotypic divergence within a land fish,
the Pacific leaping blenny. BMC Evolutionary Biology 14:97.
Mundy, N. I. (2004). Conserved genetic basis of a quantitative plumage trait involved in
mate choice. Science 303:1870–1873.
Mundy, N. I. (2005). A window on the genetics of evolution: MC1R and plumage
colouration in birds. Proceedings of the Royal Society B: Biological Sciences
272:1633–1640.
Naimi, B. (2015). usdm: Uncertainty Analysis for Species Distribution Models. R package
version 1.1-15. [Online.] Available at https://cran.r-project.org/package=usdm.
Nosil, P. (2008). Speciation with gene flow could be common. Molecular Ecology 17:2103–
2106.
Nosil, P., L. J. Harmon, and O. Seehausen (2009). Ecological explanations for
(incomplete) speciation. Trends in Ecology and Evolution 24:145–156.
Novikova, P. Y., N. Hohmann, V. Nizhynska, T. Tsuchimatsu, J. Ali, G. Muir, A.
Guggisberg, T. Paape, K. Schmid, O. M. Fedorenko, S. Holm, et al. (2016).

Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating
speciation and abundant trans-specific polymorphism. Nature Genetics 48:1077–
1082.
Ortega-Jimenez, V. M., N. Sapir, M. Wolf, E. A. Variano, and R. Dudley (2014). Into
turbulent air: size-dependent effects of von Kármán vortex streets on hummingbird
flight kinematics and energetics. Proceedings of the Royal Society B: Biological
Sciences 281.
Parra, J. L. (2010). Color evolution in the hummingbird genus Coeligena. Evolution
64:324–335.
Parra, J. L., J. V. Remsen, M. Alvarez-Rebolledo, and J. A. McGuire (2009). Molecular
phylogenetics of the hummingbird genus Coeligena. Molecular Phylogenetics and
Evolution 53:425–434.
Peele, A. M., E. H. Burtt, M. R. Schroeder, and R. S. Greenberg (2009). Dark color of the
Coastal Plain Swamp Sparrow (Melospiza georgiana nigrescens) may be an
evolutionary response to occurrence and abundance of salt-tolerant feather-
degrading bacilli in its plumage. The Auk 126:531–535.
Pfeifer, S. P., S. Laurent, V. C. Sousa, C. R. Linnen, M. Foll, L. Excoffier, H. E. Hoekstra,
and J. D. Jensen (2018). The evolutionary history of Nebraska Deer Mice: Local
adaptation in the face of strong gene flow. Molecular Biology and Evolution 35:792–
806.
Pinho, C., and J. Hey (2010). Divergence with gene flow: Models and data. Annual Review
of Ecology, Evolution, and Systematics 41:215–230.
Poelstra, J. W., N. Vijay, C. M. Bossu, H. Lantz, B. Ryll, I. Muller, V. Baglione, P.
Unneberg, M. Wikelski, M. G. Grabherr, and J. B. W. Wolf (2014). The genomic
landscape underlying phenotypic integrity in the face of gene flow in crows. Science
344:1410–1414.
Price, T. D. (1998). Sexual selection and natural selection in bird speciation. Philosophical
Transactions of the Royal Society B: Biological Sciences 353:251–260.
Price, T. D. (2007). Speciation in Birds. Roberts & Company Publishers, Greenwood
Village, Colorado.
Pritchard, J. K., M. Stephens, and P. Donnelly (2000). Inference of population structure
using multilocus genotype data. Genetics 155:945–959.
R Core Team (2017). R: A Language and Environment for Statistical Computing. [Online.]
Available at https://www.r-project.org/.

Rabiee, M., E. Sayyari, and S. Mirarab (2019). Multi-allele species reconstruction using
ASTRAL. Molecular Phylogenetics and Evolution 130:286–296.
Rambaut, A., A. J. Drummond, D. Xie, G. Baele, and M. A. Suchard (2018). Posterior
Summarization in Bayesian Phylogenetics Using Tracer 1.7. Systematic Biology
67:901–904.
Ramírez-Barahona, S., and L. E. Eguiarte (2013). The role of glacial cycles in promoting
genetic diversity in the Neotropics: The case of cloud forests during the Last Glacial
Maximum. Ecology and Evolution 3:725–738.
Reich, D., K. Thangaraj, N. Patterson, A. L. Price, and L. Singh (2009). Reconstructing
Indian population history. Nature 461:489–494.
Remsen, J. V., J. I. Areta, C. D. Cadena, S. Claramunt, A. Jaramillo, J. F. Pacheco, M. B.
Robbins, F. G. Stiles, D. F. Stotz, and K. J. Zimmer (2018). Version 1 December
2018. A classification of the bird species of South America. [Online.] Available at
http://www.museum.lsu.edu/~Remsen/SACCBaseline.htm.
Rheindt, F. E., M. K. Fujita, P. R. Wilton, and S. V. Edwards (2014). Introgression and
phenotypic assimilation in Zimmerius flycatchers (Tyrannidae): Population genetic
and phylogenetic inferences from genome-wide SNPs. Systematic Biology 63:134–
152.
Ronquist, F., M. Teslenko, P. van der Mark, D. L. Ayres, A. Darling, S. Höhna, B. Larget,
L. Liu, M. a Suchard, and J. P. Huelsenbeck (2012). MrBayes 3.2: efficient Bayesian
phylogenetic inference and model choice across a large model space. Systematic
Biology 61:539–42.
Rosenblum, E. B., C. E. Parent, E. T. Diepeveen, C. Noss, and K. Bi (2017). Convergent
Phenotypic Evolution despite Contrasting Demographic Histories in the Fauna of
White Sands. The American Naturalist 190:S44–S56.
Roulin, A., and A.-L. L. Ducrest (2013). Genetics of colouration in birds. Seminars in Cell
and Developmental Biology 24:594–608.
Schluter, D. (2001). Ecology and the origin of species. Trends in Ecology and Evolution
16:372–380.
Schluter, D. (2009). Evidence for ecological speciation and its alternative. Science
323:737–41.
Servedio, M. R. (2016). Geography, assortative mating, and the effects of sexual selection
on speciation with gene flow. Evolutionary Applications 9:91–102.
Servedio, M. R., and J. W. Boughman (2017). The role of sexual selection in local

adaptation and speciation. Annual Review of Ecology, Evolution, and Systematics
48:85–109.
Servedio, M. R., G. S. Van Doorn, M. Kopp, A. M. Frame, and P. Nosil (2011). Magic traits
in speciation: “magic” but not rare? Trends in Ecology and Evolution 26:389–397.
Shawkey, M. D., A. M. Estes, L. M. Siefferman, and G. E. Hill (2003). Nanostructure
variation predicts intraspecific in ultraviolet-blue plumage colour. Proceedings of the
Royal Society B: Biological Sciences 270:1455–1460.
Shawkey, M. D., S. R. Pillai, G. E. Hill, L. M. Siefferman, and S. R. Roberts (2007).
Bacteria as an agent for change in structural plumage color: correlational and
experimental evidence. The American Naturalist 169:S112-21.
Simpson, J. T., K. Wong, S. D. Jackman, J. E. Schein, S. J. M. Jones, and I. Birol (2009).
ABySS : A parallel assembler for short read sequence data. Genome Research
19:1117–1123.
Smith, B. T., and J. Klicka (2010). The profound influence of the late Pliocene Panamanian
uplift on the exchange, diversification, and distribution of new world birds. Ecography
33:333–342.
Smith, T. B. (1997). A role for ecotones in generating rainforest biodiversity. Science
276:1855–1857.
Sonsthagen, S. A., R. E. Wilson, R. T. Chesser, J. M. Pons, P. A. Crochet, A. Driskell, and
C. Dove (2016). Recurrent hybridization and recent origin obscure phylogenetic
relationships within the ‘white-headed’ gull (Larus sp.) complex. Molecular
Phylogenetics and Evolution 103:41–54.
Sorenson, M. D., J. C. Ast, D. E. Dimcheff, T. Yuri, and D. P. Mindell (1999). Primers for a
PCR-based approach to mitochondrial genome sequencing in birds and other
vertebrates. Molecular phylogenetics and evolution 12:105–14.
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-
analysis of large phylogenies. Bioinformatics 30:1312–1313.
Stiles, F. G. (2008). Ecomorphology and phylogeny of hummingbirds: Divergence and
convergence in adaptations to high elevations. Ornitologia Neotropical 19:511–519.
Stryjewski, K. F., and M. D. Sorenson (2017). Mosaic genome evolution in a recent and
rapid avian radiation. Nature Ecology and Evolution 1:1912–1922.
Suh, A., L. Smeds, and H. Ellegren (2015). The dynamics of incomplete lineage sorting
across the ancient adaptive radiation of neoavian birds. PLoS Biology 13:1–18.
Supple, M. A., R. Papa, H. M. Hines, W. O. McMillan, and B. A. Counterman (2015).

Divergence with gene flow across a speciation continuum of Heliconius butterflies.
BMC Evolutionary Biology 15:204.
Temeles, E. J., and W. J. Kress (2010). Mate choice and mate competition by a tropical
hummingbird at a floral resource. Proceedings of the Royal Society B: Biological
Sciences 277:1607–1613.
Theron, E., K. Hawkins, E. Bermingham, R. E. Ricklefs, and N. I. Mundy (2001). The
molecular basis of an avian plumage polymorphism in the wild: a melanocortin-1-
receptor point mutation is perfectly associated with the melanic plumage morph of the
bananaquit, Coereba flaveola. Current Biology 11:550–557.
Toews, D. P. L., S. A. Taylor, R. Vallender, A. Brelsford, B. G. Butcher, P. W. Messer, and
I. J. Lovette (2016). Plumage genes and little else distinguish the genomes of
hibridizing warblers. Current biology 26:1–6.
Uy, J. A. C., R. G. Moyle, C. E. Filardi, and Z. a Cheviron (2009). Difference in plumage
color used in species recognition between incipient species is linked to a single amino
acid substitution in the melanocortin-1 receptor. The American Naturalist 174:244–54.
Venables, W. N., and B. D. Ripley (2002). Modern Applied Statistics with S. Fourth.
Springer, New York.
Vuilleumier, F. (1969). Pleistocene speciation in birds living in the high Andes. Nature
223:1179–1180.
Wall, J. D., S. K. Kim, F. Luca, L. Carbone, A. R. Mootnick, P. J. de Jong, and A. Di
Rienzo (2013). Incomplete lineage sorting is common in extant gibbon genera. PLoS
ONE 8:1–5.
Walsberg, G. E. (1983). Avian ecological energetics. VII. Academic Press, Inc., New York.
Warren, D. L., R. E. Glor, and M. Turelli (2008). Environmental niche equivalency versus
conservatism: Quantitative approaches to niche evolution. Evolution 62:2868–2883.
Wilson, A. M., and W. Jetz (2016). Remotely sensed high-resolution global cloud dynamics
for predicting ecosystem and biodiversity distributions. PLoS Biology 14:e1002415.
Winger, B. M. (2017). Consequences of divergence and introgression for speciation in
Andean cloud forest birds. Evolution 71:1815–1831.
Winger, B. M., and J. M. Bates (2015). The tempo of trait divergence in geographic
isolation: Avian speciation across the Marañon Valley of Peru. Evolution 69:772–787.
Zhang, C., M. Rabiee, E. Sayyari, and S. Mirarab (2018). ASTRAL-III: Polynomial time
species tree reconstruction from partially resolved gene trees. BMC Bioinformatics
19:15–30.

Zhang, G., C. Li, Q. Li, B. Li, D. M. Larkin, C. Lee, J. F. Storz, A. Antunes, M. J.
Greenwold, R. W. Meredith, A. Odeen, et al. (2014). Comparative genomics reveals
insights into avian genome evolution and adaptation. Science 346:1311–1321.
Zhang, W., K. K. Dasmahapatra, J. Mallet, G. R. P. Moreira, and M. R. Kronforst (2016).
Genome-wide introgression among distantly related Heliconius butterfly species.
Genome Biology 17:25.
Zink, R. M., and J. V. Remsen (1986). Evolutionary processes and patterns of geographic
variation in birds. Current Ornithology 4:1–69.

How much is too little? – Complete mitochondrial genomes do not
distinguish phenotypically distinct lineages of Andean Coeligena
hummingbirds – Chapter 2
Developed in collaboration with Leonardo Campagna1,2, Juan Luis Parra3, and Carlos
Daniel Cadena4
1 Department of Ecology and Evolutionary Biology, Cornell University, Ithaca, New York,
USA
2 Fuller Evolutionary Biology Program, Cornell Lab of Ornithology, Cornell University,
Ithaca, New York, USA
3 Grupo de Ecología y Evolución de Vertebrados, Instituto de Biología, Universidad de
Antioquia, Calle 67 No. 53-108, Medellín, Colombia
4 Laboratorio de Biología Evolutiva de Vertebrados, Departamento de Ciencias Biológicas,
Universidad de Los Andes, Carrera 1 No. 18 A 10, Bogotá, Colombia
This chapter is under review in the Biological Journal of the Linnean Society.
Abstract
Lack of divergence in mitochondrial DNA between species with clear phenotypic
differences may be the result of low resolution of markers, incomplete lineage sorting,
introgression, or the interplay of various evolutionary mechanisms. Previous work revealed
that the Andean hummingbirds Coeligena bonapartei and C. helianthea lack genetic
divergence in the mitochondrial ND2 gene, which shows variation discordant with
coloration phenotype but consistent with geography. We sequenced and analyzed
complete mitochondrial genomes for 42 individuals of C. b. bonapartei, C. b. consita, C. h.
helianthea and C. h. tamai to assess whether patterns revealed by ND2 analyses hold
when considering the entire mitogenome, and to shed light into the evolution of these
hummingbirds. We found low genetic differentiation in mitogenomes among the four
lineages. Estimates of genetic differentiation, phylogenies and haplotype network analyses
of complete mitogenomes did not separate phenotypically distinct taxa, but were
consistent with the pattern of northern vs. southern divergence along the Cordillera
Oriental of Colombia. Mitogenomes of the nominate subspecies are indistinguishable,

suggesting incomplete lineage sorting or introgression. Mitogenomes of C. b. consita and
C. h. tamai are slightly differentiated, but they are more similar to each other than either is
to that of its respective nominate subspecies, a result also suggestive of mtDNA
introgression despite distinct phenotypic differences. Our results indicate that various
evolutionary mechanisms playing out over a complex biogeographic scenario in the
Colombian Andes drove divergence in phenotypes and mitochondrial genomes of
Coeligena hummingbirds, and lead to alternative hypotheses to be tested with whole-
genome analyses.
Resumen
La falta de divergencia en ADN mitocondrial entre especies con claras diferencias
fenotípicas puede ser el resultado de baja resolución de los marcadores, sorteo
incompleto de linajes, introgresión, o la interacción de varios mecanismos evolutivos.
Trabajos anteriores revelaron que los colibríes andinos Coeligena bonapartei y C.
helianthea carecen de divergencia en el gen mitocondrial ND2, cuya variación es
discordante con el fenotipo de la coloración, pero consistente con la geografía. En este
trabajo secuenciamos y analizamos el genoma mitocondrial completo de 42 individuos de
C. b. consita, C. b. bonapartei, C. h. helianthea y C. h. tamai para evaluar si se mantienen
los patrones presentados por los análisis con ND2 al considerar el genoma mitocondrial
completo, y para informar la evolución de estos colibríes. Encontramos baja diferenciación
genética entre los genomas mitocondriales de los cuatro linajes. Estimadores de
diferenciación genética, análisis filogenéticos y de redes de haplotipos del genoma
mitocondrial completo no separan los taxa fenotípicamente diferentes, sino que fueron
consistentes con el patrón de divergencia norte sur a lo largo de la Cordillera Oriental de
Colombia. Los genomas mitocondriales de las subespecies nominales son indistinguibles,
sugiriendo sorteo incompleto de linajes o introgresión. Los genomas mitocondriales de C.
b. consita y C. h. tamai están ligeramente diferenciados, pero son más similares entre
ellos que con sus subespecies nominales, un resultado que sugiere introgresión
mitocondrial a pesar de las diferencias fenotípicas. Nuestros resultados indican que varios
mecanismos evolutivos dirigieron la divergencia en los fenotipos y en los genomas
mitocondriales de estos Coeligena, interactuando en un complejo escenario biogeográfico
en los Andes colombianos, y conducen a hipótesis alternativas que pueden ser evaluadas
con análisis de genomas completos.

Introduction
In the early days of sequence-based molecular systematics, mitochondrial DNA (mtDNA)
was the marker of choice for most studies of population genetics, phylogenetics and
phylogeography of animals because mtDNA is a haploid non-recombinant molecule almost
free of non-coding regions, inherited via the maternal line, and abundant in tissues (Avise
et al., 1987; Galtier et al., 2009; Wilson et al., 1985). Also, mtDNA evolves largely neutrally
at a fast rate allowing one to find distinctive haplotypes among lineages (Avise et al., 1987;
Ballard and Whitlock, 2004). However, mtDNA does not always reflect the evolutionary
history of lineages owing to evolutionary and demographic processes such as selection, or
differences between paternal and maternal dispersal and gene flow (Ballard and Melvin,
2010; Edwards et al., 2005; James et al., 2017). Thus, researchers have turned to
assaying nuclear markers alongside mtDNA to study the divergence of lineages, an
approach becoming increasingly feasible with the development of sequencing
technologies allowing one to assay and analyze large numbers of genetic markers at
relatively low cost (Kraus and Wink, 2015; Oyler-McCance et al., 2016; Toews et al.,
2016). Information on genome-wide variation has not only contributed to more robust
inferences of relationships among lineages as well as insights about how evolutionary
mechanisms drive such divergence, but has also shed light on how evolutionary
mechanisms interact to shape patterns of genetic divergence across genomes (Bonnet et
al., 2017; Toews and Brelsford, 2012).
Phenotypes, nuclear genomes and mitochondrial genomes are not always equally
divergent among lineages. When divergence is mainly driven by genetic drift, mtDNA is
expected to diverge at a faster rate than nuclear DNA – and nuclear-encoded phenotypes
– because the effective population size of the former is lower (Ballard and Whitlock, 2004;
Moore, 1995). However, when selection drives divergence among populations, mtDNA
need not diverge sooner than the nuclear genome, resulting in cases where patterns of
mitochondrial and nuclear differentiation are not coincident or where phenotypic
differentiation exists with little to no mitochondrial differentiation. Furthermore,
phenotypically distinct populations may share mtDNA haplotypes because of mitochondrial
introgression due to gene flow after divergence (Irwin et al., 2009; Rheindt et al., 2011;
Toews and Brelsford, 2012).
Morphology, plumage, and songs are commonly used to compare populations and inform
the species-level taxonomy of birds (Edwards et al., 2005; Remsen, 2005). Morphological

measurements may provide evidence of barriers to gene flow (Cadena et al., 2018),
whereas visual and acoustic signals are key phenotypes for species delimitation because
they are involved in species recognition and reproductive isolation (Price, 2008; Roulin,
2004; Uy et al., 2009). Studies on Neotropical birds often show concordance in
differentiation among lineages in phenotype and mitochondrial markers (e.g. Gutiérrez-
Pinto et al., 2012; Lovette et al., 2010; Ribas et al., 2012; Sedano and Burns, 2010;
Valderrama et al., 2014; Winger and Bates, 2015), although several examples exist of
groups in which mtDNA is highly structured in distinct lineages despite little variation in
plumage (Cadena et al., 2019; Chesser et al., 2020; D’Horta et al., 2013; Valderrama et
al., 2014). Cases documenting species with marked differences in plumage coloration and
little mitochondrial genetic divergence are more scarce (Campagna et al., 2012; Lougheed
et al., 2013; Luna et al., 2017).
Among hummingbirds (Trochilidae), concordance between mtDNA divergence and overall
differences in coloration between species and populations appears to be the norm
(Chaves et al., 2007 Adelomyia; Jiménez and Ornelas, 2016 Amazilia; McGuire et al.,
2008 Trochilidae; Ornelas et al., 2014 Amazilia; Parra et al., 2009 Coeligena; Zamudio-
Beltrán and Hernández-Baños, 2018, 2015 Laprolamia and Eugenes). mtDNA divergence
often coincides with differences in coloration among hummingbirds even when phenotypic
variation is subtle, such as in the color of the crown, gorget, or tail (Benham and Witt, 2016
Metallura; Gonzalez et al., 2011 C. curvipennis; Lozano-Jaramillo et al., 2014
Antocephala; Ornelas et al., 2016 Lampornis; but see Rodríguez-Gómez and Ornelas,
2015 Amazilia; Sornoza-Molina et al., 2018 Oreotrochilus). There are, to our knowledge,
only two documented cases of hummingbirds showing lack of genetic divergence with
marked differentiation in coloration (i.e. differences in color in various plumage patches;
Eliason et al., 2020; Parra, 2010), both occurring in the high Andes. One case involves two
species of Metallura metaltails (Benham et al., 2015; García-Moreno et al., 1999, Metallura
theresiae and M. eupogon) and the other two species of starfontlets in the genus
Coeligena (Palacios et al., 2019; Parra et al., 2009) which we focus on in this study.
The Golden-bellied Starfrontlet (C. bonapartei) and the Blue-throated Starfrontlet (C.
helianthea) inhabit the Northern Andes of Colombia and Venezuela (Figure 1A). The
nominate subspecies of these species are sympatric in the southern part of their ranges in
the Cordillera Oriental, whereas subspecies C. b. consita and C. h. tamai are allopatric in
the Serranía de Perijá and Tamá Massif, respectively. These species are strikingly

different in structural plumage coloration (Eliason et al., 2020; Sosa et al., 2020): C.
bonapartei is greenish with fiery golden underparts whereas C. helianthea is blackish with
a rose belly and aquamarine rump. Despite their markedly different phenotypes, C.
bonapartei and C. helianthea are not genetically distinct in a mitochondrial gene (ND2), in
a gene involved in the melanogenesis pathway (Melanocortin 1 Receptor MC1R), nor in
regions flanking ultra-conserved elements (UCEs) across the nuclear genome (Palacios et
al., 2019). Although these hummingbirds occupy similar environments, their lack of genetic
differentiation is consistent with divergence with gene flow (Palacios et al., 2019).
Phylogenetic analyses of sequences of the ND2 mitochondrial gene also suggest that C.
b. consita and C. h. tamai are more closely related to each other than either is to their
nominate subspecies, a pattern more consistent with geography than with phenotype and
taxonomy. However, it is unclear whether lack of genetic differentiation between C.
bonapartei and C. helianthea is restricted to ND2 or if it is a general pattern across the
mitochondrial genome. Other mitochondrial markers may be more variable owing to
differences among regions in substitution rates (e.g. ND4 or the control region, Arcones et
al., 2019; Eo and DeWoody, 2010) or in selective or stochastic demographic processes
(Morales et al., 2015; Wort et al., 2017). Examining complete mitochondrial genomes
might thus reveal heretofore undetected differences between species of Coeligena.
Alternatively, if complete mitogenomes confirm lack of genetic divergence between C.
bonapartei and C. helianthea, and that relationships of lineages of these species are
inconsistent with their phenotype, then further consideration of mechanisms underlying
evolutionary divergence in mtDNA and coloration in the group would be necessary. Such
mechanisms potentially include natural and sexual selection as well as demographic
processes acting during periods of geographic isolation and contact among lineages
(Krosby and Rohwer, 2009; Morales et al., 2017; Pons et al., 2014; Toews et al., 2014).
We sequenced and assembled complete mitochondrial genomes of multiple individuals to
address the following questions: (1) Are the sequence and structure of the mitochondrial
genomes of C. bonapartei and C. helianthea like those of mitogenomes of other bird and
hummingbird species? (2) Is the lack of genetic divergence between C. bonapartei and C.
helianthea a general pattern across the mitochondrial genome? (3) Are phylogenetic
relationships of lineages of C. bonapartei and C. helianthea based on ND2 also recovered
using complete mitochondrial genomes? (4) Are different genes and regions in the
mitochondrial genome equally informative about lineage relationships? And, (5) Are there
substitutions in mitochondrial protein-coding genes among lineages of C. bonapartei and

C. helianthea involving changes between aminoacids with different funcional
characteristics which may suggest selection acting on these genes?
Materials and Methods
Samples and sequencing
We sampled 46 individuals, 23 each of C. bonapartei and C. helianthea (Supplementary
Table 1), representing subspecies C. b. bonapartei, C. b. consita, C. h. helianthea, and C.
h. tamai. Taxon identities were assigned by determination of specimens in the museum or
by geography. Because previous work indicated that populations from the Mérida
Cordillera of Venezuela often referred to C. bonapartei (subspecies C. b. eos) are
genetically divergent from other populations in the complex (Palacios et al. 2019), we did
not consider them in this study. Muscle tissue samples from voucher specimens were
obtained from the collections of the Instituto Alexander von Humboldt (IAvH) and the
Museo de Historia Natural de la Universidad de los Andes (ANDES). We employed
relatively even samples sizes of each sex and subspecies of both C. bonapartei and C.
helianthea.
We extracted total genomic DNA using a phenol/chloroform method and Phase-Lock Gel
tubes, followed by a standard cleaning protocol employing magnetic beads. We prepared
46 Illumina TruSeq Nano DNA-enriched libraries following the manufacturer’s protocol for
low-throughput configuration and 550bp insert size. We quantified the libraries using a
Qubit fluorometer. Normalizing, pooling and sequencing were done by the Genomics
Facility of the Institute of Biotechnology at Cornell University. Sequencing was performed
using two lanes of NexSeq 500 2x150 paired end. We filtered the raw data by quality
according to Illumina instructions, checked reads using Fastqc (Andrews, 2010), and
cleaned them to remove adapters using AdapterRemoval (Schubert et al., 2016).
Assembly and annotation of mitochondrial genomes
Although our sequence data contained sequences originating from both the nuclear and
mitochondrial genomes, here we focus specifically on the later. We used MITObim v.1.9.1
(Hahn et al., 2013) with default parameters to assemble complete mitochondrial genomes
from filtered reads following two alternative assembling strategies based on using different
baits: (1) two independent assemblies using as baits the complete mitochondrial genomes
of Oreotrochilus melanogaster and Heliodoxa aurescens (Genbank NC027454 and
KP853094, respectively), and (2) a third assembly using as bait the ND2 gene sequence

for each individual -or a related one- available from previous work (Palacios et al., 2019).
We expected that the first strategy would allow us to recover more complete individual
mitogenome sequences because during initial iterations, reads would map to different sites
on the reference mitogenome and this would allow extension from multiple edges. In turn,
we expected that the gene-bait strategy would enable us to identify structural changes in
genomes because it would allow extension only from the two edges of the gene, but it
would likely be susceptible to recovering incomplete sequences when reads did not
overlap, impeding continued extension.
The gene-bait strategy required multiple independent rounds of assembling. In each round
we used as bait a new fragment obtained from the final genome assembled in the previous
round. We compared the results from each strategy to determine the sequence and
structure of mitogenomes of C. bonapartei and C. helianthea. In addition, we mapped the
read-pool obtained from the complete-genome assembling strategy against the
mitogenome sequence obtained from the gene-bait strategy using the “map to reference
assemble” tool in Geneious 9.1.5 (http://www.geneious.com; Kearse et al., 2012). We
used these map-to-reference assemblies to close gaps in some sequences, to check the
number of repetitions at the end of the control region (see results), and to verify assigned
alleles in each sequence at polymorphic sites. We aligned and edited mitochondrial
genomes using ClustarO (Sievers et al., 2011) and manually in Geneious, and annotated
them using MITOS beta version (http://mitos2.bioinf.uni-leipzig.de/index.py) and Geneious.
In addition to the alignment of complete mitogenomes, for phylogenetic and population
genetic analyses described below we generated alignments of each protein-coding gene
(PCG), and a concatenated alignment of 13 PCGs (ND1, ND2, COX1, COX2, ATP8,
ATP6, COX3, ND3, ND4L, ND4, ND5, CYTB, and ND6).
Population genetic, phylogenetic, and amino-acid change analyses
Using the alignment of complete mitogenomes, we calculated nucleotide diversity (Pi) for
all sequences as a unit, and separately for C. bonapartei, C. helianthea, and for each of
the four subspecies (C. b. bonapartei, C. b. consita, C. h. helianthea, C. h. tamai). We
calculated absolute genetic divergence (Dxy) in DnaSP v6 (Rozas et al., 2017), and
relative genetic divergence (Fst) between species and among subspecies assessing
significance with 1,000 permutations using R package Hierfstat (Goudet and Jombart,
2015; R Core Team, 2017).

We examined phylogenetic relationships among individuals based on each of our
alignments using maximum-likelihood analysis and computed majority-rule consensus
trees in RAxML v8.2.12 (Stamatakis, 2014). We used the GTR+GAMMA model and
multiparametric bootstrapping stopped by the autoMRE criterion. We used mitochondrial
genomes of Oreotrochilus melanogaster and Heliodoxa aurescens (Genbank NC027454
and KP853094, respectively) as outgroups. We also built a median-joining haplotype
network (Bandelt et al., 1999) in PopArt (Leigh and Bryant, 2015) using the complete
mitogenome alignment.
Finally, we assessed whether there are fixed changes in amino-acids in proteins encoded
in the mitogenome of lineages of C. bonapartei and C. helianthea potentially suggestive of
selection. We first calculated the number and type of substitutions in each protein coding
gene in DnaSP v6 (Rozas et al., 2017). Then, for each non-synonymous substitution we
examined whether amino-acid variants were from different functional groups.
Results
Sequence and structure of mitochondrial genomes in C. bonapartei and C.
helianthea
We recovered very similar sequence assemblies using the gene-bait and the complete
mitogenome bait strategies. However, using the complete mitogenome strategy we
observed insertions in some mitogenomes not recovered with the gene-bait strategy.
Additionally, we found minor differences between assemblies obtained using the two
strategies mainly in the length and sequence of the control region. We used the read-pool
map-to-reference assemblies to resolve discrepancies between sequences from different
assemblies and to review and manually correct nucleotide assignments in variant sites.
We recovered complete mitochondrial genomes for 42 of the 46 specimens (excluding IDs
23, 24, 26 and 33 in Supplementary Table 1, which we do not consider further because the
data we obtained were of low quality), with an average coverage of 127.5x for all genomes
(Max 1,555.7, Min 11.8, see Supplementary Table 1 for details, GenBank accession
numbers MT341527 to MT341568). The size of the mitochondrial genome of C. bonapartei
and C. helianthea varied from 16,813 bp to 16,859 bp, mainly due to individual variation in
length of a repetitive motive (‘AAAC’) at the end of the control region (beginning at 16,759
bp in the alignment). The 42 sequences were identical across 16,560 bp (98.2%), showed
248 variant sites (1.5%), with 51 positions having gaps or being ambiguous (0.3%). Mean

pairwise identity was 99.7%, and total GC content was 44.8%. On average, the
mitogenome sequences of Coeligena were identical to those of O. melanogaster across
14,555 bp (86.0%) and to those of H. aurescens across 14,450 bp (85.6%). The beginning
of the control region (350 bp) was the most difficult to align between sequences of
Coeligena and those of outgroups. The mitochondrial genome structure of Coeligena
species followed the typical pattern observed in other birds including hummingbirds, with 2
ribosomal RNAs, 13 protein coding genes, 22 transfer RNAs, and the control region
(Figure 2).
Genetic divergence and clustering patterns among lineages of C. bonapartei and C.
helianthea
Across the complete mitogenome alignment including all individuals of C. bonapartei and
C. helianthea, we found only 250 mutations (two sites had 3 alleles) in 248 variable sites
(1.5% of the genome). Of these variable sites, 89 were singletons and 159 were
parsimony-informative. Nucleotide diversity was low in the complete alignment (Pi =
0.00247, SD = 0.00013). The least diverse lineage was C. b. consita (Pi = 0.00019, 9
polymorphic sites), followed by C. h. helianthea (Pi = 0.00084, 40 polymorphic sites), C. h.
tamai (Pi = 0.00124, 98 polymorphic sites), and C. b. bonapartei (Pi = 0.00254, 156
polymorphic sites). When we compared groupings based on species assignment (i.e. C.
bonapartei vs C. helianthea), we found low relative genetic divergence (Fst = 0.076, p =
0.016). However, Fst values were greater when considering the four lineages separately
(Table 1), with comparisons between lineages assigned to the same species showing
higher relative genetic divergence than those between lineages assigned to different
species (e.g. C. b. consita vs C. b. bonapartei Fst = 0.385, p-value < 0.001; C. h.
helianthea vs C. h. tamai Fst = 0.518, p < 0.001; C. b. bonapartei vs C. h. helianthea Fst =
0.083, p = 0.1). All comparisons indicated low absolute genetic divergence (Dxy),
supporting the general lack of genetic differentiation in the mitogenomes of these species
(Table 1). However, high values of relative genetic divergence (Fst) between lineages of
C. bonapartei and C. helianthea suggested genetic structure.
Phylogenetic analyses of the complete mitogenome alignment clustered all sequences of
Coeligena hummingbirds in a well-supported clade (maximum-likelihood bootstrap ML-bs
100%, Figure 1). Relationships within this clade were unresolved, with a polytomy
comprising (1) a clade grouping all sequences of C. b. consita (ML-bs 97%), (2) a clade
grouping all but one of the sequences of C. h. tamai (ML-bs 90%), and (3) the remaining

sequences (mostly of C. b. bonapartei and C. h. helianthea) scattered in smaller clades or
by themselves. Phylogenies built with other alignments (each PCG and concatenated
PCGs, Figure S1) showed lower resolution (i.e. more polytomies or lower support values).
In most phylogenies, C. b. consita and C. h. tamai were more closely related to each other
than either was to the nominate subspecies, but most support values for this grouping
were lower than 80% except in the control-region phylogeny (ML-bs 88%).
All sequences of C. b. consita clustered together in the the median-joining haplotype
network (Figure 1). All sequences but one of C. h. tamai clustered in another group which
was close to, but distinguishable from, two sequences of C. b. bonapartei (ID 12 and 15).
The remaining sequences of C. b. bonapartei, all sequences of C. h. helianthea, and the
remaining sequence of C. h. tamai (ID 40) clustered in a third group (Figure 1). The
network showed that sequences of C. b. consita and C. h. tamai are more similar to each
other than to C. b. bonapartei and C. h. helianthea. Also, two individuals of C. b.
bonapartei (ID 10 and 14) with the same haplotype were highly divergent from all other
individuals. C. b. consita was the lineage with the lowest number of haplotypes (4 among 9
individuals). In the other lineages, the number of haplotypes was similar to the number of
individuals: 12 haplotypes in C. b. bonapartei (13 individuals), 6 in C. h. helianthea (7
individuals), and 13 in C. h. tamai (13 individuals).
Based on the clustering patterns described above, we defined genetic groups for
additional analyses in which we calculated the number of substitutions and measures of
genetic divergence among groups. First, we defined (1) a northern group comprising all
sequences of C. b. consita, all sequences of C. h. tamai except ID 40, and two sequences
of C. b. bonapartei (ID 12 and 15); and (2) a southern group including most sequences of
nominate subspecies C. b. bonapartei and C. h. helianthea (except ID 10 and 14) and one
sequence of C. h. tamai (ID 40). Second, we considered separately the groups of C. b
consita and C. h. tamai (excluding ID 40). There were only 27 substitutions (0.16%) yet
high relative genetic divergence (Fst = 0.513, p-value < 0.001) between the northern and
southern groups. Likewise, there were 14 substitutions (0.083%) and genetic divergence
was high (Fst = 0.502, p-value < 0.001) between C. b. consita and C. h. tamai. The
remaining 118 parsimony-informative sites existing among all sequences corresponded to
intrapopulation diversity. Nucleotide diversity in the southern group (Pi = 0.0018) was
higher than that of C. b. consita (Pi = 0.00019) and C. h. tamai (0.00089), but comparable
to that of the northern group (Pi = 0.0012).

Given a substitution rate of 0.00256 substitutions per site per lineage per million years
(s/s/l/My) for the complete mitogenome of birds (Eo and DeWoody, 2010), we estimated
that the northern and southern groups diverged around 310,000 years ago, and that C. b.
consita and C. h. tamai diverged around 160,000 years ago. Based on 13 protein-coding
genes plus the two rRNAs and a substitution rate of 0.00164 s/s/l/My (mean rate for
Apodiformes; Arcones et al., 2019) estimates of divergence times are similar yet slightly
older: 380,000 years ago between the northern and southern groups, and 180,000 years
ago between C. b. consita and C. h. tamai.
Functional aminoacid changes
Of the total 248 variant sites, 160 were located in protein-coding genes (Table S2). The
remaining 88 variant sites were in rRNAs (6 in 12SrRNA, 20 in 16SrRNA), tRNAs (11),
inter-gene spacers (5), and the control region (46). Among the 160 variant sites in protein-
coding genes, 123 corresponded to synonymous changes and 38 to non-synonymous
changes. Most non-synonymous changes were singletons (23 sites) or varied within
populations (11 sites). Of the remaining 4 variant sites, a non-synonymous change was
shared between one individual of C. b. bonapartei and one individual of C. h. tamai (TC
position 269 in ND5). Only three non-synonymous changes corresponded to substitutions
between genetic groups. One change in ND2 and one in ND6 were fixed differences
between the northern and the southern groups (GA position 475 in ND2, and GA
position 112 in ND6). These non-synonymous substitutions do not imply any evident
functional changes because both aminoacids involved (valine and isoleucine) are aliphatic,
nonpolar, and neutral. Finally, a non-synonymous substitution between C. b. consita and
all other sequences (AG position 145 in ND4) implies a functional change in aminoacids.
Whereas C. b. bonapartei, C. h. helianthea and C. h. tamai had the aliphatic, nonpolar
alanine, C. b. consita had the hydroxyl-containing, polar threonine. Note that this change is
not between the two main mitogenome groups because C. h. tamai has the variant of the
southern mitogenenome group at this position.
Discussion
We found that the complete mitochondrial genomes of two hummingbird species differing
strikingly in phenotype, C. bonapartei and C. helianthea, are highly similar. Mitogenomes
of a sample of 42 individuals representing both species and two subspecies recognized
within each of them were 98.2% identical. Moreover, estimates of genetic differentiation

and clustering analyses of mitogenome sequences were unable to recover groups
corresponding to species, and suggested instead that mitogenomes of C. b. consita and C.
h. tamai formed distinct clusters more similar to each other than either was to
mitogenomes of the nominate subspecies C. b. bonapartei and C. h. helianthea which
were, in turn, indistinguishable from each other. These results indicate that patterns of
variation based on the ND2 gene (Palacios et al., 2019) are consistent across the
mitochondrial genome, implying that the previously documented lack of mtDNA divergence
between species does not reflect insufficient data nor atypical variation in ND2 relative to
other mitochondrial markers. Instead, patterns of variation and relationships among the
mitochondrial genomes of the four lineages are inconsistent with phenotypic variation and
current taxonomy, but seem to agree partly with geography, considering that C. b. consita
and C. h. tamai occur in the Serranía de Perijá and the north of the Cordillera Oriental
whereas both nominate subspecies occur to the south along the cordillera.
The discordance between mitochondrial genomes and coloration phenotypes in C.
bonapartei and C. helianthea can be accounted for by various evolutionary processes
which must have acted over a relatively short period of time given divergence-time
estimates for the group. Based on the ND2 gene, the clade formed by C. bonapartei and
C. helianthea diverged from C. b. eos around 310,000 years ago, and the northern and
southern clades comprising the four lineages of C. bonapartei and C. helianthea diverged
around 240,000 years ago (Palacios et al., 2019). The latter estimate is more recent than
our calculations of the divergence between the northern and southern groups at ca.
310,000 (complete mitogenome) or 380,000 years ago (PCG and rRNAs). Our estimates
of divergence times must be interpreted with caution because different factors may bias
them (Galtier et al., 2009; García-Moreno, 2004; Lovette, 2004), but they do suggest that
the divergence between the northern and southern mitogenome groups, and the
divergence between the mitogenomes of C. b. consita and C. h. tamai (160,000 estimated
through complete mitogenomes and, 180,000 years ago using the PCG and rRNAs) are
recent, i.e. happening within the past 500,000 years.
Ours is the first study in hummingbirds using complete mitochondrial genomes for a
population-level analysis of genetic structure between species and across geography we
are aware of, and few complete mitochondrial genomes of hummingbirds have been
published (Morgan-Richards et al., 2008; Prosdocimi et al., 2016; Souto et al., 2016). We
searched GenBank for complete mitochondrial genomes of closely related hummingbirds

with more than a single individual sequenced per species to compare their divergence with
the divergence we observed in Coeligena. We only found six mitogenome sequences for
three subspecies of Amazilia versicolor (A. v. versicolor KF624601, NC_024156; A. v.
milleri KP722042, NC033405; and A. v. rondoniae KP722041, NC_033404; Prosdocimi et
al., 2016) representing populations occurring over a broad geographic range. Overall,
these sequences are much more differentiated (5.1% of sites were variable) than our
entire data set (1.5%). Although this comparison is far from comprehensive, it does
support the idea that the mitogenomes of the lineages of Coeligena hummingbirds are
highly similar and their divergence is quite recent relative to other hummingbirds with
comparable data, as also indicated by analyses of individual mtDNA genes (Palacios et
al., 2019; Parra et al., 2009).
In contrast to mtDNA phylogenies, nuclear markers suggest C. b. consita was the first
branch to diverge in the group, whereas C. h. helianthea and C. h. tamai are reciprocally
monophyletic groups forming a clade sister to C. b. bonapartei (Palacios et al., 2019;
Palacios et al. unpublished). We found that complete mitogenomes of C. b. bonapartei and
C. h. helianthea are undifferentiated even though both subspecies differ strikingly in
phenotype and are also distinguishable using nuclear markers. Incomplete lineage sorting
may explain this result because the southern mitogenome group exhibited high nucleotide
diversity in comparison with C. b. consita and C. h. tamai, a pattern one would not expect
due to a recent introgression (Krosby and Rohwer, 2009). However, nuclear sorting
without mitochondrial sorting would be unlikely because the effective population size of the
latter is ¼ that of the former. Instead, then, a scenario in which one mitogenome quickly
swept through replacing the mitogenome of the other lineage and later recovered of
nucleotide diversity may explain patterns of mitogenome sharing between C. b. bonapartei
and C. h. helianthea.
The similarity in mitogenomes of C. b. consita and C. h. tamai appears more consistent
with introgression after phenotypic differentiation in isolation. Mitochondrial introgression
may often reflect selection (e.g. adaptive introgression via metabolic efficiency, Ballard and
Melvin, 2010; Toews et al., 2014), but may also be due to demographic effects or to
asymmetries between sexes in dispersal, mating behavior, and offspring production
(Harris et al., 2018; James et al., 2016; Morales et al., 2017; Rheindt et al., 2014; Toews
and Brelsford, 2012). We did not find functional changes in protein-coding genes between
the northern and the southern mitogenomes suggesting adaptation, although adaptive

changes related to substitutions in the control region (or in the 16SrRNA gen in the case of
C. h. tamai) are possible. We are unaware of differential dispersal between sexes in
Coeligena, in which dispersal and breeding biology are poorly known. Mitochondrial
introgression between C. b. consita and C. h. tamai may have been facilitated by their
geographical proximity and may have happened during a period of greater connectivity of
forests in the Pleistocene (Flantua et al., 2019; Graham et al., 2010). Then, both lineages
became isolated again and their mitogenomes diverged. The northern mitogenome may
thus have evolved within C. b. consita and introgressed into C. h. tamai in a north to south
direction, and such introgression may have further proceeded into C. b. bonapartei
explaining why individuals ID 12 and 15 have haplotypes more closely related to the
northern group.
The divergent mitogenomes of four individuals of C. b. bonapartei (ID 10, 12, 14, and 15)
were unexpected considering the similarity among all other sequences. Although
individuals ID 12 and 15 were closely related to the northern group, they shared 9 unique
variants. Individuals ID10 and ID14 shared a mitogenome haplotype which was even more
divergent (34 unique variants) sharing variants with both the northern (9) and the southern
(18) groups. We can reject hybridization with other unstudied taxa as an explanation for
these atypical mitogenomes because ND2 sequences placed these specimens within the
clade formed by C. bonapartei and C. helianthea to the exclusion of C. b. eos (Palacios et
al. 2019). These atypical sequences may instead be evidence of persistence of a relict or
a “ghost” mitochondrial lineage in C. b. bonapartei (Grandcolas et al., 2014; Zhang et al.,
2019), which may have arisen and remained in isolation in the western slope of the
Cordillera Oriental in Boyacá (Iguaque Massif and surroundings), a region where atypical
patterns in mtDNA variation have been reported in other groups (Avendaño and Donegan,
2015; Chaves et al., 2011; Chaves and Smith, 2011; Chesser et al., 2020; Guarnizo et al.,
2009). Another less likely explanation for these atypical sequences may be heteroplasmy
and mitochondrial recombination which have been recognized in vertebrates in some
cases (Piganeau et al., 2004; Rokas et al., 2003; Sammler et al., 2011).
In sum, based on our results and earlier work (Palacios et al. 2019) we hypothesize that a
plausible evolutionary scenario accounting for patterns of mtDNA and phenotypic variation
in C. bonapartei and C. helianthea is as follows. Based on comparison with the outgroup
and other related species (C. b. eos, C. lutetiae, C. orina), the most probably body
plumage coloration of the ancestor of our study clade was green with golden/orange

underparts. The first lineage to diverge was likely C. b. consita, which evolved in the
Serranía de Perijá in isolation from the ancestor of the other three lineages, retaining
features of the ancestral plumage coloration but diverging in mtDNA. A second divergence
event involved sister clades formed by C. b. bonapartei and C. helianthea (i.e. the
common ancestor of both subspecies), with the former retaining the ancestral plumage
and the latter evolving darker body coloration, rose belly, and aquamarine rump. These
two lineages diverged in phenotype while maintaining an undifferentiated mitogenome
owing to incomplete lineage sorting or introgression, except for populations of C. b.
bonapartei which became isolated in the western slope of the Cordillera Oriental and
diverged in mitogenome. Third, C. h. tamai and C. h. helianthea became isolated and
diverged slightly in phenotype. Finally, during a period of forest connectivity the
mitogenome of C. b. consita introgressed into C. h. tamai, a process followed by
subsequent isolation of these lineages resulting in some divergence in their mitogenomes.
Although this is a convoluted historical scenario, it is amenable to testing using genomic
data and demographic models (e.g. Aguillon et al., 2018; Benham and Cheviron, 2019;
Kearns et al., 2018) and other explanations for patterns of variation would appear even
more complex.
In conclusion, low genetic divergence among lineages of C. bonapartei and C. helianthea
is a general pattern across their mitochondrial genomes despite their marked phenotypic
differences. Mitogenomic variation in these lineages seems to more closely reflect
geography and demographic history than the processes shaping their phenotypes and
likely their nuclear genomes. Studying closely related lineages that diverged recently in
complex topographic scenarios, such as the system of C. bonapartei and C. helianthea,
might help to explain the different effects that evolutionary mechanisms may have in
shaping the divergence between and within genomes. Incomplete lineage sorting,
mitochondrial introgression, and demographic processes like population bottlenecks,
phases of expansion and contraction, and the persistence of relict lineages have likely
acted in this system resulting in marked discordance between phenotypes and mtDNA
variation. A natural next step to understand the processes at work in this system is to
place the results of the present study in the context of genome-wide patterns of genetic
variation.

Acknowledgments
We thank the Fundación para la promoción de la investigación y la tecnología del Banco
de la República, and the Lovette Lab at the Cornell Lab of Ornithology for financial
support. For providing tissue samples we thank the Museo de Historia Natural de la
Universidad de los Andes (ANDES) and the Instituto Alexander von Humboldt (IAvH). We
exported tissues samples to the Cornell Lab of Ornithology (Ithaca, NY) thanks to CITES
permit No. CO 41452 granted by the Ministerio de Ambiente y Desarrollo Sostenible of
Colombia. We also thank Irby J. Lovette and Bronwyn G. Butcher for facilitating laboratory
work.

Table and Figures
Table 1. Population genetic statistics and measures of genetic divergence between
C. bonapartei and C. helianthea and among groups within. Nucleotide diversity Pi is
lower in C. b. consita and C. h. tamai than in nominate subspecies. Absolute genetic
divergence Dxy is low yet relative divergence Fst is high across comparisons. Genetic
groups are derived from the clustering patterns analyses and are marked as “Gen” in the
table.
Population genetic statistics
Population # of
Seq
# of
Variants Pi
Tajima's
D
T's D p-
value
C. bonapartei 22 171 0.00249 -0.44 0.351
C. helianthea 20 133 0.00218 -0.09 0.481
C. b. consita 9 9 0.00019 -0.05 0.500
C. b. bonapartei 13 156 0.00254 -0.68 0.273
C. h. helianthea 7 40 0.00084 -0.75 0.274
C. h. tamai 13 98 0.00124 -1.54 0.057
C. b. consita Gen 9 9 0.00019 -0.05 0.500
C. h. tamai Gen 12 54 0.00089 -0.76 0.251
Northern group
Gen 23 92 0.00120 -0.76 0.242
Southern group
Gen 17 100 0.00180 -1.63 0.043
Measures of genetic divergence
Population 1 Population 2 Fst Fst p-
value Dxy
C. bonapartei C. helianthea 0.076 0.0160 0.0026
C. b. consita C. b. bonapartei 0.385 0.0010 0.0032
C. b. consita C. h. helianthea 0.764 0.0010 0.0032
C. b. consita C. h. tamai 0.402 0.0010 0.0017
C. b. bonapartei C. h. helianthea 0.083 0.1069 0.0019
C. b. bonapartei C. h. tamai 0.317 0.0010 0.0034
C. h. helianthea C. h. tamai 0.518 0.0010 0.0033
Northern group
Gen Southern group Gen 0.514 0.0010 0.0035
C. b. consita Gen C. h. tamai Gen 0.502 0.0010 0.0016

Figure 1. The maximum-likelihood phylogeny (B) and haplotype network (C) support two
main mitogenome groups in C. bonapartei and C. helianthea more related to their
geographical distribution (A) than with their taxonomic or phenotypic assignation. Note that
the mitogenomes of C. b. consita and C. h. tamai are differentiated whereas the
mitogenomes of C. b. bonapartei and C. h helianthea are indistinguishable. Numbers on
the tips of the tree, on the haplotype network and on locations in the map correspond to
individual IDs in Table S1. Colors correspond to the assigned subspecies C. b. consita
(orange), C. b. bonapartei (yellow), C. h. helianthea (light blue), and C. h. tamai (dark
blue). In the map the teal area is the region where nominate subspecies are sympatric. In
the tree, numbers on branches are ML-bootstrap values; branch lengths were set to equal.

Figure 2. The mitochondrial genome structure of Coeligena hummingbirds follows the
typical organization of birds: 22 tRNAS (pink), 2 rRNAs (raspberry), 13 protein-coding
genes PCGs (blue), and the control region (gray). Coding sequences CDS are in yellow.
Substitutions among the three genetic groups C. b. consita (orange), C. b. tamai (blue)
and the southern group (green) are represented in the inner circles (singletons and
intrapopulation variant sites are not represented). Gray boxes indicate the three non-
synonymous substitutions found, the box with black edges indicates the only substitution
involving a change between amino-acids with different functional features.

References
Aguillon, S.M., Campagna, L., Harrison, R.G., Lovette, I.J., 2018. A flicker of hope:
Genomic data distinguish Northern Flicker taxa despite low levels of divergence. Auk
135, 748–766. https://doi.org/10.1642/AUK-18-7.1
Andrews, S., 2010. FastQC: A quality control tool for high throughput sequence data.
Arcones, A., Ponti, R., Vieites, D.R., 2019. Mitochondrial substitution rates estimation for
molecular clock analyses in modern birds based on full mitochondrial genomes.
bioRxiv 855833. https://doi.org/10.1101/855833
Avendaño, J.E., Donegan, T.M., 2015. A distinctive new subspecies of Scytalopus
griseicollis (Aves, Passeriformes, Rhinocryptidae) from the northern Eastern
Cordillera of Colombia and Venezuela. Zookeys 506, 137–153.
https://doi.org/10.3897/zookeys.506.9553
Avise, J.C., Arnold, J., Ball, R.M., Bermingham, E., Lamb, T., Neigel, J.E., Reeb, C.A.,
Saunders, N.C., 1987. Intraspecific phylogeography: The mitochondrial DNA bridge
between population genetics and systematics. Annu. Rev. Ecol. Syst. 18, 489–522.
https://doi.org/10.1146/annurev.es.18.110187.002421
Ballard, J.W.O., Melvin, R.G., 2010. Linking the mitochondrial genotype to the organismal
phenotype: Invited review. Mol. Ecol. 19, 1523–1539. https://doi.org/10.1111/j.1365-
294X.2010.04594.x
Ballard, J.W.O., Whitlock, M.C., 2004. The incomplete natural history of mitochondria. Mol.
Ecol. 13, 729–744. https://doi.org/10.1046/j.1365-294X.2003.02063.x
Bandelt, H.-J., Forster, P., Röhl, A., 1999. Median-joining networks for inferring
intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48.
Benham, P.M., Cheviron, Z.A., 2019. Divergent mitochondrial lineages arose within a
large, panmictic population of the Savannah sparrow (Passerculus sandwichensis).
Mol. Ecol. 28, 1765–1783. https://doi.org/10.1111/mec.15049
Benham, P.M., Cuervo, A.M., Mcguire, J.A., Witt, C.C., 2015. Biogeography of the Andean
metaltail hummingbirds: Contrasting evolutionary histories of tree line and habitat-
generalist clades. J. Biogeogr. 42, 763–777. https://doi.org/10.1111/jbi.12452
Benham, P.M., Witt, C.C., 2016. The dual role of Andean topography in primary
divergence: Functional and neutral variation among populations of the hummingbird,
Metallura tyrianthina. BMC Evol. Biol. 16, 1–16. https://doi.org/10.1186/s12862-016-
0595-2
Bonnet, T., Leblois, R., Rousset, F., Crochet, P.A., 2017. A reassessment of explanations

for discordant introgressions of mitochondrial and nuclear genomes. Evolution (N. Y).
71, 2140–2158. https://doi.org/10.1111/evo.13296
Cadena, C.D., Pérez-Emán, J.L., Cuervo, A.M., Céspedes, L.N., Epperly, K.L., Klicka,
J.T., 2019. Extreme genetic structure and dynamic range evolution in a montane
passerine bird: implications for tropical diversification. Biol. J. Linn. Soc. 126, 487–
506. https://doi.org/10.1093/biolinnean/bly207
Cadena, C.D., Zapata, F., Jiménez, I., 2018. Issues and perspectives in species
delimitation using phenotypic data: Atlantean evolution in Darwin’s finches. Syst. Biol.
67, 181–194. https://doi.org/10.1093/sysbio/syx071
Campagna, L., Benites, P., Lougheed, S.C., Lijtmaer, D.A., Di Giacomo, A.S., Eaton, M.D.,
Tubaro, P.L., 2012. Rapid phenotypic evolution during incipient speciation in a
continental avian radiation. Proc. R. Soc. B Biol. Sci. 279, 1847–1856.
https://doi.org/10.1098/rspb.2011.2170
Chaves, J.A., Pollinger, J.P., Smith, T.B., LeBuhn, G., 2007. The role of geography and
ecology in shaping the p hylogeography of the speckled hummingbird (Adelomyia
melanogenys) in Ecuador. Mol. Phylogenet. Evol. 43, 795–807.
https://doi.org/http://dx.doi.org/10.1016/j.ympev.2006.11.006
Chaves, J.A., Smith, T.B., 2011. Evolutionary patterns of diversification in the Andean
hummingbird genus Adelomyia. Mol. Phylogenet. Evol. 60, 207–218.
https://doi.org/10.1016/j.ympev.2011.04.007
Chaves, J.A., Weir, J.T., Smith, T.B., 2011. Diversification in Adelomyia hummingbirds
follows Andean uplift. Mol. Ecol. 20, 4564–4576. https://doi.org/10.1111/j.1365-
294X.2011.05304.x
Chesser, R.T., Isler, M.L., Cuervo, A.M., Cadena, C.D., Galen, S.C., Lane, D.F., Hosner,
P.A., 2020. Conservative plumage masks extraordinary phylogenetic diversity in the
Grallaria rufula (Rufous Antpitta) complex of the humid Andes. Auk In press.
D’Horta, F.M., Cuervo, A.M., Ribas, C.C., Brumfield, R.T., Miyaki, C.Y., 2013. Phylogeny
and comparative phylogeography of Sclerurus (Aves: Furnariidae) reveal constant
and cryptic diversification in an old radiation of rain forest understorey specialists. J.
Biogeogr. 40, 37–49. https://doi.org/10.1111/j.1365-2699.2012.02760.x
Edwards, S. V., Kingan, S.B., Calkins, J.D., Balakrishnan, C.N., Jennings, W.B., Swanson,
W.J., Sorenson, M.D., 2005. Speciation in birds: Genes, geography, and sexual
selection. Proc. Natl. Acad. Sci. 102, 6550–6557.
https://doi.org/10.1073/pnas.0501846102

Eliason, C.M., Maia, R., Parra, J.L., Shawkey, M.D., 2020. Signal evolution and
morphological complexity in hummingbirds (Aves: Trochilidae). Evolution (N. Y). 1–
12. https://doi.org/10.1111/evo.13893
Eo, S.H., DeWoody, J.A., 2010. Evolutionary rates of mitochondrial genomes correspond
to diversification rates and to contemporary species richness in birds and reptiles.
Proc. R. Soc. B Biol. Sci. 277, 3587–3592. https://doi.org/10.1098/rspb.2010.0965
Flantua, S.G.A., O’Dea, A., Onstein, R.E., Giraldo, C., Hooghiemstra, H., 2019. The
flickering connectivity system of the north Andean páramos. J. Biogeogr. 1808–1825.
https://doi.org/10.1111/jbi.13607
Galtier, N., Nabholz, B., GlÉmin, S., Hurst, G.D.D., 2009. Mitochondrial DNA as a marker
of molecular diversity: A reappraisal. Mol. Ecol. 18, 4541–4550.
https://doi.org/10.1111/j.1365-294X.2009.04380.x
García-Moreno, J., 2004. Is there a universal mtDNA clock for birds? J. Avian Biol. 35,
465–468. https://doi.org/10.1111/j.0908-8857.2004.03316.x
García-Moreno, J., Arctander, P., Fjeldså, J., 1999. Strong Diversification at the treeline
among Metallura hummingbirds. Auk 116, 702–711.
González, C., Ornelas, J.F., Gutiérrez-Rodríguez, C., Gonzalez, C., Ornelas, J.F.,
Gutierrez-Rodriguez, C., 2011. Selection and geographic isolation influence
hummingbird speciation: Genetic, acoustic and morphological divergence in the
wedge-tailed sabrewing (Campylopterus curvipennis). BMC Evol. Biol. 11, 38.
https://doi.org/10.1186/1471-2148-11-38
Goudet, J., Jombart, T., 2015. HIERFSTAT, a package for R to compute and test
hierarchical F-statistics.
Graham, C.H., Silva, N., Velásquez-Tibatá, J., 2010. Evaluating the potential causes of
range limits of birds of the Colombian Andes. J. Biogeogr. 37, 1863–1875.
https://doi.org/10.1111/j.1365-2699.2010.02356.x
Grandcolas, P., Nattier, R., Trewick, S., 2014. Relict species: A relict concept ? Trends
Ecol. Evol. 29, 655–663. https://doi.org/10.1016/j.tree.2014.10.002
Guarnizo, C.E., Amézquita, A., Bermingham, E., 2009. The relative roles of vicariance
versus elevational gradients in the genetic differentiation of the high Andean tree frog,
Dendropsophus labialis. Mol. Phylogenet. Evol. 50, 84–92.
https://doi.org/10.1016/j.ympev.2008.10.005
Gutiérrez-Pinto, N., Cuervo, A.M., Miranda, J., Pérez-Emán, J.L., Brumfield, R.T., Cadena,
C.D., 2012. Non-monophyly and deep genetic differentiation across low-elevation

barriers in a Neotropical montane bird (Basileuterus tristriatus; Aves: Parulidae). Mol.
Phylogenet. Evol. 64, 156–65. https://doi.org/10.1016/j.ympev.2012.03.011
Hahn, C., Bachmann, L., Chevreux, B., 2013. Reconstructing mitochondrial genomes
directly from genomic next-generation sequencing reads - A baiting and iterative
mapping approach. Nucleic Acids Res. 41. https://doi.org/10.1093/nar/gkt371
Harris, R.B., Alström, P., Ödeen, A., Leaché, A.D., 2018. Discordance between genomic
divergence and phenotypic variation in a rapidly evolving avian genus (Motacilla).
Mol. Phylogenet. Evol. 120, 183–195. https://doi.org/10.1016/j.ympev.2017.11.020
Irwin, D.E., Rubtsov, A.S., Panov, E.N., 2009. Mitochondrial introgression and
replacement between yellowhammers (Emberiza citrinella) and pine buntings
(Emberiza leucocephalos) (Aves: Passeriformes). Biol. J. Linn. Soc. 98, 422–438.
https://doi.org/10.1111/j.1095-8312.2009.01282.x
James, J., Castellano, D., Eyre-Walker, A., 2017. DNA sequence diversity and the
efficiency of natural selection in animal mitochondrial DNA. Heredity (Edinb). 118, 88–
95. https://doi.org/10.1038/hdy.2016.108
James, J.E., Piganeau, G., Eyre-Walker, A., 2016. The rate of adaptive evolution in animal
mitochondria. Mol. Ecol. 25, 67–78. https://doi.org/10.1111/mec.13475
Jiménez, R.A., Ornelas, J.F., 2016. Historical and current introgression in a Mesoamerican
hummingbird species complex: A biogeographic perspective. PeerJ 2016.
https://doi.org/10.7717/peerj.1556
Kearns, A.M., Restani, M., Szabo, I., Schrøder-nielsen, A., Kim, J.A., Richardson, H.M.,
Marzluff, J.M., Fleischer, R.C., Johnsen, A., Omland, K.E., 2018. Genomic evidence
of speciation reversal in ravens. Nat. Commun. 9. https://doi.org/10.1038/s41467-
018-03294-w
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S.,
Cooper, A., Markowitz, S., Duran, C., Thierer, T., Ashton, B., Meintjes, P.,
Drummond, A., 2012. Geneious Basic: An integrated and extendable desktop
software platform for the organization and analysis of sequence data. Bioinformatics
28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kraus, R.H.S., Wink, M., 2015. Avian genomics: Fledging into the wild! J. Ornithol. 156,
851–865. https://doi.org/10.1007/s10336-015-1253-y
Krosby, M., Rohwer, S., 2009. A 2000 km genetic wake yields evidence for northern
glacial refugia and hybrid zone movement in a pair of songbirds. Proc. R. Soc. B Biol.
Sci. 276, 615–621. https://doi.org/10.1098/rspb.2008.1310

Leigh, J.W., Bryant, D., 2015. POPART: Full-feature software for haplotype network
construction. Methods Ecol. Evol. 6, 1110–1116. https://doi.org/10.1111/2041-
210X.12410
Lougheed, S.C., Campagna, L., Dávila, J.A., Tubaro, P.L., Lijtmaer, D.A., Handford, P.,
2013. Continental phylogeography of an ecologically and morphologically diverse
Neotropical songbird, Zonotrichia capensis. BMC Evol. Biol. 13.
https://doi.org/10.1186/1471-2148-13-58
Lovette, I.J., 2004. Mitochondrial dating and mixed support for the “2% rule” in birds. Auk
121, 1–6. https://doi.org/10.2307/4090049
Lovette, I.J., Pérez-Emán, J.L., Sullivan, J.P., Banks, R.C., Fiorentino, I., Córdoba-
Córdoba, S., Echeverry-Galvis, M., Barker, F.K., Burns, K.J., Klicka, J., Lanyon, S.M.,
Bermingham, E., 2010. A comprehensive multilocus phylogeny for the wood-warblers
and a revised classification of the Parulidae (Aves). Mol. Phylogenet. Evol. 57, 753–
770. https://doi.org/10.1016/j.ympev.2010.07.018
Lozano-Jaramillo, M., Rico-Guevara, A., Cadena, C.D., 2014. Genetic differentiation, niche
divergence, and the origin and maintenance of the disjunct distribution in the
blossomcrown Anthocephala floriceps (Trochilidae). PLoS One 9.
https://doi.org/10.1371/journal.pone.0108345
Luna, L.W., Rêgo, P.S. do, Sampaio, I., Schneider, H., Carneiro, L.S., Araripe, J., de Girão
e Silva, W.A., Souza, T.O., 2017. Molecular data and distribution dynamics indicate a
recent and incomplete separation of manakins species of the genus Antilophia (Aves:
Pipridae) in response to Holocene climate change. J. Avian Biol. 48, 1177–1188.
https://doi.org/10.1111/jav.01378
McGuire, J.A., Witt, C.C., Remsen, J. V., Dudley, R., Altshuler, D.L., 2008. A higher-level
taxonomy for hummingbirds. J. Ornithol. 150, 155–165.
https://doi.org/10.1007/s10336-008-0330-x
Moore, W.S., 1995. Inferring phylogenies from mtDNA variation: Mitochondrial-gene trees
versus nuclear-gene trees. Evolution (N. Y). 49, 718. https://doi.org/10.2307/2411136
Morales, H.E., Pavlova, A., Joseph, L., Sunnucks, P., 2015. Positive and purifying
selection in mitochondrial genomes of a bird with mitonuclear discordance. Mol. Ecol.
24, 2820–2837. https://doi.org/10.1111/mec.13203
Morales, H.E., Sunnucks, P., Joseph, L., Pavlova, A., 2017. Perpendicular axes of
differentiation generated by mitochondrial introgression. Mol. Ecol. 26, 3241–3255.
https://doi.org/10.1111/mec.14114

Morgan-Richards, M., Trewick, S.A., Bartosch-Härlid, A., Kardailsky, O., Phillips, M.J.,
McLenachan, P.A., Penny, D., 2008. Bird evolution: Testing the Metaves clade with
six new mitochondrial genomes. BMC Evol. Biol. 8, 1–12.
https://doi.org/10.1186/1471-2148-8-20
Ornelas, J.F., González, C., Espinosa de los Monteros, A., Rodríguez-Gómez, F., García-
Feria, L.M., 2014. In and out of Mesoamerica: Temporal divergence of Amazilia
hummingbirds pre-dates the orthodox account of the completion of the Isthmus of
Panama. J. Biogeogr. 41, 168–181. https://doi.org/10.1111/jbi.12184
Ornelas, J.F., González, C., Hernández-Baños, B.E., García-Moreno, J., 2016. Molecular
and iridescent feather reflectance data reveal recent genetic diversification and
phenotypic differentiation in a cloud forest hummingbird. Ecol. Evol. 6, 1104–1127.
https://doi.org/10.1002/ece3.1950
Oyler-McCance, S.J., Oh, K.P., Langin, K.M., Aldridge, C.L., 2016. A field ornithologist’s
guide to genomics: Practical considerations for ecology and conservation. Auk 133,
626–648. https://doi.org/10.1642/AUK-16-49.1
Palacios, C., Garcia-R, S., Parra, J.L., Cuervo, A.M., Stiles, F.G., McCormack, J.E.,
Cadena, C.D., 2019. Shallow evolutionary divergence between two Andean
hummingbirds: Speciation with gene flow? Auk 136. https://doi.org//10.1101/249755
Parra, J.L., 2010. Color evolution in the hummingbird genus Coeligena. Evolution (N. Y).
64, 324–335. https://doi.org/10.1111/j.1558-5646.2009.00827.x
Parra, J.L., Remsen, J. V., Alvarez-Rebolledo, M., McGuire, J.A., 2009. Molecular
phylogenetics of the hummingbird genus Coeligena. Mol. Phylogenet. Evol. 53, 425–
434. https://doi.org/10.1016/j.ympev.2009.07.006
Piganeau, G., Gardner, M., Eyre-Walker, A., 2004. A broad survey of recombination in
animal mitochondria. Mol. Biol. Evol. 21, 2319–2325.
https://doi.org/10.1093/molbev/msh244
Pons, J.M., Sonsthagen, S., Dove, C., Crochet, P.A., 2014. Extensive mitochondrial
introgression in North American Great Black-backed Gulls (Larus marinus) from the
American Herring Gull (Larus smithsonianus) with little nuclear DNA impact. Heredity
(Edinb). 112, 226–239. https://doi.org/10.1038/hdy.2013.98
Price, T.D., 2008. Speciation in Birds. Roberts & Company Publishers, Greenwood Village,
Colorado.
Prosdocimi, F., Souto, H.M., Ruschi, P.A., Furtado, C., Jennings, W.B., 2016. Complete
mitochondrial genome of the versicoloured emerald hummingbird Amazilia versicolor,

a polymorphic species. Mitochondrial DNA 27, 3214–3215.
https://doi.org/10.3109/19401736.2015.1007352
R Core Team, 2017. R: A Language and environment for statistical computing.
Remsen, J. V., 2005. Pattern, process, and rigor meet classification. Auk 122, 403.
https://doi.org/10.1642/0004-8038(2005)122[0403:pparmc]2.0.co;2
Rheindt, F.E., Fujita, M.K., Wilton, P.R., Edwards, S. V., 2014. Introgression and
phenotypic assimilation in Zimmerius flycatchers (Tyrannidae): Population genetic
and phylogenetic inferences from genome-wide SNPs. Syst. Biol. 63, 134–152.
https://doi.org/10.1093/sysbio/syt070
Rheindt, F.E., Székely, T., Edwards, S. V, Lee, P.L.M., Burke, T., Kennerley, P.R.,
Bakewell, D.N., Alrashidi, M., Kosztolányi, A., Weston, M. a, Liu, W.-T., Lei, W.-P.,
Shigeta, Y., Javed, S., Zefania, S., Küpper, C., 2011. Conflict between genetic and
phenotypic differentiation: The evolutionary history of a “lost and rediscovered”
shorebird. PLoS One 6, e26995. https://doi.org/10.1371/journal.pone.0026995
Ribas, C.C., Aleixo, A., Nogueira, A.C.R., Miyaki, C.Y., Cracraft, J., 2012. A
palaeobiogeographic model for biotic diversification within Amazonia over the past
three million years. Proc. R. Soc. B Biol. Sci. 279, 681–689.
https://doi.org/10.1098/rspb.2011.1120
Rodríguez-Gómez, F., Ornelas, J.F., 2015. At the passing gate: Past introgression in the
process of species formation between Amazilia violiceps and A. Viridifrons
hummingbirds along the Mexican Transition Zone. J. Biogeogr. 42, 1305–1318.
https://doi.org/10.1111/jbi.12506
Rokas, A., Ladoukakis, E., Zouros, E., 2003. Animal mitochondrial DNA recombination
revisited. Trends Ecol. Evol. 18, 411–417. https://doi.org/10.1016/S0169-
5347(03)00125-3
Roulin, A., 2004. The evolution, maintenance and adaptive function of genetic colour
polymorphism in birds. Biol. Rev. Camb. Philos. Soc. 79, 815–48.
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J.C., Guirao-Rico, S., Librado, P., Ramos-
Onsins, S.E., Sanchez-Gracia, A., 2017. DnaSP 6: DNA sequence polymorphism
analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302.
https://doi.org/10.1093/molbev/msx248
Sammler, S., Bleidorn, C., Tiedemann, R., 2011. Full mitochondrial genome sequences of
two endemic Philippine hornbill species (Aves: Bucerotidae) provide evidence for
pervasive mitochondrial DNA recombination. BMC Genomics 12.

https://doi.org/10.1186/1471-2164-12-35
Schubert, M., Lindgreen, S., Orlando, L., 2016. AdapterRemoval v2: rapid adapter
trimming, identification, and read merging. BMC Res. Notes 9, 88.
https://doi.org/10.1186/s13104-016-1900-2
Sedano, R.E., Burns, K.J., 2010. Are the Northern Andes a species pump for Neotropical
birds? Phylogenetics and biogeography of a clade of Neotropical tanagers (Aves:
Thraupini). J. Biogeogr. 37, 325–343. https://doi.org/10.1111/j.1365-
2699.2009.02200.x
Sievers, F., Wilm, A., Dineen, D., Gibson, T.J., Karplus, K., Li, W., Lopez, R., McWilliam,
H., Remmert, M., Söding, J., Thompson, J.D., Higgins, D.G., 2011. Fast, scalable
generation of high-quality protein multiple sequence alignments using Clustal Omega.
Mol. Syst. Biol. 7, 539. https://doi.org/10.1038/msb.2011.75
Sornoza-Molina, F., Freile, J.F., Nilsson, J., Krabbe, N., Bonaccorso, E., 2018. A striking,
critically endangered, new species of hillstar (Trochilidae: Oreotrochilus) from the
southwestern Andes of Ecuador. Auk 135, 1146–1171. https://doi.org/10.1642/auk-
18-58.1
Sosa, J., Parra, J.L., Stavenga, D.G., Giraldo, M.A., 2020. Sexual dichromatism of the
Blue-throated Starfrontlet, Coeligena helianthea, hummingbird plumage. J. Ornithol.
161, 289–296. https://doi.org/10.1007/s10336-019-01709-z
Souto, H.M., Ruschi, P.A., Furtado, C., Jennings, W.B., Prosdocimi, F., 2016. The
complete mitochondrial genome of the ruby-topaz hummingbird Chrysolampis
mosquitus through Illumina sequencing. Mitochondrial DNA 27, 769–770.
https://doi.org/10.3109/19401736.2014.915533
Stamatakis, A., 2014. RAxML version 8: A tool for phylogenetic analysis and post-analysis
of large phylogenies. Bioinformatics 30, 1312–1313.
https://doi.org/10.1093/bioinformatics/btu033
Toews, D.P.L., Brelsford, A., 2012. The biogeography of mitochondrial and nuclear
discordance in animals. Mol. Ecol. 21, 3907–3930. https://doi.org/10.1111/j.1365-
294X.2012.05664.x
Toews, D.P.L., Campagna, L., Taylor, S.A., Balakrishnan, C.N., Baldassarre, D.T., Deane-
Coe, P.E., Harvey, M.G., Hooper, D.M., Irwin, D.E., Judy, C.D., Mason, N.A.,
McCormack, J.E., McCracken, K.G., Oliveros, C.H., Safran, R.J., Scordato, E.S.C.,
Stryjewski, K.F., Tigano, A., Uy, J.A.C., Winger, B.M., 2016. Genomic approaches to
understanding population divergence and speciation in birds. Auk 133, 13–30.

https://doi.org/10.1642/AUK-15-51.1
Toews, D.P.L., Mandic, M., Richards, J.G., Irwin, D.E., 2014. Migration, mitochondria, and
the yellow-rumped warbler. Evolution (N. Y). 68, 241–255.
https://doi.org/10.1111/evo.12260
Uy, J.A.C., Moyle, R.G., Filardi, C.E., 2009. Plumage and song differences mediate
species recognition between incipient Flycatcher species of the Solomon Islands.
Evolution (N. Y). 63, 153–164. https://doi.org/10.1111/j.1558-5646.2008.00530.x
Valderrama, E., Pérez-Emán, J.L., Brumfield, R.T., Cuervo, A.M., Cadena, C.D., 2014.
The influence of the complex topography and dynamic history of the montane
Neotropics on the evolutionary differentiation of a cloud forest bird (Premnoplex
brunnescens, Furnariidae). J. Biogeogr. 41, 1533–1546.
https://doi.org/10.1111/jbi.12317
Wilson, A.C., Kathleen, M., Higuchi, R.G., Stephen, R., Prager, E.M., 1985. Mitochondrial
DNA and two perspectives on evolutionary genetics. Biol. J. Linn. Soc. 26, 375–400.
Winger, B.M., Bates, J.M., 2015. The tempo of trait divergence in geographic isolation:
Avian speciation across the Marañon Valley of Peru. Evolution (N. Y). 69, 772–787.
https://doi.org/10.1111/evo.12607
Wort, E.J.G., Fenberg, P.B., Williams, S.T., 2017. Testing the contribution of individual
genes in mitochondrial genomes for assessing phylogenetic relationships in
Vetigastropoda. J. Molluscan Stud. 83, 123–128.
https://doi.org/10.1093/mollus/eyw044
Zamudio-Beltrán, L.E., Hernández-Baños, B.E., 2018. Genetic and morphometric
divergence in the Garnet-Throated Hummingbird Lamprolaima rhami (Aves:
Trochilidae). PeerJ 2018, 1–22. https://doi.org/10.7717/peerj.5733
Zamudio-Beltrán, L.E., Hernández-Baños, B.E., 2015. A multilocus analysis provides
evidence for more than one species within Eugenes fulgens (Aves: Trochilidae). Mol.
Phylogenet. Evol. 90, 80–84. https://doi.org/10.1016/j.ympev.2015.04.024
Zhang, D., Tang, L., Cheng, Y., Hao, Y., Xiong, Y., Song, G., Qu, Y., Rheindt, F.E., Alstro,
P., Jia, C., Lei, F., 2019. “ Ghost Introgression ” as a cause of deep mitochondrial
divergence in a bird species complex. Mol. Biol. Evol. 36, 2375–2386.
https://doi.org/10.1093/molbev/msz170

Seeking gold and roses – Genomic differentiation and evolution of lineages
of Andean Coeligena Hummingbirds – Chapter 3
Developed in collaboration with Leonardo Campagna1,2, Juan Luis Parra3, and Carlos
Daniel Cadena4
1 Department of Ecology and Evolutionary Biology, Cornell University, Ithaca, New York,
USA
2 Fuller Evolutionary Biology Program, Cornell Lab of Ornithology, Cornell University,
Ithaca, New York, USA
3 Grupo de Ecología y Evolución de Vertebrados, Instituto de Biología, Universidad de
Antioquia, Calle 67 No. 53-108, Medellín, Colombia
4 Laboratorio de Biología Evolutiva de Vertebrados, Departamento de Ciencias Biológicas,
Universidad de Los Andes, Carrera 1 No. 18 A 10, Bogotá, Colombia
Introduction
Uncovering the genetic basis of phenotypes is crucial for understanding the role that
different evolutionary mechanisms play in driving divergence between species. Genomic
comparisons of closely related species are especially useful to inform about the role of
mechanisms acting to produce evolutionary diversification, and to uncover information on
candidate genes and genomic regions potentially involved in phenotypic differentiation. In
particular, highly differentiated genomic regions between species showing little overall
genetic differentiation may play a major role in establishing phenotypic differences relevant
to speciation even if populations experience gene flow (Ellegren et al., 2012; Feder, Egan,
& Nosil, 2012). However, highly differentiated genomic regions may also be the result of
processes associated to genomic architecture, linked selection, and variation in
recombination rates among regions (Burri, 2017; Seehausen et al., 2014). In turn, low
differentiation between species in genomic regions may be a transient phenomenon if
such a pattern reflects common ancestry (i.e. incomplete lineage sorting Wang et al.,
2018), or may be maintained owing to recurrent gene flow (i.e. introgression Martin &
Jiggins, 2017; Ottenburghs et al., 2017). Given such complexity and dynamism of the
genomic landscape of differentiation between species, more genomic data of better quality

are necessary to move forward in our understanding of how mutation, drift, gene flow, and
selection shape the origin and evolution of species.
Genomic studies of birds have blossomed over recent years (Kraus & Wink, 2015; Zhang
et al., 2014). Since the first sequencing of the chicken genome in 2004 (ICGSC
International Chicken Genome Sequencing Consortium, 2004), several group initiatives
like the Avian Phylogenomics Project and the B10K project (Genome 10K, 2009; Koepfli,
Paten, & Brien, 2015) as well as the work of many others have provided us with one of the
largest and better quality genomic data sets for any animal group existing to date (Rhie et
al., 2020; Zhang et al., 2014), with more than 500 genomics projects for more than 180
bird species currently registered in Genbank [https://www.ncbi.nlm.nih.gov/bioproject/].
The development of avian genomic resources has been instrumental in connecting theory
with data relevant to understanding evolution and speciation (Edwards et al., 2005; Toews,
Campagna, et al., 2016). For example, genomic studies of hybridizing bird species have
enabled recognizing mechanisms of isolation leading to hybrid speciation (Hermansen,
Haas, Trier, & Bailey, 2014), and have provided insights into genomic regions prone to and
resistant to introgression, the direction and magnitude of introgression, and discrepancies
or correspondence in spatial patterns of genetic and morphological variation informative of
mechanisms involved in speciation (Billerman, Cicero, Bowie, & Carling, 2019;
Ottenburghs, 2020; Walsh, Shriver, Olsen, & Kovach, 2016). Comparisons of avian
genomes have also helped to understand structural and functional features of genome
evolution such as genomes size, chromosomal structure, gene synteny, recombination
rates, and the role of transposable elements in the stability of genome structure (Kapusta
& Suh, 2017; Kawakami et al., 2017; Van Doren et al., 2017; Zhang et al., 2014). An
emerging pattern from genomic comparisons of many avian systems is that sexual
chromosomes are often highly differentiated between species, a pattern likelyy related to
both structural features of genomes, and to the presence of genes involved in phenotypic
differentiation and evolving under selection (Batttey, 2020; Ellegren et al., 2012; Sigeman
et al., 2019).
Avian genomic studies have also provided information on genes and genomic regions
candidates to be the genetic basis of a variety of phenotypic traits. Examples include gene
regions associated with the evolution of flightlessness (Burga et al., 2017; Sackton et al.,
2019), the origin of distinct reproductive morphs (Küpper et al., 2015; Tuttle et al., 2016)
and sexual dichromatism (Gazda et al., 2020), distinct migratory behaviors (Toews, Taylor,

Streby, Kramer, & Lovette, 2019) and various genes and regulatory regions involved in bill
(Bosse et al., 2017; Lamichhaney et al., 2015b, 2016; Yusuf et al., 2020). Among the
many avian traits being the focus of genomic analyses, coloration has figured prominently
among studies seeking to understand the genetic basis of phenotypes (Orteu & Jiggins,
2020).
Birds exhibit a remarkable diversity of colors (Stoddard & Prum, 2011) and plumage
coloration is central for avian communication, playing an important role in species
recognition, reproductive state signaling, sexual selection, and reproductive isolation
(Price, 2008; Roulin, 2004). In birds, colors may be produced by pigments found across
many taxa such as melanins and carotenoids (the latter recruited from diet), and also by
more specialized and taxonomically restricted pigments such as porphyrins, pterins,
spheniscins and psittacofulvins (Hill & McGraw, 2006). Alternatively, colors may be the
result of the interaction between light and the structure of feathers (i.e. structural
coloration, Eliason, Maia, Parra, & Shawkey, 2020; Orteu & Jiggins, 2020). Genomic
studies of different avian systems have found associations between variation in coding
sequences and regulatory regions of genes involved in the melanin pathway as important
for phenotypic differences involving melanin coloration (Campagna et al., 2017; Cooper &
Uy, 2017; Delmore et al., 2016; Poelstra et al., 2014; San-jose, Ducret, & Ducrest, 2017;
Toews, Taylor, et al., 2016). Although carotenoid pigments are obtained from the diet,
genomic studies have found that plumage variation involving such pigments is related to
genes involved in carotenoid processing (Brelsford, Toews, & Irwin, 2017; Lopes et al.,
2016; Mundy et al., 2016). Little is known about the genetic underpinnings of other
pigment types, though progress is being made through genomic comparisons such as
those helping to identify a gene related to the expression of psittacofulvins (Cooke et al.,
2017).
In contrast to pigment-based coloration, the genetic basis of structural coloration in birds
remains essentially unknown (Orteu & Jiggins, 2020). Structural coloration is considered a
complex trait involving several morphological, physiological, and developmental
components which in birds interact to produce the nanostructure of feathers (Eliason et al.,
2020). Variation in the quantity and organization of layers of keratin, air space, and
melanosomes (organelles with the pigment melanin) in feathers is linked to diversity of
structural colors, but the interaction between such components is still not fully understood
(Orteu & Jiggins, 2020). Because structural coloration is a complex trait, it is expected to

have a polygenic basis, with involvement of several genes and regulatory factors, including
both genes affecting feather nanostructure and those involved in the melanin metabolic
pathway. The polygenic base of structural coloration has been supported on other animal
systems (Brien et al., 2019), but studies on the topic focused on birds are lacking. Among
birds, hummingbirds stand out for their remarkable diversity in iridescent plumage colors
produced by structural coloration (Eliason et al., 2020). Here, we studied a system of
closely related species of Andean hummingbirds to characterize their genomic landscape
of differentiation and search for candidate genomic regions related to structural color likely
involved in the speciation processes.
The Golden-bellied Starfrontlet (Coeligena bonapartei) and the Blue-throated Starfrontlet
(Coeligena helianthea) are recently diverged species differing strikingly in their coloration.
C. bonapartei males are greenish with fiery golden underparts, whereas C. helianthea
males are blackish with rose bellies and aquamarine rumps. Females are paler than
males, but still distinct. Despite the marked differences in coloration between species
which may partly reflect melanin content (as well as structural differences, Eliason et al.,
2020; Sosa, Parra, Stavenga, & Giraldo, 2020), they exhibit no differences in the coding
sequence of the Melanocortin-1 receptor (MC1R) (Palacios et al., 2019), a gene involved
in the metabolic pathway of melanin responsible for color variation in many vertebrate
taxa. These hummingbird species comprise four subspecies: C. b. consita, C. b.
bonapartei, C. h. helianthea and C. h. tamai, which diverged from their closest relative (C.
b. eos) less than 500,000 years ago (Palacios et al., 2019; Palacios et al. under review)
and are sympatric in part of the Cordillera Oriental in the Colombian Andes where different
phenotypes persist (Figure 1 A). Patterns of genetic variation among these lineages in
individual mitochondrial genes, complete mitochondrial genomes, and a set of ultra-
conserved elements did not match patterns of plumage variation, suggesting speciation in
the group may have proceeded in the face of gene flow, with phenotypic divergence
potentially maintained by selection on loci related to traits involved in mating or ecological
adaptation (Palacios et al., 2019; Palacios et al. under review). Predictions of such
divergence-with-gene flow hypothesis of speciation have not been examined based on
genome-wide patterns of genetic variation.
To further examine the history of speciation of C. bonapartei and C. helianthea clarify, the
evolutionary relationships among its four lineages, and to look for candidate genes related
to differences in the structural coloration of these hummingbirds, we adopted a population

genomic approach in which we sequenced complete draft genomes of 46 individuals. We
addressed the following questions: (1) Are the four lineages in C. bonapartei and C.
helianthea distinguishable with whole-genome data? (2) What are the evolutionary
relationships among lineages of these hummingbirds as inferred based on complete
genomes? (3) How is the landscape of genomic differentiation among the four lineages of
Coeligena? (4) Are there candidate genes associated with coloration differences in these
hummingbirds? And (5) Are patterns of genomic differentiation between Coeligena
hummingbirds consistent with introgression and selection (i.e. divergence with gene flow)
as suggested by previous analyses based on mitochondrial data? By answering these
questions our study furthers our understanding of the evolution of Coeligena
hummingbirds, and sheds light on the role of different evolutionary mechanisms as drivers
of divergence and speciation.
Materials and Methods
Samples, Genome sequencing and assembly
We obtained tissue samples (muscle) from 46 voucher specimens of C. bonapartei and C.
helianthea from the collections at Instituto Alexander von Humboldt (IAvH), Museo de
Historia Natural de la Universidad de los Andes (ANDES), and Instituto de Ciencias
Naturales de la Universidad Nacional de Colombia (ICN-Aves). Taxon identities were
assigned by museum curators according to phenotype and geography. We sampled 9
individuals of C. b. consita, 12 C. b. bonapartei, 10 C. h. helianthea and 15 C. h. tamai,
and relatively even samples of each sex (Table S1).
We used a custom phenol/chloroform method coupled with Phase-Lock Gel tubes and a
magnetic beads cleaning protocol to extract total genomic DNA from samples. We
followed the manufacturer’s protocol to prepare Illumina TruSeq Nano DNA-enriched
libraries for low-throughput configuration and 550bp insert size per sample. Libraries were
quantified with a Qubit fluorometer. Normalizing, pooling and sequencing of the libraries
were done by the Genomics Facility of the Institute of Biotechnology of Cornell University.
Two lanes of NexSeq 500 2x150 paired end were used for sequencing. We filtered raw
reads by quality following Illumina recommendations, checked reads using Fastqc
(Andrews, 2010), and cleaned adapters using AdapterRemoval (Schubert, Lindgreen, &
Orlando, 2016). We estimated average coverage according to the retained reads after
filtered and considering the total genome size as 1.1 Gb estimated for the Anna’s

Hummingbird (Calypte anna) (Zhang B; Li, C; Gilbert,M.T.P.; Mello, C.V.; Jarvis, E.D.; The
Avian Genome Consortium; Wang,J;, 2014).
We used two reference genomes to assemble our data: that of C. anna provided by Erich
Jarvis’ Lab at Rockefeller University (Rhie et al., 2020), and that of the Black-breasted
Hillstar (Oreotrochilus melanogaster) provided by Christopher Witt’s Lab at the University
of New Mexico (umpubl. Data). We employed both reference genomes because the
genome of C. anna is robustly assembled and annotated; O. melanogaster is closely
related to Coeligena hummingbirds (McGuire, Witt, Remsen, Dudley, & Altshuler, 2008)
but its genome is partially assembled and not annotated. Because the percentage of
alignment reads against to the genome of O. melanogaster was higher than to the genome
of C. anna (see results) we proceeded using the genome of O. melanogaster as reference.
We used Bowtie (Langmead & Salzberg, 2012), Samtools (Li, 2011; Li & Durbin, 2009),
and Picardtools (MIT, 2017) to index and align the filtered reads to each reference genome
and marked duplicates. We used Genome Analysis Toolkit GATK (McKenna et al., 2010)
to call the variants per individual and then join them in a single vcf file. We filtered this vcf
file to get sets of single nucleotide polymorphisms (SNPs) files for analyses.
Phylogenetic and Genomic Population Analyses
We assessed whether lineages of Coeligena (i.e. species and subspecies defined based
on plumage coloration) are distinguishable with genomic data using a biallelic SNPs data
set to perform a principal component analysis with the R package SNPRelate (R Core
Team, 2017; Zheng et al., 2012). We also examined phylogenetic relationships among
individuals and determined whether lineages formed clades. We used the biallelic SNPs
data set to build a maximum-likelihood phylogenetic tree with the SVDquartets method
(Chifman & Kubatko, 2014, 2015) in PAUP* V4 (Swofford, 2003), using 100 bootstrap
replicates to assess nodal support, and including O. melanogaster as outgroup.
We characterized the genomic landscape of differentiation among lineages of Coeligena
by calculating Fst values in non-overlapping 25 kb windows across the genome for all
comparisons (i.e. between species and subspecies) using VCFtools 0.1.14 (Danecek et
al., 2011). To identify candidate genes potentially involved in phenotypic differences
between the lineages of Coeligena we initially focused on Fst comparisons between
lineages differing in plumage phenotype, i.e. C. bonapartei vs. C. helianthea. We defined
outlier windows as those with mean Fst values four times the standard deviation above the

genomic mean. However, our finding of phylogenetic relationships inconsistent with
phenotypic variation (see below) provided a unique opportunity to identify candidate
regions via comparisons involving pairs of close relatives being either different (C. b.
bonapartei and C. h. helianthea) or similar in coloration phenotype (C. h. helianthea and C.
h. tamai). Because Fst comparisons between all lineages revealed peaks of differentiation
in broadly the same regions (see results), we sought to identify outlier windows between
lineages with different coloration phenotype while controlling by genomic differentiation
between lineages with similar coloration phenotype. Therefore, we focused on examining
genomic differentiation between C. b. bonapartei and C. h. helianthea (whith strikingly
different coloration) relative to that between C. h. helianthea and C. h. tamai (whit similar
coloration). For this comparison, we Z-transformed Fst values (Z-Fst = (Fst value – Fst
mean)/Fst standard deviation) and then calculated Delta Z-Fst (Z-Fst) as the difference of
Z-Fst values between comparisons (Vijay et al., 2016). We selected outlier windows as
those with Z-Fst values at the 99th percentile. To identify loci in outliers windows we used
BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and considered the first three best blast
hits. We then grouped the obtained loci according functional categories based on
information from the NCBI (https://www.ncbi.nlm.nih.gov/gene/).
Results
Genome sequencing and assembly
We recovered genomes with an average coverage of 3.9X (range 2.7- 6.5X, Table S1).
We excluded 3 individuals from subsequent analyses due to low sequencing or assembly
quality (ID 23, 24 and 33 in Table S1). Our reads assembled on average 91% against the
genome of C. anna and 97% against the genome of O. melanogaster. Thus, we conducted
subsequent analyses with 43 individuals and assemblies which used the genome of O.
melanogaster as reference. The version we used of this reference genome was
assembled in 434,731 scaffolds with scaffold N50 and N90 of 3.5 Mb and 183.8 kb, and a
scaffold L50 of 87. We obtained a total of 7.3 million SNPs and 6.3 million biallelic SNPs
after filtering. Considering the size of the genome of O. melanogaster (1.17 Gb) and the
percentage of it covered by our reads assemblies (97%) we estimated the size of the draft
genomes of Coeligena hummingbirds is roughly 1.14 Gb. Considering the total number of
SNPs we obtained as the variable proportion of the genome among the lineages of
Coeligena, these hummingbirds may differ in approximately 0.064% of their genomes.

Genomic differentiation and phylogenetic relationships among lineages
Individuals from each lineage (i.e. subspecies) clustered together in principal component
analyses (PCA, Figure 1 B). The first principal component (PC1) explained 12.44% of
genetic variation and mainly separated C. b. consita from the other three lineages,
whereas PC2 explained 5.79% of genetic variation and mainly separated C. b. bonapartei
from both lineages of C. helianthea. We ran a second PCA excluding C. b. consita and
found that PC1 explained 7.36% of genetic variation and separated individuals of C. b.
bonapartei from lineages of C. helianthea. PC2 explained 4.81% of genetic variation and
mainly separated C. h. helianthea from C. h. tamai. Individuals referred to C. b. bonapartei
appeared to cluster in two different groups which agree with their geographical distribution,
with one group from localities in the north and the other from localities in the south of this
taxon’s range (Figure 1 A).
Phylogenetic analysis clustered all individuals of Coeligena in a clade with 100% bootstrap
support (Figure 1 C). Within this clade, individuals of each lineage clustered together in
well-supported clades (100%) yet phylogenetic relationships inferred with complete
genomes were not consistent with taxonomy. Despite being considered a subspecies of C.
bonapartei, C. b. consita was not sister to C. b. bonapartei, but rather was sister to a
strongly supported clade including C. b. bonapartei and a clade formed by C. h. helianthea
and C. h. tamai. As in the PCA analyses, C. b. bonapartei consisted of two distinct
geographic groups (both with 100% bootstrap support).
Mean Fst values averaged across the genome also showed that C. b. consita is the most
genetically differentiated lineage in the group (Fst values 0.239, 0.297, and 0.240 against
C. bonapartei, C. h. helianthea, and C. h. tamai, respectively). Fst values among the other
three lineages were lower and generally similar to each other (0.078 and 0.083 between C.
b. bonapartei and C. h. helianthea and C. h. tamai respectively, and 0.054 between the
two lineages of C. helianthea).
Genomic landscape of differentiation and candidate genes for phenotypic
differentiation
Mean Fst values estimated for 25-kb windows across the genome were unevenly
distributed among comparisons (Figure S1). Manhattan plots showed dozens of regions
with high Fst values indicating marked differentiation among lineages across multiple
scaffolds (Figure 1 D and Figure S2). Several of the Fst peaks were common across

comparison and many mapped to the Z-chromosome (e.g. Scaffolds 19, 71, 75, 107, and
117 in Figure 1 D, see below). Scaffolds mapping to the Z-chromosome were the most
differentiated between C. h. helianthea and C. h. tamai, two lineages showing similar
coloration and exhibiting the lowest average genomic differentiation.
Comparisons of mean Fst values from 25 kb windows between C. bonapartei (both
subspecies with similar phenotype) and C. helianthea (both subspecies) revealed 149
outlier windows (i.e. with mean Fst values four standard deviations above the genome Fst
mean, Figure 1 D). However, given our interest in finding candidate loci associated with
plumage variation and the overall patterns genomic differentiation and phylogenetic
relationships among lineages we uncovered, we focused on identifying windows showing
strong differentiation between C. b. bonapartei and C. h. helianthea (with different
coloration phenotype) but low differentiation between C. h. helianthea and C. h. tamai (with
similar coloration phenotype). Z-Fst values revealed 174 outlier windows (above the 99th
percentile value) in 79 scaffolds across the genome (Figure 2). We found that 43 windows
were identified as distinct in both the Fst outlier’s analysis comparing C. bonapartei and C.
helianthea and int the Z-Fst analysis comparing phenotypically distinct and
phenotypically similar taxa. These shared windows appear especially promising in terms of
containing candidate genes underlying differences in coloration between lineages.
Of the 79 scaffolds showing outlier windows identified using Z-Fst, 32 were small (<500
bp), thus involved a single outlier window. Among the longer scaffolds (>25 kb), 27
scaffolds showed a single outlier window, and 20 included two or more outlier windows
(Table S2). We were able to identify most of the latter 20 scaffolds because BLAST
mapped most of their windows to the same chromosome in the genome of C. anna. Most
scaffolds (9) mapped to the Z-chromosome (including a total of 47 outlier windows), 3
scaffolds mapped to chromosome 12 (12 total outlier windows), and each of 5 scaffolds
mapped to chromosomes 6, 4, 5A, 4B and 11 (with 14, 8, 6, 4 and 3 outlier windows
respectively). We were unable to identify 3 scaffolds because windows inside them
mapped to different chromosomes, including scaffold 135 (ID in the ordered by size) which
was the one with the highest number of outlier windows (17).
Within the total 174 outlier windows, we found 119 loci which we considered potential
candidates involved in phenotypic differences between C. b. bonapartei and C. h.
helianthea. 75 of these genes were related to nine main functional categories, 25 genes
were scattered in other functional categories, 11 loci were unknown or uncharacterized,

and 8 loci (which we called the usual suspects) mapped to different windows and scaffolds
but with little coverage (Figure 2 and Table S3). The functional categories to which the 119
loci most often corresponded were cell growth, cell differentiation, cell cycle, apoptosis and
autophagy, including transcription factors, homeobox genes, ubiquitin related genes, zinc
finger genes, and members of the Rho and Ras gene family. Other, less frequent
categories were those of genes involved in brain, neurons and neuronal signaling, genes
related to thyroid hormones and a GABA receptor gene. Outlier windows also included
genes related to functional categories such as endothelial cells, glycolysis, collagen,
immune cells, mitochondria-related, epithelia development, and melanin.
Discussion
Relationships among Coeligena lineages and mito-nuclear discordance
Complete genome analyses clearly allowed us to distinguish four lineages of Coeligena
hummingbirds, two of which are treated as subspecies of C. bonapartei and two as
subspecies of C. helianthea based on coloration. However, in contrast to current taxonomy
and to overall coloration similarities we found that C. b. consita is the most genetically
distinct lineage in the complex, and phylogenetic analyses revealed this taxon is not sister
to C. b. bonapartei but rather is the first branch to diverge in the group. Because previous
work showed that C. b. eos, a taxon with coloration similar to that of C. bonapartei, is the
outgroup of the four lineages in which we focused in this study (Palacios et al., 2019),
orange-golden-green plumage coloration is the ancestral state in this clade, whereas the
blackish-rose-aquamarine coloration of C. helianthea is derived.
Despite the marked similarity in plumage between C. b. bonapartei and C. b. consita,
genomic differentiation between them and between C. b. consita and the two lineages of
C. helianthea is quite high relative to genomic differentiation reported in other recently
diverged avian systems (Campagna et al., 2017; Poelstra et al., 2014; Vijay et al., 2016).
Such marked genomic divergence is striking given previous work in which mitochondrial
genomes (and some nuclear loci) did not distinguish lineages of Coeligena corresponding
to species or subspecies (Palacios et al., 2019; Palacios et al. in review). Moreover,
mitogenomes suggested that C. b. consita is more closely related to C. h. tamai than to C.
b. bonapartei, which in turn was indistinguishable from C. h. helianthea in its mitochondrial
genome, indicating that mtDNA variation better reflects geography than taxonomy and
plumage (Palacios et al. in review). These results contrast with our finding that complete

nuclear genomes recover subspecies of C. helianthea as sister groups differentiated from
both C. b. bonapartei and C. b. consita, implying a case of mito-nuclear discordance likely
caused by gene flow and introgression. Although our biallelic SNPs data set produced a
strongly supported phylogeny resolving the relationships among lineages of Coeligena,
phylogenetic analyses focused on independent windows across the genome would be
useful to identify discordant regions, whereas demographic analyses may inform about the
role of incomplete lineage sorting, gene flow, and introgression in the history of the group
(Lamichhaney et al., 2015a; Ottenburghs, 2020).
Landscape of genomic differentiation among Coeligena lineages
An emerging pattern from examining the landscape of differentiation among Coeligena
lineages is that, overall, the same genetic regions seem to show peaks of differentiation in
all comparisons. Because such regions were observed to exhibit divergence both in
comparisons involving phenotypically similar and phenotypically distinct lineages, their
patterns of variation most likely reflect effects of genomic architecture (e.g. regions
standing out in all comparisons may experience low recombination) or regions responding
to the same selective pressures (Vijay et al., 2016). Given such pattern, the Z-Fst
analysis was especially relevant for identifying candidate regions exhibiting high
differentiation between lineages with different phenotypes but low differentiation between
lineages with similar phenotypes. However, many outlier windows identified in the Z-Fst
analysis are in the same scaffolds where we detected peaks of differentiation, such as
those mapping to sexual chromosomes. Elevated values of genetic differentiation in sexual
chromosomes have been reported in many other studies of avian genomics (Batttey, 2020;
Campagna et al., 2017; Elgvin et al., 2017; Ellegren et al., 2012; Poelstra et al., 2014;
Sigeman et al., 2019; Toews, Taylor, et al., 2016), reflecting their smaller effective
population size relative to autosomes, low recombination rates and high linkage
disequilibrium, selection on sex-linked genes, and Haldane´s rule (Irwin, 2018). Recent
work suggest high levels of differentiation between species in sexual chromosomes may
also reflect linked selection (Burri, 2017; Kawakami et al., 2017), effects of sexual
selection (i.e. variance in male reproductive success, Batttey, 2020); and female-biased
gene flow (Lamichhaney et al., 2020). In Coeligena, high levels of differentiation in sexual
chromosomes may be related to any of the above scenarios and thus are not necessarily
coupled to differences in coloration

Candidate genes for phenotypic differences between C. b. bonapartei and C.
helianthea
Because C. b. bonapartei is the sister clade of C. helianthea, because taxa are sympatric
in part of their ranges in areas where both coloration phenotypes persist without current
evidence of intermediates (Palacios et al., 2019), and because they diverged so recently,
lineages in this group are an ideal system to search for candidate genes for the genetic
basis of structural coloration. In agreement with the expectation of structural coloration
being polygenic given all morphological, physiological, and developmental components
involved (Eliason et al., 2020), we found several genes in outlier windows that may be
candidates for having a role in feather development considering their known functions, yet
pointing out direct associations between specific variants and phenotypes is not yet
possible. Nonetheless, we describe the type of candidate genes we uncovered and how
might they relate to coloration due to their possible influence on feather development and
pigmentation.
First, consistent with the hypothesis that feathers showing different structural colors may
undergo different genetic programs of development, we found that several genes involved
in cell growth and cell differentiation (e.g. EMB, GAB3, LIN54, LTBP2, RAD51B, and
BTG1-like), as well as homeobox genes (e.g. CDX1, CDX4, and NKX2-3) differed between
lineages of Coeligena with distinct plumage coloration. More specifically, some of the
candidate genes we identified (e.g. FRS2 and RCAN2) are related to fibroblast growth
factor genes which play an important role in the development of the feather buds
(Rouzankina, Abate-Shen, & Niswander, 2004). In addition, Coeligena lineages differed in
genes involved in endothelial and epithelial cell development (e.g. PTGER4, FAT1,
HS3ST1-like), which may reflect that feather development involves the interaction of
epidermal and dermal cells (Yu et al., 2004). Other functional categories including
candidate genes in Coeligena were those related to programed cellular death (i.e.
apoptosis) and degradation of cellular components (i.e. autophagy) such as SHISA5,
ATG10 and ATG7; given that cell degeneration is relevant in feather morphogenesis
(Alibardi, 2018), and that hummingbirds show hollow melanosomes in the nanostructure of
their feathers (Eliason, Bitton, & Shawkey, 2013; Eliason et al., 2020) such processes may
strongly influence the configuration of structural colors. Finally, we found two genes related
to melanin as candidates for phenotypic differences: CSPG4, a cell surface proteoglycan
involved in melanoma tumors in humans (Tang, Lord, Stallcup, & Whitelock, 2018; X.

Wang et al., 2010), and SLC45A2, a gene associated with color variation in other animals
systems including birds (Abolins-Abols et al., 2018; Domyan et al., 2014; Gunnarsson et
al., 2007). Given that SLC45A2 has a role in vesicle sorting in melanocytes and variation
on the d allele of in this gene is associated to lighter vs. darker plumages in pigeons
(Domyan et al., 2014). All the loci above are the first candidates towards establishing the
genetic basis of structural coloration in birds, but functional evidence of their role in feather
development and pigment deposition is largely lacking. Detailed analyses of the genetic
variation on these genes coupled to larger population studies (i.e. GWAS) will help to
refine whether they are or not involved in variation of structural coloration. In addition,
expression analyses (transcriptomes and real time PCR) will help to clarify the functional
role that candidate genes may have in to produce structural coloration phenotypes. In
particular, SLC45A2 appears to be a promising candidate to be involved in the production
of darker plumages in C. helianthea, a hypothesis to be later tasted with a functional
genomic approach.
Speciation and the landscape of differentiation in Coeligena hummingbirds
Understanding what drove speciation between C. bonapartei and C. helianthea was the
original question that motivated this and our previous studies on this system (Palacios et
al., 2019; Palacios et al. in review). Because coloration is a major trait distinguishing these
hummingbirds and plumage coloration has a main role in avian communication and
species recognition, we hypothesize that the evolution of a novel coloration phenotype in
C. helianthea caused partly by changes in structural properties of feathers (Eliason et al.,
2020; Sosa et al., 2020) was involved in the process of speciation in these hummingbirds.
Because C. bonapartei and C. helianthea diverged recently and are sympatric in part of
their range, their speciation likely proceeded while maintaining high rates of gene flow, and
analyses of variation in the mitochondrial genome provided evidence consistent with such
a divergence-with-gene-flow model of speciation (Palacios et al., 2019). Our findings of
mito-nuclear discordance revealed by full genome analyses showing relationships and
patterns of differentiation between these lineages differing from those inferred from mtDNA
support the idea that they have experienced gene flow.
Under the divergence-with-gene-flow model of speciation (Fitzpatrick, Gerberich,
Kronenberger, Angeloni, & Funk, 2015), selection is thought to counteract the
homogenizing effects of gene flow, a process often resulting in genomic landscapes of
divergence in which genes under selection show marked differences between species

relative to a genomic background of low divergence. Regarding the role of color
differences in avian speciation, evidence of such genomic landscapes in which regions
controlling pigmentation stand out as exceptionally divergent is mounting across various
taxa (Campagna et al., 2017; Poelstra et al., 2014; Stryjewski & Sorenson, 2017; Toews,
Brelsford, Grossen, Milá, & Irwin, 2016), but we are unaware of similar patterns in cases
involving structural coloration. We hypothesize that selective pressures acting on the novel
phenotype of C. helianthea were likely drivers of speciation and maintain C. bonapartei
and C. helianthea as distinct in the face of gene flow, but this is not manifested in a
genomic landscape with overt peaks of differentiation containing genes related to
coloration because of the likely polygenic basis of structural colors. Our set of candidate
loci should aid future studies on the genetic basis of speciation in Coeligena via functional
analyses. Also, the genomes we generated are a valuable resource one may use to link
genetic variants with the structural properties of feathers involved in the production of
colors.
In addition to offering clues about processes involved in speciation, the landscape of
genomic divergence in Coeligena lineages undoubtedly also reflects their likely complex
biogeographic history. The topography of the Andes and climatic fluctuations during the
Pleistocene (Flantua, O’Dea, Onstein, Giraldo, & Hooghiemstra, 2019) likely resulted in
episodic contractions and expansions of the mountain forest inhabited by lineages of
Coeligena. Thus, changes in population size and population fragmentation during the
history of these lineages should have also affected their landscape of genomic
differentiation. Such genomic landscape thus reflects the interplay of evolutionary
mechanism including high levels of gene flow, episodes involving variable levels of drift
during periods of isolation, and selective pressures possibly coupled to a novel coloration
phenotype in C. helianthea. Such interplay of evolutionary mechanisms is likely common
to other systems in which species evolve and are maintained in the face of gene flow
(Lamichhaney et al., 2015b).
Acknowledgments
We thank the Fundación para la promoción de la investigación y la tecnología del Banco
de la República, the Facultad de Ciencias de la Universidad de los Andes and the Lovette
Lab at Cornell Lab of Ornithology for financial support. We thank the Instituto Alexander
von Humboldt, the Museo de Historia Natural de la Universidad de los Andes, and the
Instituto de Ciencias Naturales de la Universidad Nacional de Colombia for providing

tissue samples. We exported tissue samples thanks to the CITES permit No. CO41452
granted by the Ministerio de Ambiente y Desarrollo Sostenible of Colombia. We thank
Christopher Witt and his lab, and Erich Jarvis and his lab for provided us with reference
genomes. We thank Irby J. Lovette and Bronwyn G. Butcher for facilitating laboratory
work. We thank Silvia Restrepo, Andrew J. Crawford, Daniel Cadena’s lab members, and
anonymous reviewers, whose valuable comments helped to improve this work.

Figures
Figure 1. Principal component analyses PCA (B), phylogenetic analysis (C), and
Manhattan plots showing divergence across the genome (D) indicate that C. b. consita is
the most genetically differentiated lineage in the group although all are genetically
distinguishable. PCAs and the SVDquartets phylogeny based on complete genomes
distinguish all four lineages as well as two additional two groups within C. b. bonapartei
coinciding with geography as shown in (A). Colors on the map, PCA plots and the

phylogeny correspond to C. b. consita (orange), C. b. bonapartei (yellow), C. h. helianthea
(light blue), and C. h. tamai (dark blue); the hatched green area on the map is the region
where C. b. bonapartei and C. h. helianthea are sympatric. Numbers on the map and on
the phylogeny correspond to individuals ID in Table S1. Numbers on branches are
bootstrap values; branch lengths were set to equal. In (D), dots are mean Fst values from
25-kb windows for different pairwise comparisons, showing several regions with high Fst
values and sharing of such divergent regions across comparisons. Outlier windows (with
mean Fst values 4 SD above the mean) are highlighted in the first comparison with blue
dots. Hummingbird illustrations by Jesús de Orion.

Figure 2. Z-Fst values comparing divergence across the genome between a pair of taxa
with different coloration (C. b. bonapartei and C. h. helianthea) and a pair with similar
coloration (C. h. helianthea and C. h. tamai) identified174 outlier windows (99th percentile,
blue dots) in 79 scaffolds across the genome. 119 loci identified in these windows are
grouped in 12 functional categories (colored ellipses at the bottom), and examples of these
loci with their location are shown on top of the Manhattan plot, with colored lines
corresponding to functional categories. Scaffolds that mapped to known chromosomes in
the genome of C. anna are pointed out. 43 outlier windows were shared between C.
bonapartei and C. helianthea Fst analysis and the Z-Fst outlier analysis (unfilled blue
dots).

References
Abolins-Abols, M., Kornobis, E., Ribeca, P., Wakamatsu, K., Peterson, M. P., Ketterson, E.
D., & Milá, B. (2018). Differential gene regulation underlies variation in melanic
plumage coloration in the dark-eyed junco (Junco hyemalis). Molecular Ecology,
27(22), 4501–4515. doi: 10.1111/mec.14878
Alibardi, L. (2018). Transmission electron microscopic and immunohistochemical
observations of resting follicles of feathers in chicken show massive cell degeneration.
Anatomical Science International, 93(4), 548–558. doi: 10.1007/s12565-018-0449-7
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data.
Retrieved from https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Batttey, C. J. (2020). Evidence of linked selection on the Z chromosome of hybridizing
hummingbirds. Evolution, 74(4), 699–804. doi: 10.1111/evo.13888
Billerman, S. M., Cicero, C., Bowie, R. C. K., & Carling, M. D. (2019). Phenotypic and
genetic introgression across a moving woodpecker hybrid zone. Molecular Biology
Reports, 28(November 2017), 1692–1708. doi: 10.1111/mec.15043
Bosse, M., Spurgin, L. G., Laine, V. N., Cole, E. F., Firth, J. A., Gienapp, P., … Slate, J.
(2017). Recent natural selection causes adaptive evolution of an avian polygenic trait.
Science, 358(6361), 365–368. doi: 10.1126/science.aal3298
Brelsford, A., Toews, D. P. L., & Irwin, D. E. (2017). Admixture mapping in a hybrid zone
reveals loci associated with avian feather coloration. Proceedings of the Royal Society
B: Biological Sciences, 284. doi: 10.1098/rspb.2017.1106
Brien, M. N., Enciso-Romero, J., Parnell, A. J., Salazar, P. A., Morochz, C., Chalá, D., …
Nadeau, N. J. (2019). Phenotypic variation in Heliconius erato crosses shows that
iridescent structural colour is sex-linked and controlled by multiple genes. Interface
Focus, 9(1). doi: 10.1098/rsfs.2018.0047
Burga, A., Wang, W., Ben-David, E., Wolf, P. C., Ramey, A. M., Verdugo, C., … Kruglyak,
L. (2017). A genetic signature of the evolution of loss of flight in the Galapagos
cormorant. Science, 356(6341). doi: 10.1126/science.aal3345
Burri, R. (2017). Linked selection, demography and the evolution of correlated genomic
landscapes in birds and beyond. Molecular Ecology, 26(15), 3853–3856. doi:
10.1111/mec.14167
Campagna, L., Repenning, M., Silveira, L. F., Suertegaray Fontana, C., Tubaro, Pablo, L.,
& Lovette, I. J. (2017). Repeated divergent selection on pigmentation genes in a rapid
finch radiation. Science Advances, 3(5), e1602404. doi: 10.1126/sciadv.1602404

Chifman, J., & Kubatko, L. (2014). Quartet inference from SNP data under the coalescent
model. Bioinformatics, 30(23), 3317–3324. doi: 10.1093/bioinformatics/btu530
Chifman, J., & Kubatko, L. (2015). Identifiability of the unrooted species tree topology
under the coalescent model with time-reversible substitution processes, site-specific
rate variation, and invariable sites. Journal of Theoretical Biology, 374, 35–47. doi:
10.1016/j.jtbi.2015.03.006
Cooke, T. F., Fischer, C. R., Wu, P., Jiang, T., Xie, K. T., Kuo, J., … Bustamante, C. D.
(2017). Article genetic mapping and biochemical basis of yellow feather pigmentation
in budgerigars. Cell, 171(2), 427-432.e21. doi: 10.1016/j.cell.2017.08.016
Cooper, E. A., & Uy, J. A. C. (2017). Genomic evidence for convergent evolution of a key
trait underlying divergence in island birds. Molecular Ecology, 26(14), 3760–3774. doi:
10.1111/mec.14116
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., … Durbin,
R. (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158.
doi: 10.1093/bioinformatics/btr330
Delmore, K. E., Toews, D. P. L., Germain, R. R., Owens, G. L., Irwin, D. E., Delmore, K.
E., … Irwin, D. E. (2016). The Genetics of Seasonal Migration and Plumage Report
The Genetics of Seasonal Migration and Plumage Color. Current Biology, 1–7. doi:
10.1016/j.cub.2016.06.015
Domyan, E. T., Guernsey, M. W., Kronenberg, Z., Krishnan, S., Boissy, R. E., Vickrey, A.
I., … Shapiro, M. D. (2014). Epistatic and Combinatorial Effects of Pigmentary Gene
Mutations in the Domestic Pigeon. Current Biology : CB, 24(4), 459–464. doi:
10.1016/j.cub.2014.01.020
Edwards, S. V., Kingan, S. B., Calkins, J. D., Balakrishnan, C. N., Jennings, W. B.,
Swanson, W. J., & Sorenson, M. D. (2005). Speciation in birds: Genes, geography,
and sexual selection. Proceedings of the National Academy of Sciences,
102(Supplement 1), 6550–6557. doi: 10.1073/pnas.0501846102
Elgvin, T. O., Trier, C. N., Tørresen, O. K., Hagen, I. J., Lien, S., Nederbragt, A. J., …
Sætre, G. P. (2017). The genomic mosaicism of hybrid speciation. Science Advances,
3(6), 1–16. doi: 10.1126/sciadv.1602996
Eliason, C. M., Bitton, P. P., & Shawkey, M. D. (2013). How hollow melanosomes affect
iridescent colour production in birds. Proceedings of the Royal Society B: Biological
Sciences, 280(1767), 15. doi: 10.1098/rspb.2013.1505

Eliason, C. M., Maia, R., Parra, J. L., & Shawkey, M. D. (2020). Signal evolution and
morphological complexity in hummingbirds (Aves: Trochilidae). Evolution, 1–12. doi:
10.1111/evo.13893
Ellegren, H., Smeds, L., Burri, R., Olason, P. I., Backström, N., Kawakami, T., … Wolf, J.
B. W. (2012). The genomic landscape of species divergence in Ficedula flycatchers.
Nature, 491(7426), 756–760. doi: 10.1038/nature11584
Feder, J. L., Egan, S. P., & Nosil, P. (2012). The genomics of speciation-with- gene-flow.
Trends in Genetics, 28(7), 342–350. doi: 10.1016/j.tig.2012.03.009
Fitzpatrick, S. W., Gerberich, J. C., Kronenberger, J. A., Angeloni, L. M., & Funk, W. C.
(2015). Locally adapted traits maintained in the face of high gene flow. Ecology
Letters, 18(1), 37–47. doi: 10.1111/ele.12388
Flantua, S. G. A., O’Dea, A., Onstein, R. E., Giraldo, C., & Hooghiemstra, H. (2019). The
flickering connectivity system of the north Andean páramos. Journal of Biogeography,
(March), 1808–1825. doi: 10.1111/jbi.13607
Gazda, M. A., Araújo, P. M., Lopes, R. J., Toomey, M. B., Andrade, P., Afonso, S., …
Carneiro, M. (2020). A genetic mechanism for sexual dichromatism in birds. Science
(New York, N.Y.), 368(6496), 1270–1274. doi: 10.1126/science.aba0803
Genome 10K, C. of S. (2009). Genome 10K : A Proposal to Obtain Whole-Genome
Sequence for 10 000 Vertebrate Species. Journal of Heredity, 100(6), 659–674. doi:
10.1093/jhered/esp086
Gunnarsson, U., Hellström, A. R., Tixier-Boichard, M., Minvielle, F., Bed’hom, B., Ito, S., …
Andersson, L. (2007). Mutations in SLC45A2 cause plumage color variation in chicken
and Japanese quail. Genetics, 175(2), 867–877. doi: 10.1534/genetics.106.063107
Hermansen, J. O. S., Haas, F., Trier, C. N., & Bailey, R. I. (2014). Hybrid speciation
through sorting of parental incompatibilities in Italian sparrows. Molecular Ecology, 1–
12. doi: 10.1111/mec.12910
Hill, G. E., & McGraw, K. J. (2006). Bird coloration. Volume I. Mechanisms and
measurements. Cambridge: Harvard University Press.
ICGSC International Chicken Genome Sequencing Consortium. (2004). Sequence and
comparative analysis of the chicken genome provide unique perspectives on
vertebrate evolution. Nature, 432(7018), 695–716. doi: 10.1038/nature03154
Irwin, D. E. (2018). Sex chromosomes and speciation in birds and other ZW systems.
Molecular Ecology, 27(19), 3831–3851. doi: 10.1111/mec.14537

Kapusta, A., & Suh, A. (2017). Evolution of bird genomes—a transposon’s-eye view.
Annals of the New York Academy of Sciences, 1389(1), 164–185. doi:
10.1111/nyas.13295
Kawakami, T., Mugal, C. F., Suh, A., Nater, A., Burri, R., Smeds, L., & Ellegren, H. (2017).
Whole-genome patterns of linkage disequilibrium across flycatcher populations clarify
the causes and consequences of fine-scale recombination rate variation in birds.
Molecular Ecology, 26(16), 4158–4172. doi: 10.1111/mec.14197
Koepfli, K., Paten, B., & Brien, S. J. O. (2015). The Genome 10K Project : A Way Forward.
doi: 10.1146/annurev-animal-090414-014900
Kraus, R. H. S., & Wink, M. (2015). Avian genomics: Fledging into the wild! Journal of
Ornithology, 156(4), 851–865. doi: 10.1007/s10336-015-1253-y
Küpper, C., Stocks, M., Risse, J. E., Dos Remedios, N., Farrell, L. L., McRae, S. B., …
Burke, T. (2015). A supergene determines highly divergent male reproductive morphs
in the ruff. Nature Genetics, 48(1), 79–83. doi: 10.1038/ng.3443
Lamichhaney, S., Berglund, J., Almén, M. S., Maqbool, K., Grabherr, M., Martinez-Barrio,
A., … Andersson, L. (2015a). Evolution of Darwin’s finches and their beaks revealed
by genome sequencing. Nature, 518(7539), 371–375. doi: 10.1038/nature14181
Lamichhaney, S., Berglund, J., Almén, M. S., Maqbool, K., Grabherr, M., Martinez-Barrio,
A., … Andersson, L. (2015b). Evolution of Darwin’s finches and their beaks revealed
by genome sequencing. Nature. doi: 10.1038/nature14181
Lamichhaney, S., Han, F., Berglund, J., Wang, C., Almén, M. S., Webster, M. T., …
Andersson, L. (2016). A beak size locus in Darwin’s finches facilitated character
displacement during a drought. Science, 352(6284), 470–474. doi:
10.1126/science.aad8786
Lamichhaney, S., Han, F., Webster, M. T., Grant, B. R., Grant, P. R., & Andersson, L.
(2020). Female-biased gene flow between two species of Darwin’s finches. Nature
Ecology and Evolution. doi: 10.1038/s41559-020-1183-9
Langmead, B., & Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat
Methods, 9(4), 357–359. doi: 10.1038/nmeth.1923
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association
mapping and population genetical parameter estimation from sequencing data.
Bioinformatics, 27(21), 2987–2993. doi: 10.1093/bioinformatics/btr509

Li, H., & Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler
transform. Bioinformatics (Oxford, England), 25(14), 1754–1760. doi:
10.1093/bioinformatics/btp324
Lopes, R. J., Johnson, J. D., Toomey, M. B., Hill, G. E., Corbo, J. C., Carneiro, M., …
Araujo, P. M. (2016). Genetic Basis for Red Coloration in Birds Report Genetic Basis
for Red Coloration in Birds. Current Biology, 26, 1427–1434. doi:
10.1016/j.cub.2016.03.076
Martin, S. H., & Jiggins, C. D. (2017). Interpreting the genomic landscape of introgression.
Current Opinion in Genetics and Development, 47, 69–74. doi:
10.1016/j.gde.2017.08.007
McGuire, J. A., Witt, C. C., Remsen, J. V., Dudley, R., & Altshuler, D. L. (2008). A higher-
level taxonomy for hummingbirds. Journal of Ornithology, 150(1), 155–165. doi:
10.1007/s10336-008-0330-x
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., …
DePristo, M. a. (2010). The Genome Analysis Toolkit: a MapReduce framework for
analyzing next-generation DNA sequencing data. Genome Research, 20(9), 1297–
1303. doi: 10.1101/gr.107524.110
MIT, and H. B. I. (2017). Picard. Retrieved from http://broadinstitute.github.io/picard/
Mundy, N. I., Stapley, J., Bennison, C., Tucker, R., Twyman, H., Kim, K.-W., … Slate, J.
(2016). Red Carotenoid Coloration in the Zebra Finch Is Controlled by a Cytochrome
P450 Gene Cluster. Current Biology, 26(11), 1435–1440. doi:
10.1016/j.cub.2016.04.047
Orteu, A., & Jiggins, C. D. (2020). The genomics of coloration provides insights into
adaptive evolution. Nature Reviews Genetics. doi: 10.1038/s41576-020-0234-z
Ottenburghs, J. (2020). Ghost Introgression : Spooky Gene Flow in the Distant Past.
BioEssays, 2000012, 1–5. doi: 10.1002/bies.202000012
Ottenburghs, J., Kraus, R. H. S., van Hooft, P., van Wieren, S. E., Ydenberg, R. C., &
Prins, H. H. T. (2017). Avian introgression in the genomic era. Avian Research, 8(1),
30. doi: 10.1186/s40657-017-0088-z
Palacios, C., Garcia-R, S., Parra, J. L., Cuervo, A. M., Stiles, F. G., McCormack, J. E., &
Cadena, C. D. (2019). Shallow evolutionary divergence between two Andean
hummingbirds: Speciation with gene flow? The Auk, 136(4). doi: /10.1101/249755

Poelstra, J. W., Vijay, N., Bossu, C. M., Lantz, H., Ryll, B., Muller, I., … Wolf, J. B. W.
(2014). The genomic landscape underlying phenotypic integrity in the face of gene
flow in crows. Science, 344(6190), 1410–1414. doi: 10.1126/science.1253226
Price, T. D. (2008). Speciation in Birds. Greenwood Village, Colorado: Roberts &
Company Publishers.
R Core Team. (2017). R: A Language and environment for statistical computing. Retrieved
from https://www.r-project.org/
Rhie, A., Mccarthy, S. A., Fedrigo, O., Damas, J., Formenti, G., London, S. E., …
Friedrich, S. R. (2020). Towards complete and error-free genome assemblies of all
vertebrate species. 1–56.
Roulin, A. (2004). The evolution, maintenance and adaptive function of genetic colour
polymorphism in birds. Biological Reviews of the Cambridge Philosophical Society,
79(4), 815–848. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15682872
Rouzankina, I., Abate-Shen, C., & Niswander, L. (2004). Dlx genes integrate positive and
negative signals during feather bud development. Developmental Biology, 265(1),
219–233. doi: 10.1016/j.ydbio.2003.09.023
Sackton, T. B., Grayson, P., Cloutier, A., Hu, Z., Liu, J. S., Wheeler, N. E., … Edwards, S.
V. (2019). Convergent regulatory evolution and loss of flight in paleognathous birds.
Science, 364(6435), 74–78. doi: 10.1126/science.aat7244
San-jose, L. M., Ducret, V., & Ducrest, A. (2017). Beyond mean allelic effects : A locus at
the major color gene MC1R associates also with differing levels of phenotypic and
genetic (co)variance for coloration in barn owls. Evolution, 71(10), 2469–2483. doi:
10.1111/evo.13343
Schubert, M., Lindgreen, S., & Orlando, L. (2016). AdapterRemoval v2: rapid adapter
trimming, identification, and read merging. BMC Research Notes, 9(1), 88. doi:
10.1186/s13104-016-1900-2
Seehausen, O., Butlin, R. K., Keller, I., Wagner, C. E., Boughman, J. W., Hohenlohe, P.
A., … Widmer, A. (2014). Genomics and the origin of species. Nature Reviews.
Genetics, 15(3), 176–192. doi: 10.1038/nrg3644
Sigeman, H., Ponnikas, S., Chauhan, P., Dierickx, E., Brooke, M. D. L., Hansson, B., …
Hansson, B. (2019). Repeated sex chromosome evolution in vertebrates supported by
expanded avian sex chromosomes. Proceedings of the Royal Society B: Biological
Sciences, 286.

Sosa, J., Parra, J. L., Stavenga, D. G., & Giraldo, M. A. (2020). Sexual dichromatism of the
Blue-throated Starfrontlet, Coeligena helianthea, hummingbird plumage. Journal of
Ornithology, 161(1), 289–296. doi: 10.1007/s10336-019-01709-z
Stoddard, M. C., & Prum, R. O. (2011). How colorful are birds? Evolution of the avian
plumage color gamut. Behavioral Ecology, 22(5), 1042–1052. doi:
10.1093/beheco/arr088
Stryjewski, K. F., & Sorenson, M. D. (2017). Mosaic genome evolution in a recent and
rapid avian radiation. Nature Ecology and Evolution, 1(12), 1912–1922. doi:
10.1038/s41559-017-0364-7
Swofford, D. L. (2003). PAUP* Phylogenetic Analysis Using Parsimony (*and Other
Methods). Sunderland, MA: Sinauer Associates.
Tang, F., Lord, M. S., Stallcup, W. B., & Whitelock, J. M. (2018). Cell surface chondroitin
sulphate proteoglycan 4 (CSPG4) binds to the basement membrane heparan sulphate
proteoglycan, perlecan, and is involved in cell adhesion. Journal of Biochemistry,
163(5), 399–412. doi: 10.1093/jb/mvy008
Toews, D. P. L., Brelsford, A., Grossen, C., Milá, B., & Irwin, D. E. (2016). Genomic
variation across the Yellow-rumped Warbler species complex. The Auk, 133(4), 698–
717. doi: 10.1642/AUK-16-61.1
Toews, D. P. L., Campagna, L., Taylor, S. A., Balakrishnan, C. N., Baldassarre, D. T.,
Deane-Coe, P. E., … Winger, B. M. (2016). Genomic approaches to understanding
population divergence and speciation in birds. The Auk, 133(1), 13–30. doi:
10.1642/AUK-15-51.1
Toews, D. P. L., Taylor, S. A., Streby, H. M., Kramer, G. R., & Lovette, I. J. (2019).
Selection on VPS13A linked to migration in a songbird. Proceedings of the National
Academy of Sciences of the United States of America, 116(37), 18272–18274. doi:
10.1073/pnas.1909186116
Toews, D. P. L., Taylor, S. A., Vallender, R., Brelsford, A., Butcher, B. G., Messer, P. W.,
& Lovette, I. J. (2016). Plumage genes and little else distinguish the genomes of
hibridizing warblers. Current Biology, 26, 1–6. doi: 10.1016/j.cub.2016.06.034
Tuttle, E. M., Bergland, A. O., Korody, M. L., Brewer, M. S., Newhouse, D. J., Minx, P., …
Balakrishnan, C. N. (2016). Divergence and functional degradation of a sex
chromosome-like supergene. Current Biology, 26(3), 344–350. doi:
10.1016/j.cub.2015.11.069

Van Doren, B. M., Campagna, L., Helm, B., Illera, J. C., Lovette, I. J., & Liedvogel, M.
(2017). Correlated patterns of genetic diversity and differentiation across an avian
family. Molecular Ecology, 26, 3982–3997. doi: 10.1111/mec.14083
Vijay, N., Bossu, C. M., Poelstra, J. W., Weissensteiner, M. H., Suh, A., Kryukov, A. P., &
Wolf, J. B. W. (2016). Evolution of heterogeneous genome differentiation across
multiple contact zones in a crow species complex. Nature Communications, 7, 1–10.
doi: 10.1038/ncomms13195
Walsh, J., Shriver, W. G., Olsen, B. J., & Kovach, A. I. (2016). Differential introgression
and the maintenance of species boundaries in an advanced generation avian hybrid
zone. BMC Evolutionary Biology, 16(1), 65. doi: 10.1186/s12862-016-0635-y
Wang, K., Lenstra, J. A., Liu, L., Hu, Q., Ma, T., Qiu, Q., & Liu, J. (2018). Incomplete
lineage sorting rather than hybridization explains the inconsistent phylogeny of the
wisent. Communications Biology, 1(169). doi: 10.1038/s42003-018-0176-6
Wang, X., Wang, Y., Yu, L., Sakakura, K., Visus, C., Schwab, J. H., … Ferrone, S. (2010).
CSPG4 in Cancer: Multiple Roles. Current Molecular Medicine, 10(4), 419–429. doi:
10.2174/156652410791316977
Yu, M., Yue, Z., Wu, P., Wu, D. Y., Mayer, J. A., Medina, M., … Chuong, C. M. (2004).
The developmental biology of feather follicles. International Journal of Developmental
Biology, 48(2–3), 181–191. doi: 10.1387/ijdb.15272383
Yusuf, L., Heatley, M. C., Palmer, J. P. G., Barton, H. J., Cooney, C. R., & Gossmann, T. I.
(2020). Noncoding regions underpin avian bill shape diversification at
macroevolutionary scales. Genome Research, 30(4), 553–565. doi:
10.1101/gr.255752.119
Zhang B; Li, C; Gilbert,M.T.P.; Mello, C.V.; Jarvis, E.D.; The Avian Genome Consortium;
Wang,J;, G. L. (2014). Genomic data of the Anna’s Hummingbird (Calypte anna).
Retrieved from http://dx.doi.org/10.5524/101004
Zhang, G., Li, C., Li, Q., Li, B., Larkin, D. M., Lee, C., … Froman, D. P. (2014).
Comparative genomics reveals insights into avian genome evolution and adaptation.
Science, 346(6215), 1311–1320. doi: 10.1126/science.1251385
Zheng, X., Levine, D., Shen, J., Gogarten, S., Laurie, C., & Weir, B. (2012). A High-
performance Computing Toolset for Relatedness and Principal Component Analysis of
SNP Data. Bioinformatics, 28(24), 3326–3328. doi: 10.1093/bioinformatics/bts606

Supplementary Material
Figure S1. Distribution of mean Fst values from 25 kb windows across the genome were
different for each comparison.

Table S1. Individuals information and sequencing coverage. Paper-ID (identification
number) through the paper. Sample ID corresponds to the tissue sample ID, to the
specimen voucher ID or to the collector’s ID. Specimen voucher corresponds to the skin ID
in collections or to collector’s ID. All samples are from Colombia. Sex F = females and M =
males.
Paper ID
Sample ID Species Subspecies Sex Specimen voucher
Latitude and Longitude
Sequencing Coverage (X)
1 ANDES-T1287 C. bonapartei consita F ICNAves36833 10.3669 -72.8975 3.5
2 ANDES-T1288 C. bonapartei consita F ICNAves36820 10.3669 -72.8975 3.6
3 ANDES-T1289 C. bonapartei consita M ICNAves36819 10.3669 -72.8975 3.8
4 ANDES-T1290 C. bonapartei consita M ICNAves36841 10.3669 -72.8975 4.0
5 ANDES-T1291 C. bonapartei consita F ICNAves36818 10.3669 -72.8975 2.7
6 ANDES-T1292 C. bonapartei consita M ICNAves36822 10.3669 -72.8975 4.1
7 IAvH-CT8567 C. bonapartei consita F ICNAves37116 10.3669 -72.8975 3.6
8 IAvH-CT8473 C. bonapartei consita M ICNAves37115 10.3640 -72.9474 3.4
9 IAvH-CT8503 C. bonapartei consita F ICNAves37104 10.3640 -72.9474 3.6
10 IAvH-CT00017312 C. bonapartei bonapartei M IAvH15365 5.8643 -73.1305 4.2
11 IAvH-CT00004191 C. bonapartei bonapartei M IAvH12581 5.7297 -73.4628 3.3
12 IAvH-CT4188 C. bonapartei bonapartei M IAvH12578 5.7297 -73.4628 4.2
13 IAvH-CT6966 C. bonapartei bonapartei F IAvH14196 5.7066 -73.4601 4.1
14 IAvH-CT6973 C. bonapartei bonapartei M IAvH14203 5.7046 -73.4572 4.0
15 IAvH-CT00002277 C. bonapartei bonapartei M IAvH12299 5.6394 -73.4872 4.9
16 IAvH-CT2265 C. bonapartei bonapartei F IAvH12290 5.6394 -73.4872 4.2
17 IAvH-CT2271 C. bonapartei bonapartei F IAvH12292 5.6394 -73.4872 3.3
18 ICN-Aves34450 C. bonapartei bonapartei M ICNAves34450 4.9333 -74.1833 3.7
19 ANDES-T2006 C. bonapartei bonapartei F JLPV74 4.9290 -74.1121 2.9
20 DCP01 C. bonapartei bonapartei F DCP01 4.8817 -74.4267 4.7
21 IAvH-CT00006791 C. bonapartei bonapartei M IAvH13986 4.6271 -74.3076 3.7
22 IAvH-CT6802 C. bonapartei bonapartei F IAvH13997 4.6271 -74.3076 4.2
23 Andes-BT 402 C. helianthea helianthea M ICNAves36409 7.0724 -72.9380 NA
24 IAvH-CT-11225 C. helianthea helianthea F IAvH8398 7.3042 -72.3711 NA
25 IAvH-CT18134 C. helianthea helianthea M ICNAves38141 5.2819 -73.3608 3.0
26 IAvH-CT-2530 C. helianthea helianthea F IAvH12633 4.4939 -73.6925 4.5
27 IAvH-CT00002569 C. helianthea helianthea F IAvH12682 4.7036 -73.8511 3.7
28 IAvH-CT2599 C. helianthea helianthea M IAvH12719 4.7036 -73.8511 6.5
29 IAvH-CT2601 C. helianthea helianthea F IAvH12722 4.6900 -73.8558 4.5
30 ANDES-T813 C. helianthea helianthea ? FGS4129 4.6667 -74.0330 3.8
31 IAvH-CT00002504 C. helianthea helianthea M IAvH12590 4.4939 -73.6925 3.9
32 ANDES-T70 C. helianthea helianthea F ICNAves36307 4.3213 -73.7768 4.2

33 Andes-BT 1126 C. helianthea tamai M IAvH 14908 7.4181 -72.4431 NA
34 ANDES-T1127 C. helianthea tamai F IAvH14906 7.4181 -72.4431 2.9
35 ANDES-T1128 C. helianthea tamai F IAvH14899 7.4181 -72.4431 5.2
36 ANDES-T1129 C. helianthea tamai F IAvH14897 7.4181 -72.4431 5.0
37 ANDES-T1130 C. helianthea tamai M IAvH14885 7.4181 -72.4431 3.4
38 ANDES-T1131 C. helianthea tamai F IAvH14884 7.4181 -72.4431 3.9
39 ANDES-T916 C. helianthea tamai M IAvH14818 7.4181 -72.4431 3.3
40 ANDES-T931 C. helianthea tamai F IAvH14836 7.4181 -72.4431 4.3
41 IAvH-CT11474 C. helianthea tamai M IAvH14915 7.4181 -72.4431 3.8
42 IAvH-CT11511 C. helianthea tamai F IAvH14912 7.4181 -72.4431 4.9
43 ANDES-T933 C. helianthea tamai M IAvH14964 7.4032 -72.4415 3.1
44 ANDES-T940 C. helianthea tamai M IAvH14971 7.4032 -72.4415 3.4
45 ANDES-T170 C. helianthea tamai M IAvHA8406 7.3042 -72.3711 3.2
46 ANDES-T1570 C. helianthea tamai M ICNAves37550 6.7308 -72.7956 3.3
Table S2. Twenty scaffolds showed more than two outliers windows identified in Z-Fst
analyses, and 18 of these scaffolds mapped to known chromosomes in the genome of C.
anna.
Ordered by size Scaffold ID
Mapped to Scaffold ID in the reference genome
No. of outlier windows
3 Chrom 4 scaffold17 8
4 Chrom 5A scaffold62 6
13 Chrom 6 scaffold90 14
19 Chrom Z scaffold68 4
71 Chrom Z scaffold297 3
75 Chrom Z scaffold385 8
107 Chrom Z scaffold715 5
115 Chrom 4B scaffold313 4
117 Chrom Z scaffold45 6
135 None scaffold101 17
140 Chrom 11 scaffold396 3
153 Chrom Z scaffold243 3
175 Chrom Z scaffold645 3
203 Chrom Z scaffold108 13
217 Chrom 12 scaffold238 2
246 None scaffold14 2
303 Chrom 12 scaffold344 2
320 Chrom Z scaffold361 2
338 None scaffold100 2
416 Chrom 12 scaffold159 8
Table S3 Using BLAST we found 119 loci in the 174 outliers windows identified in Z-Fst
analyses. We grouped loci in categories related to their available function information.

Main Functional Category Functional Category 1 Functional Category 2 Gene
Brain* Thyroid hormones action
DIO2
Brain* Neuronal proliferation Tumor DUSP26
Brain* Heart and Neurons Sour taste HCN1
Brain* ? Brain? RPP25
Brain* Sodium
SLC6A2
Brain* Brain
SNCAIP
Brain* Brain
STON2
Brain* Brain Lysosomal TMEM175
Brain* Gaba sensitivity
GABA""
Cell growth** Cell growth Rho ARHGAP27
Cell growth** Autophagy Ubiquitin ATG10
Cell growth** Autophagy Transport vacuole ATG7
Cell growth** Transcription factors
BATF
Cell growth** Cytoskeletal structure Rho CCM2
Cell growth** Transcription factors Homeobox CDX4
Cell growth** Mitosis
CENPP
Cell growth** Cell growth Membrane EMB
Cell growth** Calcium Extracellular matrix FBLN2
Cell growth** Phosphorylation-dependent ubiquitination
Ubiquitin FBXO4
Cell growth** Cell motility Rho FGD3
Cell growth** Cell growth Macrophage differentiation GAB3
Cell growth** Immune cell differentiation Ubiquitin ITCH
Cell growth** Cell growth Cell cycle genes LIN54
Cell growth** Cell growth Extracellular matrix LTBP2
Cell growth** Mitosis Kinase MOB3B
Cell growth** Cell growth Heart and brain NISCH
Cell growth** Transcription factors Homeobox NKX2-3
Cell growth** Osteoblast differentiation Heart OGN
Cell growth** Zinc finger Cell growth PRDM5
Cell growth** Endothelial cells Epithelia development PTGER4
Cell growth** Ras Tumor RAB5A
Cell growth** Cell growth Cell cycle genes RAD51B
Cell growth** Ras Tumor? RASAL2
Cell growth** Ubiquitin
RNF180
Cell growth** Rho Cell growth RTKN
Cell growth** Apoptosis
SHISA5
Cell growth** Cell growth
Wnt-7a
Cell growth** Cell growth Cell cycle genes BTG1-like""
Cell growth** Transcription factors Homeobox CDX-1
Cell growth** Zinc finger Cell growth KLF5""
Cell growth** Zinc finger
LOC115599778
Cell growth** ? Ras LOC103534149
Collagen Chondrogenesis Collagen ASPN
Collagen Collagen
COL14A1
Collagen Collagen Nephritis COL4A5
Collagen Collagen Fibroblast FRS2
Collagen Collagen
LOC115598664
Endothelial cells Endothelial cells Kidney ESM1
Endothelial cells Endothelial cells Calcium FLVCR2
Endothelial cells Endothelial cells Transcription factor GPBP1
Endothelial cells Endothelial cells
NDNF
Endothelial cells Endothelial cells Fibroblast RCAN2

Endothelial cells Endothelial cells Scavenger STAB1
Endothelial cells Blood coagulation
TFPI
Endothelial cells Endothelial cells Apoptosis TNFSF15
Endothelial cells Endothelial cells
LOC115599371
Epithelia development Epithelia development
FAT1
Epithelia development Epithelia development
LOC113982597
Epithelia development ? Kinase LOC109370333
Glycolysis, Sweet taste, Insulin Transcription factors Insulin ISL1
Glycolysis, Sweet taste, Insulin Oligosaccharide processing pathway
MOGS
Glycolysis, Sweet taste, Insulin Supply of D-mannose derivatives
MPI
Glycolysis, Sweet taste, Insulin Glycolysis Fat PDPR
Glycolysis, Sweet taste, Insulin Sweet taste
REEP2
Glycolysis, Sweet taste, Insulin Diabetes Heart disease LOC103535721
Immune cells B cell development and activation
BLNK
Immune cells pre-B and pre-T lymphocytes
DNTT
Immune cells Gamma interferon receptor
IFNGR1
Immune cells B cell and T cells
LRRC23
Melanin Melanoma
SLC45A2
Melanin Melanoma
CSPG4
Mitochondria Dehydrogenase Mitochondria ALDH7A1
Mitochondria Mitochondria Calcium MICU3
Mitochondria Mitochondria
MRPS30
Mitochondria Mitochondria
QRSL1
Others Transport across membranes Cholesterol ABCA1
Others DNA damage
ATRIP
Others ? Kidney CTXN3
Others Nitric oxide generation
DDAH1
Others Kinase Hypospadias DGKK
Others Heat shock protein
DNAJC15
Others Heat shock protein Protein folding DNAJC28
Others Extracellular matrix Fat ECM2
Others Membrane Tumor ENTPD1
Others Fat
FAM219B
Others Prenyltransferases Cisteine residue FNTA
Others Endoplasmic reticulum Protein folding GPX8
Others DNA damage
IPPK
Others Kinase Signal transduction MAP3K1
Others Vesicular transport
NIPSNAP3A
Others Nuclear pore
NUP210
Others Fat? Calcium? PLA2G15
Others Transport across membranes Sodium SLC12A2
Others Nanophthalmos
TMEM98
Others Development Head TRABD2A
Others Protein-Protein intreaction
WBP1
Others Ovalbumin
LOC103527411
Others ? Tumor LOC103535886
Others ? Tumor? LOC113982579
Others Antioxidant defense
LOC115598252
The usual suspects
CGGBP1
The usual suspects
CSDC2
The usual suspects
GGPS1
The usual suspects
JMJD4
The usual suspects
PITPNC1

The usual suspects
TMED8
The usual suspects
TRMT1L
The usual suspects
LOC103531782
Unknown and uncharacterized ? X-inactivation CHIC1
Unknown and uncharacterized ?
CZH5orf51
Unknown and uncharacterized ?
ENDOD1
Unknown and uncharacterized ?
GPATCH2L
Unknown and uncharacterized ? ? TTC33
Unknown and uncharacterized ? ? TWSG1
Unknown and uncharacterized ?
LOC103534192
Unknown and uncharacterized ? ? LOC108497351
Unknown and uncharacterized ? ? LOC115598456
Unknown and uncharacterized ? ? LOC115599873
Unknown and uncharacterized ? ? LOC115600101
*Brain, neurons, and neuronal signaling
**Cell growth, differentiation, cell cycle, apoptosis and autophagy

Conclusions
Evolution and speciation are ongoing processes, and they are happening right now. I
began this project considering C. bonapartei and C. helianthea as two species but found
that they comprise at least four recently diverged lineages: C. b. eos, C. b. consita, C. b.
bonapartei and C. helianthea. My work revealed that the evolution of these lineages has
been shaped by the interplay among evolutionary mechanisms whereby drift in the
absence of gene flow has likely driven the differentiation of the allopatric C. b. eos and C.
b. consita, whereas selection in the face of gene flow has driven the differentiation
between C. b. bonapartei and C. helianthea. I found remarkably low differentiation in the
mitochondrial genomes of C. b. consita, C. b. bonapartei, C. h. helianthea and C. h. tamai
which is consistent with their recent divergence and with introgression. Whole genome
analyses, however, readily distinguished lineages and allow me to identify peaks in the
genomic landscape of differentiation between C. b. bonapartei and C. helianthea
containing candidate genes potentially associated with their phenotypic differences and
presumably involved in their speciation. My analyses revealed that the genetic basis of the
differences in coloration between these hummingbirds is likely polygenic, with various
genes and regulatory regions influencing phenotypes through feather development. Thus,
patterns in gene expression, metabolic pathways, and developmental changes during
feather growth involved in differences in structural coloration between these hummingbirds
are yet to be characterized in detail. Such work would be facilitated with the access to
references genome of high quality for these species that would complement my inferences
based on draft genomes for multiple individuals to better understand differentiation among
Coeligena lineages.
My findings lend support to the hypothesis that natural selection, sexual selection, or both
have acted to drive and maintain phenotypic divergence on the hummingbirds I studied,
but unravelling the specific mechanisms through which selection is acting requires further
investigation. Testing whether coloration is adapted to microclimatic environments to
enhance signal efficacy, addressing the preferences of females for different coloration
phenotypes, or studying how coloration is linked to other traits like feeding or breeding
behaviors will require spending time in the field. My work in genomics calls for basic
studies on natural history of these species. My study, the first to generate a data set
including complete genomes of multiple individuals in hummingbirds, allowed me to gain

great insight into their evolutionary history and, more broadly, on how species form in
megadiverse tropical mountains where historical climate changes may have isolated and
reconnected populations throughout their divergence. However, my research has only
opened the door to understanding the mechanisms underlying speciation and to identify
the genetic basis of phenotypic differentiation in young systems evolving in the complex
biogeographical scenario of the South American Andes.






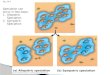

![V. SPECIATION A. Allopatric Speciation B. Parapatric Speciation (aka Local or Progenitor - Derivative) C. Adaptive Radiation D. Sympatric Speciation [Polyploidy]](https://img.pdfslide.us/doc/110x75/56649d3f5503460f94a186e2/v-speciation-a-allopatric-speciation-b-parapatric-speciation-aka-local.jpg)




















