Embed Size (px)
Citation preview

Revista Latinoamericana de Metalurgia y Materiales, Vol. 22, N° 2,2002,40 - 51
EVIDENCIAS ESPECTROSCOPICAS DE LA DEGRADACIÓNHIDROLÍTICA DE LA POLI(p-DIOXANONA)
M. A. Sabino 1, O. Nuñez 2, A. J. Müller 1
1. Grupo de Polímeros USB, Departamento Ciencia de los Materiales;2. Departamento de Procesos y Sistemas.
Universidad Simón Bolívar, Apartado 89000, Caracas 1080-A, Venezuela
Resumen
En este trabajo se estudió la degradación hidrolítica de un polímero biodegradable y bioabsorbible, la Poli (p-Dioxanona) oPPDX. La PPDX es un poliéster-éter alifático con gran potencial biomédico, debido a su estructura molecular y propiedadesadecuadas para utilizarse como material de osteosíntesis. La PPDX se degrada hidrolíticamente originando subproductos dedegradación de bajo peso molecular, atóxicos que pueden ser metabolizados o bioabsorbidos por el organismo. Se utilizaronmonofilamentos de PPDX que se degradaron hidrolíticamente en una solución buffer fosfato yagua a 37°C durante 12 semanas,y se utilizaron técnicas de caracterización como Espectroscopia Infrarrojo por Transformada de Fourier (FfIR) y Espectrometríade masa (EM). A través de FTIR, se demostró que durante el período de degradación se evidencia la disminución de grupos 0-H libres, producto de la posible asociación de estos grupos a los productos resultantes de las rupturas de enlaces. También seevidenció la posible formación de estructuras intermediarias o complejas, mediante asociación de grupos, donde puedenintervenir los grupos fosfatos presentes en el medio de hidrólisis. Además, como consecuencia de los rompimientos del grupoéster presente en la estructura de la PPDX, se observan variaciones de absorbancia de las bandas asociadas a los gruposcarbonilos dada la aparición del grupo ácido carboxílico. Estas evidencias fueron verificadas mediante Espectrometría de Masasdonde se determinó que uno de los subproductos de la degradación de la PPDX corresponde al ácido 2-hidroxi-etoxi- etanoico.
Palabras claves: Poli (p-dioxanona), degradación, hidrálisis, espectroscopia, biomateriales
Abstract
In this work, the hydrolytic degradation of a biodegradable and bioabsorbable polymer, Poly(p-dioxanone) or PPDX, wasstudied. PPDX is an alifatic polyester-ether with potential for biomedical applications since its molecular structure and propertiesare adequate to be used as osteosynthesis material. PPDX degrades hydrolytically originating byproducts of low molecularweight that are non-toxic and that can be metabolize or bioabsorbed by the body. PPDX monofilaments were hydrolyticallydegraded in a phosphate buffer solution and in water at 37°C during 12 weeks, Fourier Transform Infrared Spectroscopy (FTIR)and Mass spectrometry (MS) were employed to characterize the degradation products. FTIR results showed that during thedegradation period a decrease in the number of free O-H groups was recorded, a possible product of the association of thesegroups to tbose produced by degradation. It was also found that intermediary or complex structures were formed by groupassociation where a contribution of the phosphate groups present in the hydrolysis medium is probable. Furthermore, as aconsequence of the ester bond cleavage present in PPDX, a variation in the absorbance of bands associated to carbonyl groupsare observed in view of the formation of carboxylic acid groups. These evidences were verified by Mass Spectrometry, whoseresults indicated that one of the degradation byproducts of PPDX is 2-hydroxy-etoxi-ethanoic acid.
Keywords: Polytp-dioxanone), degradation, hydrolysis, spectroscopy, biomaterials
1. Introducción ampliamente como material biodegradable. Si este polímerose compara con otros poliésteres biodegradables como elPoli(ácido Glicólico) PGA Yel Poli(ácido L-láctico) PLLA, sepuede concluir que la presencia de un enlace éter y de un -CH2- adicional le confieren gran flexibilidad. Adicionalmente,y debido a que presenta menor concentración de gruposéster debe presentar una menor velocidad de degradaciónvía hidrólisis, 16 cual conduciría a una mayor retención desus propiedades en función del tiempo [2-5]. Es por lo tanto
El medio fisiológico humano (H20 extracelular) reúne lascondiciones apropiadas para que se puedan producir confacilidad los procesos hidrolíticos. Para ello el polímero debeposeer enlaces hidrolíticamente inestables para que elproceso de biodegradación se produzca en un tiemporazonable y el-mismo pueda llevarse a cabo en condicionesde pHfisiológico (entre 7 y 7.4) [1]. LaPPDX se ha utilizado

M. A. Sabina y col. /Revista Latinoamericana de Metalurgia y Materiales
de gran importancia práctica el poder determinar lasevidencias de la ocurrencia del proceso de degradaciónhidrolítica, siendo éste el momento a partir del cual el materialpresentará una modificación (o deterioro) importante de suspropiedades iniciales.
2. Metodología Experimental
2.1. Materiales
En la experiencia se trabajó con un monofilamento dePoli(p-Dioxanona) PPDX Johnson & Johnson (marcaEthicon), con un diámetro promedio de 1mm. La muestra seencontraba esterilizada y empacada al vacío. Para el procesode degradación in vitro de las muestras, se utilizó como mediode hidrólisis agua destilada la cual presentó un pH inicial de7.2 Ytranscurridas las dos primeras semanas el pH descendióhasta un valor de 4.5 donde se mantuvo durante el resto deltiempo para una duración total de 16 semanas. El otro mediode hidrólisis utilizado fue una solución buffer fostato 0.2 M(a base de fosfato de sodio hidrogenado Na2HP04y fosfatode potasio di-hidrogenado-Kfí.Ptj.) la cual presentó un pHinicial de 7.4, donde otro grupo de monofilamentos fueroncolocados.
2.2. Espectroscopia Infrarroja. FTIR
La experiencia consistió en preparar películas porevaporación de solvente realizando la disolución de lasmuestras en una solución fenol/tetracloroetano (2:3) ygarantizando el proceso de evaporación dentro de un hornoal vacío a temperatura ambiente. Para los ensayos se utilizó
un Espectrofotómetro Infrarrojo por TransformadasFourier FTIR marca Nicolet, modelo Magna 750. La resol -empleada fue de 4 cm', el número de barridos igual a 30 ydetector DTGS KBr con beamsplitter de KBr. Paradeterminación de la constante de velocidad k, de acucon el método de velocidades iniciales [6] se obtuviespectros infrarrojos para muestras extraídas día a día dla primera semana de degradación en ambos mediPreviamente, se obtuvo una curva de calibración cofinalidad de establecer la relación [-COO-]/ Absorbancia,importante recalcar que la determinación de la absorbestuvo centrada en la banda de 1750 cm' atribuible ~absorción de los ésteres alifáticosf-Ce=O); y con el fin -evitar los problemas de espesor entre las diferentes pelíse estableció la relación entre la banda atribuible alcarbonilo y la banda atribuible a la vibración de alargamidel grupo C-O-C, que en el caso de éteres alifáticosencuentra alrededor de 1125-1150 cm:' debido al alargamieacasimétrico.
2.3. Espectrometría de Masas
Para poder realizar los análisis por la técnica GC-MSChromatoghaphy-Mass Spectrometry") fue nece ariderivatización de los subproductos de la degrada -hidrolítica del polímero, para así garantizar que los mi:smesno fuesen retenidos en la columna del espectrómetro de[7]. En base a esto, se empleó N-t-butildimetilsililimi(TBD MSIM), cuya formula molecular y su estructura quími::zes:C(CH3)3-Si(CH3)2NCH=NCH=CH (Figura 1), y cuya acci ~derivatizante se esquematiza en la Figura 2.
CH]I s+CH1 CH &-
I Muestra ~o: +CH1- C-CH1 Ir J
I / N ----. I Muestra ~6- Si-XI CH1-Si-N(dH I - I I
CH1H CH1
CH] 1ICH3- C-CH] CH1
I / N II
Muestra ~O-Sí-CHJ +HXCHJ-Si-NU II - CH1
CH3 Fig. 2. Esquema de la reacción de derivatización
Fig. 1. Estructura molecular de TBDMSIM

42 Revista Latinoamericana de Metalurgia y Materiales, Vol. 22, N° 2, 2002.
En un ambiente estéril las muestras de PPDX virgen, en formade discos con diámetro de 20 mm y un espesor aproximadode 0.5 rnm (obtenidos a partir de moldeo por comprensión delos monofilamentos desde el fundido y enfriadasbruscamente), fueron introducidas en tubos de ensayo quecontenían 50 rnL de la solución buffer fosfato, y dichos tubosse introdujeron en un baño termostatizado a una temperaturade 37°C durante un periodo de 16 semanas. La solución bufferfue filtrada (004 um) antes de ser usada, y se tomaron alícuotasdurante períodos de 4 semanas, los cuales fueronsubsecuentemente analizadas, con el fin de hacerseguimiento a los productos de degradación que se presentanen función del tiempo. Sin embargo, este seguimiento nopudo ser posible debido a que las alícuotas correspondientesa la 4ta, 8va y 12va semana no presentaron indiciosdetectables por el equipo de los subproductos dedegradación de bajo peso molecular, pero si se obtuvo unabuena resolución para las alícuotas tomadas al final, es decir,luego de 16 semanas de degradación donde se habíanformado las especies de más bajo peso molecular.
Derivatización
Las muestras o alícuotas tomadas luego del tiempo referido(16 semanas) se empleó para realizar su derivatización enpresencia del reactivo TBDMSIM. La muestra tomada (1 rnL)de la solución buffer, contenedora de los subproductos de ladegradación hidrolítica de la PPDX, fue puesta en un tubode ensayo, se le añadió 1 mg de ácido ascórbico con el fin deevitar la oxidación de los ácidos orgánicos presentes en lasolución; luego fue acidificada a pH 2-3 con 50 ul, de HCL,luego fue extraída tres veces con acetato de etilo (gradoanálisis 99.9% Merck). Seguidamente se añadió sulfato desodio como desecante para recoger el agua presente. Elextracto fue combinado y posteriormente evaporado conNitrógeno (gas)" El sustrato en forma de polvo que quedó en elfondo del tubo, fue secado en una estufa al vacío atemperatura ambiente por 4 horas, en espera de suderivatización.
Para determinar las condiciones óptimas de laderivatización, se procedió de la siguiente manera: en vialescon capacidad de 1 rnL se añadieron 0.10 rnL de iso-octano ydiferentes proporciones (entre 5-50 ul.) del reactivoTBDMSIM, y se evaluaron las siguientes condiciones de
o//,
OH -R- C-OH + 2 TBDMSIM
tiempo y temperatura, 30 minutos a 60°C. Al inyectar lasdiferentes mezclas de iso-octano y TBDMSIM, se encontróque con 20 ul. del derivatizante fue posible elucidar los ácidosformados.
Una vez determinado que las condiciones óptimas para laderivatización corresponden a 20 ul. de TBDMSIM, y 30 mina 60°C, se procedió a mezclar 1 mg de los subproductos de ladegradación de la PPDX previamente secos con la cantidadcorrespondiente del derivatizante y 0.1 rnL de iso-octano.
La Figura 3, muestra la formación del derivatizado ter-butil-dimetilsilil (TBDMS) de los grupos ácidos y alcoholobtenidos de la hidrólisis.
La mezcla mencionada fue inyectada automáticamente enun Cromatografo de Gases (HP 5890, serie-2) acoplado a unEspectrómetro de Masa (HP 5972) (GC-MS), el cual cuentacon un inyector en modo split-splitless, y una columna capilarWCOTdesílica(CP-Sil-19CB,Chrompack,25mx0.32rnmI.D.).Se utilizó Helio como gas de arrastre. Las muestras fueronintroducidas en el inyector split a 250°C; para esto, latemperatura inicial delhomo fue 40°C por un tiempo de 1min, luego fue llevada hasta 200°C a una velocidad de 10°CImin y mantenida a esa temperatura por 8 minutos, para luegoalcanzar la temperatura final. La fragmentación de las especiesse realizó a través de impacto electrónico. El resto de lascondiciones operacionales fueron las siguientes: el tiempode corrida fue de 68 minutos; inyección/masa: 20-500 ¡.tUmin; segundos por barrido: 1.0; voltaje 1600 V; temperaturade transferencia 250°C; defecto de masa 30 rnmu/1 OOamu;yel modo sean: full. El método de adquisición de datos fue através del programa HP CHEM 5890 column flow control.
3. Resultados
3.1. EspectroscODia lnfrarroia. FTIR
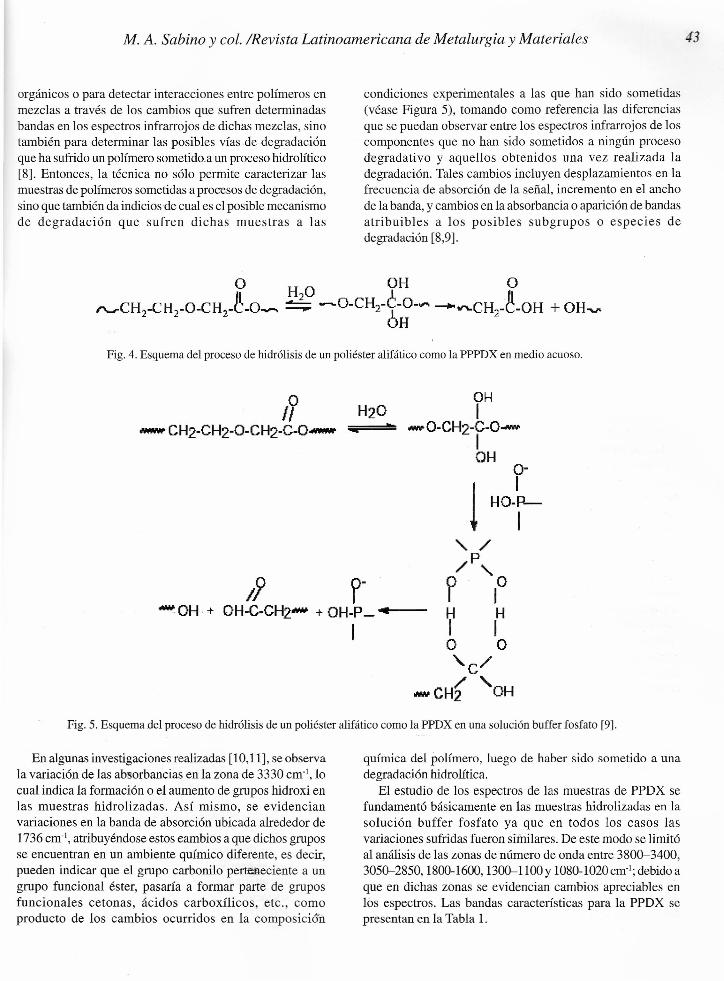
El análisis infrarrojo es una herramienta importante paraidentificar los cambios en la composición química luego deestar sometida la PPDX a una degradación hidrolítica,manifestándose dichos cambios en variaciones en lasregiones donde se presenta la absorción de bandasatribuibles a grupos hidróxi, carbonilos, éteres o ésteres(Figura 4).
La Espectroscopia Infrarrojo no sólo puede sersatisfactoriamente aplicada para caracterizar compuestos
CH3 CH] O CH] C~I 1 11 I I
---+- CH)- C- Si - O - R- C- O - Sí- C-CH]I I I ICH3 CH] CH3 CH3
Fig. 3. Derivatización de ácidos y alcoholes productos de la degradación hidrolítica de un poliéster alifático.
M. A. Sabino y col. /Revista Latinoamericana de Metalurgia y Materiales 43
orgánicos o para detectar interacciones entre polímeros enmezclas a través de los cambios que sufren determinadasbandas en los espectros infrarrojos de dichas mezclas, sinotambién para determinar las posibles vías de degradaciónque ha sufrido un polímero sometido. a un proceso hidrolítico[8]. Entonces, la técnica no sólo permite caracterizar lasmuestras de polímeros sometidas a procesos de degradación,sino que también da indicios de cual es el posible mecanismode degradación que sufren dichas muestras a las
condiciones experimentales a las que han sido sometidas(véase Figura 5), tomando como referencia las diferenciasque se puedan observar entre los espectros infrarrojos de loscomponentes que no han sido sometidos a ningún procesodegradativo y aquellos obtenidos una vez realizada ladegradación. Tales cambios incluyen desplazamientos en lafrecuencia de absorción de la señal, incremento en el anchode la banda, y cambios en la absorbancia o aparición de bandasatribuibles a los posibles subgrupos o especies dedegradación [8,9].
Fig. 4. Esquema del proceso de hidrólisis de un poliéster alifático como la PPPDX en medio acuoso.
o OH11 H20 I
_ CH2-CH2-0-CH2-C-O- :;¡¡__¡::==te.~ -O-CH2-C-O-IOH
Ir¡ p-- OH.+ OH-C-CH2- + OH-P_ ..•••••1---
I
Fig. 5. Esquema del proceso de hidrólisis de un poliéster alifático como la PPDX en una solución buffer fosfato [9].
En algunas investigaciones realizadas [10,11], se observala variación de las absorbancias en la zona de 3330 cm", locual indica la formación o el aumento de grupos hidroxi enlas muestras hidrolizadas. Así mismo, se evidencianvariaciones en la banda de absorción ubicada alrededor de1736 cm', atribuyéndose estos eambios a que dichos gruposse encuentran en un ambiente químico diferente, es decir,pueden indicar que el grupo carbonilo perteneciente a ungrupo funcional éster, pasaría a formar parte de gruposfuncionales cetonas, ácidos carboxílicos, etc., comoproducto de los cambios ocurridos en la composición
química del polírnero, luego de haber sido sometido a unadegradación hidrolítica.
El estudio de los espectros de las muestras de PPDX sefundamentó básicamente en las muestras hidrolizadas en lasolución buffer fosfato ya que en todos los casos lasvariaciones sufridas fueron similares. De este modo se limitóal análisis de las zonas de número de onda entre 3800---3400,3050---2850,1800-1600, 1300---1100y 1080-1020 cm'; debido aque en dichas zonas se evidencian cambios apreciables enlbs espectros. Las bandas características para la PPDX sepresentan en la Tabla 1.

44 Revista Latinoamericana de Metalurgia y Materiales, Vol. 22, N° 2, 2002.
Tabla l. Bandas de absorbancia IR características para la PPDX.
N° onda (cm") Grupo Observaciones
3620-3610 -OH Alargamiento de grupos -OH libre y asociados
3460 -OH Alargamiento de enlaces O-H
2960 -CH3 Oscilación de grupos metilos terminales
2920 -CHTCHT Oscilación de grupos etilenos
l740 -C=O Alargamiento del grupo carbonilo asociado al éster
1135 -CO-C- Vibración antisimétrica asociada a éteres alifáticos
1060 -CO- Alargamiento de los grupos asociados a un éster
Al apreciar la Figura 6, de manera general se evidencia, alincrementarse el tiempo de degradación, un engrosamientode la banda entre 3700- 3400 cm:', lo cual es característico a lapresencia de grupos hidroxi -OH pertenecientes a ácidoscarboxílicos y alcoholes, ésta banda también podría indicarla presencia de hidroperóxidos debido a que se ha reportadoque estos son relativamente estables a temperaturasmoderadas [12]. En esta misma Figura, en la región alrededorde 3615 crn', atribuible a los grupos O-H libres, se tiene quedicha banda muestra una tendencia a su desaparición, locual puede ser producto del proceso degradativo y de laposible asociación de estos grupos a los productosresultantes de las rupturas de enlace debido a la degradaciónhidrolítica, por lo que los O-H libres pueden pasar a formarparte de carbonos terciarios o carbonos asociados. Esteargumento es corroborado por el incremento de la banda deabsorbancia en 3460 cm:', que de acuerdo a la literaturacorresponde a impedimentos del grupo O-H [13].
Así mismo, en la Figura 7, la banda ubicada hacia 1240cm' muestra un comportamiento similar al visto para la bandade 3615 cm", en donde para la muestra virgen se observa lapresencia de un pico intenso y agudo, el cual se va debilitandoen el tiempo debido a que, como se expresó anteriormente,durante el proceso degradativo se promueven interaccionesentre cadenas más fuertes que las iniciales, disminuyendodicha banda.
De acuerdo a la literatura [13], la presencia de una bandahacia 1240 cm' se debe a la disposición ecuatorial del R-OHen acetatos, ya que, si la configuración fuese axial sepresentarían múltiples bandas en dicha región. Con relacióna lo expresado, se ha encontrado en la literatura [9], que ladesaparición de esta banda ubicada en 1240 crrr' se debe al
acoplamiento en el plano del grupo O-H enlazado al carboniloy sus homólogos, pudiéndose formar estructuras como lamostrada en la Figura 5 anterior, y lo cual se evidencia en laFigura 7.
Lo anteriormente expuesto se aprecia adicionalmente porel desdoblamiento que sufre el pico ubicado en 1050 cm:' endos bandas bien diferenciadas como se muestra en la Figura8. Como es sabido, la banda que señala el alargamiento delC-O en alcoholes primarios se presenta entre 1064-1030 cm',y la correspondiente a los ésteres de los alcoholessecundarios se presenta entre 1070-1100 cm': por lo que sepodría pensar que la banda de absorbancia a 1050 cm'corresponde a grupos O-H primarios, mientras que la bandaubicada a 1070 cm', es atribuida a O-H secundarios, lo cualparece indicar que se debe a la ruptura al azar de enlacesinducida por la hidrólisis, siendo entonces posible que seorigine este tipo de sustitución secundaria [13].
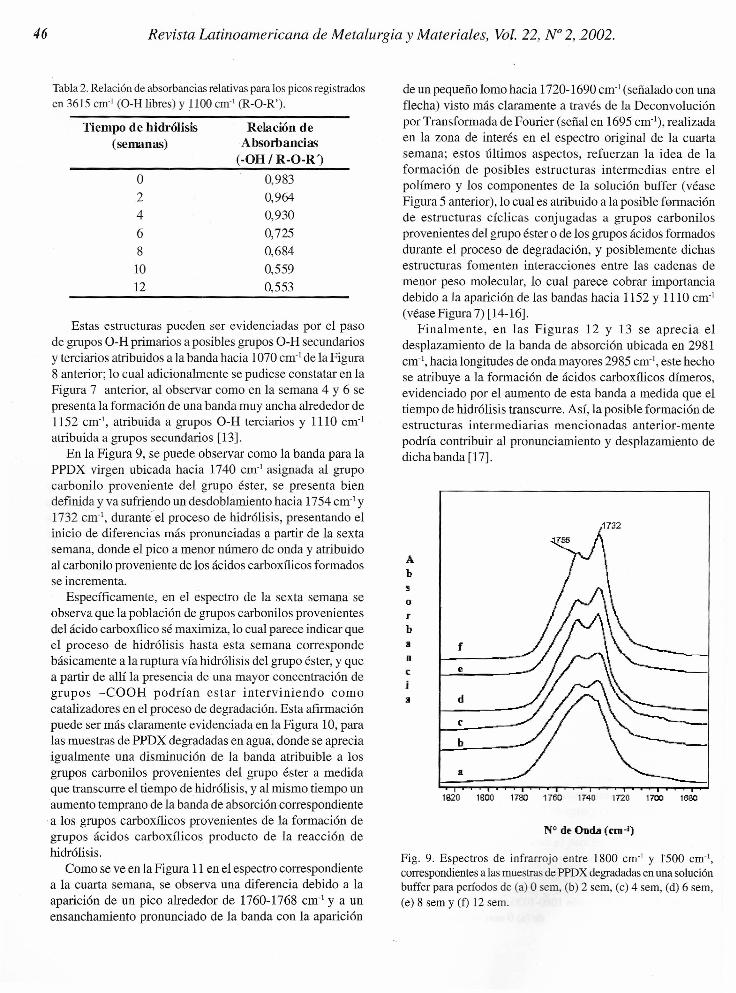
La disminución de grupos terminales libres se pudodeterminar cuantitativamente mediante la relación deabsorbancias relativas entre los picos en 3615 cm:'correspondientes a O-H libres y 1135 cm', correspondienteal grupo éter (R-O-R'), obteniéndose que el valor disminuyecon el tiempo de degradación, debido a la desaparición delos grupos O-H libres a medida que transcurre el tiempo dehidrólisis (véase Tabla 2). La misma tendencia se obtienepara las muestras hidrolizadas en agua y en la solución bufferfosfato.
Parece posible que durante el proceso de degradación yde ruptura al azar de enlaces vía hidrólisis, se obtenganestructuras intermediarias o complejas, las cuales se formanpor la asociación de grupos (puentes de hidrógeno, etc.).

M. A. Sabino y col. /Revista Latinoamericana de Metalurgia y Materiales 45
Absorbaneia
3800 3700 34003600 3500N° de Onda (cm")
Fig. 6. Espectros FTIR, región 3800-3400cm', correspondientes a las muestras de PPDX degradadas en una solución buffer para períodosde (a) O sem, (b) 2 sem, (c) 4 sem, (d) 6 sem, (e) 8 sem y (f) 12 sem.
1200
N° de Onda (cm")
Fig. 7. Espectros FTIR, región 1300-1100cm", correspondientes a las muestras de PPDX degradadas en una solución buffer para períodosde (a) Osem, (b) 2sem, (e) 4 sem, (d) 6 sem, (e) 8 sem y (f) 12 sem.
Absorbaneia
1300 1250 1150 1100
Absorbi:Ineia
[al
1080 1060 1040ND de Onda (cm")
1020
Fig. 8. Espectros FTIR, región 1080-1020cm-1, correspondientes a las muestras de PPDX degradadas en una solución buffer para períodosde (a) O sem, (b) 2 sem, (e) 4 sem, (d) 6 sem, (e) 8 sem y (f) 12 sem.
46 Revista Latinoamericana de Metalurgia y Materiales, Vol. 22, N° 2,2002.
Tabla 2. Relación de absorbancias relativas para los picos registradosen 3615 cm' (O-H libres) y 1100 crn' (R-O-R').
Tiempo de hidrólisis(semanas)
Relación deAbsorbancias
(-OH I R-O-R')
o24
681012
0,9830,9640,9300,7250,6840,5590,553
Estas estructuras pueden ser evidenciadas por el pasode grupos O-H primarios a posibles grupos O-H secundariosy terciarios atribuidos a la banda hacia 1070 cm' de la Figura8 anterior; lo cual adicionalmente se pudiese constatar en laFigura 7 anterior, al observar como en la semana 4 y 6 sepresenta la formación de una banda muy ancha alrededor de1152 cm", atribuida a grupos O-H terciarios y 1110 cm"atribuida a grupos secundarios [13].
En la Figura 9, se puede observar como la banda para laPPDX virgen ubicada hacia 1740 cm:' asignada al grupocarbonilo proveniente del grupo éster, se presenta biendefinida y va sufriendo un desdoblamiento hacia 1754 cm' y1732 cm', durante-el proceso de hidrólisis, presentando elinicio de diferencias más pronunciadas a partir de la sextasemana, donde el pico a menor número de onda y atribuidoal carbonilo proveniente de los ácidos carboxílicos formadosse incrementa.
Específicamente, en el espectro de la sexta semana seobserva que la población de grupos carbonilos provenientesdel ácido carboxílico sé maxirniza, lo cual parece indicar queel proceso de hidrólisis hasta esta semana correspondebásicamente a la ruptura vía hidrólisis del grupo éster, y quea partir de allí la presencia de una mayor concentración degrupos -COOH podrían estar interviniendo comocatalizadores en el proceso de degradación. Esta afirmaciónpuede ser más claramente evidenciada en la Figura 10, paralas muestras de PPDX degradadas en agua, donde se apreciaigualmente una disminución de la banda atribuible a losgrupos carbonilos provenientes del grupo éster a medidaque transcurre el tiempo de hidrólisis, y al mismo tiempo unaumento temprano de la banda de absorción correspondiente
.a los grupos carboxílicos provenientes de la formación degrupos ácidos carboxílicos producto de la reacción dehidrólisis.
Como se ve en la Figura 11 en el espectro correspondientea la cuarta semana, se observa una diferencia debido a laaparición de un pico alrededor de 1760-1768 cm' y a unensanchamiento pronunciado de la banda con la aparición
de un pequeño lomo hacia 1720-1690 cm' (señalado con unaflecha) visto más claramente a través de la Deconvoluciónpor Transformada de Fourier (señal en 1695 cm"), realizadaen la zona de interés en el espectro original de la cuartasemana; estos últimos aspectos, refuerzan la idea de laformación de posibles estructuras intermedias entre elpolímero y los componentes de la solución buffer (véaseFigura 5 anterior), lo cual es atribuido a la posible formaciónde estructuras cíclicas conjugadas a grupos carbonilosprovenientes del grupo éster o de los grupos ácidos formadosdurante el proceso de degradación, y posiblemente dichasestructuras fomenten interacciones entre las cadenas demenor peso molecular, lo cual parece cobrar importanciadebido a la aparición de las bandas hacia 1152 y 1110 cm'!(véase Figura 7) [14-16].
Finalmente, en las Figuras 12 y 13 se aprecia eldesplazamiento de la banda de absorción ubicada en 2981cm:', hacia longitudes de onda mayores 2985 cm:', este hechose atribuye a la formación de ácidos carboxílicos dímeros,evidenciado por el aumento de esta banda a medida que eltiempo de hidrólisis transcurre. Así, la posible formación deestructuras intermediarias mencionadas anterior-mentepodría contribuir al pronunciamiento y desplazamiento dedicha banda [17].
Ab
a
e
orba fne
a d
b
1820 1800 1780 1760 1740 1720 1700 1680
N° de Onda (cm-1)
Fig. 9. Espectros de infrarrojo entre 1800 cm' y 1500 cm',correspondientes a las muestras de PPDX degradadas en una soluciónbuffer para períodos de (a) Osem, (b) 2 sem, (e) 4 sem, (d) 6 sem,(e) 8 sem y (f) 12 sem.

M. A. Sabino y col. /Revista Latinoamericana de Metalurgia y Materiales
A Ab bs
sor ob ra
bD de ai e na
b e
aa
18101&101100 11&11110 1181 11á1114() 1T.lCJ1120 1110 111X11SQD 16&1
N° de Onda (eDIl)
Fig. 10. Espectros FTIR, región 1800-1500 cm", correspondientesa las muestras de PPDX degradadas en agua para períodos dedegradación de (a) Osem, (b) 4 sem, (e) 8 sem, y (d) 12 sem.
.\bs if,Q
riE!'1
b
idi
¡b'l
i "
3100 3050 3000 2950 2000\0 de Onda (cm .1)
Fig. 12. Espectros de infrarrojo entre 3100 cm'! y 2850 cm',correspondientes a las muestras de PPDX degradadas en una soluciónbuffer para períodos de (a) Osem, (b) 2 sem, (c) 4 sem, (d) 6 sem,(e) 8 sem y {f) 12 sem.
3.2. Esvectrometría de Masas
Cuando un compuesto tiene baja volatilidad o cuando su. masa padre no puede determinarse, podría existir la posibilidadde preparar un derivado adecuado. El derivado seleccionadodebe proporcionar una volatilidad mejorada, un modo
b
47
N° de Onda (cnr')Fig.l1. Espectros FTIR, región 1640 - 1850 cm', correspondientesa las muestras de PPDX degradadas en agua para períodos dedegradación de (a) Osem; (b) 4 sem. El espectro intermedio es unaDeconvolución FT del espectro (b).
JfDeconvoludón
FT
a
Y'lCI 1B<1D 1Bla lID! 118D 111!C1 1740 112D 1100 111BD 111BD 184(J •
t\hsorh¡¡
'11
e
N<' lIl' onda (l'Ill,I)
Fig. 13. Espectros FTIR, región 3100-2850 cm" correspondientesa las muestras de PPDX degradadas en agua para períodos dedegradación de (a) Osem, (b) 4 sem, (e) 8 sem y (f) 12 sem.
predecible de escisión, un modelo de fragmentaciónsimplificado o la estabilidad incrementada del ión padre.
Los compuestos que contienen varios grupos polares,pueden tener una volatilidad bastante baja, por ejemplo,arninoácidos, ácidos carboxílicos, etc., es por ello que estoscompuestos son selecciones evidentes y efectivas para ser

48
retención para los productos de la degradación de la PPDXhidrolizada en la solución buffer fosfato, vemos que los picosintensos alrededor de 13 y 16 min., corresponden a lasespecies que se forman a partir de los iones provenientes delderivatizante y de los grupos fosfatos, como por ejemplo:Silanol, trimetil-fosfato; como se aprecia en la Figura 14.
Es importante destacar que las muestras de PPDX en formade discos sometidas a la degradación hidrolítica en lasolución buffer, fueron preparadas de esa manera con el finde garantizar el mayor porcentaje de fase amorfa y que ladegradación ocurriera casi en su totalidad, a pesar de ello laintensidad de los picos para las primeras semanas dedegradación no permitieron obtener cromatogramas conbuena resolución, sino hasta la semana 16 donde sepresentaron especies de bajo peso molecular detectablespor el equipo. El proceso de degradación a tiemposprolongados permite una mejor definición de los gruposácidos que se van formando, y es por ello que en esta Figura14, se puede apreciar que después de la semana 16, laintensidad de los picos alrededor de 6, 23 Y 39 minutos deretención son pronunciados. Como se señaló anteriormente,los picos observados a 13.5 y 15.8 minutos corresponden alas especies que se forman a partir de los iones provenientesdel derivatizante.
Revista Latinoamericana de [etahirvia -~ tueriales, Vol. 22, N° 2, 2002.
tratados químicamente con el fin de aumentar su volatilidady que puedan así proporcionar picos característicos.Seguramente el uso de los derivados de Trimetilsilil con elfin de enmascarar los grupos OH y -COOH provenientes dela hidrólisis de la PPDX son lo suficientemente volátiles parapasar a través de las columnas del cromatógrafo de gases(sin quedar atrapadas en las paredes de la misma) y darseñales características de los productos que se han formado.Es importante considerar que para el caso de losderivatizados, el pico del ión molecular [M]+7podría no estarsiempre presente, pero el pico [M-15]+ debido a la escisiónde uno de los enlaces Si-CH3 siempre resultará notable.
La columna de sílica utilizada en el experimento presentauna alta capacidad de separar los compuestos polares, debidoa la intensidad de los picos determinados por el detector, sinembargo parte de los compuestos no derivatizados pudieronbien ser absorbidos durante la línea de inyección, o bien, porla columna una vez que fueron transferidos a la misma. Espor ello que las alícuotas provenientes de las solucionesdonde se realizó la degradación in vitro de la PPDX fuerondebidamente derivatizadas de manera de mejorar laseparación y evitar la retención de las especies en el sistemaGC-MS. El compuesto TBDMSIM fue seleccionado parahacer la derivatización debido a que este compuesto puedeser aplicado para todos aquellos compuestos que tienen lacapacidad de donar protones (como por ejemplo, ácidosdecarboxílicos, hidroxi-ácidos, etc.) y que el productoobtenido al reaccionar el TBDMSIM con la muestra, es decir,el derivatizado terbutildimetil-silil (TBDMS) tiene una altaestabilidad hidrolítica y excelentes propiedades para losanálisis GC-MS [8, 18, 19].
Un pico cromatográfico simple sin cola, generalmente seobtiene para los derivatizados TBDMS separados encolumnas como la utilizada en este tipo de experimento. Comose señalo al principio, una característica de los derivatizadoses que no presentan iones moleculares y que el ión [M-15]+se debe a la perdida del grupo terbutil, el cual es, sin embargo,muy intenso, y entonces permite la fácil identificación de losgrupos ácidos. Asimismo, el ión con rn/z=73 (siendo rn/z larelación masa/carga del ión) correspondiente al grupo
_(CH3)3Si+es de una alta intensidad, y también proviene delderivatizado, Otros iones prominentes que serán apreciadosa valores rn/z 75, 115, 147 Y189 corresponden a los grupos:
(a) HO+=Si(C~)2(b) [(C~\C](C~)2Si+(e) (C~)3SiO+=Si(C~)2(d) [(C~\C](C~)2SiO+=Si(C~)2
formados respectivamente. Asimismo, si observamos elcromatograma general GC- MS en función de los tiempos de
A11UNDANeI,\
15.84
-13:90!
38.95
5.95 23.60rr-~~~ I .. -
..10 20 30 40 50
~tiempoFig. 14. Cromatogramas GC- MS para los productos de degradaciónde la PPDX derivatizados en función del tiempo de hidrólisis, luegode 16 semanas del proceso degradativo.
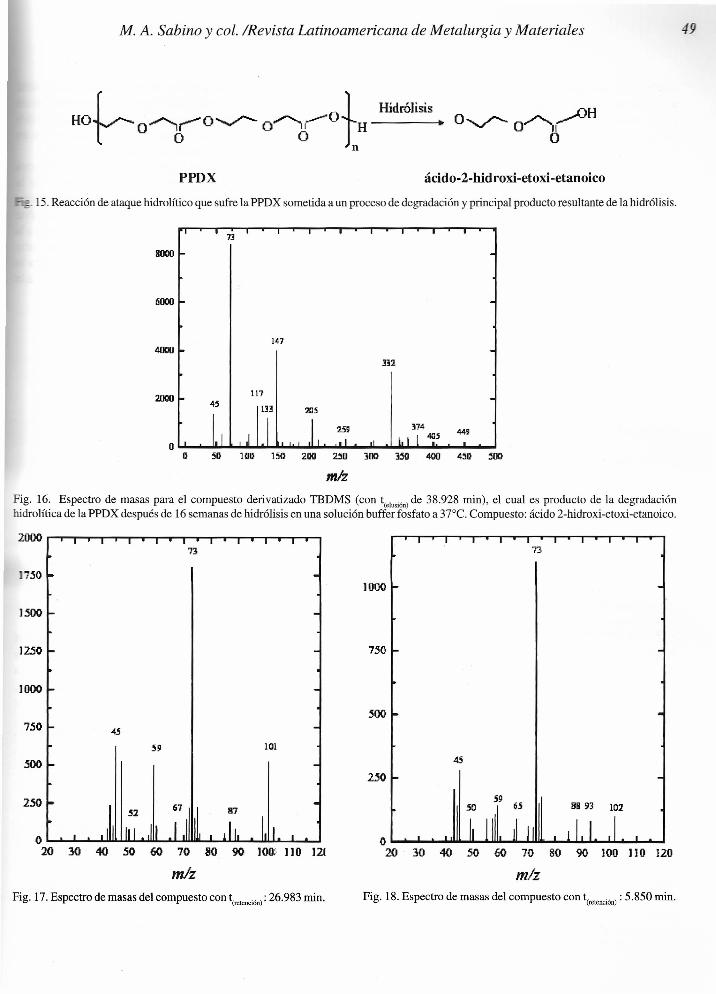
En la Figura 15, se presenta el esquema de la reacción deformación de la principal especie hidroxi-ácida que resultade la hidrólisis de la PPDX, y que de acuerdo a los resultadosobtenidos en el cromatograma de la Figura 14 anterior,corresponde al pico ubicado a los 38.928 minutos de retenciónen el GC-MS, y cuyo espectro de masas se mostrará másadelante en la Figura 16.
M. A. Sabina y col. /Revista Latinoamericana de Metalurgia y Materiales 49
r _ J HidrólisisHOlO&O~OyO[H---" O~O~Hn
PPDX
- _ 15. Reacción de ataque hidrolítico que sufre la PPDX sometida a un proceso de degradación y principal producto resultante de la hidrólisis.
ácido-2-hidroxi-etoxi-etanoico
7]
6000
1474000
]32-
2000 11745 1]] 205
44~
OO 50 100 150 200 250 300 350 400 450 500
mh
8000
Fig. 16. Espectro de masas para el compuesto derivatizado TBDMS (con t(eluSión)de 38.928 min), el cual es producto de la degradaciónbidrolítica de la PPDX después de 16 semanas de hidrólisis en una solución buffer fosfato a 37°C. Compuesto: ácido 2-hidroxi-etoxi-etanoico.
200073
1750
1500
1250
1000
750 45
59 IDI
500
250
OL....l.---L...&.....LJ.ULJ.JL.I.....t.J.LLL....LL.I20 30 40 50 60 70 80 90 JOO JJO J21
mlzFig. 17. Espectro de masas del compuesto con t(retención):26.983 mino
73
]000
750
500
45
250
.59
o~~~~~~~~.w~~~~~~~~20 30 40 50 60 70 80 90 100 Jl O 120
m/zFig. 18. Espectro de masas del compuesto con t(retenciÓn): 5.850 mino

50 Revista Latinoamericana de Metalurgia y Materiales, Vol. 22, N° 2, 2002.
1)(119,103)
2) (205, 17)
3) (191,31)
4)(177,45)
5) (161,61)
6)(133,89)
7) (147,75)
8) (163,59)
9) = 2) (205,17)Fig. 19.Diagrama de las posibles fragmentaciones que puede sufrir la PPDX o un oligómero producto de la
degradaciónhidrolítica del polímero.
Haciendo un seguimiento, al pico alrededor de los 39 min.,al aumentar el tiempo de degradación se incrementa laconcentración del ácido formado en el medio dehidrólisis ypor lo tanto la intensidad de la banda. Asimismo, seidentificaron otros picos, los cuales se corresponden a otrosgrupos ácidos que también se forman a partir del progresode la degradación de los productos inicialmente formados,lo cual conduce a la posterior obtención de especies de másbajo peso molecular. Entonces, el espectro de masas delprincipal compuesto derivado de la degradación hidrolíticade laPPDX se mostró en la Figura 16 anterior, y los espectrosde masa de los diferentes compuestos y su correspondienteasignación mlz e intensidad, pueden ser vistos en las Figuras17 y 18. La abundancia relativa de estos compuestos y losiones provenientes de las fragmentaciones sufridas (mlz), seresumen en la Tabla 3.
2 ~ 4 6 1
4. Conclusiones
Los biomateriales poliméricos presentan una serie deventajas (sobre otros materiales como los metales o lasbiocerámicas) que suponen la fabricación de dispositivos yaparatos ortopédicos que sustituyan parcial o totalmente alos miembros del aparato locomotor o simplemente permitanservir de sostén en la reparación de fracturas óseas. En estesentido, la posibilidad de que el polímero sea degradado,ofrece la oportunidad de que pueda conseguirse un procesode curación óptimo, con la recuperación de la funcionalidaddel sistema fisiológico y por ende del tejido afectado,mientras el polímero se degrada mediante un mecanismopuramente hidrolítico, el cual da lugar a la fragmentación delpolímero en cadenas macromoleculares (debido a que elpolímero posee un enlace hidrolíticaraente inestable),originando productos de degradación de bajo peso molecular,atóxicos y que pueden ser metabolizados o bioabsorbidospor el organismo.
A través de FTIR, se demostró que durante el período de
Un esquema de las posibles fragmentaciones que podríasufrir la molécula de la PPDX, se presenta esquemáticamenteen la Figura 19, Yallí mismo se muestran los fragmentos mlzque resultarían de dicha fragmentación y que aparecen enlos espectros de masa señalados en las Figuras 16-18.
Es importante destacar, que la Figura 16, muestraclaramente que los fragmentos originados a partir de ladegradación hidrolítica de la PPDX conduce mayormente ala formación del ácido-2 hidroxi- etoxi-etanoico, y que éstecompuesto al seguirse degradando vía hidrólisis, induce a lageneración de especies de muy bajo peso molecular entrelas que se encuentra el monómero, como 10 manifiesta laFigura 18, donde losfragmentosparecen corresponder a lafragmentación del compuesto dioxano.
Fragmento(mlz)
9
+n
degradación se evidencia la disminución de grupos Q-Hlibres, producto de la posible asociación de estos grupos alos productos resultantes de las rupturas de enlaces; y laposible formación de estructuras intermediarias o complejas,mediante asociación de grupos, donde pueden intervenirlos grupos fosfatos presentes en el medio de hidrólisis.Además, como consecuencia de los rompimientos del grupoéster presente en la estructura de la PPDX, se observanvariaciones de absorbancia de las bandas asociadas a losgrupos carbonilos dada la aparición del grupo ácidocarboxílico.
Estas evidencias fueron verificadas medianteEspectrometría de Masas donde se determinó que uno delos subproñuctos de la degradación de la PPDX correspondeal ácido 2-hidroxi-etoxi- etanoico.
A través de.FTIR,se. demostró que durante el período dedegradación se evidencia la disminución de grupos Q-Hlibres, producto de la posible asociación de estos grupos alos productos resultantes de las rupturas de enlaces; y la

M. A. Sabina y col. /Revista Latinoamericana de Metalurgia y Materiales
Tabla 3. Tiempos de retención e iones.prominentes obtenidos delos espectros de masa de los derivatizados (Figuras 16, 17 Y 18) dela hidrólisis de la PPDX.
Tíempo de..retencion (min)
m/z Intensidadrelativa (%)
38.928
455g7389103117133147157169189205215243259304332345359374405449
13410025201144121
143132
3933212
26.983
455259677387101
3643031005
28
5.850
455059-'66738893102
5110167100875
posible formación de estructuras intermediarias o complejas,mediante asociación de grupos, donde pueden intervenirlos grupos fosfatos presentes en el medio de hidrólisis.Además, como consecuencia de los rompimientos del grupoéster presente en la estructura de la PPDX, se observanvariaciones de absorbancia de las bandas asociadas a losgrupos carbonilos dada la aparición del grupo ácidocarboxílico. Estas evidencias fueron verificadas medianteEspectrometría de Masas donde se determinó que uno delos subproductos de la degradación de la PPDX correspondeal ácido 2-hidroxi-etoxi- etanoico.
Adicionalmente, se ha reportado en publicacionesanteriores [20-23], que el peso molecular viscosimétrieodecrece linealmente en función del tiempo de hidrólisis,debido a que la degradación hidrolítica comienza en lasregiones amorfas, en donde los segmentos de enlaces de
cadena, las terminaciones de cadena libres, y las cadenasplegadas son degradadas en fragmentos; por lo que a medidaque la degradación procede, el peso molecular disminuye, ysufre una caída considerable una vez que la mayor cantidadde zona amorfa ha sido degradada hidrolíticamente y se hadado comienzo al ataque a las zonas cristalinas.
Referencias
1. P. Craig, J.A. Williams, K.W.Davis, A.D.Magoun.,Surgery. Gynecology & Obstetrics, 141,(1975) 1
2. P. Tormala, J. Vasenius, S. Vainionpaa, J. Laiho, T.Pohjonen, P. Rokkanen, J. Biomedical MaterialResearch, 21, (1991) 1
3. J. Schitz, J.O. Hollinger, Clinic. Orthopedics & RelatedResearch,237,(1988)245
4. J.A. Ray, N. Doddi, D. Regula, J.A. Wiliams, A. Melveger,Surgery Gynecology Obstetrics, 153, (1981) 497
5. J. San Roman, Rev. Plást. Mod., 413, (1990) 689.6. W. Jencks, Catalysis in Chemistry and Enzymology,
Me Graw-Hill, Inc, New York (1969) 457. T. Iwata, Y. Doi, F. Kokubu, Sh. Teramachi,
Macromolecules, 32, (1999) 8325.8. 1.Rehman, E. Andrews, R. Smith, J. of Materials Science,
7,(1996) 179. L. Lu, C. Garcia, A.G. Mikos, J. Biomed. Mat. Res., 46,
(1999)23610. C.e. Chu, N.D. Campbell, J. of Biomedical Materials
Research, 16, (1982) 41711. G. Farrow, D. Ravens, Polymer, 3, (1962) 1712. B.J. PoI, P. Wachem, L. Van der Does, A. Bantjes, J.
Biomed. Mat. Res., 32, (1996) 32113. K. Nakanissi, H.S. Fhilippa, Infrared absorption
spectroscopy, Holden-Day, 2da. Edition, San Francisco,(1977) 17
14. G. Grimandi, P. Weiss, F. Millot, N. Passuti, J. Biomed.Mater. Res., 39, (1998) 660
15. P.Weiss, M. Lapkowski, R. Legeros, J. Bouler, J. Mater.Sci. Mater Med., 10, (1997) 621
16. G. Meijs, S. McCarthy, E. Rizzardo, Y. Chen, J. Biomed.Mat. Res. 27 ,(1993) 345
17. U. Edlmund, A.C.Albertsson, J. Polym. Sci. A. PolymChem., 37, (1999) 1877
18. E. King, S. Robinson, R. Cameron, Polym. Int., 48, (1995)915
19. X. Qu, A. Wirsen, A.e. Albertsson, J. Appl. Polym. Sci.,74, (1999) 3186
20. M.A. Sabino, J.L. Feijoo, AJ. Müller, Macromol. Chem.Phys.,201 (2000) 2687
21. M.A. Sabino, S. Gonzalez, L. Márquez, J.L. Feijoo;Polym.Deg. & Stab., 69, (2000)-209
22. M.A. Sabino, J.L. Feijoo, A.J. Müller, Polym. Deg. &Stab., 73, (2001) 541
23. M.A. Sabino, G. Ronca, L. Sabater, A.J. Müller, Polym.Bulletin, 48, (2002) 291
51





























![UNIVERSIDAD DE BURGOS - COnnecting REpositories · gases con derivatización; HPLC; de par iónico con fase estacionaria polar o apolar) [3, 5, 16, 18] o la extracción con disolventes](https://img.pdfslide.us/doc/110x75/5eb484e39a59033ccd5eade0/universidad-de-burgos-connecting-repositories-gases-con-derivatizacin-hplc.jpg)