Embed Size (px)
Citation preview

EVIDENCE FOR MULTILAYER NANOSCALE ENZYME
ACTIVE SITES
A Dissertation Presented
By
Heather R. Brodkin
To
The Department of Chemistry and Chemical Biology
in partial fulfillment of the requirements
For the degree of
Doctor of Philosophy
in the field of
Chemistry
Northeastern University Boston, Massachusetts
January, 2009
1

© 2009
Heather R. Brodkin
ALL RIGHTS RESERVED
2

EVIDENCE FOR MULTILAYER NANOSCALE ENZYME
ACTIVE SITES
By
Heather R. Brodkin
ABSTRACT OF DISSERTATION
Submitted in partial fulfillment of the requirements for the degree of Doctor of
Philosophy in Chemistry in the Department of Chemistry and Chemical Biology in the
Graduate School of Arts and Sciences of Northeastern University,
Boston, Massachusetts
January, 2009
3

Abstract of Dissertation
One of the most fundamental questions in biochemistry today is how enzymes work.
Most often this discussion focuses on the amino acid residues in direct contact with the
reactive metal or reacting substrate within the three-dimensional (3D) structure of the
protein. What is very rarely mentioned is the influence that remote residues have on
enzyme catalysis. Remote residues refer to those residues which are, or are farther from,
second-nearest neighbors to the reactive metal or reacting substrate molecule.
The literature has scarce information pertaining to the importance of these second- and
third-shell residues in enzyme catalysis. The idea of the involvement of residues located
in outer coordination spheres in catalysis was first introduced by Leatherbarrow, Fersht
and Winter when the concept of site-directed mutagenesis was introduced. It was
discovered that mutations made to residues located far from the reaction site resulted in
proteins with reduced catalytic rate. In some cases; however, these mutations resulted in
proteins whose catalytic rate was increased. It was at this time that the term ‘protein
engineering’ was coined.
While limited studies have been performed to understand the role of remote residues in
enzyme catalysis, a thorough investigation of the importance of second- and third-shell
residues has not been performed. In this thesis, two different computational methods,
THEMATICS and Evolutionary Trace (ET), based on two very different types of input,
are used to identify functionally important residues in the first-, second- and third-shells
4

of an enzyme. It is shown that both of these methods predict residues in the second- and
third- shells to be important. Once the concept of remote residue involvement in enzyme
catalysis has been established theoretically, the focus shifts to one particular enzyme, Co-
type nitrile hydratase from Pseudomonas putida, for which both THEMATICS and ET
predict a multilayer active site. First, the x-ray crystal structure of the wild type enzyme
and its kinetic properties are reported. A kinetic analysis of single point mutations is
presented for five second- and third-shell residues that were predicted computationally to
be functionally important. Additionally, crystal structures are presented for four of the
mutants. It is shown that for some of the mutants there are small, local structural
differences which may explain the effects on catalytic rate, however, for others, no
structural differences are observed compared to wild type. For these examples, it is
proposed that the differences are due primarily to electrostatic effects. While no
unequivocal explanation emerges at this stage for why these residues in the outer
coordination spheres influence catalysis, this work makes a strong case for the concept
that enzyme active sites are built in multiple layers. It is suggested that computational
approaches, and the concept of multilayer active sites introduced herein, can help to
guide protein engineering efforts.
5

Acknowledgements
First and foremost I would like to offer my sincerest gratitude to my advisor, Dr Mary Jo
Ondrechen, who has supported me throughout my thesis with her patience and
knowledge while allowing me the room to work in my own way. I attribute the level of
work achieved to her encouragement without which this thesis would not have been
completed. I would like to thank all of the members of my thesis committee, all of whom
have been instrumental during this process with their expertise and guidance.
Specifically, I would like to thank Dr. Ira Krull for his guidance and friendship
throughout this entire process. He has truly been an inspiration. A special thanks goes to
Dr. Graham Jones who also has been instrumental throughout my time at Northeastern.
Finally, thank you to Dr. Penny Beuning who has always been there to answer any
question I have had.
Thank you to all the members of the THEMATICS group, past and present. I appreciate
the support. I would like to especially thank Dr. Leo Murga for all of his guidance and
scientific input, and Ying Wei, Wenxu Tong and Terry Yang for their friendship.
I want to say a huge thank you to all the graduate students at Northeastern University for
their support, knowledge and guidance. Specifically, I would like to thank Jim Glick and
Susie Schiavo. You both have been not only good friends, but true colleagues; a rare
attribute. I could not have survived this process without you both and I value our
friendship immensely.
A special thank you goes to Dr. Vouros at Northeastern University for the unlimited use
of his HPLCs. This work would not have been completed without access to his lab, and
for that I am truly grateful.
6

All of the experimental work for this thesis was performed at Brandeis University and I
am indebted to Dr. Dagmar Ringe for providing the opportunity to feel truly at home in
the Brandeis labs. She has taught me a great deal and I look forward to working with her
in the future. I have learned a great deal from many of the students and post docs there,
and am truly fortunate to have had the opportunity to work with Dr. Walter Novak on my
projects. He has been a true inspiration, and this work would not have been completed
without his help.
An additional thank you goes to all my professors at Framingham State College, without
whom, I would never have become a scientist. Specifically, I would like to thank Dr.
Eames, Dr. Russell, Dr. Simonson and Dr. Allen.
Finally, I owe a huge THANK YOU to my family and friends who have put up with me
throughout the last 5 ½ years. These are special times and I will never forget you all for
the love and support. Leeanne and Laura, all I can say is thank you for everything, I love
you all. A special thanks to John Shostak for listening and to Father Joe for his
inspiration. Motley, Nathalie, and Soulmate, thank you for the unconditional love. Lolita
V. Hall, thank you for the best cat whiskers ever. Adam, I love you and I thought for sure
you would become a doctor before me. Paul, no matter what, I know you would be there
for me. Dad, I love you always and miss you more than words can say. Mom, you are the
best friend a girl could ask for and Dad reminds me of that every day in my dreams. This
is dedicated to you. Thank you!
This work was supported by the National Science Foundation under grants MCB-
0517292, MCB-0843603, and DGE-0504331. An IGERT Traineeship, funded by the
National Cancer Institute and administered by the National Science Foundation,
supported a part of my doctoral education and is gratefully acknowledged.
7

Table of Contents
ABSTRACT OF DISSERTATION ................................................................................................ 4
ACKNOWLEDGEMENTS ............................................................................................................ 6
TABLE OF CONTENTS ................................................................................................................ 8
LIST OF TABLES ........................................................................................................................ 11
LIST OF FIGURES....................................................................................................................... 14
LIST OF ABBREVIATIONS AND SYMBOLS.......................................................................... 21
CHAPTER 1 - INTRODUCTION ................................................................................................ 23
1.1 WHY THE NEED FOR ENZYMES?.......................................................................................... 24
1.2 BACKGROUND ON SECOND- AND THIRD-SHELL RESIDUE INVOLVEMENT IN CATALYSIS .. 28
1.3 DIRECTED EVOLUTION ........................................................................................................ 29
1.4 RATIONAL PROTEIN DESIGN ............................................................................................... 31
1.5 DISADVANTAGES OF DIRECTED EVOLUTION AND RATIONAL PROTEIN DESIGN METHODS33
1.6 COMPUTATIONAL APPROACHES TO THE IDENTIFICATION OF FUNCTIONAL RESIDUES ...... 33
1.7 OVERVIEW OF THESIS.......................................................................................................... 34
1.8 THESIS CHAPTERS ............................................................................................................... 35
1.9 REFERENCES........................................................................................................................ 39
CHAPTER 2 - EVIDENCE FOR REMOTE RESIDUE INVOLVEMENT IN CATALYSIS;
ARE ENZYME ACTIVE SITES BUILT IN MULTIPLE LAYERS? ......................................... 42
2.1 INTRODUCTION.................................................................................................................... 43
2.2 MATERIALS AND METHODS ................................................................................................ 46
2.3 RESULTS AND DISCUSSION.................................................................................................. 48
2.3.1 Experimental Design ................................................................................................... 48
8

2.3.2 THEMATICS and ET: Identification of Residues and Predictions by Shell............... 52
2.3.3 Metalloenzymes........................................................................................................... 57
2.3.4 Non-Metalloenzymes................................................................................................... 71
2.4 SUMMARY OF RESULTS ....................................................................................................... 80
2.5 CONSERVATIVE VERSUS NONCONSERVATIVE MUTATIONS................................................ 81
2.6 CONCLUSIONS ..................................................................................................................... 83
2.7 SUPPLEMENTAL TABLES ..................................................................................................... 85
2.8 REFERENCES...................................................................................................................... 114
CHAPTER 3 - STRUCTURAL AND KINETIC ANALYSIS OF WILD TYPE CO-TYPE
NITRILE HYDRATASE FROM PSEUDOMONAS PUTIDA ................................................. 122
3.1 INTRODUCTION.................................................................................................................. 123
3.2 MATERIALS AND METHODS .............................................................................................. 133
3.3 RESULTS AND DISCUSSION................................................................................................ 137
3.4 INTRODUCTION TO MICHAELIS-MENTEN KINETICS.......................................................... 149
3.5 CONCLUSIONS ................................................................................................................... 166
3.6 REFERENCES...................................................................................................................... 168
CHAPTER 4 - EVIDENCE FOR PARTICIPATION OF REMOTE RESIDUES IN THE
CATALYTIC ACTIVITY OF CO-TYPE NITRILE HYDRATASE FROM PSEUDOMONAS
PUTIDA - A KINETIC AND CRYSTAL STRUCTURE ANALYSIS ..................................... 171
4.1 INTRODUCTION.................................................................................................................. 172
4.2 MATERIALS AND METHODS .............................................................................................. 174
4.3 RESULTS AND DISCUSSION................................................................................................ 180
4.3.1 αAsp164Asn .............................................................................................................. 200
4.3.2 αGlu168Gln ............................................................................................................... 201
4.3.3 βGlu56Gln ................................................................................................................. 203
9

4.3.4 βHis71Leu ................................................................................................................. 205
4.3.5 βTyr215Phe ............................................................................................................... 207
4.4 CONCLUSIONS ................................................................................................................... 214
4.5 REFERENCES...................................................................................................................... 216
CHAPTER 5 - CONCLUSIONS, FUTURE WORK AND FUTURE DIRECTIONS ............... 218
5.1 CONCLUSIONS ................................................................................................................... 219
5.2 FUTURE WORK .................................................................................................................. 222
5.3 FUTURE DIRECTIONS – COLLABORATIONS ....................................................................... 225
SUPPLEMENTAL CHAPTER 1 - COMPUTATIONALLY GUIDED PROTEIN-SPECIFIC
LABELING WITH NANOPARTICLES - A TEST CASE USING HER2................................ 228
SUPPLEMENTAL CHAPTER.1 INTRODUCTION .......................................................................... 229
SUPPLEMENTAL CHAPTER.2 MATERIALS AND METHODS ...................................................... 232
SUPPLEMENTAL CHAPTER.3 RESULTS AND DISCUSSION........................................................ 235
Supplementa1 Chapter.3.1 4-3-3 σ ..................................................................................... 236
Supplemental Chapter.3.2 HER2........................................................................................ 240
SUPPLEMENTAL CHAPTER.4 FUTURE WORK .......................................................................... 248
SUPPLEMENTAL CHAPTER.5 CONCLUSIONS............................................................................ 250
SUPPLEMENTAL CHAPTER.6 REFERENCES.............................................................................. 251
CURRICULUM VITAE ............................................................................................................. 254
10

List of Tables
Table 1-1: Rate constants for uncatalyzed reactions (k ), turnover numbers (k ), catalytic efficiencies (k /K ), and rate enhancements (k /k ) for water consuming reactions at 25 ºC.
non cat
cat M cat non4 ............................................................................................................ 26
Table 2-1: Metallo- and Non-metalloenzyme Test Set..................................................... 51 Table 2-2: THEMATICS results for five metallo and non-metalloenzymes, alkaline phosphatase, carbonic anhydrase II, mandelate racemase, triosephosphate isomerase and tyrosyl-tRNA synthetase. (bold = annotated catalytic residues, italics = annotated ligand or metal binding residues, underlined = those residues that have been experimentally mutated, ND = no residues identified by THEMATICS). ................................................ 55 Table 2-3: ET results for five metallo and non-metalloenzymes, alkaline phosphatase, carbonic anhydrase II, mandelate racemase, triosephosphate isomerase and tyrosyl-tRNA synthetase. (bold = annotated catalytic residues, italics = annotated ligand or metal binding residues, underlined = those residues that have been experimentally mutated). . 56 Table S-1: THEMATICS predicted residues for metallo enzyme test set. Residues in bold are known catalytic residues (i.e. those residues directly involved in the chemistry of the protein), and italics indicates a ligand or metal binding residue. ND = no residues predicted for that shell. ..................................................................................................... 85 Table S-2: Evolutionary Trace predicted residues for metallo enzyme test set. Residues in bold are known catalytic residues (i.e. those residues directly involved in the chemistry of the protein), and italics indicates a ligand or metal binding residue. NC = conservations score not calculated; ND = no residues predicted for that shell. ...................................... 88 Table S-3: THEMATICS predicted residues for non-metallo enzyme test set. Residues in bold are known catalytic residues (i.e. those residues directly involved in the chemistry of the protein), and italics indicates a ligand or metal binding residue. ND = no residues predicted for that shell. ..................................................................................................... 95 Table S-4: Evolutionary Trace predicted residues for non-metallo enzyme test set. Residues in bold are known catalytic residues (i.e. those residues directly involved in the chemistry of the protein), and italics indicates a ligand or metal binding residue. NC = conservations score not calculated; ND = no residues predicted for that shell. ............... 98 Table S-5: Experimental mutations to Alkaline Phosphatase (AP) and their effect on k for residues identified by THEMATICS and/or ET. (+ = increase in catalytic activity, - = decrease in catalytic activity). All mutations cited were carried out in Tris buffer.
cat
....... 104 Table S-6: Experimental mutations to human Carbonic Anhydrase II and their effect on k for residues identified by THEMATICS and/or ET. (+ = increase in hydrolytic activity, - = decrease in hydrolytic activity). Only CO hydration was considered.
cat
2 ...... 106
11

Table S-7: Experimental mutations to Mandelate Racemase and their effect on k for residues identified by THEMATICS and/or ET. (+ = increase in catalytic activity, - = decrease in catalytic activity).
cat
......................................................................................... 109 Table S-8: Experimental mutations to Triosephosphate Isomerase and their effect on k for residues identified by THEMATICS and/or ET. (+ = increase in catalytic activity, - = decrease in catalytic activity).
cat
......................................................................................... 110 Table S-9: Experimental mutations to Tyrosyl tRNA Synthetase and their effect on k for residues identified by THEMATICS and/or ET. (+ = increase in catalytic activity, - = decrease in catalytic activity). indicates catalytic effect for step 1, the formation of the adenylate intermediate, indicates catalytic effect for step 2, the formation of tyrosyl t-RNA.
cat
1
2
............................................................................................................................... 112 Table 3-1: Overview of experimental mutations made to both Co- and Fe-type nitrile hydratases........................................................................................................................ 131 Table 3-2: Data collection and refinement statistics for wild type ppNHase. ................ 140 Table 3-3: Kinetics results comparing wild type ppNHase to dissolved ppNHase crystals and wild type ppNHase with the addition of 10% polyacrylate. .................................... 145 Table 3-4: Kinetics results for ppNHase at pH 5.7, 6.7, 7.2, 7.5 and 8.5. The results at pH 6.7 represent an n=3, while only an n=2 was run at all other pH values. ....................... 162 Table 3-5: Kinetics overview for numerous Co- and Fe-type nitrile hydratases for the hydrolysis of methacrylonitrile at room temperature at pH approximately 7.2. – refers to values not found in the literature. ................................................................................... 165 Table 4-1: Primers designed for site-directed mutagenesis. Only forward primers are listed. Mutated codons are shown in boldface. ............................................................... 176 Table 4-2: THEMATICS predictions of functional sites for wild type NHase from Pseudonocardia thermophila (PDB ID: 1IRE ), wild type NHase from Pseudomonas putida, and five NHase mutants from Pseudomonas putida. Predicted residues are listed by shell with average normalized conservation scores for each coordination shell. Bold face refers to residues predicted by THEMATICS which are annotated in the CSA as catalytic residues; italics refers to residues predicted by THEMATICS which are found in LPC to be metal binding or ligand binding residues; those residues which are in both bold face and italics refers to THEMATICS positives which are both annotated in the CSA as catalytic residues and annotated in LPC as binding residues.
1
........................................ 182 Table 4-3: Evolutionary Trace functional site predictions for wild type NHase from Pseudonocardia thermophila (PDB ID: 1IRE ). Predicted residue sequence numbers are listed by shell with average normalized conservation scores for each coordination shell. Bold face refers to residues predicted by ET which are annotated in the CSA as catalytic
1
12

residues; italics refers to residues predicted by ET which are found in LPC to be metal binding or ligand binding residues; those residues which are both in bold face and italics refers to ET positives which are both annotated in the CSA as catalytic residues and annotated in LPC as binding residues............................................................................. 183 Table 4-4: Kinetics results for the conversion of n-Valeronitrile to n-Valeramide for wild type NHase from Pseudomonas putida and five NHase mutants from Pseudomonas putida at pH 5.8, 6.7, 7.2, 7.5 and 8.5. pH 6.7 represents an n = 3 and therefore standard deviations are included. All other pH values represent an n = 2, and therefore no standard deviations are included. .................................................................................................. 193 Table 4-5: Data collection and refinement statistics for wild type ppNHase and four mutant proteins................................................................................................................ 199 Table A-1: THEMATICS results 14-3-3 σ and HER2. .................................................. 236
13

List of Figures
Figure 1-1: Rate constants and half-lives of biological reactions proceeding spontaneously in water in the absence of enzyme.1 .......................................................... 25 Figure 2-1: Reaction mechanism for alkaline phosphatase. In the first step, Ser102 is phosphorylated giving a phosphoseryl intermediate. In the second step, this intermediate is hydrolyzed to give a non-covalent enzyme-phosphate complex. In the presence of a phosphate acceptor such as Tris, the enzyme shows transphosphorylation activity and transfers a phosphate to the alcohol to form a phosphate monoester.
31
30........................... 59 Figure 2-2: Cartoon representation of active site of alkaline phosphatase (PBD ID: 1ALK ) including metal binding residues in the first-shell known to be functionally important and the catalytic residue, Ser102. Grey spheres = zinc ions and green sphere = magnesium ion. refers to first-shell residues identified by THEMATICS and ET; refers to first-shell residues identified by ET only. Note that Ser102 and Thr155 will not be found by the THEMATICS method as they are non-ionizable residues.
20
a b
......................... 60 Figure 2-3: Residues involved in interaction with the first-shell residue, Asp153 for alkaline phosphatase (PDB ID: 1ALK ) including the THEMATICS positive residues in the second- and third-shell. Grey spheres = zinc ions, green sphere = magnesium ion and red cross = water. refers to THEMATICS and ET positive residue in the first-shell; refers to residues identified by ET in the second-shell; refers to residues identified by THEMATICS and ET in the second-shell; refers to a third-shell residue identified by THEMATICS and ET. The Asp153Gly/Asp330Asn double mutant resulted in a 40-fold increase in catalytic rate.
20
a b
c
d
25 ................................................................................................ 62 Figure 2-4: Reaction mechanism for carbonic anhydrase II. Zinc-bound hydroxide acts as the nucleophile to attack CO to form a zinc-bound bicarbonate intermediate. This intermediate is then displaced by a water molecule creating a zinc-H O form. In the rate determining step, the zinc-bound hydroxide is regenerated through the transfer of a proton to the solvent facilitated by the active site histidine, His64, which acts as a proton shuttle.
38
2
2
36,42........................................................................................................................ 64 Figure 2-5: Cartoon representation of known active site and metal binding residues for carbonic anhydrase II (PDB ID: 1CA2 ) in addition to select second-shell residues known to be functionally important. Grey sphere = zinc. refers to first-shell residues identified by THEMATICS and ET; refers to second-sell residues identified by THEMATICS and ET; refers to additional first-shell residues identified by only ET; refers to second-shell residues identified by only ET.
21
a
b
c d
...................................................... 65 Figure 2-6: Cartoon representation of select second- and third-shell residues located in the hydrophobic face of the active site pocket predicted by THEMATICS and/or ET for carbonic anhydrase II (PDB ID: 1CA2 ). The three zinc coordinating histidine residues are included for orientation. Grey sphere = zinc. refers to first-shell residues predicted by THEMATICA and ET; refers to a second-shell residue predicted by both
21
a
b
14

THEMATICS and ET; refers to second-shell residues predicted by ET. The Leu198Arg, Leu198Pro and Leu203Arg mutations resulted in at least one order of magnitude decrease in the catalytic rate of CO hydrolysis.
c
250 .......................................................................... 67
Figure 2-7: Reaction mechanism for mandelate racemase. His297 abstracts the α-proton to generate an intermediate, and Lys166 protonates the opposite face of the intermediate to produce the inverted product.
58
56,57 ................................................................................. 69 Figure 2-8: Cartoon representation of active site and metal binding residues known to be functionally important for mandelate racemase predicted by THEMATICS and/or ET (PDB ID: 2MNR ). Second-shell residues predicted by THEMATICS and/or ET are also shown. Purple sphere = Mn. refers to first-shell residues predicted by THEMATICS and ET; refers to first-shell residue identified only by ET; refers to a second-shell residue identified by THEMATICS and ET; refers to a second-shell residue predicted only by THEMATICS. The single Asp270Asn mutation results in a 10 -fold decrease in catalysis for both (R)- and (S)- mandelate substrates, while the single mutant His297Asn and the double mutant His297Lys/Asp270Asn result in complete loss of activity with both (R)- and (S)- mandelate substrates.
22
a
b c
d
59 4
56 60
...................................................... 69 Figure 2-9: Reaction mechanism for trisosephosphate isomerase. A proton is abstracted from DHAP by the catalytic base Glu165 which causes the formation of an enediol/endiolate intermediate. His95 acts as the catalytic acid.
23
63 ................................... 73 Figure 2-10: Select set of known functionally important residues for triosephosphate isomerase from yeast in the ‘open’ form (PDB ID:1YPI ). refers to first-shell residues predicted by both THEMATICS and ET; refers to second-shell residues predicted by THEMATICS and ET; refers to the third-shell residue predicted by THEMATICS and ET; refers to first-shell residues identified only by ET; refers to second-shell residues identified only by ET; refers to third-shell residues identified only by ET.
66 a
b
c
d e
f ................... 74 Figure 2-11: Select set of known functionally important residues for triosephosphate isomerase from yeast in the ‘closed’ form (PDB ID:2YPI ). refers to first-shell residues predicted by both THEMATICS and ET; refers to second-shell residues predicted by THEMATICS and ET; refers to the third-shell residue predicted by THEMATICS and ET; refers to first-shell residues identified only by ET; refers to second-shell residues identified only by ET; refers to third-shell residues identified only by ET. In the closed structure, Glu129 flips in toward Trp168. Mutations to two hinge residues, Tyr164Phe and Glu129Gln, result in a 2-fold and a 30-fold decrease in catalytic rate.
66 a
b
c
d e
f
64................... 74 Figure 2-12: Reaction mechanism for the formation of tyrosyl-adenylate from tyrosine and ATP for tyrosyl t-RNA synthetase. Residues from tyrosyl t-RNA synthetase that H-bond with the intermediate are shown for clarity.
1
67.......................................................... 77 Figure 2-13: Active site of tyrosyl-tRNA synthetase (PDB ID: 1TYD ) showing first- and second-shell residues in contact with the tyrosine. Red = tyrosine. refers to first-shell residues identified by THEMATICS and ET; refers to first-shell residue identified
24
a
b
15

by only THEMATICS; refers to first-shell residues identified only by ET; refers to the second-shell residues identified by THEMATICS and ET; refers to a third-shell residues identified only by ET; those residues with no superscripts are known to be in the active site, but are not identified by either THEMATICS or ET. Mutation of His45 to Gly results in a 250-fold decrease in catalytic rate indicating this second-shell residue is necessary to stabilize and orient the catalytic residue His48.
c d
e
1.......................................... 79 Figure 3-1: Sequence alignment of four Co-type Nitrile Hydratases (NHase) and four Fe-type NHases. Known functional residues are highlighted in yellow. refers to Co-type nitrile hydratases; refers to Fe-type nitrile hydratases; refers to the Co-type nitrile hydratase from Pseudomonas putida determined in this thesis from x-ray crystallography.
1
2 3
......................................................................................................................................... 126 Figure 3-2: Proposed reaction mechanisms for ppNHase.11........................................... 128 Figure 3-3: Cartoon diagram of active site of nitrile hydratase from Pseudonocardia thermophila (PDB ID: 1UGP ) shown in wall-eyed stereo view. All atoms are shown in CPK coloring; pink sphere = cobalt. Black dashed lines show atoms coordinating to the metal, green dashed lines refer to hydrogen bonds between the arginine residues and the cysteines, and the magenta dashed line refers to interactions between the binding residue, Tyr68, and the bound inhibitor, butanoic acid.
19
............................................................... 130 Figure 3-4: Crystal forms identified for ppNHase (clockwise from upper left: hexagonal plates, rods, needles, rods). ............................................................................................. 138 Figure 3-5: Typical diffraction pattern observed for wild type ppNHase. ..................... 139 Figure 3-6: Superposition of ppNHase and ptNHase structures. ppNHase and ptNHase α-subunits are in red and yellow and β-subunits are in blue and green, respectively. RMSD for the α subunits is 0.7 Å over 177 residues for the α subunit and 0.9 Å over 183 residues for the β subunits. The arrowed line in the left panel indicates the difference in the loop region between the α5 and α6 helices. The active site cobalt is enlarged and shown in pink. The two glycerol molecules associated with each dimer are rendered as ball and stick and shown in CPK coloring. The N- and C- termini are labeled.15 .......... 142 Figure 3-7: Active site of wild type ppNHase shown as wall-eyed stereo. Atom coloring is CPK. Black dotted line indicates coordinating atoms to the cobalt. ........................... 143 Figure 3-8: Comparison of Co-cyano-cobalamin (left panel) with active site of non-corrinoid Co-type nitrile hydratase (right panel). In the right panel, the active site of ppNHase (magenta) is superimposed with Co-cyano-cobalamin (red). Sphere = cobalt.......................................................................................................................................... 144 Figure 3-9: Superimposed active sites of nitrile hydratase from Pseudomonas putida (vide supra) (grey CPK coloring) and Pseudonocardia thermophila (PBD ID: 1IRE ) (magenta CPK coloring). Pink sphere = cobalt. (P. putida numbering)
16
......................... 144
16

Figure 3-10: Electron density of the cobalt site in ppNHase prior to the incorporation of the cysteine oxidation. Atom coloring is in CPK. The 2F -F map is rendered at 1.5 σ and is shown in blue. The F -F difference map is rendered at 4.5 σ and is shown in green.
o c
o c15 ............................................................................................................................ 147
Figure 3-11: FT-ICR mass spectrum of A and B chain of wild type ppNHase. Top inset panel shows the deconvoluted spectrum of the A chain for the +11 ion with the observed mass and the bottom inset panel shows the deconvoluted spectrum of the B chain for the +11 ion with the observed mass...................................................................................... 148 Figure 3-12: Enzymatic reaction obeying Michaelis-Menten kinetics for wild type ppNHase.......................................................................................................................... 152 Figure 3-13: Lineweaver-Burk plot for wild type ppNHase at pH 6.7. This plot shows a straight line, with a K of 1.86 mM and V of 0.908. Note that this method has greater error than the nonlinear regression used in this thesis and therefore there are differences between the kinetics constants from this plot and those in Table 3-3.
M max
40......................... 154 Figure 3-14: Typical HPLC spectra for blank and standard, n-Valeramide. The x-axis is time in minutes and the y-axis is absorbance at 210 nm in mAU. In panels A-D, the circled area represents the peak of interest, n-Valeramide. Panel A shows the spectrum for the blank, 100 mM HEPES and 10% 0.3 N HCl and 2 mM βME. Notice there are no peaks in the black circle. Panel B shows the spectrum of n-Valeramide at 7.8 μg/mL, panel C shows the spectrum of n-Valeramide at 30 μg/mL, and panel D shows the spectrum of n-Valeramide at 125 μg/mL. Note that there is no variation in retention time; all peaks are at 7.2 minutes. ............................................................................................ 157 Figure 3-15: Typical HPLC spectra for blank and kinetics analysis with 0.625 mM n-Valeronitrile at time points 40 and 60 min. The x-axis is time in minutes and the y-axis is absorbance at 210 nm in mAU. In panels A-C, the circled area represents the peak of interest, the product, n-Valeramide. Panel A shows the spectrum for the blank, 100 mM HEPES and 10% 0.3 N HCl and 2 mM βME. Notice there are no peaks in the black circle. Panel B shows the spectrum of the formation of n-Valeramide (approximately 5.0 μg/mL) at 40 min., and panel C shows the spectrum of the formation of n-Valeramide (approximately 9.0 μg/mL) at 60 min. Note that there is no variation in retention time; all peaks are at 7.2 minutes. ................................................................................................. 158 Figure 3-16: Typical HPLC spectra for blank and kinetics analysis with 5.0 mM n-Valeronitrile at time points 40 and 60 min. The x-axis is time in minutes and the y-axis is absorbance at 210 nm in mAU. In panels A-C, the circled area represents the peak of interest, the product, n-Valeramide. Panel A shows the spectrum for the blank, 100 mM HEPES and 10% 0.3 N HCl and 2 mM βME. Notice there are no peaks in the black circle. Panel B shows the spectrum of the formation of n-Valeramide (approximately 12 μg/mL) at 40 min., and panel C shows the spectrum of the formation of n-Valeramide (approximately 19 μg/mL) at 60 min. Note that there is no variation in retention time; all peaks are at 7.2 minutes. ................................................................................................. 159
17

Figure 3-17: Typical HPLC spectra for blank and kinetics analysis with 40 mM n-Valeronitrile at time points 40 and 60 min. The x-axis is time in minutes and the y-axis is absorbance at 210 nm in mAU. In panels A-C, the circled area represents the peak of interest, the product, n-Valeramide. Panel A shows the spectrum for the blank, 100 mM HEPES and 10% 0.3 N HCl and 2 mM βME. Notice there are no peaks in the black circle. Panel B shows the spectrum of the formation of n-Valeramide (approximately 25 μg/mL) at 40 min., and panel C shows the spectrum of the formation of n-Valeramide (approximately 19 μg/mL) at 60 min. Note that there is no variation in retention time; all peaks are at 7.2 minutes. ................................................................................................. 160 Figure 3-18: Sample plot for the calculation of K and V using Solver in Excel. The measured curves are shown in pink and the calculated curves are shown in blue. For this curve, the K was calculated to be 6.73 mM and V was calculated to be 0.939 μg/mL/min. The sum of squares was calculated to be .0043.
M max
M max
......................................... 161 Figure 3-19: pH profile for wild type ppNHase. pH is plotted in the x-axis and k (min ) is plotted on the y-axis. pH values tested were 5.8, 6.7, 7.2, 7.5 and 8.5. All measurements were made in 100 mM HEPES and 2 mM βME. Note that there were insufficient data points collected in the pH range 5.5 to 6.7, so the inflection point was approximated from the literature. Error bars are shown and represent variability in the measurements (i.e. one standard deviation above and below the mean).
cat-1
....................... 163 Figure 4-1: Superimposed active sites of nitrile hydratase from Pseudomonas putida (Chapter 3) (grey CPK coloring) and Pseudonocardia thermophila (PBD ID: 1IRE ) (magenta CPK coloring). Sphere = cobalt. (P. putida numbering).
1
............................... 186 Figure 4-2: Sequence alignment of four Co-type Nitrile Hydratases (NHase) and four Fe-type NHases. Known functional residues are highlighted in yellow. Residues chosen for second- and third-shell mutations are highlighted in red. refers to Co-type nitrile hydratases; refers to Fe-type nitrile hydratases; refers to the Co-type nitrile hydratase from Pseudomonas putida determined in this thesis from x-ray crystallography.
1
2 3
......... 188 Figure 4-3: Active site of wild type ppNHase (Chapter 3) superimposed with wild type ptNHase (PDB ID: 1IRE ) including second- and third-shell residues chosen for mutation. Active site residues for P. putida are shown in grey CPK coloring; active site residues for P. thermophila are shown in magenta CPK coloring. The residues chosen for site-directed mutagenesis studies for P. putida are shown in light blue CPK coloring, and the residues chosen for site-directed mutagenesis studies for P. thermophila are shown in dark blue CPK coloring. The selected residues for mutation are highlighted with red circles for clarity. Pink spheres = cobalt. (P. putida numbering).
1
.................................. 189 Figure 4-4: CD spectrum comparing wild type ppNHase to the αAsp164Asn mutant. The curves superimpose well indicating the protein is folded correctly................................ 190 Figure 4-5: Typical standard curve for kinetics experiments. R values were always greater than 0.99.
2
............................................................................................................. 191
18

Figure 4-6: Lineweaver-Burk plot for wild type ppNHase at pH 6.7. This plot shows a straight line, with a K of 1.86 mM and V of 0.908. Note that this method has greater error than the nonlinear regression used in this thesis and therefore there are differences between the kinetics constants from this plot and those in Table 3-3.
M max
25......................... 192 Figure 4-7:A-F MM curves for wild type and all five mutant proteins at pH 6.7. Error bars are shown and represent variability in the measurements (i.e. one standard deviation above and below the mean). (n=3) for wild type and all five mutants. .......................... 195 Figure 4-8: pH profile for WT and mutant ppNHase proteins. pH is plotted in the x-axis and k (min ) is plotted on the y-axis. pH values tested were 5.8, 6.7, 7.2, 7.5 and 8.5. All measurements were made in 100 mM HEPES and 2 mM βME. Note that there were insufficient data points collected in the pH range 5.5 to 6.7, so the inflection point was approximated from the literature. Error bars are shown and represent variability in the measurements (i.e. one standard deviation above and below the mean).
cat-1
....................... 196 Figure 4-9: Expanded view for four of the ppNHase mutant enzymes, α Asp164Asn, β Glu56Gln, β His71Leu and β Tyr215Phe. pH is plotted in the x-axis and k (min ) is plotted on the y-axis. pH values tested were 5.8, 6.7, 7.2, 7.5 and 8.5. All measurements were made in 100 mM HEPES and 2 mM βME. The symbols and colors are the same as in Figure 4-8 for clarity. Error bars are shown and represent variability in the measurements (i.e. one standard deviation above and below the mean).
cat-1
....................... 197 Figure 4-10: (A) Active site of wild type ppNHase. (B) Active site of αGlu168Gln ppNHase. In the mutant structure, residue 168 has flipped out of salt bridge distance of βArg52 and forms an H-bond with the backbone oxygen atom of βVal169. (purple sphere = cobalt). ......................................................................................................................... 203 Figure 4-11: (A) Active site of wild type ppNHase. (B) Active site of βGlu56Gln ppNHase. Wild type and mutant structures are essentially the same. (purple sphere = cobalt, red sphere = water).............................................................................................. 205 Figure 4-12: (A) Active site of wild type ppNHase. (B) Active site of βHis71Leu ppNHase. Wild type and mutant structures are essentially the same, with a slight movement in one of the waters (w1). (purple sphere = cobalt, red sphere = water). ..... 207 Figure 4-13: (A), (C) Active site of wild type ppNHase. (B), (D) Active site of βTyr215Phe ppNHase. Wild type and mutant structures are essentially the same in panels A and B. However, panels C and D show a lengthening in the salt bridge distance between αGlu168 and βArg52, shown as red dotted lines. (purple sphere = cobalt). .... 210 Figure A-1: A ribbon diagram of 14-3-3 sigma (PBD ID: 1YZ5 ). The THEMATICS predicted residues for the known catalytic and/or binding residues are shown in green CPK coloring, while the THEMATICS predicted residues for the dimer interface are shown in pink CPK coloring. Note there are two sites colored green, one for each subunit.
15
......................................................................................................................................... 238
19

Figure A-2: Surface view of the dimer interface predicted by THEMATICS for 14-3-3σ.......................................................................................................................................... 239 Figure A-3: Representative compounds identified through molecular docking for 14-3-3 σ from the Zinc database (http://zinc.docking.org/). All compounds identified are drug-like compounds. .............................................................................................................. 239 Figure A-4: Crystal structure of human HER2 labeled by domain (PDB ID: 1N8Z ). (A) Crystal structure of human HER2 without Herceptin (magenta). Domains I-IV are labeled. (B) Crystal structure of human HER2 (magenta) complexed with Herceptin (green and blue). Domains I-IV are labeled as is the Herceptin antibody.
12
..................... 242 Figure A-5: Surface display of HER2 (PDB ID: 1N8Z ) (magenta = ECD HER2, blue and green = Herceptin). Arrows point to the two THEMATICS predicted sites (site 1, blue and site 2, grey), and the known antibody binding site in red.
12
............................... 243 Figure A-6: Representative set of compounds identified through molecular docking for site 1 for human HER2 from zinc database of drug-like compounds (http://zinc.docking.org/). ............................................................................................... 245 Figure A-7: Representative set of compounds identified through molecular docking for site 2 for human HER2 from zinc database of drug-like compounds (http://zinc.docking.org/). ............................................................................................... 246 Figure A-8: Representative small molecules docked into site 1. Left panel = zinc ID # 331908, Right panel = zinc ID # 1231760...................................................................... 247 Figure A-9: Representatives small molecules docked into site 2. Left panel = zinc ID # 218583, Right panel = zinc ID # 1302657...................................................................... 247
20
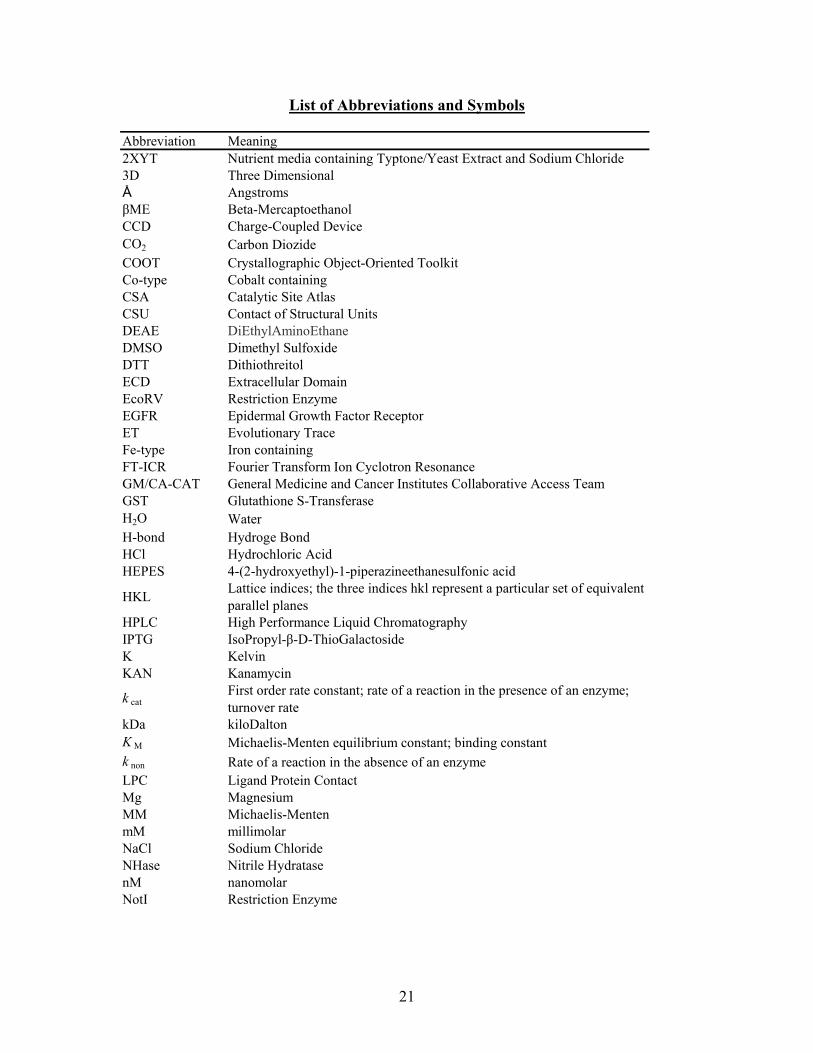
List of Abbreviations and Symbols
Abbreviation Meaning2XYT Nutrient media containing Typtone/Yeast Extract and Sodium Chloride3D Three DimensionalÅ AngstromsβME Beta-MercaptoethanolCCD Charge-Coupled DeviceCO2 Carbon DiozideCOOT Crystallographic Object-Oriented ToolkitCo-type Cobalt containingCSA Catalytic Site AtlasCSU Contact of Structural UnitsDEAE DiEthylAminoEthaneDMSO Dimethyl SulfoxideDTT DithiothreitolECD Extracellular DomainEcoRV Restriction EnzymeEGFR Epidermal Growth Factor ReceptorET Evolutionary TraceFe-type Iron containingFT-ICR Fourier Transform Ion Cyclotron Resonance GM/CA-CAT General Medicine and Cancer Institutes Collaborative Access TeamGST Glutathione S-TransferaseH2O WaterH-bond Hydroge BondHCl Hydrochloric AcidHEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HKLLattice indices; the three indices hkl represent a particular set of equivalent parallel planes
HPLC High Performance Liquid ChromatographyIPTG IsoPropyl-β-D-ThioGalactoside K KelvinKAN Kanamycin
k catFirst order rate constant; rate of a reaction in the presence of an enzyme; turnover rate
kDa kiloDaltonK M Michaelis-Menten equilibrium constant; binding constantk non Rate of a reaction in the absence of an enzymeLPC Ligand Protein ContactMg MagnesiumMM Michaelis-MentenmM millimolarNaCl Sodium ChlorideNHase Nitrile HydratasenM nanomolarNotI Restriction Enzyme
21

NP NanoparticleºC degrees CelciusPBD Protein Data BankPBS Phosphate Buffered SalinePCR Polymerase Chain ReactionpDEST 17 Ampicillin resistant 6-His tag vectorpDEST15 Ampicillin resistant GST-tag vectorPEG Poly(ethylene glycol)pENTR/TEV/D-TOPO
KAN resistant Entry Vector that contains a Tobacco Etch Virus (TEV) recognition site for TEV protease dependant cleavage
PHENIX Python-based Hierarchical EnviroNment for Integrated XtallographyppNHase Nitrile Hydratase from Pseudomonas putidaptNHase Nitrile Hydratase from Pseudonocardia thermohpilaPVDF Polyvinylidene FluorideR cryst ∑║Fobs│- │Fcalc║ / ∑│Fobs│REFMAC Crystallography refinement tool
R freecalculated same as Rcryst but for a test set comprising reflections mot used in the refinement
RIPL Receptor Interacting ProteinR merge ∑ │I - <I>│ / ∑ <I>RMSD Root Mean Square DeviationSDS-PAGE Sodium Sodecyl Sulfate Polyacrylamide Gel Electrophoresis
SNAPStatistical de-isotope algorithm, makes use of signal/noise and goodness threshold values
STE buffer Sodium Chloride–Tris–ethylenediaminetetraacetic acid (EDTA)THEMATICS Theoretical Microscopic Titration CurvesTris Buffer, 2-Amino-2-hydroxymethyl-propane-1,3-diol
Vmax maximum enzyme velocity; velocity at maximum substrate concentration
Zn Zincα Chain A in a multimeric proteinβ Chain B in a multimeric proteinμM micromolar
22

Chapter 1
Introduction
23

1.1 Why the Need for Enzymes?
In the absence of enzymes, biological reactions take place very slowly, if at all.1 This
slow progress of reactions in the absence of some sort of catalyst provides a standard by
which the catalytic power of existing enzymes can be compared. However, most
biological reactions proceed so slowly in the absence of an enzyme that their uncatalyzed
rates in water have never been measured. Examples of rate constants and half-lives of
biological reactions proceeding spontaneously in water in the absence of enzyme are
shown in Figure 1-1.1 Recent experiments have shown that many reactions which
proceed spontaneously in solution in the absence of a catalyst can be increased by
temperature. The basic rule of thumb has been that reactions double in rate for every 10
ºC increase in temperature.2 It has recently been shown that reactions can actually
increase by a factor of 10 or more for every 10 ºC increase in temperature. For example,
the decarboxylation of orotidine 5'-phosphate increased by a factor of 12.5 as the
temperature increased from 20 to 30 ºC.3 This allows the study of biological reactions in
neutral solutions in sealed tubes at high temperatures, using the Arrhenius plot to
extrapolate to room temperature.1
24

Figure 1-1: Rate constants and half-lives of biological reactions proceeding spontaneously in water in the absence of enzyme.1
25
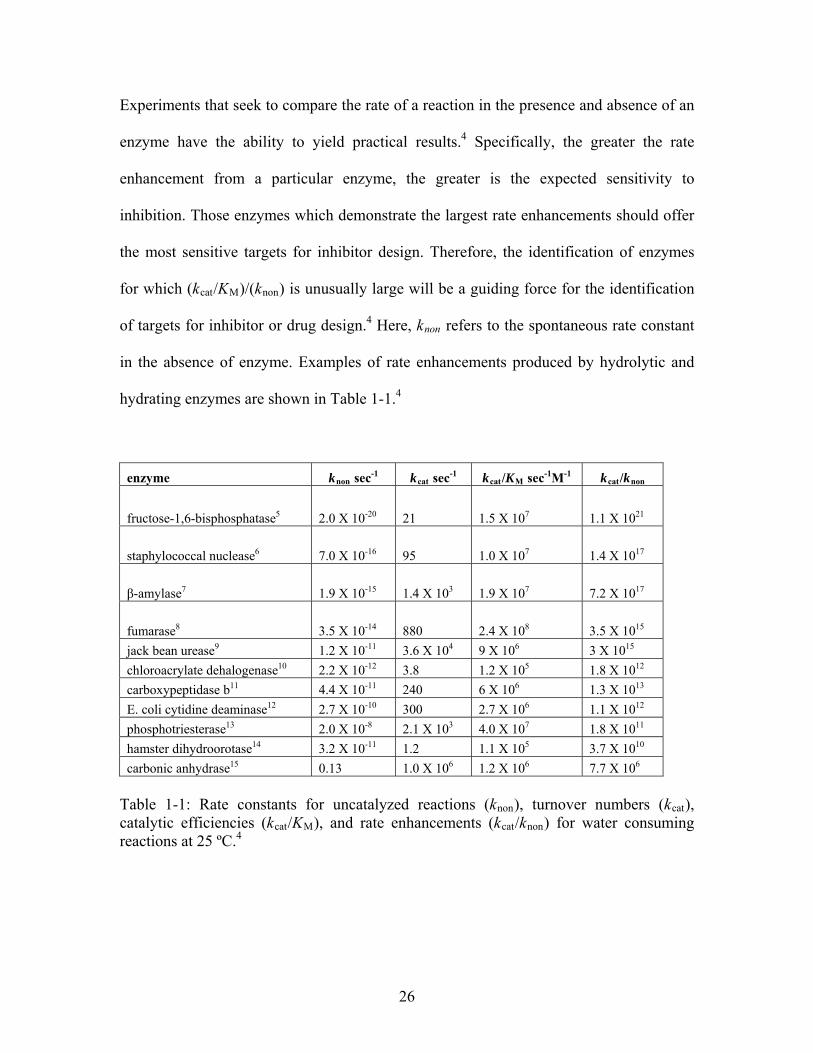
Experiments that seek to compare the rate of a reaction in the presence and absence of an
enzyme have the ability to yield practical results.4 Specifically, the greater the rate
enhancement from a particular enzyme, the greater is the expected sensitivity to
inhibition. Those enzymes which demonstrate the largest rate enhancements should offer
the most sensitive targets for inhibitor design. Therefore, the identification of enzymes
for which (kcat/KM)/(knon) is unusually large will be a guiding force for the identification
of targets for inhibitor or drug design.4 Here, knon refers to the spontaneous rate constant
in the absence of enzyme. Examples of rate enhancements produced by hydrolytic and
hydrating enzymes are shown in Table 1-1.4
enzyme knon sec-1 kcat sec-1 kcat/KM sec-1M-1 kcat/knon
fructose-1,6-bisphosphatase5 2.0 X 10-20 21 1.5 X 107 1.1 X 1021
staphylococcal nuclease6 7.0 X 10-16 95 1.0 X 107 1.4 X 1017
β-amylase7 1.9 X 10-15 1.4 X 103 1.9 X 107 7.2 X 1017
fumarase8 3.5 X 10-14 880 2.4 X 108 3.5 X 1015
jack bean urease9 1.2 X 10-11 3.6 X 104 9 X 106 3 X 1015
chloroacrylate dehalogenase10 2.2 X 10-12 3.8 1.2 X 105 1.8 X 1012
carboxypeptidase b11 4.4 X 10-11 240 6 X 106 1.3 X 1013
E. coli cytidine deaminase12 2.7 X 10-10 300 2.7 X 106 1.1 X 1012
phosphotriesterase13 2.0 X 10-8 2.1 X 103 4.0 X 107 1.8 X 1011
hamster dihydroorotase14 3.2 X 10-11 1.2 1.1 X 105 3.7 X 1010
carbonic anhydrase15 0.13 1.0 X 106 1.2 X 106 7.7 X 106 Table 1-1: Rate constants for uncatalyzed reactions (knon), turnover numbers (kcat), catalytic efficiencies (kcat/KM), and rate enhancements (kcat/knon) for water consuming reactions at 25 ºC.4
26

In many cases, it is possible to carry out reactions at a faster rate in the absence of an
enzyme by using either harsh conditions (acids or bases) or metal catalysts. However, in
these cases, the desired product may not be produced, or the reaction still may not
proceed at a rate comparable to the enzyme catalyzed reaction. One example involves the
conversion of nitriles to amides. Hydrolysis of nitriles to amides is important not only in
the laboratory but also has industrial applications.16 Currently, about 30 kilotons of
acrylamide are produced each year using the enzyme nitrile hydratase.17 The use of nitrile
hydratase for this reaction is one of the most successful applications of “green
chemistry”. Producing acrylamide this way bypasses many issues associated with the
chemical production of acrylamide, including higher costs and more side products such
as acrylic acid and polymerized acrylamide. Due to its industrial importance, the
production of amides has been the focus of many studies. It has been shown that the
uncatalyzed reaction has a half-life of approximately 106 hours.16 The reaction can be
catalyzed by various acids and bases, but these methods require harsh conditions and give
low yields. Additionally, further hydrolysis of an amide to carboxylic acid and ammonia
cannot be avoided because the reaction is faster than hydration.16 Therefore, this is an
undesirable route for the formation of amides. These harsh conditions can be avoided
through the use of metal catalysts, which also have the advantage of being highly
selective. In the case of the production of amides with metal catalysts, the reaction
conditions are such that the carboxylic acid will not be formed. Specifically, a palladium
(II) catalyst has been used for the production of acrylamide with an observed rate of 0.60
h-1 in water.16 While the reaction did in fact proceed with the use of the palladium
catalyst, the rate was substantially lower than that observed with the enzyme catalyzed
27

reaction (specific activity 76 U/ml).17 This provides an interesting example where the
uncatalyzed reaction of a nitrile to an amide will not proceed, but the enzyme catalyzed
reaction using nitrile hydratase allows for the large scale production of an industrial
product.
1.2 Background on Second- and Third-shell Residue Involvement in Catalysis
One of the most fundamental questions in biochemistry today is how enzymes work.
Most often this discussion focuses on the amino acid residues in direct contact with the
reactive metal or reacting substrate within the three-dimensional (3D) structure of the
protein. What is very rarely mentioned is the influence that remote residues have on
enzyme catalysis. Remote residues refer to those residues which are, or are farther from,
second-nearest neighbors to the reactive metal or reacting substrate molecule. In this
study, these second or even third nearest neighbors are called second- and third-shell
residues. The literature has scarce information pertaining to the importance of these
second- and third-shell residues in enzyme catalysis. The idea of the involvement of
residues located in outer coordination spheres in catalysis was first introduced by
Leatherbarrow, Fersht and Winter when the concept of site-directed mutagenesis was
introduced.18 It was discovered that mutations made to residues located far from the
reaction site resulted in proteins with reduced catalytic rate. In some cases; however,
these mutations resulted in proteins whose catalytic rate was increased. It was at this time
that the term ‘protein engineering’ was coined.19
Enzymes have evolved through time to be powerful biocatalysts with a high degree of
specificity and fast catalytic rate.20 These characteristics have allowed enzymes to be
28

used in industrial processes, where they often perform better than man-made catalysts.
However, stability issues and the production of unwanted by-products have limited the
scope of their industrial applications. In order for enzymes to reach their full potential as
industrial biocatalysts, efforts are underway to improve them through protein
engineering, using both rational-protein design and directed evolution techniques.21 The
concept of protein engineering has been around for 25 years, and was first used on
tyrosyl-tRNA synthetase and β-lactamase.22-24 Site-directed mutagenesis allows the
substitution of specific amino acids in proteins and has been the guiding force for truly
understanding protein structure-function relationships. Rational protein design takes
advantage of the 3D structural information about proteins obtained through x-ray
crystallography or homology modeling, while directed evolution relies solely on the
principles of mutation and selection without regard to protein structure-function
relationships. While both methods have proved successful, each has advantages and
disadvantages.
1.3 Directed Evolution
Mutations that Affect Activity
One of the advantages of directed evolution techniques is the ability to identify
functionally important residues that are not necessarily obvious from the 3D structure,
particularly residues not in direct contact with the substrate or inhibitor. Those residues in
direct contact with the ligand may be thought of as the first shell of the protein’s site of
interaction. Residues that are not in the first shell but are in direct contact with one or
more first-shell residues may then be thought of as second-shell residues, and so on. For
29

present purposes, residues in the second shell and beyond are called remote residues. The
activity of human carbonic anhydrase II on the ester substrate 2-naphthyl acetate was
increased 40-fold through three rounds of mutagenesis, selection and recombination.25
Specifically, a mutant containing three amino acid substitutions at positions Ala65,
Asp110 and Thr200 was reported. Ala65 is a remote residue adjacent to His64, the proton
shuttle residue; Thr200 is a known coordinating ligand to CO2; and Asp110 is a surface
residue. The single Ala65Val mutation resulted in a 3-fold increase in activity, the single
Thr200Ala mutation resulted in a 10-fold increase in catalysis and the single Asp110Asn
had no impact on catalysis. However, the triple mutation showed an additive effect on
catalysis.
The flavoenzyme vanillyl-alcohol oxidase was subjected to random mutagenesis to
generate mutants with enhanced reactivity to creosol (2-methoxy-4-methylphenol).
Specifically, four mutants were identified with a 40-fold increase in catalytic rate where
the point mutations were located outside of the presumptive active site. X-ray crystal
structures of both wild type and mutant proteins demonstrated that this altered efficiency
was not due to mis-folded protein as all structures were superimposable. Finally, a mutant
metallo-β-lactamase was discovered through directed evolution that resulted in an
enzyme with increased hydrolytic efficiency toward cephalexin.26 This mutant contained
four amino acid substitutions, two in the second coordination sphere of the metal ion, and
two far removed from the annotated active site.
30

Mutations that Affect Specificity
Examples of remote residue involvement in specificity have been reported in the directed
evolution literature.21 In the directed evolution of E. coli D-sialic acid aldolase to L-3-
deoxy-manno-2-octulosonic acid aldolase, changes in eight amino acids, all of which are
located outside the first-shell, were necessary to produce a mutant enzyme with a 1000-
fold increase in specificity for the unnatural sugar substrate, L- D-3-deoxy-manno-2-
octulosonic acid.27 Directed evolution techniques were also used to alter the specificity of
aspartate aminotransferase.28 A mutant enzyme containing 17 amino acid substitutions
resulted in a 2.1 X 106-fold increase in the catalytic rate for the non-native valine
substrate.29 Interestingly, only one of the mutated residues was in contact with the
substrate; all others were remote residues.
1.4 Rational Protein Design
Mutations that Affect Activity
Much attention has been paid to metalloenzymes using rational protein design methods,
focusing on metal coordinating ligands and residues located in the second shell around
the metal ion, i.e. residues which are H-bonded to the metal coordinating residues. In the
metalloenzymes alkaline phosphatase (AP) and mandelate racemase (MR), distinctive
patterns have been observed in both the first, (i.e. directly coordinating the metal) second,
and third layers of residues around the metal ions, suggesting that second- and third-shell
residues are important to the chemical properties of the metal ion.30,31 Mutations of single
residues in both the second and third shells in AP have been demonstrated to both
decrease or increase catalytic rates, depending on the mutation21-24. While in MR, a major
31

change in catalytic rate was observed for enzyme containing a second-shell mutation.
Asp270 is a second-shell residue that forms hydrogen bonds with the catalytic His297.
The single, conservative mutation of Asp270 to Asn results in a 104-fold decrease in
enzyme activity compared to wild type for both (R)- and (S)- mandelate substrates32.
These results suggest that second-shell residues are important in catalysis, at least in
some cases.30,31 Clearly, residues distant from the active site can have an important effect
on catalysis and should be considered in enzyme design.
Mutations that Affect Specificity
Outside of the directed evolution literature, studies on second-shell residues are limited.
Some second-shell point mutations have been made through rational protein design
techniques to better understand the functionality of the proteins of interest. An early
example of this is found in the efforts to impart chymotrypsin specificity onto trypsin.
The active sites of these two proteases are nearly identical, but trypsin cleaves the peptide
bond on the C-terminal side of positively charged residues (Arg, Lys) and chymotrypsin
cleaves on the C-terminal side of hydrophobic residues (Tyr, Phe, Trp, Met, Leu).
Asp189 in the binding pocket of trypsin was thought to be responsible for the recognition
of positively charged residues; however, the rational mutation at this single site was
insufficient to confer chymotrypsin specificity. Rather, 16 mutations were required to
engineer chymotrypsin specificity onto trypsin, including residues not in direct contact
with the substrate.33-35
32

1.5 Disadvantages of Directed Evolution and Rational Protein Design Methods
While both directed evolution and rational-protein design techniques have been
successful in identifying important residues located outside of the active site, these
techniques are not simple enough to be used to study proteins broadly. Directed evolution
can be time consuming, requires a high-throughput selection method to be feasible, and
relies on sufficient sampling of sequence space to yield positive results.2 In rational-
protein design methods, the correct identification of residues located outside the active
site that may affect protein function based solely on structure-function relationships poses
a difficult problem. In the absence of some form of guidance, there are just too many
residues outside the active site to consider in the design of an enzyme with altered or
improved function. Therefore, the development of computational approaches that are able
to identify important second-shell residues would prove valuable in protein design efforts
and may yield new information about how enzymes catalyze reactions with such
efficiency and specificity.
1.6 Computational Approaches to the Identification of Functional Residues
Evolutionary Trace (ET) is a sequence alignment-based method used to identify residues
that are statistically likely to be under some form of evolutionary pressure, and therefore
are considered structurally or functionally important.36-39 The ET method consistently
identifies not only the first-shell residues but also many more residues outside of the first-
shell of the active site. Thus, ET clusters are generally quite large and, while correctly
indentifying active site residues with a high success rate, precision tends to be very low
with high false positive rates. Rational techniques may benefit further from more precise
33

computational methods that identify smaller clusters of residues that are more tractable
for mutagenesis experiments. THEMATICS (THEoretical Microscopic TItration CurveS)
is an electrostatics-based method that utilizes only the 3D structure and no sequence
information to identify active site residues; catalytically important residues are identified
by their perturbed theoretical titration curves.33-37 Identified residues have been
demonstrated to be reliable predictors of annotated active sites, and since THEMATICS
is quite selective, these clusters tend to be much smaller. It has recently been suggested
that THEMATICS may be able to identify important second shell residues.40
1.7 Overview of Thesis
While limited studies have been performed to understand the role of remote residues in
enzyme catalysis, a thorough investigation of the importance of second- and third-shell
residues has not been performed. In this thesis, two very different computational
methods, THEMATICS33-37 and Evolutionary Trace (ET)36-39, will be used to identify
functionally important residues in the first-, second- and third-shells of an enzyme. Due
to the large number of residues in these shells, we chose to use computational methods in
an attempt to focus on those residues which are theoretically predicted to be important.
Once the concept of remote residue involvement in enzyme catalysis has been
introduced, the focus will shift to one particular enzyme, Co-type nitrile hydratase from
Pseudomonas putida, for which both THEMATICS and ET predict a multilayer active
site. A kinetic analysis of single point mutations will be presented for five second- and
third-shell residues that were predicted computationally to be functionally important.
Additionally, crystal structures will be presented for four of the mutants. This work does
34

not unequivocally explain why these residues in the outer coordination spheres influence
catalysis, but makes a strong argument for the concept that enzyme active sites are built
in multiple layers. It will be suggested that computational approaches, and the concept of
multilayer nanoscale active sites introduced herein, can help to guide protein engineering
efforts.
1.8 Thesis Chapters
Chapter 2 - Evidence for Remote Residue Involvement in Catalysis; Are Enzyme Active
Sites Built in Multiple Layers?
In chapter 2, the predictions of THEMATICS and ET are examined to identify residues
predicted to be important in the first-, second-, and third-shells for a test set of 39
metallo- and non-metalloenzymes. For this study, first-shell refers to those residues in
direct contact with a bound substrate or metal ion; second-shell residues are those
residues in direct contact with first-shell residues; third-shell refers to those residues in
direct contact with second-shell residues. The residues identified by these methods are
compared with experimental mutagenesis data from the literature. These results show that
both THEMATICS and ET predict functionally important residues not only in the first-
shell of an interaction site, but also residues located in interaction spheres beyond the
first-shell. Using data obtained from the literature, we find that those residues identified
by THEMATICS and ET in the second and third interaction spheres, for a few cases, are
reported to have substantial effects on protein function. This study suggests that a
combination of computational tools, including THEMATICS, may be used to guide the
rational study of second- and third-shell residues with respect to protein function.
35

Chapter 3 - Structural and Kinetic Analysis of Wild Type Co-type Nitrile Hydratase from
Pseudomonas putida
In chapter 3, the first known structure of the enantioselective Co-type nitrile hydratase
from Pseudomonas putida NRRL-18668 (ppNHase) is presented to 2.1 Å, in addition to a
full kinetic analysis of the wild type protein at five different pH values. This chapter will
provide a comprehensive overview of both Co-type and Fe-type nitrile hydratases,
including experimental mutations made. Additionally, a brief introduction to Michaelis-
Menten kinetics will be presented.
Chapter 4 - Evidence for Participation of Remote Residues in the Catalytic Activity of
Co-type Nitrile Hydratase from Pseudomonas putida – A Crystal Structure and Kinetic
Analysis
In chapter 4, a systematic approach to the mutation of second- and third-shell residues
specifically in hopes of understanding their role in enzyme catalysis is undertaken. In this
chapter, the enzymatic effect of five second- and third-shell mutants predicted by
THEMATICS and ET for Co-type nitrile hydratase from Pseudomonas putida are
reported. The mutations include αAsp164Asn, αGlu168Gln, βGlu56Gln, βHis71Leu,
βTyr215Phe (P. putida numbering) where α and β designate the two subunits of the
protein. It will be demonstrated experimentally through site-directed mutagenesis studies
that these second- and third-shell residues, predicted theoretically by THEMATICS and
ET, are functionally important with each one contributing to the catalytic rate of this
protein. In addition, the crystal structures of four of the mutants, αGlu168Gln,
36

βGlu56Gln, βHis71Leu, βTyr215Phe (P. putida numbering), are presented. The kinetic
analysis of these mutants versus wild type will demonstrate the functional importance of
second- and third-shell residues on catalysis for ppNHase. The kinetic analysis alone was
not sufficient to explain why the decreased catalytic rates were observed. It was
suggested in chapter 3 that there could be numerous reasons why these second- and third-
shell mutations affect catalytic rate and include 1) local rotations or side chain shifts, 2)
shifts in hydrogen-bonding (H-bonding) networks, 3) changes in the electric field in the
active site, and/or 4) quantum mechanical (QM) effects. This chapter, focusing on the
crystal structures, may help explain the catalytic effects through structural changes.
Chapter 5 - Conclusions and Future Work
This thesis presents a systematic approach to computationally identifying functional
residues located in the outer coordination spheres of enzymes (i.e. beyond the active site
or first-shell). What is most striking is that two completely different types of theoretical
methods both support multilayer active sites. Additionally, experimental mutagenesis was
performed on the enzyme Co-type nitrile hydratase from Pseudomonas putida. Kinetic
and crystallographic studies both support the concept of multilayer active sites.
Understanding how nature designs enzyme active sites is a fundamental question in
enzymology with implications for protein engineering. The present results suggest that
computational methods could help guide the identification of functionally important
second- and/or third-shell residues and can serve as a useful guide for rational protein
design studies.
37

The present work has lead to new collaborations in protein engineering and a new project
funded by the National Science Foundation to continue the investigation of the
importance of remote residues in enzyme catalysis. These new projects are described
briefly in the concluding chapter.
Appendix 1 - Computationally Guided Protein-Specific Labeling with Nanoparticles – A
Test Case Using Her2
The opportunity to work on a nanomedicine related project stemmed from my fellowship
as an IGERT trainee. Since this work was not related to remote residue involvement in
enzyme catalysis, I have chosen to include this work as Appendix 1. This work focused
on the use of THEMATICS to predict previously unidentified binding sites for disease
marker proteins of known 3D structure. Following the identification of a few candidate
proteins, 14-3-3 σ41 and the extracellular domain of HER242, approximately 100,000
compounds from the zinc database (http://zinc.docking.org/) were docked into the
predicted sites to identify a set of small molecule candidates that may bind specifically to
the targets. After careful analysis of both systems, we chose to continue work with
HER2. The project is now at the point to begin experimental work to test these identified
small molecules for affinity to the target protein. While only the first stages of this project
have been completed, it has been brought to a point where a future student could continue
the work. The concept holds promise as a novel medical diagnostic methodology and as a
new approach to targeted drug delivery.
38

1.9 References 1. Wolfenden, R. (2003). Thermodynamic and extrathermodynamic requirements of
enzyme catalysis. Biophys Chem 105, 559-572. 2. Harcourt, A. V. (1867). On the Observation of the course of Chemical Change. J.
Chem. Soc. 20, 460-492. 3. Radzicka, A. & Wolfenden, R. (1995). A proficient enzyme. Science 267, 90-93. 4. Wolfenden, R. (2006). Degrees of difficulty of water-consuming reactions in the
absence of enzymes. Chem Rev 106, 3379-3396. 5. Kelley, N., Giroux, E. L., Lu, G. & Kantrowitz, E. R. (1996). Glutamic acid
residue 98 is critical for catalysis in pig kidney fructose-1,6-bisphosphatase. Biochem Biophys Res Commun 219, 848-852.
6. Serpersu, E. H., Shortle, D. & Mildvan, A. S. (1987). Kinetic and magnetic resonance studies of active-site mutants of staphylococcal nuclease: factors contributing to catalysis. Biochemistry 26, 1289-1300.
7. Balls, A. K., Walden, M. K. & Thompson, R. R. (1948). A crystalline beta-amylase from sweet potatoes. J Biol Chem 173, 9-19.
8. Brant, D. A., Barnett, L. B., & Alberty, R. A. (1963). The Temperature Dependence of the Steady State Kinetic Parameters of the Fumarase Reaction. J. Am. Chem. Soc. 85, 2204-2209.
9. Laidler, K. J., & Hoare, J. P. (1950). The Molecular Kinetics of the Urea-Urease System. III. Heats and Entropies of Complex Formation and Reaction. J. Am. Chem. Soc. 72, 2489-2494.
10. Horvat, C. M. & Wolfenden, R. V. (2005). A persistent pesticide residue and the unusual catalytic proficiency of a dehalogenating enzyme. Proc Natl Acad Sci U S A 102, 16199-16202.
11. Radzicka, A., & Wolfenden, R. J. (1996). Rates of Uncatalyzed Peptide Bond Hydrolysis in Neutral Solution and the Transition State Affinities of Proteases. J. Am. Chem. Soc. 118, 6105-6109.
12. Snider, M. J., Gaunitz, S., Ridgway, C., Short, S. A. & Wolfenden, R. (2000). Temperature effects on the catalytic efficiency, rate enhancement, and transition state affinity of cytidine deaminase, and the thermodynamic consequences for catalysis of removing a substrate "anchor". Biochemistry 39, 9746-9753.
13. Dumas, D. P., Caldwell, S. R., Wild, J. R. & Raushel, F. M. (1989). Purification and properties of the phosphotriesterase from Pseudomonas diminuta. J Biol Chem 264, 19659-19665.
14. Huang, D. T., Kaplan, J., Menz, R. I., Katis, V. L., Wake, R. G., Zhao, F., Wolfenden, R. & Christopherson, R. I. (2006). Thermodynamic analysis of catalysis by the dihydroorotases from hamster and Bacillus caldolyticus, as compared with the uncatalyzed reaction. Biochemistry 45, 8275-8283.
15. Steiner, H., Jonsson, B. H. & Lindskog, S. (1975). The catalytic mechanism of carbonic anhydrase. Hydrogen-isotope effects on the kinetic parameters of the human C isoenzyme. Eur J Biochem 59, 253-259.
16. Kaminskaia, N. V. K., N. M. (1996). Nitrile hydration catalyzed by palladium(II) complexes. Dalton Trans., 3677-3686.
39

17. Kobayashi, M., Nagasawa, T. & Yamada, H. (1992). Enzymatic synthesis of acrylamide: a success story not yet over. Trends Biotechnol 10, 402-408.
18. Leatherbarrow, R. J., Fersht, A. R. & Winter, G. (1985). Transition-state stabilization in the mechanism of tyrosyl-tRNA synthetase revealed by protein engineering. Proc Natl Acad Sci U S A 82, 7840-7844.
19. Brannigan, J. A. & Wilkinson, A. J. (2002). Protein engineering 20 years on. Nat Rev Mol Cell Biol 3, 964-970.
20. Kaur, J. & Sharma, R. (2006). Directed evolution: an approach to engineer enzymes. Crit Rev Biotechnol 26, 165-199.
21. Johannes, T. W. & Zhao, H. (2006). Directed evolution of enzymes and biosynthetic pathways. Curr Opin Microbiol 9, 261-267.
22. Winter, G., Fersht, A. R., Wilkinson, A. J., Zoller, M. & Smith, M. (1982). Redesigning enzyme structure by site-directed mutagenesis: tyrosyl tRNA synthetase and ATP binding. Nature 299, 756-758.
23. Sigal, I. S., Harwood, B. G. & Arentzen, R. (1982). Thiol-beta-lactamase: replacement of the active-site serine of RTEM beta-lactamase by a cysteine residue. Proc Natl Acad Sci U S A 79, 7157-7160.
24. Dalbadie-McFarland, G., Cohen, L. W., Riggs, A. D., Morin, C., Itakura, K. & Richards, J. H. (1982). Oligonucleotide-directed mutagenesis as a general and powerful method for studies of protein function. Proc Natl Acad Sci U S A 79, 6409-6413.
25. Gould, S. M. & Tawfik, D. S. (2005). Directed evolution of the promiscuous esterase activity of carbonic anhydrase II. Biochemistry 44, 5444-5452.
26. Tomatis, P. E., Rasia, R. M., Segovia, L. & Vila, A. J. (2005). Mimicking natural evolution in metallo-beta-lactamases through second-shell ligand mutations. Proc Natl Acad Sci U S A 102, 13761-13766.
27. Hsu, C.-C., Hong, Z., Wada, M., Franke, D. & Wong, C.-H. (2005). Directed evolution of D-sialic acid aldolase to L-3-deoxy-manno-2-octulosonic acid (L-KDO) aldolase. Proc Natl Acad Sci USA 102, 9122-9126.
28. Oue, S., Okamoto, A., Yano, T. & Kagamiyama, H. (1999). Redesigning the substrate specificity of an enzyme by cumulative effects of the mutations of non-active site residues. J Biol Chem 274, 2344-2349.
29. van den Heuvel, R. H., van den Berg, W. A., Rovida, S. & van Berkel, W. J. (2004). Laboratory-evolved vanillyl-alcohol oxidase produces natural vanillin. J Biol Chem 279, 33492-33500.
30. Karlin, S., Zhu, Z.-Y. & Karlin, K. D. (1997). The extended environment of mononuclear metal centers in protein structures. Proc Natl Acad Sci USA 94, 14225-14230.
31. Karlin, S. & Zhu, Z.-Y. (1997). Classification of mononuclear zinc metal sites in protein structures. Proc Natl Acad Sci USA 94, 14231-14236.
32. Schafer, S. L., Barrett, W. C., Kallarakal, A. T., Mitra, B., Kozarich, J. W., Gerlt, J. A., Clifton, J. G., Petsko, G. A. & Kenyon, G. L. (1996). Mechanism of the reaction catalyzed by mandelate racemase: structure and mechanistic properties of the D270N mutant. Biochemistry 35, 5662-5669.
33. Graf, L., Craik, C. S., Patthy, A., Roczniak, S., Fletterick, R. J. & Rutter, W. J. (1987). Selective alteration of substrate specificity by replacement of aspartic
40

acid-189 with lysine in the binding pocket of trypsin. Biochemistry 26, 2616-2623.
34. Perona, J. J., Hedstrom, L., Rutter, W. J. & Fletterick, R. J. (1995). Structural origins of substrate discrimination in trypsin and chymotrypsin. Biochemistry 34, 1489-1499.
35. Venekei, I., Szilagyi, L., Graf, L. & Rutter, W. J. (1996). Attempts to convert chymotrypsin to trypsin. FEBS Lett 379, 143-147.
36. Lichtarge, O., Bourne, H. R. & Cohen, F. E. (1996). An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol 257, 342-358.
37. Lichtarge, O., Sowa, M. E. & Philippi, A. (2002). Evolutionary traces of functional surfaces along G protein signaling pathway. Methods Enzymol 344, 536-556.
38. Madabushi, S., Yao, H., Marsh, M., Kristensen, D. M., Philippi, A., Sowa, M. E. & Lichtarge, O. (2002). Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J Mol Biol 316, 139-154.
39. Yao, H., Kristensen, D. M., Mihalek, I., Sowa, M. E., Shaw, C., Kimmel, M., Kavraki, L. & Lichtarge, O. (2003). An accurate, sensitive, and scalable method to identify functional sites in protein structures. J Mol Biol 326, 255-261.
40. Murga, L. F., Ondrechen, M. J. & Ringe, D. (2008). Prediction of Interaction Sites from Apo 3D Structures When the Holo Conformation is Different. Proteins 72, 980-992.
41. Benzinger, A., Popowicz, G. M., Joy, J. K., Majumdar, S., Holak, T. A. & Hermeking, H. (2005). The crystal structure of the non-liganded 14-3-3sigma protein: insights into determinants of isoform specific ligand binding and dimerization. Cell Res 15, 219-227.
42. Cho, H. S., Mason, K., Ramyar, K. X., Stanley, A. M., Gabelli, S. B., Denney, D. W., Jr. & Leahy, D. J. (2003). Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756-760.
43. Ericsson, U. B., Hallberg, B. M., Detitta, G. T., Dekker, N. & Nordlund, P. (2006). Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem 357, 289-298.
44. Velazquez-Campoy, A. & Freire, E. (2006). Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat Protoc 1, 186-191.
45. Okochi, M., Nomura, T., Zako, T., Arakawa, T., Iizuka, R., Ueda, H., Funatsu, T., Leroux, M. & Yohda, M. (2004). Kinetics and binding sites for interaction of the prefoldin with a group II chaperonin: contiguous non-native substrate and chaperonin binding sites in the archaeal prefoldin. J Biol Chem 279, 31788-31795.
41

Chapter 2
Evidence for Remote Residue Involvement in Catalysis; Are Enzyme Active Sites Built in
Multiple Layers?
42

2.1 Introduction
It is commonly assumed that all of the residues participating in enzymatic catalysis and
substrate recognition are in close proximity to the substrate when bound. However, recent
directed evolution and protein engineering studies have suggested that residues more
distant than those in direct contact with the substrate may have a profound effect on both
catalysis and substrate specificity.1,2 In this study, we refer to residues interacting directly
with the substrate as first-shell residues, residues interacting directly with one or more
first-shell residues as second-shell residues, and finally, residues interacting with one or
more second-shell residues as third-shell residues. All residues thus defined as first-,
second- or third-shell belong to what we define as an interaction sphere. To date, no
specific studies on the effects of second- and third-shell residues on catalysis have been
performed. It is likely that such studies have been impeded by the inability to select
candidate residues in the second and third shells that may be important for catalysis.
Indeed the systematic mutagenesis of all second- and third-shell residues in an enzyme
may not be feasible simply because of the sheer number of residues involved in these
shells. Therefore, we have examined the ability of THEMATICS3-6, a structure based
method which identifies functionally important residues based on charge perturbations, to
identify the second- and third-shell residues that may be functionally important in
catalysis. We then compare these results with the sequence-based Evolutionary Trace
(ET) method which identifies residues based on sequence conservation.7-9 While
experimental evidence for the participation of residues outside the first shell is scattered,
the computational and bioinformatics evidence paints a more unified picture. Most
43

strikingly, two completely different types of theoretical methods, which will now be
described, both support multilayer active sites.
THEMATICS4 (THEoretical Microscopic TItration CurveS) is a theoretical
computational approach for the identification of the active sites of proteins, requiring
only the 3D structure as input. THEMATICS calculations are based on predicted titration
curve shapes determined computationally from a Poisson-Boltzmann procedure. Active
site residues are identified by abnormal or “perturbed’ theoretical titration curves. It has
been demonstrated that spatial clusters of these perturbed residues are reliable predictors
of active sites and/or binding sites. Most residues identified by THEMATICS are
documented in the literature as catalytically important or important in substrate binding,
as determined experimentally, principally by site-directed mutagenesis.
Evolutionary Trace Report Method7 (ET) is a computational method which determines
the functional importance of residues in a protein based on the rate of evolution of a
residue at a particular site. A multiple sequence alignment and a phylogenetic tree are
required as input, and evolutionary relationships between sequence homologues are
explored. The Evolutionary Trace (ET) method assigns a “functional importance” score
to each residue through correlation of the evolutionary variability with points of
divergence in the phylogenetic tree. When these scores are placed on the 3D structure of
the protein, the top-scoring residues are observed to cluster together spatially and these
clusters are used to generate functional site predictions.
44

In this study, the ability of THEMATICS (Theoretical Microscopic Titration Curves) to
predict functionally important remote residues is studied by comparing computational
predictions with experimental mutagenesis data from the literature. These results are also
compared with the sequence-based Evolutionary Trace (ET) method. Our results indicate
that both THEMATICS and ET predict functionally important residues located in
interacting spheres beyond the first-shell, and we find that those residues identified by
THEMATICS and ET in the second and third interaction spheres, for a few cases, are
reported to have substantial effects on protein function. Based on the nature of the
methods, ET predictions in all interaction spheres are very large, while THEMATICS
predictions are shown to be highly selective. Although ET identifies a larger number of
residues, not all have been shown to be functionally important. It will be shown that
THEMATICS predicts more specific, precise sites, especially in the first- and second-
shell; many of these residues are known in the literature to be functionally important.
This study suggests that a combination of computational tools, including THEMATICS,
may be used to guide the rational study of second- and third-shell residues with respect to
protein function.
45

2.2 Materials and Methods
Protein Test Set
A total of 39 proteins were included in the test set; 20 metalloenzymes and 19 non-
metalloenzymes. In addition, the proteins represent all six of the enzyme E.C. classes.
This test set includes a variety of quaternary structures. The metalloenzyme structures
include 1EBF, 1NID, 2JCW, 1PUD, 1FUG, 2PHK, 1HXQ, 1ALK, 1AMP, 1MMQ,
1BQQ, 1CA2, 1B57, 1AHJ, 1IRE, 1MNR, 1PYM, 1MUC, 1HGS and 1DGS. The non-
metallo protein structures include 2HDH, 1A4S, 1A4I, 1GET, 1AKM, 1MLA, 1KZL,
1UOK, 9PAP, 1APY, 1DBT, 1QFE, 1AB8, 1B73, 1TPH, 1REQ, 1TYD, 12AS, 1DEA.
Full names and enzyme classes are given in Table 2-1.
Computational Methods
All methods were run as previously described, and default parameters were used, except
where stated. The protein structures used as the input data for all calculations were
downloaded from the Protein Data Bank (PDB, http://www.rcsb.org/pdb/). Coordinates
for all the proteins in the test set were analyzed by Theoretical Microscopic Titration
Curves (THEMATICS )4,5,10-12 using the method of Wei6, except that a cut-off of 0.96
was used. Structures with missing atoms were fixed using swiss-pdb viewer. Substrate,
inhibitor, and water molecules, cofactors, and salts that are co-crystallized with the
proteins were not included in the THEMATICS analysis. In addition, in cases where the
biological unit is a homo-multimer, calculations were run on the monomer and multimer,
but only the monomer results are included in the results. In cases where the proteins are
hetero-multimers, THEMATICS calculations were run on the full biological unit.
46

Evolutionary Trace Report Maker (ET,
http://mammoth.bcm.tmc.edu/report_maker/index.html) 7-9,13 analysis was performed as
provided. The Catalytic Site Atlas (CSA, http://www.ebi.ac.uk/thornton-
srv/databases/CSA/) was used to identify the literature annotated catalytic residues. First-
shell residues, those residues in contact with a bound ligand or metal ion, were identified
using Ligand Protein Contact (LPC, http://bip.weizmann.ac.il/oca-bin/lpccsu); second-
and third-shell residues, those in contact with a given residue, were determined using
Contacts of Structural Units (CSU, (http://bip.weizmann.ac.il/oca-bin/lpccsu)14,15. Those
residues in direct contact with the first-shell residues were considered second-shell and
those residues in direct contact with second-shell residues were considered third-shell.
Conservation Surface-Mapping (Consurf, http://consurf.tau.ac.il/)16-18 was performed
using the default values. Once the first-, second- and third-shell residues were identified
from THEMATICS and ET, normalized conservation scores were determined for each of
those residues. The normalized scores were then averaged for each interaction shell.
Experimental mutagenesis data for each of the five test cases was obtained from the
BRENDA enzyme database19 (http://www.brenda-enzymes.info/) and the Protein Mutant
Database (http://www.genome.ad.jp/dbget-bin/www_bfind?pmd).
47

2.3 Results and Discussion
2.3.1 Experimental Design
In order to examine the ability of THEMATICS and ET to identify functionally important
first-, second- and third-shell residues, each method was run on a test set of 39 enzymes,
20 metallo- and 19 non-metalloenzymes (Table 2-1). All the proteins chosen have well
described catalytic mechanisms and annotated active sites. Results for five enzymes,
bacterial alkaline phosphatase (AP), human carbonic anhydrase isoform II (CAII),
mandelate racemase from Pseudomonas putida (MR), triosephosphate isomerase
from Gallus gallus (TIM), and tyrosyl-tRNA synthetase from Bacillus
stearothermophilus (TyrRS), are discussed in detail. These five enzymes were chosen to
represent a diverse set of well studied enzymatic reactions, which all require proton
transfer (i.e. ionizable residues). Redox reactions were not studied as it is very difficult to
computationally model the oxidation states of metals. The catalytic site atlas (CSA) was
used to identify catalytic residues, Ligand Protein Contacts (LPC) was used to identify
ligand or metal binding residues and Contacts of Structural Units (CSU) was used to
identify residues as second- or third-shell for each set of predicted residues.15 On average,
second-shell residues are approximately 5-10 Å from the catalytic center of the protein,
while third-shell residues are approximately 10-15 Å from the catalytic center. For 17 out
of the 39 proteins in the test set, THEMATICS identified residues beyond the third-shell,
while ET always identified residues beyond the third-shell. These residues are not
discussed further in this chapter.
48

ET was run using the default parameters.7 THEMATICS was run as previously described,
but modified to use a statistical cut-off of 0.96 rather 0.99.6 The statistical cut-off of 0.99
was previously determined to maximize performance in the selection of CSA-annotated
residues.6 This method computes metrics of anomalous titration behavior, μ3 and μ4, and
selects ionizable residues with metrics more than one standard deviation above the mean
for all ionizable residues in the protein. As nearly all CSA-annotated residues are found
in the first-shell, this study necessitated a lower statistical cut-off in order to increase the
number of residues predicted outside the first-shell. When a statistical cut-off of 0.96 is
used, the top 4% of residues with the highest metrics are excluded in the calculation of
the mean and standard deviation. Residues with metrics more than one standard deviation
above the mean (THEMATICS positive), including any in the top 4%, are considered
outliers and are analyzed further. The outliers located within 9 Å of at least one other
outlier constitute the THEMATICS predictions. Each residue in the THEMATICS
positive and ET positive groups of residues was then identified as belonging to the first-,
second- or third-shell.
Normalized conservation scores were obtained with Consurf16 and then averaged for each
interaction shell. A normalized conservation score is calculated so that the average score
for all residues in a protein is zero and the standard deviation is one. The more negative
the normalized conservation score, the more conserved the residue, and the more positive
the score, the more variable is the residue in the protein. A normalized conservation score
of -1.000 corresponds to a residue that is more conserved than the average by one
standard deviation, a score of -2.000 corresponds to a residue that is more conserved by
49

two standard deviations, and so on. By design, ET identifies functional residues based on
conservation through evolution; therefore, the sets of residues identified by this method
automatically have a high average conservation score. We suggest that the average
conservation scores are useful as a guide to compare the conservation of those residues
identified by THEMATICS, a method that uses no sequence-based information at all.
50
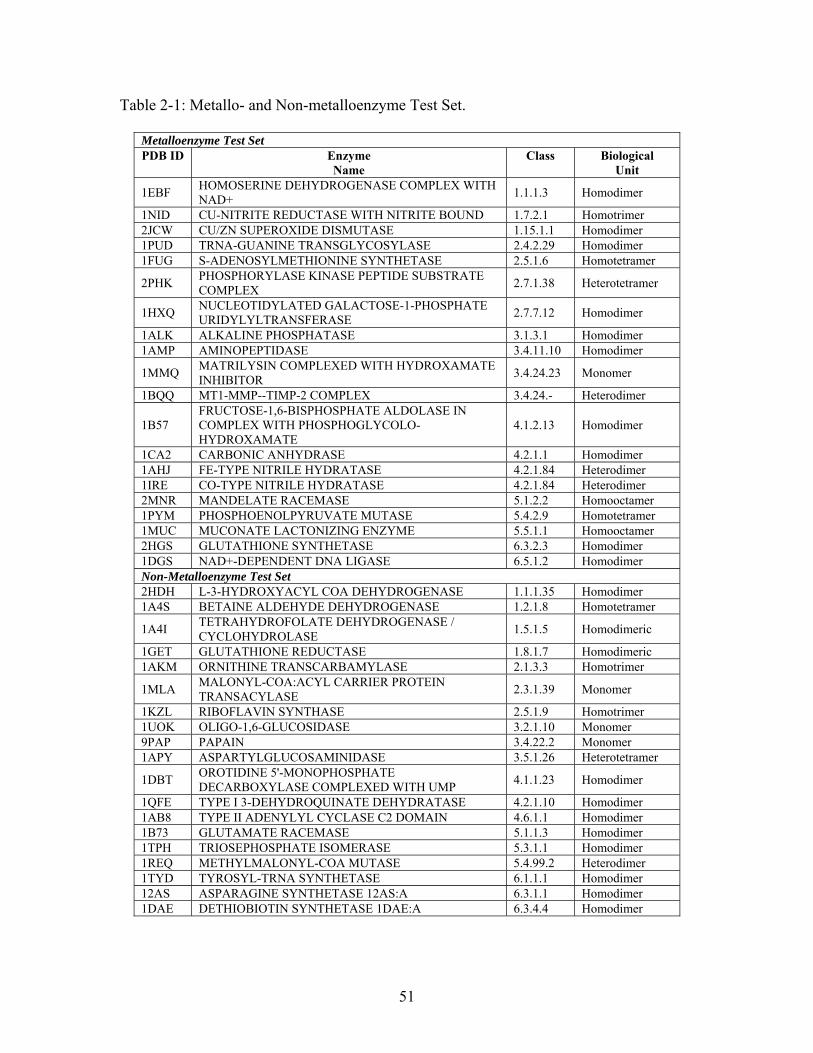
Table 2-1: Metallo- and Non-metalloenzyme Test Set.
Metalloenzyme Test Set PDB ID Enzyme
Name Class Biological
Unit
1EBF HOMOSERINE DEHYDROGENASE COMPLEX WITH NAD+
1.1.1.3 Homodimer
1NID CU-NITRITE REDUCTASE WITH NITRITE BOUND 1.7.2.1 Homotrimer 2JCW CU/ZN SUPEROXIDE DISMUTASE 1.15.1.1 Homodimer 1PUD TRNA-GUANINE TRANSGLYCOSYLASE 2.4.2.29 Homodimer 1FUG S-ADENOSYLMETHIONINE SYNTHETASE 2.5.1.6 Homotetramer
2PHK PHOSPHORYLASE KINASE PEPTIDE SUBSTRATE COMPLEX
2.7.1.38 Heterotetramer
1HXQ NUCLEOTIDYLATED GALACTOSE-1-PHOSPHATE URIDYLYLTRANSFERASE
2.7.7.12 Homodimer
1ALK ALKALINE PHOSPHATASE 3.1.3.1 Homodimer 1AMP AMINOPEPTIDASE 3.4.11.10 Homodimer
1MMQ MATRILYSIN COMPLEXED WITH HYDROXAMATE INHIBITOR
3.4.24.23 Monomer
1BQQ MT1-MMP--TIMP-2 COMPLEX 3.4.24.- Heterodimer
1B57 FRUCTOSE-1,6-BISPHOSPHATE ALDOLASE IN COMPLEX WITH PHOSPHOGLYCOLO-HYDROXAMATE
4.1.2.13 Homodimer
1CA2 CARBONIC ANHYDRASE 4.2.1.1 Homodimer 1AHJ FE-TYPE NITRILE HYDRATASE 4.2.1.84 Heterodimer 1IRE CO-TYPE NITRILE HYDRATASE 4.2.1.84 Heterodimer 2MNR MANDELATE RACEMASE 5.1.2.2 Homooctamer 1PYM PHOSPHOENOLPYRUVATE MUTASE 5.4.2.9 Homotetramer 1MUC MUCONATE LACTONIZING ENZYME 5.5.1.1 Homooctamer 2HGS GLUTATHIONE SYNTHETASE 6.3.2.3 Homodimer 1DGS NAD+-DEPENDENT DNA LIGASE 6.5.1.2 Homodimer Non-Metalloenzyme Test Set 2HDH L-3-HYDROXYACYL COA DEHYDROGENASE 1.1.1.35 Homodimer 1A4S BETAINE ALDEHYDE DEHYDROGENASE 1.2.1.8 Homotetramer
1A4I TETRAHYDROFOLATE DEHYDROGENASE / CYCLOHYDROLASE
1.5.1.5 Homodimeric
1GET GLUTATHIONE REDUCTASE 1.8.1.7 Homodimeric 1AKM ORNITHINE TRANSCARBAMYLASE 2.1.3.3 Homotrimer
1MLA MALONYL-COA:ACYL CARRIER PROTEIN TRANSACYLASE
2.3.1.39 Monomer
1KZL RIBOFLAVIN SYNTHASE 2.5.1.9 Homotrimer 1UOK OLIGO-1,6-GLUCOSIDASE 3.2.1.10 Monomer 9PAP PAPAIN 3.4.22.2 Monomer 1APY ASPARTYLGLUCOSAMINIDASE 3.5.1.26 Heterotetramer
1DBT OROTIDINE 5'-MONOPHOSPHATE DECARBOXYLASE COMPLEXED WITH UMP
4.1.1.23 Homodimer
1QFE TYPE I 3-DEHYDROQUINATE DEHYDRATASE 4.2.1.10 Homodimer 1AB8 TYPE II ADENYLYL CYCLASE C2 DOMAIN 4.6.1.1 Homodimer 1B73 GLUTAMATE RACEMASE 5.1.1.3 Homodimer 1TPH TRIOSEPHOSPHATE ISOMERASE 5.3.1.1 Homodimer 1REQ METHYLMALONYL-COA MUTASE 5.4.99.2 Heterodimer 1TYD TYROSYL-TRNA SYNTHETASE 6.1.1.1 Homodimer 12AS ASPARAGINE SYNTHETASE 12AS:A 6.3.1.1 Homodimer 1DAE DETHIOBIOTIN SYNTHETASE 1DAE:A 6.3.4.4 Homodimer
51

2.3.2 THEMATICS and ET: Identification of Residues and Predictions by Shell
The THEMATICS and ET results for all proteins in the test set are included as
supplemental tables (S1-S4) and include average normalized conservation scores for each
interaction shell. As expected, clusters predicted by THEMATICS are substantially
smaller than those predicted by ET. On average, THEMATICS predictions for this test
set constitute approximately 2.6% of the protein, while ET predictions for this test set
account for approximately 22% of the protein. THEMATICS identifies most of the
annotated first-shell residues although it is limited to ionizable residues, while ET most
often identifies all known first-shell residues plus a large number of apparent false
positives. In addition, the predictions of both methods usually include residues located in
the second- and third-shells.
For 37 of the 39 enzymes in the test set, THEMATICS makes a correct active site or first-
shell prediction. The two cases where THEMATICS fails include papain and aspartyl-
glucosaminidase (Table S3). Aspartyl-glucosaminidase uses a catalytic threonine, which
THEMATICS will not find as it is treated as non-ionizable, and THEMATICS does not
identify the catalytic cysteine of papain. For the 37 other cases, the prediction includes at
least some of the annotated, active site residues (catalytic or substrate binding residues),
with an average of 4.1 annotated, active site residues in the first-shell predicted per
subunit. For 31 of the enzymes, THEMATICS predicts at least one residue in the second
shell of an annotated binding site with an average of 2.0 second-shell residues predicted.
At least one third-shell residue is predicted for 19 of the enzymes and for these, the
average number of predicted third-shell residues is 1.3. Furthermore, the predicted
52

second- and third-shell residues on the average tend to be almost as well conserved as the
first-shell residues according to Consurf. The average normalized conservation scores are
-1.2, -1.0, and -0.85, for the first-, second-, and third-shell residues that are predicted by
THEMATICS, respectively. Thus, for most enzymes in our test set, THEMATICS
successfully predicts the active site and also identifies a few second- and third- shell
residues that are generally well conserved.
For all 39 proteins in the test set, the ET predictions contain residues in each of the first
three shells. ET predicts an average of 13.3 residues in the first-shell, 21.7 in the second-
shell, and 18.8 in the third-shell per subunit. By the very nature of the ET method, one
would expect that the predicted residues have high average conservation scores. Indeed,
for the 39 enzymes, the average conservation scores are -1.1, -0.9, and -0.9 for the
predicted residues in the first-, second- and third-shells, respectively.
53

Comparison of THEMATICS and ET for Metallo-enzymes Alkaline Phosphatase,
Carbonic Anhydrase II and Mandelate Racemase and Non-Metallo Enzymes
Triosephosphate Isomerase and Tyrosyl-tRNA Synthetase
There are a few cases in the experimental literature for which the roles of some of the
second- and third-shell residues have been studied, allowing for the comparison of
THEMATICS and ET predictions with experimental data. Three metalloenzymes and
two non-metalloenzymes were selected for this comparison and are now described in
detail. For these enzymes, we have compiled experimental mutagenesis data found in the
literature, including the effect on catalytic efficiency. The metalloenzymes include
bacterial alkaline phosphatase (AP), human carbonic anhydrase isoform II (CAII) and
mandelate racemase (MR) from Pseudomonas putida. The non-metalloenzymes include
triosephosphate isomerase (TIM) from Gallus gallus, and tyrosyl-tRNA synthetase
(TyrRS) from Bacillus stearothermophilus. The THEMATICS and ET results for these
five proteins are shown in Tables 2-2 and 2-3, respectively. Residues identified as
catalytic from the CSA are shown in boldface and those residues identified by LPC as
substrate- or metal-binding residues are italicized. Those residues for which experimental
mutagenesis data are available are underlined. Only those residues identified by
THEMATICS and/or ET are included. Supplemental tables S5-S9 provide a complete
summary of all experimental mutations made to AP, CAII, MR, TIM and TyrRS
respectively.
54
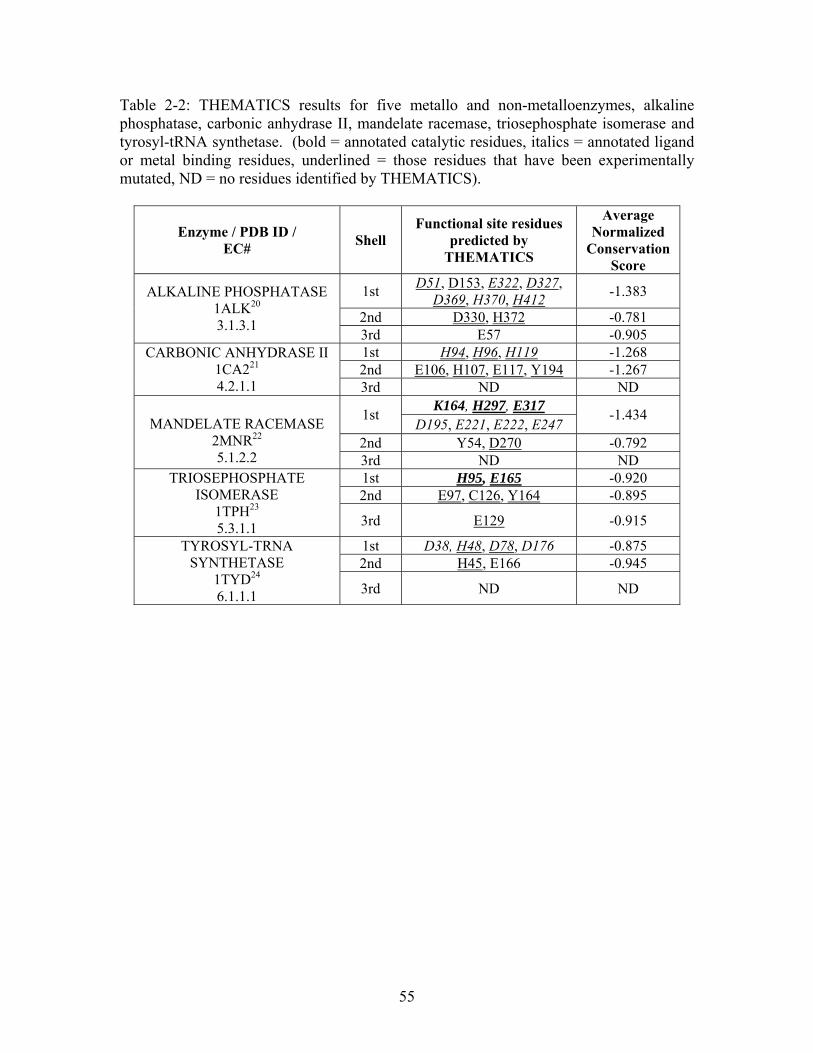
Table 2-2: THEMATICS results for five metallo and non-metalloenzymes, alkaline phosphatase, carbonic anhydrase II, mandelate racemase, triosephosphate isomerase and tyrosyl-tRNA synthetase. (bold = annotated catalytic residues, italics = annotated ligand or metal binding residues, underlined = those residues that have been experimentally mutated, ND = no residues identified by THEMATICS).
Enzyme / PDB ID / EC#
Shell Functional site residues
predicted by THEMATICS
Average Normalized
Conservation Score
1st D51, D153, E322, D327,
D369, H370, H412 -1.383
2nd D330, H372 -0.781
ALKALINE PHOSPHATASE 1ALK20 3.1.3.1
3rd E57 -0.905 1st H94, H96, H119 -1.268 2nd E106, H107, E117, Y194 -1.267
CARBONIC ANHYDRASE II 1CA221 4.2.1.1 3rd ND ND
K164, H297, E317 1st
D195, E221, E222, E247 -1.434
2nd Y54, D270 -0.792
MANDELATE RACEMASE
2MNR22 5.1.2.2 3rd ND ND
1st H95, E165 -0.920 2nd E97, C126, Y164 -0.895
TRIOSEPHOSPHATE ISOMERASE
1TPH23 5.3.1.1 3rd E129 -0.915
1st D38, H48, D78, D176 -0.875 2nd H45, E166 -0.945
TYROSYL-TRNA SYNTHETASE
1TYD24 6.1.1.1 3rd ND ND
55
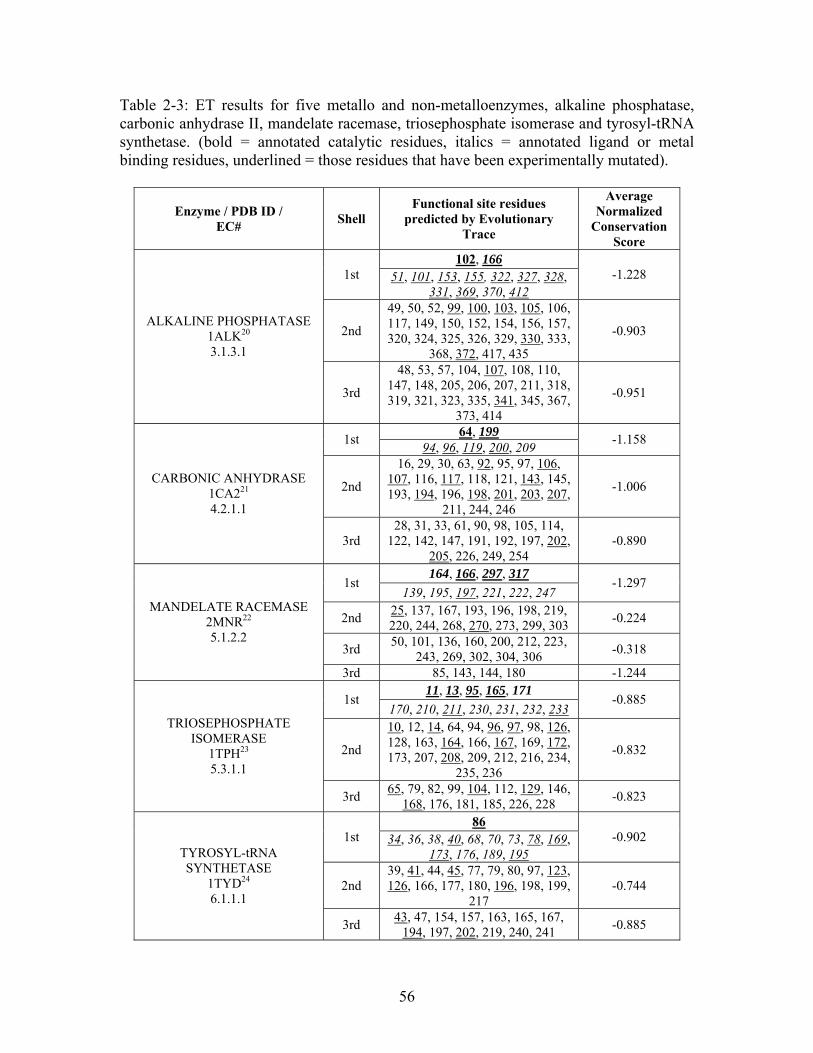
Table 2-3: ET results for five metallo and non-metalloenzymes, alkaline phosphatase, carbonic anhydrase II, mandelate racemase, triosephosphate isomerase and tyrosyl-tRNA synthetase. (bold = annotated catalytic residues, italics = annotated ligand or metal binding residues, underlined = those residues that have been experimentally mutated).
Enzyme / PDB ID / EC#
Shell Functional site residues
predicted by Evolutionary Trace
Average Normalized
Conservation Score
102, 166 1st 51, 101, 153, 155, 322, 327, 328,
331, 369, 370, 412 -1.228
2nd
49, 50, 52, 99, 100, 103, 105, 106, 117, 149, 150, 152, 154, 156, 157, 320, 324, 325, 326, 329, 330, 333,
368, 372, 417, 435
-0.903 ALKALINE PHOSPHATASE
1ALK20 3.1.3.1
3rd
48, 53, 57, 104, 107, 108, 110, 147, 148, 205, 206, 207, 211, 318, 319, 321, 323, 335, 341, 345, 367,
373, 414
-0.951
64, 199 1st
94, 96, 119, 200, 209 -1.158
2nd
16, 29, 30, 63, 92, 95, 97, 106, 107, 116, 117, 118, 121, 143, 145, 193, 194, 196, 198, 201, 203, 207,
211, 244, 246
-1.006 CARBONIC ANHYDRASE
1CA221 4.2.1.1
3rd 28, 31, 33, 61, 90, 98, 105, 114,
122, 142, 147, 191, 192, 197, 202, 205, 226, 249, 254
-0.890
164, 166, 297, 317 1st
139, 195, 197, 221, 222, 247 -1.297
2nd 25, 137, 167, 193, 196, 198, 219, 220, 244, 268, 270, 273, 299, 303
-0.224
3rd 50, 101, 136, 160, 200, 212, 223,
243, 269, 302, 304, 306 -0.318
MANDELATE RACEMASE 2MNR22 5.1.2.2
3rd 85, 143, 144, 180 -1.244 11, 13, 95, 165, 171
1st 170, 210, 211, 230, 231, 232, 233
-0.885
2nd
10, 12, 14, 64, 94, 96, 97, 98, 126, 128, 163, 164, 166, 167, 169, 172, 173, 207, 208, 209, 212, 216, 234,
235, 236
-0.832
TRIOSEPHOSPHATE ISOMERASE
1TPH23 5.3.1.1
3rd 65, 79, 82, 99, 104, 112, 129, 146,
168, 176, 181, 185, 226, 228 -0.823
86 1st 34, 36, 38, 40, 68, 70, 73, 78, 169,
173, 176, 189, 195 -0.902
2nd 39, 41, 44, 45, 77, 79, 80, 97, 123, 126, 166, 177, 180, 196, 198, 199,
217 -0.744
TYROSYL-tRNA SYNTHETASE
1TYD24 6.1.1.1
3rd 43, 47, 154, 157, 163, 165, 167,
194, 197, 202, 219, 240, 241 -0.885
56

2.3.3 Metalloenzymes
Bacterial Alkaline Phosphatase - (PDB ID: 1ALK20)
Bacterial alkaline phosphatase (AP) is currently used in indirect ELISA assays. It is
conjugated to a secondary antibody and the substrate, p-Nitrophenyl phosphate (pNPP),
is used as a chromogenic substrate for colorimetric detection. The enzyme has the
potential to be used for applications in other diagnostic tests; however, its turnover rate is
considered too slow.25 Therefore AP has been widely studied using both directed
evolution and rational-protein design approaches.25 AP is a dimeric metalloenzyme
containing two zinc atoms and one magnesium atom per monomer and it catalyzes the
reversible hydrolysis of phosphomonoesters to yield inorganic phosphate plus an alcohol.
In the crystal structure (PDB ID: 1ALK20), there is an inorganic phosphate ion bound to
the two zinc atoms (Zn1 and Zn2), and to the guanidinium group of Arg166. Additional
residues that coordinate to Zn1 are Asp327, His331 and His412. The residues that
coordinate to Zn2 are Asp51, Asp369 and His370, while Asp51, Thr155 and Glu322
coordinate the Mg ion.26 The active site region of the protein includes Asp101, Ser102,
Ala103, the three metal atoms and Arg166.20 There is a hydrogen bond network that
includes hydrogen bonds between Asp101 and Arg166 and between Arg 166 and a water
molecule.27 Lys328 interacts with the phosphate group in the active center through a
water mediated hydrogen bond. This water is also involved in a hydrogen bond with
Asp327, which is a bidentate ligand of Zn1.28 Finally, Asp153 serves as an indirect ligand
to the Mg ion through a water mediated interaction.25
57

The reaction proceeds via a two step mechanism (Figure 2-1),29 where in the first step,
Ser102 is phosphorylated giving a phosphoseryl intermediate. In the second step, this
intermediate is hydrolyzed to give a non-covalent enzyme-phosphate complex. In the
presence of a phosphate acceptor such as Tris, the enzyme shows transphosphorylation
activity and transfers a phosphate to the alcohol to form a phosphate monoester.30 Figure
2-2 shows a cartoon representation of the active site and metal binding residues in
addition to the catalytic serine.
58
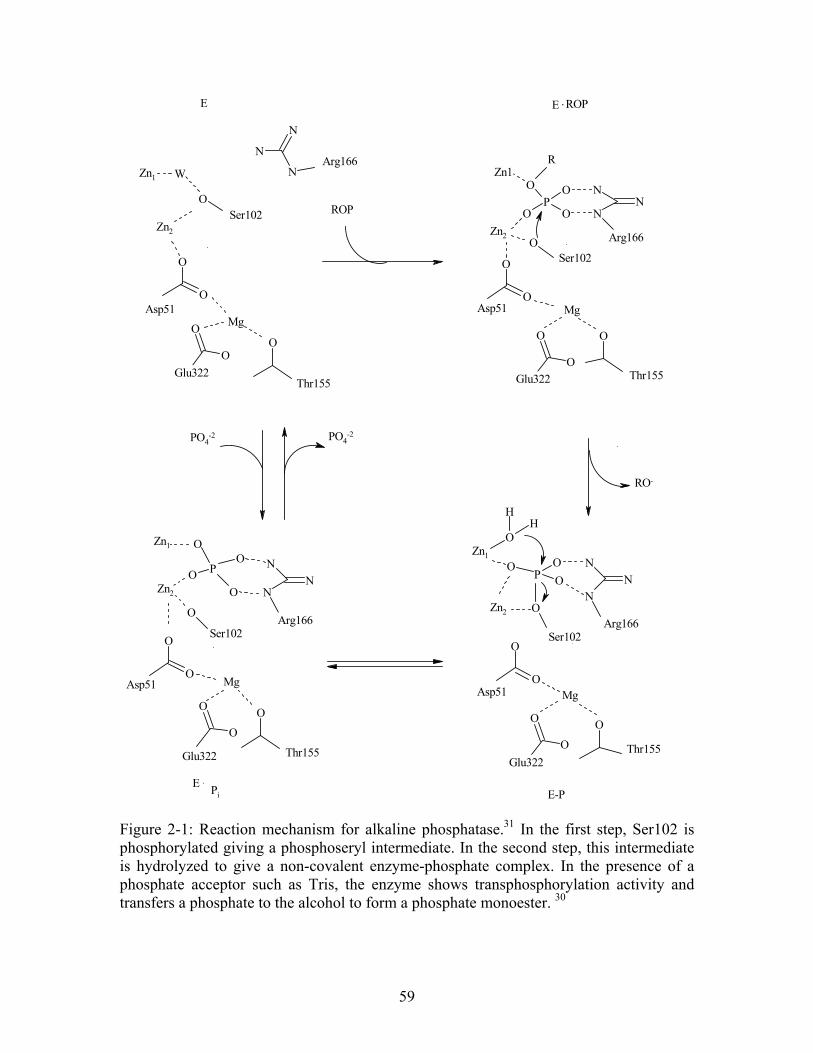
N
N
N
O
O
OO
O
O P
O
NN
NO
O
O
OO
O
PO
O
N
N
N
O
HH
O
O
O
O
O
O
O
PO
O
O
O N
NNO
O
O
O
O
OO
O
O
PO4-2
Arg166
Mg
Zn2
Glu322Thr155
PO4-2
Zn2
Arg166
Asp51
Asp51
Thr155Glu322
W
Zn1
Zn1
Arg166Zn2
Zn1
Ser102
Asp51
Glu322Thr155
Zn1
Zn2
R
ROP
Arg166
Ser102
Ser102
Ser102
Asp51
Thr155Glu322
MgMg
Mg
RO-
E . Pi E-P
E E . ROP
Figure 2-1: Reaction mechanism for alkaline phosphatase.31 In the first step, Ser102 is phosphorylated giving a phosphoseryl intermediate. In the second step, this intermediate is hydrolyzed to give a non-covalent enzyme-phosphate complex. In the presence of a phosphate acceptor such as Tris, the enzyme shows transphosphorylation activity and transfers a phosphate to the alcohol to form a phosphate monoester. 30
59

Arg166b
His412a
His370a Thr155
b
Asp369a
PO4 Ser102b
Asp153a
His331b
Asp327a
Asp51a
Glu322a
Figure 2-2: Cartoon representation of active site of alkaline phosphatase (PBD ID: 1ALK20) including metal binding residues in the first-shell known to be functionally important and the catalytic residue, Ser102. Grey spheres = zinc ions and green sphere = magnesium ion. a refers to first-shell residues identified by THEMATICS and ET; b refers to first-shell residues identified by ET only. Note that Ser102 and Thr155 will not be found by the THEMATICS method as they are non-ionizable residues.
Two interesting mutations, designed specifically to increase the catalytic activity of AP,
are associated with the first-shell residue Asp153 and the second-shell residue Asp330.
Asp153 is involved in an ion pair interaction with Lys328, and in a water-mediated
interaction with the Mg ion.25 Asp 153 also serves to position the catalytic residue,
Arg166 (Figure 2-3). Using rational protein design methods, four point mutations were
made at the Asp153 position in an attempt to more clearly understand the role of Asp153
and to increase the catalytic efficiency of the protein.32-35 Three of the four mutations,
Asp153Gly, Asp153Glu, and Asp153Ala, resulted in an increased catalytic rate. Given
these results, Muller et al. searched for additional mutations that may increase the
turnover rate to a level comparable to that of mammalian enzymes, while allowing the
protein to retain its high thermostability.25 It had been shown that mutations in the active
60

site that resulted in increased catalytic rate always caused a substantial decrease in
thermostability.32 They assumed that while mutations to residues in the active site most
often had a negative effect on catalysis, residues outside the catalytic site may also
influence catalysis. They sought to identify residues located outside the site of catalysis
which would inactivate the protein. The goal then was to reactivate the enzyme by
random mutational analysis while maintaining the correct protein conformation. Using
directed evolution approaches, they discovered that the mutation of Asp330 to Asn,
which is located 12Å from the center of the catalytic pocket, causes an almost 3-fold
increase in activity compared to the wild type enzyme.25 While this single mutation,
Asp330Asn, has only a small increase in catalytic efficiency, the double mutant
Asp153Gly/Asp330Asn resulted in a 40-fold increase in activity while still maintaining
thermostability.25 In this example, both rational protein design and directed evolution
methodologies were combined to achieve a faster, stable enzyme. Additionally, this
double mutant was as active as mammalian AP’s (i.e. the kcat and KM values were
essentially the same as bovine intestinal phosphatase).
61

Arg166b
Asp153a
PO4
His372c
Lys328b
Glu57d
Asp330c
Figure 2-3: Residues involved in interaction with the first-shell residue, Asp153 for alkaline phosphatase (PDB ID: 1ALK20) including the THEMATICS positive residues in the second- and third-shell. Grey spheres = zinc ions, green sphere = magnesium ion and red cross = water. a refers to THEMATICS and ET positive residue in the first-shell; b refers to residues identified by ET in the second-shell; c refers to residues identified by THEMATICS and ET in the second-shell; d refers to a third-shell residue identified by THEMATICS and ET. The Asp153Gly/Asp330Asn double mutant resulted in a 40-fold increase in catalytic rate.25
THEMATICS and ET identify most of the known phosphate and metal binding residues;
however, ET also identifies the catalytic residues Ser102 and Arg166, where
THEMATICS does not. THEMATICS, in the form used here, does not identify serine
and other non-ionizable residues. In addition to the first-shell residues, the second shell
residues identified by THEMATICS are Asp330 and His372. ET also identifies Asp330
and His372 in addition to 24 other residues in the second-shell. THEMATICS finds one
third-shell residue, Glu57, which has not been experimentally investigated, and ET
62

identifies 21 third-shell residues, including two, Thr107 and Glu341, for which there are
experimental mutagenesis data.
Human Carbonic Anhydrase Isoform II - (PBD ID: 1CA221)
Human carbonic anhydrase isoform II (CAII) is an excellent model for the study of
protein structure-function relationships in protein-zinc binding sites. It is one of the most
efficient biological catalysts in addition to being one of the few enzymes whose catalytic
efficiency approaches the limit of diffusion control.36 This is only the case with isoform
II.37 It has been extensively studied with respect to hydrogen bond networks and the
involvement of remote residues in catalysis. It also serves as a good model for the de
novo design of metal binding sites in other systems based on the numerous protein
engineering lessons learned from this system.38 In addition, efforts are underway to
utilize CAII as a metal ion biosensor to quantify trace metals in biological media for
toxicological and environmental monitoring.39,40
Human CAII is a zinc dependent metallo-enzyme that catalyzes the reversible hydration
of carbon dioxide.41 There are seven distinct forms of this protein (known as isozymes I-
VII), and the substrate and zinc binding sites are conserved among all isoforms.38 The
hydrolysis reaction of carbon dioxide to yield the bicarbonate ion and a proton occurs via
a two-step mechanism (Figure 2-4). In short, a zinc-bound hydroxide ion acts as the
nucleophile to attack CO2 to form a zinc-bound bicarbonate intermediate. This
intermediate is then displaced by a water molecule creating a zinc-H2O form. In the rate
63

determining step, the zinc-bound hydroxide is regenerated through the transfer of a
proton to the solvent facilitated by the active site histidine, His64, which acts as a proton
shuttle.36,42
O
C
O
O
HH
O
CO
O
C
O O
O
H
O
HH
O
H
-
His119
His96
Zn+2
His94 His119
His96
Zn+2
-
Zn+2
His96
His119His94
-H2O HCO3
-
His94
His96
Zn+2
Zn+2
His94 His119
His96
-
CO2
H+ to buffer through proton shuttleresidue His64
His94 His119
Figure 2-4: Reaction mechanism for carbonic anhydrase II.38 Zinc-bound hydroxide acts as the nucleophile to attack CO2 to form a zinc-bound bicarbonate intermediate. This intermediate is then displaced by a water molecule creating a zinc-H2O form. In the rate determining step, the zinc-bound hydroxide is regenerated through the transfer of a proton to the solvent facilitated by the active site histidine, His64, which acts as a proton shuttle. 36,42
In the crystal structure of human CAII (PDB ID: 1CA221), the active site comprises a
cleft that is about 15 Å deep with the zinc ion at the bottom. The zinc ion is tetrahedrally
coordinated with the side chains of His94, His96 and His119 and a hydroxide ion. These
zinc binding histidines participate in hydrogen bonds with second-shell residues Gln92,
Asn244 and Glu117, respectively. The active site cavity of CAII is amphiphilic; i.e. one
side is dominated by hydrophobic residues and the other side by mostly hydrophilic
64

residues.43 Thr199 accepts a hydrogen bond from the zinc-bound hydroxide ion and
donates a hydrogen bond to the side chain of Glu106. Thr199 also helps to orient the
hydroxide ion in order for nucleophilic attack on the substrate, CO244, and is a
coordinating residue to the substrate.45 Thr200 is also a coordinating residue to the CO2
substrate.46 Some of the highly conserved hydrophobic residues in the active site cavity
include Val121, Val143, Leu198 and Trp209 (Figure 2-5).
His119a
Glu106b
Thr200c
His94a
Val121d
Val143d
Leu198d
Glu117b
His107b
Thr199c
His96a
His64c
Asn244d
Tyr194b
Trp209c
Gln92d
Figure 2-5: Cartoon representation of known active site and metal binding residues for carbonic anhydrase II (PDB ID: 1CA221) in addition to select second-shell residues known to be functionally important. Grey sphere = zinc. a refers to first-shell residues identified by THEMATICS and ET; b refers to second-sell residues identified by THEMATICS and ET; c refers to additional first-shell residues identified by only ET; d refers to second-shell residues identified by only ET.
Most of the mutations to CAII were made through rational protein design techniques to
better understand zinc metallo-enzymes. Numerous mutations have also been made to
remote residues in CAII to better understand the folding and stability of the protein,47-49
as well as metal and substrate specificity.39,46 Directed evolution studies were undertaken
65

in an effort to understand the functional role of the residues comprising the hydrophobic
face of the active site,50 specifically in the region between Asp190 and Ile210, in addition
to probing the esterase activity of CAII.51 The residues in this region that were identified
by THEMATICS and/or ET include four second-shell residues, Tyr194, Leu198, Pro201,
and Leu203, and two third-shell residues, Pro202 and Leu204 (Figure 2-6). Many of the
mutations had no effect on CO2 hydration; however, mutagenesis at two positions
produced interesting results. The Leu198Pro and Leu198Arg mutations produced a
substantial decrease in activity (25 X and 50 X, respectively). In addition, the Leu203Arg
mutation resulted in a decrease in activity (10 X), while the Leu203Phe resulted in an
increase in catalytic activity (2.5 X). The conclusion of this study highlighted the fact that
this hydrophobic face is extremely plastic and can accommodate amino acid substitutions
of varying size, charge and hydrophobicity.
66
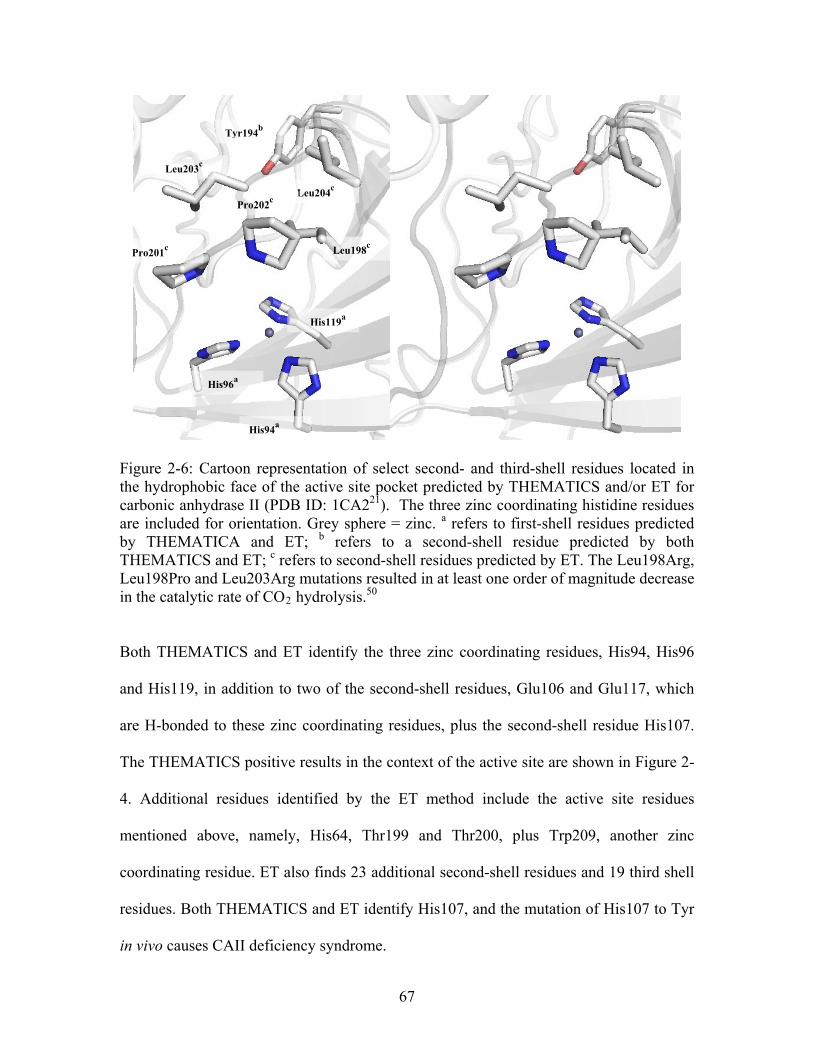
Tyr194b
Leu203c
Leu204c
Pro202c
Leu198c
Pro201c
His119a
His96a
His94a
Figure 2-6: Cartoon representation of select second- and third-shell residues located in the hydrophobic face of the active site pocket predicted by THEMATICS and/or ET for carbonic anhydrase II (PDB ID: 1CA221). The three zinc coordinating histidine residues are included for orientation. Grey sphere = zinc. a refers to first-shell residues predicted by THEMATICA and ET; b refers to a second-shell residue predicted by both THEMATICS and ET; c refers to second-shell residues predicted by ET. The Leu198Arg, Leu198Pro and Leu203Arg mutations resulted in at least one order of magnitude decrease in the catalytic rate of CO2 hydrolysis.50
Both THEMATICS and ET identify the three zinc coordinating residues, His94, His96
and His119, in addition to two of the second-shell residues, Glu106 and Glu117, which
are H-bonded to these zinc coordinating residues, plus the second-shell residue His107.
The THEMATICS positive results in the context of the active site are shown in Figure 2-
4. Additional residues identified by the ET method include the active site residues
mentioned above, namely, His64, Thr199 and Thr200, plus Trp209, another zinc
coordinating residue. ET also finds 23 additional second-shell residues and 19 third shell
residues. Both THEMATICS and ET identify His107, and the mutation of His107 to Tyr
in vivo causes CAII deficiency syndrome.
67

Mandelate Racemase from Pseudomonas putida – (PDB ID: 2MNR22)
Mandelate racemase (MR) has been studied as a model for enzymes which catalyze the
rapid carbon-hydrogen bond cleavage of carbon acids with high pKa’s.52 In addition, its
structural relationship to muconate lactonizing enzyme (MLE) has been studied in an
attempt to better understand the evolution of superfamilies of enzymes, i.e.
structure/function relationships between enzyme families.53 MR and MLE are structurally
similar enzymes that catalyze different overall reactions. Other structurally related
proteins in this superfamily have recently been discovered; thus MR is a member of a
superfamily with high functional diversity.54
MR catalyzes the interconversion of the (R) and (S) enantiomers of mandelic acid via
abstraction of a proton from the α-carbon atom.55-57 It is a divalent cation-dependent
protein, and in the crystal structure (PDB ID: 2MNR22), the Mn 2+ ion is coordinated by
Asp195, Glu221, Glu222 and Glu247.22 Residues that are near the metal ion and a bound
sulfate ion are Ser139, Lys164, Asn197,52 and Glu31755. Mutations have not been made
at Ser139, Lys164, Asp195, Glu221 and Glu247, but according to the Catalytic Site Atlas
(CSA), these residues are necessary for catalytic function of the protein. The MR reaction
proceeds via a two base mechanism (Figure 2-7), where one residue abstracts the α-
proton to generate an intermediate, and the other residue protonates the opposite face of
the intermediate to produce the inverted product.56 His297 acts as the (R)- specific
acid/base catalyst,56 while Lys166 acts as the (S)- specific acid/base catalyst.57 A cartoon
diagram showing the catalytic and metal binding residues is shown in Figure 2-8.
68
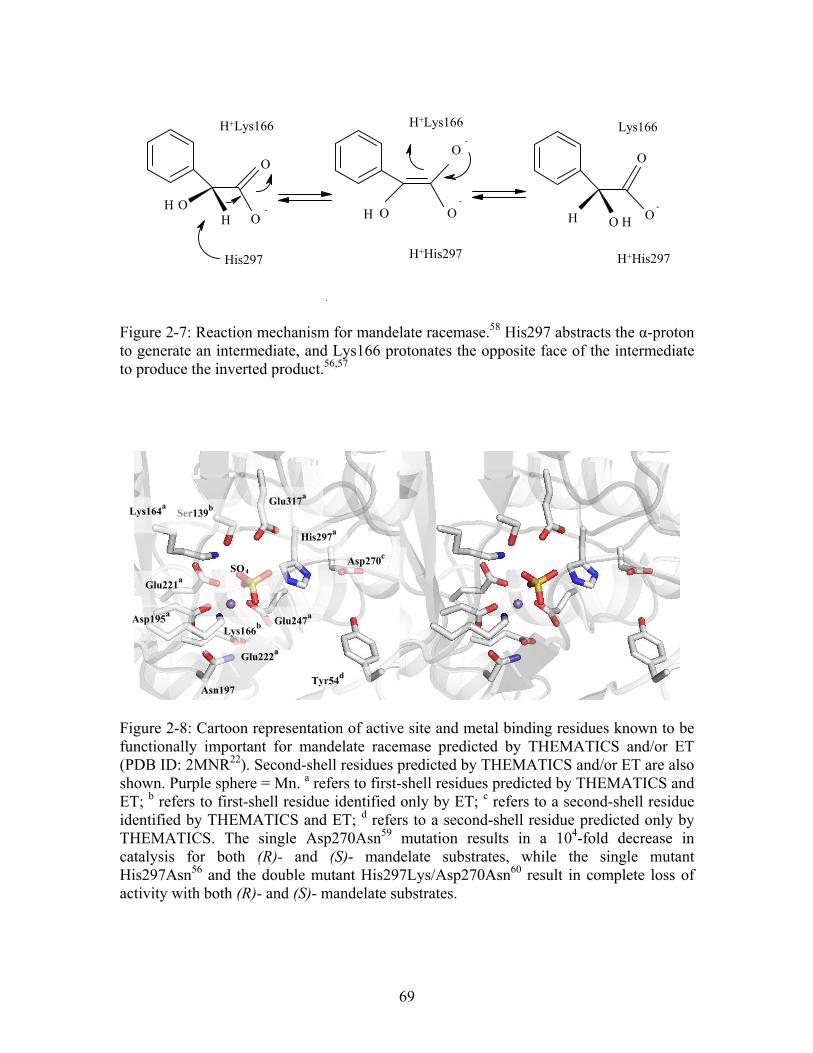
O
O
HOH
O
OOH
O
OOH H-
His297
H+Lys166
-
-
H+His297
H+Lys166
-
Lys166
H+His297
Figure 2-7: Reaction mechanism for mandelate racemase.58 His297 abstracts the α-proton to generate an intermediate, and Lys166 protonates the opposite face of the intermediate to produce the inverted product.56,57
Glu317a
Ser139b Lys164
a
7a
His29
Glu247a
Lys166b
Asn197
SO4
Glu221a
Tyr54d
Glu222a
Asp195a
Asp270c
Figure 2-8: Cartoon representation of active site and metal binding residues known to be functionally important for mandelate racemase predicted by THEMATICS and/or ET (PDB ID: 2MNR22). Second-shell residues predicted by THEMATICS and/or ET are also shown. Purple sphere = Mn. a refers to first-shell residues predicted by THEMATICS and ET; b refers to first-shell residue identified only by ET; c refers to a second-shell residue identified by THEMATICS and ET; d refers to a second-shell residue predicted only by THEMATICS. The single Asp270Asn59 mutation results in a 104-fold decrease in catalysis for both (R)- and (S)- mandelate substrates, while the single mutant His297Asn56 and the double mutant His297Lys/Asp270Asn60 result in complete loss of activity with both (R)- and (S)- mandelate substrates.
69

One mutation study of MR involves the interaction between His297 and the second-shell
residue Asp270. Asp270 forms a hydrogen bond with the catalytic base, His297, and
therefore is believed to affect its orientation (Figure 2-8).59 The single mutation
Asp270Asn results in a 104- fold decrease in enzyme activity compared to wild type for
both (R)- and (S)- mandelate substrates.59 The Asn270 side chain in the mutant structure
is superimposable on the Asp270 side chain of the wild type structure, and the remainder
of the two structures are identical except that the side chain of the catalytic His297 is
“rotated and displaced toward the binding site” in the mutant structure.59 The authors
argue that Asp270 is necessary to impart the correct pKa to the catalytic His297.59 The
single His297Asn56 mutation and the double mutation, His297Lys /Asp270Asn60, results
in complete loss of enzyme activity with both (R) - and (S)- mandelate substrates. His297
and Asp270 therefore function as a catalytic dyad as shown by the Asp270Asn mutant
data.
THEMATICS and ET both identify the metal and/or SO4 coordinating ligands, Lys164,
Asp195, Glu221, Glu222 and Glu247, the catalytic base, His297, and the substrate
coordinating ligand Glu317 (Figure 2-8). In addition to the first-shell residues, both
THEMATICS and ET find second-shell residues. THEMATICS identifies Tyr54 and
Asp270. While ET also predicts Asp270, it does not identify Tyr54. ET additionally
identifies residues in close proximity to the metal and sulfate ions; namely, Ser139 and
Asn197, both of which are considered non-ionizable residues. ET also predicts the second
catalytic base Lys166. No third-shell residues are identified for MR by THEMATICS,
70

but ET predicts 12 second-shell residues in addition to Asp270; among these additional
residues, there is experimental data for only one: Ala25.
2.3.4 Non-Metalloenzymes
Triosephosphate Isomerase from Gallus gallus – (PDB ID: 1TPH23)
The triosephosphate isomerase (TIM) 3D-structure is termed a classic TIM-barrel fold,
where eight parallel β-strands on the inside of the protein are surrounded by eight α-
helices on the outside.61 This fold, first noted in TIM, is the most common enzyme fold in
the Protein Data Bank, and has members in all major enzyme classification classes but
one (no ligases are known to date). It has therefore become one of the most widely
studied protein folds. In all TIM-barrel enzymes, the active site is at the C-terminal end
of the β-strands.
TIM catalyzes the interconversion of dihydroxyacetone phosphate (DHAP) and D-
glyceraldehyde 3-phosphate (GAP) in the glycolytic pathway. In most organisms, TIM
exists as a homodimer and is only active in that state.62 There is a major conformational
change between the unliganded (‘open’ conformation) and the liganded (‘closed’
conformation).63 There is a mobile loop region of approximately 11 conserved residues,
from ~ 166 – 176, that leaves the active site open to solvent in the unliganded form and
moves approximately 7Å to close over the substrate in the liganded form.64,65 In this
conserved loop region, residues 170 – 173 provide new H-bonds to the phosphate group
of the substrate in the closed form. The purpose of this loop is twofold; one to ensure
proper turnover of substrate to product and two to stabilize the reaction intermediate.
71

The active site of the enzyme is located at the bottom of a deep polar pocket. In the open
conformation, there is a water molecule bound at the bottom of the cleft that is H-bonded
to the catalytic residues Asn11 and His95.63 When a ligand is bound, this water is
displaced and the ligand H-bonds with backbone nitrogen atoms of Gly171, Ser211,
Gly232 and Gly233 to stabilize the ligand-protein complex. The H-bonds with Gly171
and Ser211 are possible because in the unliganded form, the NH group of Gly212 points
into the core of the active site, but is rotated outward in the liganded form. Lys13 is the
only positively charged residue at the active site. Asnll, Gly210, Ser211, Leu230,
Va1231, Gly232, and Gly233 are all residues which make up the core of the active site
and enclose the substrate.23
The reaction for TIM proceeds through a three step mechanism (Figure 2-9).63 In the first
step, a proton is abstracted from C1 of the ligand by the catalytic residue Glu165
producing a negative charge on O2 which is stabilized by interactions with His95. His95
then transfers a proton from O1 to O2 in the second step. Finally, Glu165 transfers a
proton back to C2. In summary, Glu165 and His95 are the catalytic residues responsible
for proton transfer between two carbon atoms and two oxygen atoms respectively.
72
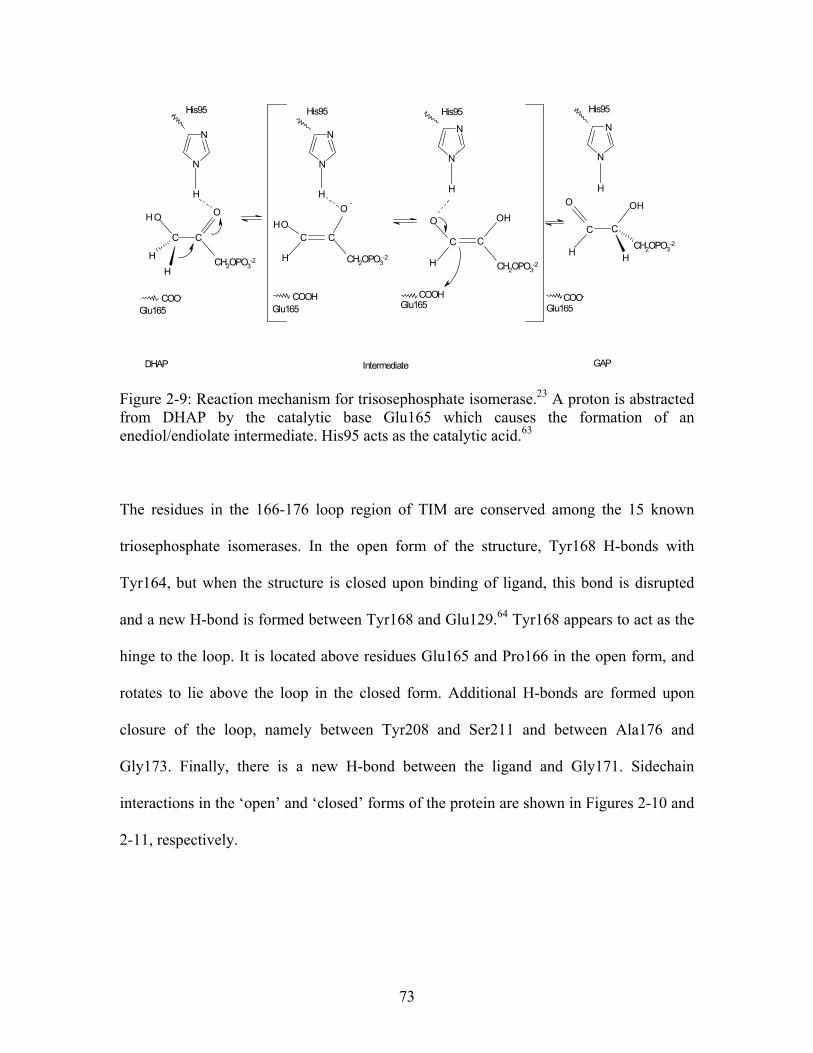
N
N
H
O
CC
OH
H
H
H
O
H
OHC C
N
N
N
N
H
O
C C
H
HO
N
N
H
C C
OH
H
O
HCH2OPO3-2
His95
COO-
Glu165
-
CH2OPO3-2
His95
COOHGlu165
His95
CH2OPO3-2
COOHGlu165
His95
CH2OPO3-2
Glu165COO-
DHAP Intermediate GAP Figure 2-9: Reaction mechanism for trisosephosphate isomerase.23 A proton is abstracted from DHAP by the catalytic base Glu165 which causes the formation of an enediol/endiolate intermediate. His95 acts as the catalytic acid.63
The residues in the 166-176 loop region of TIM are conserved among the 15 known
triosephosphate isomerases. In the open form of the structure, Tyr168 H-bonds with
Tyr164, but when the structure is closed upon binding of ligand, this bond is disrupted
and a new H-bond is formed between Tyr168 and Glu129.64 Tyr168 appears to act as the
hinge to the loop. It is located above residues Glu165 and Pro166 in the open form, and
rotates to lie above the loop in the closed form. Additional H-bonds are formed upon
closure of the loop, namely between Tyr208 and Ser211 and between Ala176 and
Gly173. Finally, there is a new H-bond between the ligand and Gly171. Sidechain
interactions in the ‘open’ and ‘closed’ forms of the protein are shown in Figures 2-10 and
2-11, respectively.
73

Gly232d
Ala212e
His95a
Cys126b
Glu97b
Glu165a
Gly233
d
Ser211d
Gly173e
Gly171
d
Trp168f
Glu129c
Pro166e
Tyr208e
Tyr164b
Ala176f
Figure 2-10: Select set of known functionally important residues for triosephosphate isomerase from yeast in the ‘open’ form (PDB ID:1YPI66). a refers to first-shell residues predicted by both THEMATICS and ET; b refers to second-shell residues predicted by THEMATICS and ET; c refers to the third-shell residue predicted by THEMATICS and ET; d refers to first-shell residues identified only by ET; e refers to second-shell residues identified only by ET; f refers to third-shell residues identified only by ET.
Gly173e
PGA Glu165
a Ala212
e
Gly233d
Gly171d
Ser211d
Trp168f
Tyr164b
Tyr208e
Pro166e
Ala176f
Glu129c
Cys126b His95
a
Glu97b
Figure 2-11: Select set of known functionally important residues for triosephosphate isomerase from yeast in the ‘closed’ form (PDB ID:2YPI66). a refers to first-shell residues predicted by both THEMATICS and ET; b refers to second-shell residues predicted by THEMATICS and ET; c refers to the third-shell residue predicted by THEMATICS and ET; d refers to first-shell residues identified only by ET; e refers to second-shell residues identified only by ET; f refers to third-shell residues identified only by ET. In the closed structure, Glu129 flips in toward Trp168. Mutations to two hinge residues, Tyr164Phe and Glu129Gln, result in a 2-fold and a 30-fold decrease in catalytic rate.64
Gly232d
74

One conservative second-shell mutation and one conservative third-shell mutation have
been made to triosephosphate isomerase, Tyr164Phe and Glu129Gln respectively, to
study the stability of the open and closed conformations.64 These are residues which are
H-bonded to the hinge Tyr168, one in the open form and one in the closed form,
respectively (Figures 2-10 and 2-11). The Tyr164Phe mutation was designed to remove
the H-bonding capabilities between Tyr168 and Tyr164. It was postulated that this
mutation would destabilize the open conformation, creating a protein in which the loop
was always partially closed. This then should inhibit the ligand from getting into the
active site pocket and decrease catalytic rate. In fact, this was not the case, and the rate
constant only decreased by a factor of 2. The Glu129Gln mutation was made in an
attempt to destabilize the ‘closed’ conformation of the protein whereby the removal of
the H-bond between Tyr168 and Glu129 would cause the ligand to be held less tightly in
place. This was the case as the Glu129Gln mutation resulted in a 30-fold decrease in the
catalytic rate. Other mutations were made to those residues creating important H-bonds in
the ‘closed’ form, and these also resulted in decreased rates.64 It was concluded that
stability of the ‘closed’ form of the protein is most important for the catalytic function of
triosephosphate isomerase.
THEMATICS and ET identify the catalytic residues His95 and Glu165, while ET
additionally finds the annotated catalytic and ligand binding residues Asn11, Lys13,
Ile170, Gly171, Gly210, Ser211, Leu230, Val231, Gly232 and Gly233. Both of the
methods identify the second shell residues Glu97, Cys126 and Tyr164; ET also finds 22
additional second shell residues, including Pro166, Gly173 and Tyr208 mentioned above.
75

One third-shell residue found by both THEMATICS and ET is Glu129, while ET
identifies 13 additional third-shell residues, including Tyr168 and Ala176 mentioned
above.
Tyrosyl-tRNA Synthetase from Bacillus stearothermophilus - (PDB ID: 1TYD ) 24
One of the first proteins mutated using site-directed mutagenesis was tyrosyl-tRNA
synthetase (TyrRS), the enzyme responsible for the attachment of tyrosine to its cognate
tRNA. The accurate incorporation of the cognate tRNA is essential for the translation
of genetic code. The study of TyrRS allowed enzymologists to finally answer questions
about the types of interactions involved in catalysis and substrate binding. The reaction
proceeds via a two step mechanism (Figure 2-12), where tyrosine is first activated by a
molecule of ATP to form tyrosyl adenylate followed by the transfer to tyrosyl tRNA.
The carboxylate group of the tyrosine acts as a nucleophile and the pyrophosphate from
the activating ATP is the leaving group.
1,67
68
68
67
1
76
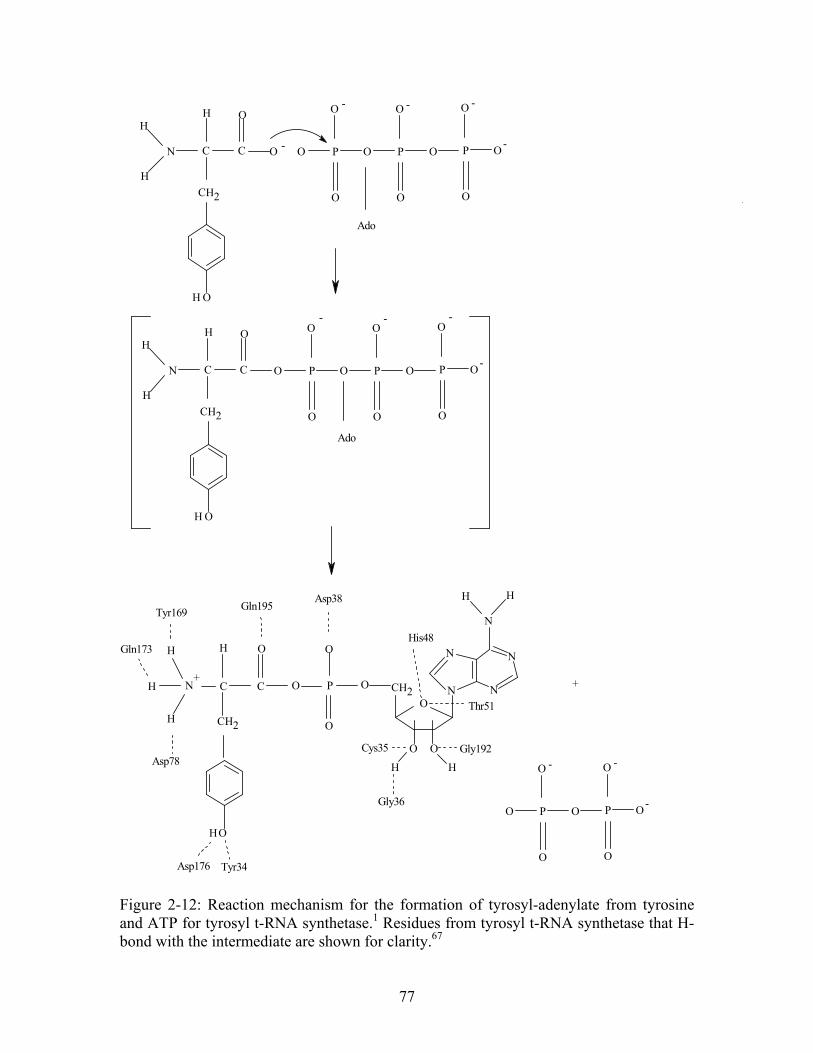
O
O O
H H
N
N
N
N
N
HH
OP
O
O
OC
O
C
H
CH
H
H
H
OH
N CH
OH
CH
C C
O
ON
HH
H
O P
O
O
O
P
O
O
O
P
O
O
O
OH
CH
C C
O
N
HH
H
O P
O
O
O
P
O
O
O
P
O
O
O
O P
O
O
O
P
O
O
O
Ado
His48
Thr51
Asp38
Asp78
Asp176
Cys35 Gly192
Gly36
Tyr34
Tyr169
Gln173
Gln195
+
2
2
2
-
- - -
-
Ado
2
- - -
-
+
- -
-
Figure 2-12: Reaction mechanism for the formation of tyrosyl-adenylate from tyrosine and ATP for tyrosyl t-RNA synthetase.1 Residues from tyrosyl t-RNA synthetase that H-bond with the intermediate are shown for clarity.67
77

The crystal structure of TyrRS (PDB ID: 1TYD ) shows that the molecule has three
domains, an α/β domain consisting of 5 parallel and 1 anti-parallel β-strands, a helical
domain, and a disordered domain. It is composed of two identical subunits, each having
a complete active site. However, only one active site is functional in solution per dimer.
The tyrosine binding site lies at the bottom of a deep cleft. At the bottom of this cleft are
two residues, Tyr34 and Asp176 which form H-bonds with the phenolic hydroxyl group
of the tyrosine side chain. Through random mutagenesis studies, it was discovered that
Arg86 was crucial for catalysis by altering the free energy profile of the reaction. These
residues bind to the tyrosyl adenylate intermediate. Those residues which form H-bonds
with the tyrosine substrate include Tyr34, Gly36, Asp38, Thr40, His48, Leu68, Gly70,
Thr73, Asp78, Tyr169, Gln173, Asp176, Gln189 and Gln195. Those residues that form
H-bonds with the substrate intermediate, tyrosyl adenylate, are Tyr34, Cys35, Gly36,
Asp38, His48, Thr51 (Ser51), Asp78, Tyr169, Gln173, Gln192, Asp194 and Gln195.
Finally, an H-bond between His48 and a second-shell residue, His45, stabilizes the
substrate intermediate (Figure 2-13).
24
24
69
70
24,71
24
78

His45d
Glu166d
Asp38a
TYR
Asp78a
His48b
Tyr169c
Asp194e
Gln195c
Gly192 Cys35
Tyr34c
Asp176a
Gly36c
Gln173c
Ser51
Figure 2-13: Active site of tyrosyl-tRNA synthetase (PDB ID: 1TYD24) showing first- and second-shell residues in contact with the tyrosine. Red = tyrosine. a refers to first-shell residues identified by THEMATICS and ET; b refers to first-shell residue identified by only THEMATICS; c refers to first-shell residues identified only by ET; d refers to the second-shell residues identified by THEMATICS and ET; e refers to a third-shell residues identified only by ET; those residues with no superscripts are known to be in the active site, but are not identified by either THEMATICS or ET. Mutation of His45 to Gly results in a 250-fold decrease in catalytic rate indicating this second-shell residue is necessary to stabilize and orient the catalytic residue His48.1
To probe the importance of the second-shell residue His45 with respect to its role in
stabilizing the substrate intermediate, site-directed mutagenesis studies were performed
whereby His was replaced by Gly. His45 is H-bonded to His48, a residue which is H-
bonded to the tyrosyl adenylate intermediate (Figure 2-13). This mutation was expected
to remove the H-bonding capabilities of His45 resulting in an enzyme with decreased
catalytic activity and decreased binding of pyrophosphate. The resultant enzyme had a
250-fold decrease in catalytic rate and a 4-fold weakening of the ATP binding constant.
Therefore, the authors asserted that His45 is a stabilizing residue needed to keep His48 in
position for the reaction to proceed.
1
79

THEMATICS and ET both identify the ligand binding residues Asp38, Asp78 and
Asp176. THEMATICS additionally finds another ligand binding residue, His48, while
ET identifies 10 of the other ligand binding residues. Both methods also identify second-
shell residues His45 and Glu166, while ET finds 15 other second-shell residues. Finally,
ET identifies 13 third-shell residues, whereas THEMATICS finds none.
2.4 Summary of Results
THEMATICS predicts at least one annotated active site residue in the first-shell for 37 of
the 39 proteins; at least one second-shell residue is predicted by THEMATICS for 31 of
the proteins; and at least one third-shell residue is predicted by THEMATICS in 19 of the
39 proteins in the test set. THEMATICS identifies an average of 4.1 residues in the first-
shell, 2.0 residues in the second-shell and 1.3 residues in the third-shell. For all 39
proteins in the test set, the ET predictions include second- and third-shell residues as well
as first-shell residues. ET identifies an average of 13.3 residues in the first-shell, an
average of 21.7 residues in the second-shell, and an average of 18.8 residues in the third-
shell. Each method identified additional residues that did not belong to either the first,
second or third interaction spheres, and these residues were excluded from this analysis.
However, of the 39 proteins in the test set, THEMATICS identified additional residues
for 18 of the proteins with an average of 1.7 residues. On the other hand, ET found
additional residues for all 39 proteins in the test set with an average of 21.1 residues. This
translates to the fact that ET predictions include approximately 22% of the protein, while
THEMATICS predictions include only 2.6% of the protein. The THEMATICS method is
thus highly selective and is a useful computational method for the identification of
80

specific functionally important residues in the first, second and third interaction spheres
of a protein.
Based on the nature of the ET method, it is expected that the predicted residues are well
conserved, which they are, with average normalized conservation scores of -1.1, -0.90
and -0.90 for the first-, second- and third-shells, respectively. Comparison of
conservation scores with THEMATICS indicates that the THEMATICS predicted
residues are also well conserved; -1.2, -1.0 and -0.85 for the first-, second- and third-
shells, respectively. At this point we are unable to measure the efficacy (i.e recall and
specificity) of either THEMATICS or ET in the prediction of functionally important
second- and third-shell residues, simply because there are not enough experimental
examples available in the literature to use for validation purposes.
2.5 Conservative versus Nonconservative Mutations
While the experimental mutation data cited in this paper relate only differences in k , we
feel it is important to note that non-conservative mutations may have an effect on the
proper or productive binding of substrate, and as a result affect the catalytic efficiency of
the enzyme. The difficulty therefore lies in the interpretation of the data. For example,
two second-shell mutations to Glu117 in human carbonic anhydrase II were analyzed to
understand the functional importance of the hydrogen bond network of the zinc ligand
His119 by characterizing the catalytic efficiency of two mutants. The Glu117Ala mutant
resulted in essentially no change in catalytic rate, but an approximately 3-fold increase in
cat
81

K , while the Glu117Asp mutant also resulted in essentially no change in catalytic rate,
but a 2-fold increase in K . The results indicate that this indirect ligand may stabilize
the transition state for CO hydration, and the non-conservative mutations result in
structural changes in the active site. The conservative Glu117Gln mutation was not made,
so it is difficult to interpret the results.
M
M72
2
On the other hand, three second-shell mutations to CAII at the 106 position, Glu106Ala,
Glu106Gln and Glu106Asp were examined, and include both non-conservative and
conservative mutations. The resultant mutants have an 1100-fold decrease and an 850-
fold decrease in k for the Glu106Ala and Glu106Gln mutants respectively, while the
Glu106Asp mutation has no effect on CO hydration compared to wild type. The K for
unmodified CAII is approximately 10 mM, while the K ’s for the three mutants
mentioned above are 0.59 mM, 0.040 mM and 6.0 mM respectively. Glu106 acts as an H-
bond acceptor with the hydroxyl group of Thr199, and acts to keep Thr199 in the correct
orientation for catalysis. Mutation to alanine removes not only the H-bonding
capabilities, but also removes the charge. The result of this mutation is a large decrease in
k , with a 16-fold decrease in K . Mutation to Gln results in the loss of the negative
charge and causes an 850-fold decrease in k and a 250-fold decrease in K . Finally,
mutation to Asp, which retains the negative charge, does not result in any large change in
either k or K . The crystal structure reveals that the H-bond between residue 106 and
199 is intact. The protein backbone has moved only slightly to accommodate the slightly
smaller side chain.
cat
2 M
M
cat M
cat M
cat M
82

Computational analysis of experimental mutagenesis data is complicated by the choice of
mutation. Mutagenesis of small residues (e.g. Gly or Ala to His or Lys) to large residues
or large residues to small residues can have a substantial impact on the substrate binding
environment and catalytic efficiency of the enzyme, and may create a false impression of
the functional importance of the position. We suggest that in a study such as this, where
we are looking at the impact of second- and third-shell residues on catalysis, that isosteric
mutations may be the most informative as they are the least likely to lead to large
structural perturbations.
2.6 Conclusions
In this chapter, the ability of THEMATICS to identify a subset of remote residues has
been demonstrated and these results have been compared with the sequence-based ET
method. The residues identified by these methods were compared with experimental
mutagenesis data from the literature, and our results indicate that both THEMATICS and
ET predict functionally important residues not only in the first-shell of an interaction site,
but also residues located in interaction spheres beyond the first.
While experimental mutational data do exist in the rational protein design and directed
evolution literature for second- and third-shell residues in addition to first-shell, no
systematic studies have been undertaken to explore their significance in relation to
catalytic efficiency. This study was the first systematic approach to computationally
identifying functional residues located in the outer interaction spheres of enzymes (i.e.
beyond the nominal active site or first-shell). What is most striking is that two completely
83

different types of theoretical methods both support multilayer active sites. Understanding
how nature designs enzyme active sites is a fundamental question in enzymology with
implications for protein engineering. The present results suggest that computational
methods could help guide the identification of functional second- and/or third-shell
residues and can serve as a useful guide for rational protein design studies.
Having established the concept of multilayer active sites, the following chapters begin a
systematic experimental study into one specific enzyme, Co-type nitrile hydratase from
Pseudomonas putida (ppNHase). The following chapter, chapter 3, will be an
introduction to the family of nitrile hydratase enzymes and an introduction to Michaelis-
Menten kinetics. In addition, the first known structure of nitrile hydratase from
Pseudomonas putida will be presented, followed by a full structural and kinetic analysis
of wild type ppNHase. A comprehensive catalytic analysis of all Co-type and Fe-type
nitrile hydratases from the literature will be also presented.
84
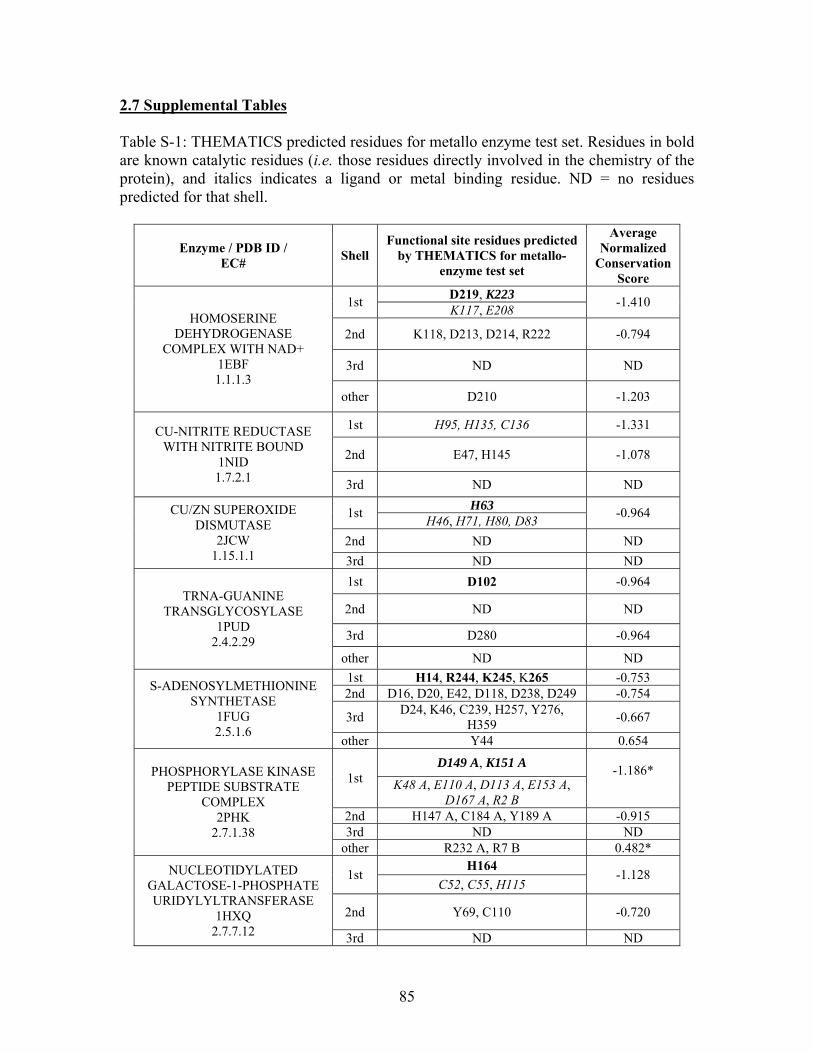
2.7 Supplemental Tables Table S-1: THEMATICS predicted residues for metallo enzyme test set. Residues in bold are known catalytic residues (i.e. those residues directly involved in the chemistry of the protein), and italics indicates a ligand or metal binding residue. ND = no residues predicted for that shell.
Enzyme / PDB ID / EC#
Shell Functional site residues predicted
by THEMATICS for metallo- enzyme test set
Average Normalized
Conservation Score
D219, K223 1st
K117, E208 -1.410
2nd K118, D213, D214, R222 -0.794
3rd ND ND
HOMOSERINE DEHYDROGENASE
COMPLEX WITH NAD+ 1EBF 1.1.1.3
other D210 -1.203
1st H95, H135, C136 -1.331
2nd E47, H145 -1.078
CU-NITRITE REDUCTASE WITH NITRITE BOUND
1NID 1.7.2.1 3rd ND ND
H63 1st
H46, H71, H80, D83 -0.964
2nd ND ND
CU/ZN SUPEROXIDE DISMUTASE
2JCW 1.15.1.1 3rd ND ND
1st D102 -0.964
2nd ND ND
3rd D280 -0.964
TRNA-GUANINE TRANSGLYCOSYLASE
1PUD 2.4.2.29
other ND ND
1st H14, R244, K245, K265 -0.753 2nd D16, D20, E42, D118, D238, D249 -0.754
3rd D24, K46, C239, H257, Y276,
H359 -0.667
S-ADENOSYLMETHIONINE SYNTHETASE
1FUG 2.5.1.6
other Y44 0.654
D149 A, K151 A 1st K48 A, E110 A, D113 A, E153 A,
D167 A, R2 B
-1.186*
2nd H147 A, C184 A, Y189 A -0.915
3rd ND ND
PHOSPHORYLASE KINASE PEPTIDE SUBSTRATE
COMPLEX 2PHK
2.7.1.38 other R232 A, R7 B 0.482*
H164 1st
C52, C55, H115 -1.128
2nd Y69, C110 -0.720
NUCLEOTIDYLATED GALACTOSE-1-PHOSPHATE URIDYLYLTRANSFERASE
1HXQ 2.7.7.12 3rd ND ND
85
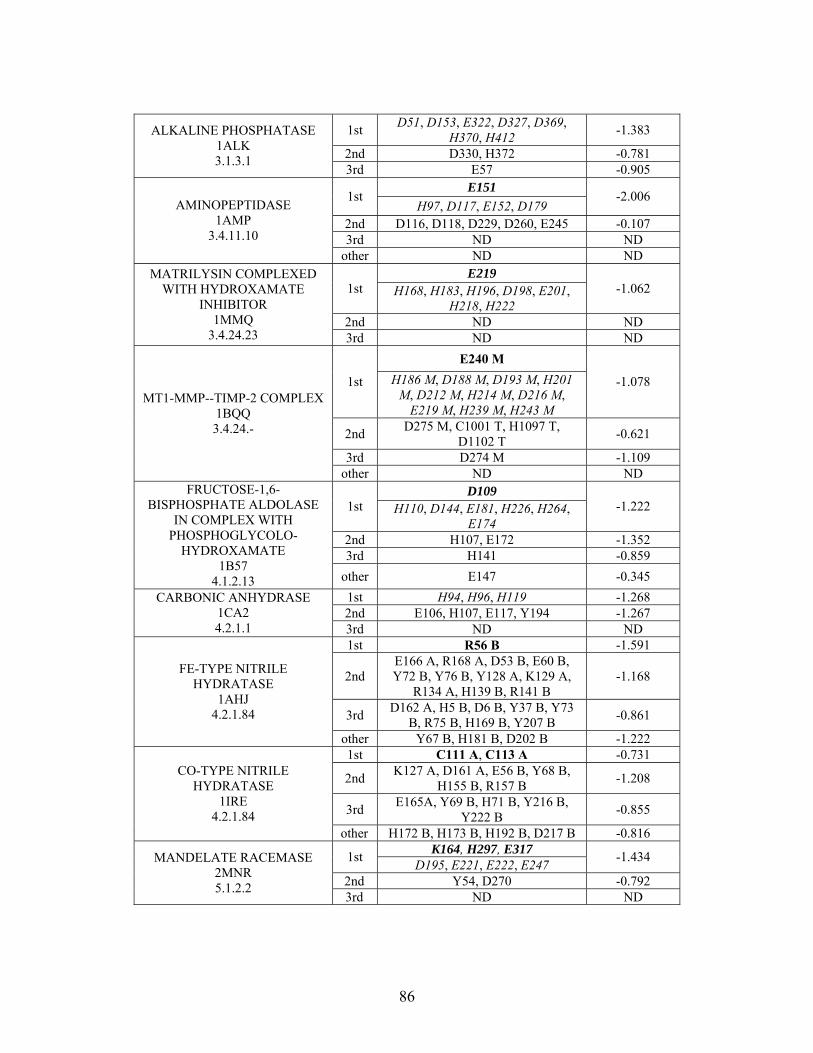
1st D51, D153, E322, D327, D369,
H370, H412 -1.383
2nd D330, H372 -0.781
ALKALINE PHOSPHATASE 1ALK 3.1.3.1
3rd E57 -0.905 E151
1st H97, D117, E152, D179
-2.006
2nd D116, D118, D229, D260, E245 -0.107 3rd ND ND
AMINOPEPTIDASE 1AMP
3.4.11.10
other ND ND E219
1st H168, H183, H196, D198, E201, H218, H222
-1.062
2nd ND ND
MATRILYSIN COMPLEXED WITH HYDROXAMATE
INHIBITOR 1MMQ
3.4.24.23 3rd ND ND
E240 M
1st H186 M, D188 M, D193 M, H201 M, D212 M, H214 M, D216 M,
E219 M, H239 M, H243 M
-1.078
2nd D275 M, C1001 T, H1097 T,
D1102 T -0.621
3rd D274 M -1.109
MT1-MMP--TIMP-2 COMPLEX 1BQQ 3.4.24.-
other ND ND D109
1st H110, D144, E181, H226, H264, E174
-1.222
2nd H107, E172 -1.352 3rd H141 -0.859
FRUCTOSE-1,6-BISPHOSPHATE ALDOLASE
IN COMPLEX WITH PHOSPHOGLYCOLO-
HYDROXAMATE 1B57
4.1.2.13 other E147 -0.345
1st H94, H96, H119 -1.268 2nd E106, H107, E117, Y194 -1.267
CARBONIC ANHYDRASE 1CA2 4.2.1.1 3rd ND ND
1st R56 B -1.591
2nd E166 A, R168 A, D53 B, E60 B, Y72 B, Y76 B, Y128 A, K129 A,
R134 A, H139 B, R141 B -1.168
3rd D162 A, H5 B, D6 B, Y37 B, Y73
B, R75 B, H169 B, Y207 B -0.861
FE-TYPE NITRILE HYDRATASE
1AHJ 4.2.1.84
other Y67 B, H181 B, D202 B -1.222 1st C111 A, C113 A -0.731
2nd K127 A, D161 A, E56 B, Y68 B,
H155 B, R157 B -1.208
3rd E165A, Y69 B, H71 B, Y216 B,
Y222 B -0.855
CO-TYPE NITRILE HYDRATASE
1IRE 4.2.1.84
other H172 B, H173 B, H192 B, D217 B -0.816 K164, H297, E317
1st D195, E221, E222, E247
-1.434
2nd Y54, D270 -0.792
MANDELATE RACEMASE 2MNR 5.1.2.2
3rd ND ND
86
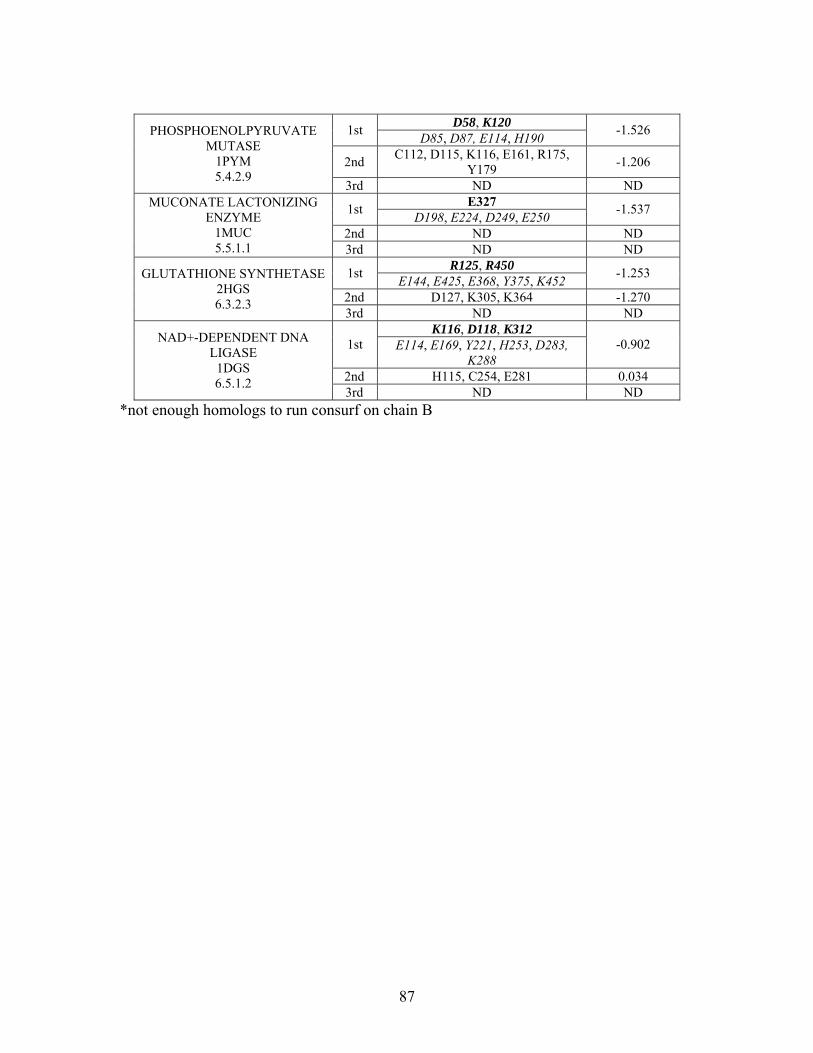
D58, K120
1st D85, D87, E114, H190
-1.526
2nd C112, D115, K116, E161, R175,
Y179 -1.206
PHOSPHOENOLPYRUVATE MUTASE
1PYM 5.4.2.9
3rd ND ND E327
1st D198, E224, D249, E250
-1.537
2nd ND ND
MUCONATE LACTONIZING ENZYME
1MUC 5.5.1.1 3rd ND ND
R125, R450 1st
E144, E425, E368, Y375, K452 -1.253
2nd D127, K305, K364 -1.270
GLUTATHIONE SYNTHETASE 2HGS 6.3.2.3
3rd ND ND K116, D118, K312
1st E114, E169, Y221, H253, D283, K288
-0.902
2nd H115, C254, E281 0.034
NAD+-DEPENDENT DNA LIGASE
1DGS 6.5.1.2
3rd ND ND *not enough homologs to run consurf on chain B
87
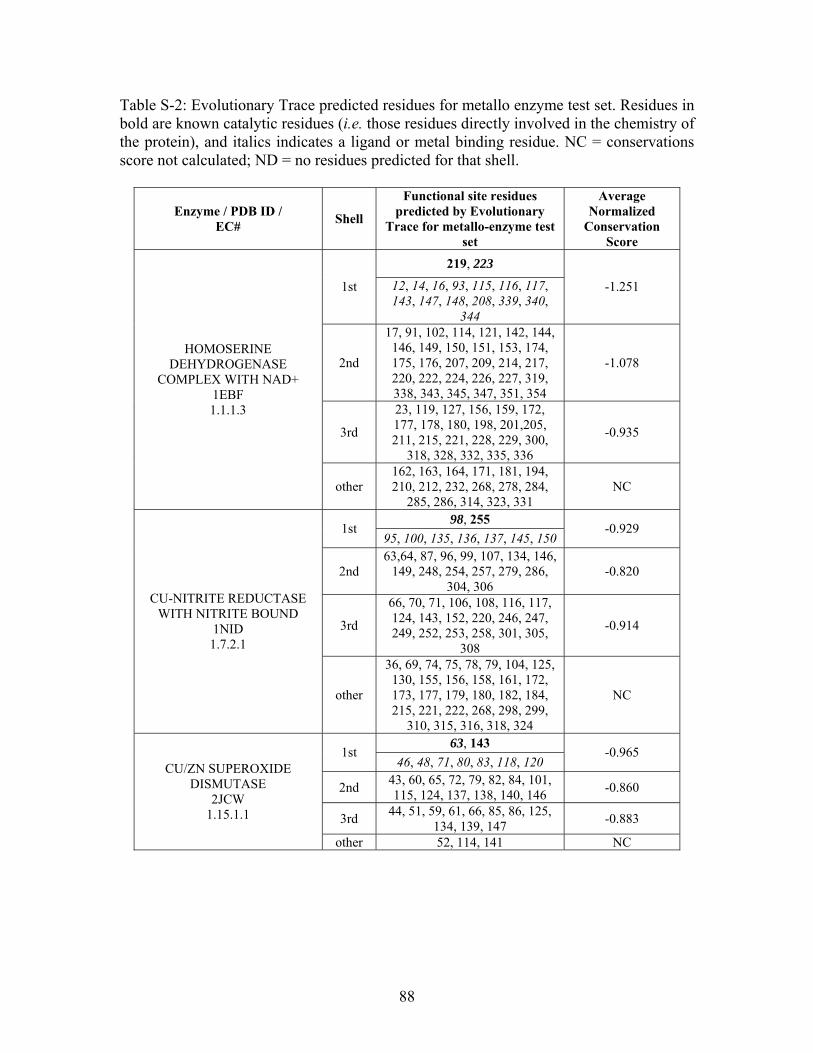
Table S-2: Evolutionary Trace predicted residues for metallo enzyme test set. Residues in bold are known catalytic residues (i.e. those residues directly involved in the chemistry of the protein), and italics indicates a ligand or metal binding residue. NC = conservations score not calculated; ND = no residues predicted for that shell.
Enzyme / PDB ID / EC#
Shell
Functional site residues predicted by Evolutionary
Trace for metallo-enzyme test set
Average Normalized
Conservation Score
219, 223
1st 12, 14, 16, 93, 115, 116, 117, 143, 147, 148, 208, 339, 340,
344
-1.251
2nd
17, 91, 102, 114, 121, 142, 144, 146, 149, 150, 151, 153, 174, 175, 176, 207, 209, 214, 217, 220, 222, 224, 226, 227, 319, 338, 343, 345, 347, 351, 354
-1.078
3rd
23, 119, 127, 156, 159, 172, 177, 178, 180, 198, 201,205, 211, 215, 221, 228, 229, 300,
318, 328, 332, 335, 336
-0.935
HOMOSERINE DEHYDROGENASE
COMPLEX WITH NAD+ 1EBF 1.1.1.3
other 162, 163, 164, 171, 181, 194, 210, 212, 232, 268, 278, 284,
285, 286, 314, 323, 331 NC
98, 255 1st
95, 100, 135, 136, 137, 145, 150 -0.929
2nd 63,64, 87, 96, 99, 107, 134, 146,
149, 248, 254, 257, 279, 286, 304, 306
-0.820
3rd
66, 70, 71, 106, 108, 116, 117, 124, 143, 152, 220, 246, 247, 249, 252, 253, 258, 301, 305,
308
-0.914
CU-NITRITE REDUCTASE WITH NITRITE BOUND
1NID 1.7.2.1
other
36, 69, 74, 75, 78, 79, 104, 125, 130, 155, 156, 158, 161, 172, 173, 177, 179, 180, 182, 184, 215, 221, 222, 268, 298, 299,
310, 315, 316, 318, 324
NC
63, 143 1st
46, 48, 71, 80, 83, 118, 120 -0.965
2nd 43, 60, 65, 72, 79, 82, 84, 101, 115, 124, 137, 138, 140, 146
-0.860
3rd 44, 51, 59, 61, 66, 85, 86, 125,
134, 139, 147 -0.883
CU/ZN SUPEROXIDE DISMUTASE
2JCW 1.15.1.1
other 52, 114, 141 NC
88
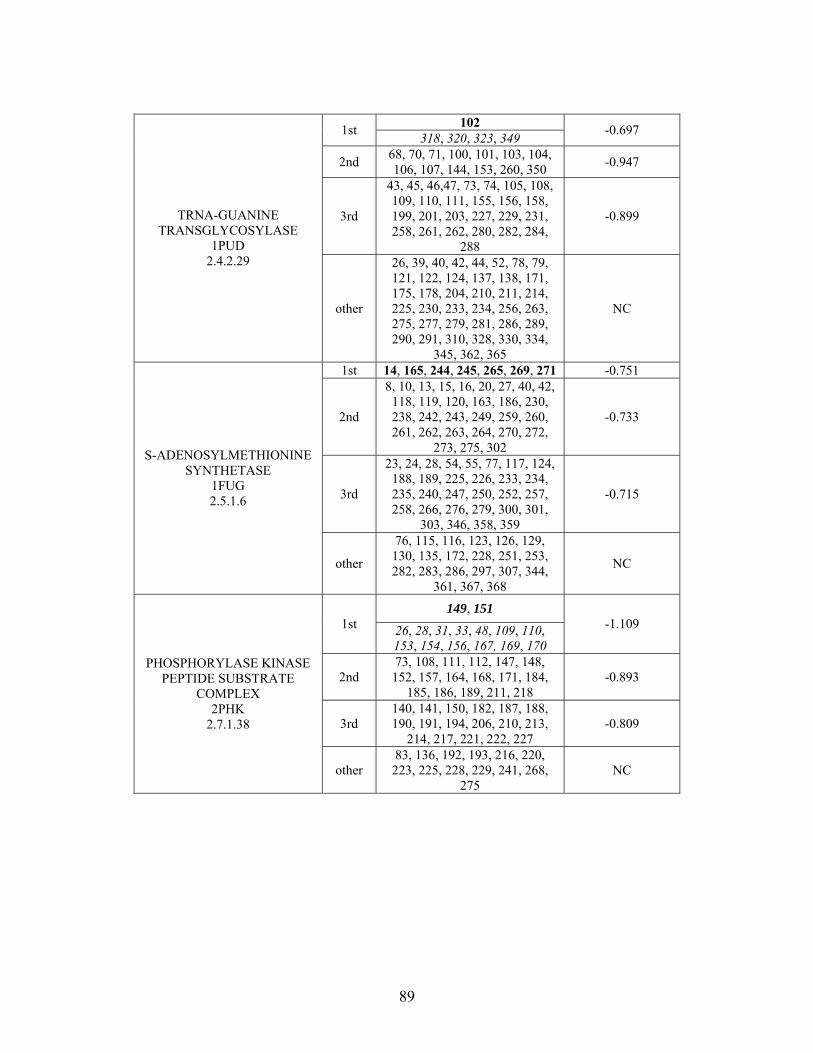
102
1st 318, 320, 323, 349
-0.697
2nd 68, 70, 71, 100, 101, 103, 104, 106, 107, 144, 153, 260, 350
-0.947
3rd
43, 45, 46,47, 73, 74, 105, 108, 109, 110, 111, 155, 156, 158, 199, 201, 203, 227, 229, 231, 258, 261, 262, 280, 282, 284,
288
-0.899 TRNA-GUANINE TRANSGLYCOSYLASE
1PUD 2.4.2.29
other
26, 39, 40, 42, 44, 52, 78, 79, 121, 122, 124, 137, 138, 171, 175, 178, 204, 210, 211, 214, 225, 230, 233, 234, 256, 263, 275, 277, 279, 281, 286, 289, 290, 291, 310, 328, 330, 334,
345, 362, 365
NC
1st 14, 165, 244, 245, 265, 269, 271 -0.751
2nd
8, 10, 13, 15, 16, 20, 27, 40, 42, 118, 119, 120, 163, 186, 230, 238, 242, 243, 249, 259, 260, 261, 262, 263, 264, 270, 272,
273, 275, 302
-0.733
3rd
23, 24, 28, 54, 55, 77, 117, 124, 188, 189, 225, 226, 233, 234, 235, 240, 247, 250, 252, 257, 258, 266, 276, 279, 300, 301,
303, 346, 358, 359
-0.715
S-ADENOSYLMETHIONINE SYNTHETASE
1FUG 2.5.1.6
other
76, 115, 116, 123, 126, 129, 130, 135, 172, 228, 251, 253, 282, 283, 286, 297, 307, 344,
361, 367, 368
NC
149, 151 1st 26, 28, 31, 33, 48, 109, 110,
153, 154, 156, 167, 169, 170
-1.109
2nd 73, 108, 111, 112, 147, 148, 152, 157, 164, 168, 171, 184,
185, 186, 189, 211, 218 -0.893
3rd 140, 141, 150, 182, 187, 188, 190, 191, 194, 206, 210, 213,
214, 217, 221, 222, 227 -0.809
PHOSPHORYLASE KINASE PEPTIDE SUBSTRATE
COMPLEX 2PHK
2.7.1.38
other 83, 136, 192, 193, 216, 220, 223, 225, 228, 229, 241, 268,
275 NC
89
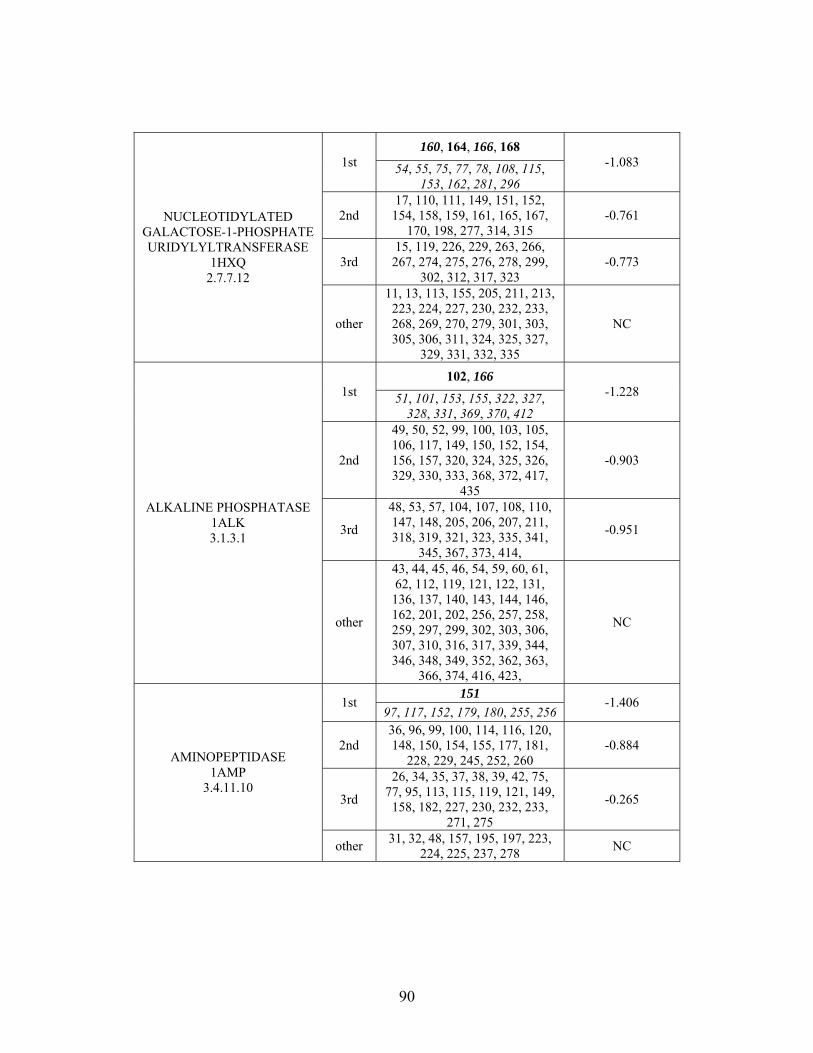
160, 164, 166, 168 1st 54, 55, 75, 77, 78, 108, 115,
153, 162, 281, 296
-1.083
2nd 17, 110, 111, 149, 151, 152, 154, 158, 159, 161, 165, 167,
170, 198, 277, 314, 315 -0.761
3rd 15, 119, 226, 229, 263, 266, 267, 274, 275, 276, 278, 299,
302, 312, 317, 323 -0.773
NUCLEOTIDYLATED GALACTOSE-1-PHOSPHATE URIDYLYLTRANSFERASE
1HXQ 2.7.7.12
other
11, 13, 113, 155, 205, 211, 213, 223, 224, 227, 230, 232, 233, 268, 269, 270, 279, 301, 303, 305, 306, 311, 324, 325, 327,
329, 331, 332, 335
NC
102, 166 1st 51, 101, 153, 155, 322, 327,
328, 331, 369, 370, 412
-1.228
2nd
49, 50, 52, 99, 100, 103, 105, 106, 117, 149, 150, 152, 154, 156, 157, 320, 324, 325, 326, 329, 330, 333, 368, 372, 417,
435
-0.903
3rd
48, 53, 57, 104, 107, 108, 110, 147, 148, 205, 206, 207, 211, 318, 319, 321, 323, 335, 341,
345, 367, 373, 414,
-0.951
ALKALINE PHOSPHATASE 1ALK 3.1.3.1
other
43, 44, 45, 46, 54, 59, 60, 61, 62, 112, 119, 121, 122, 131, 136, 137, 140, 143, 144, 146, 162, 201, 202, 256, 257, 258, 259, 297, 299, 302, 303, 306, 307, 310, 316, 317, 339, 344, 346, 348, 349, 352, 362, 363,
366, 374, 416, 423,
NC
151 1st
97, 117, 152, 179, 180, 255, 256 -1.406
2nd 36, 96, 99, 100, 114, 116, 120, 148, 150, 154, 155, 177, 181,
228, 229, 245, 252, 260 -0.884
3rd
26, 34, 35, 37, 38, 39, 42, 75, 77, 95, 113, 115, 119, 121, 149,
158, 182, 227, 230, 232, 233, 271, 275
-0.265
AMINOPEPTIDASE 1AMP
3.4.11.10
other 31, 32, 48, 157, 195, 197, 223,
224, 225, 237, 278 NC
90
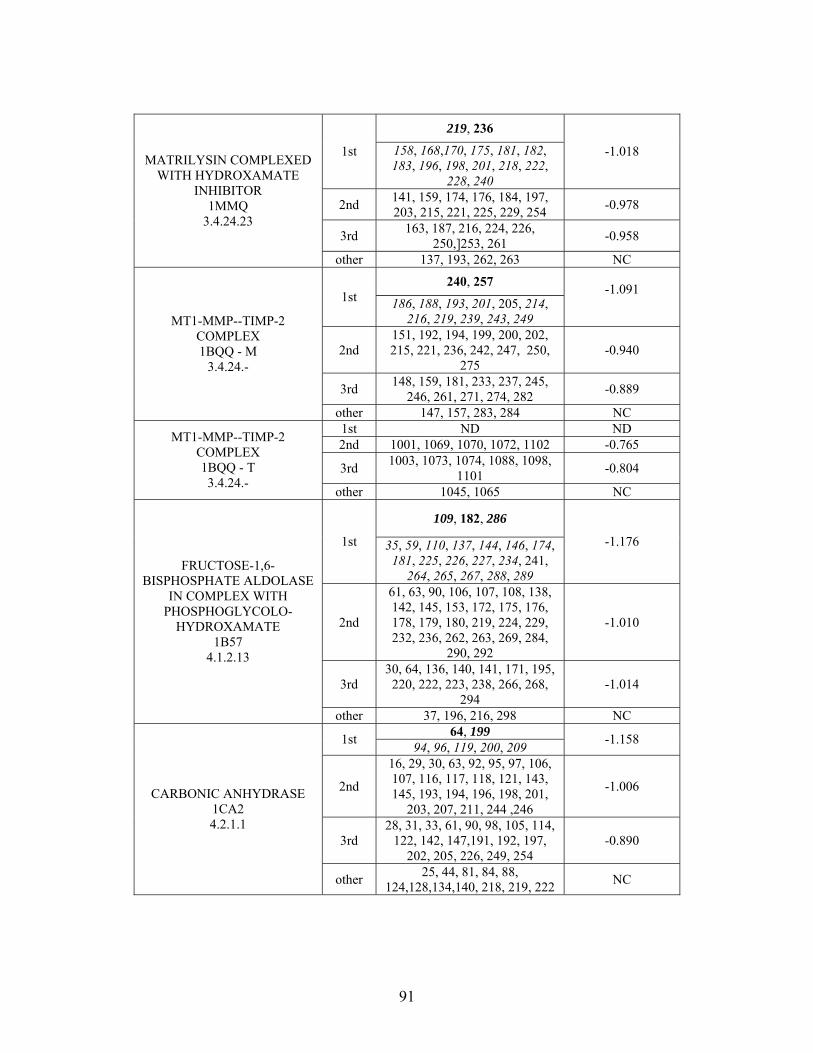
219, 236
1st 158, 168,170, 175, 181, 182, 183, 196, 198, 201, 218, 222,
228, 240
-1.018
2nd 141, 159, 174, 176, 184, 197, 203, 215, 221, 225, 229, 254
-0.978
3rd 163, 187, 216, 224, 226,
250,]253, 261 -0.958
MATRILYSIN COMPLEXED WITH HYDROXAMATE
INHIBITOR 1MMQ
3.4.24.23
other 137, 193, 262, 263 NC
240, 257 1st 186, 188, 193, 201, 205, 214,
216, 219, 239, 243, 249
-1.091
2nd 151, 192, 194, 199, 200, 202, 215, 221, 236, 242, 247, 250,
275 -0.940
3rd 148, 159, 181, 233, 237, 245,
246, 261, 271, 274, 282 -0.889
MT1-MMP--TIMP-2 COMPLEX 1BQQ - M
3.4.24.-
other 147, 157, 283, 284 NC 1st ND ND 2nd 1001, 1069, 1070, 1072, 1102 -0.765
3rd 1003, 1073, 1074, 1088, 1098,
1101 -0.804
MT1-MMP--TIMP-2 COMPLEX 1BQQ - T 3.4.24.-
other 1045, 1065 NC
109, 182, 286
1st 35, 59, 110, 137, 144, 146, 174, 181, 225, 226, 227, 234, 241,
264, 265, 267, 288, 289
-1.176
2nd
61, 63, 90, 106, 107, 108, 138, 142, 145, 153, 172, 175, 176, 178, 179, 180, 219, 224, 229, 232, 236, 262, 263, 269, 284,
290, 292
-1.010
3rd 30, 64, 136, 140, 141, 171, 195,
220, 222, 223, 238, 266, 268, 294
-1.014
FRUCTOSE-1,6-BISPHOSPHATE ALDOLASE
IN COMPLEX WITH PHOSPHOGLYCOLO-
HYDROXAMATE 1B57
4.1.2.13
other 37, 196, 216, 298 NC 64, 199
1st 94, 96, 119, 200, 209
-1.158
2nd
16, 29, 30, 63, 92, 95, 97, 106, 107, 116, 117, 118, 121, 143, 145, 193, 194, 196, 198, 201,
203, 207, 211, 244 ,246
-1.006
3rd 28, 31, 33, 61, 90, 98, 105, 114,
122, 142, 147,191, 192, 197, 202, 205, 226, 249, 254
-0.890
CARBONIC ANHYDRASE 1CA2 4.2.1.1
other 25, 44, 81, 84, 88,
124,128,134,140, 218, 219, 222 NC
91
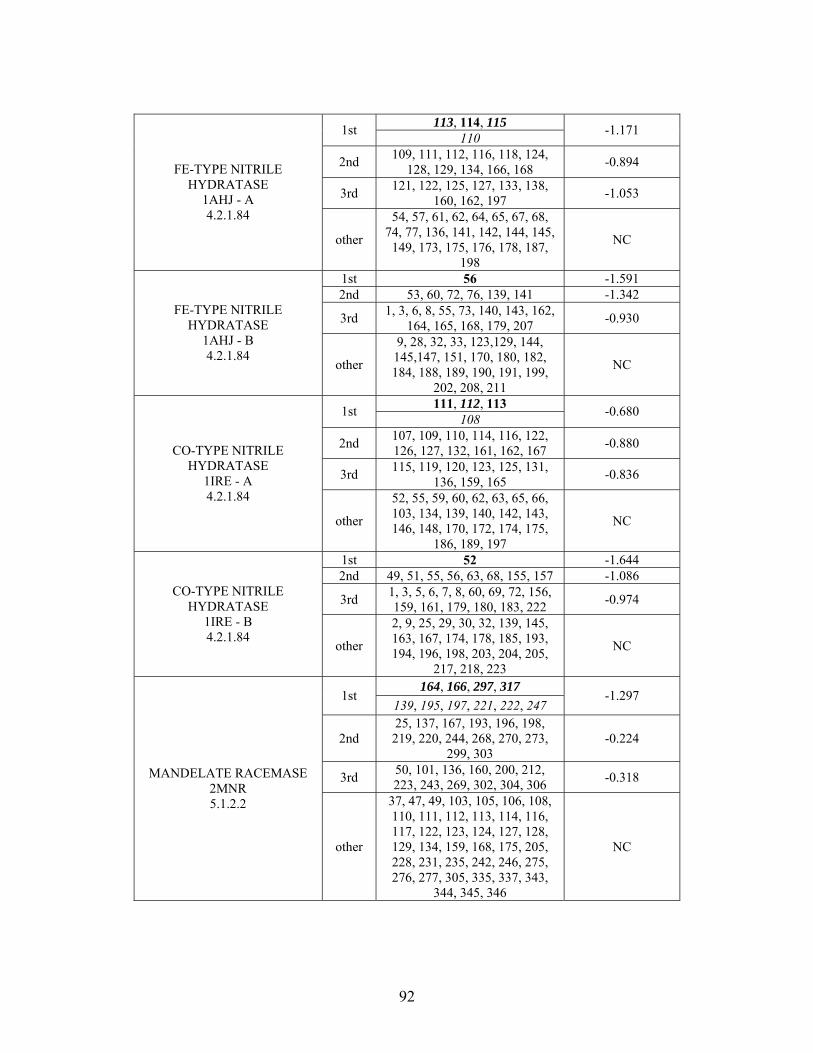
113, 114, 115
1st 110
-1.171
2nd 109, 111, 112, 116, 118, 124,
128, 129, 134, 166, 168 -0.894
3rd 121, 122, 125, 127, 133, 138,
160, 162, 197 -1.053
FE-TYPE NITRILE HYDRATASE
1AHJ - A 4.2.1.84
other
54, 57, 61, 62, 64, 65, 67, 68, 74, 77, 136, 141, 142, 144, 145,
149, 173, 175, 176, 178, 187, 198
NC
1st 56 -1.591 2nd 53, 60, 72, 76, 139, 141 -1.342
3rd 1, 3, 6, 8, 55, 73, 140, 143, 162,
164, 165, 168, 179, 207 -0.930
FE-TYPE NITRILE HYDRATASE
1AHJ - B 4.2.1.84
other
9, 28, 32, 33, 123,129, 144, 145,147, 151, 170, 180, 182, 184, 188, 189, 190, 191, 199,
202, 208, 211
NC
111, 112, 113 1st
108 -0.680
2nd 107, 109, 110, 114, 116, 122, 126, 127, 132, 161, 162, 167
-0.880
3rd 115, 119, 120, 123, 125, 131,
136, 159, 165 -0.836
CO-TYPE NITRILE HYDRATASE
1IRE - A 4.2.1.84
other
52, 55, 59, 60, 62, 63, 65, 66, 103, 134, 139, 140, 142, 143, 146, 148, 170, 172, 174, 175,
186, 189, 197
NC
1st 52 -1.644 2nd 49, 51, 55, 56, 63, 68, 155, 157 -1.086
3rd 1, 3, 5, 6, 7, 8, 60, 69, 72, 156, 159, 161, 179, 180, 183, 222
-0.974 CO-TYPE NITRILE
HYDRATASE 1IRE - B 4.2.1.84
other
2, 9, 25, 29, 30, 32, 139, 145, 163, 167, 174, 178, 185, 193, 194, 196, 198, 203, 204, 205,
217, 218, 223
NC
164, 166, 297, 317 1st
139, 195, 197, 221, 222, 247 -1.297
2nd 25, 137, 167, 193, 196, 198, 219, 220, 244, 268, 270, 273,
299, 303 -0.224
3rd 50, 101, 136, 160, 200, 212, 223, 243, 269, 302, 304, 306
-0.318 MANDELATE RACEMASE 2MNR 5.1.2.2
other
37, 47, 49, 103, 105, 106, 108, 110, 111, 112, 113, 114, 116, 117, 122, 123, 124, 127, 128, 129, 134, 159, 168, 175, 205, 228, 231, 235, 242, 246, 275, 276, 277, 305, 335, 337, 343,
344, 345, 346
NC
92
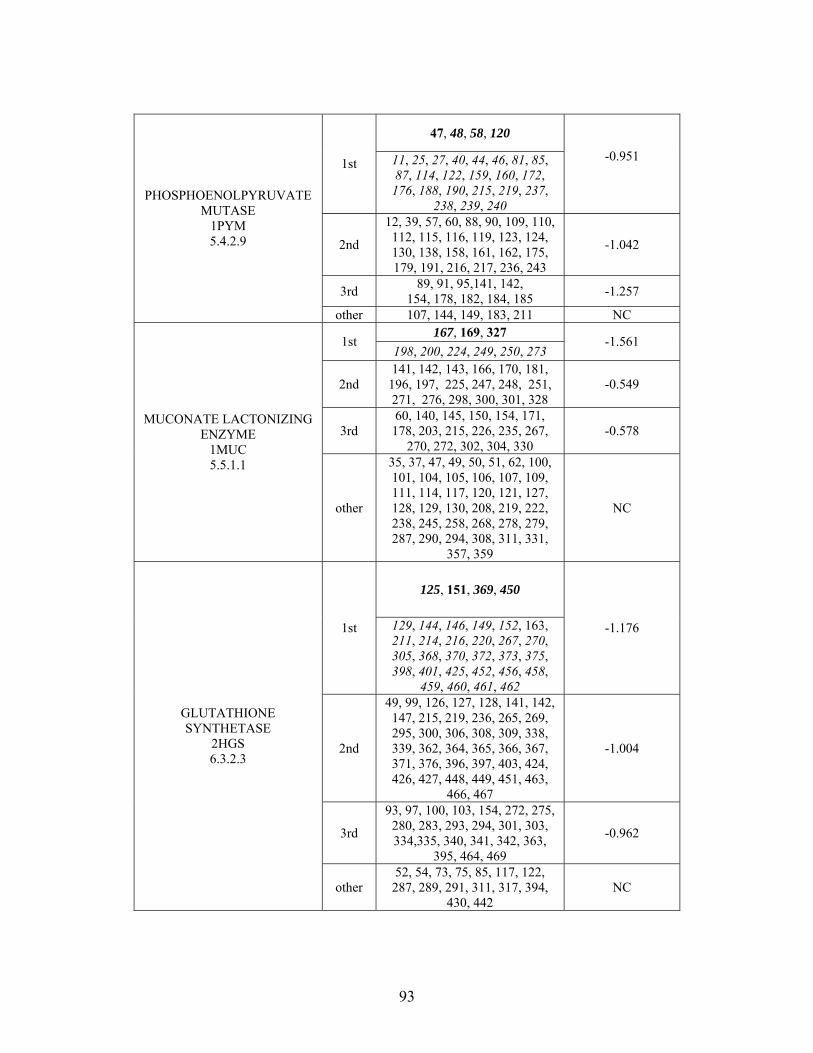
47, 48, 58, 120
1st 11, 25, 27, 40, 44, 46, 81, 85, 87, 114, 122, 159, 160, 172, 176, 188, 190, 215, 219, 237,
238, 239, 240
-0.951
2nd
12, 39, 57, 60, 88, 90, 109, 110, 112, 115, 116, 119, 123, 124, 130, 138, 158, 161, 162, 175, 179, 191, 216, 217, 236, 243
-1.042
3rd 89, 91, 95,141, 142,
154, 178, 182, 184, 185 -1.257
PHOSPHOENOLPYRUVATE MUTASE
1PYM 5.4.2.9
other 107, 144, 149, 183, 211 NC 167, 169, 327
1st 198, 200, 224, 249, 250, 273
-1.561
2nd 141, 142, 143, 166, 170, 181,
196, 197, 225, 247, 248, 251, 271, 276, 298, 300, 301, 328
-0.549
3rd 60, 140, 145, 150, 154, 171, 178, 203, 215, 226, 235, 267,
270, 272, 302, 304, 330 -0.578
MUCONATE LACTONIZING ENZYME
1MUC 5.5.1.1
other
35, 37, 47, 49, 50, 51, 62, 100, 101, 104, 105, 106, 107, 109, 111, 114, 117, 120, 121, 127, 128, 129, 130, 208, 219, 222, 238, 245, 258, 268, 278, 279, 287, 290, 294, 308, 311, 331,
357, 359
NC
125, 151, 369, 450
1st 129, 144, 146, 149, 152, 163, 211, 214, 216, 220, 267, 270, 305, 368, 370, 372, 373, 375, 398, 401, 425, 452, 456, 458,
459, 460, 461, 462
-1.176
2nd
49, 99, 126, 127, 128, 141, 142, 147, 215, 219, 236, 265, 269, 295, 300, 306, 308, 309, 338, 339, 362, 364, 365, 366, 367, 371, 376, 396, 397, 403, 424, 426, 427, 448, 449, 451, 463,
466, 467
-1.004
3rd
93, 97, 100, 103, 154, 272, 275, 280, 283, 293, 294, 301, 303, 334,335, 340, 341, 342, 363,
395, 464, 469
-0.962
GLUTATHIONE SYNTHETASE
2HGS 6.3.2.3
other 52, 54, 73, 75, 85, 117, 122, 287, 289, 291, 311, 317, 394,
430, 442 NC
93
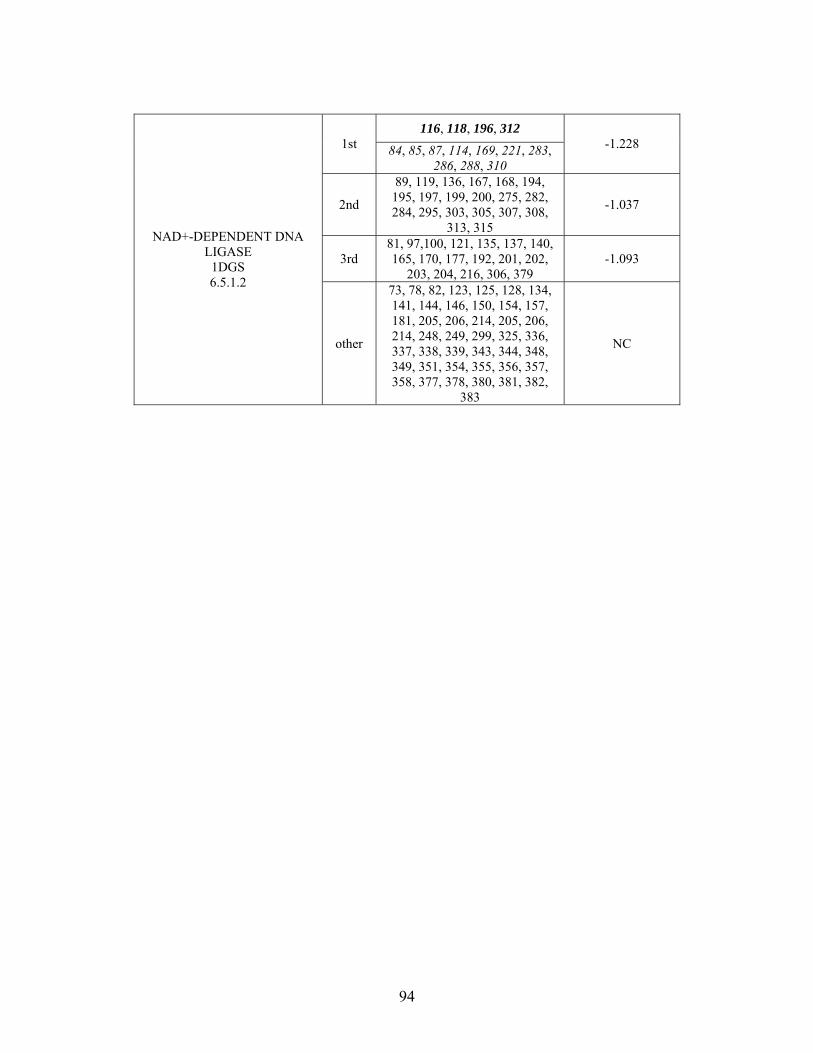
116, 118, 196, 312 1st 84, 85, 87, 114, 169, 221, 283,
286, 288, 310
-1.228
2nd
89, 119, 136, 167, 168, 194, 195, 197, 199, 200, 275, 282, 284, 295, 303, 305, 307, 308,
313, 315
-1.037
3rd 81, 97,100, 121, 135, 137, 140, 165, 170, 177, 192, 201, 202,
203, 204, 216, 306, 379 -1.093
NAD+-DEPENDENT DNA LIGASE
1DGS 6.5.1.2
other
73, 78, 82, 123, 125, 128, 134, 141, 144, 146, 150, 154, 157, 181, 205, 206, 214, 205, 206, 214, 248, 249, 299, 325, 336, 337, 338, 339, 343, 344, 348, 349, 351, 354, 355, 356, 357, 358, 377, 378, 380, 381, 382,
383
NC
94
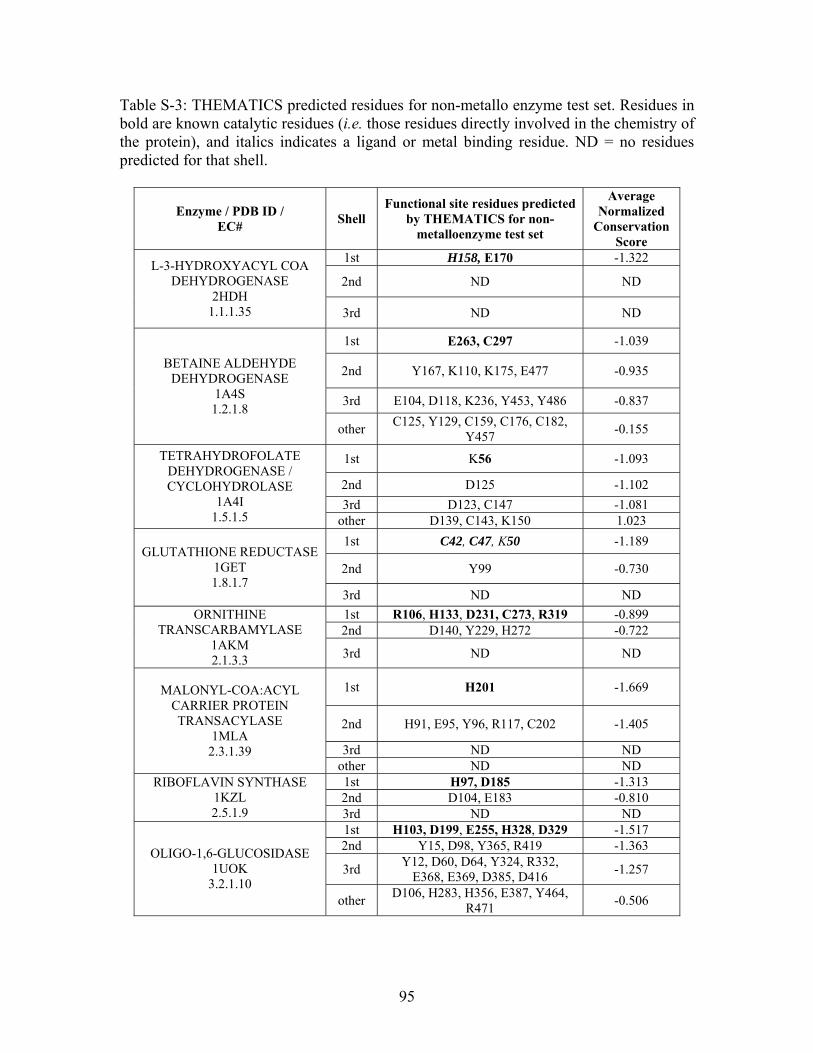
Table S-3: THEMATICS predicted residues for non-metallo enzyme test set. Residues in bold are known catalytic residues (i.e. those residues directly involved in the chemistry of the protein), and italics indicates a ligand or metal binding residue. ND = no residues predicted for that shell.
Enzyme / PDB ID / EC#
Shell Functional site residues predicted
by THEMATICS for non- metalloenzyme test set
Average Normalized
Conservation Score
1st H158, E170 -1.322
2nd ND ND L-3-HYDROXYACYL COA
DEHYDROGENASE 2HDH
1.1.1.35 3rd ND ND
1st E263, C297 -1.039
2nd Y167, K110, K175, E477 -0.935
3rd E104, D118, K236, Y453, Y486 -0.837
BETAINE ALDEHYDE DEHYDROGENASE
1A4S 1.2.1.8
other C125, Y129, C159, C176, C182,
Y457 -0.155
1st K56 -1.093
2nd D125 -1.102
3rd D123, C147 -1.081
TETRAHYDROFOLATE DEHYDROGENASE / CYCLOHYDROLASE
1A4I 1.5.1.5 other D139, C143, K150 1.023
1st C42, C47, K50 -1.189
2nd Y99 -0.730 GLUTATHIONE REDUCTASE
1GET 1.8.1.7
3rd ND ND
1st R106, H133, D231, C273, R319 -0.899 2nd D140, Y229, H272 -0.722
ORNITHINE TRANSCARBAMYLASE
1AKM 2.1.3.3 3rd ND ND
1st H201 -1.669
2nd H91, E95, Y96, R117, C202 -1.405
3rd ND ND
MALONYL-COA:ACYL CARRIER PROTEIN TRANSACYLASE
1MLA 2.3.1.39
other ND ND 1st H97, D185 -1.313 2nd D104, E183 -0.810
RIBOFLAVIN SYNTHASE 1KZL 2.5.1.9 3rd ND ND
1st H103, D199, E255, H328, D329 -1.517 2nd Y15, D98, Y365, R419 -1.363
3rd Y12, D60, D64, Y324, R332,
E368, E369, D385, D416 -1.257
OLIGO-1,6-GLUCOSIDASE 1UOK
3.2.1.10 other
D106, H283, H356, E387, Y464, R471
-0.506
95
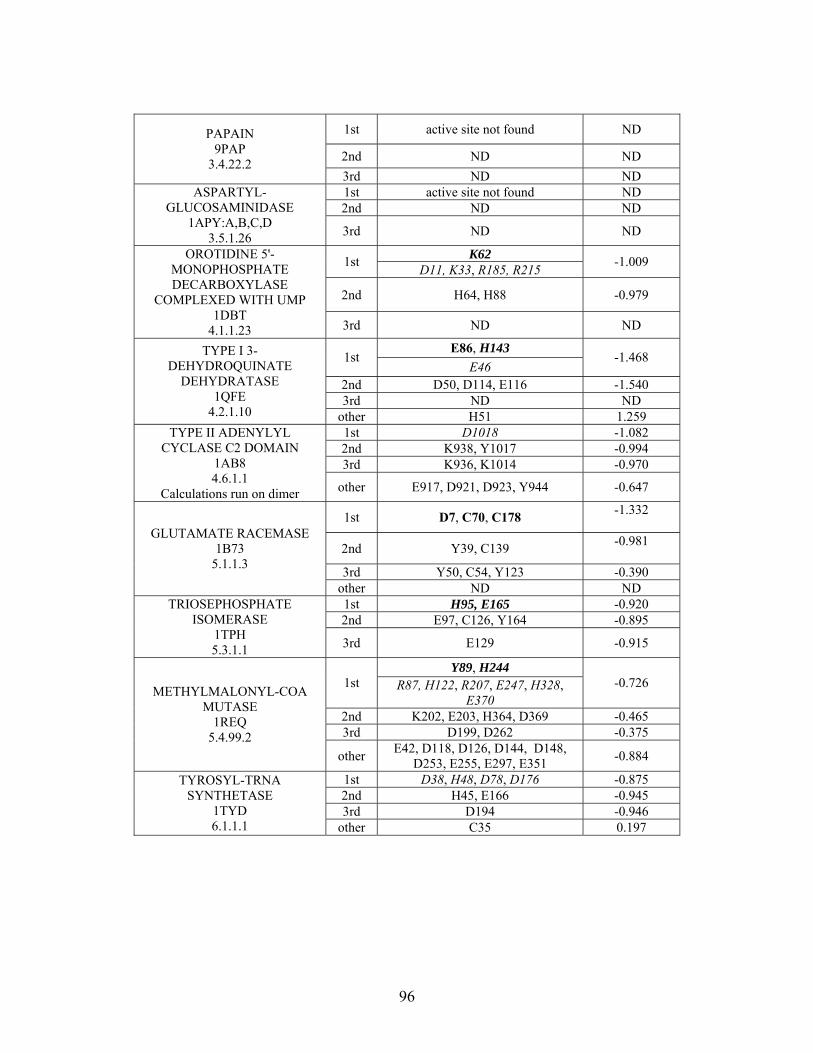
1st active site not found ND
2nd ND ND
PAPAIN 9PAP
3.4.22.2 3rd ND ND 1st active site not found ND 2nd ND ND
ASPARTYL-GLUCOSAMINIDASE
1APY:A,B,C,D 3rd ND ND 3.5.1.26
K62 1st
D11, K33, R185, R215 -1.009
2nd H64, H88 -0.979
OROTIDINE 5'-MONOPHOSPHATE DECARBOXYLASE
COMPLEXED WITH UMP 1DBT
3rd ND ND 4.1.1.23 E86, H143
1st E46
-1.468
2nd D50, D114, E116 -1.540 3rd ND ND
TYPE I 3-DEHYDROQUINATE
DEHYDRATASE 1QFE
4.2.1.10 other H51 1.259 1st D1018 -1.082 2nd K938, Y1017 -0.994 3rd K936, K1014 -0.970
TYPE II ADENYLYL CYCLASE C2 DOMAIN
1AB8 4.6.1.1
Calculations run on dimer other E917, D921, D923, Y944 -0.647
1st D7, C70, C178 -1.332
2nd Y39, C139 -0.981
3rd Y50, C54, Y123 -0.390
GLUTAMATE RACEMASE 1B73
5.1.1.3
other ND ND 1st H95, E165 -0.920 2nd E97, C126, Y164 -0.895
TRIOSEPHOSPHATE ISOMERASE
1TPH 5.3.1.1 3rd E129 -0.915
Y89, H244 1st R87, H122, R207, E247, H328,
E370 -0.726
2nd K202, E203, H364, D369 -0.465 3rd D199, D262 -0.375
METHYLMALONYL-COA MUTASE
1REQ 5.4.99.2
other E42, D118, D126, D144, D148,
D253, E255, E297, E351 -0.884
1st D38, H48, D78, D176 -0.875 2nd H45, E166 -0.945 3rd D194 -0.946
TYROSYL-TRNA SYNTHETASE
1TYD 6.1.1.1 other C35 0.197
96
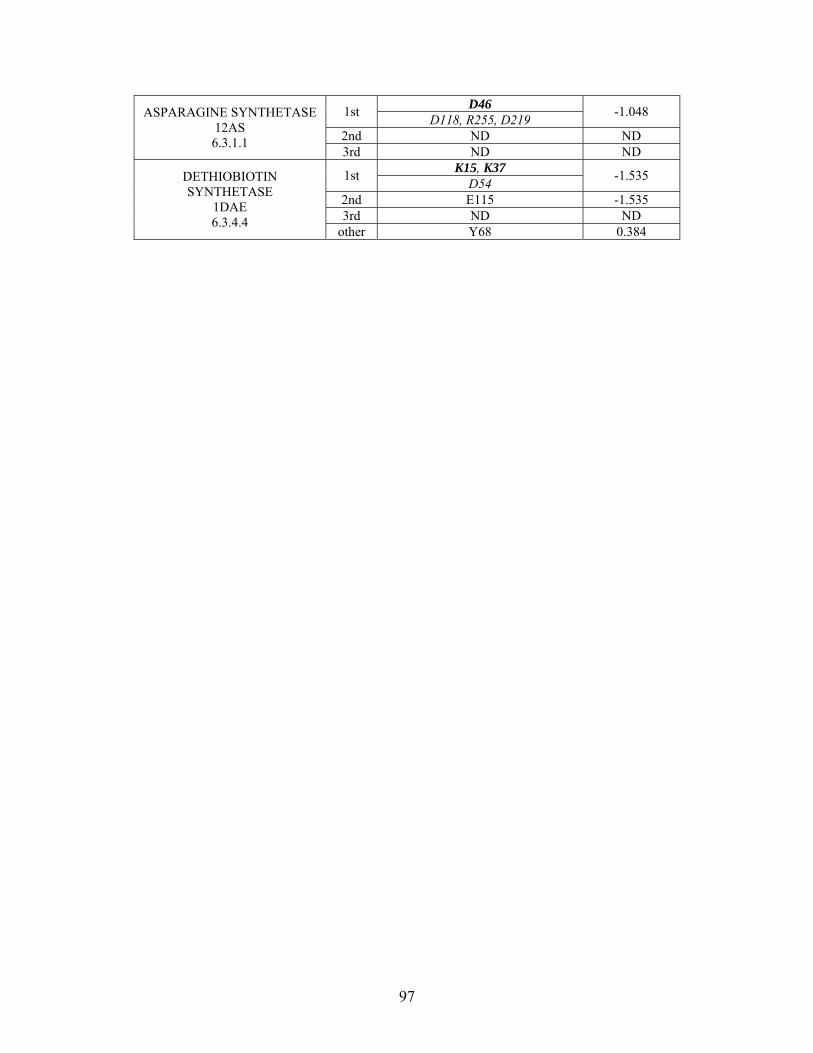
D46 1st
D118, R255, D219 -1.048
2nd ND ND
ASPARAGINE SYNTHETASE 12AS 6.3.1.1
3rd ND ND K15, K37
1st D54
-1.535
2nd E115 -1.535 3rd ND ND
DETHIOBIOTIN SYNTHETASE
1DAE 6.3.4.4
other Y68 0.384
97
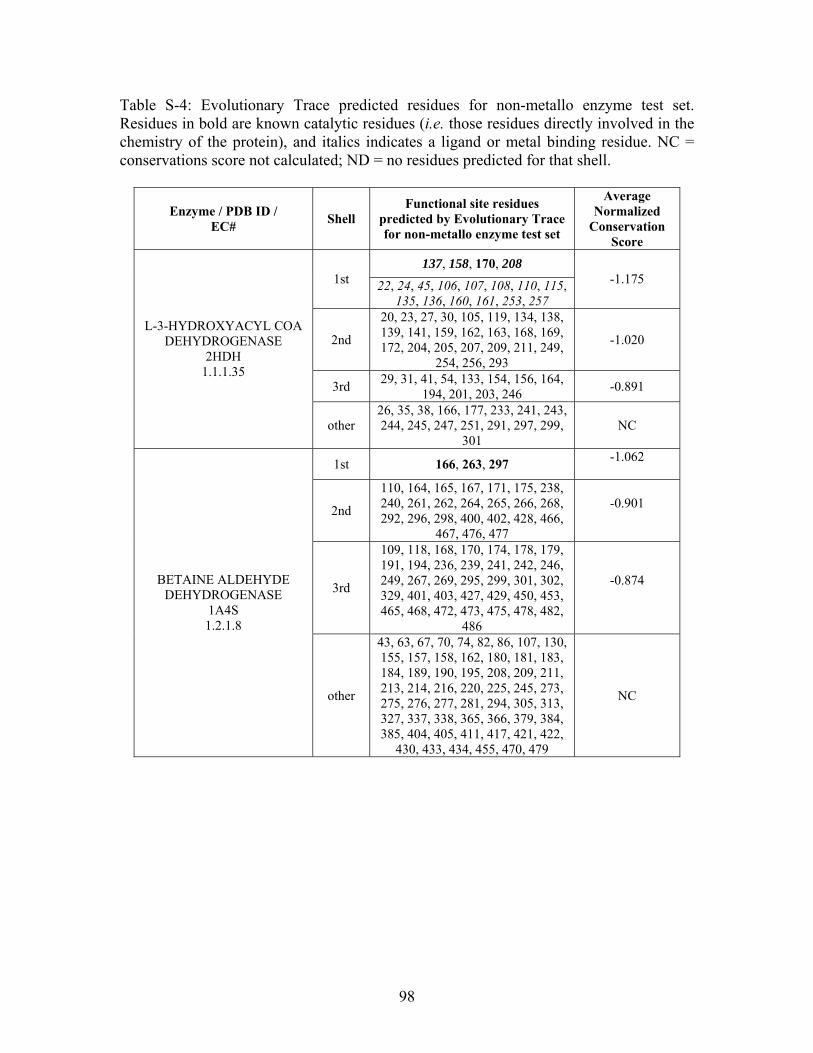
Table S-4: Evolutionary Trace predicted residues for non-metallo enzyme test set. Residues in bold are known catalytic residues (i.e. those residues directly involved in the chemistry of the protein), and italics indicates a ligand or metal binding residue. NC = conservations score not calculated; ND = no residues predicted for that shell.
Enzyme / PDB ID / EC#
Shell Functional site residues
predicted by Evolutionary Trace for non-metallo enzyme test set
Average Normalized
Conservation Score
137, 158, 170, 208 1st 22, 24, 45, 106, 107, 108, 110, 115,
135, 136, 160, 161, 253, 257
-1.175
2nd
20, 23, 27, 30, 105, 119, 134, 138, 139, 141, 159, 162, 163, 168, 169, 172, 204, 205, 207, 209, 211, 249,
254, 256, 293
-1.020
3rd 29, 31, 41, 54, 133, 154, 156, 164,
194, 201, 203, 246 -0.891
L-3-HYDROXYACYL COA DEHYDROGENASE
2HDH 1.1.1.35
other 26, 35, 38, 166, 177, 233, 241, 243, 244, 245, 247, 251, 291, 297, 299,
301 NC
1st 166, 263, 297 -1.062
2nd
110, 164, 165, 167, 171, 175, 238, 240, 261, 262, 264, 265, 266, 268, 292, 296, 298, 400, 402, 428, 466,
467, 476, 477
-0.901
3rd
109, 118, 168, 170, 174, 178, 179, 191, 194, 236, 239, 241, 242, 246, 249, 267, 269, 295, 299, 301, 302, 329, 401, 403, 427, 429, 450, 453, 465, 468, 472, 473, 475, 478, 482,
486
-0.874
BETAINE ALDEHYDE DEHYDROGENASE
1A4S 1.2.1.8
other
43, 63, 67, 70, 74, 82, 86, 107, 130, 155, 157, 158, 162, 180, 181, 183, 184, 189, 190, 195, 208, 209, 211, 213, 214, 216, 220, 225, 245, 273, 275, 276, 277, 281, 294, 305, 313, 327, 337, 338, 365, 366, 379, 384, 385, 404, 405, 411, 417, 421, 422,
430, 433, 434, 455, 470, 479
NC
98
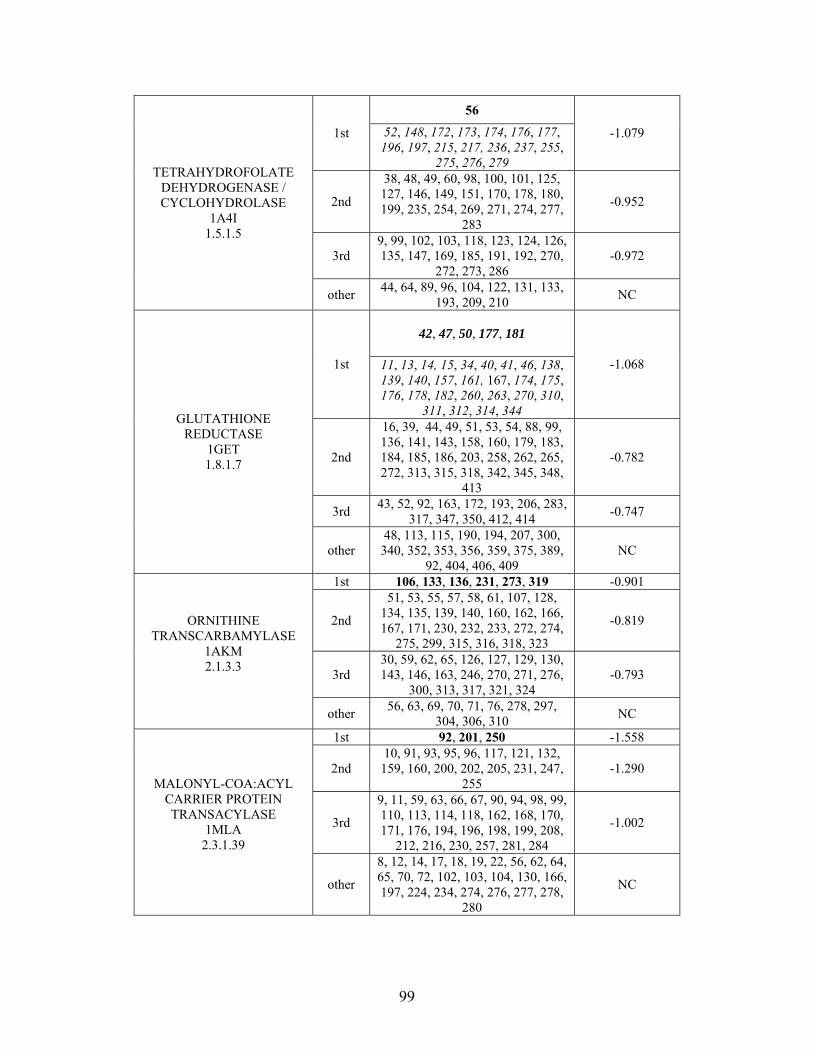
56
1st 52, 148, 172, 173, 174, 176, 177, 196, 197, 215, 217, 236, 237, 255,
275, 276, 279
-1.079
2nd
38, 48, 49, 60, 98, 100, 101, 125, 127, 146, 149, 151, 170, 178, 180, 199, 235, 254, 269, 271, 274, 277,
283
-0.952
3rd 9, 99, 102, 103, 118, 123, 124, 126, 135, 147, 169, 185, 191, 192, 270,
272, 273, 286 -0.972
TETRAHYDROFOLATE DEHYDROGENASE / CYCLOHYDROLASE
1A4I 1.5.1.5
other 44, 64, 89, 96, 104, 122, 131, 133,
193, 209, 210 NC
42, 47, 50, 177, 181
1st 11, 13, 14, 15, 34, 40, 41, 46, 138, 139, 140, 157, 161, 167, 174, 175, 176, 178, 182, 260, 263, 270, 310,
311, 312, 314, 344
-1.068
2nd
16, 39, 44, 49, 51, 53, 54, 88, 99, 136, 141, 143, 158, 160, 179, 183, 184, 185, 186, 203, 258, 262, 265, 272, 313, 315, 318, 342, 345, 348,
413
-0.782
3rd 43, 52, 92, 163, 172, 193, 206, 283,
317, 347, 350, 412, 414 -0.747
GLUTATHIONE REDUCTASE
1GET 1.8.1.7
other 48, 113, 115, 190, 194, 207, 300, 340, 352, 353, 356, 359, 375, 389,
92, 404, 406, 409 NC
1st 106, 133, 136, 231, 273, 319 -0.901
2nd
51, 53, 55, 57, 58, 61, 107, 128, 134, 135, 139, 140, 160, 162, 166, 167, 171, 230, 232, 233, 272, 274,
275, 299, 315, 316, 318, 323
-0.819
3rd 30, 59, 62, 65, 126, 127, 129, 130, 143, 146, 163, 246, 270, 271, 276,
300, 313, 317, 321, 324 -0.793
ORNITHINE TRANSCARBAMYLASE
1AKM 2.1.3.3
other 56, 63, 69, 70, 71, 76, 278, 297,
304, 306, 310 NC
1st 92, 201, 250 -1.558
2nd 10, 91, 93, 95, 96, 117, 121, 132, 159, 160, 200, 202, 205, 231, 247,
255 -1.290
3rd
9, 11, 59, 63, 66, 67, 90, 94, 98, 99, 110, 113, 114, 118, 162, 168, 170, 171, 176, 194, 196, 198, 199, 208,
212, 216, 230, 257, 281, 284
-1.002
MALONYL-COA:ACYL CARRIER PROTEIN TRANSACYLASE
1MLA 2.3.1.39
other
8, 12, 14, 17, 18, 19, 22, 56, 62, 64, 65, 70, 72, 102, 103, 104, 130, 166, 197, 224, 234, 274, 276, 277, 278,
280
NC
99
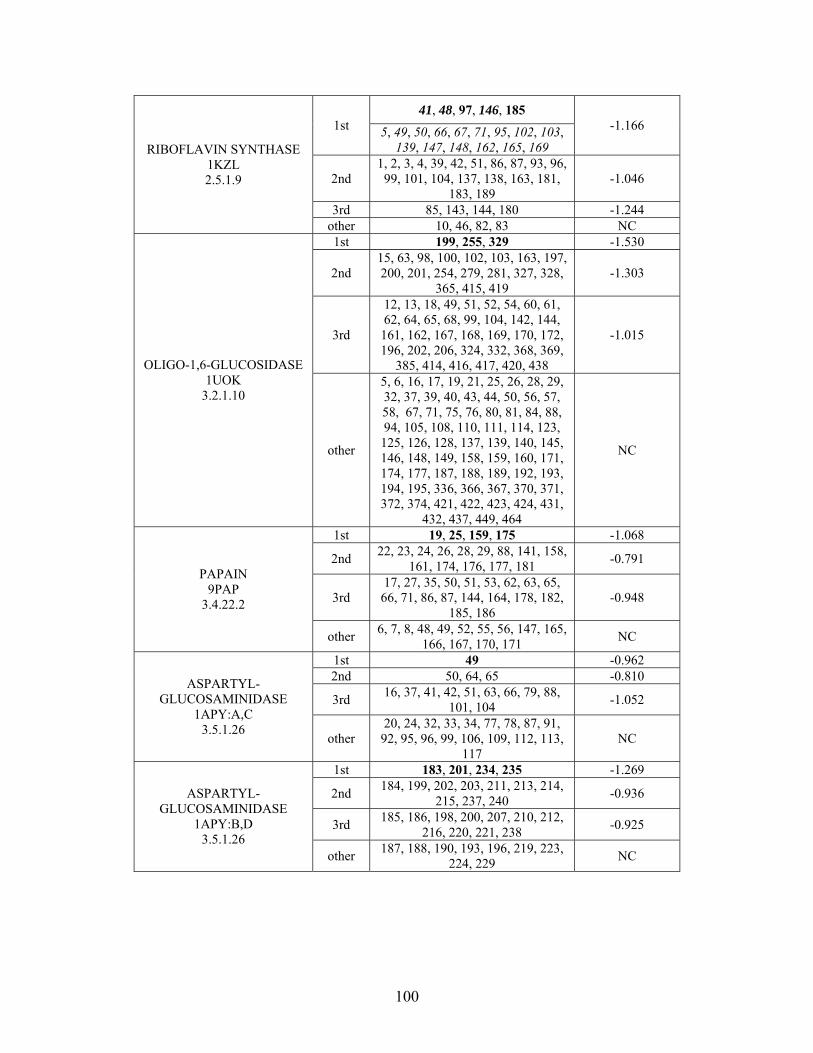
41, 48, 97, 146, 185 1st 5, 49, 50, 66, 67, 71, 95, 102, 103,
139, 147, 148, 162, 165, 169
-1.166
2nd 1, 2, 3, 4, 39, 42, 51, 86, 87, 93, 96, 99, 101, 104, 137, 138, 163, 181,
183, 189 -1.046
3rd 85, 143, 144, 180 -1.244
RIBOFLAVIN SYNTHASE 1KZL 2.5.1.9
other 10, 46, 82, 83 NC 1st 199, 255, 329 -1.530
2nd 15, 63, 98, 100, 102, 103, 163, 197, 200, 201, 254, 279, 281, 327, 328,
365, 415, 419 -1.303
3rd
12, 13, 18, 49, 51, 52, 54, 60, 61, 62, 64, 65, 68, 99, 104, 142, 144, 161, 162, 167, 168, 169, 170, 172, 196, 202, 206, 324, 332, 368, 369,
385, 414, 416, 417, 420, 438
-1.015
OLIGO-1,6-GLUCOSIDASE 1UOK
3.2.1.10
other
5, 6, 16, 17, 19, 21, 25, 26, 28, 29, 32, 37, 39, 40, 43, 44, 50, 56, 57, 58, 67, 71, 75, 76, 80, 81, 84, 88, 94, 105, 108, 110, 111, 114, 123, 125, 126, 128, 137, 139, 140, 145, 146, 148, 149, 158, 159, 160, 171, 174, 177, 187, 188, 189, 192, 193, 194, 195, 336, 366, 367, 370, 371, 372, 374, 421, 422, 423, 424, 431,
432, 437, 449, 464
NC
1st 19, 25, 159, 175 -1.068
2nd 22, 23, 24, 26, 28, 29, 88, 141, 158,
161, 174, 176, 177, 181 -0.791
3rd 17, 27, 35, 50, 51, 53, 62, 63, 65, 66, 71, 86, 87, 144, 164, 178, 182,
185, 186 -0.948
PAPAIN 9PAP
3.4.22.2
other 6, 7, 8, 48, 49, 52, 55, 56, 147, 165,
166, 167, 170, 171 NC
1st 49 -0.962 2nd 50, 64, 65 -0.810
3rd 16, 37, 41, 42, 51, 63, 66, 79, 88,
101, 104 -1.052
ASPARTYL-GLUCOSAMINIDASE
1APY:A,C 3.5.1.26
other 20, 24, 32, 33, 34, 77, 78, 87, 91, 92, 95, 96, 99, 106, 109, 112, 113,
117 NC
1st 183, 201, 234, 235 -1.269
2nd 184, 199, 202, 203, 211, 213, 214,
215, 237, 240 -0.936
3rd 185, 186, 198, 200, 207, 210, 212,
216, 220, 221, 238 -0.925
ASPARTYL-GLUCOSAMINIDASE
1APY:B,D 3.5.1.26
other 187, 188, 190, 193, 196, 219, 223,
224, 229 NC
100
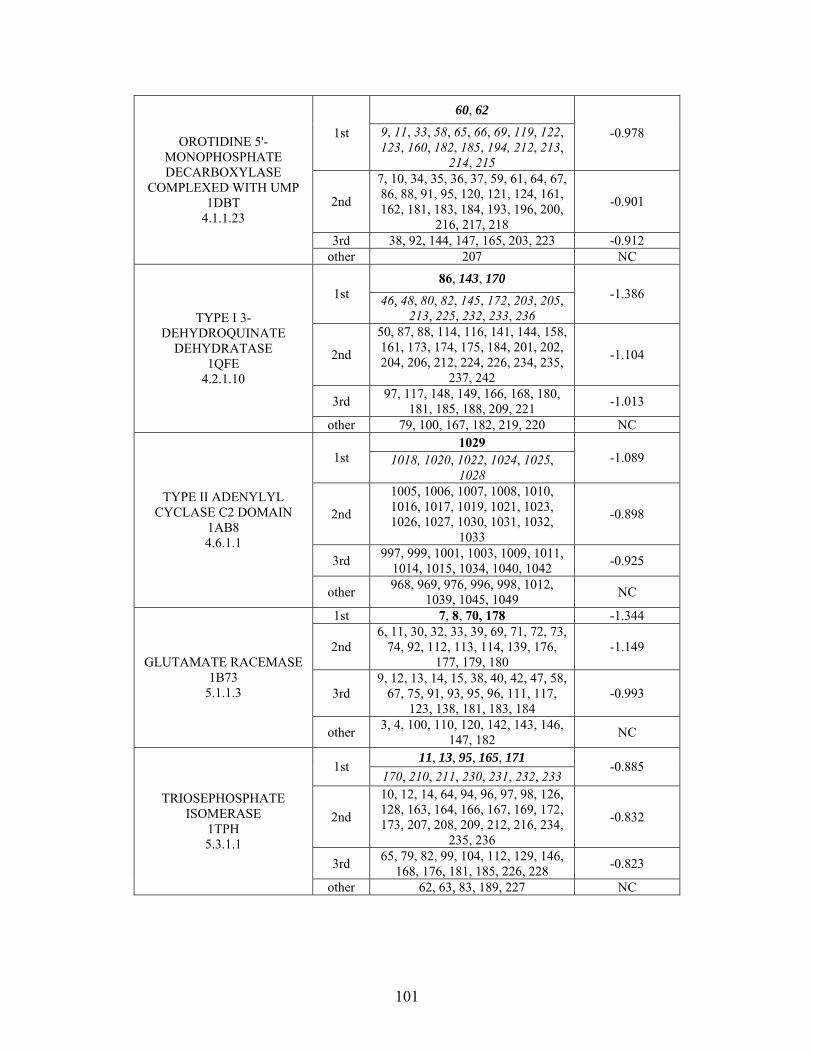
60, 62
1st 9, 11, 33, 58, 65, 66, 69, 119, 122, 123, 160, 182, 185, 194, 212, 213,
214, 215
-0.978
2nd
7, 10, 34, 35, 36, 37, 59, 61, 64, 67, 86, 88, 91, 95, 120, 121, 124, 161, 162, 181, 183, 184, 193, 196, 200,
216, 217, 218
-0.901
3rd 38, 92, 144, 147, 165, 203, 223 -0.912
OROTIDINE 5'-MONOPHOSPHATE DECARBOXYLASE
COMPLEXED WITH UMP 1DBT
4.1.1.23
other 207 NC
86, 143, 170 1st 46, 48, 80, 82, 145, 172, 203, 205,
213, 225, 232, 233, 236
-1.386
2nd
50, 87, 88, 114, 116, 141, 144, 158, 161, 173, 174, 175, 184, 201, 202, 204, 206, 212, 224, 226, 234, 235,
237, 242
-1.104
3rd 97, 117, 148, 149, 166, 168, 180,
181, 185, 188, 209, 221 -1.013
TYPE I 3-DEHYDROQUINATE
DEHYDRATASE 1QFE
4.2.1.10
other 79, 100, 167, 182, 219, 220 NC 1029
1st 1018, 1020, 1022, 1024, 1025, 1028
-1.089
2nd
1005, 1006, 1007, 1008, 1010, 1016, 1017, 1019, 1021, 1023, 1026, 1027, 1030, 1031, 1032,
1033
-0.898
3rd 997, 999, 1001, 1003, 1009, 1011,
1014, 1015, 1034, 1040, 1042 -0.925
TYPE II ADENYLYL CYCLASE C2 DOMAIN
1AB8 4.6.1.1
other 968, 969, 976, 996, 998, 1012,
1039, 1045, 1049 NC
1st 7, 8, 70, 178 -1.344
2nd 6, 11, 30, 32, 33, 39, 69, 71, 72, 73,
74, 92, 112, 113, 114, 139, 176, 177, 179, 180
-1.149
3rd 9, 12, 13, 14, 15, 38, 40, 42, 47, 58,
67, 75, 91, 93, 95, 96, 111, 117, 123, 138, 181, 183, 184
-0.993
GLUTAMATE RACEMASE 1B73
5.1.1.3
other 3, 4, 100, 110, 120, 142, 143, 146,
147, 182 NC
11, 13, 95, 165, 171 1st
170, 210, 211, 230, 231, 232, 233 -0.885
2nd
10, 12, 14, 64, 94, 96, 97, 98, 126, 128, 163, 164, 166, 167, 169, 172, 173, 207, 208, 209, 212, 216, 234,
235, 236
-0.832
3rd 65, 79, 82, 99, 104, 112, 129, 146,
168, 176, 181, 185, 226, 228 -0.823
TRIOSEPHOSPHATE ISOMERASE
1TPH 5.3.1.1
other 62, 63, 83, 189, 227 NC
101
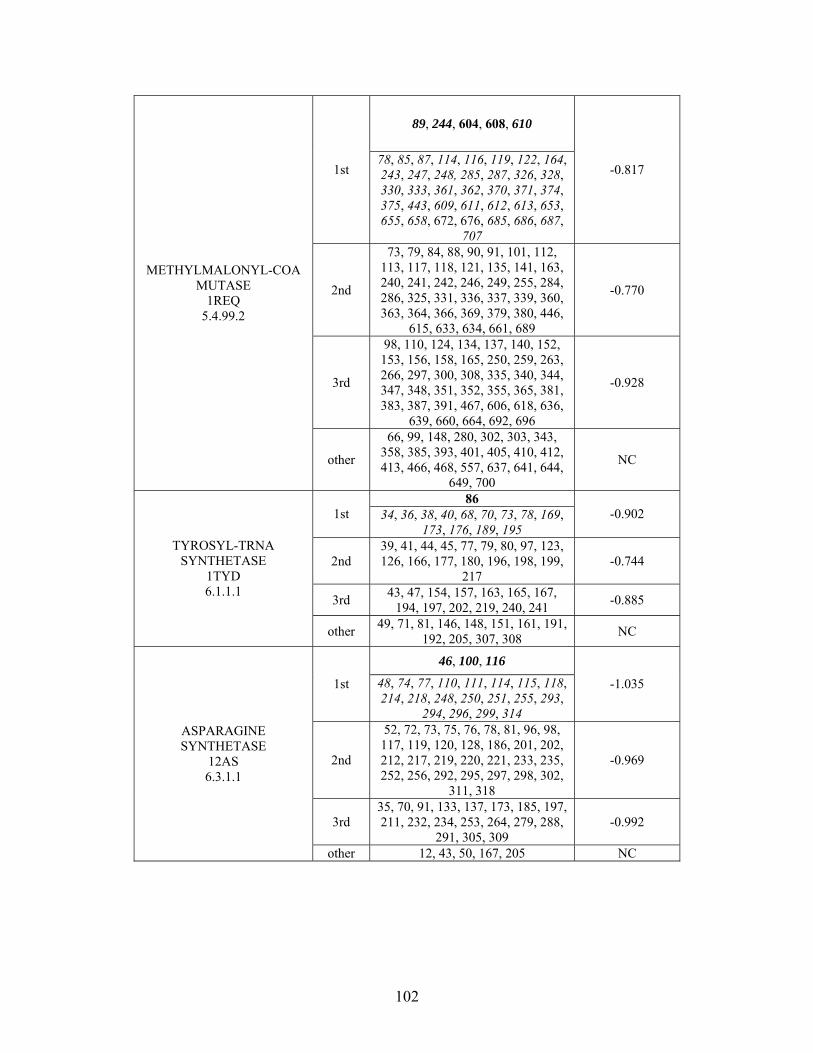
89, 244, 604, 608, 610
1st 78, 85, 87, 114, 116, 119, 122, 164, 243, 247, 248, 285, 287, 326, 328, 330, 333, 361, 362, 370, 371, 374, 375, 443, 609, 611, 612, 613, 653, 655, 658, 672, 676, 685, 686, 687,
707
-0.817
2nd
73, 79, 84, 88, 90, 91, 101, 112, 113, 117, 118, 121, 135, 141, 163, 240, 241, 242, 246, 249, 255, 284, 286, 325, 331, 336, 337, 339, 360, 363, 364, 366, 369, 379, 380, 446,
615, 633, 634, 661, 689
-0.770
3rd
98, 110, 124, 134, 137, 140, 152, 153, 156, 158, 165, 250, 259, 263, 266, 297, 300, 308, 335, 340, 344, 347, 348, 351, 352, 355, 365, 381, 383, 387, 391, 467, 606, 618, 636,
639, 660, 664, 692, 696
-0.928
METHYLMALONYL-COA MUTASE
1REQ 5.4.99.2
other
66, 99, 148, 280, 302, 303, 343, 358, 385, 393, 401, 405, 410, 412, 413, 466, 468, 557, 637, 641, 644,
649, 700
NC
86 1st 34, 36, 38, 40, 68, 70, 73, 78, 169,
173, 176, 189, 195 -0.902
2nd 39, 41, 44, 45, 77, 79, 80, 97, 123, 126, 166, 177, 180, 196, 198, 199,
217 -0.744
3rd 43, 47, 154, 157, 163, 165, 167,
194, 197, 202, 219, 240, 241 -0.885
TYROSYL-TRNA SYNTHETASE
1TYD 6.1.1.1
other 49, 71, 81, 146, 148, 151, 161, 191,
192, 205, 307, 308 NC
46, 100, 116
1st 48, 74, 77, 110, 111, 114, 115, 118, 214, 218, 248, 250, 251, 255, 293,
294, 296, 299, 314
-1.035
2nd
52, 72, 73, 75, 76, 78, 81, 96, 98, 117, 119, 120, 128, 186, 201, 202, 212, 217, 219, 220, 221, 233, 235, 252, 256, 292, 295, 297, 298, 302,
311, 318
-0.969
3rd 35, 70, 91, 133, 137, 173, 185, 197, 211, 232, 234, 253, 264, 279, 288,
291, 305, 309 -0.992
ASPARAGINE SYNTHETASE
12AS 6.3.1.1
other 12, 43, 50, 167, 205 NC
102
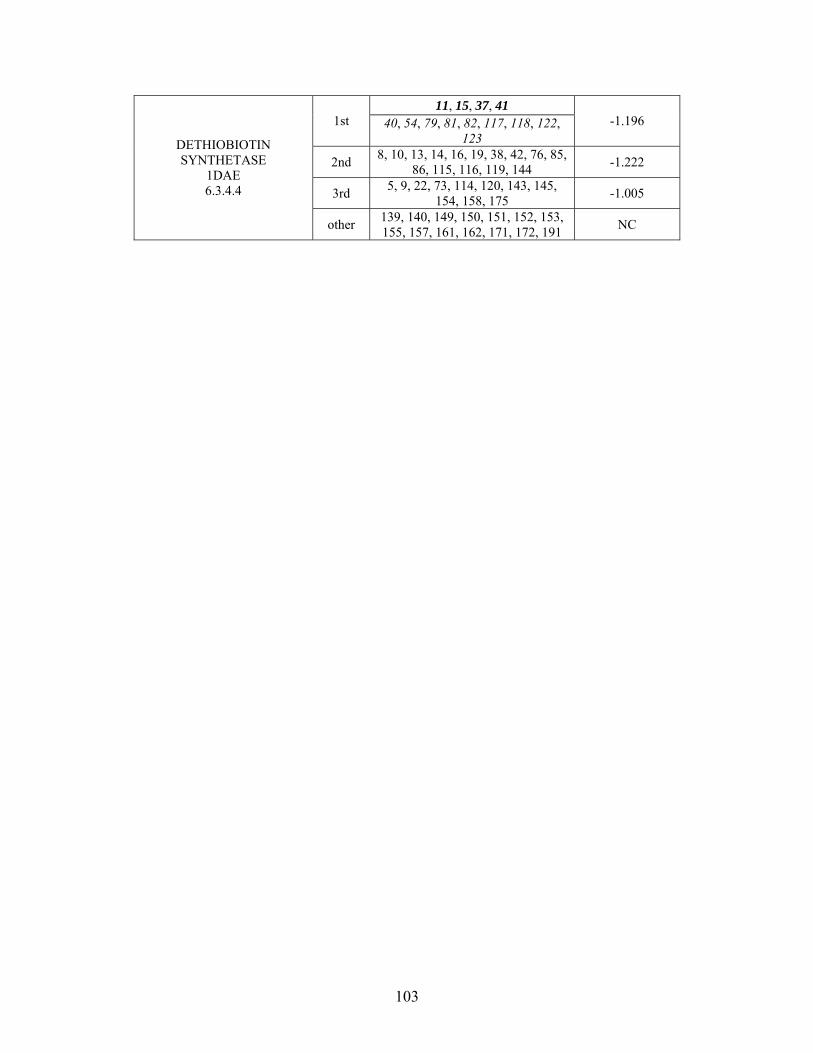
11, 15, 37, 41 1st 40, 54, 79, 81, 82, 117, 118, 122,
123 -1.196
2nd 8, 10, 13, 14, 16, 19, 38, 42, 76, 85,
86, 115, 116, 119, 144 -1.222
3rd 5, 9, 22, 73, 114, 120, 143, 145,
154, 158, 175 -1.005
DETHIOBIOTIN SYNTHETASE
1DAE 6.3.4.4
other 139, 140, 149, 150, 151, 152, 153, 155, 157, 161, 162, 171, 172, 191
NC
103
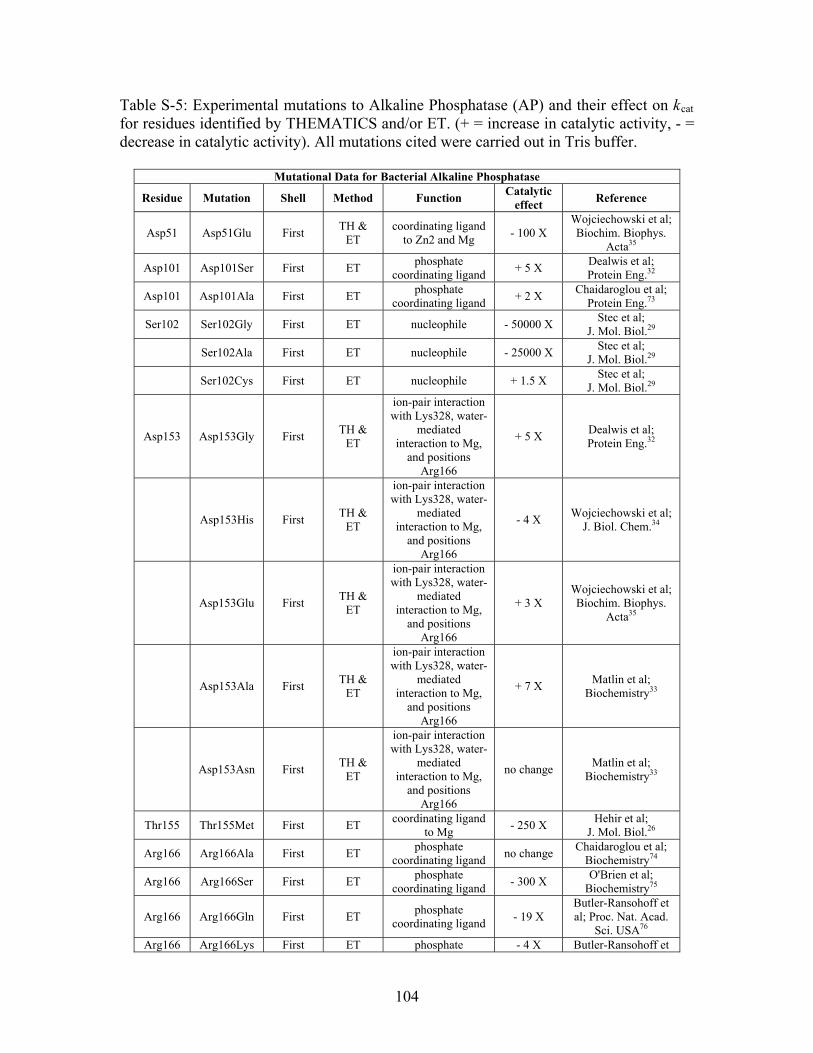
Table S-5: Experimental mutations to Alkaline Phosphatase (AP) and their effect on kcat for residues identified by THEMATICS and/or ET. (+ = increase in catalytic activity, - = decrease in catalytic activity). All mutations cited were carried out in Tris buffer.
Mutational Data for Bacterial Alkaline Phosphatase
Residue Mutation Shell Method Function Catalytic
effect Reference
Asp51 Asp51Glu First TH &
ET coordinating ligand
to Zn2 and Mg - 100 X
Wojciechowski et al; Biochim. Biophys.
Acta35
Asp101 Asp101Ser First ET phosphate
coordinating ligand + 5 X
Dealwis et al; Protein Eng.32
Asp101 Asp101Ala First ET phosphate
coordinating ligand + 2 X
Chaidaroglou et al; Protein Eng.73
Ser102 Ser102Gly First ET nucleophile - 50000 X Stec et al;
J. Mol. Biol.29 Ser102Ala First ET nucleophile - 25000 X
Stec et al; J. Mol. Biol.29
Ser102Cys First ET nucleophile + 1.5 X Stec et al;
J. Mol. Biol.29
Asp153 Asp153Gly First TH &
ET
ion-pair interaction with Lys328, water-
mediated interaction to Mg,
and positions Arg166
+ 5 X Dealwis et al; Protein Eng.32
Asp153His First TH &
ET
ion-pair interaction with Lys328, water-
mediated interaction to Mg,
and positions Arg166
- 4 X Wojciechowski et al;
J. Biol. Chem.34
Asp153Glu First TH &
ET
ion-pair interaction with Lys328, water-
mediated interaction to Mg,
and positions Arg166
+ 3 X Wojciechowski et al; Biochim. Biophys.
Acta35
Asp153Ala First TH &
ET
ion-pair interaction with Lys328, water-
mediated interaction to Mg,
and positions Arg166
+ 7 X Matlin et al;
Biochemistry33
Asp153Asn First TH &
ET
ion-pair interaction with Lys328, water-
mediated interaction to Mg,
and positions Arg166
no change Matlin et al;
Biochemistry33
Thr155 Thr155Met First ET coordinating ligand
to Mg - 250 X
Hehir et al; J. Mol. Biol.26
Arg166 Arg166Ala First ET phosphate
coordinating ligand no change
Chaidaroglou et al; Biochemistry74
Arg166 Arg166Ser First ET phosphate
coordinating ligand - 300 X
O'Brien et al; Biochemistry75
Arg166 Arg166Gln First ET phosphate
coordinating ligand - 19 X
Butler-Ransohoff et al; Proc. Nat. Acad.
Sci. USA76 Arg166 Arg166Lys First ET phosphate - 4 X Butler-Ransohoff et
104
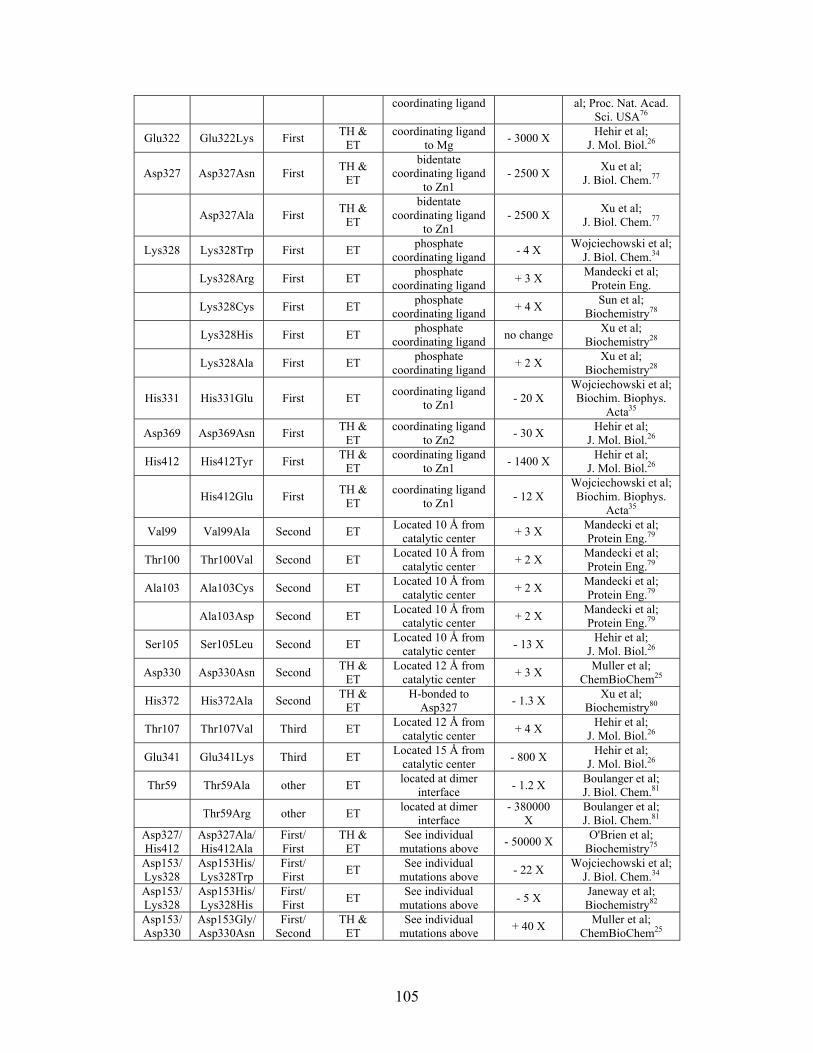
coordinating ligand al; Proc. Nat. Acad. Sci. USA76
Glu322 Glu322Lys First TH &
ET coordinating ligand
to Mg - 3000 X
Hehir et al; J. Mol. Biol.26
Asp327 Asp327Asn First TH &
ET
bidentate coordinating ligand
to Zn1 - 2500 X
Xu et al; J. Biol. Chem.77
Asp327Ala First TH &
ET
bidentate coordinating ligand
to Zn1 - 2500 X
Xu et al; J. Biol. Chem.77
Lys328 Lys328Trp First ET phosphate
coordinating ligand - 4 X
Wojciechowski et al; J. Biol. Chem.34
Lys328Arg First ET phosphate
coordinating ligand + 3 X
Mandecki et al; Protein Eng.
Lys328Cys First ET phosphate
coordinating ligand + 4 X
Sun et al; Biochemistry78
Lys328His First ET phosphate
coordinating ligand no change
Xu et al; Biochemistry28
Lys328Ala First ET phosphate
coordinating ligand + 2 X
Xu et al; Biochemistry28
His331 His331Glu First ET coordinating ligand
to Zn1 - 20 X
Wojciechowski et al; Biochim. Biophys.
Acta35
Asp369 Asp369Asn First TH &
ET coordinating ligand
to Zn2 - 30 X
Hehir et al; J. Mol. Biol.26
His412 His412Tyr First TH &
ET coordinating ligand
to Zn1 - 1400 X
Hehir et al; J. Mol. Biol.26
His412Glu First TH &
ET coordinating ligand
to Zn1 - 12 X
Wojciechowski et al; Biochim. Biophys.
Acta35
Val99 Val99Ala Second ET Located 10 Å from
catalytic center + 3 X
Mandecki et al; Protein Eng.79
Thr100 Thr100Val Second ET Located 10 Å from
catalytic center + 2 X
Mandecki et al; Protein Eng.79
Ala103 Ala103Cys Second ET Located 10 Å from
catalytic center + 2 X
Mandecki et al; Protein Eng.79
Ala103Asp Second ET Located 10 Å from
catalytic center + 2 X
Mandecki et al; Protein Eng.79
Ser105 Ser105Leu Second ET Located 10 Å from
catalytic center - 13 X
Hehir et al; J. Mol. Biol.26
Asp330 Asp330Asn Second TH &
ET Located 12 Å from
catalytic center + 3 X
Muller et al; ChemBioChem25
His372 His372Ala Second TH &
ET H-bonded to
Asp327 - 1.3 X
Xu et al; Biochemistry80
Thr107 Thr107Val Third ET Located 12 Å from
catalytic center + 4 X
Hehir et al; J. Mol. Biol.26
Glu341 Glu341Lys Third ET Located 15 Å from
catalytic center - 800 X
Hehir et al; J. Mol. Biol.26
Thr59 Thr59Ala other ET located at dimer
interface - 1.2 X
Boulanger et al; J. Biol. Chem.81
Thr59Arg other ET located at dimer
interface - 380000
X Boulanger et al; J. Biol. Chem.81
Asp327/His412
Asp327Ala/His412Ala
First/ First
TH & ET
See individual mutations above
- 50000 X O'Brien et al;
Biochemistry75 Asp153/Lys328
Asp153His/Lys328Trp
First/ First
ET See individual
mutations above - 22 X
Wojciechowski et al; J. Biol. Chem.34
Asp153/Lys328
Asp153His/Lys328His
First/ First
ET See individual
mutations above - 5 X
Janeway et al; Biochemistry82
Asp153/ Asp330
Asp153Gly/ Asp330Asn
First/ Second
TH & ET
See individual mutations above
+ 40 X Muller et al;
ChemBioChem25
105
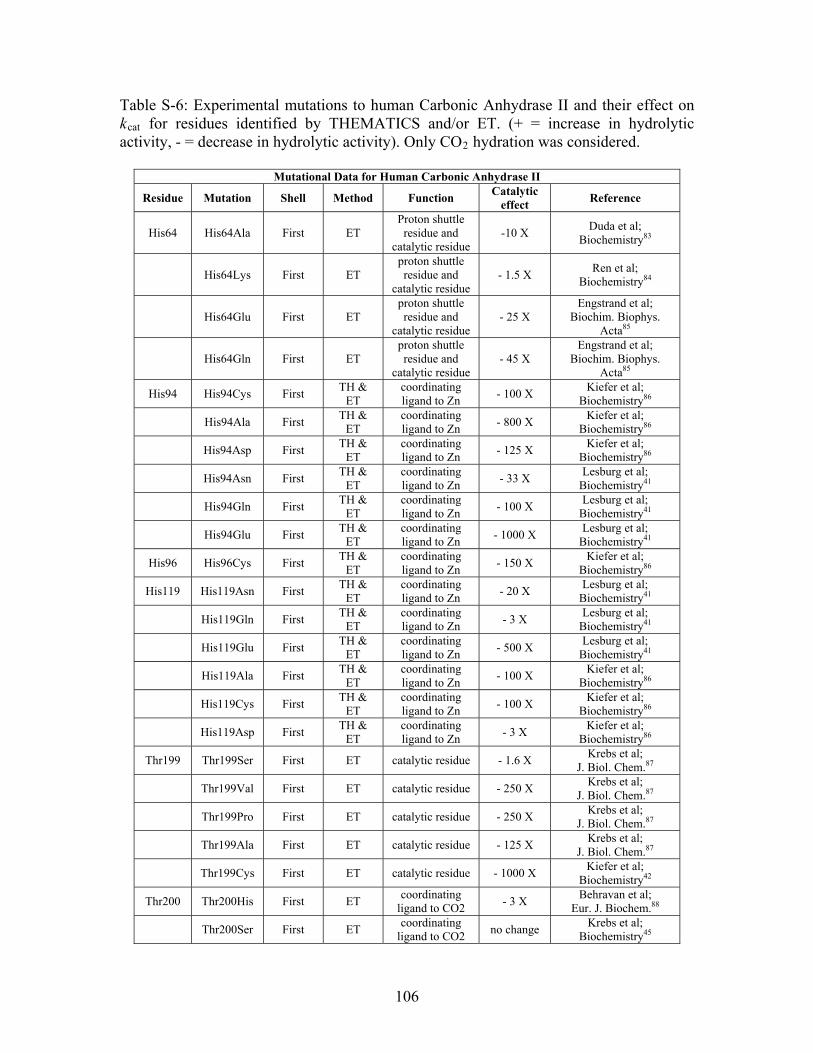
Table S-6: Experimental mutations to human Carbonic Anhydrase II and their effect on kcat for residues identified by THEMATICS and/or ET. (+ = increase in hydrolytic activity, - = decrease in hydrolytic activity). Only CO2 hydration was considered.
Mutational Data for Human Carbonic Anhydrase II
Residue Mutation Shell Method Function Catalytic
effect Reference
His64 His64Ala First ET Proton shuttle residue and
catalytic residue -10 X
Duda et al; Biochemistry83
His64Lys First ET proton shuttle residue and
catalytic residue - 1.5 X
Ren et al; Biochemistry84
His64Glu First ET proton shuttle residue and
catalytic residue - 25 X
Engstrand et al; Biochim. Biophys.
Acta85
His64Gln First ET proton shuttle residue and
catalytic residue - 45 X
Engstrand et al; Biochim. Biophys.
Acta85
His94 His94Cys First TH &
ET coordinating ligand to Zn
- 100 X Kiefer et al;
Biochemistry86
His94Ala First TH &
ET coordinating ligand to Zn
- 800 X Kiefer et al;
Biochemistry86
His94Asp First TH &
ET coordinating ligand to Zn
- 125 X Kiefer et al;
Biochemistry86
His94Asn First TH &
ET coordinating ligand to Zn
- 33 X Lesburg et al;
Biochemistry41
His94Gln First TH &
ET coordinating ligand to Zn
- 100 X Lesburg et al;
Biochemistry41
His94Glu First TH &
ET coordinating ligand to Zn
- 1000 X Lesburg et al;
Biochemistry41
His96 His96Cys First TH &
ET coordinating ligand to Zn
- 150 X Kiefer et al;
Biochemistry86
His119 His119Asn First TH &
ET coordinating ligand to Zn
- 20 X Lesburg et al;
Biochemistry41
His119Gln First TH &
ET coordinating ligand to Zn
- 3 X Lesburg et al;
Biochemistry41
His119Glu First TH &
ET coordinating ligand to Zn
- 500 X Lesburg et al;
Biochemistry41
His119Ala First TH &
ET coordinating ligand to Zn
- 100 X Kiefer et al;
Biochemistry86
His119Cys First TH &
ET coordinating ligand to Zn
- 100 X Kiefer et al;
Biochemistry86
His119Asp First TH &
ET coordinating ligand to Zn
- 3 X Kiefer et al;
Biochemistry86
Thr199 Thr199Ser First ET catalytic residue - 1.6 X Krebs et al;
J. Biol. Chem.87 Thr199Val First ET catalytic residue - 250 X
Krebs et al; J. Biol. Chem.87
Thr199Pro First ET catalytic residue - 250 X Krebs et al;
J. Biol. Chem.87 Thr199Ala First ET catalytic residue - 125 X
Krebs et al; J. Biol. Chem.87
Thr199Cys First ET catalytic residue - 1000 X Kiefer et al;
Biochemistry42
Thr200 Thr200His First ET coordinating
ligand to CO2 - 3 X
Behravan et al; Eur. J. Biochem.88
Thr200Ser First ET coordinating
ligand to CO2 no change
Krebs et al; Biochemistry45
106
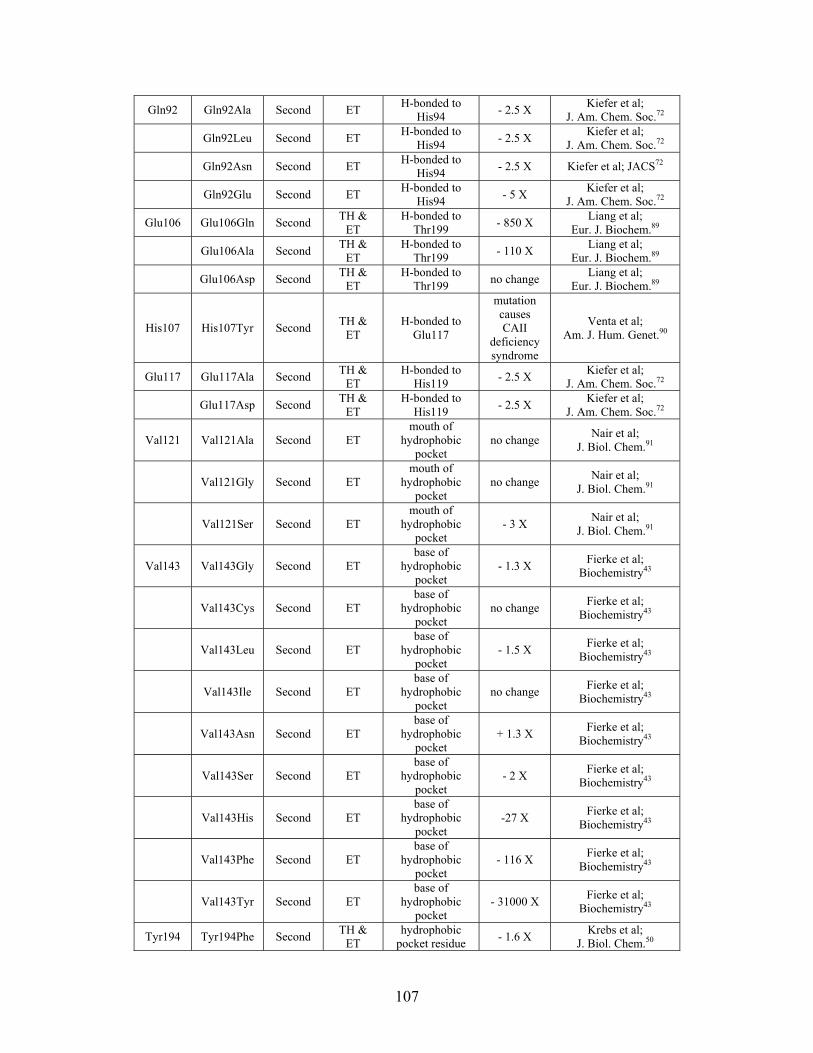
Gln92 Gln92Ala Second ET H-bonded to
His94 - 2.5 X
Kiefer et al; J. Am. Chem. Soc.72
Gln92Leu Second ET H-bonded to
His94 - 2.5 X
Kiefer et al; J. Am. Chem. Soc.72
Gln92Asn Second ET H-bonded to
His94 - 2.5 X Kiefer et al; JACS72
Gln92Glu Second ET H-bonded to
His94 - 5 X
Kiefer et al; J. Am. Chem. Soc.72
Glu106 Glu106Gln Second TH &
ET H-bonded to
Thr199 - 850 X
Liang et al; Eur. J. Biochem.89
Glu106Ala Second TH &
ET H-bonded to
Thr199 - 110 X
Liang et al; Eur. J. Biochem.89
Glu106Asp Second TH &
ET H-bonded to
Thr199 no change
Liang et al; Eur. J. Biochem.89
His107 His107Tyr Second TH &
ET H-bonded to
Glu117
mutation causes CAII
deficiency syndrome
Venta et al; Am. J. Hum. Genet.90
Glu117 Glu117Ala Second TH &
ET H-bonded to
His119 - 2.5 X
Kiefer et al; J. Am. Chem. Soc.72
Glu117Asp Second TH &
ET H-bonded to
His119 - 2.5 X
Kiefer et al; J. Am. Chem. Soc.72
Val121 Val121Ala Second ET mouth of
hydrophobic pocket
no change Nair et al;
J. Biol. Chem.91
Val121Gly Second ET mouth of
hydrophobic pocket
no change Nair et al;
J. Biol. Chem.91
Val121Ser Second ET mouth of
hydrophobic pocket
- 3 X Nair et al;
J. Biol. Chem.91
Val143 Val143Gly Second ET base of
hydrophobic pocket
- 1.3 X Fierke et al;
Biochemistry43
Val143Cys Second ET base of
hydrophobic pocket
no change Fierke et al;
Biochemistry43
Val143Leu Second ET base of
hydrophobic pocket
- 1.5 X Fierke et al;
Biochemistry43
Val143Ile Second ET base of
hydrophobic pocket
no change Fierke et al;
Biochemistry43
Val143Asn Second ET base of
hydrophobic pocket
+ 1.3 X Fierke et al;
Biochemistry43
Val143Ser Second ET base of
hydrophobic pocket
- 2 X Fierke et al;
Biochemistry43
Val143His Second ET base of
hydrophobic pocket
-27 X Fierke et al;
Biochemistry43
Val143Phe Second ET base of
hydrophobic pocket
- 116 X Fierke et al;
Biochemistry43
Val143Tyr Second ET base of
hydrophobic pocket
- 31000 X Fierke et al;
Biochemistry43
Tyr194 Tyr194Phe Second TH &
ET hydrophobic
pocket residue - 1.6 X
Krebs et al; J. Biol. Chem.50
107
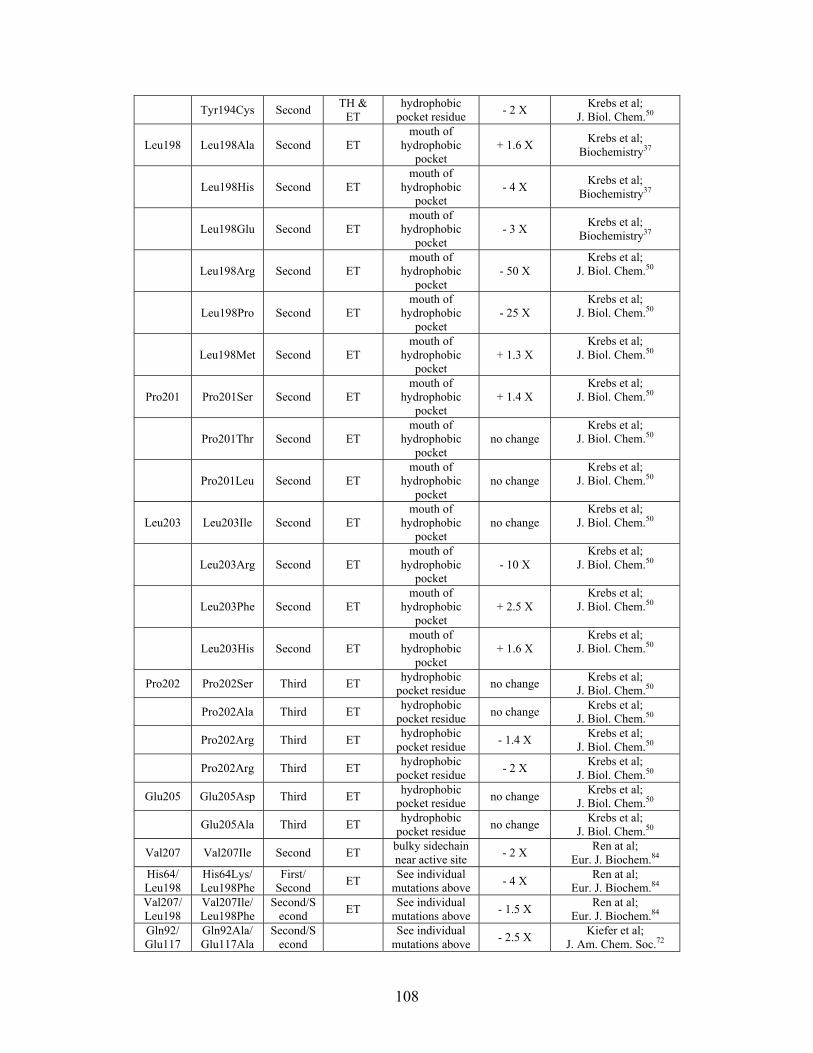
Tyr194Cys Second TH &
ET hydrophobic
pocket residue - 2 X
Krebs et al; J. Biol. Chem.50
Leu198 Leu198Ala Second ET mouth of
hydrophobic pocket
+ 1.6 X Krebs et al;
Biochemistry37
Leu198His Second ET mouth of
hydrophobic pocket
- 4 X Krebs et al;
Biochemistry37
Leu198Glu Second ET mouth of
hydrophobic pocket
- 3 X Krebs et al;
Biochemistry37
Leu198Arg Second ET mouth of
hydrophobic pocket
- 50 X Krebs et al;
J. Biol. Chem.50
Leu198Pro Second ET mouth of
hydrophobic pocket
- 25 X Krebs et al;
J. Biol. Chem.50
Leu198Met Second ET mouth of
hydrophobic pocket
+ 1.3 X Krebs et al;
J. Biol. Chem.50
Pro201 Pro201Ser Second ET mouth of
hydrophobic pocket
+ 1.4 X Krebs et al;
J. Biol. Chem.50
Pro201Thr Second ET mouth of
hydrophobic pocket
no change Krebs et al;
J. Biol. Chem.50
Pro201Leu Second ET mouth of
hydrophobic pocket
no change Krebs et al;
J. Biol. Chem.50
Leu203 Leu203Ile Second ET mouth of
hydrophobic pocket
no change Krebs et al;
J. Biol. Chem.50
Leu203Arg Second ET mouth of
hydrophobic pocket
- 10 X Krebs et al;
J. Biol. Chem.50
Leu203Phe Second ET mouth of
hydrophobic pocket
+ 2.5 X Krebs et al;
J. Biol. Chem.50
Leu203His Second ET mouth of
hydrophobic pocket
+ 1.6 X Krebs et al;
J. Biol. Chem.50
Pro202 Pro202Ser Third ET hydrophobic
pocket residue no change
Krebs et al; J. Biol. Chem.50
Pro202Ala Third ET hydrophobic
pocket residue no change
Krebs et al; J. Biol. Chem.50
Pro202Arg Third ET hydrophobic
pocket residue - 1.4 X
Krebs et al; J. Biol. Chem.50
Pro202Arg Third ET hydrophobic
pocket residue - 2 X
Krebs et al; J. Biol. Chem.50
Glu205 Glu205Asp Third ET hydrophobic
pocket residue no change
Krebs et al; J. Biol. Chem.50
Glu205Ala Third ET hydrophobic
pocket residue no change
Krebs et al; J. Biol. Chem.50
Val207 Val207Ile Second ET bulky sidechain near active site
- 2 X Ren at al;
Eur. J. Biochem.84 His64/ Leu198
His64Lys/ Leu198Phe
First/ Second
ET See individual
mutations above - 4 X
Ren at al; Eur. J. Biochem.84
Val207/Leu198
Val207Ile/ Leu198Phe
Second/Second
ET See individual
mutations above - 1.5 X
Ren at al; Eur. J. Biochem.84
Gln92/ Glu117
Gln92Ala/ Glu117Ala
Second/Second
See individual
mutations above - 2.5 X
Kiefer et al; J. Am. Chem. Soc.72
108
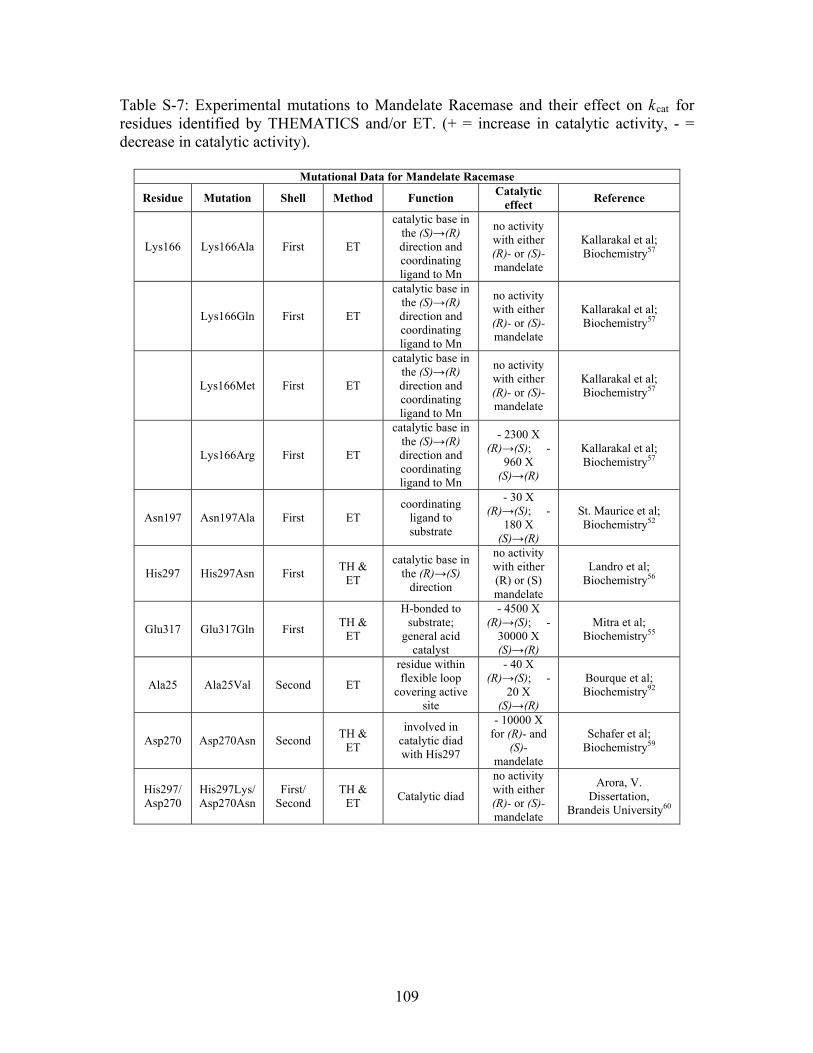
Table S-7: Experimental mutations to Mandelate Racemase and their effect on kcat for residues identified by THEMATICS and/or ET. (+ = increase in catalytic activity, - = decrease in catalytic activity).
Mutational Data for Mandelate Racemase
Residue Mutation Shell Method Function Catalytic
effect Reference
Lys166 Lys166Ala First ET
catalytic base in the (S)→(R) direction and coordinating ligand to Mn
no activity with either (R)- or (S)- mandelate
Kallarakal et al; Biochemistry57
Lys166Gln First ET
catalytic base in the (S)→(R) direction and coordinating ligand to Mn
no activity with either (R)- or (S)- mandelate
Kallarakal et al; Biochemistry57
Lys166Met First ET
catalytic base in the (S)→(R) direction and coordinating ligand to Mn
no activity with either (R)- or (S)- mandelate
Kallarakal et al; Biochemistry57
Lys166Arg First ET
catalytic base in the (S)→(R) direction and coordinating ligand to Mn
- 2300 X (R)→(S); -
960 X (S)→(R)
Kallarakal et al; Biochemistry57
Asn197 Asn197Ala First ET coordinating
ligand to substrate
- 30 X (R)→(S); -
180 X (S)→(R)
St. Maurice et al; Biochemistry52
His297 His297Asn First TH &
ET
catalytic base in the (R)→(S)
direction
no activity with either (R) or (S) mandelate
Landro et al; Biochemistry56
Glu317 Glu317Gln First TH &
ET
H-bonded to substrate;
general acid catalyst
- 4500 X (R)→(S); -
30000 X (S)→(R)
Mitra et al; Biochemistry55
Ala25 Ala25Val Second ET
residue within flexible loop
covering active site
- 40 X (R)→(S); -
20 X (S)→(R)
Bourque et al; Biochemistry92
Asp270 Asp270Asn Second TH &
ET
involved in catalytic diad with His297
- 10000 X for (R)- and
(S)- mandelate
Schafer et al; Biochemistry59
His297/ Asp270
His297Lys/ Asp270Asn
First/ Second
TH & ET
Catalytic diad
no activity with either (R)- or (S)- mandelate
Arora, V. Dissertation,
Brandeis University60
109
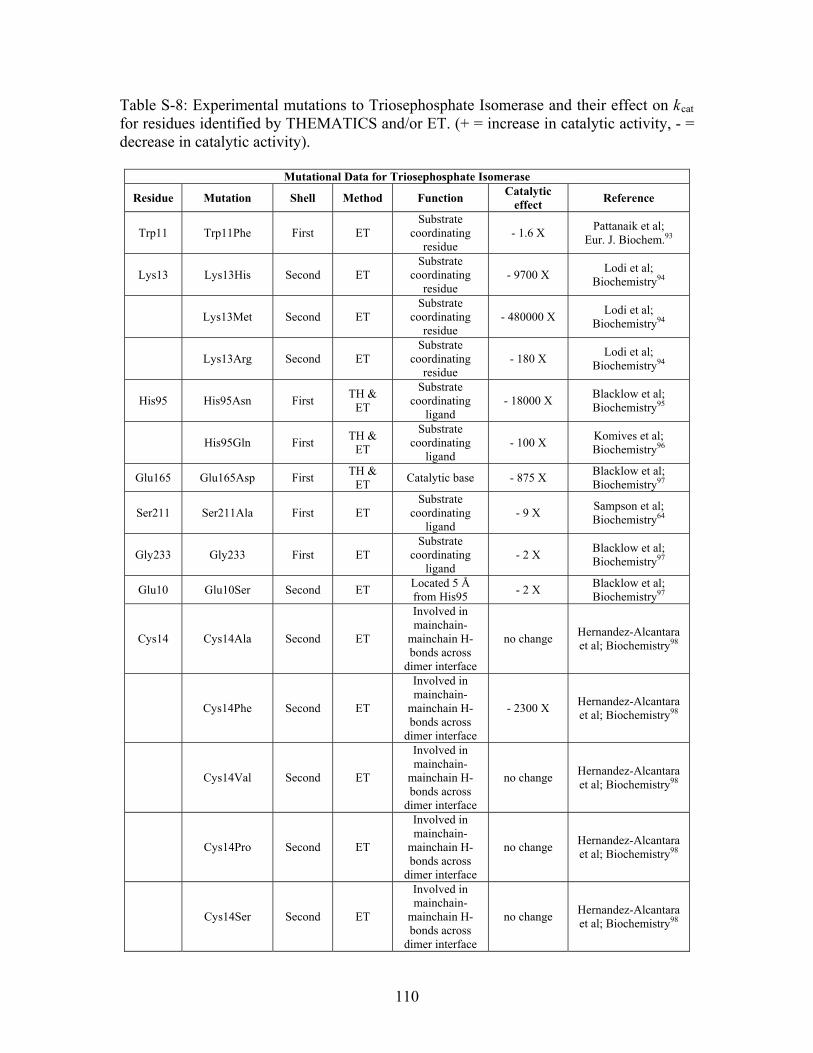
Table S-8: Experimental mutations to Triosephosphate Isomerase and their effect on kcat for residues identified by THEMATICS and/or ET. (+ = increase in catalytic activity, - = decrease in catalytic activity).
Mutational Data for Triosephosphate Isomerase
Residue Mutation Shell Method Function Catalytic
effect Reference
Trp11 Trp11Phe First ET Substrate
coordinating residue
- 1.6 X Pattanaik et al;
Eur. J. Biochem.93
Lys13 Lys13His Second ET Substrate
coordinating residue
- 9700 X Lodi et al;
Biochemistry94
Lys13Met Second ET Substrate
coordinating residue
- 480000 X Lodi et al;
Biochemistry94
Lys13Arg Second ET Substrate
coordinating residue
- 180 X Lodi et al;
Biochemistry94
His95 His95Asn First TH &
ET
Substrate coordinating
ligand - 18000 X
Blacklow et al; Biochemistry95
His95Gln First TH &
ET
Substrate coordinating
ligand - 100 X
Komives et al; Biochemistry96
Glu165 Glu165Asp First TH &
ET Catalytic base - 875 X
Blacklow et al; Biochemistry97
Ser211 Ser211Ala First ET Substrate
coordinating ligand
- 9 X Sampson et al; Biochemistry64
Gly233 Gly233 First ET Substrate
coordinating ligand
- 2 X Blacklow et al; Biochemistry97
Glu10 Glu10Ser Second ET Located 5 Å from His95
- 2 X Blacklow et al; Biochemistry97
Cys14 Cys14Ala Second ET
Involved in mainchain-
mainchain H-bonds across
dimer interface
no change Hernandez-Alcantara et al; Biochemistry98
Cys14Phe Second ET
Involved in mainchain-
mainchain H-bonds across
dimer interface
- 2300 X Hernandez-Alcantara et al; Biochemistry98
Cys14Val Second ET
Involved in mainchain-
mainchain H-bonds across
dimer interface
no change Hernandez-Alcantara et al; Biochemistry98
Cys14Pro Second ET
Involved in mainchain-
mainchain H-bonds across
dimer interface
no change Hernandez-Alcantara et al; Biochemistry98
Cys14Ser Second ET
Involved in mainchain-
mainchain H-bonds across
dimer interface
no change Hernandez-Alcantara et al; Biochemistry98
110
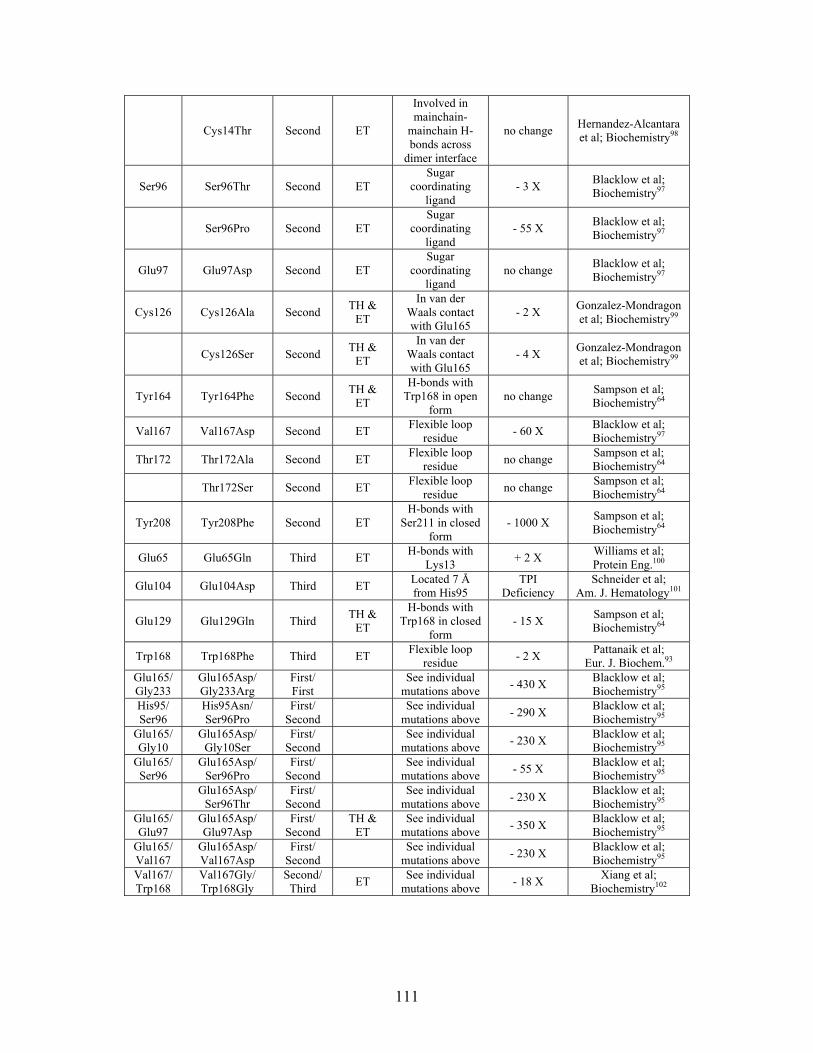
Cys14Thr Second ET
Involved in mainchain-
mainchain H-bonds across
dimer interface
no change Hernandez-Alcantara et al; Biochemistry98
Ser96 Ser96Thr Second ET Sugar
coordinating ligand
- 3 X Blacklow et al; Biochemistry97
Ser96Pro Second ET Sugar
coordinating ligand
- 55 X Blacklow et al; Biochemistry97
Glu97 Glu97Asp Second ET Sugar
coordinating ligand
no change Blacklow et al; Biochemistry97
Cys126 Cys126Ala Second TH &
ET
In van der Waals contact with Glu165
- 2 X Gonzalez-Mondragon et al; Biochemistry99
Cys126Ser Second TH &
ET
In van der Waals contact with Glu165
- 4 X Gonzalez-Mondragon et al; Biochemistry99
Tyr164 Tyr164Phe Second TH &
ET
H-bonds with Trp168 in open
form no change
Sampson et al; Biochemistry64
Val167 Val167Asp Second ET Flexible loop
residue - 60 X
Blacklow et al; Biochemistry97
Thr172 Thr172Ala Second ET Flexible loop
residue no change
Sampson et al; Biochemistry64
Thr172Ser Second ET Flexible loop
residue no change
Sampson et al; Biochemistry64
Tyr208 Tyr208Phe Second ET H-bonds with
Ser211 in closed form
- 1000 X Sampson et al; Biochemistry64
Glu65 Glu65Gln Third ET H-bonds with
Lys13 + 2 X
Williams et al; Protein Eng.100
Glu104 Glu104Asp Third ET Located 7 Å from His95
TPI Deficiency
Schneider et al; Am. J. Hematology101
Glu129 Glu129Gln Third TH &
ET
H-bonds with Trp168 in closed
form - 15 X
Sampson et al; Biochemistry64
Trp168 Trp168Phe Third ET Flexible loop
residue - 2 X
Pattanaik et al; Eur. J. Biochem.93
Glu165/Gly233
Glu165Asp/ Gly233Arg
First/ First
See individual
mutations above - 430 X
Blacklow et al; Biochemistry95
His95/ Ser96
His95Asn/ Ser96Pro
First/ Second
See individual
mutations above - 290 X
Blacklow et al; Biochemistry95
Glu165/Gly10
Glu165Asp/ Gly10Ser
First/ Second
See individual
mutations above - 230 X
Blacklow et al; Biochemistry95
Glu165/Ser96
Glu165Asp/ Ser96Pro
First/ Second
See individual
mutations above - 55 X
Blacklow et al; Biochemistry95
Glu165Asp/
Ser96Thr First/
Second
See individual mutations above
- 230 X Blacklow et al; Biochemistry95
Glu165/Glu97
Glu165Asp/ Glu97Asp
First/ Second
TH & ET
See individual mutations above
- 350 X Blacklow et al; Biochemistry95
Glu165/Val167
Glu165Asp/ Val167Asp
First/ Second
See individual
mutations above - 230 X
Blacklow et al; Biochemistry95
Val167/Trp168
Val167Gly/ Trp168Gly
Second/Third
ET See individual
mutations above - 18 X
Xiang et al; Biochemistry102
111
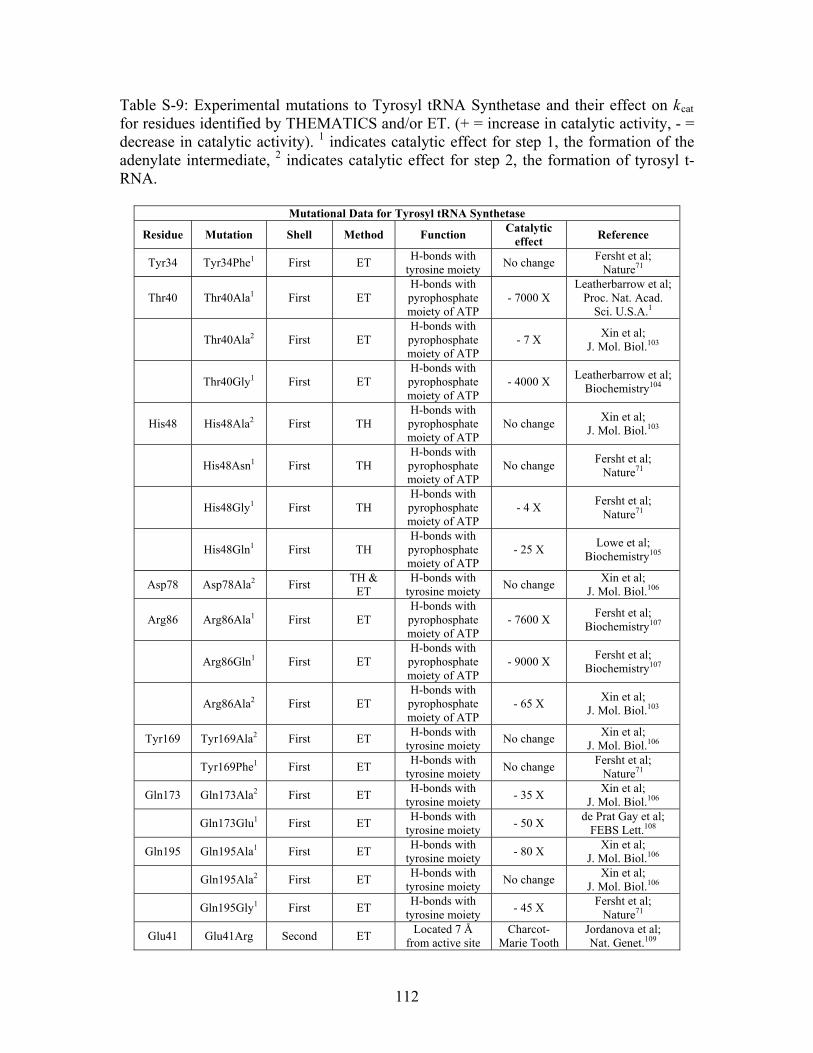
Table S-9: Experimental mutations to Tyrosyl tRNA Synthetase and their effect on kcat for residues identified by THEMATICS and/or ET. (+ = increase in catalytic activity, - = decrease in catalytic activity). 1 indicates catalytic effect for step 1, the formation of the adenylate intermediate, 2 indicates catalytic effect for step 2, the formation of tyrosyl t-RNA.
Mutational Data for Tyrosyl tRNA Synthetase
Residue Mutation Shell Method Function Catalytic
effect Reference
Tyr34 Tyr34Phe1 First ET H-bonds with
tyrosine moiety No change
Fersht et al; Nature71
Thr40 Thr40Ala1 First ET H-bonds with pyrophosphate moiety of ATP
- 7000 X Leatherbarrow et al;
Proc. Nat. Acad. Sci. U.S.A.1
Thr40Ala2 First ET H-bonds with pyrophosphate moiety of ATP
- 7 X Xin et al;
J. Mol. Biol.103
Thr40Gly1 First ET H-bonds with pyrophosphate moiety of ATP
- 4000 X Leatherbarrow et al;
Biochemistry104
His48 His48Ala2 First TH H-bonds with pyrophosphate moiety of ATP
No change Xin et al;
J. Mol. Biol.103
His48Asn1 First TH H-bonds with pyrophosphate moiety of ATP
No change Fersht et al;
Nature71
His48Gly1 First TH H-bonds with pyrophosphate moiety of ATP
- 4 X Fersht et al;
Nature71
His48Gln1 First TH H-bonds with pyrophosphate moiety of ATP
- 25 X Lowe et al;
Biochemistry105
Asp78 Asp78Ala2 First TH &
ET H-bonds with
tyrosine moiety No change
Xin et al; J. Mol. Biol.106
Arg86 Arg86Ala1 First ET H-bonds with pyrophosphate moiety of ATP
- 7600 X Fersht et al;
Biochemistry107
Arg86Gln1 First ET H-bonds with pyrophosphate moiety of ATP
- 9000 X Fersht et al;
Biochemistry107
Arg86Ala2 First ET H-bonds with pyrophosphate moiety of ATP
- 65 X Xin et al;
J. Mol. Biol.103
Tyr169 Tyr169Ala2 First ET H-bonds with
tyrosine moiety No change
Xin et al; J. Mol. Biol.106
Tyr169Phe1 First ET H-bonds with
tyrosine moiety No change
Fersht et al; Nature71
Gln173 Gln173Ala2 First ET H-bonds with
tyrosine moiety - 35 X
Xin et al; J. Mol. Biol.106
Gln173Glu1 First ET H-bonds with
tyrosine moiety - 50 X
de Prat Gay et al; FEBS Lett.108
Gln195 Gln195Ala1 First ET H-bonds with
tyrosine moiety - 80 X
Xin et al; J. Mol. Biol.106
Gln195Ala2 First ET H-bonds with
tyrosine moiety No change
Xin et al; J. Mol. Biol.106
Gln195Gly1 First ET H-bonds with
tyrosine moiety - 45 X
Fersht et al; Nature71
Glu41 Glu41Arg Second ET Located 7 Å
from active site Charcot-
Marie Tooth Jordanova et al; Nat. Genet.109
112
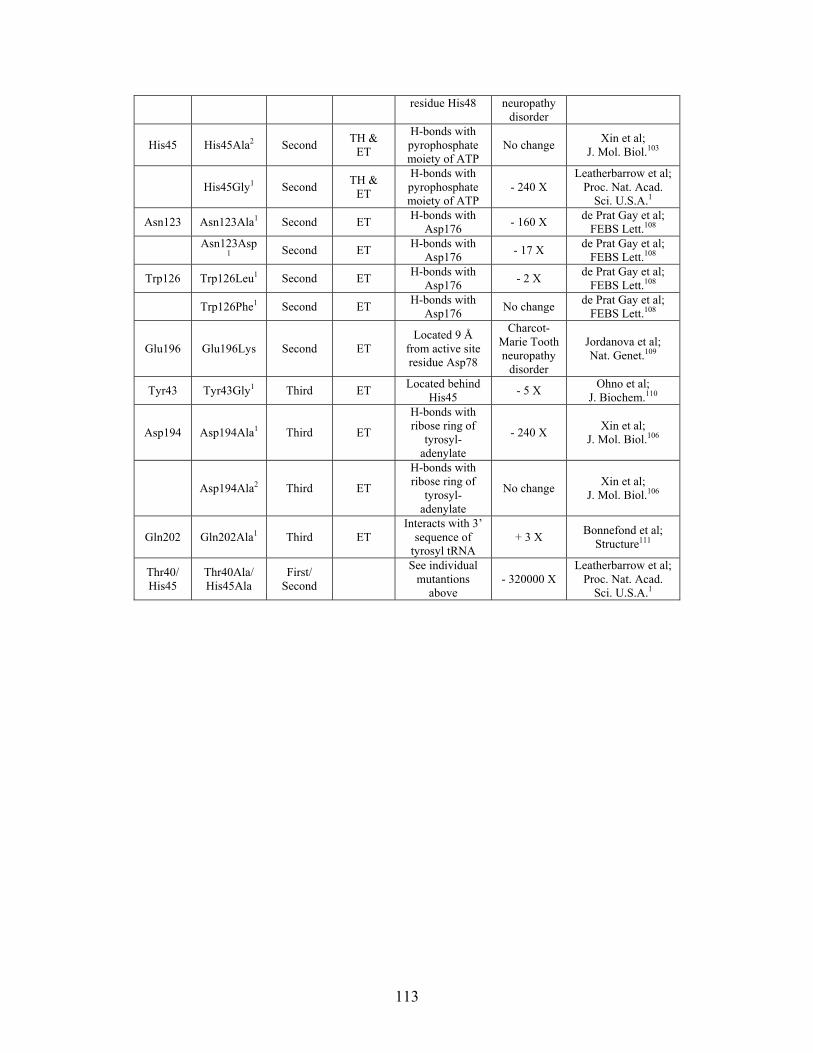
residue His48 neuropathy disorder
His45 His45Ala2 Second TH &
ET
H-bonds with pyrophosphate moiety of ATP
No change Xin et al;
J. Mol. Biol.103
His45Gly1 Second TH &
ET
H-bonds with pyrophosphate moiety of ATP
- 240 X Leatherbarrow et al;
Proc. Nat. Acad. Sci. U.S.A.1
Asn123 Asn123Ala1 Second ET H-bonds with
Asp176 - 160 X
de Prat Gay et al; FEBS Lett.108
Asn123Asp
1 Second ET H-bonds with
Asp176 - 17 X
de Prat Gay et al; FEBS Lett.108
Trp126 Trp126Leu1 Second ET H-bonds with
Asp176 - 2 X
de Prat Gay et al; FEBS Lett.108
Trp126Phe1 Second ET H-bonds with
Asp176 No change
de Prat Gay et al; FEBS Lett.108
Glu196 Glu196Lys Second ET Located 9 Å
from active site residue Asp78
Charcot-Marie Tooth neuropathy
disorder
Jordanova et al; Nat. Genet.109
Tyr43 Tyr43Gly1 Third ET Located behind
His45 - 5 X
Ohno et al; J. Biochem.110
Asp194 Asp194Ala1 Third ET
H-bonds with ribose ring of
tyrosyl-adenylate
- 240 X Xin et al;
J. Mol. Biol.106
Asp194Ala2 Third ET
H-bonds with ribose ring of
tyrosyl-adenylate
No change Xin et al;
J. Mol. Biol.106
Gln202 Gln202Ala1 Third ET Interacts with 3’
sequence of tyrosyl tRNA
+ 3 X Bonnefond et al;
Structure111
Thr40/ His45
Thr40Ala/ His45Ala
First/ Second
See individual
mutantions above
- 320000 X Leatherbarrow et al;
Proc. Nat. Acad. Sci. U.S.A.1
113

2.8 References
1. Leatherbarrow, R. J., Fersht, A. R. & Winter, G. (1985). Transition-state
stabilization in the mechanism of tyrosyl-tRNA synthetase revealed by protein engineering. Proc Natl Acad Sci U S A 82, 7840-7844.
2. Johannes, T. W. & Zhao, H. (2006). Directed evolution of enzymes and biosynthetic pathways. Curr Opin Microbiol 9, 261-267.
3. Murga, L. F., Wei, Y. & Ondrechen, M. J. (2007). Computed Protonation Properties: Unique Capabilities for Protein Functional Site Prediction. Genome Informatics 19, 107-118.
4. Ondrechen, M. J., J.G. Clifton and D. Ringe. (2001). THEMATICS: A simple computational predictor of enzyme function from structure. Proc. Natl. Acad. Sci. (USA) 98, 12473-12478.
5. Ondrechen, M. J. (2004). Identification of functional sites based on prediction of charged group behavior. In Current Protocols in Bioinformatics (Baxevanis, A. D., Davison, D. B., Page, R. D. M., Petsko, G. A., Stein, L. D. & Stormo, G. D., eds.), pp. 8.6.1 - 8.6.10. John Wiley & Sons, Hoboken, N.J.
6. Wei, Y., Ko, J., Murga, L. & Ondrechen, M. J. (2007). Selective prediction of Interaction sites in protein structures with THEMATICS. BMC Bioinformatics 8, 119.
7. Lichtarge, O., Bourne, H. R. & Cohen, F. E. (1996). An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol 257, 342-358.
8. Madabushi, S., Yao, H., Marsh, M., Kristensen, D. M., Philippi, A., Sowa, M. E. & Lichtarge, O. (2002). Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J Mol Biol 316, 139-154.
9. Yao, H., Kristensen, D. M., Mihalek, I., Sowa, M. E., Shaw, C., Kimmel, M., Kavraki, L. & Lichtarge, O. (2003). An accurate, sensitive, and scalable method to identify functional sites in protein structures. J Mol Biol 326, 255-261.
10. Ondrechen, M. J. (2002). THEMATICS as a tool for functional genomics. Genome Informatics 13, 563-564.
11. Ondrechen, M. J., L.F. Murga, J.G. Clifton and D. Ringe. (2003). Prediction of Protein Function with THEMATICS. Currents in Computational Molecular Biology, 21-22.
12. Ondrechen, M. J., Clifton, J. G. & D. Ringe. (2001). An Electrostatic Model for the Active Sites of Three Different Vitamin B6 Dependent Enzymes (submitted).
13. Lichtarge, O., Sowa, M. E. & Philippi, A. (2002). Evolutionary traces of functional surfaces along G protein signaling pathway. Methods Enzymol 344, 536-556.
14. Sobolev, V., Eyal, E., Gerzon, S., Potapov, V., Babor, M., Prilusky, J. & Edelman, M. (2005). SPACE: a suite of tools for protein structure prediction and analysis based on complementarity and environment. Nucleic Acids Res 33, W39-43.
15. Sobolev, V., Sorokine, A., Prilusky, J., Abola, E. E. & Edelman, M. (1999). Automated analysis of interatomic contacts in proteins. Bioinformatics 15, 327-332.
114

16. Armon, A., Graur, D. & Ben-Tal, N. (2001). ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J Mol Biol 307, 447-463.
17. Glaser, F., Pupko, T., Paz, I., Bell, R. E., Bechor-Shental, D., Martz, E. & Ben-Tal, N. (2003). ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163-164.
18. Landau, M., Mayrose, I., Rosenberg, Y., Glaser, F., Martz, E., Pupko, T. & Ben-Tal, N. (2005). ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33, W299-302.
19. Chang, A., Scheer, M., Grote, A., Schomburg, I. & Schomburg, D. (2009). BRENDA, AMENDA and FRENDA the enzyme information system: new content and tools in 2009. Nucleic Acids Res 37, D588-592.
20. Kim, E. E. & Wyckoff, H. W. (1991). Reaction mechanism of alkaline phosphatase based on crystal structures. Two-metal ion catalysis. J Mol Biol 218, 449-464.
21. Eriksson, A. E., Jones, T. A. & Liljas, A. (1988). Refined structure of human carbonic anhydrase II at 2.0 A resolution. Proteins 4, 274-282.
22. Neidhart, D. J., Howell, P. L., Petsko, G. A., Powers, V. M., Li, R. S., Kenyon, G. L. & Gerlt, J. A. (1991). Mechanism of the reaction catalyzed by mandelate racemase. 2. Crystal structure of mandelate racemase at 2.5-A resolution: identification of the active site and possible catalytic residues. Biochemistry 30, 9264-9273.
23. Zhang, Z., Sugio, S., Komives, E. A., Liu, K. D., Knowles, J. R., Petsko, G. A. & Ringe, D. (1994). Crystal structure of recombinant chicken triosephosphate isomerase-phosphoglycolohydroxamate complex at 1.8-A resolution. Biochemistry 33, 2830-2837.
24. Brick, P., Bhat, T. N. & Blow, D. M. (1989). Structure of tyrosyl-tRNA synthetase refined at 2.3 A resolution. Interaction of the enzyme with the tyrosyl adenylate intermediate. J Mol Biol 208, 83-98.
25. Muller, B. H., Lamoure, C., Le Du, M. H., Cattolico, L., Lajeunesse, E., Lemaitre, F., Pearson, A., Ducancel, F., Menez, A. & Boulain, J. C. (2001). Improving Escherichia coli alkaline phosphatase efficacy by additional mutations inside and outside the catalytic pocket. Chembiochem 2, 517-523.
26. Hehir, M. J., Murphy, J. E. & Kantrowitz, E. R. (2000). Characterization of heterodimeric alkaline phosphatases from Escherichia coli: an investigation of intragenic complementation. J Mol Biol 304, 645-656.
27. Coleman, J. E. (1992). Structure and mechanism of alkaline phosphatase. Annu Rev Biophys Biomol Struct 21, 441-483.
28. Xu, X. & Kantrowitz, E. R. (1991). A water-mediated salt link in the catalytic site of Escherichia coli alkaline phosphatase may influence activity. Biochemistry 30, 7789-7796.
29. Stec, B., Hehir, M. J., Brennan, C., Nolte, M. & Kantrowitz, E. R. (1998). Kinetic and X-ray structural studies of three mutant E. coli alkaline phosphatases: insights into the catalytic mechanism without the nucleophile Ser102. J Mol Biol 277, 647-662.
115

30. Holtz, K. M. & Kantrowitz, E. R. (1999). The mechanism of the alkaline phosphatase reaction: insights from NMR, crystallography and site-specific mutagenesis. FEBS Lett 462, 7-11.
31. Stec, B., Holtz, K. M. & Kantrowitz, E. R. (2000). A revised mechanism for the alkaline phosphatase reaction involving three metal ions. J Mol Biol 299, 1303-1311.
32. Dealwis, C. G., Chen, L., Brennan, C., Mandecki, W. & Abad-Zapatero, C. (1995). 3-D structure of the D153G mutant of Escherichia coli alkaline phosphatase: an enzyme with weaker magnesium binding and increased catalytic activity. Protein Eng 8, 865-871.
33. Matlin, A. R., Kendall, D. A., Carano, K. S., Banzon, J. A., Klecka, S. B. & Solomon, N. M. (1992). Enhanced catalysis by active-site mutagenesis at aspartic acid 153 in Escherichia coli alkaline phosphatase. Biochemistry 31, 8196-8200.
34. Wojciechowski, C. L. & Kantrowitz, E. R. (2002). Altering of the metal specificity of Escherichia coli alkaline phosphatase. J Biol Chem 277, 50476-50481.
35. Wojciechowski, C. L. & Kantrowitz, E. R. (2003). Glutamic acid residues as metal ligands in the active site of Escherichia coli alkaline phosphatase. Biochim Biophys Acta 1649, 68-73.
36. Christianson, D. W. & Cox, J. D. (1999). Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes. Annu Rev Biochem 68, 33-57.
37. Krebs, J. F., Rana, F., Dluhy, R. A. & Fierke, C. A. (1993). Kinetic and spectroscopic studies of hydrophilic amino acid substitutions in the hydrophobic pocket of human carbonic anhydrase II. Biochemistry 32, 4496-4505.
38. Christianson, D. W. F., C. A. (1996). Carbonic Anhydrase: Evolution of the Zinc Binding Site by Nature and Design. Acc. Chem. Rev. 29, 331-339.
39. Hunt, J. A., Ahmed, M. & Fierke, C. A. (1999). Metal binding specificity in carbonic anhydrase is influenced by conserved hydrophobic core residues. Biochemistry 38, 9054-9062.
40. Thompson, R. B. J., E. R. (1993). Enzyme-Based Fiber Optic Zinc Biosensor. Anal. Chem. 65, 730-734.
41. Lesburg, C. A., Huang, C., Christianson, D. W. & Fierke, C. A. (1997). Histidine --> carboxamide ligand substitutions in the zinc binding site of carbonic anhydrase II alter metal coordination geometry but retain catalytic activity. Biochemistry 36, 15780-15791.
42. Kiefer, L. L., Krebs, J. F., Paterno, S. A. & Fierke, C. A. (1993). Engineering a cysteine ligand into the zinc binding site of human carbonic anhydrase II. Biochemistry 32, 9896-9900.
43. Fierke, C. A., Calderone, T. L. & Krebs, J. F. (1991). Functional consequences of engineering the hydrophobic pocket of carbonic anhydrase II. Biochemistry 30, 11054-11063.
44. Ippolito, J. A. & Christianson, D. W. (1993). Structure of an engineered His3Cys zinc binding site in human carbonic anhydrase II. Biochemistry 32, 9901-9905.
45. Krebs, J. F., Fierke, C. A., Alexander, R. S. & Christianson, D. W. (1991). Conformational mobility of His-64 in the Thr-200----Ser mutant of human carbonic anhydrase II. Biochemistry 30, 9153-9160.
116

46. Elleby, B., Sjoblom, B. & Lindskog, S. (1999). Changing the efficiency and specificity of the esterase activity of human carbonic anhydrase II by site-specific mutagenesis. Eur J Biochem 262, 516-521.
47. Aronsson, G., Martensson, L. G., Carlsson, U. & Jonsson, B. H. (1995). Folding and stability of the N-terminus of human carbonic anhydrase II. Biochemistry 34, 2153-2162.
48. Persson, M., Hammarstrom, P., Lindgren, M., Jonsson, B. H., Svensson, M. & Carlsson, U. (1999). EPR mapping of interactions between spin-labeled variants of human carbonic anhydrase II and GroEL: evidence for increased flexibility of the hydrophobic core by the interaction. Biochemistry 38, 432-441.
49. Andersson, D., Freskgard, P. O., Jonsson, B. H. & Carlsson, U. (1997). Formation of local native-like tertiary structures in the slow refolding reaction of human carbonic anhydrase II as monitored by circular dichroism on tryptophan mutants. Biochemistry 36, 4623-4630.
50. Krebs, J. F. & Fierke, C. A. (1993). Determinants of catalytic activity and stability of carbonic anhydrase II as revealed by random mutagenesis. J Biol Chem 268, 948-954.
51. Gould, S. M. & Tawfik, D. S. (2005). Directed evolution of the promiscuous esterase activity of carbonic anhydrase II. Biochemistry 44, 5444-5452.
52. St Maurice, M. & Bearne, S. L. (2000). Reaction intermediate analogues for mandelate racemase: interaction between Asn 197 and the alpha-hydroxyl of the substrate promotes catalysis. Biochemistry 39, 13324-13335.
53. Hasson, M. S., Schlichting, I., Moulai, J., Taylor, K., Barrett, W., Kenyon, G. L., Babbitt, P. C., Gerlt, J. A., Petsko, G. A. & Ringe, D. (1998). Evolution of an enzyme active site: the structure of a new crystal form of muconate lactonizing enzyme compared with mandelate racemase and enolase. Proc Natl Acad Sci U S A 95, 10396-10401.
54. Babbitt, P. C. & Gerlt, J. A. (1997). Understanding enzyme superfamilies. Chemistry As the fundamental determinant in the evolution of new catalytic activities. J Biol Chem 272, 30591-30594.
55. Mitra, B., Kallarakal, A. T., Kozarich, J. W., Gerlt, J. A., Clifton, J. G., Petsko, G. A. & Kenyon, G. L. (1995). Mechanism of the reaction catalyzed by mandelate racemase: importance of electrophilic catalysis by glutamic acid 317. Biochemistry 34, 2777-2787.
56. Landro, J. A., Kallarakal, A. T., Ransom, S. C., Gerlt, J. A., Kozarich, J. W., Neidhart, D. J. & Kenyon, G. L. (1991). Mechanism of the reaction catalyzed by mandelate racemase. 3. Asymmetry in reactions catalyzed by the H297N mutant. Biochemistry 30, 9274-9281.
57. Kallarakal, A. T., Mitra, B., Kozarich, J. W., Gerlt, J. A., Clifton, J. G., Petsko, G. A. & Kenyon, G. L. (1995). Mechanism of the reaction catalyzed by mandelate racemase: structure and mechanistic properties of the K166R mutant. Biochemistry 34, 2788-2797.
58. Babbitt, P. C., Mrachko, G. T., Hasson, M. S., Huisman, G. W., Kolter, R., Ringe, D., Petsko, G. A., Kenyon, G. L. & Gerlt, J. A. (1995). A functionally diverse enzyme superfamily that abstracts the alpha protons of carboxylic acids. Science 267, 1159-1161.
117

59. Schafer, S. L., Barrett, W. C., Kallarakal, A. T., Mitra, B., Kozarich, J. W., Gerlt, J. A., Clifton, J. G., Petsko, G. A. & Kenyon, G. L. (1996). Mechanism of the reaction catalyzed by mandelate racemase: structure and mechanistic properties of the D270N mutant. Biochemistry 35, 5662-5669.
60. Arora, V. (2001). Structural Enzymology, Ligand Recognition and Mechanistic Pathways. Dissertation, Brandeis University.
61. Wierenga, R. K. (2001). The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett 492, 193-198.
62. Espinoza-Fonseca, L. M. & Trujillo-Ferrara, J. G. (2004). Exploring the possible binding sites at the interface of triosephosphate isomerase dimer as a potential target for anti-tripanosomal drug design. Bioorg Med Chem Lett 14, 3151-3154.
63. Wierenga, R. K., Borchert, T. V. & Noble, M. E. (1992). Crystallographic binding studies with triosephosphate isomerases: conformational changes induced by substrate and substrate-analogues. FEBS Lett 307, 34-39.
64. Sampson, N. S. & Knowles, J. R. (1992). Segmental movement: definition of the structural requirements for loop closure in catalysis by triosephosphate isomerase. Biochemistry 31, 8482-8487.
65. Knowles, J. R. (1991). Enzyme catalysis: not different, just better. Nature 350, 121-124.
66. Lolis, E., Alber, T., Davenport, R. C., Rose, D., Hartman, F. C. & Petsko, G. A. (1990). Structure of yeast triosephosphate isomerase at 1.9-A resolution. Biochemistry 29, 6609-6618.
67. Winter, G., Fersht, A. R., Wilkinson, A. J., Zoller, M. & Smith, M. (1982). Redesigning enzyme structure by site-directed mutagenesis: tyrosyl tRNA synthetase and ATP binding. Nature 299, 756-758.
68. Brannigan, J. A. & Wilkinson, A. J. (2002). Protein engineering 20 years on. Nat Rev Mol Cell Biol 3, 964-970.
69. Ward, W. H., Jones, D. H. & Fersht, A. R. (1987). Effects of engineering complementary charged residues into the hydrophobic subunit interface of tyrosyl-tRNA synthetase. Appendix: Kinetic analysis of dimeric enzymes that reversibly dissociate into inactive subunits. Biochemistry 26, 4131-4138.
70. Fersht, A. R. (1987). Dissection of the structure and activity of the tyrosyl-tRNA synthetase by site-directed mutagenesis. Biochemistry 26, 8031-8037.
71. Fersht, A. R., Shi, J. P., Knill-Jones, J., Lowe, D. M., Wilkinson, A. J., Blow, D. M., Brick, P., Carter, P., Waye, M. M. & Winter, G. (1985). Hydrogen bonding and biological specificity analysed by protein engineering. Nature 314, 235-238.
72. Kiefer, L. L., Paterno, S. A. & Fierke, C. A. (1995). Hydrogen Bond Network in the Metal Binding Site of Carbonic Anhydrase Enhances Zinc Affinity and Catalytic Efficiency. J. Am. Chem. Soc. 117, 6831-6837.
73. Chaidaroglou, A. & Kantrowitz, E. R. (1989). Alteration of aspartate 101 in the active site of Escherichia coli alkaline phosphatase enhances the catalytic activity. Protein Eng 3, 127-132.
74. Chaidaroglou, A., Brezinski, D. J., Middleton, S. A. & Kantrowitz, E. R. (1988). Function of arginine-166 in the active site of Escherichia coli alkaline phosphatase. Biochemistry 27, 8338-8343.
118

75. O'Brien, P. J. & Herschlag, D. (2001). Functional interrelationships in the alkaline phosphatase superfamily: phosphodiesterase activity of Escherichia coli alkaline phosphatase. Biochemistry 40, 5691-5699.
76. Butler-Ransohoff, J. E., Kendall, D. A. & Kaiser, E. T. (1988). Use of site-directed mutagenesis to elucidate the role of arginine-166 in the catalytic mechanism of alkaline phosphatase. Proc Natl Acad Sci U S A 85, 4276-4278.
77. Xu, X. & Kantrowitz, E. R. (1992). The importance of aspartate 327 for catalysis and zinc binding in Escherichia coli alkaline phosphatase. J Biol Chem 267, 16244-16251.
78. Sun, L., Martin, D. C. & Kantrowitz, E. R. (1999). Rate-determining step of Escherichia coli alkaline phosphatase altered by the removal of a positive charge at the active center. Biochemistry 38, 2842-2848.
79. Mandecki, W., Shallcross, M. A., Sowadski, J. & Tomazic-Allen, S. (1991). Mutagenesis of conserved residues within the active site of Escherichia coli alkaline phosphatase yields enzymes with increased kcat. Protein Eng 4, 801-804.
80. Xu, X., Qin, X. Q. & Kantrowitz, E. R. (1994). Probing the role of histidine-372 in zinc binding and the catalytic mechanism of Escherichia coli alkaline phosphatase by site-specific mutagenesis. Biochemistry 33, 2279-2284.
81. Boulanger, R. R., Jr. & Kantrowitz, E. R. (2003). Characterization of a monomeric Escherichia coli alkaline phosphatase formed upon a single amino acid substitution. J Biol Chem 278, 23497-23501.
82. Janeway, C. M., Xu, X., Murphy, J. E., Chaidaroglou, A. & Kantrowitz, E. R. (1993). Magnesium in the active site of Escherichia coli alkaline phosphatase is important for both structural stabilization and catalysis. Biochemistry 32, 1601-1609.
83. Duda, D., Tu, C., Qian, M., Laipis, P., Agbandje-McKenna, M., Silverman, D. N. & McKenna, R. (2001). Structural and kinetic analysis of the chemical rescue of the proton transfer function of carbonic anhydrase II. Biochemistry 40, 1741-1748.
84. Ren, X. L., Jonsson, B. H. & Lindskog, S. (1991). Some properties of site-specific mutants of human carbonic anhydrase II having active-site residues characterizing carbonic anhydrase III. Eur J Biochem 201, 417-420.
85. Engstrand, C., Forsman, C., Liang, Z. & Lindskog, S. (1992). Proton transfer roles of lysine 64 and glutamic acid 64 replacing histidine 64 in the active site of human carbonic anhydrase II. Biochim Biophys Acta 1122, 321-326.
86. Kiefer, L. L. & Fierke, C. A. (1994). Functional characterization of human carbonic anhydrase II variants with altered zinc binding sites. Biochemistry 33, 15233-15240.
87. Krebs, J. F., Ippolito, J. A., Christianson, D. W. & Fierke, C. A. (1993). Structural and functional importance of a conserved hydrogen bond network in human carbonic anhydrase II. J Biol Chem 268, 27458-27466.
88. Behravan, G., Jonsson, B. H. & Lindskog, S. (1991). Fine tuning of the catalytic properties of human carbonic anhydrase II. Effects of varying active-site residue 200. Eur J Biochem 195, 393-396.
119

89. Liang, Z., Xue, Y., Behravan, G., Jonsson, B. H. & Lindskog, S. (1993). Importance of the conserved active-site residues Tyr7, Glu106 and Thr199 for the catalytic function of human carbonic anhydrase II. Eur J Biochem 211, 821-827.
90. Venta, P. J., Welty, R. J., Johnson, T. M., Sly, W. S. & Tashian, R. E. (1991). Carbonic anhydrase II deficiency syndrome in a Belgian family is caused by a point mutation at an invariant histidine residue (107 His----Tyr): complete structure of the normal human CA II gene. Am J Hum Genet 49, 1082-1090.
91. Nair, S. K., Calderone, T. L., Christianson, D. W. & Fierke, C. A. (1991). Altering the mouth of a hydrophobic pocket. Structure and kinetics of human carbonic anhydrase II mutants at residue Val-121. J Biol Chem 266, 17320-17325.
92. Bourque, J. R. & Bearne, S. L. (2008). Mutational analysis of the active site flap (20s loop) of mandelate racemase. Biochemistry 47, 566-578.
93. Pattanaik, P., Ravindra, G., Sengupta, C., Maithal, K., Balaram, P. & Balaram, H. (2003). Unusual fluorescence of W168 in Plasmodium falciparum triosephosphate isomerase, probed by single-tryptophan mutants. Eur J Biochem 270, 745-756.
94. Lodi, P. J., Chang, L. C., Knowles, J. R. & Komives, E. A. (1994). Triosephosphate isomerase requires a positively charged active site: the role of lysine-12. Biochemistry 33, 2809-2814.
95. Blacklow, S. C. & Knowles, J. R. (1990). How can a catalytic lesion be offset? The energetics of two pseudorevertant triosephosphate isomerases. Biochemistry 29, 4099-4108.
96. Komives, E. A., Chang, L. C., Lolis, E., Tilton, R. F., Petsko, G. A. & Knowles, J. R. (1991). Electrophilic catalysis in triosephosphate isomerase: the role of histidine-95. Biochemistry 30, 3011-3019.
97. Blacklow, S. C., Liu, K. D. & Knowles, J. R. (1991). Stepwise improvements in catalytic effectiveness: independence and interdependence in combinations of point mutations of a sluggish triosephosphate isomerase. Biochemistry 30, 8470-8476.
98. Hernandez-Alcantara, G., Garza-Ramos, G., Hernandez, G. M., Gomez-Puyou, A. & Perez-Montfort, R. (2002). Catalysis and stability of triosephosphate isomerase from Trypanosoma brucei with different residues at position 14 of the dimer interface. Characterization of a catalytically competent monomeric enzyme. Biochemistry 41, 4230-4238.
99. Gonzalez-Mondragon, E., Zubillaga, R. A., Saavedra, E., Chanez-Cardenas, M. E., Perez-Montfort, R. & Hernandez-Arana, A. (2004). Conserved cysteine 126 in triosephosphate isomerase is required not for enzymatic activity but for proper folding and stability. Biochemistry 43, 3255-3263.
100. Williams, J. C., Zeelen, J. P., Neubauer, G., Vriend, G., Backmann, J., Michels, P. A., Lambeir, A. M. & Wierenga, R. K. (1999). Structural and mutagenesis studies of leishmania triosephosphate isomerase: a point mutation can convert a mesophilic enzyme into a superstable enzyme without losing catalytic power. Protein Eng 12, 243-250.
101. Schneider, A., Westwood, B., Yim, C., Prchal, J., Berkow, R., Labotka, R., Warrier, R. & Beutler, E. (1995). Triosephosphate isomerase deficiency: repetitive occurrence of point mutation in amino acid 104 in multiple apparently unrelated families. Am J Hematol 50, 263-268.
120

102. Xiang, J., Jung, J. Y. & Sampson, N. S. (2004). Entropy effects on protein hinges: the reaction catalyzed by triosephosphate isomerase. Biochemistry 43, 11436-11445.
103. Xin, Y., Li, W., Dwyer, D. S. & First, E. A. (2000). Correlating amino acid conservation with function in tyrosyl-tRNA synthetase. J Mol Biol 303, 287-298.
104. Leatherbarrow, R. J. & Fersht, A. R. (1987). Investigation of transition-state stabilization by residues histidine-45 and threonine-40 in the tyrosyl-tRNA synthetase. Biochemistry 26, 8524-8528.
105. Lowe, D. M., Fersht, A. R., Wilkinson, A. J., Carter, P. & Winter, G. (1985). Probing histidine-substrate interactions in tyrosyl-tRNA synthetase using asparagine and glutamine replacements. Biochemistry 24, 5106-5109.
106. Xin, Y., Li, W. & First, E. A. (2000). Stabilization of the transition state for the transfer of tyrosine to tRNA(Tyr) by tyrosyl-tRNA synthetase. J Mol Biol 303, 299-310.
107. Fersht, A. R., Knill-Jones, J. W., Bedouelle, H. & Winter, G. (1988). Reconstruction by site-directed mutagenesis of the transition state for the activation of tyrosine by the tyrosyl-tRNA synthetase: a mobile loop envelopes the transition state in an induced-fit mechanism. Biochemistry 27, 1581-1587.
108. de Prat Gay, G., Duckworth, H. W. & Fersht, A. R. (1993). Modification of the amino acid specificity of tyrosyl-tRNA synthetase by protein engineering. FEBS Lett 318, 167-171.
109. Jordanova, A., Irobi, J., Thomas, F. P., Van Dijck, P., Meerschaert, K., Dewil, M., Dierick, I., Jacobs, A., De Vriendt, E., Guergueltcheva, V., Rao, C. V., Tournev, I., Gondim, F. A., D'Hooghe, M., Van Gerwen, V., Callaerts, P., Van Den Bosch, L., Timmermans, J. P., Robberecht, W., Gettemans, J., Thevelein, J. M., De Jonghe, P., Kremensky, I. & Timmerman, V. (2006). Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet 38, 197-202.
110. Ohno, S., Yokogawa, T. & Nishikawa, K. (2001). Changing the amino acid specificity of yeast tyrosyl-tRNA synthetase by genetic engineering. J Biochem 130, 417-423.
111. Bonnefond, L., Frugier, M., Touze, E., Lorber, B., Florentz, C., Giege, R., Sauter, C. & Rudinger-Thirion, J. (2007). Crystal structure of human mitochondrial tyrosyl-tRNA synthetase reveals common and idiosyncratic features. Structure 15, 1505-1516.
121

Chapter 3
Structural and Kinetic Analysis of Wild Type Co-type Nitrile Hydratase from
Pseudomonas putida
122

3.1 Introduction
Nitrile hydratases (E.C. 4.2.1.84, NHases) are a class of enzymes that have evolved to
utilize low-spin Co(III) to catalyze the hydrolysis of a wide variety of nitrile substrates to
their corresponding amides.
N
O
NH2
This is unusual since cobalt is utilized in only a handful of metalloenzymes, and when it
is used, it is primarily present in the corrinoid center of cobalamin.1-3 In cobalamin
containing enzymes, the Co(III) form is inactive and low-spin Co(III) substitution is
inert.1,2,4 The reactivity of these low-spin Co(III) NHases has led to their prevalent use as
a biocatalyst used for the industrial production of commodity chemicals.5 Specifically,
NHase is currently being used to produce acrylamide on the kiloton scale each year.6
Prior to the use of NHase as a biocatalyst, chemical methods were used which included
copper salts as the catalyst. These chemical methods were expensive and inefficient,
consumed a great deal of energy and produced unwanted byproducts. NHases were
considered superior as catalysts due to the mild reaction conditions, high yields, absence
of byproducts and the possibility for stereoselectivity. Originally, Fe-type NHases were
used in this process, but in the last decade it has been determined that Co-type NHase
from R. rhodococcus J1 is a much better catalyst due to its better efficiency 7 and broad
specificity toward both aromatic and aliphatic substrates.8
In addition to the use of NHase in the production of acrylamide, it is also being used in
the production of nicotinamide and 5-cyanovaleramide, a starting material for the
123

synthesis of a herbicide from DuPont, azafenidin.9 As with the acrylamide production,
the advantages of using NHases in these processes include low energy consumption, less
waste, high efficiency and product purity. Finally, NHases have been employed in the
removal of nitrile compounds from wastewater and soils.10
NHases are bacterial heterodimeric metalloenzymes, each subunit (α and β) having a
molecular weight of around 23 kDa, and are found containing either cobalt or iron. The
subunits do not show homology to each other, but each of these subunits does show a
high degree of homology among all known NHase’s (Figure 3-1).
124

α Subunit
P. Putida3 -----------------------MGQSHTHDHHHDGYQAPPED------- 20
P. thermophila1 ------------------------MTENILRKSDEEIQKEIT-------- 18
Rho. rhodochrous1 ------------------------TAHNPVQGTLPRSNEEIA-------- 18
Therm. Bac. Sm.1 -----------------------MAIEQKLMDDHHEVDPRFPHHHPRPQS 27
Rho. sp. R3122 -------------------------MSVTIDHTTENAAPAQAA------- 18
Comamonas testosterone2 -----------------------MGQSHTHDHHHDGYQAPPED------- 20
Bradyrhizobium japonicum2 MQPIPWPDVSRVFASTRPGFWDYLPSMSDHHHHHDHDHSELSE------- 43
Pseudomonas chlororaphis2 --------------------------STSISTTATPSTPG---------- 14
P. Putida3 -IALRVKALESLLIEKGLVDPAAMDLVVQTYEHKVGPRNGAKVVAKAWVD 69
P. thermophila1 ---ARVKALESMLIEQGILTTSMIDRMAEIYENEVGPHLGAKVVVKAWTD 65
Rho. rhodochrous1 ---ARVKAMEAILVDKGLISTDAIDHMSSVYENEVGPQLGAKIVARAWVD 65
Therm. Bac. Sm.1 FWEARAKALESLLIEKRLLSSDAIERVIKHYEHELGPMNGAKVVAKAWTD 77
Rho. sp. R3122 -VSDRAWALFRALDGKGLVPDGYVEGWKKTFEEDFSPRRGAELVARAWTD 67
Comamonas testosterone2 -IALRVKALESLLIEKGLVDPAAMDLVVQTYEHKVGPRNGAKVVAKAWVD 69
Bradyrhizobium japonicum2 -TELRVRALETILTEKGYVEPAALDAIIQAYETRIGPHNGARVVAKAWTD 92
Pseudomonas chlororaphis2 ---ERAWALFQVLKSKELIPEGYVEQLTQLMAHDWSPENGARVVAKAWVD 61
P. Putida3 PAYKARLLADGTAGIAELGFSGVQGEDMVILENTPAVHNVFVCTLCSCYP 119
P. thermophila1 PEFKKRLLADGTEACKELGIGGLQGEDMMWVENTDEVHHVVVCTLXSXYP 115
Rho. rhodochrous1 PEFKQRLLTDATSACREMGVGGMQGEEMVVLENTGTVHNMVVCTLCSCYP 115
Therm. Bac. Sm.1 PEFKQRLLEDPETVLRELGYFGLQGEHIRVVENTDTVHNVVVCTLCSCYP 127
Rho. sp. R3122 PEFRQLLLTDGTAAVAQYGYLGPQGEYIVAVEDTPTLKNVIVCSLCSCTA 117
Comamonas testosterone2 PAYKARLLADGTAGIAELGFSGVQGEDMVILENTPAVHNVVVCTLCSCYP 119
Bradyrhizobium japonicum2 PAFKQALLEDGSKAIGTLGHVSRVGDHLVVVENTPQRHNMVVCTLCSCYP 142
Pseudomonas chlororaphis2 PQFRALLLKDGTAACAQFGYTGPQGEYIVALEDTPGVKNVIVCSLCSCTN 111
P. Putida3 WPTLGLPPAWYKAAPYRSRMVSDPRGVL-AEFGLVIPANKEIRVWDTTAE 168
P. thermophila1 WPVLGLPPNWFKEPQYRSRVVREPRQLLKEEFGFEVPPSKEIKVWDSSSE 165
Rho. rhodochrous1 WPVLGLPPNWYKYPAYRARAVRDPRGVL-AEFGYTPDPDVEIRIWDSSAE 164
Therm. Bac. Sm.1 WPLLGLPPSWYKEPAYRSRVVKEPRKVL-QEFGLDLPDSVEIRVWDSSSE 176
Rho. sp. R3122 WPILGLPPTWYKSFEYRARVVREPRKVL-SEMGTEIASDIEIRVYDTTAE 166
Comamonas testosterone2 WPTLGLPPAWYKAPPYRSRMVSDPRGVL-AEFGLVIPA-KEIRVWDTTAE 167
Bradyrhizobium japonicum2 WEMLGLPPVWYKAAPYRSRAVKDPRGVL-ADFGVALPKDIEVRVWDSTAE 191
Pseudomonas chlororaphis2 WPVLGLPPEWYKGFEFRARLVREGRTVL-RELGTELPSDTVIKVWDTSAE 160
P. Putida3 LRYMVLPERPAGTEAYSEEQLAELVTRDSMIGTGLPTQP-TPSH- 211
P. thermophila1 MRFVVLPQRPAGTDGWSEEELATLVTRESMIG----VEPAKAV-- 204
Rho. rhodochrous1 LRYWVLPQRPAGTENFTEEQLADLVTRDSLIGVSVPTTPSKA--- 206
Therm. Bac. Sm.1 VRFMVLPQRPEGTEGMTEEELAQIVTRDSMIGVAK-VQPPKVIQE 220
Rho. sp. R3122 TRYMVLPQRPAGTEGWSQEQLQEIVTKDCLIGVAIPQVPTV---- 207
Comamonas testosterone2 LRYMVLPERPAGTEAYSEEQLAELVTRDSMIGTGLPIQP-TPSH- 210
Bradyrhizobium japonicum2 TRFLVLPMRPGGTEGWSEEQLAELVTRDSMIGTGFPKTPGAPS-- 234
Pseudomonas chlororaphis2 SRYLVLPQRPEGSEHMSEEQLQQLVTKDVLIGVALPRVG------ 199
125

β Subunit
P. putida3 MNGIHDTGGAHGYG----PVYREPNEPVFRYDWEKTVMSLLPALLAN--G 44
P. thermophila1 MNGVYDVGGTDGLG----PINRPADEPVFRAEWEKVAFAMFPATFRA--G 44
Rho. rhodochrous1 MDGIHDLGGRAGLG----PIKPESDEPVFHSDWERSVLTMFPAMALA--G 44
Therm. Bac. Sm.1 MNGIHDVGGMDGFG--KIMYVKEEEDTYFKHDWERLTFGLVAGCMAQGLG 48
Rho. sp. R3122 --------------------------------------------------
Comamonas testosterone2 MNGIHDTGGAHGYG----PVYREPNEPVFRYDWEKTVMSLFPALFAN--G 44
Bradyrhizobium japonicum2 MNGVHDMGGMDGFG----KVEPEPNEPMFHEEWESRVLAMVRA-MGA-AG 44
Pseudomonas chlororaphis2 MDGFHDLGGFQGFGKVPHTINSLSYKQVFKQDWEHLAYSLMFVGVDQ-LK 49
P. putida3 NFNLD-EFRHSIERMGPAHYLEGTYYEHWLHVFENLLVEKGVLTATEVAT 93
P. thermophila1 FMGLD-EFRFGIEQMNPAEYLESPYYWHWIRTYIHHGVRTGKIDLEELER 93
Rho. rhodochrous1 AFNLD-QFRGAMEQIPPHDYLTSQYYEHWMHAMIHHGIEAGIFDSDELDR 93
Therm. Bac. Sm.1 MKAFD-EFRIGIEKMRPVDYLTSSYYGHWIATVAYNLLETGVLDEKELED 97
Rho. sp. R3122 -------------RMEPRHYMMTPYYERYVIGVATLMVEKGILTQDELES 37
Comamonas testosterone2 NFNLD-EFRHGIERMNPIDYLKGTYYEHWIHSIETLLVEKGVLTATELAT 93
Bradyrhizobium japonicum2 AFNID-TSRFYRETLPPDVYLSSSYYKKWFLGLEEMLIEKGYLTREEVAA 93
Pseudomonas chlororaphis2 KFSVD-EVRHAVERLDVRQHVGTQYYERYIIATATLLVETGVITQAELDQ 98
P. putida3 G-KAASGKTATP-------VLTPAIVDGLLSTGASAAREEGARARFAVGD 135
P. thermophila1 RTQYYRENPDAPLPEHEQKPELIEFVNQAVYGGLPASREVDRPPKFKEGD 143
Rho. rhodochrous1 RTQYYMDHPDDTTPTR-QDPQLVETISQLITHGADYRRPTDTEAAFAVGD 142
Therm. Bac. Sm.1 RTQAFMEKPDTKIQRW-ENPKLVKVVEKALLEGLSPVREVSSFPRFEVGE 146 Rho. sp. R3122 --------------------LAGGPFPLSRPSESEGRPAPVETTTFEVGQ 67
Comamonas testosterone2 G-KAS-GKTATP-------VLTPAIVDGLLSTGASAAREEGARARFAVGD 134
Bradyrhizobium japonicum2 GHAIQPAKALKHGK------FDLANVERVMVRGK-FARPAPAPAKFNIGD 136
Pseudomonas chlororaphis2 --------------------ALGSHFKLANPAHATGRPAITGRPPFEVGD 128
P. putida3 KVR-----VLNKNPVGHTRMPRYTRGKVG-TVVIDHGVFVTPDTAAHGKG 179
P. thermophila1 -VVRFS----TASPKGHARRARYVRGKTG-TVVKHHGAYIYPDTAGNGLG 187
Rho. rhodochrous1 KVIVRS----DASPNTHTRRAGYVRGRVG-EVVATHGAYVFPDTNALGAG 187
Therm. Bac. Sm.1 RIK-----TRNIHPTGHTRFPRYVRDKYG-VIEEVYGAHVFPDDAAHRKG 190
Rho. sp. R3122 RVR-----VRDEYVPGHIRMPAYCRGRVGTISHRTTEKWPFPDAIGHGRN 112
Comamonas testosterone2 KVR-----VLNKNPVGHTRMPRYTRGKVG-TVVIDHGVFVTPDTAAHGKG 178
Bradyrhizobium japonicum2 RVR-----AKNIHPATHTRLPRYVRGHVG-VVELNHGCHVFPDSAAMELG 180
Pseudomonas chlororaphis2 RVV-----VRDEYVAGHIRMPAYVRGKEGVVLHRTSEQWPFPDAIGHGDL 173
P. putida3 EH-PQHVYTVSFTSVELWGQDASSPKDTIRVDLWDDYLEPA-------- 219
P. thermophila1 EC-PEHLYTVRFTAQELWG-PEGDPNSSVYYDCWEPYIELVDT------ 228
Rho. rhodochrous1 ES-PEHLYTVRFSATELWG-EPAAPNVVNHIDVFEPYLLPA-------- 226
Therm. Bac. Sm.1 EN-PQYLYRVRFDAEELWG---VKQNDSVYIDLWEGYLEPVSH------ 229
Rho. sp. R3122 DAGEEPTYHVKFAAEELFG--SDTDGGSVVVDLFEGYLEPAA------- 152
Comamonas testosterone2 EH-PQHVYTVSFTSVELWGQDASSPKDTIRVDLWDDYLEPA-------- 218
Bradyrhizobium japonicum2 EN-PQWLYTVVFEGSDLWG-ADGDPTSKVSIDAFEPYLDLA-------- 219
Pseudomonas chlororaphis2 SAAHQPTYHVEFRVKDLWG--DAADDGYVVVDLFESYLDKAPGAQAVNA 220
Figure 3-1: Sequence alignment of four Co-type Nitrile Hydratases (NHase) and four Fe-type NHases. Known functional residues are highlighted in yellow. 1 refers to Co-type nitrile hydratases; 2 refers to Fe-type nitrile hydratases; 3 refers to the Co-type nitrile hydratase from Pseudomonas putida determined in this thesis from x-ray crystallography.
126

Despite the industrial importance of NHases and many structural, kinetic and theoretical
studies, many questions linger about the catalytic mechanism. The proposed mechanisms
for NHases can be divided into three main categories (Figure 3-2).11 In mechanism 1, the
nitrile binds directly to the metal ion, displacing a water molecule in the sixth coordinate
position. The nitrile carbon atom is then subjected to nucleophilic attack by a nearby
water molecule. In mechanism 2, the hydroxide ion bound to the metal directly performs
a nucleophilic attack on the nitrile carbon atom. In mechanism 3, the hydroxide ion
bound to the metal activates a second water, which carries out the nucleophilic attack on
the nitrile carbon atom. Mechanism 1, the first-shell mechanism, could feasibly be
distinguished from the second-shell mechanisms 2 and 3, if the binding mode of nitrile
substrates could be determined. While no crystal structures are currently available with
nitrile substrate bound, NHase has been co-crystallized with the inhibitor n-butanoic
acid.12 In these structures, n-butanoic acid displaces the sixth-ligand water or hydroxide
ion to coordinate the cobalt. Unfortunately, the electronic character of nitriles and
carboxylates are quite different and it is difficult to draw mechanistic conclusions from
these structures.
Two computational studies have been performed to investigate the potential substrate
binding modes in the immediate vicinity of the cobalt. Desai and Zimmer performed
Monte Carlo simulations of bromoxynil and acrylonitrile on an Fe-type NHase in which
only mechanism 1, where the metal ion directly binds nitrile nitrogen of the substrate,
was considered.13 This study found that bromoxynil has room in the active site when
coordinated to the metal ion, and suggests that the inability of Fe-type NHase to catalyze
127

the hydration of bromoxynil may be due to the small active site entrance. Peplowski, et
al. examined the binding modes of small aliphatic and aromatic nitriles to ptNHase using
docking.14 In this study, only mechanisms 2 and 3 were considered, as the sixth-position
coordinated water was retained in the active site. The authors noted the preference for
different binding sites between aliphatic and aromatic nitriles.
Figure 3-2: Proposed reaction mechanisms for ppNHase.11
Molecular docking to the active site of NHase may yield insight into its catalytic
mechanism. Recently, Peplowski, et al. studied the docking of several small aliphatic and
128

aromatic substrates to the NHase from P. thermophila JCM 3095 (ptNHase), indicating
the feasibility of the approach.14 Novak, et al. explored the possible binding modes of
large chiral substrates in an attempt to sort out the catalytic mechanism of ppNHase.15
Through docking studies, it was suggested that either mechanism 2 or 3 was most
plausible based on amide oxygen to cobalt distances in addition to previously published
studies.3,16
Crystal structures have been reported for Co-type NHase from Pseudonocardia
thermophila,16 Bacillus smithii,17 and Bacillus sp. rapc8 (PDB ID: 2PDD), and for Fe-
type nitrile hydratase from Rhodococcus erythropolis (PDB ID: 2CZ6), Rhodococcus sp.
r31211 and Rhodococcus sp. N-77118. The active site of NHase consists of three cysteine
residues, αCys108, αCys111 and αCys113 and one serine residue, αSer112, on the α
subunit which coordinate the metal ion, and two arginine residues, βArg52 and βArg157,
on the β subunit (Figure 3-3) (Pseudonocardia thermophila numbering).16 The ligands to
the cobalt atom are three sulfur atoms from the cysteine residues (αCys108, αCys111 and
αCys113), two main chain nitrogen atoms from αSer112 and αCys113, and a water
oxygen atom.16 The two arginine residues are thought to hydrogen-bond to the cysteine
residues that coordinate the metal ion. These arginine residues therefore appear to
stabilize the claw setting in the active site. Additionally, there is a tyrosine residue,
βTyr68, on the β subunit which is involved in ligand binding.
129

βArg52
βArg157
Figure 3-3: Cartoon diagram of active site of nitrile hydratase from Pseudonocardia thermophila (PDB ID: 1UGP19) shown in wall-eyed stereo view. All atoms are shown in CPK coloring; pink sphere = cobalt. Black dashed lines show atoms coordinating to the metal, green dashed lines refer to hydrogen bonds between the arginine residues and the cysteines, and the magenta dashed line refers to interactions between the binding residue, Tyr68, and the bound inhibitor, butanoic acid.
Mutations to the known active site and ligand binding residues for Co-type and Fe-type
NHase have been made (Table 3-1). In particular, the three cysteine residues which are
known to be ligands to the metal center, have been mutated in single, double and triple
forms.20 αCys111Ala, αCys113Ala, αCys108Ala/αCys111Ala, and
αCys111Ala/αCys113Ala mutations exhibited no activity. αCys108Ala,
αCys108Ala/αCys113Ala, and αCys108Ala/αCys111Ala/αCys113Ala resulted in no
protein expression according to SDS-PAGE gel. Additionally, mutations have been made
to the tyrosine residue thought to be involved in ligand binding.19 The Tyr68Phe mutation
results in a 125-fold decrease in kcat and a 10-fold increase in KM with aliphatic
substrates. It is believed that the –OH group forms hydrogen bonds with the ligand.
Removal of this functional group affects binding and subsequently activity. Finally, the
two arginine residues have previously been mutated in the Fe-type NHase.21,22 These
mutants exhibited either sharply decreased enzymatic activity or no activity.
αCys111
Butanoic Acid
αCys113
αSer112
αCys108βTyr68
130
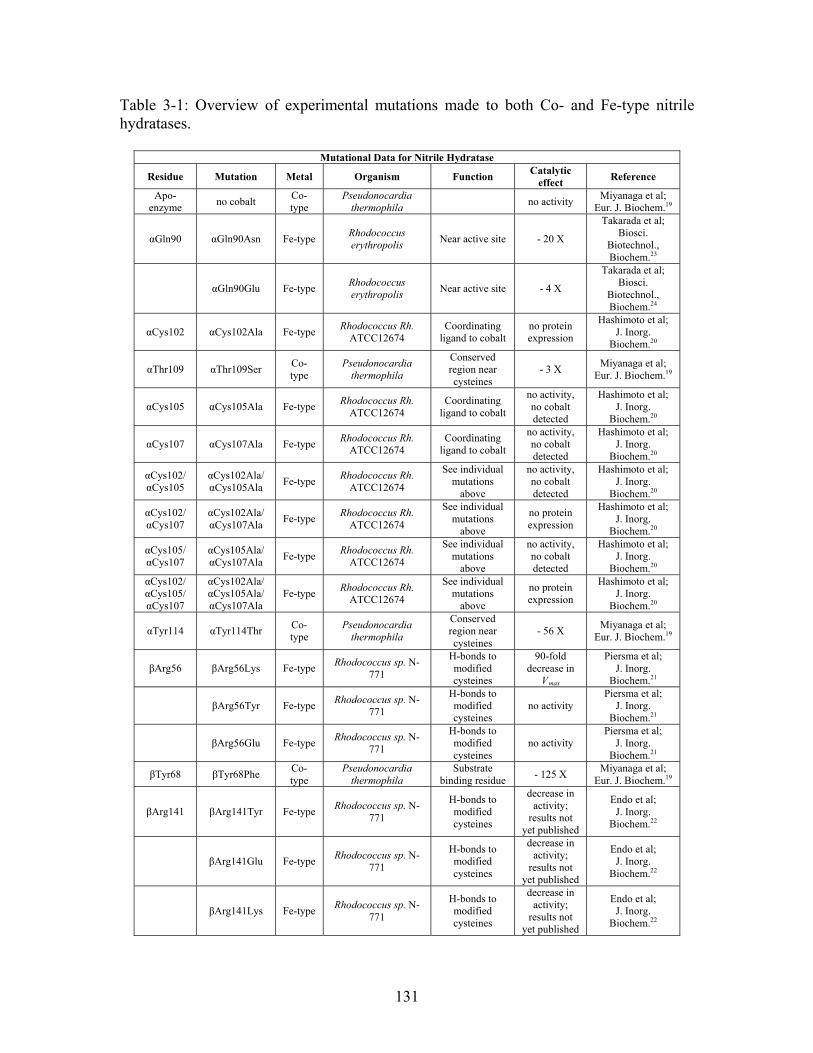
Table 3-1: Overview of experimental mutations made to both Co- and Fe-type nitrile hydratases.
Mutational Data for Nitrile Hydratase
Residue Mutation Metal Organism Function Catalytic
effect Reference
Apo-enzyme
no cobalt Co-type
Pseudonocardia thermophila
no activity Miyanaga et al;
Eur. J. Biochem.19
αGln90 αGln90Asn Fe-type Rhodococcus erythropolis
Near active site - 20 X
Takarada et al; Biosci.
Biotechnol., Biochem.23
αGln90Glu Fe-type Rhodococcus erythropolis
Near active site - 4 X
Takarada et al; Biosci.
Biotechnol., Biochem.24
αCys102 αCys102Ala Fe-type Rhodococcus Rh.
ATCC12674 Coordinating
ligand to cobalt no protein expression
Hashimoto et al; J. Inorg.
Biochem.20
αThr109 αThr109Ser Co-type
Pseudonocardia thermophila
Conserved region near cysteines
- 3 X Miyanaga et al;
Eur. J. Biochem.19
αCys105 αCys105Ala Fe-type Rhodococcus Rh.
ATCC12674 Coordinating
ligand to cobalt
no activity, no cobalt detected
Hashimoto et al; J. Inorg.
Biochem.20
αCys107 αCys107Ala Fe-type Rhodococcus Rh.
ATCC12674 Coordinating
ligand to cobalt
no activity, no cobalt detected
Hashimoto et al; J. Inorg.
Biochem.20
αCys102/ αCys105
αCys102Ala/ αCys105Ala
Fe-type Rhodococcus Rh.
ATCC12674
See individual mutations
above
no activity, no cobalt detected
Hashimoto et al; J. Inorg.
Biochem.20
αCys102/ αCys107
αCys102Ala/ αCys107Ala
Fe-type Rhodococcus Rh.
ATCC12674
See individual mutations
above
no protein expression
Hashimoto et al; J. Inorg.
Biochem.20
αCys105/ αCys107
αCys105Ala/ αCys107Ala
Fe-type Rhodococcus Rh.
ATCC12674
See individual mutations
above
no activity, no cobalt detected
Hashimoto et al; J. Inorg.
Biochem.20 αCys102/ αCys105/ αCys107
αCys102Ala/ αCys105Ala/ αCys107Ala
Fe-type Rhodococcus Rh.
ATCC12674
See individual mutations
above
no protein expression
Hashimoto et al; J. Inorg.
Biochem.20
αTyr114 αTyr114Thr Co-type
Pseudonocardia thermophila
Conserved region near cysteines
- 56 X Miyanaga et al;
Eur. J. Biochem.19
βArg56 βArg56Lys Fe-type Rhodococcus sp. N-
771
H-bonds to modified cysteines
90-fold decrease in
Vmax
Piersma et al; J. Inorg.
Biochem.21
βArg56Tyr Fe-type Rhodococcus sp. N-
771
H-bonds to modified cysteines
no activity Piersma et al;
J. Inorg. Biochem.21
βArg56Glu Fe-type Rhodococcus sp. N-
771
H-bonds to modified cysteines
no activity Piersma et al;
J. Inorg. Biochem.21
βTyr68 βTyr68Phe Co-type
Pseudonocardia thermophila
Substrate binding residue
- 125 X Miyanaga et al;
Eur. J. Biochem.19
βArg141 βArg141Tyr Fe-type Rhodococcus sp. N-
771
H-bonds to modified cysteines
decrease in activity;
results not yet published
Endo et al; J. Inorg.
Biochem.22
βArg141Glu Fe-type Rhodococcus sp. N-
771
H-bonds to modified cysteines
decrease in activity;
results not yet published
Endo et al; J. Inorg.
Biochem.22
βArg141Lys Fe-type Rhodococcus sp. N-
771
H-bonds to modified cysteines
decrease in activity;
results not yet published
Endo et al; J. Inorg.
Biochem.22
131

In this study, we focused on Co-type NHase. The Co(III) coordination sphere involves a
C1-T-L-C2-S-C3 motif, where, in its most active form, cysteines C2 and C3 are oxidized
to sulfinic and sulfenic acid, respectively. The oxidation state of these cysteines is critical
for NHase activity as anaerobically expressed NHase is inactive and aerobic incubation is
required to regain activity.24 It has been demonstrated that further oxidation of the
sulfenic acid C3 to sulfinic acid results in decreased enzyme activity. The first known
structure of the enantioselective Co-type nitrile hydratase from Pseudomonas putida
NRRL-18668 (ppNHase) is presented to 2.1 Å, in addition to a full kinetic analysis of the
wild type protein at five different pHs.
132

3.2 Materials and Methods
Site-Directed Mutagenesis (SDM)
An expression plasmid for Pseudomonas putida NRRL-18668 (obtained from Mark
Payne, E.I. du Pont de Nemours and Company), which contains the genes for the α and β
subunits of NHase and for the NHase activator, P14K, was used. The DNA was
transformed into BL21 (DE3) competent cells (Stratagene, La Jolla, CA).
Protein Expression and Purification
All reagents were purchased from Fisher Scientific, Pittsburgh, PA unless otherwise
noted. The wild type ppNHase was expressed in E. coli BL21 (DE3) (Stratagene). Cells
were grown at 37 °C, in 1 L of 2XYT broth containing ampicillin (100 μg/mL). When the
A600 reached 0.8, the cells were induced by the addition of isopropyl-β-D-thiogalactoside
(IPTG) to 1 mM and cobalt chloride to 0.5 mM.25 The cells were then cultured for an
additional 3 hours at 28 °C. All subsequent manipulations were performed at 4 °C. After
cell harvesting by centrifugation, the pellet was resuspended in 40 mL of 50 mM Tris pH
8.0 and 2 mM βME (Buffer A). Cells were lysed via sonication and the suspension was
clarified by centrifugation at 10,000 × g for 40 minutes. The ppNHase-containing
supernatant was loaded onto a 60 mL DEAE anion-exchange column equilibrated in
Buffer A containing 80 mM NaCl and ppNHase was eluted with a linear gradient from 80
to 200 mM NaCl in Buffer A over 700 mL.19 Fractions containing ppNHase were pooled
and precipitated with 70% ammonium sulfate.19 After centrifugation and reconstitution in
Buffer A, the protein was loaded onto a 20 mL Phenyl Sepharose column (GE
Healthcare, Piscataway, NJ) equilibrated in Buffer A containing 0.5 M ammonium sulfate
133

and eluted with a linear gradient from 0.5 to 0 M ammonium sulfate in same buffer over
180 mL.26 Fractions containing ppNHase were pooled and concentrated using an Amicon
Ultra-15 Centrifugal Filter Unit with Ultracel-10 membrane (Millipore, Billerica, MA)
with 10 kDa nominal molecular weight limit and dialyzed 2 times (4 hours each) against
50 mM Tris pH 8.0 and 2 mM βME. The protein was loaded onto a 10 mL MonoQ
column (GE Healthcare) equilibrated in Buffer A containing 125 mM NaCl. ppNHase
was eluted with a linear gradient of 125 mM to 240 mM NaCl in Buffer A over 135
mL.19 Fractions containing ppNHase were pooled and concentrated using an Amicon
Ultra-15 Centrifugal Filter Unit with Ultracel-10 membrane (Millipore) with 10 kDa
nominal molecular weight limit and dialyzed 2 times (4 hours each) against 50 mM Tris
pH 8.0 and 2 mM βME, concentrated to ~20 mg/mL and stored at 4 °C. The
concentration of protein was determined either by the Bradford assay27, or by A280
measurement. The extinction coefficient used was 1.676 mg · mL-1 · cm-1
(http://us.expasy.org/cgi-bin/protparam).
Kinetics
NHase activity was determined by measurement of the hydration of n-Valeronitrile in a
300 μL reaction volume, containing 100 mM HEPES, 2 mM βME in an ice bath.26 The
reaction was too fast to monitor at room temperature, so the protocol was adjusted to
slow the reaction. The wild type protein was analyzed by using Michaelis-Menten (MM)
kinetics at pH 5.8, 6.7, 7.2, 7.5 and 8.5. Non-linear regression was performed in order to
obtain the MM constants, kcat and KM. Each reaction was carried out three times at pH
6.7 and two times at pH 5.8, 7.2, 7.5 and 8.5. Concentration of n-Valeronitrile was 0.625,
134

2.5, 5, 10, and 40 mM. The concentration of NHase used was 1.0 nM. The reaction was
carried out for 40 and 60 minutes in an ice bath, and stopped by the addition of 0.3 N
HCl.
The formation of n-Valeramide was monitored with either a WATERS 2690 HPLC
(Waters, Corp., Milford, MA) or an Agilent 1200 HPLC (Agilent Technologies, Santa
Clara, CA), using a Zorbax Aq reverse phase C18 column (4.6 X 150 mm) (Agilent
Technologies) at a flow rate of 1.0 mL/min.28 A standard curve was prepared every time
samples were run and ranged between 3.90 μg/mL and 250 μg/mL. Running buffers were
5 mM Potassium Phosphate pH 2.9 (A) and 100% acetonitrile (B), running at 1.0
mL/min. The product was eluted with a small gradient of 10% - 25% acetonitrile for 7
minutes. Sample run time was 14 minutes. The absorbance was measured at 210 nm.
Mass Spectrometry
Nitrile hydratase from Pseudomonas putida was desalted using Amicon Ultra-15
Centrifugal Filter Unit with Ultracel-10 membrane (Millipore, Billerica, MA) with 10
kDa nominal molecular weight limit and diluted with 10 mM ammonium bicarbonate in
HPLC grade water to 10 μM. ppNHase was directly infused using a syringe pump into an
electrospray ion source with dual ion funnel29 (Apollo II) connected to a hybrid
quadrupole Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (apex
Qe-94, Bruker Daltonics Inc., Billerica, MA). The resulting mass spectrum was
deconvoluted using Data Analysis software (version 3.4, Bruker Daltonics),
monoisotopic masses were calculated using SNAP algorithm (version 2, Bruker
135

Daltonics Inc., Billerica, MA).30 The monoisotopic masses of ppNHase α-and β-subunits
were calculated to be 24668.198 and 24008.079 Da using Isotope Pattern software
(Bruker Daltonics Inc., Billerica, MA), respectively. During mass calculations from the
mass spectrum, the existence of three charges from the Co(III) ion were considered and
three daltons were subtracted from the masses generated by the software, which assumes
all charges on the ion are due to protons.
Protein Sequencing
Purified protein was fractioned by SDS-PAGE gel, electroblotted to a PVDF membrane,
stained with Coomassie, excised and submitted to the Iowa State University Protein
Facility (Ames, IA) for Edman degradation sequencing.
Crystallization, data collection and crystallographic refinement
Crystals of ppNHase were grown at 25 ˚C by vapor diffusion in 24 well hanging drop
plates over 0.7 mL volume reservoirs using 1 + 1 µL drops. Three crystal forms were
identified from initial screens (Hampton Research, Aliso Viejo, CA), however, one of
these crystal forms failed to diffract beyond 3.5 Å and one could not be accurately
indexed due to twinning. Diffracting crystal needles were obtained using 20 mg/mL
ppNHase and a reservoir containing 22% polyacrylic acid in HEPES pH 7.5 with 20 mM
magnesium chloride and 4% acetone. Single crystals were dissected from clusters and
transferred to a solution containing 17.6% polyacrylic acid 5100 and 20% glycerol and
were flash-frozen in liquid nitrogen.
136

Data were collected at the ID-23B beamline at GM/CA-CAT (APS, Argonne, IL, USA)
at 100 K using a MARmosiac 300 CCD detector and the 10 μm mini-beam. Diffraction
images were indexed, integrated and scaled using HKL2000.31 Molecular replacement
was carried out with Phaser 32 using PDB ID: 1IRE16 as a starting model. Several rounds
of refinement and model building were performed using REFMAC 33 and COOT.34 Final
rounds of refinement, including simulated annealing and water picking were performed
using PHENIX.35
3.3 Results and Discussion
Crystal Screening
Three crystal forms were identified through initial screens for wild type ppNHase and are
shown Figure 3-4. The different forms include hexagonal plates, rods and needles. The
hexagonal plates were grown in a mother liquor of 1.4 M ammonium sulfate, 0.1 M
sodium chloride in HEPES pH 8.0. The crystals diffracted nicely, approximately 3.0 Å,
but the diffraction patterns were twinned, and we were unable to find just one lattice. In
an effort to fix the twinning issue, this condition was subjected to an additive screen
which included the addition of 72 different additives to the mother liquor (Hampton
Research, Aliso Viejo, CA). The addition of magnesium chloride (50 mM) to the drops
produced crystals in the form of rods. The rods did not diffract well (approximately 6.0
Å). After additional screening, we found a condition that produced tiny needles. While
extremely difficult to work with, the needles diffracted well (approximately 3.0 Å), and
were single-latticed.
137

Figure 3-4: Crystal forms identified for ppNHase (clockwise from upper left: hexagonal plates, rods, needles, rods).
138

Crystals and Data Collection for the Structure of Wild Type ppNHase
Final crystals of wild type (20 mg/mL) formed in 48 hours in a mother liquor of 22%
polyacrylic acid in HEPES pH 7.5 with 20mM magnesium chloride and 4% acetone. The
crystals appeared as long, thin needles. Additional growth time did not improve the size
of the crystals. A typical diffraction pattern is shown in Figure 3-5 for wild type ppNHase
where the unit cell edges measure 82 Å, 137 Å and 85 Å for a, b and c, and the unit
angles measure 90º, 92º, 90º for α, β, and γ. The data collection and refinement statistics
for wild type is shown in Table 3-2.
Figure 3-5: Typical diffraction pattern observed for wild type ppNHase.
139
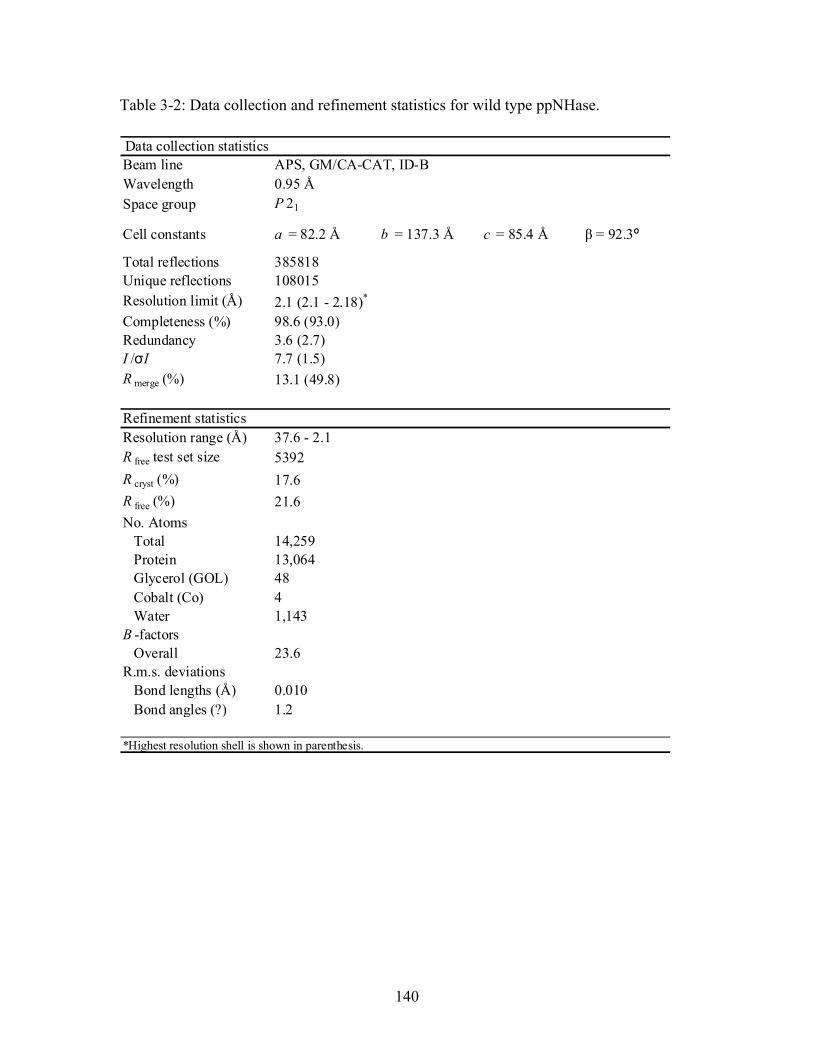
Table 3-2: Data collection and refinement statistics for wild type ppNHase.
Data collection statisticsBeam line APS, GM/CA-CAT, ID-BWavelength 0.95 ÅSpace group P 21
Cell constants a = 82.2 Å b = 137.3 Å c = 85.4 Å β = 92.3º
Total reflections 385818Unique reflections 108015Resolution limit (Å) 2.1 (2.1 - 2.18)*
Completeness (%) 98.6 (93.0)Redundancy 3.6 (2.7)I /σI 7.7 (1.5)R merge (%) 13.1 (49.8)
Refinement statisticsResolution range (Å) 37.6 - 2.1R free test set size 5392
R cryst (%) 17.6
R free (%) 21.6
No. Atoms Total 14,259 Protein 13,064 Glycerol (GOL) 48 Cobalt (Co) 4 Water 1,143B -factors Overall 23.6R.m.s. deviations Bond lengths (Å) 0.010 Bond angles (?) 1.2
*Highest resolution shell is shown in parenthesis.
140

Structure of Wild Type Nitrile Hydratase from Pseudomonas putida (ppNHase)
The crystal structure of wild type ppNHase was solved in the P21 space group. There are
four αβ heterodimers in the asymmetric unit with one cobalt ion per heterodimer. There
was no electron density for the first six or seven residues and the 16 residues of the T7-
tag and the last 4 residues of the α-subunit, while backbone density was present for the
entire β-subunit. The refined model is comprised of four copies of the α-subunit
consisting of residues 7 (or 8) – 207, each containing one cobalt ion, four copies of the β-
subunit residues 1 – 219, 1003 water molecules, and 48 glycerol molecules. The overall
structure of ppNHase is very similar to that of the other known NHase structures, both
Fe-type and Co-type, which is to be expected based on their high sequence similarity
(Figure 3-1). Superimposing ppNHase with nitrile hydratase from Pseudonocardia
thermophila (ptNHase) yields an RMSD of 0.7 Å over 177 α-carbons for the α-subunit
(out of 207), and an RMSD of 0.9 Å over 183 α-carbons for the β-subunit (out of 219)
when no atom pair distance is allowed to exceed 2.0 Å (Figure 3-6).
Despite the overall similarity among NHases, comparison of ppNHase with other Co-type
NHase structures shows large differences in the α5-loop-α6 region in the β-subunit and
in the location of the N-terminus of the α-subunit. Specifically, the β-subunit is shorter
by 8 amino acid residues causing a decrease in the size of the α5 and α6 helices.
Additionally, this causes the flexible loop of the β-subunit to be in an alternate
conformation.
141
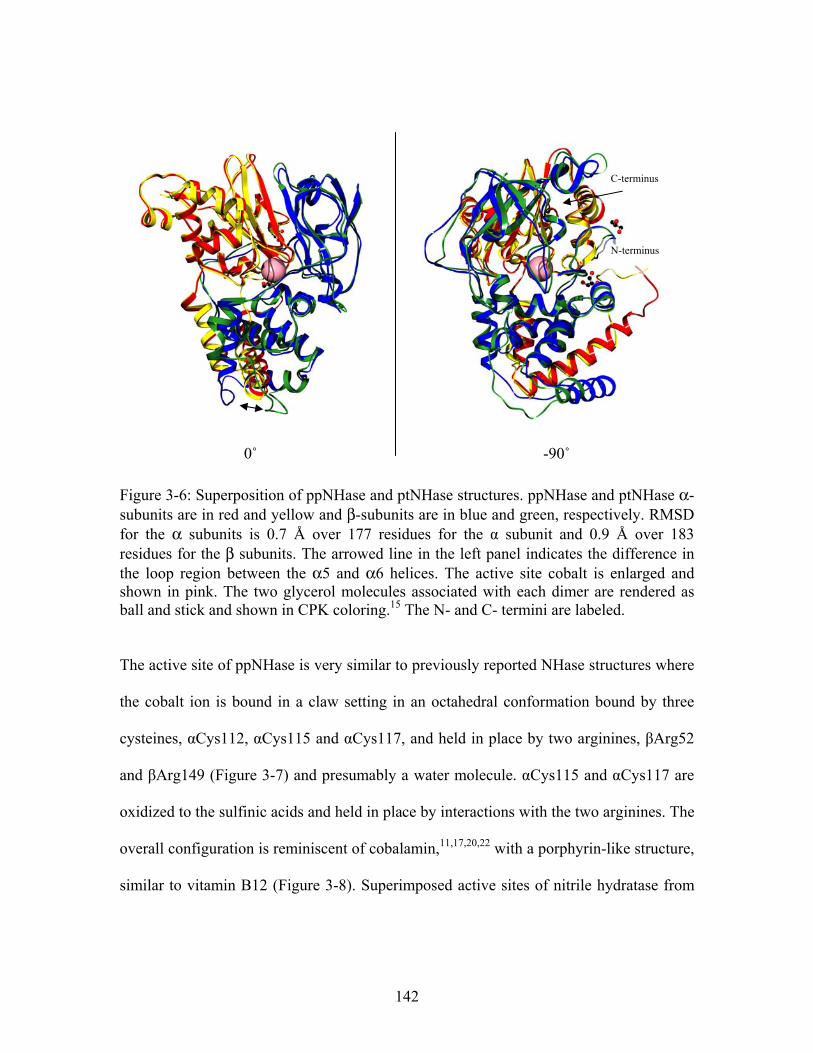
Figure 3-6: Superposition of ppNHase and ptNHase structures. ppNHase and ptNHase α-subunits are in red and yellow and β-subunits are in blue and green, respectively. RMSD for the α subunits is 0.7 Å over 177 residues for the α subunit and 0.9 Å over 183 residues for the β subunits. The arrowed line in the left panel indicates the difference in the loop region between the α5 and α6 helices. The active site cobalt is enlarged and shown in pink. The two glycerol molecules associated with each dimer are rendered as ball and stick and shown in CPK coloring.15 The N- and C- termini are labeled.
N-terminus
-90˚ 0˚
C-terminus
The active site of ppNHase is very similar to previously reported NHase structures where
the cobalt ion is bound in a claw setting in an octahedral conformation bound by three
cysteines, αCys112, αCys115 and αCys117, and held in place by two arginines, βArg52
and βArg149 (Figure 3-7) and presumably a water molecule. αCys115 and αCys117 are
oxidized to the sulfinic acids and held in place by interactions with the two arginines. The
overall configuration is reminiscent of cobalamin,11,17,20,22 with a porphyrin-like structure,
similar to vitamin B12 (Figure 3-8). Superimposed active sites of nitrile hydratase from
142
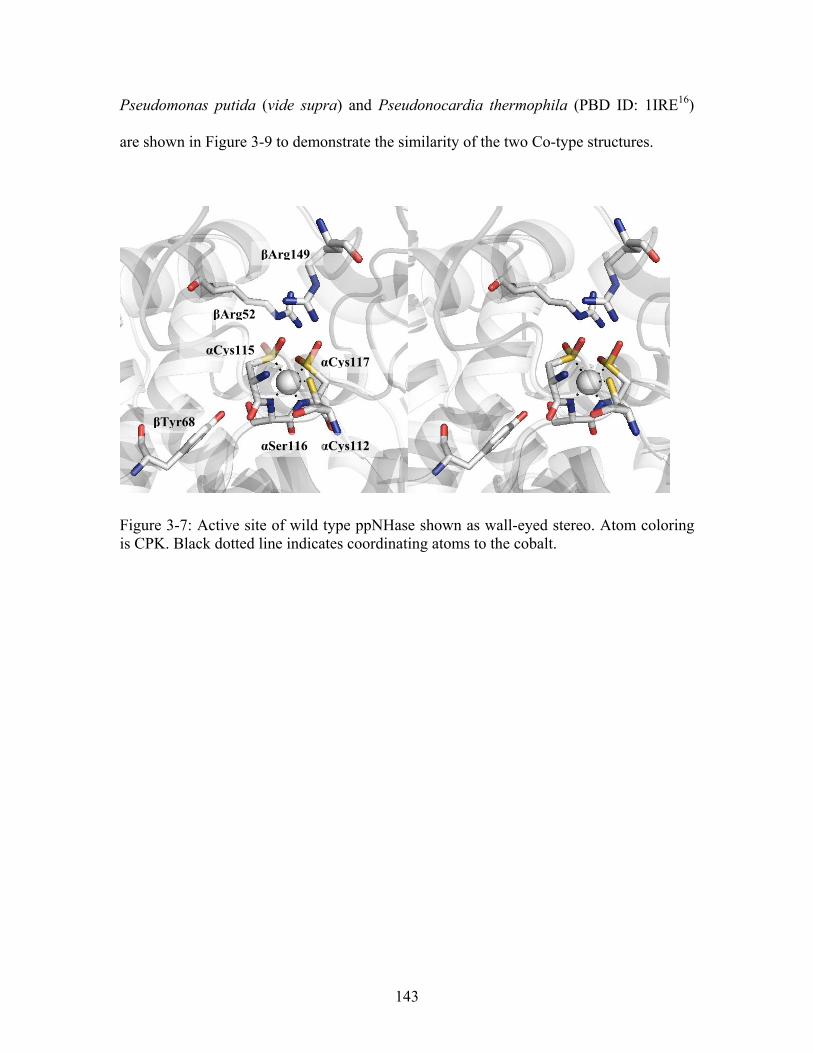
Pseudomonas putida (vide supra) and Pseudonocardia thermophila (PBD ID: 1IRE16)
are shown in Figure 3-9 to demonstrate the similarity of the two Co-type structures.
βArg149
βArg52
αCys115 αCys117
βTyr68
αCys112αSer116
Figure 3-7: Active site of wild type ppNHase shown as wall-eyed stereo. Atom coloring is CPK. Black dotted line indicates coordinating atoms to the cobalt.
143
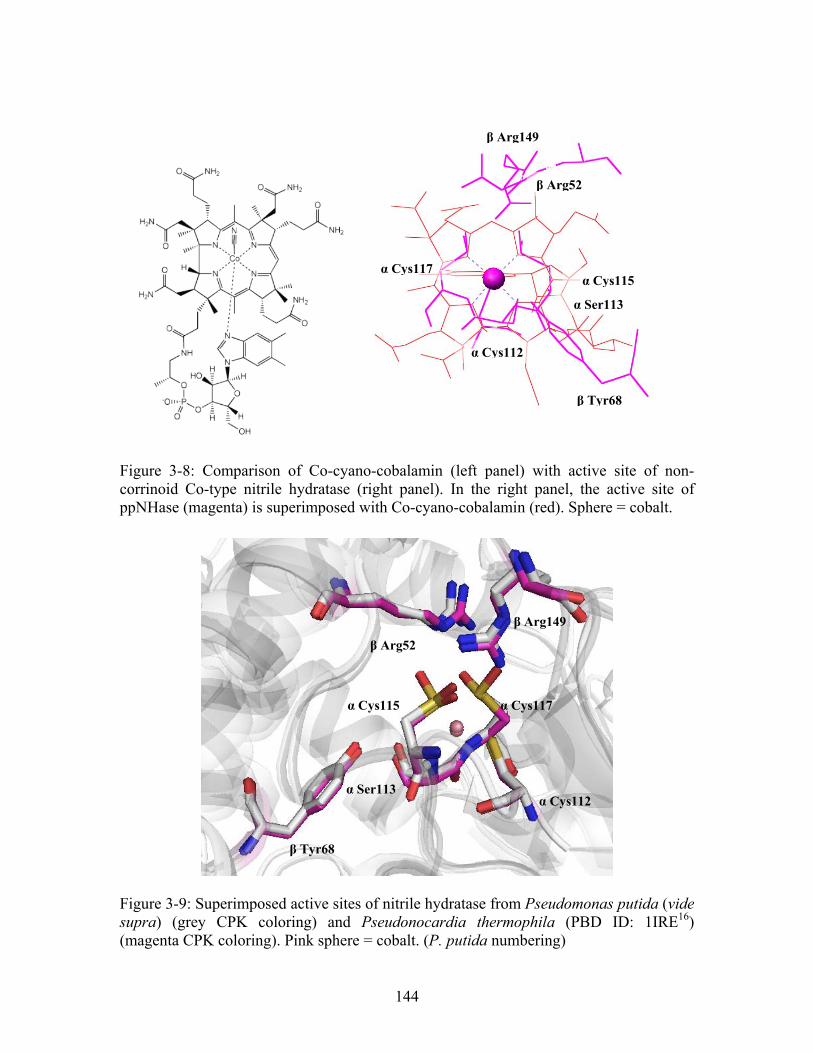
β Arg149
β Arg52
α Cys117 α Cys115
α Ser113
α Cys112
β Tyr68
Figure 3-8: Comparison of Co-cyano-cobalamin (left panel) with active site of non-corrinoid Co-type nitrile hydratase (right panel). In the right panel, the active site of ppNHase (magenta) is superimposed with Co-cyano-cobalamin (red). Sphere = cobalt.
β Arg149
β Arg52
α Cys117
α Cys112
α Cys115
α Ser113
β Tyr68
Figure 3-9: Superimposed active sites of nitrile hydratase from Pseudomonas putida (vide supra) (grey CPK coloring) and Pseudonocardia thermophila (PBD ID: 1IRE16) (magenta CPK coloring). Pink sphere = cobalt. (P. putida numbering)
144

The previously reported structures of Co-type NHase show αCys115 as a sulfinic acid
and αCys117 as a sulfenic acid.24 The wild type ppNHase structure reported here has both
cysteines oxidized to sulfinic acid in the crystal form, indicating the crystallized form is
less active than the sulfenic acid form. Wild type ppNHase crystals were dissolved in 50
mM Tris pH 8 and 2mM βME and analyzed by using Michaelis-Menten kinetics at pH
7.2. There was a 5-fold decrease in kcat compared to purified wild type protein in solution
(kcat = 4.1 min-1, KM = 16 mM) (Table 3-3). It was hypothesized that the double oxidation
of the cysteines was due to the crystallization additive polyacrylate 5100. Therefore, a
kinetic analysis was performed with the addition of 10% polyacrylate to the protein
solution resulting in a 3.5-fold decrease in kcat (kcat = 9.0 min-1, KM = 6.3 mM) compared
to wild type enzyme (kcat = 20 min-1, KM = 6.6 mM) in solution without the addition of
the polyacrylate (Table 3-3). The crystallization condition contained 22% polyacrylate
5100, but attempts to add that much caused the protein to precipitate out of solution.
9.0
Table 3-3: Kinetics results comparing wild type ppNHase to dissolved ppNHase crystals and wild type ppNHase with the addition of 10% polyacrylate.
pH 7.2
Wild Type
Dissolved Wild Type ppNHase
crystals
Wild Type ppNHase + 10%
polyacrylate
k cat (min -1) 20 4.1
K M (mM) 6.6 16 6.3
145

Recently, the crystal structure of the Fe-type NHase from R. erythropolis AJ270
(reNHase) was solved, which also shows the C2 and C3 cysteines oxidized to sulfinic
acids.36 In an attempt to determine whether this double oxidation was due to the
crystallization condition, wild type ppNHase was analyzed by FT-ICR mass
spectrometry. The data suggest that this double oxidation is due either to lengthy
exposure to air or to trace amounts of oxidizers in the crystallization condition, as
purified ppNHase prior to crystallization shows one sulfenic and one sulfinic acid based
on molecular weight. Purified wild type ppNHase has a monoisotopic α-subunit mass of
24668.140 Da, which is excellent agreement with the calculated monoisotopic mass of
24668.198 Da for T-7 tagged protein with the N-terminal methionine cleaved, a single
cobalt and three oxygen atoms (one sulfinic and one sulfenic acid modification).
Additionally, the β-subunit has a monoisotopic mass of 24008.116 Da, which is also in
excellent agreement with the calculated monoisotopic mass of 24008.079 Da. The
electron density showing the double oxidation is depicted in Figure 3-10, while the mass
spectrum is shown in Figure 3-11. To confirm the addition of the T-7 tag and the cleaved
methionine, wild type ppNHase protein was sequenced and in fact did have a cleaved
methionine confirming the protein sequence.
146

Figure 3-10: Electron density of the cobalt site in ppNHase prior to the incorporation of the cysteine oxidation. Atom coloring is in CPK. The 2Fo-Fc map is rendered at 1.5 σ and is shown in blue. The Fo-Fc difference map is rendered at 4.5 σ and is shown in
15
reen.
g
147
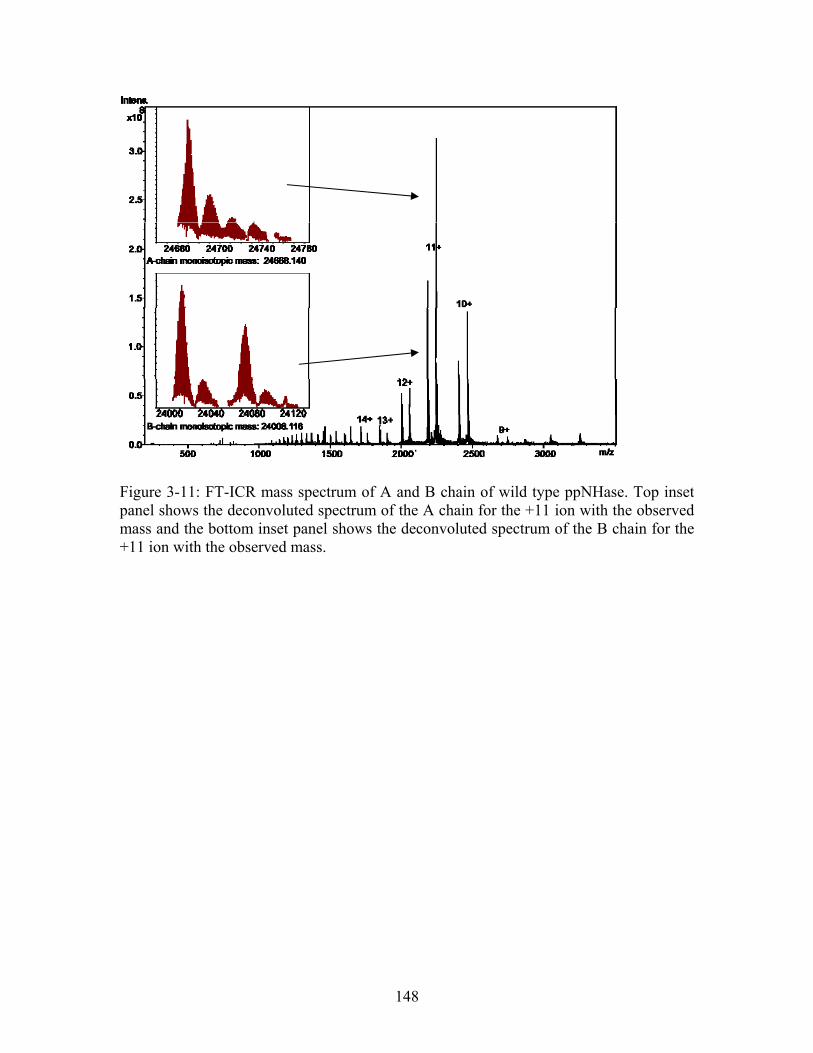
Figure 3-11: FT-ICR mass spectrum of A and B chain of wild type ppNHase. Top inset panel shows the deconvoluted spectrum of the A chain for the +11 ion with the observed mass and the bottom inset panel shows the deconvoluted spectrum of the B chain for the +11 ion with the observed mass.
148

3.4 Introduction to Michaelis-Menten Kinetics37,38
In 1903, Victor Henri proposed the idea that an enzyme (E) combines with its substrate
(S) to form an ES complex, a necessary step in enzyme catalysis. This concept was later
expanded by Leonor Michaelis and Maud Menten into a general theory of enzyme
catalysis. They suggested that an enzyme first combines with a substrate to form an
enzyme-substrate complex in a fast reversible step (equation (1)). This is followed by a
second slower step whereby the enzyme-substrate complex breaks down into free enzyme
and product (equation (2)). At early stages in the reaction, the concentration of product is
negligible, and equation (2) can be simplified to equation (3) whereby k-2 can be
ignored.37
E + S ESk1
k-1
(1)
ES E + P (2)
k2
k-2
ES E + P (3)
k2
It is assumed that the second step (equation (3)) is slower than the first step (equation (1))
and therefore limits the overall rate of the reaction. Therefore the overall rate of the
reaction, v, is proportional to the concentration of ES, the starting reagent in the second
149

step. The decomposition of the enzyme-substrate complex (ES) can be described by a
first-order rate constant, kcat.39 This is also known as the turnover number for an enzyme
catalyzed reaction (equation (4)). In this thesis, we are assuming the reaction is first-
order.
ES E + P (4)
kcat (sec -1)
where,
d(P)/dt = (kcat)(ES)
During a reaction, the enzyme exists in one of two forms; the free form, E, or the
enzyme-substrate complex, ES. The rate of a reaction is proportional to the substrate
concentration because as substrate concentration is increased, the equilibrium of equation
(1) will be pushed to the right causing the formation of more enzyme-substrate complex.
At high substrate concentrations, the reaction rate begins to slow down until a saturating
substrate concentration is reached where the rate reaches a limiting value. This reaction
rate, established at high substrate concentrations, is referred to as Vmax. When Vmax is
reached, virtually all of the enzyme is present as the enzyme-substrate complex.
150

Figure 3-12 shows the relationship between the rate of an enzymatic reaction and the
substrate concentration. The hyperbolic shape of this curve can be expressed by the
Michaelis-Menten equation (equation (5)), the basic equation of enzyme kinetics.
v = [E]0[S]kcat (5)
KM + [S]
Where, Vmax = kcat[E]0. The concentration of substrate where v = ½ Vmax is called KM,
the Michaelis-Menten constant.
Catalytic reactions are divided into two processes as seen from equations (1) and (2), and
are combined in equation (6) to interpret the kinetics of single substrate reactions.
E + S ES
k1
E + P (6)
kcat
k-1
where kcat is the first-order rate constant (turnover number) for the conversion of the
enzyme-substrate complex to the enzyme-product complex. Again, in this thesis, we are
assuming the reaction is first-order.
151

Wild Type ppNHase
0
0.2
0.4
0.6
0.8
1
0 10 20 30 40 50
n-valeronitrile concentration (mM)
rate
(u
g/m
l/min
)
Figure 3-12: Enzymatic reaction obeying Michaelis-Menten kinetics for wild type ppNHase. n-Valeronitrile concentration is plotted on the x-axis and rate is plotted on the y-axis.
In using the Michaelis-Menten equation to analyze kinetic reactions, a few assumptions
are made. First, it is assumed that the rate of the reaction is measured during a steady-
state, a period of time where the concentration of the ES complex remains constant
(d[ES]/dt = 0). Second, it is assumed that the concentration of enzyme is negligible
compared to the concentration of substrate. Finally, the assumption is made that what is
being measured is the initial rate of the reaction where changes in substrate concentration
are linear with time. Using these assumptions, it is now possible to define KM as shown
in equation (7). KM is thus defined as the substrate concentration that provides a reaction
velocity that is half of the maximum reaction velocity obtained under saturating
conditions. Additionally, KM is an apparent dissociation constant that may be treated as
the overall dissociation constant of all enzyme-bound complexes.
152

KM = k-1 + k2 (7) k1
In order to calculate the Michaelis-Menten constants, KM and Vmax, the Michaelis-
Menten equation (equation (5)) can be algebraically transformed into a form that is useful
for plotting actual data. One common transformation is to simply take the reciprocal of
both sides of the Michaelis-Menten equation to produce what is known as the
Lineweaver-Burk equation (equation (8)). A plot of 1/v versus 1/[S] yields a straight line,
where the slope = KM/Vmax, the y-intercept = 1/Vmax and the x-intercept = - 1/KM. A
Lineweaver-Burk plot (double-reciprocal plot) for wild type ppNHase at pH 6.7 is shown
in Figure 3-13.
1 = KM 1 + 1 (8)
v Vmax [S] Vmax
153
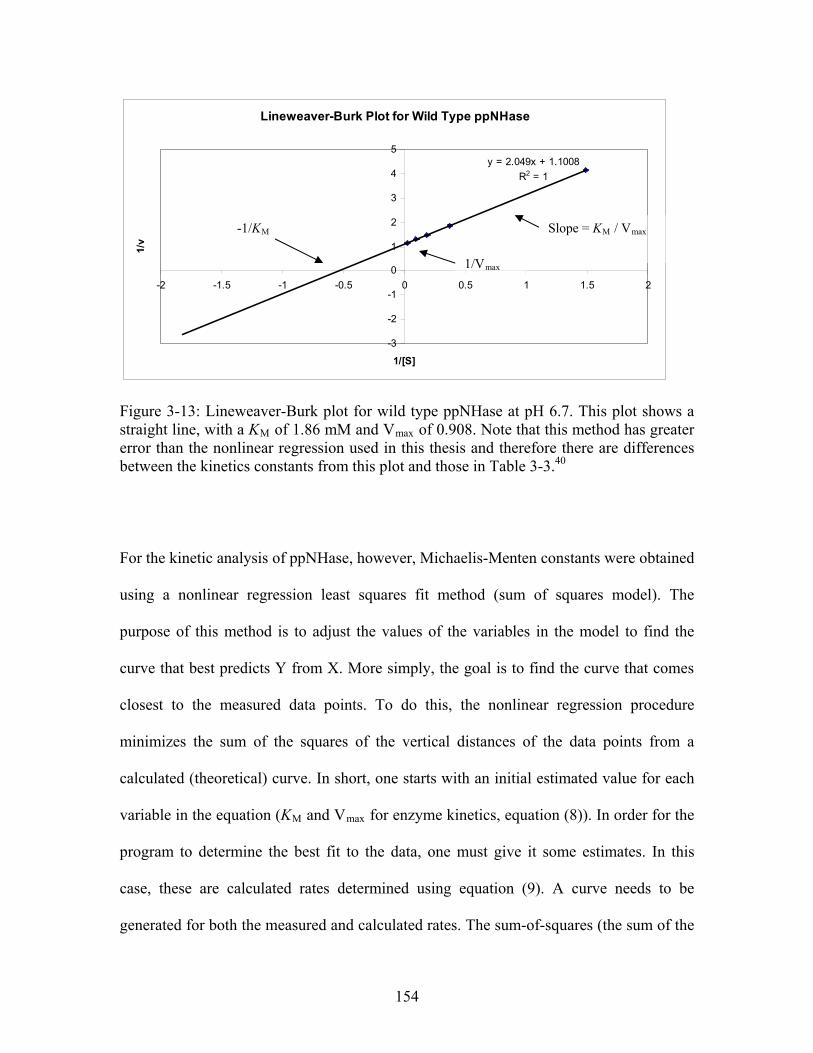
Lineweaver-Burk Plot for Wild Type ppNHase
y = 2.049x + 1.1008
R2 = 1
-3
-2
-1
0
1
2
3
4
5
-2 -1.5 -1 -0.5 0 0.5 1 1.5 2
1/[S]
1/v
1/Vmax
Slope = K / VM max -1/KM
Figure 3-13: Lineweaver-Burk plot for wild type ppNHase at pH 6.7. This plot shows a straight line, with a KM of 1.86 mM and Vmax of 0.908. Note that this method has greater error than the nonlinear regression used in this thesis and therefore there are differences between the kinetics constants from this plot and those in Table 3-3.40
For the kinetic analysis of ppNHase, however, Michaelis-Menten constants were obtained
using a nonlinear regression least squares fit method (sum of squares model). The
purpose of this method is to adjust the values of the variables in the model to find the
curve that best predicts Y from X. More simply, the goal is to find the curve that comes
closest to the measured data points. To do this, the nonlinear regression procedure
minimizes the sum of the squares of the vertical distances of the data points from a
calculated (theoretical) curve. In short, one starts with an initial estimated value for each
variable in the equation (KM and Vmax for enzyme kinetics, equation (8)). In order for the
program to determine the best fit to the data, one must give it some estimates. In this
case, these are calculated rates determined using equation (9). A curve needs to be
generated for both the measured and calculated rates. The sum-of-squares (the sum of the
154

squares of the vertical distances of the points from the curve) is calculated according to
equation (10). The variables, KM and Vmax, are adjusted to make the calculated curve
come closer to the data points (measured curve). Excel then uses an algorithm to adjust
the variables until the adjustments make virtually no difference in the sum-of-squares.
Calculated Rate = Vmax [S] (9)
KM + [S]
Sum of Squares = (10)
[∑ (measured rate at each [S]) – (calculated rate at each [S])]2
Rates were obtained by measuring the formation of product (n-Valeramide) over time at
five different enzyme concentrations using an end-point method. Enzyme and substrate
were mixed, and the reaction was stopped at different time points by the addition of
hydrochloric acid. The amount of product formed was measured by HPLC. Figure 3-14
shows typical HPLC spectra obtained for the blank (kinetics buffer, 100 mM HEPES,
10% 0.3N HCl, 2 mM βME) and the standard, n-Valeramide, at three concentrations.
Figures 13-15 – 13-17 show the HPLC spectra for a typical kinetics analysis for wild type
ppNHase (1 nM) at three concentrations of substrate (0.625 mM, 5.0 mM and 40 mM) for
time points 40 and 60 min. By plotting the concentration of product formed versus time,
rates at each substrate concentration were obtained by determining the slope of each line.
155

The Michaelis-Menten constants, KM and Vmax, were obtained using equations (9) and
(10), where the measured rates were compared to calculated rates by varying the values
of KM and Vmax. These calculations were performed using Solver in Excel. The kcat
values were calculated by equation (11). A sample plot for ppNHase is shown in Figure
3-18, showing measured (pink) and calculated (blue) curves.
kcat = Vmax (11) [E]
156

A
B
C
D
Figure 3-14: Typical HPLC spectra for blank and standard, n-Valeramide. The x-axis is time in minutes and the y-axis is absorbance at 210 nm in mAU. In panels A-D, the circled area represents the peak of interest, n-Valeramide. Panel A shows the spectrum for the blank, 100 mM HEPES and 10% 0.3 N HCl and 2 mM βME. Notice there are no peaks in the black circle. Panel B shows the spectrum of n-Valeramide at 7.8 μg/mL, panel C shows the spectrum of n-Valeramide at 30 μg/mL, and panel D shows the spectrum of n-Valeramide at 125 μg/mL. Note that there is no variation in retention time; all peaks are at 7.2 minutes.
157

A
B
C
Figure 3-15: Typical HPLC spectra for blank and kinetics analysis with 0.625 mM n-Valeronitrile at time points 40 and 60 min. The x-axis is time in minutes and the y-axis is absorbance at 210 nm in mAU. In panels A-C, the circled area represents the peak of interest, the product, n-Valeramide. Panel A shows the spectrum for the blank, 100 mM HEPES and 10% 0.3 N HCl and 2 mM βME. Notice there are no peaks in the black circle. Panel B shows the spectrum of the formation of n-Valeramide (approximately 5.0 μg/mL) at 40 min., and panel C shows the spectrum of the formation of n-Valeramide (approximately 9.0 μg/mL) at 60 min. Note that there is no variation in retention time; all peaks are at 7.2 minutes.
158

B
A
C
Figure 3-16: Typical HPLC spectra for blank and kinetics analysis with 5.0 mM n-Valeronitrile at time points 40 and 60 min. The x-axis is time in minutes and the y-axis is absorbance at 210 nm in mAU. In panels A-C, the circled area represents the peak of interest, the product, n-Valeramide. Panel A shows the spectrum for the blank, 100 mM HEPES and 10% 0.3 N HCl and 2 mM βME. Notice there are no peaks in the black circle. Panel B shows the spectrum of the formation of n-Valeramide (approximately 12 μg/mL) at 40 min., and panel C shows the spectrum of the formation of n-Valeramide (approximately 19 μg/mL) at 60 min. Note that there is no variation in retention time; all peaks are at 7.2 minutes.
159

B
A
C
Figure 3-17: Typical HPLC spectra for blank and kinetics analysis with 40 mM n-Valeronitrile at time points 40 and 60 min. The x-axis is time in minutes and the y-axis is absorbance at 210 nm in mAU. In panels A-C, the circled area represents the peak of interest, the product, n-Valeramide. Panel A shows the spectrum for the blank, 100 mM HEPES and 10% 0.3 N HCl and 2 mM βME. Notice there are no peaks in the black circle. Panel B shows the spectrum of the formation of n-Valeramide (approximately 25 μg/mL) at 40 min., and panel C shows the spectrum of the formation of n-Valeramide (approximately 19 μg/mL) at 60 min. Note that there is no variation in retention time; all peaks are at 7.2 minutes.
160
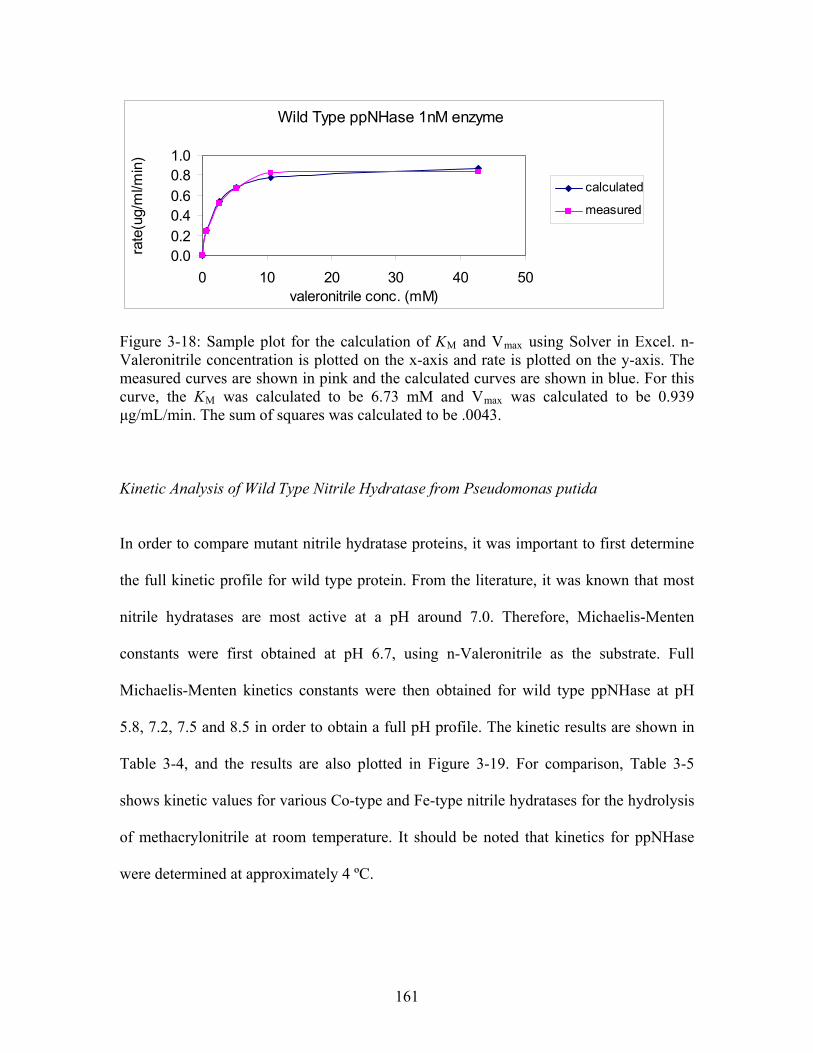
Wild Type ppNHase 1nM enzyme
0.00.2
0.40.6
0.81.0
0 10 20 30 40 50valeronitrile conc. (mM)
rate
(ug/
ml/m
in)
calculated
measured
Figure 3-18: Sample plot for the calculation of KM and Vmax using Solver in Excel. n-Valeronitrile concentration is plotted on the x-axis and rate is plotted on the y-axis. The measured curves are shown in pink and the calculated curves are shown in blue. For this curve, the KM was calculated to be 6.73 mM and Vmax was calculated to be 0.939 μg/mL/min. The sum of squares was calculated to be .0043.
Kinetic Analysis of Wild Type Nitrile Hydratase from Pseudomonas putida
In order to compare mutant nitrile hydratase proteins, it was important to first determine
the full kinetic profile for wild type protein. From the literature, it was known that most
nitrile hydratases are most active at a pH around 7.0. Therefore, Michaelis-Menten
constants were first obtained at pH 6.7, using n-Valeronitrile as the substrate. Full
Michaelis-Menten kinetics constants were then obtained for wild type ppNHase at pH
5.8, 7.2, 7.5 and 8.5 in order to obtain a full pH profile. The kinetic results are shown in
Table 3-4, and the results are also plotted in Figure 3-19. For comparison, Table 3-5
shows kinetic values for various Co-type and Fe-type nitrile hydratases for the hydrolysis
of methacrylonitrile at room temperature. It should be noted that kinetics for ppNHase
were determined at approximately 4 ºC.
161

Table 3-4: Kinetics results for ppNHase at pH 5.7, 6.7, 7.2, 7.5 and 8.5. The results at pH 6.7 represent an n=3, while only an n=2 was run at all other pH values.
pH 5.8 pH 6.7 pH 7.2 pH 7.5 pH 8.5
k cat (min -1 ) 2.0 20 20 13 13
std dev k cat (min -1) 0.35
K M (mM) 13 6.6 11 7.8 9.0
std. dev. K M (mM) 0.91
k cat (min -1) WT pH 7.2/ k cat (min -1) WT at different pH 10 1.0 1.0 1.5 1.6
kcat and KM for Wild Type ppNHase
Wild type protein is most active at pH 7.2 with a kcat of 20 min-1 and a KM of 11 mM.
There is a 10-fold decrease in kcat when the pH is lowered to 5.8, no change at pH 6.7,
and a 1.5-fold decrease when the pH is raised to both 7.5 and 8.5 (Table 3-4). As shown
in Figure 3-13, the enzyme reaches maximum kcat at pH 7.2, and decreases with both a
decrease and an increase in pH. The rate tails off at pH 7.5 and remains essentially
constant at pH 8.5. There is no significant change in KM at all pH values tested within the
error of the assay; the value remains constant at around 11 mM.
162
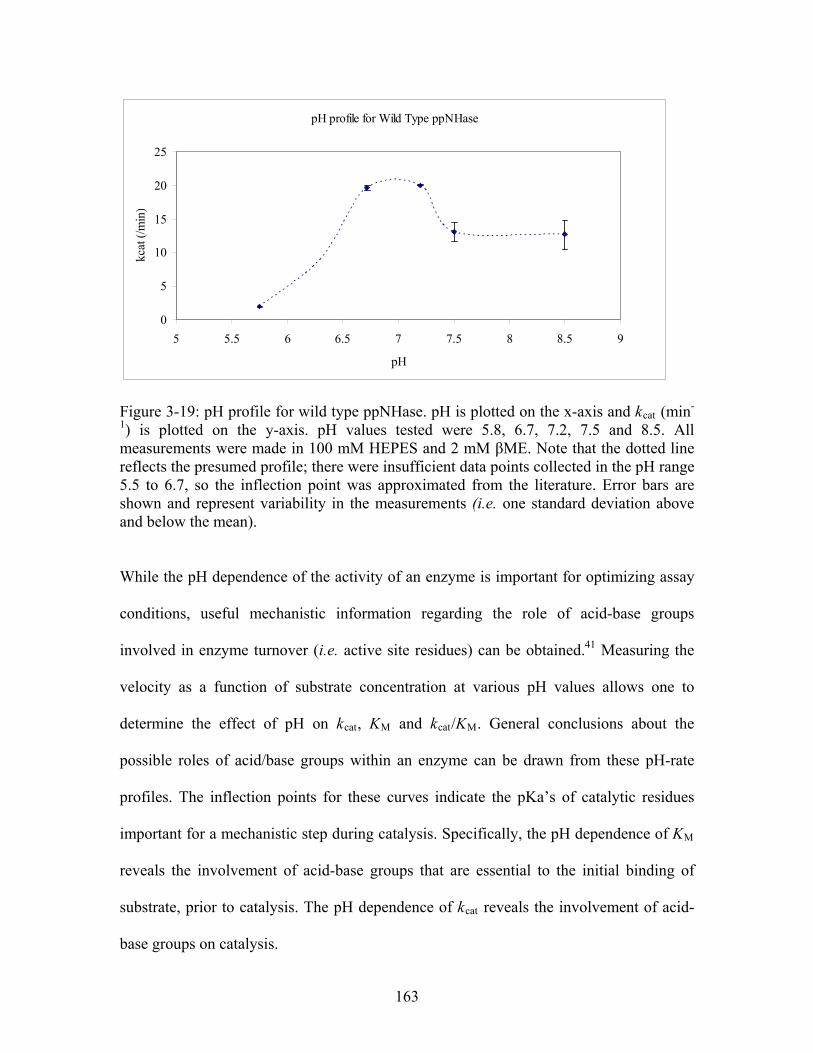
pH profile for Wild Type ppNHase
0
5
10
15
20
25
5 5.5 6 6.5 7 7.5 8 8.5 9
pH
kcat
(/m
in)
Figure 3-19: pH profile for wild type ppNHase. pH is plotted on the x-axis and kcat (min-
1) is plotted on the y-axis. pH values tested were 5.8, 6.7, 7.2, 7.5 and 8.5. All measurements were made in 100 mM HEPES and 2 mM βME. Note that the dotted line reflects the presumed profile; there were insufficient data points collected in the pH range 5.5 to 6.7, so the inflection point was approximated from the literature. Error bars are shown and represent variability in the measurements (i.e. one standard deviation above and below the mean). While the pH dependence of the activity of an enzyme is important for optimizing assay
conditions, useful mechanistic information regarding the role of acid-base groups
involved in enzyme turnover (i.e. active site residues) can be obtained.41 Measuring the
velocity as a function of substrate concentration at various pH values allows one to
determine the effect of pH on kcat, KM and kcat/KM. General conclusions about the
possible roles of acid/base groups within an enzyme can be drawn from these pH-rate
profiles. The inflection points for these curves indicate the pKa’s of catalytic residues
important for a mechanistic step during catalysis. Specifically, the pH dependence of KM
reveals the involvement of acid-base groups that are essential to the initial binding of
substrate, prior to catalysis. The pH dependence of kcat reveals the involvement of acid-
base groups on catalysis.
163

For the pH dependence curve shown in Figure 3-19 for ppNHase, there is an inflection
point around pH 7.3 indicating that there is a residue in the active site with a pKa close to
7.3 which is important for catalysis and acts as an acid. At this point, it is difficult to
draw conclusions as to what that particular residue may be. The pH vs. kcat curve for wild
type does not appear to have an inflection point in the acidic side. This however was due
to the fact that there are a limited number of kinetics points at pH values between 5.8 and
6.7; additional points would be needed to obtain the correct inflection. Kinetics results
from the literature do in fact have this inflection point; therefore a presumed inflection
point was added to the pH curve. (Figure 3-19). Based on this added approximated data,
there is an inflection point around pH 6.4 indicating there is an essential active site
residue with a pKa around 6.4. The pKa of cysteine sulfenic acid is approximately 6.1,
suggesting that αCys117 in ppNHase is necessary for catalysis. This is the singly
oxidized cysteine of the wild type ppNHase protein in solution. Additionally, there was
no pH effect on KM, suggesting there are no acid-base groups in the active site (first-
shell) that are essential for binding. Interestingly, for ppNHase, the known ligand binding
residue, βTyr68, is a second-shell residue, and therefore may not be affected by pH
changes.
164
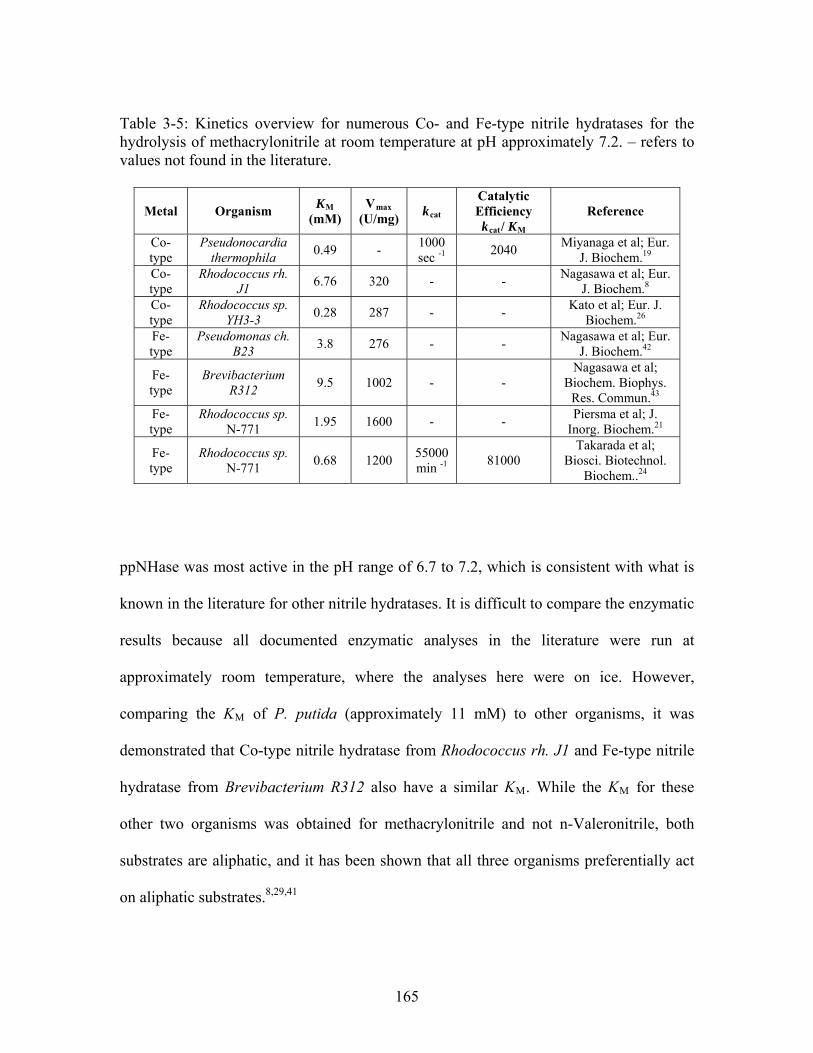
Table 3-5: Kinetics overview for numerous Co- and Fe-type nitrile hydratases for the hydrolysis of methacrylonitrile at room temperature at pH approximately 7.2. – refers to values not found in the literature.
Metal Organism KM
(mM) Vmax
(U/mg) kcat
Catalytic Efficiency kcat/ KM
Reference
Co-type
Pseudonocardia thermophila
0.49 - 1000 sec -1
2040 Miyanaga et al; Eur.
J. Biochem.19 Co-type
Rhodococcus rh. J1
6.76 320 - - Nagasawa et al; Eur.
J. Biochem.8 Co-type
Rhodococcus sp. YH3-3
0.28 287 - - Kato et al; Eur. J.
Biochem.26 Fe-type
Pseudomonas ch. B23
3.8 276 - - Nagasawa et al; Eur.
J. Biochem.42
Fe-type
Brevibacterium R312
9.5 1002 - - Nagasawa et al;
Biochem. Biophys. Res. Commun.43
Fe-type
Rhodococcus sp. N-771
1.95 1600 - - Piersma et al; J.
Inorg. Biochem.21
Fe-type
Rhodococcus sp. N-771
0.68 1200 55000 min -1
81000 Takarada et al;
Biosci. Biotechnol. Biochem..24
ppNHase was most active in the pH range of 6.7 to 7.2, which is consistent with what is
known in the literature for other nitrile hydratases. It is difficult to compare the enzymatic
results because all documented enzymatic analyses in the literature were run at
approximately room temperature, where the analyses here were on ice. However,
comparing the KM of P. putida (approximately 11 mM) to other organisms, it was
demonstrated that Co-type nitrile hydratase from Rhodococcus rh. J1 and Fe-type nitrile
hydratase from Brevibacterium R312 also have a similar KM. While the KM for these
other two organisms was obtained for methacrylonitrile and not n-Valeronitrile, both
substrates are aliphatic, and it has been shown that all three organisms preferentially act
on aliphatic substrates.8,29,41
165

3.5 Conclusions
Chapter 3 presented the structure of the enantioselective NHase from P. putida to 2.1 Å.
The structure reveals global similarity to other NHases except for large differences in the
α5-loop-α6 region in the β-subunit and in the location of the N-terminus of the α-
subunit. In addition, a full kinetic profile of ppNHase was obtained. It was determined
that the enzyme was most active in the pH range between 6.7 and 7.2, with a kcat of 20
min-1 and a KM of 6.6 mM at pH 6.7 and a KM of 11 mM at pH 7.2. The kcat decreased at
high and low pH values while the KM remained essentially unaffected.
Now that the wild type structure of nitrile hydratase from Pseudomonas putida has been
solved, the following chapter begins a systematic mutational study of second- and third-
shell residues for ppNHase. The computational data presented in chapter 2, providing
evidence that second- and third-shell residues are functionally important, was the guiding
force for this study. Four second- and third-shell mutant NHase structures will be
presented, in addition to a complete kinetic analysis. These four mutants include,
αGlu168Gln, βGlu56Gln, βHis71Leu and βTyr215Phe (P. putida numbering).
Additionally, kinetic results will be presented for a fifth second-shell mutant,
αAsp164Asn, for which no structure was solved. It will be shown that these second- and
third-shell mutations do affect the kcat of the protein compared to wild type. In some of
the cases, it will be shown that there are local structural changes which help explain these
results. However, there are cases where no obvious structural changes occur, making an
argument that the decrease in kcat is due to other effects. There are many possible
proposed mechanisms for these effects which include 1) local rotations or side chain
166

shifts, 2) shifts in hydrogen-bonding (H-bonding) networks, 3) changes in the electric
field in the active site, or 4) quantum mechanical effects. Chapter 4 will provide some
answers to these questions.
167

3.6 References
1. Kovacs, J. A. (2004). Synthetic analogues of cysteinate-ligated non-heme iron and
non-corrinoid cobalt enzymes. Chem Rev 104, 825-848. 2. Lippard, S. J. & Berg, J. M. (1994). Principles of Bioinorganic Chemistry,
University Science, Mill Valley, CA. 3. Kobayashi, M. & Shimizu, S. (1999). Cobalt proteins. Eur J Biochem 261, 1-9. 4. Shearer, J., Kung, I. Y., Lovell, S., Kaminsky, W. & Kovacs, J. A. (2001). Why is
there an "inert" metal center in the active site of nitrile hydratase? Reactivity and ligand dissociation from a five-coordinate Co(III) nitrile hydratase model. J Am Chem Soc 123, 463-468.
5. Nagasawa, T. (1989). Microbial transformations of nitriles. Trends Biotechnol 7, 153-158.
6. Komeda, H., Kobayashi, M. & Shimizu, S. (1996). A novel gene cluster including the Rhodococcus rhodochrous J1 nhlBA genes encoding a low molecular mass nitrile hydratase (L-NHase) induced by its reaction product. J Biol Chem 271, 15796-15802.
7. Kobayashi, M., Nagasawa, T. & Yamada, H. (1992). Enzymatic synthesis of acrylamide: a success story not yet over. Trends Biotechnol 10, 402-408.
8. Nagasawa, T., Takeuchi, K. & Yamada, H. (1991). Characterization of a new cobalt-containing nitrile hydratase purified from urea-induced cells of Rhodococcus rhodochrous J1. Eur J Biochem 196, 581-589.
9. Cowan, D. A., Cameron, R. A. & Tsekoa, T. L. (2003). Comparative biology of mesophilic and thermophilic nitrile hydratases. Adv Appl Microbiol 52, 123-158.
10. Kobayashi, M. & Shimizu, S. (1998). Metalloenzyme nitrile hydratase: structure, regulation, and application to biotechnology. Nat Biotechnol 16, 733-736.
11. Huang, W., Jia, J., Cummings, J., Nelson, M., Schneider, G. & Lindqvist, Y. (1997). Crystal structure of nitrile hydratase reveals a novel iron centre in a novel fold. Structure 5, 691-699.
12. Miyanaga, A., Fushinobu, S., Ito, K., Shoun, H. & Wakagi, T. (2004). Mutational and structural analysis of cobalt-containing nitrile hydratase on substrate and metal binding. Eur J Biochem 271, 429-438.
13. Desai, L. V. & Zimmer, M. (2004). Substrate selectivity and conformational space available to bromoxynil and acrylonitrile in iron nitrile hydratase. Dalton Trans, 872-877.
14. Peplowski, L., Kubiak, K. & Nowak, W. (2007). Insights into catalytic activity of industrial enzyme Co-nitrile hydratase. Docking studies of nitriles and amides. J Mol Model 13, 725-730.
15. Novak, W. R. P., Brodkin, H., Milne, A. C., Goldberg, I. G., Karabacak, M., Payne, M. S., Agar, J. N., Ondrechen, M. J., Petsko, G. A., & Ringe, D. (2009). Crystal structure of the enantioselective nitrile hydratase from Pseudomonas putida: mechanistic insights from docking studies. In preparation.
16. Miyanaga, A., Fushinobu, S., Ito, K. & Wakagi, T. (2001). Crystal structure of cobalt-containing nitrile hydratase. Biochem Biophys Res Commun 288, 1169-1174.
168

17. Hourai, S., Miki, M., Takashima, Y., Mitsuda, S. & Yanagi, K. (2003). Crystal structure of nitrile hydratase from a thermophilic Bacillus smithii. Biochem Biophys Res Commun 312, 340-345.
18. Nagashima, S., Nakasako, M., Dohmae, N., Tsujimura, M., Takio, K., Odaka, M., Yohda, M., Kamiya, N. & Endo, I. (1998). Novel non-heme iron center of nitrile hydratase with a claw setting of oxygen atoms. Nat Struct Biol 5, 347-351.
19. Miyanaga, A., Fushinobu, S., Ito, K., Shoun, H. & Wakagi, T. (2004). Mutational and structural analysis of cobalt-containing nitrile hydratase on substrate and metal binding. Eur. J. Biochem. 271, 429-438.
20. Hashimoto, Y., Sasaki, S., Herai, S., Oinuma, K., Shimizu, S. & Kobayashi, M. (2002). Site-directed mutagenesis for cysteine residues of cobalt-containing nitrile hydratase. J Inorg Biochem 91, 70-77.
21. Piersma, S. R., Nojiri, M., Tsujimura, M., Noguchi, T., Odaka, M., Yohda, M., Inoue, Y. & Endo, I. (2000). Arginine 56 mutation in the beta subunit of nitrile hydratase: importance of hydrogen bonding to the non-heme iron center. J Inorg Biochem 80, 283-288.
22. Endo, I., Nojiri, M., Tsujimura, M., Nakasako, M., Nagashima, S., Yohda, M. & Odaka, M. (2001). Fe-type nitrile hydratase. J Inorg Biochem 83, 247-253.
23. Takarada, H., Kawano, Y., Hashimoto, K., Nakayama, H., Ueda, S., Yohda, M., Kamiya, N., Dohmae, N., Maeda, M. & Odaka, M. (2006). Mutational study on alphaGln90 of Fe-type nitrile hydratase from Rhodococcus sp. N771. Biosci Biotechnol Biochem 70, 881-889.
24. Murakami, T., Nojiri, M., Nakayama, H., Odaka, M., Yohda, M., Dohmae, N., Takio, K., Nagamune, T. & Endo, I. (2000). Post-translational modification is essential for catalytic activity of nitrile hydratase. Protein Sci 9, 1024-1030.
25. Wu, S., Fallon, R. D. & Payne, M. S. (1997). Over-production of stereoselective nitrile hydratase from Pseudomonas putida 5B in Escherichia coli: activity requires a novel downstream protein. Appl Microbiol Biotechnol 48, 704-708.
26. Kato, Y., Tsuda, T. & Asano, Y. (1999). Nitrile hydratase involved in aldoxime metabolism from Rhodococcus sp. strain YH3-3 purification and characterization. Eur J Biochem 263, 662-670.
27. Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248-254.
28. Fallon, R. D., Stieglitz, B. & Turner Jr., I. (1997). A Pseudomonas putida capable of stereoselective hydrolysis of nitriles. Appl. Microbiol. Biotechnol. 47, 156-161.
29. Kim, T., Tolmachev, A. V., Harkewicz, R., Prior, D. C., Anderson, G., Udseth, H. R. & Smith, R. D. (2000). Design and implementation of a new electrodynamic ion funnel. Anal Chem 72, 2247-2255.
30. Karabacak, N. M., Li, L., Tiwari, A., Hayward, L. J., Hong, P., Easterling, M. L. & Agar, J. N. (2008). Sensitive and specific identification of wild-type and variant proteins from 8 to 669 kDa using top-down mass spectrometry. Mol Cell Proteomics.
31. Otwinowski, Z. & Minor, W. (1997). Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 276, 307-326.
169

32. Mccoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). Phaser crystallographic software. Journal of Applied Crystallography 40, 658-674.
33. Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D-Biological Crystallography 53, 240-255.
34. Emsley, P. & Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallographica Section D-Biological Crystallography 60, 2126-2132.
35. Adams, P. D., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallographica Section D-Biological Crystallography 58, 1948-1954.
36. Song, L., Wang, M., Shi, J., Xue, Z., Wang, M. X. & Qian, S. (2007). High resolution X-ray molecular structure of the nitrile hydratase from Rhodococcus erythropolis AJ270 reveals posttranslational oxidation of two cysteines into sulfinic acids and a novel biocatalytic nitrile hydration mechanism. Biochem Biophys Res Commun 362, 319-324.
37. Lehninger, A. L., Nelson, D. L. & Cox, M. M. (1993). Principles of Biochemistry. Second edit, Worth Publishers, Inc.
38. Fersht, A. R. (1998). Structure and Mechanism in Protein Science: A guide to Enzyme Catalysis and Protein Folding (Julet, M. R., Ed.), W. H. Freeman and Company.
39. Wolfenden, R. (2006). Degrees of difficulty of water-consuming reactions in the absence of enzymes. Chem Rev 106, 3379-3396.
40. Motulsky, H. C., A. (2004). Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting (GraphPad Software, I., Ed.), Oxford University Press, New York.
41. Copeland, R. A. (2000). Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. Second edit (Wiley-VCH, Ed.), John Wiley and Sons, Inc.
42. Nagasawa, T., Nanba, H., Ryuno, K., Takeuchi, K. & Yamada, H. (1987). Nitrile hydratase of Pseudomonas chlororaphis B23. Purification and characterization. Eur J Biochem 162, 691-698.
43. Nagasawa, T., Ryuno, K. & Yamada, H. (1986). Nitrile hydratase of Brevibacterium R312--purification and characterization. Biochem Biophys Res Commun 139, 1305-1312.
170

Chapter 4
Evidence for Participation of Remote Residues in the Catalytic Activity of Co-type Nitrile
Hydratase from Pseudomonas putida – A Kinetic and Crystal Structure Analysis
171

4.1 Introduction
Enzyme active sites have evolved distinct electrostatic and chemical properties which
facilitate catalysis and substrate recognition. Do these properties arise solely from
residues immediately surrounding the reacting substrate molecule, or do the next-nearest
neighbors, the “second-shell” residues located behind the first layer, contribute also? An
abundance of experimental evidence has established the importance of particular residues
in the catalytic and recognition capabilities of enzymes. Typically, the residues that have
been studied are, in the active form of the enzyme, in direct contact with the reacting
substrate or bound metal ion. These residues may be considered to be in the “first
interaction sphere” of the bound substrate molecule or bound metal ion. Computational
evidence from chapter 2, in addition to a limited set of previous experimental data,
suggest that remote residues, particularly those in the “second- and third-shell” around
the reacting substrate, also contribute to catalytic activity. We chose to use two
completely different computational methods to identify residues in the first, second, and
third interaction spheres to provide evidence that enzyme active sites are built in multiple
layers. We examined the predictions of THEMATICS, a structure based method which
identifies functionally important residues based on charge perturbations, and ET which
identifies functionally important residues based on sequence conservation and
evolutionary pressure. Based on the nature of the methods, ET predictions in all three
interaction spheres are very large, while THEMATICS predictions are shown to be
highly selective. Although ET identifies a larger number of residues, not all have been
shown to be functionally important. It was shown (Chapter 2) that THEMATICS predicts
specific residues, especially in the first- and second-shell, which are known in the
172

literature to be functionally important. In this case, second-shell refers to those residues
which are in direct contact with residues in the first interaction sphere, and third-shell
refers to those residues which are in direct contact with residues in the second interaction
sphere. The combination of theoretical and limited experimental evidence suggests that
enzyme active sites are nanoscale entities built in multiple layers, and residues beyond
the first-shell of the enzyme active site are important for catalysis and/or substrate
binding.
Chapter 3 presented the crystal structure and kinetic analysis of Co-type nitrile hydratase
from Pseudomonas putida. This was one enzyme for which THEMATICS and ET both
predicted a multilayer active site. To date, there have been no systematic approaches to
mutating second- and third-shell residues specifically to understand their role in enzyme
catalysis. In this chapter, the enzymatic effect of five second- and third-shell mutations
predicted by THEMATICS and ET for Co-type nitrile hydratase from Pseudomonas
putida will be reported. The mutations include αD164N, αE168Q, βE56Q, βH71L, and
βY215F (P. putida numbering). It will be demonstrated experimentally, through site-
directed mutagenesis studies, that these second- and third-shell residues, predicted
theoretically by THEMATICS and ET, are functionally important with each one
contributing to the catalytic efficiency of this protein. Further, the crystal structures are
reported for four of the mutant ppNHases, αGlu168Gln, βGlu56Gln, βHis71Leu,
βTyr215Phe (P. putida numbering). It was suggested in Chapter 3 that there could be
numerous reasons why these second- and third-shell mutations affect kcat and include 1)
local rotations or side chain shifts, 2) shifts in hydrogen-bonding (H-bonding) networks,
173

3) changes in the electric field in the active site, and/or 4) quantum mechanical effects.
This chapter, focusing on both the kinetics and the crystal structures may help explain the
catalytic effects through structural changes.
4.2 Materials and Methods
Computational Methods
All methods were run as previously described, and default parameters were used, except
where stated. The nitrile hydratase protein structure from Pseudonocardia thermophila
(PDB ID: 1IRE1) was downloaded from the Protein Data Bank (PDB,
http://www.rcsb.org/pdb/). The data for the nitrile hydratase structures from
Pseudomonas putida were collected at the ID-23B beamline at GM/CA-CAT (APS,
Argonne, IL, USA) and solved using REFMAC2, COOT3 and PHENIX4. Coordinates for
all the nitrile hydratase proteins, wild type and mutants, were analyzed by Theoretical
Microscopic Titration Curves (THEMATICS )5-8 using the method of Wei et al.9, except
that a cut-off of 0.96 was used instead of 0.99. Structures with missing atoms were fixed
in swiss-pdb viewer. Substrate, inhibitor, and water molecules, cofactors, and salts that
crystallized with the proteins were not included in the THEMATICS analysis. Since the
protein of interest is a hetero-multimer, THEMATICS calculations were run on the full
biological unit. Evolutionary Trace Report Marker (ET,
http://mammoth.bcm.tmc.edu/report_maker/index.html)10-12 analysis was performed as
provided. The Catalytic Site Atlas (CSA, http://www.ebi.ac.uk/thornton-
srv/databases/CSA/) was used to identify the literature annotated catalytic residues. First-
shell residues, those residues in contact with a bound ligand or metal ion, were identified
174

using Ligand Protein Contact (LPC, http://bip.weizmann.ac.il/oca-bin/lpccsu); second-
and third-shell residues, those in contact with a given first- or second-shell residue,
respectively, were determined using Contacts of Structural Units (CSU)
(http://bip.weizmann.ac.il/oca-bin/lpccsu)13,14. Those residues in direct contact with the
first-shell residues were considered second-shell, and those residues in direct contact with
second-shell residues, were considered third-shell. Conservation Surface-Mapping
(Consurf, http://consurf.tau.ac.il/)15-17 was performed using the default values. Once the
first-, second- and third-shell residues were identified from THEMATICS and ET,
normalized conservation scores were determined for each of those residues. The
normalized scores were then averaged for each coordination shell.
Experimental Methods
Site-Directed Mutagenesis (SDM)
An expression plasmid for Pseudomonas putida NRRL-18668 (obtained from Mark
Payne, E.I. du Pont de Nemours and Company), which contains the genes for the α and β
subunits of NHase and for the NHase activator, P14K, was used for mutagenesis.18 Site-
directed mutagenesis was carried out using Stratagene’s Quikchange II Site Directed
Mutagenesis Kit (Stratagene, La Jolla, CA). The primers used are shown in Table 4-1.
Mutated codons are shown in bold face. PCR amplification was performed per the
manufacturer’s instructions. The mutated DNA was sequenced at Genewiz (Genewiz,
Inc., South Plainfield, NJ). After confirmation of successful construction of the intended
mutation, the DNA was transformed into BL21 (DE3) competent cells (Stratagene).
175

Table 4-1: Primers designed for site-directed mutagenesis. Only forward primers are listed. Mutated codons are shown in boldface.
Mutant Primer
αAsp164Asn 5’CCCGCCAACAAGGAAATCCGCGTCTGGAACACCACGGCCGAATTG-3’
αGlu168Gln 5’-GTCTGGGACACCACGGCCCAATTGCGCTACATG GTGCTG-3’
βGlu56Gln 5’GAATTTCGGCATTCGATCCAGCGAATGGGCCCGGCCCAC-3’
βHis71Leu 5’GCCCACTATCTGGAGGGAACCTACTACGAACTCTGGCTTCATGTCTTTG
AGAACCTGCTGGTC-3’
βTyr215Phe 5’-CGCGTCGACTTGTGGGATGACTTCCTGGAGCCAGAGTGA-3’
Protein Expression and Purification
All reagents were purchased from Fisher Scientific, Pittsburgh, PA unless otherwise
noted. The wild type ppNHase was expressed in E. coli BL21 (DE3) (Stratagene). Cells
were grown at 37 °C, in 1 L of 2XYT broth containing ampicillin (100 μg/mL). When the
A600 reached 0.8, the cells were induced by the addition of isopropyl-β-D-thiogalactoside
(IPTG) to 1 mM and cobalt chloride to 0.5 mM.18 The cells were then cultured for an
additional 3 hours at 28 °C. All subsequent manipulations were performed at 4 °C. After
cell harvesting by centrifugation, the pellet was resuspended in 40 mL of 50 mM Tris pH
8.0 and 2 mM βME (Buffer A). Cells were lysed via sonication and the suspension was
clarified by centrifugation at 10,000 × g for 40 minutes. The ppNHase-containing
supernatant was loaded onto a 60 mL DEAE anion-exchange column equilibrated in
Buffer A containing 80 mM NaCl and ppNHase was eluted with a linear gradient from 80
to 200 mM NaCl in Buffer A over 700 mL.19 Fractions containing ppNHase were pooled
and precipitated with 70% ammonium sulfate.19 After centrifugation and reconstitution of
176

the ppNHase containing pellet in Buffer A, the protein was loaded onto a 20 mL Phenyl
Sepharose column (GE Healthcare, Piscataway, NJ) equilibrated in Buffer A containing
0.5 M ammonium sulfate and eluted with a linear gradient from 0.5 to 0 M ammonium
sulfate in same buffer over 180 mL.20 Fractions containing ppNHase were pooled and
concentrated using an Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-10
membrane (Millipore, Billerica, MA) with 10 kDa nominal molecular weight limit and
dialyzed 2 times (4 hours each) against 50 mM Tris pH 8.0 and 2 mM βME. The protein
was loaded onto a 10 mL MonoQ column (GE Healthcare) equilibrated in Buffer A
containing 125 mM NaCl. ppNHase was eluted with a linear gradient of 125 mM to 240
mM NaCl in Buffer A over 135 mL.19 Fractions containing ppNHase were pooled and
concentrated using an Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-10
membrane (Millipore) with 10 kDa nominal molecular weight limit and dialyzed 2 times
(4 hours each) against 50 mM Tris pH 8.0 and 2 mM βME, concentrated to ~20 mg/mL
and stored at 4 °C.
It was discovered that the αAsp164Asn mutant protein was slightly destabilized by the
mutation, and tended to dissociate into monomers when subjected to high salt as
evidenced by the disappearance of one protein band in the SDS PAGE gel. The
purification protocol was adjusted slightly by the removal of the ammonium sulfate
precipitation and phenyl sepharose column. After the DEAE column, the protein was
concentrated and only subjected to the Mono-Q column. The protein was clean enough
for further work as evidenced by the SDS PAGE gel. The concentration of all proteins
177

was determined either by the Bradford assay21, or by A280 measurement. The extinction
coefficient used was 1.676 mg · mL-1 · cm-1 (http://us.expasy.org/cgi-bin/protparam).
Kinetics
NHase activity was determined by measurement of the hydration of n-Valeronitrile in a
300 μL reaction volume, containing 100mM HEPES and 2 mM βME in an ice bath.20
Because the reaction was too fast to monitor at room temperature, the protocol was
adjusted and the reaction was run on ice to slow down the rate of turnover. The mutant
proteins were analyzed by using Michaelis-Menten (MM) kinetics at pH 5.8, 6.7, 7.2, 7.5
and 8.5. Non-linear regression was performed in order to obtain the MM constants, kcat
and KM. Bis-Tris buffer was used at pH 5.8 for the βGlu56Gln mutant and Tris buffer
was used at pH 8.5 for the α Asp164Asn mutant. Each buffer had an effect on the
kinetics; therefore, correction factors were obtained. For the β Glu56Gln mutant, kinetic
data were obtained at pH 6.7 in HEPES and Bis-Tris and the difference in kcat (9 X more
active in Bis-Tris) was applied to the value obtained in Bis-Tris at pH 5.8. For the
αAsp164Asn mutant, kinetic data were obtained at pH 7.5 in HEPES and Tris and the
difference in kcat (9 X more active in Tris) was applied to the value obtained in Tris at
pH 8.5. All other mutants were analyzed in HEPES at all pH values tested. Each reaction
was run three times at pH 6.7 and two times at pH 5.8, 7.2, 7.5 and 8.5. Initial
concentrations of n-Valeronitrile were 0.63, 2.5, 5.0, 10, and 40 mM. The concentration
of mutant ppNHase varied depending upon the mutation and the pH. The concentration of
enzyme was adjusted so that the concentration of product formed was within the range of
178

the standard curve; the range was 6 nM to 60 nM. The reaction was carried out for 40 and
60 minutes in an ice bath, and stopped by the addition of 0.3 N HCl.
The formation of n-Valeramide was monitored with either a WATERS 2690 HPLC
(Waters, Corp., Milford, MA) or an Agilent 1200 HPLC (Agilent Technologies, Santa
Clara, CA), using a Zorbax Aq reverse phase C18 column (4.6 X 150 mm) (Agilent
Technologies, Santa Clara, CA) at a flow rate of 1.0 mL /min.22 A standard curve was
prepared every time samples were run and ranged between 3.90 μg/mL and 250 μg/mL.
Running buffers were 5 mM Potassium Phosphate pH 2.9 (A) and 100% acetonitrile (B),
running at 1.0 mL/min. The product was eluted with a shallow gradient of 10% - 25% B
for 7 minutes. Sample run time was 14 minutes. The absorbance was measured at 210
nm.
Crystallization, data collection and crystallographic refinement
Crystals of ppNHase were grown at 25 ˚C by vapor diffusion in 24 well hanging drop
plates over 0.7 mL volume reservoirs using 1 + 1 µL drops. Three crystal forms were
identified from initial screens (Hampton Research, Aliso Viejo, CA), however, one of
these crystal forms failed to diffract beyond 3.5 Å and one could not be accurately
indexed due to twinning. Diffracting crystal needles were obtained using 20 mg/mL
ppNHase and a reservoir containing 22% polyacrylic acid in HEPES pH 7.5 with 20 mM
magnesium chloride and 4% acetone. Single crystals were dissected from clusters and
transferred to a solution containing 17.6% polyacrylic acid 5100 and 20% glycerol and
179

were flash-frozen in liquid nitrogen. In order to obtain larger, single crystals of the
βGlu56Gln mutant, initial crystal forms were used to seed new drops.
Data were collected at both the ID-23B and ID-23D beamlines at GM/CA-CAT (APS,
Argonne, IL, USA) at 100 K using a MARmosiac 300 CCD detector and the 10 μm mini-
beam. Diffraction images were indexed, integrated and scaled using HKL2000.23
Molecular replacement was carried out with Phaser24 using PDB ID: 1IRE1 as a starting
model. Several rounds of refinement and model building were performed using
REFMAC 2 and COOT.3 Final rounds of refinement, including simulated annealing and
water picking were performed using PHENIX.4
4.3 Results and Discussion
Theoretical Results
THEMATICS and Evolutionary Trace (ET)
THEMATICS and ET calculations were initially run on wild type NHase from
Pseudonocardia thermophila (PBD ID: 1IRE1) because there was no known structure of
Co-type nitrile hydratase from Pseudomonas putida. Once the structures were solved for
wild type ppNHase and the mutant proteins, THEMATICS calculations were run on the
proteins from Pseudomonas putida. For purposes of this study, first-shell residues are
defined as those which are in direct contact with a metal ion or substrate molecule,
second-shell residues as those which make direct contact with first-shell residues, and
third-shell residues as those which make direct contact with second-shell residues. Ligand
180

protein contacts (LPC) was used to identify ligand or metal binding residues, and contacts
of structural units (CSU) was used to identify residues as second- or third-shell for each
cluster.13 The THEMATICS results are shown in Table 4-2 and the ET results are shown
in Table 4-3. ET was not run on any of the NHase proteins from Pseudomonas putida
because the online server only accepts structures from the protein data bank and no
significant differences in ET predictions were expected.
181
Table 4-2: THEMATICS predictions of functional sites for wild type NHase from Pseudonocardia thermophila (PDB ID: 1IRE1), wild type NHase from Pseudomonas putida, and five NHase mutants from Pseudomonas putida. Predicted residues are listed by shell with average normalized conservation scores for each coordination shell. Bold face refers to residues predicted by THEMATICS which are annotated in the CSA as catalytic residues; italics refers to residues predicted by THEMATICS which are found in LPC to be metal binding or ligand binding residues; those residues which are in both bold face and italics refers to THEMATICS positives which are both annotated in the CSA as catalytic residues and annotated in LPC as binding residues.
Enzyme Shell Functional site residues predicted by
THEMATICS for NHase proteins
Average Normalized
Conservation Score
C111 A, C113 A 1st
C108 A -0.731
2nd K127 A, D161 A, E56 B, Y68 B, H155 B,
R157 B -1.208
3rd E165A, Y69 B, H71 B, Y216 B, Y222 B -0.855
Co-TYPE NITRILE HYDRATASE
Pseudonocardia thermophila PDB ID: 1IRE1
other H172 B, H173 B, H192 B, D217 B -0.816
C111 A, C113 A, R52 B 1st
C108 A -1.205
2nd Y114 A, Y126 A, K127 A, D160 A, D49
B, H53 B, E56 B, Y68 B, H147 B, R149 B -0.985
3rd E164 A, H5 B, Y69 B, H71 B -1.086
Co-TYPE NITRILE HYDRATASE
Pseudomonas putida
other D210 B -1.418
C111 A, C113 A, R52 B 1st
C108 A -1.207
2nd Y114 A, Y126 A, K127 A, D160 A, H53
B, E56 B, Y68 B, H147 B, R149 B -0.918
3rd M1 B, H5 B, Y69 B, H71 B -1.307
Co-TYPE NITRILE HYDRATASE
α E168Q Pseudomonas putida
other E30 B, D210 B -1.481
C111 A, C113 A, R52 B 1st
CYS 108 A -1.214
2nd Y114 A, Y126 A, K127 A, D160 A, D49
B, H53 B, Y68 B, H147 B, R149 B -0.936
3rd E164 A, Y69 B, H71 B -1.159
Co-TYPE NITRILE HYDRATASE β Glu56Gln
Pseudomonas putida
other H5 B, D6 B, E22 B, E30 B, E70 B, D210 B -0.978
C111 A, C113 A, R52 B 1st
C108 A -1.216
2nd Y114 A, Y126 A, K127 A, D160 A, D49
B, H53 B, E56 B, Y68 B, H147 B, R149 B -1.005
3rd E164 A, H5 B, Y69 B -1.021
Co-TYPE NITRILE HYDRATASE β His71Leu
Pseudomonas putida
other D210 B -1.466
C111 A, C113 A, R52 B 1st
C108 A -1.214
2nd Y114 A, Y126 A, K127 A, D160 A, D49
B, H53 B, E56 B, Y68 B, H147 B, R149 B -0.910
3rd E164 A, H5 B, TYR 69 B, H71 B -1.100
Co-TYPE NITRILE HYDRATASE β Tyr215Phe
Pseudomonas putida
other D210 B -1.459
182
Table 4-3: Evolutionary Trace functional site predictions for wild type NHase from Pseudonocardia thermophila (PDB ID: 1IRE1). Predicted residue sequence numbers are listed by shell with average normalized conservation scores for each coordination shell. Bold face refers to residues predicted by ET which are annotated in the CSA as catalytic residues; italics refers to residues predicted by ET which are found in LPC to be metal binding or ligand binding residues; those residues which are both in bold face and italics refers to ET positives which are both annotated in the CSA as catalytic residues and annotated in LPC as binding residues.
Enzyme Shell Functional site residues
predicted by ET for NHase proteins
Average Normalized
Conservation Score
111, 112, 113 1st
108 -0.680
2nd 107, 109, 110, 114, 116, 122, 126, 127, 132, 161, 162, 167
-0.880
3rd 115, 119, 120, 123, 125, 131,
136, 159, 165 -0.836
Co-TYPE NITRILE HYDRATASE
1IRE1 - A 4.2.1.84
other
52, 55, 59, 60, 62, 63, 65, 66, 103, 134, 139, 140, 142, 143, 146, 148, 170, 172, 174, 175,
186, 189, 197
NA
1st 52 -1.644 2nd 49, 51, 55, 56, 63, 68, 155, 157 -1.086
3rd 1, 3, 5, 6, 7, 8, 60, 69, 72, 156, 159, 161, 179, 180, 183, 222
-0.974 Co-TYPE NITRILE
HYDRATASE 1IRE1 - B 4.2.1.84
other
2, 9, 25, 29, 30, 32, 139, 145, 163, 167, 174, 178, 185, 193, 194, 196, 198, 203, 204, 205,
217, 218, 223
NA
The sequence identity between Pseudonocardia thermophila and Pseudomonas putida is
approximately 58% for the alpha subunit and 43% for the beta subunit; this degree of
sequence identity is sufficient to use the Pseudonocardia thermophila structure as a good
model for the identification of important remote residues for ppNHase. THEMATICS
was run using Wei, et al.’s method9, modified to use a statistical cutoff of 0.96, rather
than 0.99. We note that the statistical cut-off of 0.99 was determined by Wei, et al.9 to be
the value that maximizes performance in the selection of residues annotated in the
Catalytic Site Atlas (CSA). However, nearly all CSA-annotated residues are in the first
183

shell. Thus, the statistical cut-off of 0.96 was used here in order to increase the number of
residues predicted outside the first-shell. Wei, et al.’s method9 computes metrics of
anomalous titration behavior, μ3 and μ4, and selects ionizable residues with metrics more
than one standard deviation above the average for all ionizable residues in the protein.
When the statistical cut-off is 0.96, the top 4% of residues with the highest metrics are
excluded in the calculation of the mean and standard deviation. Residues with metrics
more than one standard deviation above the mean are then clustered using a 9Å cut-off.
Evolutionary trace identifies a larger number of residues using the default parameters;
thus ET predictions were made without modification. Normalized conservation scores for
each of the residues identified in the clusters as first-, second- or third-shell were
obtained with Consurf and then averaged for each shell. A normalized conservation score
is calculated so that the average score for all residues in a protein is zero and the standard
deviation is one. The more negative the normalized conservation score, the more
conserved is the residue and the more positive the score, the more variable is the residue.
A normalized conservation score of -1.000 corresponds to a residue that has a score that
is more conserved than the average by one standard deviation. By design, ET identifies
functional residues based on conservation through evolution; therefore, the set of residues
identified by this method automatically have a high average conservation score. The
average conservation scores are useful as a guide to determine and compare the
conservation of those residues identified by THEMATICS, a method that uses no
sequence-based information at all.
184

THEMATICS and ET both identify the three cobalt coordinating cysteine residues for
both NHase from Pseudonocardia thermophila and Pseudomonas putida, in addition to
the ligand binding tyrosine residue and one of the arginine residues, βArg157 (equivalent
to βArg149 in ppNHase), known to H-bond to αCys111 and αCys113 (equivalent to
αCys115 and αCys117 in ppNHase) (Figure 4-1). THEMATICS additionally identifies
the catalytic residue βArg52 for NHase from Pseudomonas putida, while ET also
identifies this arginine residue for NHase from Pseudonocardia thermophila. The major
difference between the predictions for the two organisms is that two third shell residues,
βTyr216 (equivalent to βVal209 in ppNHase) and βTyr222 (equivalent to βTyr215 in
ppNHase), identified by THEMATICS and ET for NHase from Pseudonocardia
thermophila, were not predicted for NHase from Pseudomonas putida. Based on these
data and the conservation of all residues identified, five residues were chosen for site-
directed mutagenesis studies. Figure 4-2 shows the sequence alignment from Chapter 3,
highlighting the second- and third-shell residues chosen for mutagenesis in addition to the
known functionally important active site, metal binding and ligand binding residues.
Specifically, the second-shell resides chosen were αAsp164 and βGlu56, and the third-
shell residues chosen were αGlu168, βHis71 and βTyr215 (P. putida numbering) (Figure
4-3).
185
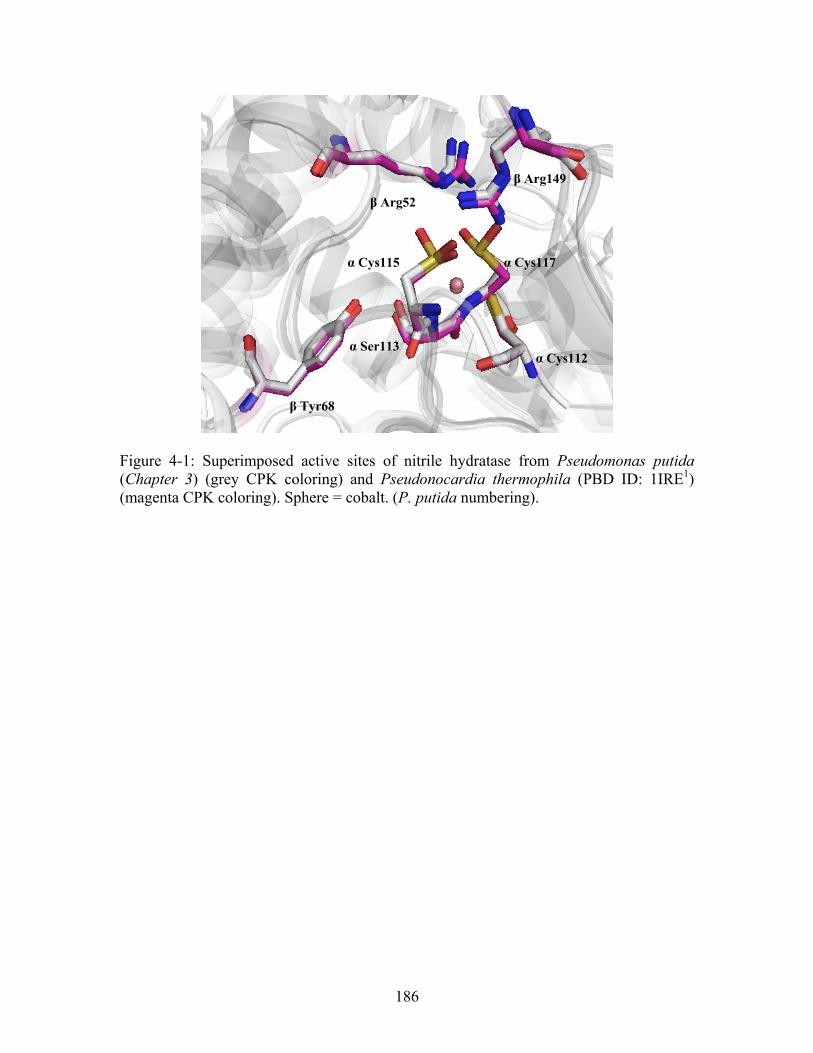
β Arg149
β Arg52
α Cys117 α Cys115
α Ser113 α Cys112
β Tyr68
Figure 4-1: Superimposed active sites of nitrile hydratase from Pseudomonas putida (Chapter 3) (grey CPK coloring) and Pseudonocardia thermophila (PBD ID: 1IRE1) (magenta CPK coloring). Sphere = cobalt. (P. putida numbering).
186

α Subunit
P. Putida3 -----------------------MGQSHTHDHHHDGYQAPPED------- 20
P. thermophila1 ------------------------MTENILRKSDEEIQKEIT-------- 18
Rho. rhodochrous1 ------------------------TAHNPVQGTLPRSNEEIA-------- 18
Therm. Bac. Sm.1 -----------------------MAIEQKLMDDHHEVDPRFPHHHPRPQS 27
Rho. sp. R3122 -------------------------MSVTIDHTTENAAPAQAA------- 18
Comamonas testosterone2 -----------------------MGQSHTHDHHHDGYQAPPED------- 20
Bradyrhizobium japonicum2 MQPIPWPDVSRVFASTRPGFWDYLPSMSDHHHHHDHDHSELSE------- 43
Pseudomonas chlororaphis2 --------------------------STSISTTATPSTPG---------- 14
P. Putida3 -IALRVKALESLLIEKGLVDPAAMDLVVQTYEHKVGPRNGAKVVAKAWVD 69
P. thermophila1 ---ARVKALESMLIEQGILTTSMIDRMAEIYENEVGPHLGAKVVVKAWTD 65
Rho. rhodochrous1 ---ARVKAMEAILVDKGLISTDAIDHMSSVYENEVGPQLGAKIVARAWVD 65
Therm. Bac. Sm.1 FWEARAKALESLLIEKRLLSSDAIERVIKHYEHELGPMNGAKVVAKAWTD 77
Rho. sp. R3122 -VSDRAWALFRALDGKGLVPDGYVEGWKKTFEEDFSPRRGAELVARAWTD 67
Comamonas testosterone2 -IALRVKALESLLIEKGLVDPAAMDLVVQTYEHKVGPRNGAKVVAKAWVD 69
Bradyrhizobium japonicum2 -TELRVRALETILTEKGYVEPAALDAIIQAYETRIGPHNGARVVAKAWTD 92
Pseudomonas chlororaphis2 ---ERAWALFQVLKSKELIPEGYVEQLTQLMAHDWSPENGARVVAKAWVD 61
P. Putida3 PAYKARLLADGTAGIAELGFSGVQGEDMVILENTPAVHNVFVCTLCSCYP 119
P. thermophila1 PEFKKRLLADGTEACKELGIGGLQGEDMMWVENTDEVHHVVVCTLXSXYP 115
Rho. rhodochrous1 PEFKQRLLTDATSACREMGVGGMQGEEMVVLENTGTVHNMVVCTLCSCYP 115
Therm. Bac. Sm.1 PEFKQRLLEDPETVLRELGYFGLQGEHIRVVENTDTVHNVVVCTLCSCYP 127
Rho. sp. R3122 PEFRQLLLTDGTAAVAQYGYLGPQGEYIVAVEDTPTLKNVIVCSLCSCTA 117
Comamonas testosterone2 PAYKARLLADGTAGIAELGFSGVQGEDMVILENTPAVHNVVVCTLCSCYP 119
Bradyrhizobium japonicum2 PAFKQALLEDGSKAIGTLGHVSRVGDHLVVVENTPQRHNMVVCTLCSCYP 142
Pseudomonas chlororaphis2 PQFRALLLKDGTAACAQFGYTGPQGEYIVALEDTPGVKNVIVCSLCSCTN 111
P. Putida3 WPTLGLPPAWYKAAPYRSRMVSDPRGVL-AEFGLVIPANKEIRVWDTTAE 168
P. thermophila1 WPVLGLPPNWFKEPQYRSRVVREPRQLLKEEFGFEVPPSKEIKVWDSSSE 165
Rho. rhodochrous1 WPVLGLPPNWYKYPAYRARAVRDPRGVL-AEFGYTPDPDVEIRIWDSSAE 164
Therm. Bac. Sm.1 WPLLGLPPSWYKEPAYRSRVVKEPRKVL-QEFGLDLPDSVEIRVWDSSSE 176
Rho. sp. R3122 WPILGLPPTWYKSFEYRARVVREPRKVL-SEMGTEIASDIEIRVYDTTAE 166
Comamonas testosterone2 WPTLGLPPAWYKAPPYRSRMVSDPRGVL-AEFGLVIPA-KEIRVWDTTAE 167
Bradyrhizobium japonicum2 WEMLGLPPVWYKAAPYRSRAVKDPRGVL-ADFGVALPKDIEVRVWDSTAE 191
Pseudomonas chlororaphis2 WPVLGLPPEWYKGFEFRARLVREGRTVL-RELGTELPSDTVIKVWDTSAE 160
P. Putida3 LRYMVLPERPAGTEAYSEEQLAELVTRDSMIGTGLPTQP-TPSH- 211
P. thermophila1 MRFVVLPQRPAGTDGWSEEELATLVTRESMIG----VEPAKAV-- 204
Rho. rhodochrous1 LRYWVLPQRPAGTENFTEEQLADLVTRDSLIGVSVPTTPSKA--- 206
Therm. Bac. Sm.1 VRFMVLPQRPEGTEGMTEEELAQIVTRDSMIGVAK-VQPPKVIQE 220
Rho. sp. R3122 TRYMVLPQRPAGTEGWSQEQLQEIVTKDCLIGVAIPQVPTV---- 207
Comamonas testosterone2 LRYMVLPERPAGTEAYSEEQLAELVTRDSMIGTGLPIQP-TPSH- 210
Bradyrhizobium japonicum2 TRFLVLPMRPGGTEGWSEEQLAELVTRDSMIGTGFPKTPGAPS-- 234
Pseudomonas chlororaphis2 SRYLVLPQRPEGSEHMSEEQLQQLVTKDVLIGVALPRVG------ 199
187

β Subunit
P. putida3 MNGIHDTGGAHGYG----PVYREPNEPVFRYDWEKTVMSLLPALLAN--G 44
P. thermophila1 MNGVYDVGGTDGLG----PINRPADEPVFRAEWEKVAFAMFPATFRA--G 44
Rho. rhodochrous1 MDGIHDLGGRAGLG----PIKPESDEPVFHSDWERSVLTMFPAMALA--G 44
Therm. Bac. Sm.1 MNGIHDVGGMDGFG--KIMYVKEEEDTYFKHDWERLTFGLVAGCMAQGLG 48
Rho. sp. R3122 --------------------------------------------------
Comamonas testosterone2 MNGIHDTGGAHGYG----PVYREPNEPVFRYDWEKTVMSLFPALFAN--G 44
Bradyrhizobium japonicum2 MNGVHDMGGMDGFG----KVEPEPNEPMFHEEWESRVLAMVRA-MGA-AG 44
Pseudomonas chlororaphis2 MDGFHDLGGFQGFGKVPHTINSLSYKQVFKQDWEHLAYSLMFVGVDQ-LK 49
P. putida3 NFNLD-EFRHSIERMGPAHYLEGTYYEHWLHVFENLLVEKGVLTATEVAT 93
P. thermophila1 FMGLD-EFRFGIEQMNPAEYLESPYYWHWIRTYIHHGVRTGKIDLEELER 93
Rho. rhodochrous1 AFNLD-QFRGAMEQIPPHDYLTSQYYEHWMHAMIHHGIEAGIFDSDELDR 93
Therm. Bac. Sm.1 MKAFD-EFRIGIEKMRPVDYLTSSYYGHWIATVAYNLLETGVLDEKELED 97
Rho. sp. R3122 -------------RMEPRHYMMTPYYERYVIGVATLMVEKGILTQDELES 37
Comamonas testosterone2 NFNLD-EFRHGIERMNPIDYLKGTYYEHWIHSIETLLVEKGVLTATELAT 93
Bradyrhizobium japonicum2 AFNID-TSRFYRETLPPDVYLSSSYYKKWFLGLEEMLIEKGYLTREEVAA 93
Pseudomonas chlororaphis2 KFSVD-EVRHAVERLDVRQHVGTQYYERYIIATATLLVETGVITQAELDQ 98
P. putida3 G-KAASGKTATP-------VLTPAIVDGLLSTGASAAREEGARARFAVGD 135
P. thermophila1 RTQYYRENPDAPLPEHEQKPELIEFVNQAVYGGLPASREVDRPPKFKEGD 143
Rho. rhodochrous1 RTQYYMDHPDDTTPTR-QDPQLVETISQLITHGADYRRPTDTEAAFAVGD 142
Therm. Bac. Sm.1 RTQAFMEKPDTKIQRW-ENPKLVKVVEKALLEGLSPVREVSSFPRFEVGE 146 Rho. sp. R3122 --------------------LAGGPFPLSRPSESEGRPAPVETTTFEVGQ 67
Comamonas testosterone2 G-KAS-GKTATP-------VLTPAIVDGLLSTGASAAREEGARARFAVGD 134
Bradyrhizobium japonicum2 GHAIQPAKALKHGK------FDLANVERVMVRGK-FARPAPAPAKFNIGD 136
Pseudomonas chlororaphis2 --------------------ALGSHFKLANPAHATGRPAITGRPPFEVGD 128
P. putida3 KVR-----VLNKNPVGHTRMPRYTRGKVG-TVVIDHGVFVTPDTAAHGKG 179
P. thermophila1 -VVRFS----TASPKGHARRARYVRGKTG-TVVKHHGAYIYPDTAGNGLG 187
Rho. rhodochrous1 KVIVRS----DASPNTHTRRAGYVRGRVG-EVVATHGAYVFPDTNALGAG 187
Therm. Bac. Sm.1 RIK-----TRNIHPTGHTRFPRYVRDKYG-VIEEVYGAHVFPDDAAHRKG 190
Rho. sp. R3122 RVR-----VRDEYVPGHIRMPAYCRGRVGTISHRTTEKWPFPDAIGHGRN 112
Comamonas testosterone2 KVR-----VLNKNPVGHTRMPRYTRGKVG-TVVIDHGVFVTPDTAAHGKG 178
Bradyrhizobium japonicum2 RVR-----AKNIHPATHTRLPRYVRGHVG-VVELNHGCHVFPDSAAMELG 180
Pseudomonas chlororaphis2 RVV-----VRDEYVAGHIRMPAYVRGKEGVVLHRTSEQWPFPDAIGHGDL 173
P. putida3 EH-PQHVYTVSFTSVELWGQDASSPKDTIRVDLWDDYLEPA-------- 219
P. thermophila1 EC-PEHLYTVRFTAQELWG-PEGDPNSSVYYDCWEPYIELVDT------ 228
Rho. rhodochrous1 ES-PEHLYTVRFSATELWG-EPAAPNVVNHIDVFEPYLLPA-------- 226
Therm. Bac. Sm.1 EN-PQYLYRVRFDAEELWG---VKQNDSVYIDLWEGYLEPVSH------ 229
Rho. sp. R3122 DAGEEPTYHVKFAAEELFG--SDTDGGSVVVDLFEGYLEPAA------- 152
Comamonas testosterone2 EH-PQHVYTVSFTSVELWGQDASSPKDTIRVDLWDDYLEPA-------- 218
Bradyrhizobium japonicum2 EN-PQWLYTVVFEGSDLWG-ADGDPTSKVSIDAFEPYLDLA-------- 219
Pseudomonas chlororaphis2 SAAHQPTYHVEFRVKDLWG--DAADDGYVVVDLFESYLDKAPGAQAVNA 220
Figure 4-2: Sequence alignment of four Co-type Nitrile Hydratases (NHase) and four Fe-type NHases. Known functional residues are highlighted in yellow. Residues chosen for second- and third-shell mutations are highlighted in red. 1 refers to Co-type nitrile hydratases; 2 refers to Fe-type nitrile hydratases; 3 refers to the Co-type nitrile hydratase from Pseudomonas putida determined in this thesis from x-ray crystallography.
188
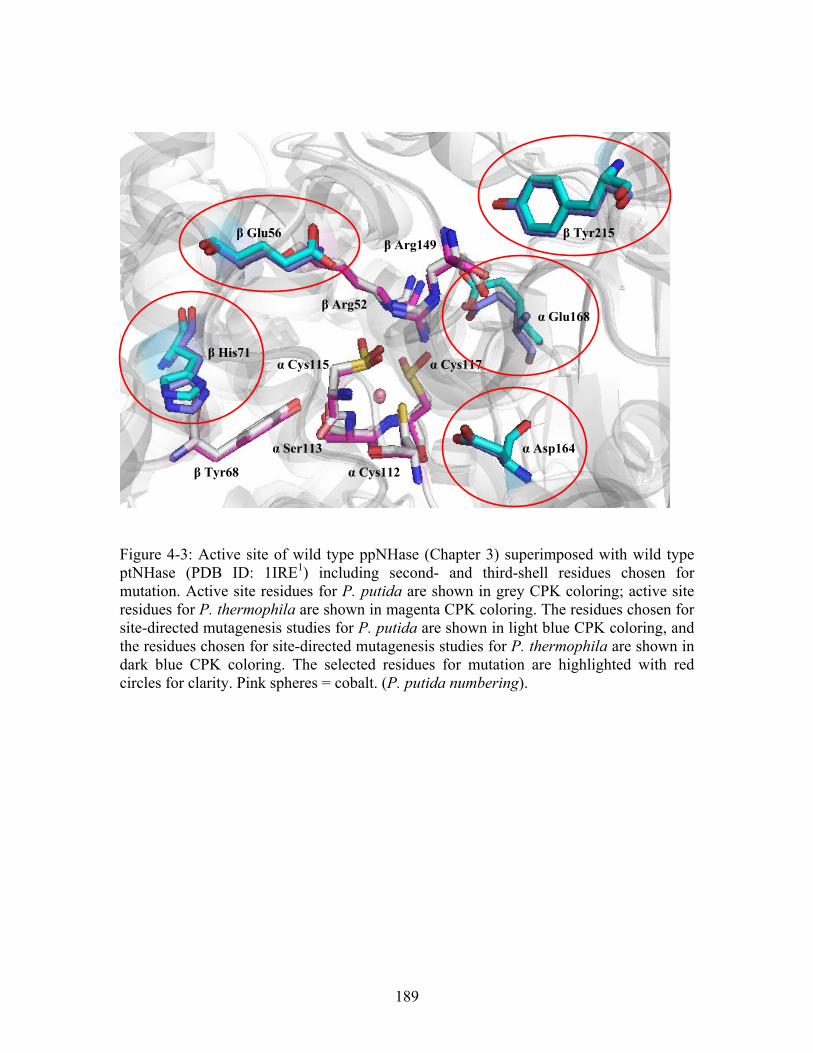
α Cys117
β Arg149
α Glu168
β Tyr215
α Cys115
β Arg52
β Glu56
β His71
α Asp164 α Ser113
β Tyr68 α Cys112
Figure 4-3: Active site of wild type ppNHase (Chapter 3) superimposed with wild type ptNHase (PDB ID: 1IRE1) including second- and third-shell residues chosen for mutation. Active site residues for P. putida are shown in grey CPK coloring; active site residues for P. thermophila are shown in magenta CPK coloring. The residues chosen for site-directed mutagenesis studies for P. putida are shown in light blue CPK coloring, and the residues chosen for site-directed mutagenesis studies for P. thermophila are shown in dark blue CPK coloring. The selected residues for mutation are highlighted with red circles for clarity. Pink spheres = cobalt. (P. putida numbering).
189
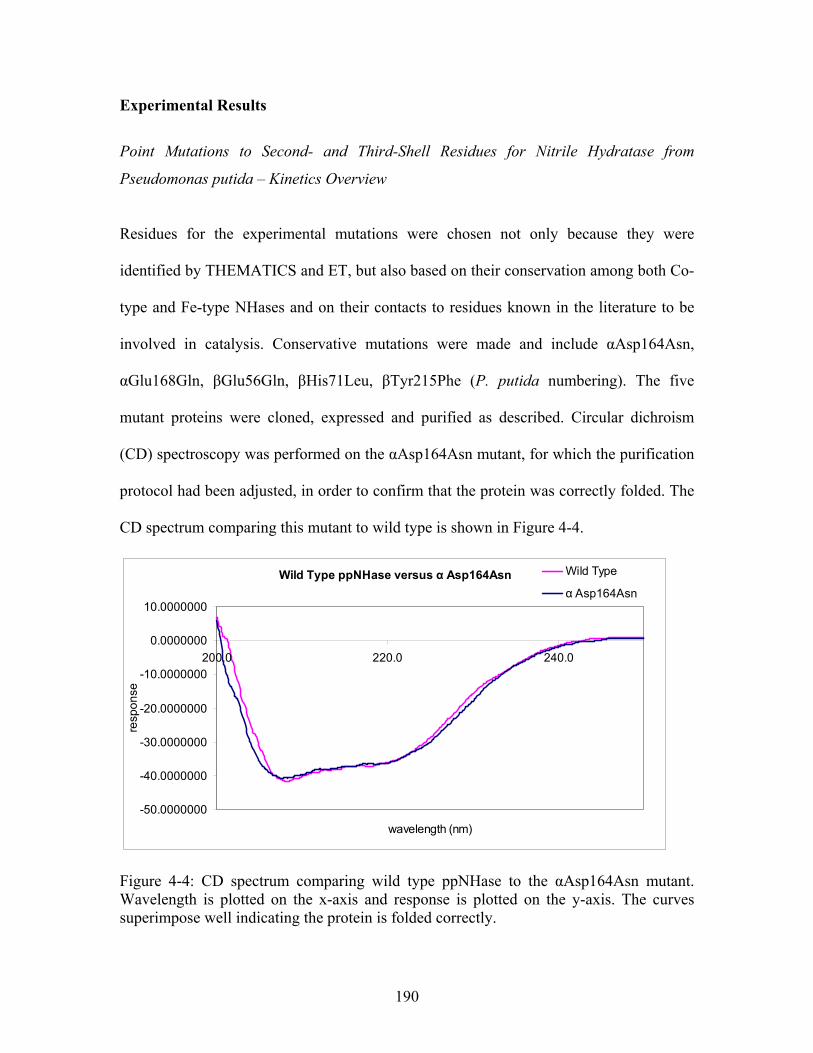
Experimental Results
Point Mutations to Second- and Third-Shell Residues for Nitrile Hydratase from
Pseudomonas putida – Kinetics Overview
Residues for the experimental mutations were chosen not only because they were
identified by THEMATICS and ET, but also based on their conservation among both Co-
type and Fe-type NHases and on their contacts to residues known in the literature to be
involved in catalysis. Conservative mutations were made and include αAsp164Asn,
αGlu168Gln, βGlu56Gln, βHis71Leu, βTyr215Phe (P. putida numbering). The five
mutant proteins were cloned, expressed and purified as described. Circular dichroism
(CD) spectroscopy was performed on the αAsp164Asn mutant, for which the purification
protocol had been adjusted, in order to confirm that the protein was correctly folded. The
CD spectrum comparing this mutant to wild type is shown in Figure 4-4.
Wild Type ppNHase versus α Asp164Asn
-50.0000000
-40.0000000
-30.0000000
-20.0000000
-10.0000000
0.0000000
10.0000000
200.0 220.0 240.0
wavelength (nm)
resp
on
se
Wild Type
α Asp164Asn
Figure 4-4: CD spectrum comparing wild type ppNHase to the αAsp164Asn mutant. Wavelength is plotted on the x-axis and response is plotted on the y-axis. The curves superimpose well indicating the protein is folded correctly.
190
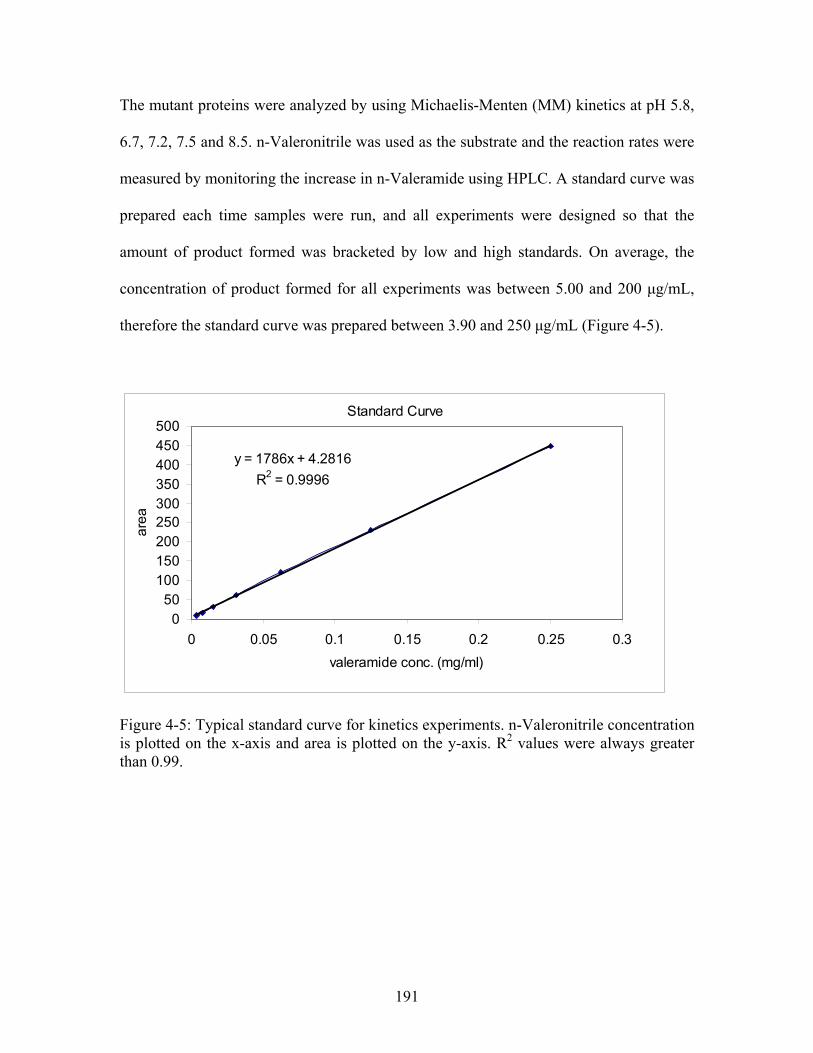
The mutant proteins were analyzed by using Michaelis-Menten (MM) kinetics at pH 5.8,
6.7, 7.2, 7.5 and 8.5. n-Valeronitrile was used as the substrate and the reaction rates were
measured by monitoring the increase in n-Valeramide using HPLC. A standard curve was
prepared each time samples were run, and all experiments were designed so that the
amount of product formed was bracketed by low and high standards. On average, the
concentration of product formed for all experiments was between 5.00 and 200 μg/mL,
therefore the standard curve was prepared between 3.90 and 250 μg/mL (Figure 4-5).
Standard Curve
y = 1786x + 4.2816
R2 = 0.9996
050
100150200250300350400450500
0 0.05 0.1 0.15 0.2 0.25 0.3
valeramide conc. (mg/ml)
are
a
Figure 4-5: Typical standard curve for kinetics experiments. n-Valeronitrile concentration is plotted on the x-axis and area is plotted on the y-axis. R2 values were always greater than 0.99.
191
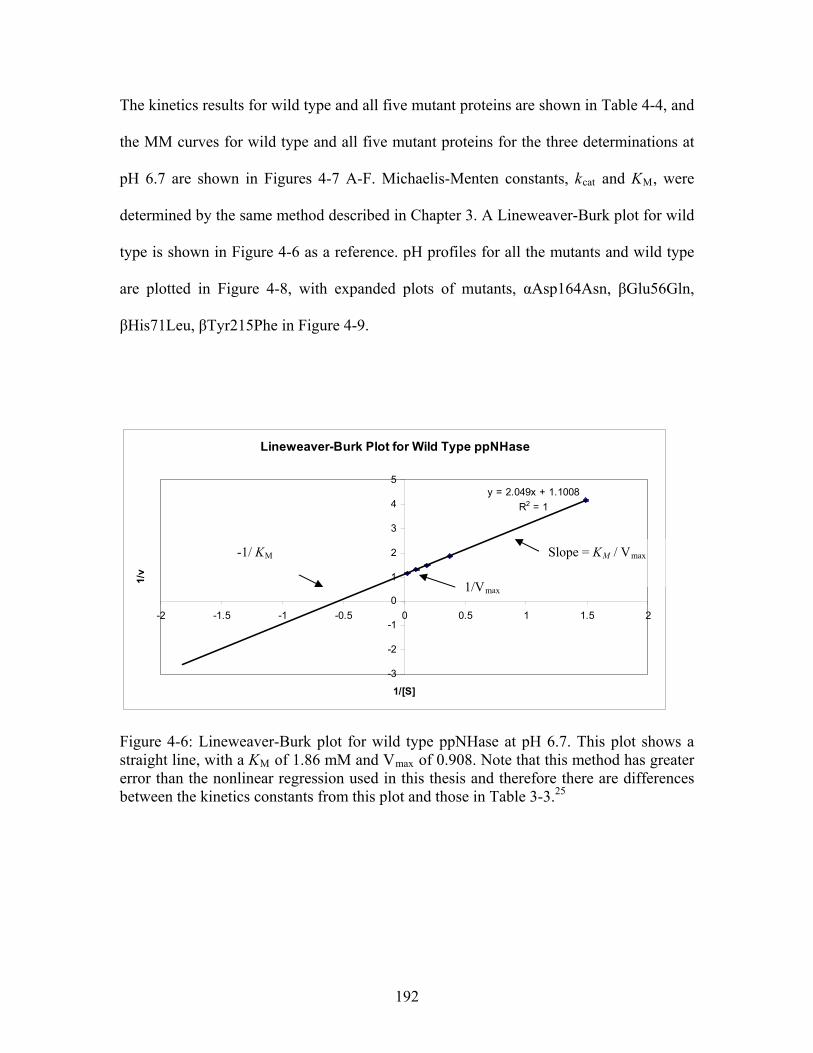
The kinetics results for wild type and all five mutant proteins are shown in Table 4-4, and
the MM curves for wild type and all five mutant proteins for the three determinations at
pH 6.7 are shown in Figures 4-7 A-F. Michaelis-Menten constants, kcat and KM, were
determined by the same method described in Chapter 3. A Lineweaver-Burk plot for wild
type is shown in Figure 4-6 as a reference. pH profiles for all the mutants and wild type
are plotted in Figure 4-8, with expanded plots of mutants, αAsp164Asn, βGlu56Gln,
βHis71Leu, βTyr215Phe in Figure 4-9.
Lineweaver-Burk Plot for Wild Type ppNHase
y = 2.049x + 1.1008
R2 = 1
-3
-2
-1
0
1
2
3
4
5
-2 -1.5 -1 -0.5 0 0.5 1 1.5 2
1/[S]
1/v
1/Vmax
Slope = KM / Vmax -1/ KM
Figure 4-6: Lineweaver-Burk plot for wild type ppNHase at pH 6.7. This plot shows a straight line, with a KM of 1.86 mM and Vmax of 0.908. Note that this method has greater error than the nonlinear regression used in this thesis and therefore there are differences between the kinetics constants from this plot and those in Table 3-3.25
192

Table 4-4: Kinetics results for the conversion of n-Valeronitrile to n-Valeramide for wild type NHase from Pseudomonas putida and five NHase mutants from Pseudomonas putida at pH 5.8, 6.7, 7.2, 7.5 and 8.5. pH 6.7 represents an n = 3 and therefore standard deviations are included. All other pH values represent an n = 2, and therefore no standard deviations are included.
pH
5.8
Wil
d T
ypeα
Asp
164A
snα
Glu
168G
ln β
Glu
56G
lnβ
His
71L
euβ
Tyr
215P
he
kca
t (m
in -1
)2.
00.
251.
60.
170.
620.
12
KM
(m
M)
1317
2312
3527
kca
t (m
in -1
) W
T /
kca
t (m
in -1
) m
utan
t1.
07.
71.
211
3.1
17p
H 6
.7W
ild
Typ
eα
Asp
164A
snα
Glu
168G
ln β
Glu
56G
lnβ
His
71L
euβ
Tyr
215P
he
kca
t (m
in -1
)20
0.27
3.7
0.20
1.2
2.4
std
dev
kca
t (m
in -1
)0.
350.
017
0.25
0.02
80.
120.
22
KM
(m
M)
6.6
1.8
4.5
1510
2.7
std
dev
KM
(m
M)
0.91
0.56
1.3
3.9
1.9
0.87
kca
t (m
in -1
) W
T /
kca
t (m
in -1
) m
utan
t1.
072
5.4
9616
8.1
pH
7.2
Wil
d T
ypeα
Asp
164A
snα
Glu
168G
ln β
Glu
56G
lnβ
His
71L
euβ
Tyr
215P
he
kca
t (m
in -1
)20
0.38
6.4
0.29
1.5
1.7
KM
(m
M)
112.
210
8.6
137.
1
kca
t (m
in -1
) W
T /
kca
t (m
in -1
) m
utan
t1.
052
3.1
7013
12p
H 7
.5W
ild
Typ
eα
Asp
164A
snα
Glu
168G
ln β
Glu
56G
lnβ
His
71L
euβ
Tyr
215P
he
kca
t (m
in -1
)13
0.18
6.5
0.28
2.2
1.3
KM
(m
M)
7.8
2.0
1013
158.
7
kca
t (m
in -1
) W
T /
kca
t (m
in -1
) m
utan
t1.
072
2.0
466.
19.
8p
H 8
.5W
ild
Typ
eα
Asp
164A
snα
Glu
168G
ln β
Glu
56G
lnβ
His
71L
euβ
Tyr
215P
he
kca
t (m
in -1
)13
0.01
87.
00.
162.
11.
2
KM
(m
M)
9.0
4.8
7.8
1415
7.0
kca
t (m
in -1
) W
T /
kca
t (m
in -1
) m
utan
t1.
07.
0E+
021.
878
6.0
11
193
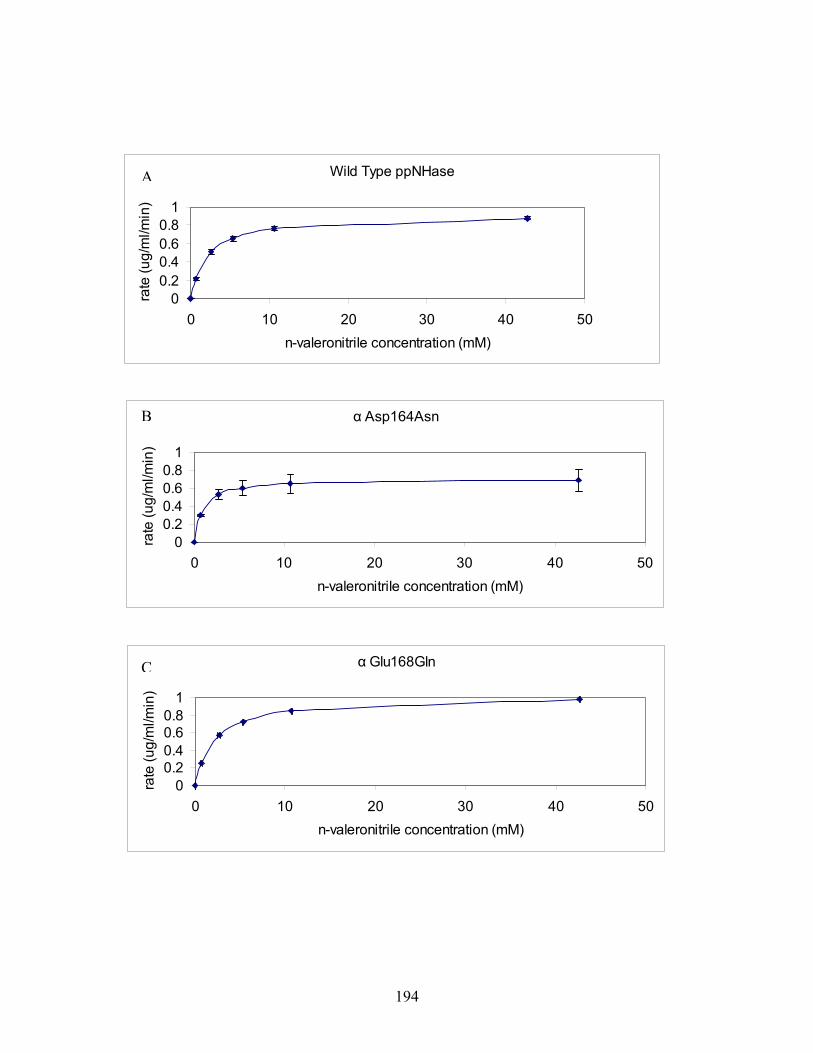
Wild Type ppNHase
00.20.40.60.8
1
0 10 20 30 40 50
n-valeronitrile concentration (mM)
rate
(u
g/m
l/min
)
α Asp164Asn
00.20.40.60.8
1
0 10 20 30 40 50
n-valeronitrile concentration (mM)
rate
(u
g/m
l/min
)
α Glu168Gln
00.20.40.60.8
1
0 10 20 30 40 50
n-valeronitrile concentration (mM)
rate
(u
g/m
l/min
)
C
B
A
194
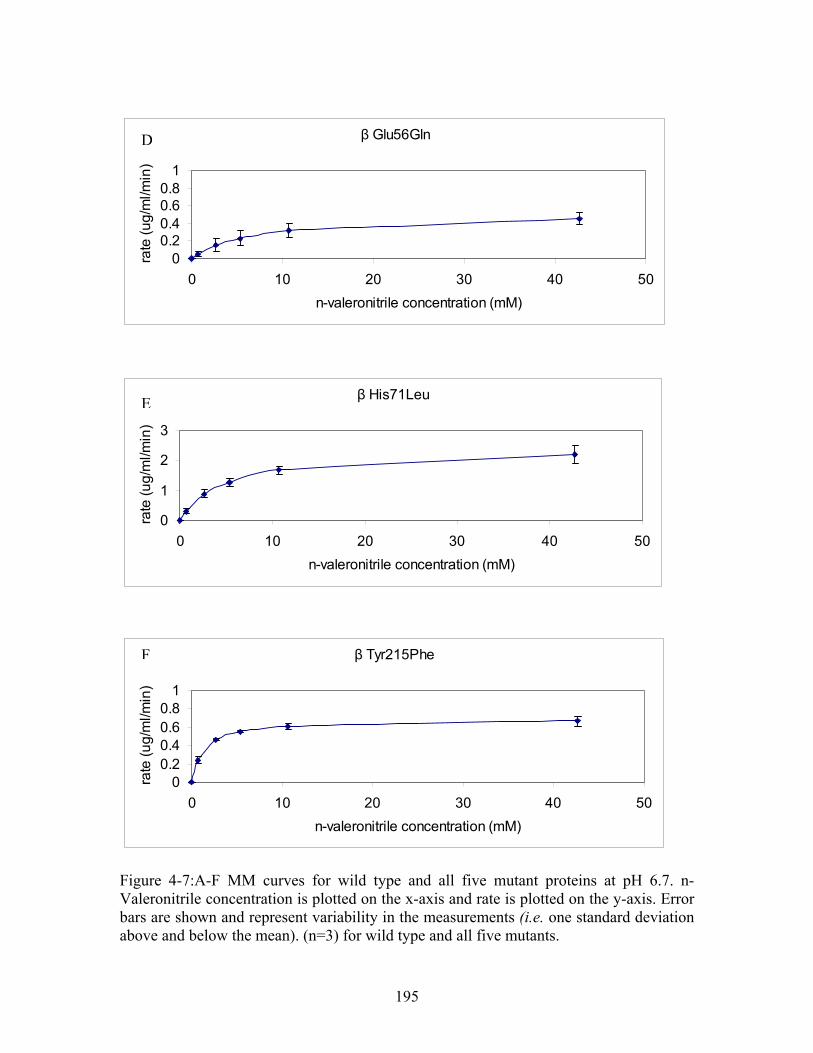
β Glu56Gln
00.20.40.60.8
1
0 10 20 30 40 50
n-valeronitrile concentration (mM)
rate
(u
g/m
l/min
)
β His71Leu
0
1
2
3
0 10 20 30 40 50
n-valeronitrile concentration (mM)
rate
(u
g/m
l/min
)
β Tyr215Phe
00.20.40.60.8
1
0 10 20 30 40 50
n-valeronitrile concentration (mM)
rate
(u
g/m
l/min
)
E
F
D
Figure 4-7:A-F MM curves for wild type and all five mutant proteins at pH 6.7. n-Valeronitrile concentration is plotted on the x-axis and rate is plotted on the y-axis. Error bars are shown and represent variability in the measurements (i.e. one standard deviation above and below the mean). (n=3) for wild type and all five mutants.
195
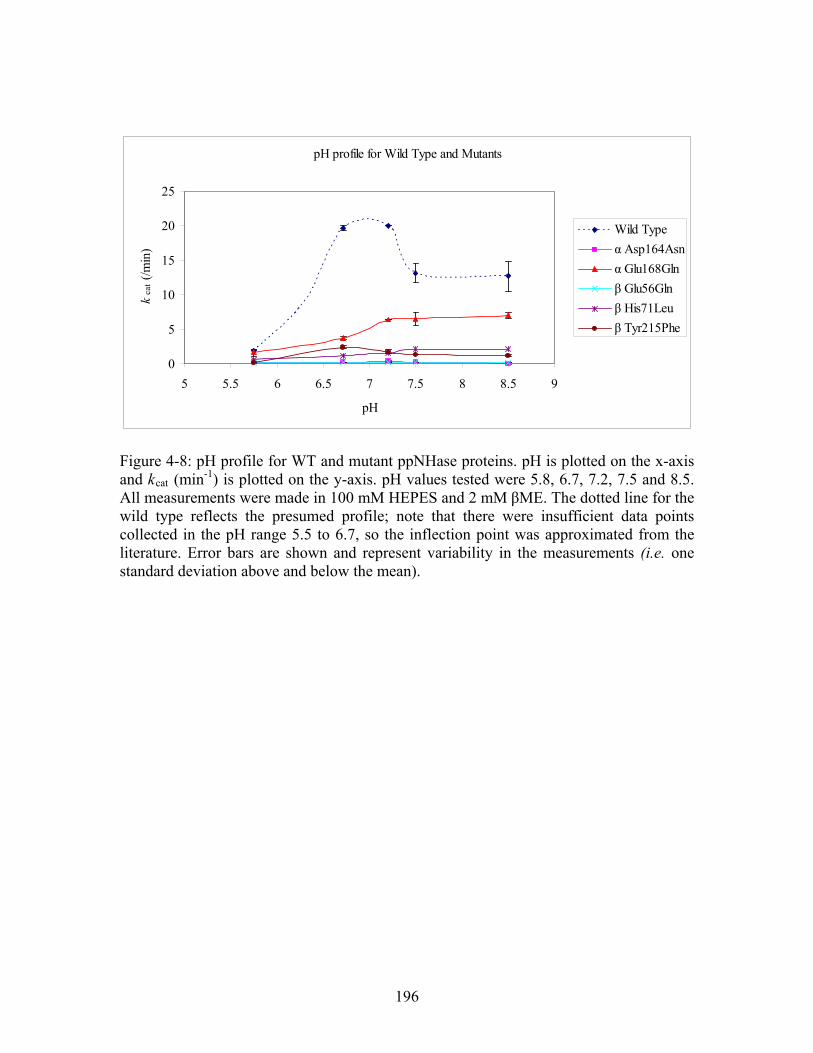
pH profile for Wild Type and Mutants
0
5
10
15
20
25
5 5.5 6 6.5 7 7.5 8 8.5 9
pH
kca
t (/m
in)
Wild Type
α Asp164Asn
α Glu168Gln
β Glu56Gln
β His71Leu
β Tyr215Phe
Figure 4-8: pH profile for WT and mutant ppNHase proteins. pH is plotted on the x-axis and kcat (min-1) is plotted on the y-axis. pH values tested were 5.8, 6.7, 7.2, 7.5 and 8.5. All measurements were made in 100 mM HEPES and 2 mM βME. The dotted line for the wild type reflects the presumed profile; note that there were insufficient data points collected in the pH range 5.5 to 6.7, so the inflection point was approximated from the literature. Error bars are shown and represent variability in the measurements (i.e. one standard deviation above and below the mean).
196
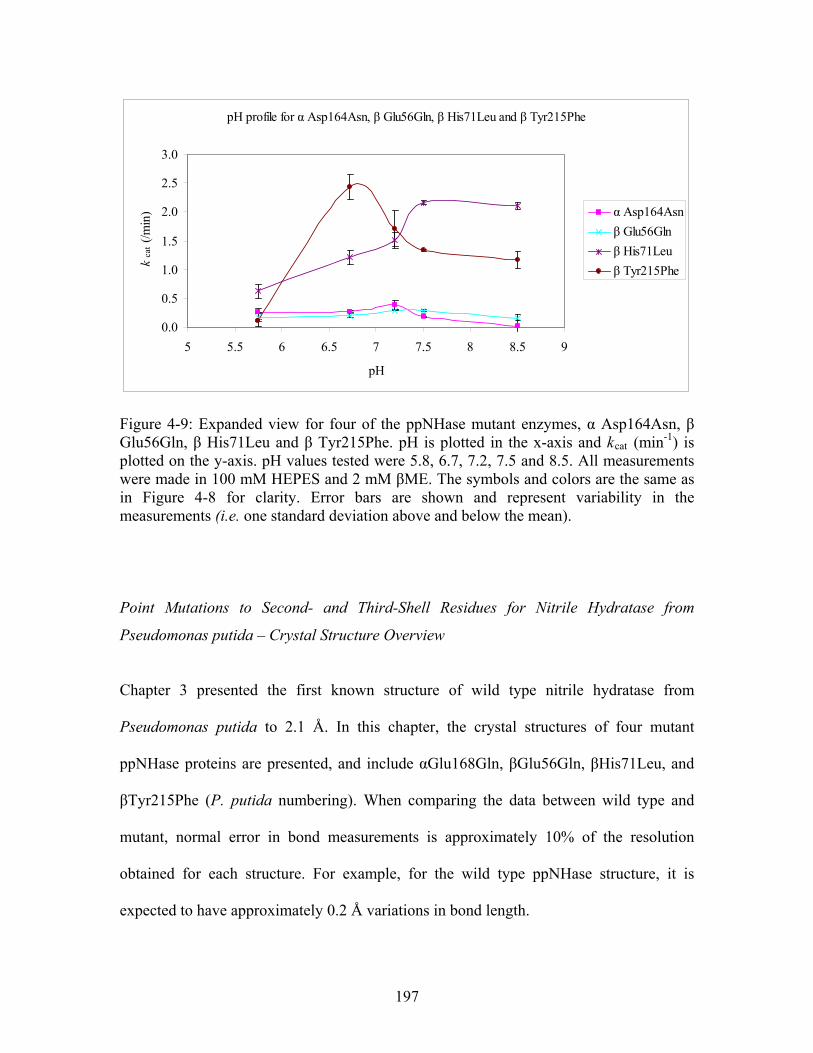
pH profile for α Asp164Asn, β Glu56Gln, β His71Leu and β Tyr215Phe
0.0
0.5
1.0
1.5
2.0
2.5
3.0
5 5.5 6 6.5 7 7.5 8 8.5 9
pH
kca
t (/m
in) α Asp164Asn
β Glu56Gln
β His71Leu
β Tyr215Phe
Figure 4-9: Expanded view for four of the ppNHase mutant enzymes, α Asp164Asn, β Glu56Gln, β His71Leu and β Tyr215Phe. pH is plotted in the x-axis and kcat (min-1) is plotted on the y-axis. pH values tested were 5.8, 6.7, 7.2, 7.5 and 8.5. All measurements were made in 100 mM HEPES and 2 mM βME. The symbols and colors are the same as in Figure 4-8 for clarity. Error bars are shown and represent variability in the measurements (i.e. one standard deviation above and below the mean).
Point Mutations to Second- and Third-Shell Residues for Nitrile Hydratase from
Pseudomonas putida – Crystal Structure Overview
Chapter 3 presented the first known structure of wild type nitrile hydratase from
Pseudomonas putida to 2.1 Å. In this chapter, the crystal structures of four mutant
ppNHase proteins are presented, and include αGlu168Gln, βGlu56Gln, βHis71Leu, and
βTyr215Phe (P. putida numbering). When comparing the data between wild type and
mutant, normal error in bond measurements is approximately 10% of the resolution
obtained for each structure. For example, for the wild type ppNHase structure, it is
expected to have approximately 0.2 Å variations in bond length.
197

Crystal screening was described in Chapter 3. The mutant proteins formed crystals under
the same conditions as wild type. Initial crystals of the β Glu56Gln mutant were
extremely small; therefore, initial crystal forms were used to streak seed drops producing
larger crystals for data collection. Diffracting crystal needles were obtained using 20
mg/mL ppNHase and a reservoir containing 22% polyacrylic acid in HEPES pH 7.5 with
20 mM magnesium chloride and 4% acetone. Single crystals (needles) were dissected
from clusters and transferred to a solution containing 17.6% polyacrylic acid 5100 and
20% glycerol in HEPES pH 7.5 and were flash-frozen in liquid nitrogen. Molecular
replacement was carried out with Phaser24 using wild type nitrile hydratase from
Pseudomonas putida as a starting model (Chapter 3). Several rounds of refinement and
model building were performed using REFMAC2 and COOT.3 Final rounds of
refinement, including simulated annealing and water picking were performed using
PHENIX.4 The data collection and refinement statistics for all four mutants are shown in
Table 4-5; wild type statistics are shown for comparison. It should be noted that all
mutant structures resemble wild type protein (Chapter 3).
198
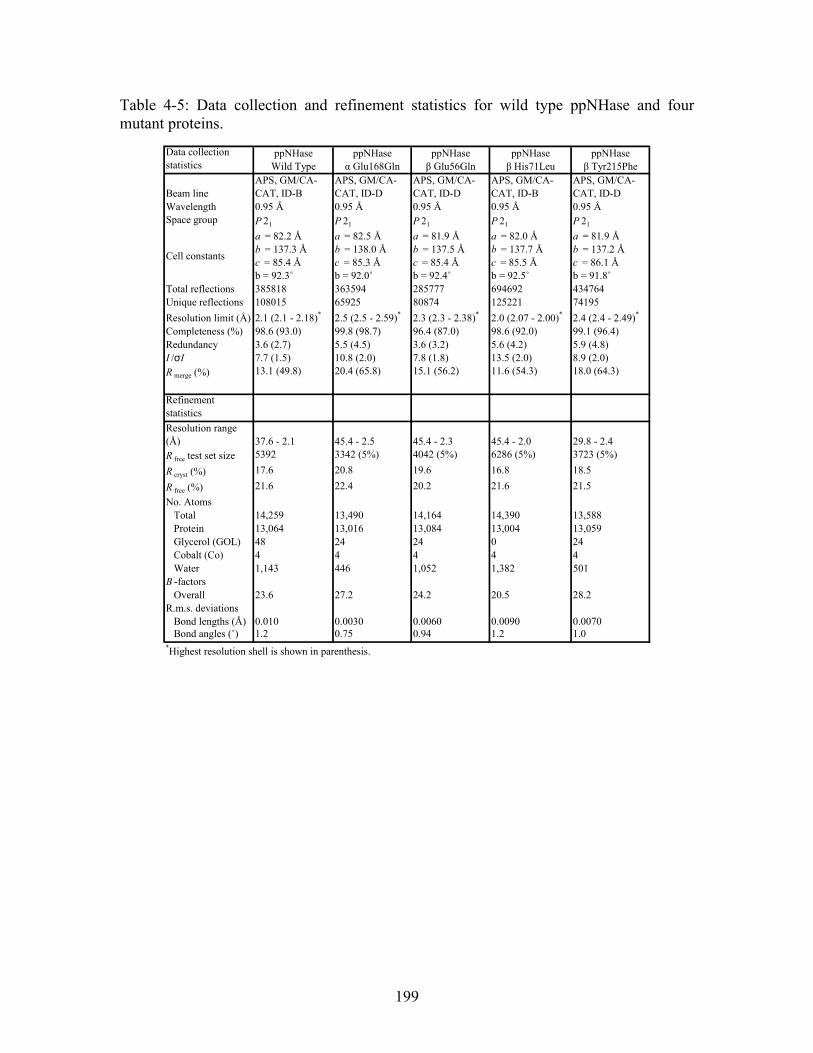
Table 4-5: Data collection and refinement statistics for wild type ppNHase and four mutant proteins.
Data collection statistics
ppNHase Wild Type
ppNHase α Glu168Gln
ppNHase β Glu56Gln
ppNHase β His71Leu
ppNHase β Tyr215Phe
Beam lineAPS, GM/CA-CAT, ID-B
APS, GM/CA-CAT, ID-D
APS, GM/CA-CAT, ID-D
APS, GM/CA-CAT, ID-B
APS, GM/CA-CAT, ID-D
Wavelength 0.95 Å 0.95 Å 0.95 Å 0.95 Å 0.95 ÅSpace group P 21 P 21 P 21 P 21 P 21
Cell constants
a = 82.2 Å b = 137.3 Å c = 85.4 Å b = 92.3˚
a = 82.5 Å b = 138.0 Å c = 85.3 Å b = 92.0˚
a = 81.9 Å b = 137.5 Å c = 85.4 Å b = 92.4˚
a = 82.0 Å b = 137.7 Å c = 85.5 Å b = 92.5˚
a = 81.9 Å b = 137.2 Å c = 86.1 Å b = 91.8˚
Total reflections 385818 363594 285777 694692 434764Unique reflections 108015 65925 80874 125221 74195
Resolution limit (Å) 2.1 (2.1 - 2.18)* 2.5 (2.5 - 2.59)* 2.3 (2.3 - 2.38)* 2.0 (2.07 - 2.00)* 2.4 (2.4 - 2.49)*
Completeness (%) 98.6 (93.0) 99.8 (98.7) 96.4 (87.0) 98.6 (92.0) 99.1 (96.4)Redundancy 3.6 (2.7) 5.5 (4.5) 3.6 (3.2) 5.6 (4.2) 5.9 (4.8)I /σI 7.7 (1.5) 10.8 (2.0) 7.8 (1.8) 13.5 (2.0) 8.9 (2.0)R merge (%) 13.1 (49.8) 20.4 (65.8) 15.1 (56.2) 11.6 (54.3) 18.0 (64.3)
Refinement statistics
Resolution range (Å) 37.6 - 2.1 45.4 - 2.5 45.4 - 2.3 45.4 - 2.0 29.8 - 2.4R free test set size 5392 3342 (5%) 4042 (5%) 6286 (5%) 3723 (5%)
R cryst (%) 17.6 20.8 19.6 16.8 18.5
R free (%) 21.6 22.4 20.2 21.6 21.5
No. Atoms Total 14,259 13,490 14,164 14,390 13,588 Protein 13,064 13,016 13,084 13,004 13,059 Glycerol (GOL) 48 24 24 0 24 Cobalt (Co) 4 4 4 4 4 Water 1,143 446 1,052 1,382 501B -factors Overall 23.6 27.2 24.2 20.5 28.2R.m.s. deviations Bond lengths (Å) 0.010 0.0030 0.0060 0.0090 0.0070 Bond angles (˚) 1.2 0.75 0.94 1.2 1.0*Highest resolution shell is shown in parenthesis.
199

Point Mutations to Second- and Third-Shell Residues for Nitrile Hydratase from
Pseudomonas putida – Kinetics and Crystal Structure Analysis
4.3.1 αAsp164Asn
kcat and KM for the αAsp164Asn ppNHase Mutant Compared to Wild Type ppNHase
Like wild type ppNHase, the αAsp164Asn mutant is also most active at pH 7.2 with a kcat
of 0.38 min-1 and a KM of 2.2 mM (Table 4-4). This is a 50-fold decrease in kcat and a 5-
fold decrease in KM compared to wild type at pH 7.2. There is an 8-fold decrease in kcat
when the pH is lowered to 5.8 compared to wild type at the same pH, while the KM
remains constant. There is a 70-fold decrease in kcat at pH 6.7 and 7.5, while there is a 5-
fold decrease in KM at both pHs compared to wild type. Finally, there is a 700-fold
decrease in kcat while KM remains essentially the same when the pH is raised to 8.5
compared to wild type.
In addition to comparing kcat and KM to wild type at the various pH values, it is also
important to compare them for the mutant itself. When the pH is lowered to 5.8 and 6.7,
there is a 1.5-fold decrease in kcat compared to the activity of this mutant at pH 7.2 where
it is most active. The KM at pH 5.8 is approximately 16.8 mM, which is an 8-fold increase
compared to pH 7.2, while the KM at pH 6.7 is the same as at pH 7.2. A 2-fold decrease
in kcat is observed when the pH is raised to 7.5 and a 20-fold decrease in kcat is observed
when the pH is raised to 8.5. The KM at both these pH values remains constant at
approximately 2.00 mM. A trend is seen where the protein reaches a maximum kcat at pH
7.2 and decreases at pH values below and above pH 7.2 (Figures 4-8 and 4-9).
200

No structure was obtained for the αAsp164Asn mutant. It was discovered that the
αAsp164Asn mutant protein was slightly destabilized by the mutation, and tended to
dissociate into monomers when subjected to high salt as evidenced by the disappearance
of one protein band in the SDS PAGE gel. It has been hypothesized that this mutation has
made crystallization difficult by slightly destabilizing the protein. No crystals formed
from initial screening, but we were able to obtain extremely small needles by seeding the
protein with other ppNHase crystals. We hope to obtain the structure of this mutant soon
by altering the crystallization conditions slightly to obtain larger crystals.
4.3.2 αGlu168Gln
kcat and KM for the αGlu168Gln ppNHase Mutant Compared to Wild Type ppNHase
The αGlu168Gln mutant is most active at pH 7.2 with a kcat of 6.4 min-1 and a KM of 9.8
mM and has the smallest effect on kcat compared to wild type at pH 7.2 (Table 4-4). This
translates to a 3-fold decrease in kcat with no change in KM compared to wild type. When
the pH is lowered to 5.8, no change in kcat is observed, but there is a 2-fold increase in
KM compared to wild type. A 5-fold decrease in kcat is seen at pH 6.7, while there is no
change in KM. Finally, when the pH is raised to both pH 7.5 and pH 8.5, there is a 2-fold
decrease in kcat compared to wild type, while again, there is no change in KM observed
compared to wild type within the error of the assay.
The pH profile for this mutant follows a different trend than that observed for wild type;
the kcat appears to plateau at pH 7.2 and does not decrease at higher pH values (Figure 4-
8). There is a 6-fold decrease in kcat when the pH is lowered to 5.8 and a 2-fold increase
201

in KM compared to the Michaelis-Menten constants at pH 7.2. When the pH is lowered to
6.7, there is a 2-fold decrease in kcat and a 2-fold decrease in KM. Again, the kcat reaches
its maximum value at pH 7.2 (approximately 6.4 min-1) and does not decrease with
increasing pH. Additionally, the KM remains constant from pH 7.2 to pH 8.5.
Crystal Structure of the αGlu168Gln ppNHase Mutant Compared to Wild Type ppNHase
The third–shell αGlu168Gln mutation results in a 3-fold decrease in kcat with no effect on
KM at pH 7.2, where wild type ppNHase is most active. A schematic diagram comparing
the active site of wild type versus the αGlu168Gln mutant is shown in Figure 4-10 A-B.
In the wild type structure, shown in Figure 4-10 A, the side chain of αGlu168 makes a
salt bridge with the annotated catalytic residue βArg52. This arginine residue in turn
interacts with the two modified cysteines. In order to test the hypothesis that remote
residues are important, we wanted to make the smallest structural change possible. The
most conservative change was to mutate the glutamate to glutamine to remove the charge.
In the mutated structure, shown in Figure 4-10 B, the side chain of αGln168 flips away
from βArg52 and breaks the salt bridge with βArg52, and forms a hydrogen bond to the
backbone oxygen atom of βVal169. Additionally, the H-bond distance between βArg52
and αCys117 has increased from 2.7 Å to 3.1 Å, while the H-bond between βArg52 and
αCys115 has essentially remained the same. The 0.4 Å difference in bond length between
wild type and the αGlu168Gln mutant is statistically significant as it is larger than 10% of
the resolution obtained for the two structures (approximately 0.2 Å). It has been shown
that βArg52 is a functionally important residue in Fe-type NHases,26 so it assumed the
same function applies to Co-type NHases as well since this arginine is completely
202

conserved among all known NHases. This arginine is known to H-bond with αCys117
and αCys112 in the active site, and seems to stabilize the known claw setting.1 The
removal of the salt bridge to βArg52 appears to destabilize the active site slightly to cause
a small decrease in kcat.
Figure 4-10: (A) Active site of wild type ppNHase. (B) Active site of αGlu168Gln ppNHase. In the mutant structure, residue 168 has flipped out of salt bridge distance of βArg52 and forms an H-bond with the backbone oxygen atom of βVal169. (purple sphere = cobalt).
4.3.3 βGlu56Gln
kcat and KM for the βGlu56Gln ppNHase Mutant Compared to Wild Type ppNHase
Like wild type ppNHase and other mutants, the βGlu56Gln mutant is most active at pH
7.2 with a kcat of 0.29 min-1 which is a 70-fold decrease compared to wild type at the
same pH, and a KM comparable to wild type (Table 4-4). There is an 11-fold decrease in
kcat at pH 5.8, a 100-fold decrease at pH 6.7, a 50-fold decrease at pH 7.5 and an 80-fold
A B
αCys115
αSer116
αGlu168
3.2Å βArg52
αCys117
αCys112
3.4Å
βVal169
2.7Å 2.5Å
3.5Å
αSer116 αCys112
αCys115
α Cys117
βArg52 βVal169
2.7Å
αGln168
3.2Å
3.1Å 3.2Å
203

decrease at pH 8.5 compared to wild type at the different pHs. The KM at all pH values is
again comparable to that observed with wild type at the various pH values.
The pH profile for this mutant is different than what has been observed for the previous
two mutants, αAsp164Asn and αGlu168Gln (Figures 4-8 and 4-9). The kcat reaches a
maximum value around pH 7.2 and decreases as the pH is lowered and raised. There is a
2-fold decrease in kcat when the pH is lowered to pH 5.8, a 1.5-fold decrease at pH 6.7, no
change in kcat at pH 7.5 and a 2-fold decrease in kcat at pH 8.5 compared to the kcat of the
mutant at pH 7.2. The KM for the βGlu56Gln mutant is essentially the same at all five pH
values (approximately 10 mM).
Crystal Structure of the βGlu56Gln ppNHase Mutant Compared to Wild Type ppNHase
The βGlu56Gln mutation had the largest decrease in kcat compared to wild type (- 70-
fold) at pH 7.2, while there was no change in the KM (Table 4-4). βGlu56 is a second-
shell residue which forms a hydrogen bond (H-bond) to both the modified cysteine,
αCys115, and to the functionally important arginine, βArg149, through a water molecule.
βArg149 H-bonds to αCys115, so any disruption in the H-bond distances would
destabilize the active site, explaining the decrease in kcat. As with the αGlu168Gln
mutant, the conservative mutation of glutamate to glutamine was made, removing just the
negative charge. Interestingly, however, the structures of both wild type and mutant
protein are the same, as are the H-bond distances (Figure 4-11 A-B). While even the
smallest change in structure was not detected, this mutant resulted in the largest decrease
204

in activity. It was hypothesized that the difference in kcat was due to electrostatic effects,
but further studies must be performed to support this.
B
βArg149
αGln56
αCys115
αSer116
αCys112
αCys117
2.6Å
3.4Å
3.1Å
2.8Å 2.7Å
A
αCys115
αCys112
βArg149
3.4Å
αGlu56
2.8Å αCys117
3.2Å 2.7Å
2.7Å
αSer116
Figure 4-11: (A) Active site of wild type ppNHase. (B) Active site of βGlu56Gln ppNHase. Wild type and mutant structures are essentially the same. (purple sphere = cobalt, red sphere = water).
4.3.4 βHis71Leu
kcat and KM for the βHis71Leu ppNHase Mutant Compared to Wild Type ppNHase
The βHis71Leu mutant is most active at pH 7.5 with a kcat of 2.2 min-1 and a KM of
approximately 15 mM (Table 4-4). This translates into a 6-fold less active protein
compared to wild type at the same pH, while the KM is 2-fold higher (7.79 min-1 vs. 14.7
min-1). When the pH is lowered to 5.8, there is a 3-fold decrease in kcat and an
approximately 3-fold increase in KM compared to wild type at the same pH. There is a
16-fold decrease in kcat when the pH is lowered to 6.7, while the KM remains essentially
constant. At pH 7.2, there is a 13-fold decrease in kcat compared to wild type at the same
205

pH with no change in the KM. Finally, when the pH is raised to 8.5, the resultant mutant
protein has a 6-fold decrease in kcat while the KM again remains constant within the error
of the assay.
The pH profile for this mutant is similar to what is observed for the αGlu168Gln mutant
in that the kcat of the mutant enzyme reaches a maximum and does not decrease when the
pH is raised; the difference being that the maximum rate for this mutant is reached at pH
7.5 and pH 7.2 for the αGlu168Gln mutant (Figures 4-8 and 4-9). The activity for this
mutant decreases almost 4-fold while the KM increases by a factor of 2 when the pH is
lowered to pH 5.8 compared to the values for the same mutant at pH 7.5. When the pH is
adjusted to both 6.7 and 7.2, the kcat decreases by a factor of 1.8 and 1.5, respectively,
while the KM remains constant. Finally, when the pH is raised to 8.5, there is no change
observed in either kcat or KM compared to the values obtained for this mutant at pH 7.5.
Crystal Structure of the βHis71Leu ppNHase Mutant Compared to Wild Type ppNHase
The third-shell βHis71Leu mutation reduced kcat 14-fold compared to wild type while the
KM was unaffected by the mutation at pH 7.2. Figure 4-12 shows the active sites of both
wild type (Figure 4-12 A) and mutant protein (Figure 4-12 B). The side chain of βHis71
is H-bonded to the main chain oxygen of αCys115 and the side chain of αSer116 through
two waters. Additionally, there is an H-bond between the side chain of βHis71 and the
side chain oxygen of αCys115 through three waters. The mutation of βHis71 to leucine
removes these H-bonding capabilities. The only other difference between the wild type
206

and mutant protein in the active site is a slight shift in a water molecule, w1 (Figure 4-
12), of approximately 0.4 Å . While this shift is significant, this in itself is not sufficient
to explain the decrease in kcat; it is therefore hypothesized that the decrease is due to
electrostatic effects.
A B
βHis71 2.7Å
3.4Å
2.9Å
3.0Å
αCys112 αSer116
αCys115 αCys117
βArg52
2.8Å
2.6Å
3.0Å
W1
βLeu71 2.8Å
3.5Å
2.5Å
2.7Å
αCys112 αSer116
αCys115 αCys117
βArg52
2.7Å
2.7Å
W1
Figure 4-12: (A) Active site of wild type ppNHase. (B) Active site of βHis71Leu ppNHase. Wild type and mutant structures are essentially the same, with a slight movement in one of the waters (w1). (purple sphere = cobalt, red sphere = water).
4.3.5 βTyr215Phe
kcat and KM for the βTyr215Phe ppNHase Mutant Compared to Wild Type ppNHase
The βTyr215Phe mutant is most active at pH 6.7 with a kcat of 2.4 min-1 and a KM of 2.7
mM (Table 4-4). Compared to wild type at the same pH, this translates to an 8-fold
decrease in kcat and a 2-fold decrease in KM. When the pH is lowered to 5.8, there is a 17-
fold decrease in kcat and a 2-fold increase in KM. As the pH is raised beyond 6.7, there is
207

a gradual decrease in kcat compared to wild type while the KM remains constant. There is
a 12-fold decrease in kcat at pH 7.2, a 10-fold decrease at pH 7.5 and an 11-fold decrease
at pH 8.5 compared to wild type at the same pH values.
The pH profile for this mutant is similar to what is seen with the αAsp164Asn mutant and
the βGlu56Gln mutant in that a maximum rate is observed which decreases with an
increase or decrease in pH (Figures 4-8 and 4-9). When the pH is lowered to 5.8, there is
a 21-fold decrease in kcat and a 10-fold increase in KM compared to the values obtained at
pH 7.2 where this mutant is most active. There is a 1.4-fold decrease in kcat and a 2-fold
decrease in KM when the pH is raised to 7.2. The kcat decreases by a factor of 1.8 and the
KM increases by a factor of 3 when the pH is raised to 7.5, and finally, the kcat decreases
2-fold and the KM increases 2-fold when the pH is raised to 8.5.
Crystal Structure of the βTyr215Phe ppNHase Mutant Compared to Wild Type ppNHase
βTyr215 is a third shell residue which is located approximately 14 Å from the cobalt ion.
The βTyr215Phe mutation resulted in 12-fold decrease in kcat with no change in KM at pH
7.2 (Table 4-4). The active sites of both wild type and mutant protein are shown in
Figures 4-13 A-D. In the wild type structure (Figure 4-13 A), the tyrosine residue is H-
bonded to the main chain nitrogen atom of βArg149 and the side chain of βAsp172.
βArg149 is a second-shell residue which H-bonds to the metal coordinating residue
αCys115 and is known to help stabilize the active site. In the mutant structure (Figure 4-
13 B), the H-bonding capabilities to βArg149 and βAsp172 are removed, but there does
not appear to be any change in the structure relative to tyrosine specifically. There are
208

very slight (0.1 – 0.2 Å) shifts in the H-bonds between βArg149 and the modified
cysteines, but they are not significant and can not explain the decrease in kcat for this
mutant. There are however, less obvious changes that have taken place that involve the
salt bridge between the third-shell residue αGlu168 and the first-shell residue βArg52
(Figures 4-13 C and 4-13 D). Specifically, there is a shift in the placement of αGlu168
causing a lengthening of this interaction by 1.0 Å, which is statistically significant. Most
often when tyrosine is mutated to phenylalanine, a water molecule will take the place of
the missing phenolic oxygen atom; this does not occur in this structure and instead there
are rearrangements of the interactions between neighboring residues. The lengthening of
the αGlu168 - βArg52 salt bridge indicates that this interaction has become less
energetically favorable. It is interesting that this change which may cause the decrease in
kcat is due to the shift in a residue which is located 10 Å from the active site and 10 Å
from the site of the mutation.
209
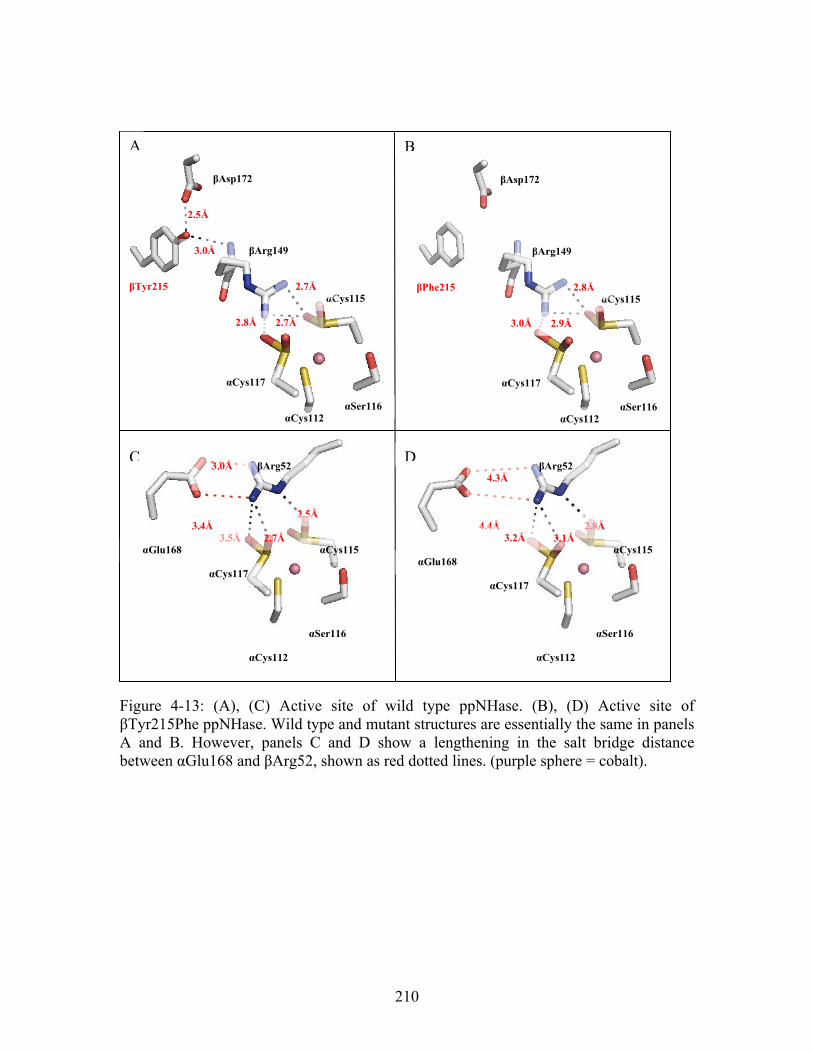
Figure 4-13: (A), (C) Active site of wild type ppNHase. (B), (D) Active site of βTyr215Phe ppNHase. Wild type and mutant structures are essentially the same in panels A and B. However, panels C and D show a lengthening in the salt bridge distance between αGlu168 and βArg52, shown as red dotted lines. (purple sphere = cobalt).
αCys117
βArg149
αCys112
αCys115
αSer116
βTyr215 2.7Å
2.8Å
βAsp172
2.7Å
3.0Å
2.5Å
βPhe215
βAsp172
αCys117
βArg149
αCys112
αCys115
αSer116
2.8Å
3.0Å 2.9Å
A B
C D
3.5Å
αCys117
βArg52
αCys112
αCys115
αSer116
2.5Å 2.7Å
3.4Å
3.0Å
αGlu168
αCys117
βArg52
αCys112
αCys115
αSer116
2.8Å 4.4Å
4.3Å
3.2Å
α
3.1Å
Glu168
210

Mechanistic Analysis
Changes in pH affect the rate (and therefore kcat) of enzyme catalyzed reactions due to the
fact that active sites are most often composed of ionizable groups which must be in the
proper protonation state to maintain the conformation of the active site, bind the
substrate, or catalyze reactions.25 The kcat as a function of pH curves shown in Figure 4-8
all show a maximum activity which declines at higher and lower pH values. This allows
one to make an educated guess about the residues involved in catalysis. These declines in
rate could be the result of the formation of an improper ionic form for a residue in the
active site necessary for catalysis.
The kcat as a function of pH curve for wild type does not appear to have an inflection
point in the acidic side. This however was due to the fact that there are a limited number
of kinetics points at pH values between 5.8 and 6.7; additional points would be needed to
obtain the correct inflection. Kinetics results from the literature do in fact have this
inflection point; therefore a presumed inflection point was added to the pH curves (Figure
4-8).
The kcat as a function of pH curve for wild type and the βTyr215Phe mutant have the
same shape, indicating that the pH-dependent properties of the functionally important
catalytic residues in the active site are unchanged by this mutation. This, however, was
not the case for the rest of the mutants. The other mutations affected the pH-dependent
properties of the catalytic residues in the active site. The αGlu168Gln mutant has lost an
ionizable group and may shift the pH-dependent properties of the catalytic residues,
211

perhaps putting them in an improper charge state for optimum catalysis. This is
evidenced by the loss of the inflection point on the basic side of the pH vs. rate curve.
This is also the case with the βHis71Leu mutant. An inflection point has been lost on the
basic side indicating that there is a residue necessary for catalysis in the active site that is
in an improper ionic state. The opposite appears to true for the αAsp164Asn mutant,
where the inflection point in the pH vs. rate curve has been lost on the acidic side. This
indicates that a residue acting as a base is in an incorrect ionic form for catalysis. Finally,
for the βGlu56Gln mutant, there are essentially no major inflection points on either the
acidic or basic sides of the pH vs. rate curve. This indicates that both the basic and acidic
catalytic residues may be in improper ionic forms. This could be a reason why this
mutant resulted in the largest decrease in kcat.
The information gained from the kcat as a function of pH curves are extremely useful for
drawing mechanistic conclusions about the ionic state of the residues involved in
catalysis. Unfortunately, it is not possible to determine exactly which residues are
affected without further studies.
212

Summary of Structural Effects
It was shown for the αGlu168Gln mutant that the catalytic effects were caused in part by
both structural differences and possibly also by electrostatic effects. The rotation of the
side chain for the glutamine residue in the mutant structure disrupted the salt bridge
interaction between αGln168 to the known catalytic residue, βArg52. The other mutant
whose structure could also explain the catalytic effects was the βTyr215Phe mutant.
While the H-bonding capabilities to the functionally important βArg149 were removed,
there were no obvious structural changes in that area. There was however, a shift in the
third-shell residue, αGlu168, which lengthened the salt bridge to βArg52 by 1.0 Å. This
suggests that the supporting role of βTyr215 in catalysis is due to a combination of
structural and electrostatic effects. The structures for the other mutants did not change,
however, making the decrease in kcat more difficult to explain. Most interesting was the
βGlu56Gln mutant which had the largest effect on kcat compared to wild type, but no
structural change. In the last mutant structure, βHis71Leu, the only structural change
observed was the shift of a water molecule in the active site, disrupting the H-bond
networks. The structures for these two mutants suggest that βGlu56 and βHis71 play a
supporting role in catalysis through electrostatic effects.
213

4.4 Conclusions
The kinetics results at pH 7.2 demonstrated a 70-fold decrease in kcat compared to wild
type for the second-shell mutation βGlu56Gln, a 3-fold decrease for the third-shell
mutant αGlu168Gln, a 13-fold decrease for the third-shell mutant βHis71Leu, a 12-fold
decrease in kcat for the third-shell mutant βTyr215Phe, and a 52-fold decrease for
αAsp164Asn. This suggests that second- and third-shell residues play a supporting role in
catalysis for NHase without drastically affecting the KM of the substrate at all pH values
tested. The point mutations in the second- and third-shell for ppNHase result in enzymes
that were still active, but have a kcat that was reduced by one or two orders of magnitude
and a KM that generally remains constant. Furthermore, while single point mutations led
to a drop in kcat by one or two orders of magnitude, collectively these residues outside the
first shell probably amount to a much larger effect on the kcat. In other words, the
composition of the second- and third-shell may be critical to the enzymatic catalytic rate.
This chapter focused on the kinetic analysis of five second- and third-shell mutant
ppNHase proteins, and the crystal structure analysis of four mutant ppNHase proteins.
Based solely on these results, experimental evidence shows that second- and third-shell
residues play a supporting role in enzyme catalysis. However, it is difficult to explain
exactly what role these predicted residues have on enzyme activity, although some clues
do emerge from the structures and pH-dependent studies.
There are many possible proposed mechanisms for these effects which include 1) local
rotations or side chain shifts, 2) shifts in hydrogen-bonding (H-bonding) networks, 3)
214

changes in the electric field in the active site, or 4) quantum mechanical effects. While
structural changes were observed for a few of the mutants, it cannot unequivocally be
stated that the kinetic effects demonstrated for second- and third-shell residues are due to
structural and/or electrostatics effects. More studies must be performed to explain the
results seen. Two computational approaches could be the use of Molecular dynamics
(MD) and Quantum Mechanical (QM) calculations which may shed some new insight
into this phenomenon. These methods may be able to detect dynamical differences in
rotameric states or changes in bond polarization. Additionally, it may be possible to
obtain information by solving for the electrostatic potentials at different time points in the
dynamics run. Quantum Mechanical – Molecular Mechanics (QM/MM) or Quantum
Mechanical – Molecular Dynamics (QM/MD) calculations treat the active site quantum
mechanically, while the rest of the proteins is treated with MM or MD. This could allow
for the computational modeling of protein-substrate complexes and transition states, and
allow one to follow computationally the step by step reaction. These approaches may
allow one to better understand exactly what residues are involved in a reaction and what
roles remote residues play. Understanding how nature designs enzyme active sites is a
fundamental question in enzymology with implications for protein engineering. The
present results suggest that computational methods could help guide the identification of
functional second- and/or third-shell residues, and for ppNHase, second-and third-shell
mutations predicted through computational techniques do have an effect on enzyme
catalysis which suggests that enzyme active sites are nanoscale entities that are built in
multiple layers.
215

4.5 References 1. Miyanaga, A., Fushinobu, S., Ito, K. & Wakagi, T. (2001). Crystal structure of
cobalt-containing nitrile hydratase. Biochem Biophys Res Commun 288, 1169-1174.
2. Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240-255.
3. Emsley, P. & Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126-2132.
4. Adams, P. D., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58, 1948-1954.
5. Ko, J., Murga, L. F., Andre, P., Yang, H., Ondrechen, M. J., Williams, R. J., Agunwamba, A. & Budil, D. E. (2005). Statistical criteria for the identification of protein active sites using Theoretical Microscopic Titration Curves. Proteins 59, 183-195.
6. Murga, L. F., Wei, Y. & Ondrechen, M. J. (2007). Computed Protonation Properties: Unique Capabilities for Protein Functional Site Prediction. Genome Informatics 19, 107-118.
7. Ondrechen, M. J., J.G. Clifton and D. Ringe. (2001). THEMATICS: A simple computational predictor of enzyme function from structure. Proc. Natl. Acad. Sci. (USA) 98, 12473-12478.
8. Ondrechen, M. J. (2004). Identification of functional sites based on prediction of charged group behavior. In Current Protocols in Bioinformatics (Baxevanis, A. D., Davison, D. B., Page, R. D. M., Petsko, G. A., Stein, L. D. & Stormo, G. D., eds.), pp. 8.6.1 - 8.6.10. John Wiley & Sons, Hoboken, N.J.
9. Wei, Y., Ko, J., Murga, L. & Ondrechen, M. J. (2007). Selective prediction of Interaction sites in protein structures with THEMATICS. BMC Bioinformatics 8, 119.
10. Lichtarge, O., Bourne, H. R. & Cohen, F. E. (1996). An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol 257, 342-358.
11. Madabushi, S., Yao, H., Marsh, M., Kristensen, D. M., Philippi, A., Sowa, M. E. & Lichtarge, O. (2002). Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J Mol Biol 316, 139-154.
12. Yao, H., Kristensen, D. M., Mihalek, I., Sowa, M. E., Shaw, C., Kimmel, M., Kavraki, L. & Lichtarge, O. (2003). An accurate, sensitive, and scalable method to identify functional sites in protein structures. J Mol Biol 326, 255-261.
13. Sobolev, V., Eyal, E., Gerzon, S., Potapov, V., Babor, M., Prilusky, J. & Edelman, M. (2005). SPACE: a suite of tools for protein structure prediction and analysis based on complementarity and environment. Nucleic Acids Res 33, W39-43.
216

14. Sobolev, V., Sorokine, A., Prilusky, J., Abola, E. E. & Edelman, M. (1999). Automated analysis of interatomic contacts in proteins. Bioinformatics 15, 327-332.
15. Armon, A., Graur, D. & Ben-Tal, N. (2001). ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J Mol Biol 307, 447-463.
16. Glaser, F., Pupko, T., Paz, I., Bell, R. E., Bechor-Shental, D., Martz, E. & Ben-Tal, N. (2003). ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163-164.
17. Landau, M., Mayrose, I., Rosenberg, Y., Glaser, F., Martz, E., Pupko, T. & Ben-Tal, N. (2005). ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33, W299-302.
18. Wu, S., Fallon, R. D. & Payne, M. S. (1997). Over-production of stereoselective nitrile hydratase from Pseudomonas putida 5B in Escherichia coli: activity requires a novel downstream protein. Appl Microbiol Biotechnol 48, 704-708.
19. Miyanaga, A., Fushinobu, S., Ito, K., Shoun, H. & Wakagi, T. (2004). Mutational and structural analysis of cobalt-containing nitrile hydratase on substrate and metal binding. Eur. J. Biochem. 271, 429-438.
20. Kato, Y., Tsuda, T. & Asano, Y. (1999). Nitrile hydratase involved in aldoxime metabolism from Rhodococcus sp. strain YH3-3 purification and characterization. Eur J Biochem 263, 662-670.
21. Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248-254.
22. Fallon, R. D., Stieglitz, B. & Turner Jr., I. (1997). A Pseudomonas putida capable of stereoselective hydrolysis of nitriles. Appl. Microbiol. Biotechnol. 47, 156-161.
23. Otwinowski, Z., & Minor, W. (1997). Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 276, 307-326.
24. Mccoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). Phaser crystallographic software. Journal of Applied Crystallography 40, 658-674.
25. Motulsky, H. C., A. (2004). Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting (GraphPad Software, I., Ed.), Oxford University Press, New York.
26. Piersma, S. R., Nojiri, M., Tsujimura, M., Noguchi, T., Odaka, M., Yohda, M., Inoue, Y. & Endo, I. (2000). Arginine 56 mutation in the beta subunit of nitrile hydratase: importance of hydrogen bonding to the non-heme iron center. J Inorg Biochem 80, 283-288.
217

Chapter 5
Conclusions, Future Work and Future Directions
218

5.1 Conclusions
The main focus of this thesis has been to explore the concept of multilayer enzymes.
Specifically, the general concept was developed theoretically and tested experimentally
with Co-type nitrile hydratase that residues located remotely from the active site
contribute in some way to the overall catalytic rate of an enzyme. For this work, these
remote residues were assigned a ‘shell’ according to their location around the active site
of the protein. First-shell residues referred to residues in direct contact with the reacting
substrate or metal ion; second-shell residues referred to residues in direct contact with
first-shell residues and third-shell residues referred to residues in direct contact with
second-shell residues. It was demonstrated through computational and experimental
methods that second- and third-shell residues are functionally important for the enzyme,
Co-type nitrile hydratase from Pseudomonas putida.
In Chapter 2, the ability of THEMATICS to identify a subset of remote residues was
demonstrated and the results were compared with the sequence-based ET method. The
residues identified by these methods were compared with experimental mutagenesis data
from the literature, and the results indicated that both THEMATICS and ET predict
functionally important residues not only in the first-shell of an interaction site, but also
residues located in coordination spheres beyond the first. This study was the first
systematic approach to computationally identifying functional residues located in the
outer interaction spheres of enzymes, i.e. beyond the first-shell, as virtually all previous
characterizations of enzyme active sites have considered only first-shell residues. What is
219

most striking is that two completely different types of theoretical methods both support
multilayer active sites.
Chapter 3 presented the first structure of the enantioselective Co-type nitrile hydratase
from Pseudomonas putida to 2.1 Å. The structure reveals global similarity to other
NHases except for large differences in the α5-loop-α6 region in the β-subunit and in the
location of the N-terminus of the α-subunit. In addition, a full kinetic profile of wild type
ppNHase was obtained. It was determined that the enzyme was most active at pH 7.2,
with a KM of 11 mM and a kcat of 20 min-1, and that this activity decreases at low and
high pH values.
Chapter 4 presented a kinetic analysis of five second- and third-shell mutant ppNHase
proteins and a crystal structure analysis of four mutant ppNHase proteins. The kinetics
results at pH 7.2 demonstrated a 50-fold decrease in kcat for the second-shell mutant
αAsp164Asn, a 3-fold decrease for the third-shell mutant αGlu168Gln, a 70-fold decrease
in kcat for the second-shell mutation βGlu56Gln, a 13-fold decrease for the third-shell
mutant βHis71Leu, and a 12-fold decrease in kcat for the third-shell mutant βTyr215Phe,
all compared to wild type. Some of these catalytic effects were explained through local
structural changes. It was shown for the αGlu168Gln mutant that the catalytic effects
were most likely due to both local structural differences and electrostatic effects. The
rotation of the side chain for the glutamine residue in the mutant structure disrupted the
salt bridge interaction between αGln168 to the known catalytic residue, βArg52. The
other mutant for which structural changes could also explain the catalytic effects was the
220

βTyr215Phe mutant. While the H-bonding capabilities to the functionally important
βArg149 were removed, there were no obvious structural changes in that area. There was,
however, a shift in the third-shell residue, αGlu168, which lengthened the salt bridge to
βArg52 by 1.0 Å. This suggests a combination of structural and electrostatic effects in the
second- and third-shell may play a role in catalysis. The structures for the other mutants
did not change making the decrease in catalytic rate more difficult to explain. Most
interesting is the βGlu56Gln mutant which had the largest effect on kcat compared to wild
type, but no structural change. In the last mutant structure, βHis71Leu, the only structural
change observed was the shift of a water molecule in the active site, disrupting the H-
bond networks. The structures for these two mutants suggest that electrostatic effects
play an important role in enzyme function and that these effects include residues outside
of the first layer of the active site.
This suggests that second- and third-shell residues play a supporting role in catalysis for
NHase without drastically affecting the binding of the substrate at all pH values tested.
Furthermore, while single point mutations lead to a drop in catalytic rate by one or two
orders of magnitude, collectively these residues outside the first shell probably amount to
a much larger effect on the catalytic rate. In other words, the composition of the second-
and third-shell may be critical to the enzymatic catalytic rate. For ppNHase, second-and
third-shell mutations predicted through computational techniques do have an effect on
enzyme catalysis which suggests that enzyme active sites are built in multiple layers.
221

5.2 Future Work
Computational Approaches
Understanding how nature designs enzyme active sites is a fundamental question in
enzymology with implications for protein engineering. The present results suggest that
computational methods could help guide the identification of functional second- and/or
third-shell residues and can serve as a useful guide for rational protein design studies.
However, a complete understanding of the effects of these remote residues is a necessary
component. There are many possible proposed mechanisms for these effects which
include 1) local rotations or side chain shifts, 2) shifts in hydrogen-bonding (H-bonding)
networks, 3) changes in the electric field in the active site, or 4) quantum mechanical
effects. Based on the data accumulated in this work, it was shown that in some cases
there are structural changes (i.e. changes in H-bond networks) that occur and can explain
differences in catalytic rates, at least in part. However, this was not always the case and
therefore other hypotheses must be made. Currently, the main hypothesis is that these
changes in catalytic rate are due to electrostatic effects or a combination of structural and
electrostatic effects. One reason for this was that THEMATICS is an electrostatics based
method, and the fact that these residues were identified by THEMATICS provides some
evidence to support this hypothesis. Additionally, it appears very reasonable that remote
residues help to provide the correct protonation properties, specifically the pKa and the
shape of the titration curve, of the ionizable residues in the first-shell. In order to truly
understand these electrostatic effects, two computational methods may prove extremely
useful, namely, Molecular dynamics (MD) and Quantum Mechanical (QM) calculations.
MD simulations would provide useful structural information that occurs as a protein
222

moves in solution. Detection of small changes in H-bond networks and rotations of side
chains may explain why residues remote to the active site affect catalysis. Additionally,
through the use of Quantum Mechanical – Molecular Mechanics (QM/MM) or Quantum
Mechanical – Molecular Dynamics (QM/MD), it will be possible to track reactions step
by step. QM/MD simulations treat the active site quantum mechanically, while the rest of
the protein is treated classically with MM or MD calculations. This will allow for the
modeling of protein-substrate complexes in addition to transition states. The goal is to
understand exactly the role of the active site residues and gain some insight into the
effects of remote residues.
The main difficulty, however, in running these calculation on Co-type ppNHase
specifically is obtaining parameters for the cobalt ion and the oxidized cysteine residues
in the active site. MD calculations have been run on this protein, however, without
constraints, the active site atoms moved drastically. It was therefore necessary to put
constraints on the active site atoms so that they could not move. These MD simulations
are currently in progress, and will be analyzed against mutant proteins if the wild type run
is successful. While this does allow one to gain information about the dynamical effects
of second- and third-shell residues on the active site, the cobalt ion and the oxidized
cysteines pose difficulties in the quantum mechanical modeling of this protein.
223

Experimental Approaches
This work presented in this thesis focused only on kcat, and did not touch upon the effects
remote residues have on specificity or on KM. While it is noted that specificity is
extremely important to consider, it was beyond the scope of this project. Interestingly, the
study on nitrile hydratase revealed no changes in KM. It would be interesting to see what
the effects of non-conservative mutations would be (i.e. substitution with alanine) on both
the catalytic rate and on the binding. It is expected that this substitution would cause a
local structural change and hence affect both kcat and KM. However, until these studies
are performed, it is difficult to draw those types of conclusions. This concept was alluded
to in chapter 2, and further work needs to be undertaken to understand all the components
involved.
Experimentally, through x-ray crystallography, it is also possible to try to understand the
role of these remote residues. Crystallizing the protein with substrate, product or inhibitor
will allow for the analysis of bound and unbound structures, and may allow one to see
differences in structure or specific residues involved in binding. Additionally, it may be
useful to crystallize this protein with transition state analogs to understand exactly which
residues are involved in the reaction and what effects remote residues may have.
This work with Co-type nitrile hydratase provided a proof of concept that computational
methods could help guide the prediction of functionally important residues located
outside of the active site. The results from this study provided more questions than
answers, but have provided evidence that enzymes are built in multiple layers. In
224

addition, work from this thesis has provided data that resulted in a grant proposal that has
been funded by the National Science Foundation for a follow up study at Northeastern
University on the role of remote residues in enzyme catalysis. This work has also
initiated a number of collaborations both within Northeastern University and beyond.
5.3 Future Directions – Collaborations
Dr. Penny Beuning, Northeastern University and the Chemical Biology Class – Second-
and Third-shell Mutations
The work presented herein was a proof of concept that THEMATICS could identify
functionally important residues in both the second- and third-shells of a protein. The next
step would be to perform these studies on numerous other systems to determine if the
same trends are seen. Currently, students in Prof. Beuning’s Chemical Biology class at
Northeastern University are studying second- and third-shell effects for the enzyme
alkaline phosphatase (AP).
Continuation of Multilayer Enzyme Active Site Investigations
Professors Beuning and Ondrechen will also study additional enzymes to start to build up
a large set of data in support of this concept. These studies will include ketosteroid
isomerase (KSI) and phosphoglucose isomerase (PGI). These two isomerases were
chosen to represent two enzymes that catalyze very similar reactions but have very
different degrees of predicted participation by residues outside the first-shell. KSI and
PGI are not metal dependent whereas the active site of AP contains metal ions. Quite
different site predictions are obtained for the two isomerases. For KSI, THEMATICS
225

predicts only first-shell residues, and while ET predicts residues in the second- and third-
shell, the predictions consist of proportionally fewer residues than for most proteins. On
the other hand, for the isomerase catalytic site of PGI, THEMATICS predicts multiple
layers; the site predicted by ET is larger than that for most proteins and includes a greater
than average fraction of residues outside the first-shell. Based on the available
information, residues outside the first-shell are less likely to be important for KSI and
more likely to be important for PGI. Thus these two isomerases represent two opposite
poles with respect to the probability of exhibiting multilayer effects and are good test
cases for the question at hand.
Dr. Penny Beuning, Northeastern University and DinB – Protein Engineering
The knowledge acquired in this thesis will be used to guide protein engineering research.
Prof. Beuning is studying the specificity of DNA polymerases to understand why some of
them replicate damaged DNA and others do not. Proposed DinB mutants will be used in
specificity studies to test whether changes in the second-shell residues can be used to
engineer changes in polymerase specificity. DinB was chosen because multilayer active
sites are predicted by both THEMATICS and ET for a homology model structure and
because of the current interest in understanding the mechanisms for the control of
substrate specificity in DNA polymerases.
226

Prof. Don Hilvert, ETH, Zürich and Chorismate Mutase vs. Isochorismate Pyruvate
Lyase – Protein Engineering
Currently, Dr. Mary Ondrechen’s THEMATICS group is giving guidance to Prof. Don
Hilvert of ETH, Zürich in his experimental studies of isochorismate pyruvate lyase (IPL)
and chorismate mutase (CM). These two enzymes have related structures and very
similar active sites, with seven out of twelve first-shell residues in common. In an attempt
to impart IPL activity onto CM, his group learned that mutating the remaining five first-
shell residues in CM to match IPL resulted in a protein with no activity for either
reaction. Changes outside the first-shell clearly are needed and the Hilvert group is in the
process of making the mutations corresponding to our predicted second-shell residues.
This dissertation has introduced the concept and provided evidence in support of
multilayer enzyme active sites. These ideas will probably prove to be important in a
number of future applications. These include the engineering of new kinds of enzymes to
catalyze reactions that do not occur in nature. The design of enzymes to catalyze the
synthesis of biofuels is one application that may be very important in the quest for
renewable energy sources. Other potential applications are in the areas of environmental
remediation, counterterrorism, and disease control. Our ability to engineer enzymes for
these important applications will be greatly enhanced by our understanding of how nature
builds catalysts. Herein evidence has been provided that nature’s catalytic sites are
multilayer, nanoscale assemblies that are larger than previously believed.
227

Supplemental Chapter 1
Computationally Guided Protein-Specific Labeling with Nanoparticles – A Test Case
Using HER2
228

Supplemental Chapter.1 Introduction
The majority of my thesis focused on the use of computational and experimental
techniques to establish the functional importance of remote residues in enzyme catalysis.
This chapter includes work that was done as part of the Integrated Graduate Education
Research and Training program in Nanomedicine at Northeastern University. The focus
of this project was the identification of previously unknown binding sites in biomarker
proteins using THEMATICS1-4 and other computational tools. The overall goal of the
project was to identify these sites with THEMATICS and geometric analysis and utilize
these sites for protein-specific labeling for imaging and diagnostic purposes. Specifically,
the plan was to use molecular docking procedures to identify small molecules to bind to
the predicted sites, and then attach nanoparticles to the best binding small molecules with
large polyethylene glycol (PEG) linkers.5 A cancer biomarker that met most of the
criteria for this project was selected. THEMATICS was used to identify a previously
unidentified binding site, and then a set of small molecules as potential binders was
identified using computational approaches (i.e. molecular docking)6. At this point, the
protein has been expressed and purified, and the project is now ready for the next stage,
the experimental binding studies.
The Integrated Graduate Education Research and Training (IGERT) Traineeship provides
an excellent opportunity to combine our experimental and computational tools of
chemical biology with nanotechnology, with a focus toward applications in
nanomedicine.7 These applications include imaging, diagnostics, drug discovery and
drug delivery. We propose to combine theoretical computational modeling of nanoscale
systems with synthesis and surface functionalization to design and characterize
229

nanostructures for biomedical applications. By integrating computational design with
experiment, we hope to provide a fast, cost effective means for the labeling of disease
marker proteins with nanoparticles using highly specific coupler ligands for biomedical
research.
THEMATICS (THEoretical Microscopic TItration CurveS) is a theoretical computational
approach for determining the active sites and binding sites of proteins, requiring only the
3D structure of the query protein as input. THEMATICS calculations are based on
predicted titration curve shapes determined computationally from a Poisson-Boltzmann
procedure. Active site residues are identified by abnormally shaped or perturbed titration
curves. It has been demonstrated that spatial clusters of these perturbed residues are
reliable predictors of an active site or binding site. Many of the residues so identified by
THEMATICS are documented in the literature as catalytically important or important in
substrate binding, as determined experimentally, principally by site-directed mutagenesis.
Recent preliminary results for hormone-receptor couples suggest that THEMATICS at
least for some cases is capable of finding the binding epitope on the surface of the
hormone alone or of the receptor alone. In addition to those sites that have been
experimentally confirmed, THEMATICS does sometimes find spatial clusters on the
surface of proteins and some of these predicted clusters are of unknown functionality.
We have argued that perturbed theoretical titration behavior results from strong
interaction between ionization events in the region of catalytic and binding sites. These
types of interactions are the source of the well known non-Henderson-Hasselbalch
titration curves exhibited by small molecule polyprotic acids. Because proteins are
230

biomacromolecular polyprotic acids, there are many such interactions. We have argued
further that nature has engineered catalytic and binding sites in proteins so that these
interactions between ionization events are especially strong. Thus, when a site on a
protein is predicted by THEMATICS, it has electrostatic properties that are especially
well suited for specific ligand binding. Some of these predicted sites do bind specific
natural ligands while others may be simply potential binding sites. We will use this
unique computational predictive tool to demonstrate that these sites can be used to design
protein-specific ligands that can be coupled to nanoparticles for labeling, imaging and
diagnostics.
The overall plan was to use THEMATICS to predict binding sites on disease marker
proteins of known 3D structure, followed by molecular docking to identify a set of small
molecule candidates that may bind specifically to the predicted site. The candidate small
molecules will be screened experimentally for affinity to the target protein using either a
thermofluor assay,8 Isothermal Titration Calorimetry (ITC)9 or Surface Plasmon
Resonance (SPR)10. Each method has its advantages and disadvantages, so experimental
method development will occur before the binding method is chosen. The molecule with
the highest affinity will be selected for further studies. We then propose to attach a gold
nanoparticle (NP) to a derivatized form of this selected small molecule via a thiol group
on a polyethylene glycol (PEG) linkage and thus attach the NP specifically to the
predicted site.5
231

Supplemental Chapter.2 Materials and Methods
Computational Methods
THEMATICS calculations
The proteins were analyzed and site predictions were made by Theoretical Microscopic
Titration Curves (THEMATICS) according to published procedures
(http://pfweb.chem.neu.edu/thematics/submit.html).1,11 The protein structures used as the
input data for the calculations were downloaded from the Protein Data Bank
(http://www.rcsb.org/pdb/). Structures with missing atoms were fixed in swiss-pdb
viewer. Substrates, water molecules, cofactors and salts that crystallized with the proteins
were not included in the electrostatic calculations. All methods were run as previously
described; however, the default parameters were adjusted to use a statistical cutoff of
0.96 instead of the default of 0.99. Residues identified as THEMATICS positives were
clustered with a 9 Å cut-off.
Computational Docking Using Glide
The protein structures were prepared for docking using the Schrödinger software package
(Schrödinger, LLC, Portland, OR). Hydrogen atoms and partial charges were added to the
protein using PPrep. The THEMATICS cluster residues were fixed during Impact
minimization (100 Truncated Newtonian cycles using the OPLS2001 force field and a
gradient coverage of 0.01). The scoring grid was generated within a 20 Å box centered on
the predicted residues. A water molecule was added to the center of the predicted
residues using Maestro. Ligand docking was performed using Glide 3.5 in Standard
232

Precision (SP) mode. The ligands used for docking were from a shortened list of the Zinc
database of ligands (http://zinc.docking.org). Results were analyzed using Glide Pose
Viewer.
Experimental Methods
Construction of Plasmids for HER2
The clone for HER2 was obtained from Dr. Dan Leahy’s group at Johns Hopkins
University School of Medicine. Primers were designed based on the gene sequence (PDB
ID: 1N8Z12) in order to amplify the HER2 gene. DMSO (5%) (Fisher Scientific,
Pittsburgh, PA) was added to the PCR reaction to prevent the primers from binding to
each other. The DNA was extracted from an agarose gel and cloned into a
pENTR/TEV/D-TOPO vector (Invitrogen, Carlsbad, CA). The TOPO cloning reaction
was then transformed into One Shot chemically competent cells (Invitrogen, Carlsbad,
CA) and plated on KAN resistant plates. The DNA from the TOPO vector was then
transformed into two types of expression vectors; pDEST 17, a 6-HIS (histidine) tag
vector and pDEST15, a GST (glutathione) tag vector (Invitrogen, Carlsbad, CA). At all
stages, the DNA was digested with EcoRV and NotI (New England Biolabs, Ipswich,
MA) to confirm the correct sequence, and at the final stage, the presence of the correct
sequence was confirmed (GENEWIZ, South Plainfield, NJ). The sequenced DNA was
then transformed into RIPL cells (Stratagene, La Jolla, CA).
233

Protein Expression and Purification of HER2
All reagents were purchased from Fisher Scientific, Pittsburgh, PA unless otherwise
noted. The extracellular domain of HER2 was expressed in E. coli RIPL cells
(Stratagene), which were grown at 37 ºC in 2XYT broth containing 100 μg/mL
ampicillin. Once the absorbance reached 0.8 at A600, the cells were induced with IPTG to
0.1 mM. The cells were cultured for an additional 4 hours at 37 ºC.13 All subsequent
manipulations were performed at 4 ºC. After harvesting the cells by centrifugation, the
pellet was resuspended in 50 mL cold STE buffer (Fisher Scientific, Pittsburgh, PA) +
100 μg/mL lysozyme and incubated on ice for 15 min.13 DTT was then added to a final
concentration of 5 mM, sarcosyl was added to a final concentration of 1.5%14 and one
protease inhibitor tablet (Roche, Branford, CT ) was added to the final mixture. The
solution was sonicated for 7.5 min at 50% power with 10 sec power bursts and 1 min wait
time between bursts. The solution was centrifuged for 20 min. The wash centrifuge cycle
was completed twice and the supernatant was saved. Triton X (Fisher Scientific,
Pittsburgh, PA) was added to the supernatant solution to a final concentration of 3%.14
This solution was then purified using glutathione sepharose 4B purification beads (GE
Healthcare, Piscataway, NJ). The beads were prepared by washing 1 mL of the 75%
slurry 3 X with 10 mL of 1X PBS. The final volume was then brought to 1.5 mL to make
a 50% slurry. After an overnight incubation, the beads were washed 3X with 10 mL of
1X PBS. The GST-tagged protein was eluted 3X with 2 mL of elution buffer (75 mM
Tris pH 8, 150 mM NaCl, 20 mM reduced glutathione, 2% N-octylglucoside (Fisher
Scientific, Pittsburgh, PA), 5 mM DTT) and placed on an orbital rotor (VWR, Arlington
Heights, IL) for 20 min each at 4 ºC.13
234

Supplemental Chapter.3 Results and Discussion
Identification of potential biomarker – THEMATICS and Molecular Docking
To identify potential protein biomarkers for this project, THEMATICS was run on
approximately ten test cases to determine if a previously unidentified binding site could
be found. Specifically, the goal was to focus on systems of medical importance where
THEMATICS identifies sites that have not been used previously for labeling, for instance
in antibody binding. Computational approaches (i.e. molecular docking) were then used
to identify small molecules which may bind to the identified site. These small molecules
could be inhibitors or ligands which can be tagged with nanoparticles for imaging
purposes. In many of the cases studied, THEMATICS identified the known catalytic
and/or binding sites, but did not identify any additional sites, for instance the matrix
metalloproteinases and prostate specific antigen (PSA). These were not ideal cases
because we wanted to demonstrate the ability of THEMATICS to identify previously
unknown binding sites. Additionally, we did not want to interfere with known binding or
catalytic sites. There were a few cases where THEMATICS did in fact identify new sites,
but they were located on the surface of the protein in shallow pockets and were therefore
not appropriate candidates for small molecule binding. These include CA-125, and
ovarian cancer target and integrins, which play an important role in cell signaling. Two
proteins were identified; however, where THEMATICS identified previously unknown
sites and appeared to be good candidates to pursue further. These include 14-3-3 σ (PBD
ID: 1YZ515) and HER2 (PDB ID: 1N8Z12). The THEMATICS predicted results are
shown in Table A-1.
235

Table A-1: THEMATICS results 14-3-3 σ and HER2.
Enzyme Cluster THEMATICS prediction
Known active site Y48, K49, R56, C96, K122, Y127, R129, Y130, E133, Y151 14-3-3 σ
PDB ID: 1YZ515 Predicted dimer interface R18, E20, D21, Y84, E91
Known antibody binding site Y36 A, H91 A, Y33 B, H35 B, R50 B, Y52 B, Y105 B, E558 C, D560 C, K593 C
Predicted site 1 D8 C, R12 C, Y28 C, E39 C, Y61 C, H415 C,
HER2 PDB ID: 1N8Z12
Predicted site 2 E299 C, E383 C, R410 C, R412 C
Supplementa1 Chapter.3.1 4-3-3 σ 14-3-3 σ (also called stratifin) is an isoform from a family of proteins known as 14-3-3.
They are 30 kDa dimeric proteins found in all eukaryotic cells,16 and have many diverse
functions including roles in signal transduction pathways and cell cycle regulation.17 14-
3-3 proteins act as chaperone molecules which can move freely from the cytoplasm to the
nucleus and vice-versa.18 There are seven distinct forms in humans which show a high
degree of sequence identity and conservation. Each monomer is formed by nine alpha
helices with anti-parallel distribution, and the inner core of each monomer is where
ligands bind.19 14-3-3 σ is unique in the sense that it forms mostly homodimers in
solution20 and is induced by the p53 tumor suppressor protein in response to DNA
damage.17 Additionally, it is the isoform most directly linked to cancer.18 Structural
analysis of 14-3-3 σ specifically reveals that there is a six to seven amino acid difference
at the dimer interface between 14-3-3 σ and the other isoforms, where five are σ-specific.
It is believed that this structural discrepancy is what allows for σ homodimerization by
236

stabilizing homodimeric interactions and destabilizing heterodimeric interactions.20 14-3-
3 σ negatively regulates the cell cycle and positively regulates p53 stability and
transcriptional activity. It is down regulated in several types of cancer, including breast,21
ovarian,22 prostate23 and lung24 cancer. This decrease in protein expression is due either
to epigenetic (i.e. transmitted from the parental genome to the next generation of cells)
silencing by methylation or to mutation of p53. There has been compelling evidence that
14-3-3 σ can act as a tumor suppressor which made it a good candidate to study further.
This, in addition to the fact that the dimer interface is specific for the σ form suggested
that this would be a great biomarker candidate to pursue further with docking studies.
THEMATICS identifies the known phosphopeptide binding pocket for 14-3-3 σ, and
additionally identifies residues located at the dimer interface (Figures A-1 and A-2).
While this may not appear to be an obvious binding site to probe, the residues along this
dimer interface are predicted to be ‘sticky’, and are therefore potentially capable of
binding a small molecule. Approximately 110,000 compounds have been docked into this
predicted site, and all of these compounds are commercially available. The top 100 best
hits have been identified, and a sampling of these compounds is shown in Figure A-3. All
of the predicted compounds are large ring structures that have the capability to span the
entire binding pocket located at the dimer interface. Additionally, since this predicted
pocket is at the dimer interface, there is plenty of space to accommodate the proposed
complex that will consist of the ligand, to which a long polyethylene glycol (PEG) linker
will be attached. The hope is that the attached linker and nanoparticle will not affect the
binding of the ligand.
237

Figure A-1: A ribbon diagram of 14-3-3 sigma (PBD ID: 1YZ515). The THEMATICS predicted residues for the known catalytic and/or binding residues are shown in green CPK coloring, while the THEMATICS predicted residues for the dimer interface are shown in pink CPK coloring. Note there are two sites colored green, one for each subunit.
238
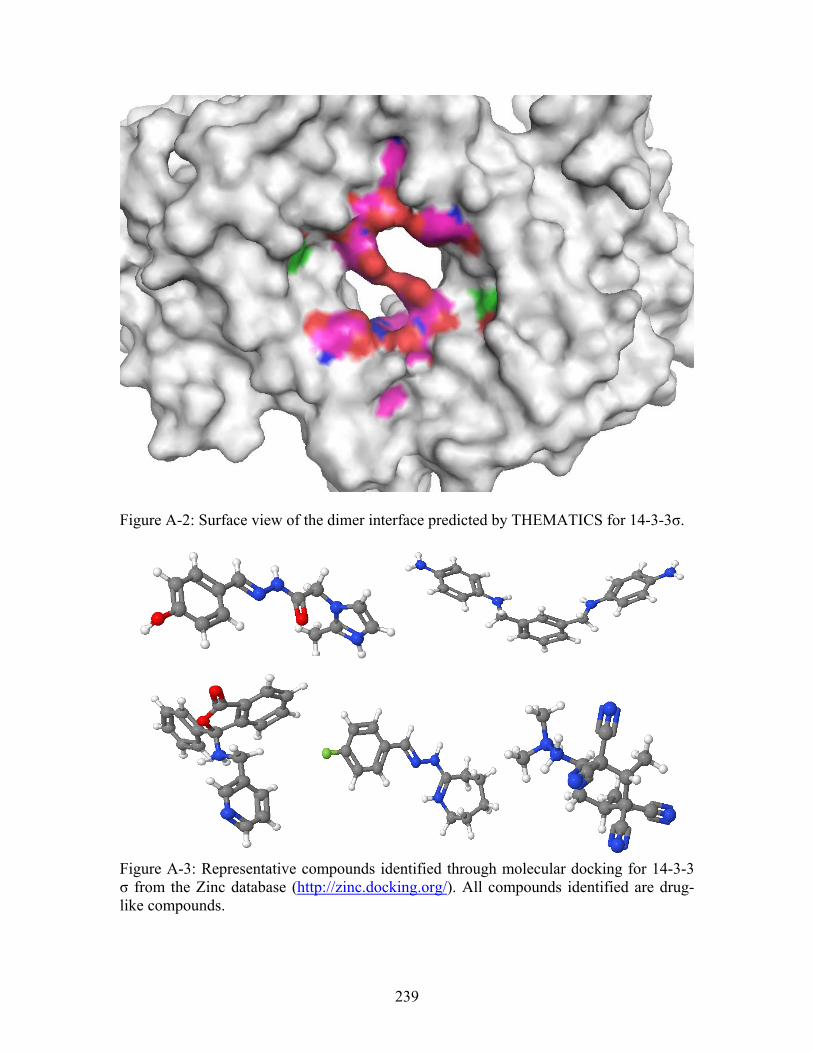
Figure A-2: Surface view of the dimer interface predicted by THEMATICS for 14-3-3σ.
Figure A-3: Representative compounds identified through molecular docking for 14-3-3 σ from the Zinc database (http://zinc.docking.org/). All compounds identified are drug-like compounds.
239

Further analysis of the compounds identified for 14-3-3 σ revealed that the molecules
were not fitting tightly into the pocket; it was just too large. Further work could have
been done to try to improve the size of the compounds or to try to dock small peptides
into the site, but it was decided to pursue another target.
Supplemental Chapter.3.2 HER2
Human epithelial growth factor 2 (HER2, neu or erbB2) encodes a 185 kDa protein that
belongs to the epidermal growth factor receptor (EGFR) family.13 It consists of three
domains, an extracellular domain (ECD), a hydrophobic transmembrane domain and an
intracellular tyrosine kinase domain. HER2 and another member of the EGFR family
form an active dimer receptor, resulting in the phosphorylation of tyrosine residues which
initiates signaling pathways leading to cell division. Overexpression of HER2 has been
observed in breast,25 ovary,25 prostate,26 colon27 and pancreatic cancers, but has been
most studied in relation to breast and ovarian cancers. In these cases, HER2 releases the
extracellular domain in the serum which has been found to be associated with metastatic
tumors.13 In breast cancer specifically, overexpression occurs in 15-30% of all cases and
predicts a significantly lower survival rate and a shorter relapse time in patients with the
lymph-node positive disease.28
Approaches are underway toward HER2 targeted therapies focusing on antibodies that
are specific to the ECD with the specific goal of killing the expressing tumor cells.
Herceptin (trastuzumab) is a human monoclonal antibody which binds with high affinity
to the ECD of HER2, thereby blocking its function in signal transduction.29 This, used in
240

conjunction with chemotherapeutics such as doxyrubicin or paclitaxel, is the current
treatment for HER2 positive breast cancer.30 Based on experimental evidence that
overexpression of HER2 plays a role in tumor formation and metastasis, studies are
underway to find an inhibitor of this receptor. To date, there is no known ligand which
binds specifically to the HER2 receptor; there are however, known ligands which bind to
other members of the EGFR family.12,31
The ECD of HER2 comprises approximately 630 amino acid residues and contains four
domains (I-IV).12 The structure of human HER2 alone and in complex with Herceptin is
shown in Figures A-4A and A-4B. Herceptin binds to the C-terminal portion of domain
IV. Structures of EGF monomers versus a ligand bound EGFR dimer shows that the
ligand binds at a site between domains I and III causing a conformational change in the
extracellular domain.31 Ligand induced dimerization allows for the normal signaling
mechanism for the erb-Bs. When unliganded, the extracellular domain of EGFR and erb-
B3 is in a closed conformation, with domain II interacting with domain IV.31 Upon ligand
binding, the structure opens and domain II points out from the rest of the molecule.
Alternately, the HER2/erb-B2 extracellular domain is always in the open confirmation.
This could explain why it is a preferred dimerization partner for erb-B1, -B3 and –B4.
This dimerization brings the two tyrosine kinase domains together, allowing
transphosphorylation of tyrosines on the C-terminal end of one erb-B and the kinase
domain of the other for signaling. Analysis of the residues comprising the known ligand
binding site for other erb-B family proteins shows that the residues comprising this site
are not conserved in erb-B2 relative to the rest of the EGF family suggesting that this site
241
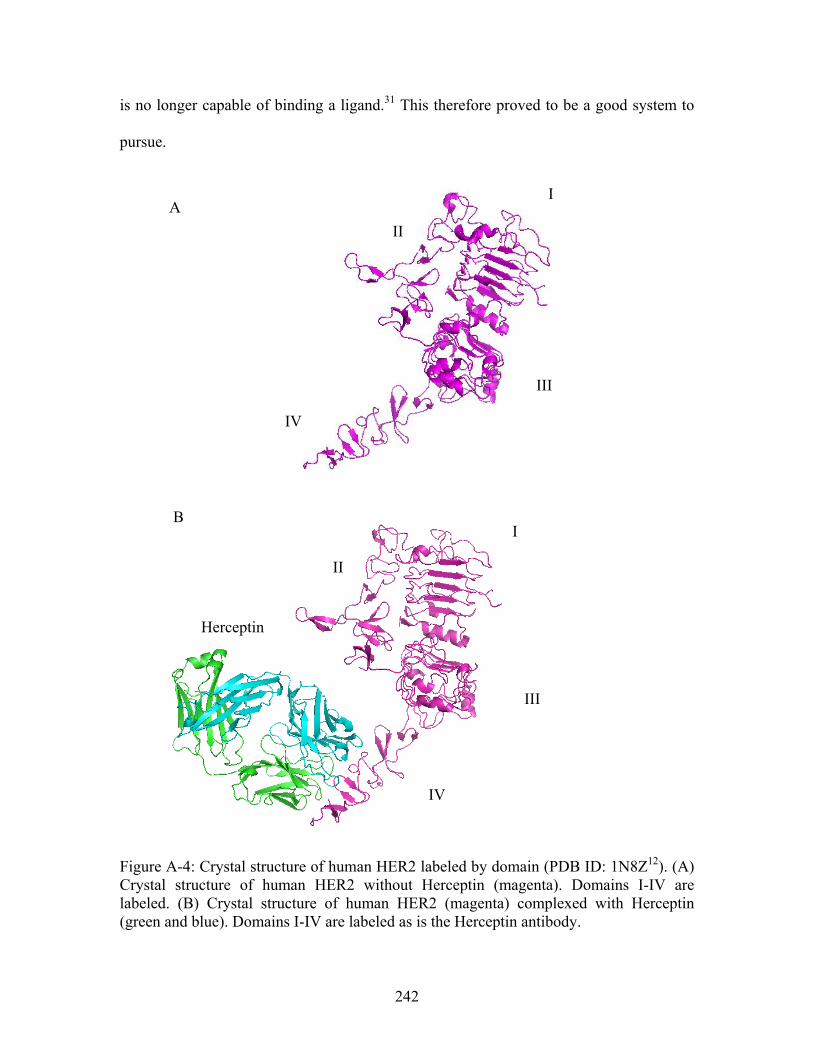
is no longer capable of binding a ligand.31 This therefore proved to be a good system to
pursue.
I A
II
III
IV
B I
II
Herceptin
III
IV
Figure A-4: Crystal structure of human HER2 labeled by domain (PDB ID: 1N8Z12). (A) Crystal structure of human HER2 without Herceptin (magenta). Domains I-IV are labeled. (B) Crystal structure of human HER2 (magenta) complexed with Herceptin (green and blue). Domains I-IV are labeled as is the Herceptin antibody.
242

THEMATICS identifies the known antibody binding site in addition to two sites of
unknown function, site 1 and site 2 (Table A-1). Figure A-5 shows a surface view of
human HER2 bound to the antibody Herceptin. Using the binding pockets predicted by
THEMATICS, molecular docking was performed using the zinc database of drug-like
compounds (http://zinc.docking.org/). Specifically, 100,000 compounds have been
docked into these two sites respectively, and some promising candidates with predicted
favorable free energy of binding have been identified.
Site 2
Site 1
Figure A-5: Surface display of HER2 (PDB ID: 1N8Z12) (magenta = ECD HER2, blue and green = Herceptin). Arrows point to the two THEMATICS predicted sites (site 1, blue and site 2, grey), and the known antibody binding site in red.
Antibody binding site
243

Shown in Figures A-6 and A-7 are representative compounds identified through docking
for sites 1 and 2 respectively. It should be noted that site 1 appears to favor smaller
molecules with phosphate, carboxylate or nitryl groups, while site 2 favors slightly larger
compounds with multiple rings. Figures A-8 and A-9 show surface diagrams of some of
the identified compounds docked into sites 1 and 2, respectively. As with the 14-3-3 σ
protein, it is important to remember that eventually a nanoparticle will be attached to
these compounds via a large PEG linker. Therefore, there must be space in the predicted
binding site for this linker without affecting the binding of the ligand. High scoring
compounds which protruded slightly out of the binding pocket were highlighted as it was
predicted that these small molecules could accommodate an attached nanoparticle
without affecting the binding of the small molecule. For example, the compound shown
in Figure A-8, right panel, would not be a good choice for this project because the
molecule slips into the hole and a large linker may interfere with the binding. While it is
possible to chemically modify the molecule, it would add more complexity to the project.
244
Figure A-6: Representative set of compounds identified through molecular docking for site 1 for human HER2 from zinc database of drug-like compounds (http://zinc.docking.org/).
245
Figure A-7: Representative set of compounds identified through molecular docking for site 2 for human HER2 from zinc database of drug-like compounds (http://zinc.docking.org/). .
246

Figure A-8: Representative small molecules docked into site 1. Left panel = zinc ID # 331908, Right panel = zinc ID # 1231760.
Figure A-9: Representatives small molecules docked into site 2. Left panel = zinc ID # 218583, Right panel = zinc ID # 1302657.
Molecular docking identified numerous compounds for both site 1 and site 2 for the
extracellular domain of human HER2. The next stage was to express and purify the
protein to begin the experimental procedures to determine if the computationally
predicted molecules do in fact bind to the protein.
247

Expression and Purification of the ECD of human HER2
HER2 is a mammalian protein and has been expressed in mammalian cells by other
research groups. However, protein expression using mammalian cells is extremely
difficult, time consuming, and expensive. Furthermore, resources are currently
unavailable. Potential collaborators were contacted in attempts to obtain this protein but
unfortunately the protein was unavailable in sufficient quantities necessary for this
project (i.e. approximately 10 mg). The genetic material was obtained from Dr. Dan
Leahy at Johns Hopkins University School of Medicine. Using experimental procedures
from the literature with a few modifications, the protein was expressed in E. coli
cells.13,32 The ECD of human HER2 contains two glycosylation sites which will not be
present through this expression protocol. We believed this was not going to be a problem
as these glycosylation sites are far from the THEMATICS predicted binding sites. After
successful expression of the protein, the ECD of HER2 was purified using glutathione-
containing affinity matrices (i.e. small columns or beads).14,33
Supplemental Chapter.4 Future Work
Based on the current expression and purification protocol, most of the protein is
expressed in an insoluble form; only approximately 1 mg of soluble protein can be
purified from 1 liter of expression broth. While this quantity will be enough for small
scale studies, it will be necessary to attempt to purify the insoluble protein on a larger
scale. While methods do exist for this process, numerous studies will need to be
performed in order to determine that the protein is folded correctly.13 We have been able
248

to denature and re-fold the insoluble portion in small amounts. One major problem is that
this protein is extremely large and extremely hydrophobic. Therefore, it is difficult to
concentrate without losing at least 75%. Since the denaturation and refolding process
requires buffers on a large scale, this is one hurdle that needs to be overcome.
Additionally, since the purification protocol uses glutathione-containing affinity matrices
for the purification step, it will be necessary to remove the GST-tag prior to performing
the binding studies. The expression vector was designed to include a TEV cleavage site
which allows the cleavage of the GST-tag with a TEV protease. This site is intact as we
have succeeded in cleaving the GST-tag on a very small scale. Additionally, it will be
important to remove the GST-tag from solution as well as the TEV protease. The TEV
protease is also tagged with GST. Therefore, after cleavage, the GST-tag and the GST-
tagged TEV protease should be purified with the use of glutathione-containing affinity
matrices. The goal is to be left with pure ECD HER2.
The next stage of the project will be to scale up the expression and purification protocol,
in addition to working out a large scale procedure to cleave the GST-tag and remove this
tag and the GST-tagged TEV protease. Following successful scale-up, the next step will
be the binding studies. There are four proposed methods to determine binding, and
include 1) a thermofluor assay using a real time PCR (Brandeis University, Waltham,
MA)8, 2) Surface Plasmon Resonance (SPR) using a Biacore system (Brandeis
University, Waltham, MA),10 3) Isothermal Titration Calorimetry (ITC)9 (MIT,
Cambridge, MA), or 4) crystallography. The starting point for the binding assays will be
249

to use the thermofluor assay as it is simple to set-up, use and analyze. Method 2 using the
Biacore system would require extensive method development and therefore may not be
the first method to try. Additionally, while ITC may provide useful information, it
requires numerous mg’s of purified protein. The main problem with techniques 1, 2 and 3
is that if binding is in fact detected, the exact area of binding will be unknown. It will
therefore be necessary to crystallize the protein/ligand complex to be completely sure the
ligand is binding to the predicted site. The protein has been crystallized as there are
structures deposited in the protein data bank. Therefore, there will be a starting point once
this stage is reached.
Supplemental Chapter.5 Conclusions
THEMATICS was used to identify two previously unknown binding sites for the
extracellular domain of the cancer biomarker, HER2. Through molecular docking studies,
small molecule ligands were identified which are predicted to bind to these sites with
favorable energies, according to Glide. Finally, the protein was expressed and purified on
a small scale. The work that has been done thus far provides a proof of concept that
THEMATICS may be able to identify binding sites of previously unknown function.
Computationally, these sites appear to bind predicted drug-like molecules. Future work
will hopefully verify that these molecules can bind with nanomolar affinities. There is a
great deal of work that needs to be done before the attachment of the nanoparticle can be
discussed.
250

Supplemental Chapter.6 References 1. Ko, J., Murga, L. F., Andre, P., Yang, H., Ondrechen, M. J., Williams, R. J.,
Agunwamba, A. & Budil, D. E. (2005). Statistical criteria for the identification of protein active sites using Theoretical Microscopic Titration Curves. Proteins 59, 183-195.
2. Murga, L. F., Wei, Y. & Ondrechen, M. J. (2007). Computed Protonation Properties: Unique Capabilities for Protein Functional Site Prediction. Genome Informatics 19, 107-118.
3. Ondrechen, M. J., J.G. Clifton and D. Ringe. (2001). THEMATICS: A simple computational predictor of enzyme function from structure. Proc. Natl. Acad. Sci. (USA) 98, 12473-12478.
4. Ondrechen, M. J., L.F. Murga, J.G. Clifton and D. Ringe. (2003). Prediction of Protein Function with THEMATICS. Currents in Computational Molecular Biology, 21-22.
5. van Vlerken, L. E., Vyas, T. K. & Amiji, M. M. (2007). Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm Res 24, 1405-1414.
6. Zhou, Z., Felts, A. K., Friesner, R. A. & Levy, R. M. (2007). Comparative performance of several flexible docking programs and scoring functions: enrichment studies for a diverse set of pharmaceutically relevant targets. J Chem Inf Model 47, 1599-1608.
7. McNeil, S. E. (2005). Nanotechnology for the biologist. J Leukoc Biol 78, 585-594.
8. Ericsson, U. B., Hallberg, B. M., Detitta, G. T., Dekker, N. & Nordlund, P. (2006). Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem 357, 289-298.
9. Velazquez-Campoy, A. & Freire, E. (2006). Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat Protoc 1, 186-191.
10. Okochi, M., Nomura, T., Zako, T., Arakawa, T., Iizuka, R., Ueda, H., Funatsu, T., Leroux, M. & Yohda, M. (2004). Kinetics and binding sites for interaction of the prefoldin with a group II chaperonin: contiguous non-native substrate and chaperonin binding sites in the archaeal prefoldin. J Biol Chem 279, 31788-31795.
11. Wei, Y., Ko, J., Murga, L. & Ondrechen, M. J. (2007). Selective prediction of Interaction sites in protein structures with THEMATICS. BMC Bioinformatics 8, 119.
12. Cho, H. S., Mason, K., Ramyar, K. X., Stanley, A. M., Gabelli, S. B., Denney, D. W., Jr. & Leahy, D. J. (2003). Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756-760.
13. Liu, X., He, Z., Zhou, M., Yang, F., Lv, H., Yu, Y. & Chen, Z. (2007). Purification and characterization of recombinant extracellular domain of human HER2 from Escherichia coli. Protein Expr Purif 53, 247-254.
14. Frangioni, J. V. & Neel, B. G. (1993). Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem 210, 179-187.
251

15. Benzinger, A., Popowicz, G. M., Joy, J. K., Majumdar, S., Holak, T. A. & Hermeking, H. (2005). The crystal structure of the non-liganded 14-3-3sigma protein: insights into determinants of isoform specific ligand binding and dimerization. Cell Res 15, 219-227.
16. Yaffe, M. B. (2002). How do 14-3-3 proteins work?-- Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett 513, 53-57.
17. Lee, M. H. & Lozano, G. (2006). Regulation of the p53-MDM2 pathway by 14-3-3 sigma and other proteins. Semin Cancer Biol 16, 225-234.
18. Mhawech, P. (2005). 14-3-3 proteins--an update. Cell Res 15, 228-236. 19. Medina, A., Ghaffari, A., Kilani, R. T. & Ghahary, A. (2007). The role of stratifin
in fibroblast-keratinocyte interaction. Mol Cell Biochem 305, 255-264. 20. Wilker, E. W., Grant, R. A., Artim, S. C. & Yaffe, M. B. (2005). A structural
basis for 14-3-3sigma functional specificity. J Biol Chem 280, 18891-18898. 21. Ferguson, A. T., Evron, E., Umbricht, C. B., Pandita, T. K., Chan, T. A.,
Hermeking, H., Marks, J. R., Lambers, A. R., Futreal, P. A., Stampfer, M. R. & Sukumar, S. (2000). High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci U S A 97, 6049-6054.
22. Mhawech, P., Benz, A., Cerato, C., Greloz, V., Assaly, M., Desmond, J. C., Koeffler, H. P., Lodygin, D., Hermeking, H., Herrmann, F. & Schwaller, J. (2005). Downregulation of 14-3-3sigma in ovary, prostate and endometrial carcinomas is associated with CpG island methylation. Mod Pathol 18, 340-348.
23. Lodygin, D., Diebold, J. & Hermeking, H. (2004). Prostate cancer is characterized by epigenetic silencing of 14-3-3sigma expression. Oncogene 23, 9034-9041.
24. Osada, H., Tatematsu, Y., Yatabe, Y., Nakagawa, T., Konishi, H., Harano, T., Tezel, E., Takada, M. & Takahashi, T. (2002). Frequent and histological type-specific inactivation of 14-3-3sigma in human lung cancers. Oncogene 21, 2418-2424.
25. Slamon, D. J., Godolphin, W., Jones, L. A., Holt, J. A., Wong, S. G., Keith, D. E., Levin, W. J., Stuart, S. G., Udove, J., Ullrich, A. & et al. (1989). Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244, 707-712.
26. Arai, Y., Yoshiki, T. & Yoshida, O. (1997). c-erbB-2 oncoprotein: a potential biomarker of advanced prostate cancer. Prostate 30, 195-201.
27. Cohen, J. A., Weiner, D. B., More, K. F., Kokai, Y., Williams, W. V., Maguire, H. C., Jr., LiVolsi, V. A. & Greene, M. I. (1989). Expression pattern of the neu (NGL) gene-encoded growth factor receptor protein (p185neu) in normal and transformed epithelial tissues of the digestive tract. Oncogene 4, 81-88.
28. Christianson, T. A., Doherty, J. K., Lin, Y. J., Ramsey, E. E., Holmes, R., Keenan, E. J. & Clinton, G. M. (1998). NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res 58, 5123-5129.
29. Eisenhauer, E. A. (2001). From the molecule to the clinic--inhibiting HER2 to treat breast cancer. N Engl J Med 344, 841-842.
30. Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., Fleming, T., Eiermann, W., Wolter, J., Pegram, M., Baselga, J. & Norton, L.
252

(2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344, 783-792.
31. Franklin, M. C., Carey, K. D., Vajdos, F. F., Leahy, D. J., de Vos, A. M. & Sliwkowski, M. X. (2004). Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317-328.
32. Yuan, C. X., Lasut, A. L., Wynn, R., Neff, N. T., Hollis, G. F., Ramaker, M. L., Rupar, M. J., Liu, P. & Meade, R. (2003). Purification of Her-2 extracellular domain and identification of its cleavage site. Protein Expr Purif 29, 217-222.
33. Smith, D. B. & Johnson, K. S. (1988). Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31-40.
253

Curriculum Vitae
Heather R. Brodkin 28 Edward Road, West Newton, MA 02465 (617) 916-5397
Education: Northeastern University Boston, Massachusetts PhD, Chemistry expected May 2009 “Evidence for Multilayer Nanoscale Enzyme Active Sites”
Masters Degree, Chemistry, Magna Cum Laude May 2004
Research: Working with Dr. Mary Ondrechen, my computational work lead to the hypothesis that, in addition to the residues in direct contact with the reacting substrate of an enzyme, residues outside this first shell of the active site can also play important roles in catalytic function. My doctoral dissertation provides computational and experimental evidence for multilayer active sites in enzymes. In particular, it is shown that residues in the “second shell” are important for enzyme reactivity and specificity.
Courses: Analytical Biotechnology, Analytical Separations, Biochemistry, Foundations of Spectroscopy, Fundamentals of Molecular Structure, Physical Methods, Principles of Mass Spectrometry, Research Skills and Ethics, Special Topics in Physical Chemistry – Molecular Modeling, Spectroscopy of Organic Compounds and Thermodynamics I.
Scientific Presentations:
• Are Enzyme Active Sites Built in Multiple Layers? – Protein Society Meeting - 2007
• Evidence for Participation of Remote Residues in the Catalytic Activity of Nitrile Hydratase – ACS 2006
• Experimental Evidence for the Functional Importance of Residues Predicted by THEMATICS – AFP 2005
• Selective tRNA Analysis Using MALDI-TOF Mass Spectrometry – ASMS 2003
• Improved Method for tRNA Analysis Using MALDI-TOF Mass Spectrometry – CNECC and NU Technology Exposition 2003
Framingham State College Framingham, Massachusetts
Bachelors Degree, Chemistry, ACS Approved, Sept. 1996 – Dec. 1999 Magna Cum Laude
• Date of Graduation: May 2000 • Compiled data and completed senior research project using NMR titled “
1H Spectral Analysis of Amino Acids and Peptides - A Biochemistry Experiment”.
254
Heather R. Brodkin 28 Edward Road, West Newton, MA 02465 (617) 916-5397
Technical Brandeis University Skills: • Stratagene QuikChange® II XL Site – Directed Mutagenesis Kit,
Protein Expression, Protein purification using FPLC with UV detection, X-ray Crystallography (HKL and Coot), Computational Docking using Glide
Northeastern University • Molecular modeling, computational chemistry & computational biology
tools, bioinformatics tools, THEMATICS • Bruker Daltonics OmniFlex MALDI-TOF with OmniFlex TOF Control,
Applied Biosystems MALDI-TOF, Bio-rad Polyacrylimide Gel Electrophoresis, Agilent 1090 and 1100 HPLC with ChemStation software and PDA - UV detection.
Alkermes, Inc. • Waters 2690 and 2695 HPLC separation module with Millenium 32
software, Dual Wavelength UV, Photodiode Array, Fluorescence, ELSD, 756 Karl Fischer Coulometer with 774 Oven Sample Processor
Technical ASMS Montreal, Canada Training: • MALDI-TOF MS: Fundamentals June 2003 and Applications Waters Milford, Massachusetts
• Alliance 2690 and 2695 HPLC Sept. 2002 Performance Maintenance
• Millenium32 Version 3.20 Software Feb. 2001 Training
SAS Boston, Massachusetts • JMP Software: Design and Feb. 2002
Analysis of Experiments Chromatography Institute of America Framingham, Massachusetts
• Normal and Reverse Phase HPLC Sept. 2001 Educational Northeastern University Achievements: • NSF-IGERT Traineeship Sept. 2005- Sept. 2008
• Chairman of colloquium committee Sept. 2007 – Sept 2008 • Vice chairman colloquium committee Sept. 2006 – Sept. 2007 • Chemistry Department, GSA committee member Sept. 2005 – Sept. 2008 • Graduate Student Assoc Travel Award, ACS SF meeting Sept. 2006
Framingham State College
• American Institute of Chemists Award April 2000 • Analytical Chemistry Award April 1999
• Polymer Chemistry Award April 1998
255

Heather R. Brodkin 28 Edward Road, West Newton, MA 02465 (617) 916-5397
Technical Brandeis University Waltham, Massachusetts Experience: Visiting Dissertation Research Scholar Jan. 2005 – Jan. 2009
• Perform Site – Directed Mutagenesis using Stratagene QuikChange® II XL Site – Directed Mutagenesis Kit to include PCR, digestion, transformation, cell lysis, etc. on Nitrile Hydratase from Pseudomonas Putida.
• Expression of wild type Nitrile Hydratase and mutant Nitrile Hydratase proteins.
• Purification of Nitrile Hydratase proteins using an FPLC with DEAE, HIC and MonoQ column chemistries.
• Exhaustive screening for crystals using numerous kits from Hampton Research and home made buffers
• X-ray Crystallography experience at Argonne National Labs, GM/CA CAT ID-B and ID-D, using both mini-beam and regular beam capabilities
• Experience with crystallography software HKL and Coot Northeastern University Boston, Massachusetts Ph.D. Candidate Research Assistant June 2003 – Jan. 2009
• Development of the hypothesis that enzyme active sites consist of multiple layers
• Use of computational and bioinformatics tools, including THEMATICS, ConSurf, & Evolutionary Trace, to predict functionally important residues
• Determine the effects of input parameters on THEMATICS results. • Explore the conservation of THEMATICS positives versus known literature
positives and also identify the conservation of ‘second shell’ residues. • Develop kinetics assay for Nitrile Hydratase using HPLC.
Teaching Assistant Mar. 2003 – May 2004 • Recitation teacher for Chemistry I and II for Biology and Pharmacy majors • Preparation of lectures and weekly quizzes; Grading of homework, quizzes
and exams • Maintain class rosters and grades for recitation, lab and class exams • Weekly tutoring sessions for all chemistry students
Alkermes, Inc. Cambridge, Massachusetts Research Associate II June 2002 – Mar. 2003
• Execute numerous stability studies for protein drugs • Design and execute research protocols • Draft reports summarizing research and GMP/GLP studies • Maintain, troubleshoot and perform both automated and direct method
moisture analysis using Karl Fisher • Anderson Cascade Impaction and Emitted Dose testing and analysis • Quarterly presentations to pulmonary formulations group
256

257
Heather R. Brodkin 28 Edward Road, West Newton, MA 02465 (617) 916-5397
Research Associate I July 2000 – June 2002
• Develop and validate both RP and SE HPLC analytical methods for the identification, separation and quantitation of protein drugs
• Develop, initiate and execute validation protocols, standard operating procedures and stability protocols
• Maintain, troubleshoot and perform both automated and direct method moisture analysis using Karl Fisher
• Accumulate and report data acquired for both research and GMP/GLP studies
• Perform daily chemical testing and analysis to support formulation optimization
• Heath and Safety Representative for pulmonary formulations group (Jan. 2002 – Mar. 2003)