Embed Size (px)
Citation preview

Neurotoxicology and Teratology, Vol. 20, No. 5, pp. 523–530, 1998© 1998 Elsevier Science Inc.
Printed in the USA. All rights reserved0892-0362/98 $19.00
1
.00
PII S0892-0362(97)00135-9
523
Ethanol Inhibition of Brain Ornithine Decarboxylase Activity in the Postnatal Rat
MARK DAVIDSON, KULDIP BEDI AND PETER WILCE
Departments of Biochemistry and Anatomy, The University of Queensland, QLD, Australia
Received 7 July 1997; Accepted 14 November 1997
DAVIDSON, M., K. BEDI AND P. WILCE.
Ethanol inhibition of brain ornithine decarboxylase activity in the postnatalrat.
NEUROTOXICOL TERATOL
20
(5) 523–530, 1998.—The purpose of this study was to determine the relationship be-tween ornithine decarboxylase activity (ODC; a marker for perturbed cell development), the blood alcohol level, and alco-hol-induced microencephaly in the developing rat brain after binge treatment with ethanol vapour. By manipulating ethanolflow we were able to adjust vapour concentrations (24–65 mg ethanol
/
l air) such that an acute exposure of ethanol vapour for3 h resulted in a range of blood alcohol levels (2.3–5.5 mg
/
ml). Acute studies showed that ethanol dose-dependently inhibitedrat hippocampal and cerebellar ODC activity at PND4–PND10. There was a significant correlation between the blood alco-hol level and degree of inhibition at all ages tested. Chronic treatment from PND4 to PND9 caused a significant decrease inboth brain to body weight ratio and in hippocampal and cerebellar ODC activities at PND10. These results indicate thatethanol-induced disruption in ODC could play a significant role in ethanol’s teratogenic effects during early postnataldevelopment. © 1998 Elsevier Science Inc.
Fetal alcohol syndrome Microencephaly Polyamine Hippocampus Cerebellum Brain development
ETHANOL is a teratogen inducing the clinically recognisedfetal alcohol syndrome (FAS) and alcohol-related birth de-fects (ARBD) (13). Mechanisms underlying the teratogenicityof ethanol have not been defined, although major hypotheseshave been developed (28,39) based on animal models, partic-ularly studies during the postnatal brain growth spurt in therat (40). The brain growth spurt, the period of most rapidbrain growth, occurs during the third prenatal trimester in hu-mans and continues into postnatal life (6). The equivalent pe-riod in the rat is the first 2 weeks of postnatal life. Rat pups ar-tificially reared and repeatedly exposed to ethanol during thevulnerable time period [postnatal day (PND) 4–10] show asignificant correlation between peak blood alcohol level(BAL) and the degree of microencephaly, neuronal loss, be-havioural hyperactivity, and impaired spatial navigation(3,40). West and his colleagues have also shown that a singlehigh dose of alcohol (BAL
5
3.6 mg
/
ml) at PND4 could causesignificant loss in regional brain weights at PND10 (4,11).
Polyamines have a complex but poorly understood regula-tory role in cell growth, proliferation, and differentiation (19).Ornithine decarboxylase (ODC) is the key regulatory enzymefor polyamine biosynthesis in the mammalian brain. Rat and
mouse brain ODC activity peaks shortly after birth andsharply declines thereafter. The postnatal rat brain is particu-larly sensitive to alterations in polyamine homeostasis. Inhibi-tion of ODC activity leads to impaired synaptic developmentand associated neurochemical and behavioural deficits (29).ODC has a short half-life (10–20 min), making it very respon-sive to conditions that perturb development or cause cell in-jury (29). Major drugs of abuse and environmental pollutantsthat cause abnormalities in neuronal development during thethird trimester equivalent have variable effects on ODC activ-ity. Specifically, cocaine suppresses ODC activity in rat fore-brain and mid
/
hind brain (26,34), lead and methylmercury ex-posure elevates ODC activity in the rat cerebellum (30,42),whereas nicotine rapidly stimulates ODC activity in the ratwith age and region dependence corresponding to the devel-opment of central nicotinic receptors (32). There are fewstudies on the effect of ethanol on the developmental profileof ODC activity in the postnatal rat. Thadani et al. (35,36)showed both inhibition and stimulation of ODC activity inrats dependent on the time of ethanol exposure. In both re-ports BALs were not indicated. We therefore determined therelationship between ODC activity, BAL, and ethanol-induced
Requests for reprints should be addressed to Mark Davidson, Department of Biochemistry, The University of Queensland, 4072, QLD, Aus-tralia. Tel:
1
61-7-33654872; Fax:
1
61-7-33654999; E-mail: [email protected]
524 DAVIDSON, BEDI AND WILCE
microencephaly at PND4–PND10 in the rat. Not all brain re-gions are equally affected by ethanol and specific populationsof developing neurons, particularly those in the hippocampusand cerebellum, are especially sensitive (4,12,23). We there-fore focussed our experiments on these two brain regions.
METHOD
Chemicals
Ornithine, pyridoxal phosphate, NAD, alcohol dehydroge-nase, and dithiothreitol (DTT) were all purchased fromSigma-Aldrich Chemical Co. (Australia). All other chemicalswere purchased from local sources and were of the highest an-alytical grade available.
Animal Experimentation
All animal experimental procedures were approved by theAnimal Experimental and Ethics Committee of The Univer-sity of Queensland. Litters (PND2) with dam (minimum num-ber of pups
5
9) were obtained from the Central AnimalBreeding House, The University of Queensland, housed atconstant temperature of 20
6
2
8
C with a 12-h light
/
dark cycleand given free access to food and water. Individual litters be-tween PND4 and PND10 (minimum number
5
9) were ran-domly divided into suckle control, separation control, and va-pour-treated groups (minimum number of pups
5
3). Bothsexes were used. Vapour treatment was routinely performedbetween 0900 and 1200 h.
Vapour Treatment—Acute Studies
Ethanol (96% w
/
v, BDH, Kilsyth, Victoria, Australia) wasdelivered via a peristaltic pump (Pharmacia Peristaltic Pump,P1), using silicone tubing, into an air-tight flask held in a 42
8
Cwater bath. A constant stream of air (10 l air
/
min) was passedthrough the flask and the ethanol vapour flowed into a 37 lchamber into which the pups were placed. The temperature inthe vapour chamber of 22
6
0.2
8
C did not differ significantlyfrom the ambient temperature. The pump speed was adjustedto vary the ethanol flow rate (0.75–1.75 ml
/
min) and createdifferent levels of ethanol vapour. The vapour concentrationreached equilibrium within 1 h and was maintained at a con-stant level for the duration of exposure. The vapour groupwas exposed for a total period of 3 h (including equilibriumtime) to a vapour concentration of between 25 and 65 mg eth-anol
/
l air (measured at the end of the 3 h exposure) to gener-ate a range of BALs. Separation controls were separated fromtheir mother and housed under similar conditions for anequivalent time period. Control pups were allowed to sucklecontinuously. At the end of the 3-h exposure pups were killed,blood collected for immediate assay, brains rapidly removed,and the hippocampi and cerebella separated, placed in liquidN
2
and stored at
2
80
8
C until used for assays.
Vapour Treatment—Chronic Studies
For chronic ethanol treatment, pups were binge ‘treated’by exposing the pups to a vapour concentration of 40 mg etha-nol
/
l air for 3 h each day from PND4 to PND9. At PND10, thepups were killed 24 h after last exposure to ethanol. The brainand body weights were recorded before the brain was dis-sected, placed in liquid N
2
, and stored at
2
80
8
C until used forassays. For these chronic studies, a separate naive group of
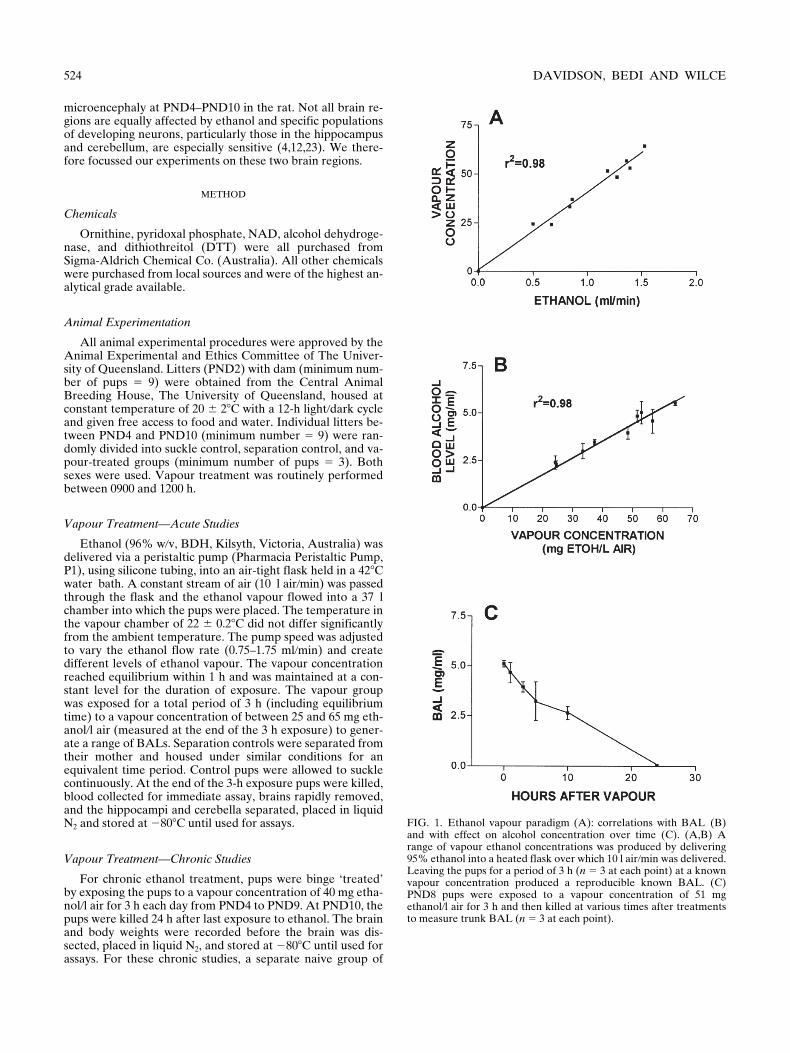
FIG. 1. Ethanol vapour paradigm (A): correlations with BAL (B)and with effect on alcohol concentration over time (C). (A,B) Arange of vapour ethanol concentrations was produced by delivering95% ethanol into a heated flask over which 10 l air/min was delivered.Leaving the pups for a period of 3 h (n 5 3 at each point) at a knownvapour concentration produced a reproducible known BAL. (C)PND8 pups were exposed to a vapour concentration of 51 mgethanol/l air for 3 h and then killed at various times after treatmentsto measure trunk BAL (n 5 3 at each point).

ETHANOL INHIBITION OF BRAIN ODC 525
RESULTS
Effect of Ethanol Inhalation Vapour on Blood Alcohol Levels and Body and Brain Weights
Figure 1 shows initial parameters that were adjusted toachieve the BAL required for the different experimental par-adigms. Although ethanol vapour concentrations were high (Fig1A, B) there was no mortality at any age. The vapour-inducedBAL showed a similar time course of elimination (Fig. 1C) tothat achieved in artificially reared pups (11), but both are dif-ferent from that of vapour-treated adult rats (7). The BAL re-mained high for 10–12 h after removal of ethanol from the airand was still detectable at 24 h (0.04
6
0.03 mg
/
ml; Fig. 1C).Daily exposure from PND4 to PND9 to ethanol vapour suffi-cient to produce a BAL of approximately 3.5 mg
/
ml resultedin a significant reduction in brain weight and body
/
brain weightratio (microencephaly; Table 1).
Ethanol Inhibition of ODC Activity: Correlation With BAL
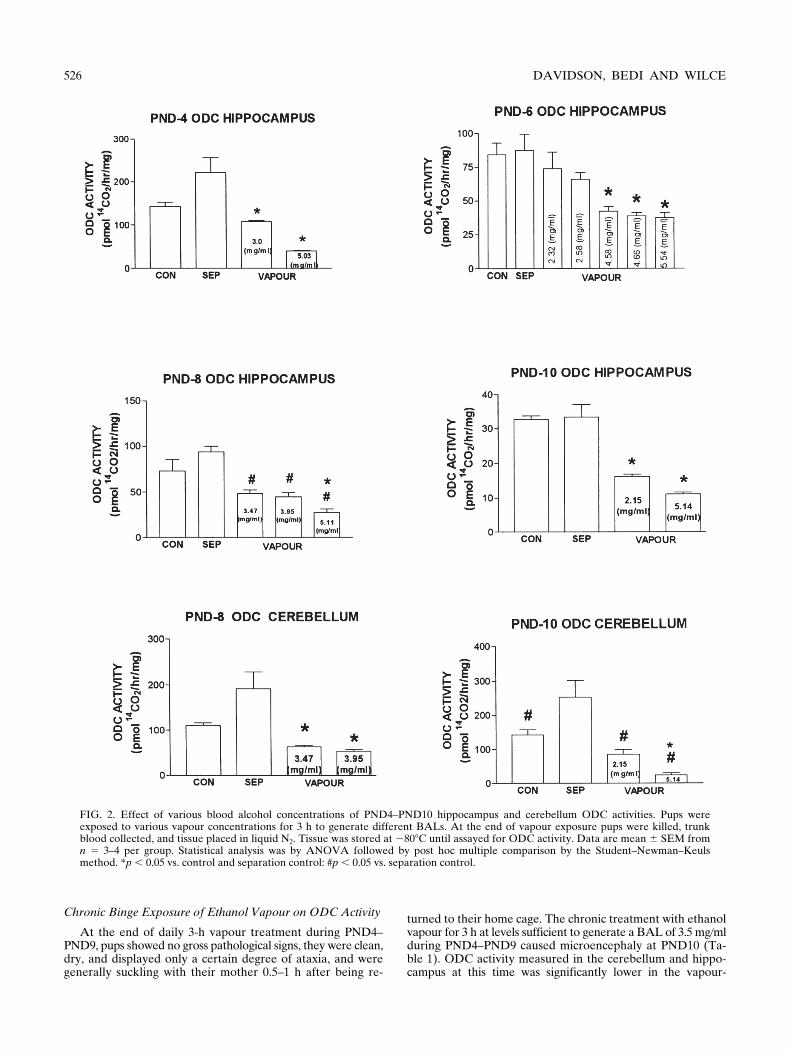
At the end of 3-h vapour treatment, pups showed no grosspathological effects. They were clean, dry, and displayed onlymild ataxia at the lower BALs (1–3 mg
/
ml), whereas at thehigher BALS (4–5 mg
/
ml) pups were noticeably sedated, le-thargic, and displayed a degree of hypothermia. There was nomortality during the exposure in any PND group. By adjust-ing ethanol concentration in the air we were able to achieve arange of BALs and then assess the effect on ODC activity atvarious ages during pup development. Figure 2 shows datafrom the hippocampi of pups at PND4, PND6, PND8, andPND10, and cerebella of groups at PND8 and PND10. Consis-tent with the reports of others (30,33,42), hippocampal ODCactivity was high at PND4 and was reduced on subsequentdays (Fig. 2). Cerebellum ODC activity was similar at PND8and PND10 (Fig. 2) (35,36). The ODC activity in the separa-tion control was higher at all PNDs but only reached signifi-cance at PND10 in the cerebellum (Fig. 2). In general, a BALof approximately 3.0 mg
/
ml significantly reduced ODC activ-ity, but with some age-related differences. Notably, a BAL of2.58 mg
/
ml did not significantly inhibit hippocampal ODC ac-tivity at PND6 yet a BAL of 2.15 mg
/
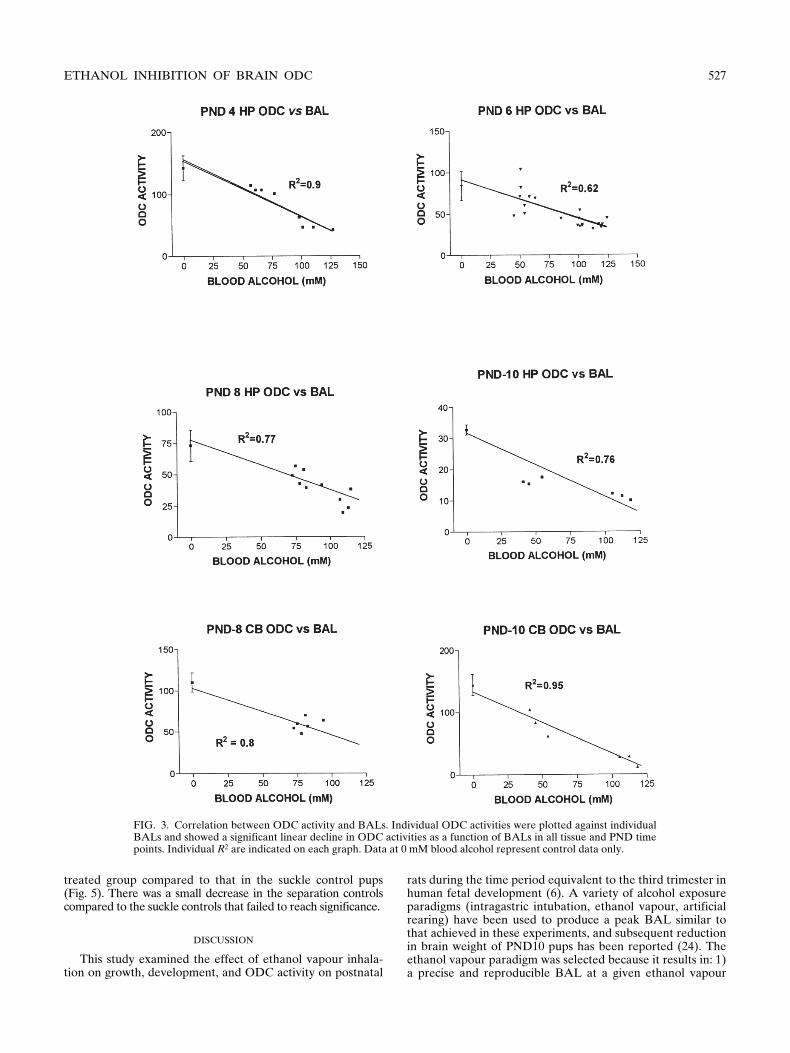
ml caused significant in-hibition at PND10 (Fig. 2). The data in Fig. 2 suggest thatODC activity is inversely related to the BAL. More detailedanalysis (Fig. 3) directly correlated ODC activity and BALs(expressed in terms of mM for clarification and comparison toFig. 4) at the different PNDs in both hippocampus and cere-bellum. There was a significant negative correlation (
p
,
0.05) in all age groups and tissues studied. This was not a di-rect effect of ethanol on enzyme activity because ethanol, upto 100 mM, added directly to the assays in vitro (naive tissue)did not inhibit activity (Fig 4).
pups was used to monitor BAL levels at the end of each dayof treatment.
Ornithine Decarboxylase Activity
Tissue was homogenised in 10 vol of 50 mM Tris-HCl, pH7.4, 0.1 mM EDTA, 5 mM DTT, and 0.1 mM pyridoxal phos-phate. After centrifugation at 20,000
3
g
for 20 min at 4
8
C,the supernatant was collected and assayed in triplicate forODC activity by trapping
14
CO
2
as described by Gaines et al. (8).Each assay (125
m
l final volume) containing 50 mM Tris-HCl,pH 7.4, 0.1 mM EDTA, 5 mM DTT, 0.1 mM pyridoxal phos-phate, 0.4 mM ornithine, and 0.125
m
Ci of
L
-[1-
14
C]ornithine(57.2 mCi
/
mmol, Du Pont, Australia) was initiated by intro-ducing up to 100
m
g of supernatant protein and incubated at37
8
C for 1 or 2 h. Gaines et al. (9) showed non-ODC-gener-ated
14
CO
2
when frozen whole kidney and liver tissue were as-sayed. DFMO (5 mM) (Merk Merril and Dow, MA) was there-fore included in some assays. Inclusion of DFMO completelyinhibited
14
CO
2
generation to the levels of assays without pro-tein, indicating that all the reaction product resulted fromODC activity. Reactions were stopped by addition of 50
m
l of50% (w
/
v) TCA and allowed to continue for a further 30 min.Scintillation fluid containing 20% (v
/
v) 2-methoxyethanol(AJAX Chemicals, Sydney, Australia) was added to filterspreviously soaked with ethanolamine
/
2-methoxyethanol (4:5)and counted in a Beckman 3801 Scintillation counter. Proteinwas assayed according to the procedure of Lowry et al. (17).
Blood Alcohol Levels and Vapour Concentration
For the determination of BAL in the acute studies, trunkblood was taken at the end of the vapour treatment and as-sayed by the enzymatic spectrophotometric method describedby Lundquist (18). To determine ethanol concentration in thechamber, triplicate 0.2-ml aliquots of vapour were taken usingan air-tight syringe. The samples were immediately injectedinto assay buffer and concentrations calculated by compari-sons with known standards (18).
Statistical Analysis
Because there was minimum of three treatment groups, alldata were expressed as mean
6
SEM and analysed by one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls method for post hoc comparisons betweengroups with the level of significance set at 0.05. Linear regres-sion analysis of ODC activity vs. BAL was performed usingPRISM software package from Graphpad (Graphpad Soft-ware Inc., San Diego, CA).
TABLE 1
EFFECT OF CHRONIC BINGE EXPOSURE OF ETHANOL VAPOUR DURING POSTNATAL DAYS 4–9 ONBODY AND BRAIN WEIGHTS AT POSTNATAL DAY 10
GroupBody weight
PND4 (g)BAL PND4–PND9
(mg/ml)Vapour mg
Ethanol/l AirBody WeightPND10 (g) Brain Weight (g) Ration (%)*
Control (5) 11.3
6
1.1 — — 20.3
6
0.5 0.999
6
0.02 4.91
6
0.2Separation (5) 11.0
6
1.4 — — 20.8
6
0.5 0.986
6
0.02 4.74
6
0.01Vapour (5) 11.8
6
1.5 3.48
6
0.6 41.9
6
3.3 20.3
6
0.9 0.88
6
0.05† 4.34
6
0.2†
All measures are mean
6
SEM.*Brain weight/body weight as a percentage.†
p
,
0.05 vs. control and separation groups: ANOVA followed by Student–Newman–Keuls post hoc multiple comparison test.
526 DAVIDSON, BEDI AND WILCE
Chronic Binge Exposure of Ethanol Vapour on ODC Activity
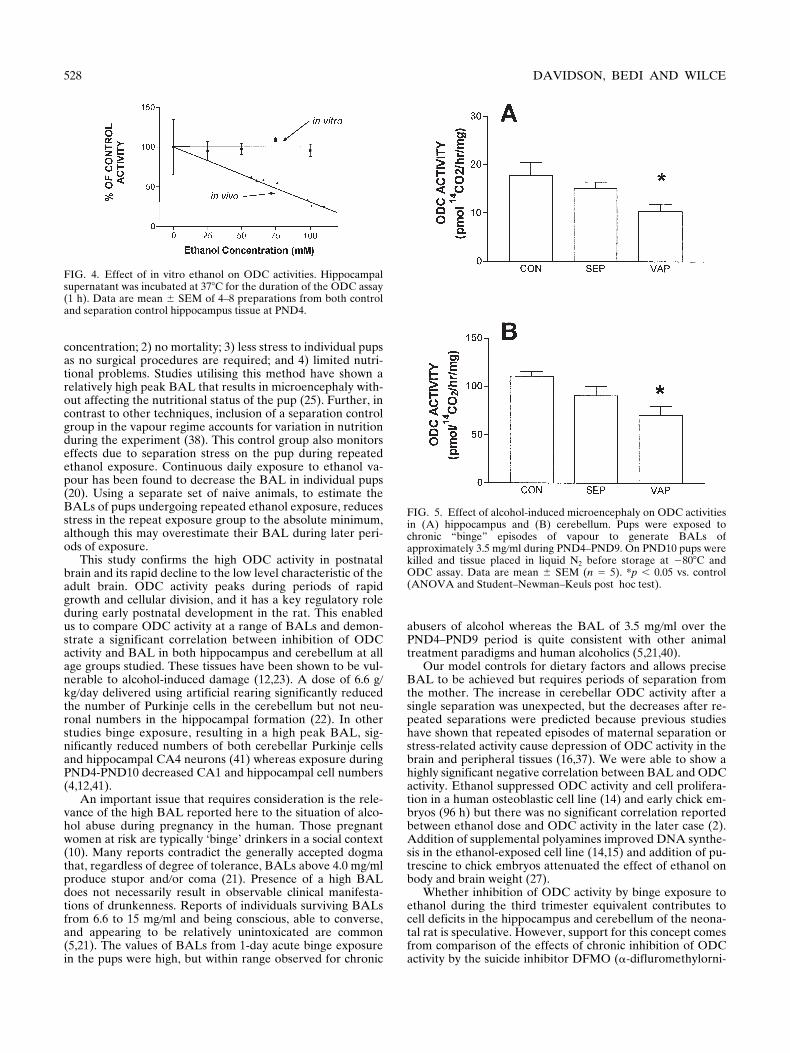
At the end of daily 3-h vapour treatment during PND4–PND9, pups showed no gross pathological signs, they were clean,dry, and displayed only a certain degree of ataxia, and weregenerally suckling with their mother 0.5–1 h after being re-
turned to their home cage. The chronic treatment with ethanolvapour for 3 h at levels sufficient to generate a BAL of 3.5 mg
/
mlduring PND4–PND9 caused microencephaly at PND10 (Ta-ble 1). ODC activity measured in the cerebellum and hippo-campus at this time was significantly lower in the vapour-
FIG. 2. Effect of various blood alcohol concentrations of PND4–PND10 hippocampus and cerebellum ODC activities. Pups wereexposed to various vapour concentrations for 3 h to generate different BALs. At the end of vapour exposure pups were killed, trunkblood collected, and tissue placed in liquid N2. Tissue was stored at 2808C until assayed for ODC activity. Data are mean 6 SEM fromn 5 3–4 per group. Statistical analysis was by ANOVA followed by post hoc multiple comparison by the Student–Newman–Keulsmethod. *p , 0.05 vs. control and separation control: #p , 0.05 vs. separation control.
ETHANOL INHIBITION OF BRAIN ODC 527
treated group compared to that in the suckle control pups(Fig. 5). There was a small decrease in the separation controlscompared to the suckle controls that failed to reach significance.
DISCUSSION
This study examined the effect of ethanol vapour inhala-tion on growth, development, and ODC activity on postnatal
rats during the time period equivalent to the third trimester inhuman fetal development (6). A variety of alcohol exposureparadigms (intragastric intubation, ethanol vapour, artificialrearing) have been used to produce a peak BAL similar tothat achieved in these experiments, and subsequent reductionin brain weight of PND10 pups has been reported (24). Theethanol vapour paradigm was selected because it results in: 1)a precise and reproducible BAL at a given ethanol vapour
FIG. 3. Correlation between ODC activity and BALs. Individual ODC activities were plotted against individualBALs and showed a significant linear decline in ODC activities as a function of BALs in all tissue and PND timepoints. Individual R2 are indicated on each graph. Data at 0 mM blood alcohol represent control data only.
528 DAVIDSON, BEDI AND WILCE
concentration; 2) no mortality; 3) less stress to individual pupsas no surgical procedures are required; and 4) limited nutri-tional problems. Studies utilising this method have shown arelatively high peak BAL that results in microencephaly with-out affecting the nutritional status of the pup (25). Further, incontrast to other techniques, inclusion of a separation controlgroup in the vapour regime accounts for variation in nutritionduring the experiment (38). This control group also monitorseffects due to separation stress on the pup during repeatedethanol exposure. Continuous daily exposure to ethanol va-pour has been found to decrease the BAL in individual pups(20). Using a separate set of naive animals, to estimate theBALs of pups undergoing repeated ethanol exposure, reducesstress in the repeat exposure group to the absolute minimum,although this may overestimate their BAL during later peri-ods of exposure.
This study confirms the high ODC activity in postnatalbrain and its rapid decline to the low level characteristic of theadult brain. ODC activity peaks during periods of rapidgrowth and cellular division, and it has a key regulatory roleduring early postnatal development in the rat. This enabledus to compare ODC activity at a range of BALs and demon-strate a significant correlation between inhibition of ODCactivity and BAL in both hippocampus and cerebellum at allage groups studied. These tissues have been shown to be vul-nerable to alcohol-induced damage (12,23). A dose of 6.6 g
/
kg
/
day delivered using artificial rearing significantly reducedthe number of Purkinje cells in the cerebellum but not neu-ronal numbers in the hippocampal formation (22). In otherstudies binge exposure, resulting in a high peak BAL, sig-nificantly reduced numbers of both cerebellar Purkinje cellsand hippocampal CA4 neurons (41) whereas exposure duringPND4-PND10 decreased CA1 and hippocampal cell numbers(4,12,41).
An important issue that requires consideration is the rele-vance of the high BAL reported here to the situation of alco-hol abuse during pregnancy in the human. Those pregnantwomen at risk are typically ‘binge’ drinkers in a social context(10). Many reports contradict the generally accepted dogmathat, regardless of degree of tolerance, BALs above 4.0 mg
/
mlproduce stupor and
/
or coma (21). Presence of a high BALdoes not necessarily result in observable clinical manifesta-tions of drunkenness. Reports of individuals surviving BALsfrom 6.6 to 15 mg
/
ml and being conscious, able to converse,and appearing to be relatively unintoxicated are common(5,21). The values of BALs from 1-day acute binge exposurein the pups were high, but within range observed for chronic
abusers of alcohol whereas the BAL of 3.5 mg
/
ml over thePND4–PND9 period is quite consistent with other animaltreatment paradigms and human alcoholics (5,21,40).
Our model controls for dietary factors and allows preciseBAL to be achieved but requires periods of separation fromthe mother. The increase in cerebellar ODC activity after asingle separation was unexpected, but the decreases after re-peated separations were predicted because previous studieshave shown that repeated episodes of maternal separation orstress-related activity cause depression of ODC activity in thebrain and peripheral tissues (16,37). We were able to show ahighly significant negative correlation between BAL and ODCactivity. Ethanol suppressed ODC activity and cell prolifera-tion in a human osteoblastic cell line (14) and early chick em-bryos (96 h) but there was no significant correlation reportedbetween ethanol dose and ODC activity in the later case (2).Addition of supplemental polyamines improved DNA synthe-sis in the ethanol-exposed cell line (14,15) and addition of pu-trescine to chick embryos attenuated the effect of ethanol onbody and brain weight (27).
Whether inhibition of ODC activity by binge exposure toethanol during the third trimester equivalent contributes tocell deficits in the hippocampus and cerebellum of the neona-tal rat is speculative. However, support for this concept comesfrom comparison of the effects of chronic inhibition of ODCactivity by the suicide inhibitor DFMO (
a
-difluromethylorni-
FIG. 4. Effect of in vitro ethanol on ODC activities. Hippocampalsupernatant was incubated at 378C for the duration of the ODC assay(1 h). Data are mean 6 SEM of 4–8 preparations from both controland separation control hippocampus tissue at PND4.
FIG. 5. Effect of alcohol-induced microencephaly on ODC activitiesin (A) hippocampus and (B) cerebellum. Pups were exposed tochronic “binge” episodes of vapour to generate BALs ofapproximately 3.5 mg/ml during PND4–PND9. On PND10 pups werekilled and tissue placed in liquid N2 before storage at 2808C andODC assay. Data are mean 6 SEM (n 5 5). *p , 0.05 vs. control(ANOVA and Student–Newman–Keuls post hoc test).

ETHANOL INHIBITION OF BRAIN ODC 529
thine). Treatment of postnatal rats with DFMO results in a re-duction in brain weight prior to a general reduction in bodyweight (31). Further, chronic postnatal ODC inhibition has apotent effect on proliferating neurons but little effect on theirpostmitotic maturation (29). Both short and prolonged post-natal inhibition of ODC activity are effective in decreasingthe size and altering the neurochemistry of the cerebellum(33). At a cellular level, prolonged treatment with DFMO re-sulted in markedly abnormal cerebellar development withgranule and Purkinje cells being irreversibly affected (1). In-terestingly, in the hippocampus, ODC inhibition did not affectthe architecture of the fascia dentate, and this is an areashown not to be altered following ethanol-induced microen-cephaly (40). Alcohol-induced reductions in cell numbers ap-
pear specific to the CA1 and CA4 areas of the tissue(12,40,41). These data indicate important similarities betweenthe effects of ODC inhibition and ethanol exposure on loss ofcell numbers in these sensitive areas.
In conclusion, these studies show a clear ethanol-inducedinhibition of ODC activity during the PND4–PND10 timeframe that is equivalent to the vulnerable human third trimes-ter. This inhibition could be a major factor leading to ethanol-induced microencephaly.
ACKNOWLEDGEMENTS
This work was supported by the National Health and Medical Re-search Council of Australia in the form of a project grant.
REFERENCES
1. Bartolome, J. V.; Schweitzer, L.; Slotkin, T. A.; Nadler, J. V.:Impaired development of cerebellar cortex in rats treated postna-tally with a-difluromethylornithine. Neuroscience 15:203–213;1985.
2. Beeker, K.; Smith, C.; Pennington, S.: Effect of cocaine, ethanolor nicotine on ornithine decarboxylase activity in early chickembryo brain. Dev. Brain Res. 69:51–57; 1992.
3. Bonthius, D. J.; West, J. R.: Blood alcohol concentration andmicroencephaly: A dose–response study in the neonatal rat. Ter-atology 37:223–231; 1988.
4. Bonthius, D. J.; West, J. R.: Alcohol-induced neuronal loss indeveloping rats: Increased brain damage with binge exposure.Alcohol. Clin. Exp. Res. 14:107–118; 1990.
5. Davis, A. R.; Lipson, A. H.: Central nervous system tolerance tohigh blood alcohol levels. Med. J. Aust. 144:9–12; 1986.
6. Dobbing, J.; Sands, J.: Comparative aspects of the brain growthspurt. Early Hum. Dev. 3:79–83; 1979.
7. Ferko, A. P.; Bobyock, E.: Rates of ethanol disappearance fromblood and hypothermia following acute and prolonged ethanolinhalation. Toxicol. Appl. Pharmacol. 50:417–427; 1979.
8. Gaines, D. W.; Friedman, L.; Braunberg, R. C.: Facilitatedmicromethod for measurement of metabolically generated 14CO2,with application to measurement of ornithine decarboxylase.Anal. Biochem. 178:52–56; 1989.
9. Gaines, D. W.; Friedman, L.; McCann, P. P.: Apparent ornithinedecarboxylase activity, measured by 14CO2 trapping, after frozenstorage of rat tissue and rat tissue supernatants. Anal. Biochem.174:88–96; 1988.
10. Gladstone, J.; Levy, M.; Nulman, I.; Koren, G.: Characteristics ofpregnant women who engage in binge alcohol consumption. Can.Med. Assoc. J. 156:789–794; 1997.
11. Goodlett, C. R.; Marcussen, B. L.; West, J. R.: A single day ofalcohol exposure during the brain growth spurt induces brainweight restriction and cerebellar Purkinje cell loss. Alcohol7:107–114; 1990.
12. Greene, P. L.; Diaz-Granados, J. L.; Amsel, A.: Blood ethanolconcentration from early postnatal exposure: Effects on memory-based learning and hippocampal neuroanatomy in infant andadult rats. Behav. Neurosci. 106:51–61; 1992.
13. Jones, K. L.; Smith, D. W.: Recognition of fetal alcohol syndromein early infancy. Lancet 2:999–1001; 1973.
14. Klein, R. F.; Carlos, A. S.: Inhibition of osteoblastic cell prolifera-tion and ornithine decarboxylase activity by ethanol. Endocrinol-ogy 136:3406–3411; 1995.
15. Klein, R. F.; Fausti, K. A.; Carlos, A. S.: Ethanol inhibits humanosteoblastic cell proliferation. Alcohol. Clin. Exp. Res. 20:572–578; 1996.
16. Lau, C.; Cameron, A. M.; Antolick, L. L.; Stanton, M. E.:Repeated maternal separation in the neonatal rat: Cellular mech-anisms contributing to brain growth sparing. J. Dev. Physiol.17:265–276; 1992.
17. Lowry, O. H.; Rosebrough, N. J.; Farr, A. L.; Randall, R. J.: Pro-
tein measurement with the folin phenol reagent. J. Biol. Chem.193:265–275; 1951.
18. Lundquist, F.: The determination of ethyl alcohol in blood andtissues. Methods Biochem. Anal. 7:217–251; 1959.
19. Marton, L. J.; Pegg, A. E.: Polyamines as targets for therapeuticintervention. Annu. Rev. Pharmacol. Toxicol. 35:55–91; 1995.
20. Pennington, S. N.; Rumbly, R. A.; Woody, D. G.: Fetal 15-hydroxyprostaglandin dehydrogenase is altered by maternal eth-anol exposure. Biol. Neonate 40:246–251; 1981.
21. Perper, J. A.; Twerski, A.; Wienand, J. W.: Tolerance at highblood alcohol concentrations: A study of 110 cases and review ofthe literature. J. Forensic Sci. 31:212–221; 1986.
22. Pierce, D. R.; Goodlet, C. R.; West, J. R.: Differential neuronalloss following early postnatal alcohol exposure. Teratology40:113–126; 1989.
23. Pierce, D. R.; West, J. R.: Differential deficits in regional braingrowth induced by postnatal alcohol. Neurotoxicol. Teratol.9:129–141; 1987.
24. Pierce, D. R.; Serbus, D. C.; Light, K. E.: Intragastric intubationof alcohol during postnatal development of rats results in selec-tive cell loss in the cerebellum. Alcohol. Clin. Exp. Res. 17:1275–1280; 1993.
25. Ryabinin, A. E.; Cole, M.; Bloom, F. E.; Wilson, M. C.: Exposureof neonatal rats to alcohol by vapor inhalation demonstratesspecificity of microencephaly and purkinje cell loss but not astro-gliosis. Alcohol. Clin. Exp. Res. 19:784–791; 1995.
26. Seidler, F. J.; Slotkin, T. A.: Prenatal cocaine and cell develop-ment in rat brain regions: Effects on ornithine decarboxylase andmacromolecules. Brain Res. Bull. 30:91–99; 1993.
27. Shibley, I. A.; Gavigan, M. D.; Pennington, S. N.: Ethanol’s effecton tissue polyamines and ornithine decarboxylase activity: A con-cise review. Alcohol. Clin. Exp. Res. 19:209–215; 1995.
28. Shoemaker, W. J.: The neurotoxicity of alcohols. Neurobehav.Toxicol. Teratol. 3:431–436; 1981.
29. Slotkin, T. A.; Bartolome, J.: Role of ornithine decarboxylaseand the polyamines in nervous system development: A review.Brain Res. Bull. 17:307–320; 1986.
30. Slotkin, T. A.; Pachman, S.; Kavlock, R. J.; Bartolome, J.: Earlybiochemical detection of adverse effects of a neurobehavorial ter-atogen: Influence of prenatal methylmercury exposure on orni-thine decarboxylase in brain and other tissues of fetal andneonatal rat. Teratology 32:195–202; 1985.
31. Slotkin, T. A.; Seidler, F. J.; Trepanier, P. A.; Whitmore, W. L.;Lerea, L.; Barnes, G. A.; Weigel, S. J.; Bartolome, J.: Ornithinedecarboxylase and polyamines in tissues of the neonatal rat:Effects of a-difluromethylornithine, a specific, irreversible inhibi-tor of ornithine decarboxylase. J. Pharmacol. Exp. Ther. 222:741–745; 1982.
32. Smith IV, W. T.; Seidler, F. J.; Slotkin, T. A.: Acute stimulation ofornithine decarboxylase in neonatal rat brain regions by nicotine: Acentral receptor-mediated process? Dev. Brain Res. 63:85–93; 1991.
33. Sparapani, M.; Virgili, M.; Caprini, M.; Facchinetti, F.; Ciani, E.;

530 DAVIDSON, BEDI AND WILCE
Contestabile, A.: Effects of gestational or neonatal treatmentwith alpha-difluromethylornithine on ornithine decarboxylaseand polyamines in developing rat brain and on adult rat neuro-chemistry. Exp. Brain Res. 108:433–440; 1996.
34. Spraggins, Y. R.; Seidler, F. J.; Slotkin, T. A.: Cocaine exacer-bates hypoxia-induced cell damage in the developing brain:Effects on ornithine decarboxylase activity and protein synthesis.Biol. Neonate 66:254–266; 1994.
35. Thadani, P. V.; Lau, C.; Slotkin, T. A.; Schanberg, S. M.: Effect ofmaternal ethanol ingestion on neonatal rat brain and heart orni-thine decarboxylase. Biochem. Pharmacol. 26:523–527; 1977.
36. Thadani, P. V.; Slotkin, T. A.; Schanberg, S. M.: Effects of late orearly postnatal ethanol exposure on ornithine decarboxylaseactivity in brain and heart of developing rats. Neuropharmacol-ogy 16:289–293; 1977.
37. Wang, S.; Bartolome, J. V.; Schanberg, S. M.: Neonatal depriva-tion of maternal touch may suppress ornithine decarboxylase viadown regulation of the proto-oncogenes c-myc and max. J. Neu-rosci. 16:836–842; 1996.
38. Wiener, S. G.; Shoemaker, W. J.; Koda, L. Y.; Bloom, F. E.:Interaction of ethanol and nutrition during gestation: Influenceon maternal and offspring development in the rat. J. Pharmacol.Exp. Ther. 216:572–579; 1981.
39. West, J. R.; Chen, W-J. A.; Pantazis, N. J.: Fetal alcohol syn-drome: The vulnerability of the developing brain and possiblemechanisms of damage. Metab. Brain Dis. 9:291–322; 1994.
40. West, J. R.; Goodlett, C. R.; Bonthius, D. J.; Pierce, D. R.:Manipulating peak blood alcohol concentrations in neonatal rats:Review of an animal model for alcohol-related developmentaleffects. Neurotoxicology 10:347–366; 1989.
41. West, J. R.; Hamre, K. M.; Cassell, M. D.: Effects of ethanolexposure during the third trimester equivalent on neuron numberin rat hippocampus and dentate gyrus. Alcohol. Clin. Exp. Res.10:190–197; 1986.
42. Zawia, N. H.; Evers, L. B.; Harry, G. J.: Developmental profilesof ornithine decarboxylase activity in the hippocampus, neocor-tex, and cerebellum: Modulation following lead exposure. Int. J.Dev. Neurosci. 12:25–30; 1994.



![Targeting ornithine decarboxylase reverses the LIN28/Let-7 ... · the LIN28/Let-7 pathway [13, 14], which is important in a number of cancers, including NB, and was recently identified](https://img.pdfslide.us/doc/110x75/5f7e699b6c944249467265c5/targeting-ornithine-decarboxylase-reverses-the-lin28let-7-the-lin28let-7-pathway.jpg)
![t e c h n ol gy Journal of Biotechnology & Biomaterials · argF proB kgd) for L-ornithine production, which could produce 4.62 g/L of L-ornithine [13]. The level of L-ornithine production](https://img.pdfslide.us/doc/110x75/5e22e2c1220ab9163b5a39e7/t-e-c-h-n-ol-gy-journal-of-biotechnology-biomaterials-argf-prob-kgd-for-l-ornithine.jpg)
























