Embed Size (px)
Citation preview

Epithelial and soft tissue tumors of the
tracheobronchial tree
Leslie Litzky, MDDepartment of Pathology and Laboratory Medicine, University of Pennsylvania Medical Center,
6 Founders Pavilion, 3400 Spruce Street, Philadelphia, PA 19104-4283, USA
Most of the tumors described in this article are rare or extraordinarily rare. In
older literature, a number of these entities were incorrectly combined together and
designated as ‘‘bronchial adenomas.’’ This article encompasses a diverse spectrum
of lesions whose pathological classification is relatively straightforward in
relationship to the normal structure and histology of the trachea and major
bronchi. An extensive review of all the literature relating to these unusual tumors
is beyond the scope of this article, but general characteristics of each tumor are
covered with particular reference to the specifics of endotracheal or endobronchial
involvement. Refinements in the pathologic classification in recent literature have
clarified the clinical presentation, therapeutic options, and prognosis of some of
these unusual tracheobronchial tumors. Special attention is directed toward the
nomenclature revisions incorporated within the 1999 World Health Organization/
International Association for the Study of Lung Cancer (WHO/IASLC) histologic
typing of lung tumors [1].
Epithelial tumors of the tracheobronchial tree—benign
Papillomas
A papilloma consists of a central core of connective tissue fronds with an
overlying epithelial surface. The vast majority of tracheobronchial papillomas
have an exophytic growth pattern, but an inverted papilloma pattern has been
reported [2]. Endoscopically and grossly, the papilloma typically appears as
sessile, cauliflower-like or verrucous excrescences that project into the tracheal
or bronchial lumen (Fig. 1). The 1999 WHO Histological Typing of Lung Tumors
subclassifies papillomas by the type of epithelium lining the fibrovascular core [1].
1052-3359/03/$ – see front matter D 2003, Elsevier Science (USA). All rights reserved.
PII: S1052 -3359 (02 )00045 -5
E-mail address: [email protected]
Chest Surg Clin N Am
13 (2003) 1–40
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–402

Squamous cell papilloma/papillomatosis
The clinicopathologic characteristics of solitary squamous papillomas should
be considered separately from cases of multiple squamous papillomas (squamous
papillomatosis). Most solitary papillomas are not preceded by papillomatosis of
the upper respiratory tract [3,4]. Tracheobronchial papillomatosis usually occurs
in the setting of prior laryngeal disease, and it frequently presents in children
(juvenile papillomatosis) or young adults. Overall, squamous papillomas of the
lung are a rare finding in recurrent respiratory papillomatosis, with a reported
incidence of 5% involving the distal trachea and less than 1% involving the
pulmonary parenchyma [5,6]. Solitary papillomas also are rare. In their series of
3937 patients undergoing bronchoscopy over an 11-year period, Shah et al
identified 53 papillomas (1.3%) [7]. Flieder et al have provided an in-depth
clinicopathologic/in situ hybridization analysis of 14 solitary papillomas in adults
along with a review of 27 cases from the literature [2]. Solitary squamous
papillomas are seen more frequently in men and generally appear in the age
range of 50 to 70 years. When smoking history was available, more than half of
patients were cigarette smokers. A few patients have been asymptomatic;
radiographic abnormalities such as a hilar mass or postobstructive pneumonia
or symptoms such as wheezing, hemoptysis, or recurrent pneumonias are
more common.
Grossly, the lesions are tan, friable, and polypoid. Mass size and gross
inspection are unreliable in excluding an invasive component within the papil-
loma. The squamous epithelium can be keratinizing or nonkeratinizing. The
nonkeratinizing subtype had been reported in older literature as ‘‘transitional’’
because of its phenotypic resemblance to urothelium [8]. A range of squamous
dysplasia can be seen, as well as in-situ or invasive carcinoma. Human papilloma
virus (HPV) has been detected in some solitary papillomas and in cases of
papillomatosis, but its presence in squamous lesions does not appear to correlate
with recurrence or malignancy [2]. HPV 6 and 11 have been found in
histologically benign papillomas [9,10], but HPV 11 also has been correlated
with malignant transformation in the setting of juvenile-onset recurrent respi-
ratory papillomatosis [11]. HPV 16 and 18 have been reported in cases of
papillomas associated with carcinoma [9,10], but the relatively small numbers of
cases in the literature make it difficult to comment on the strength of this
association and the relative risk of malignant transformation. Nevertheless, in all
circumstances complete excision of a solitary papilloma is recommended to
exclude an invasive malignancy and to prevent recurrence.
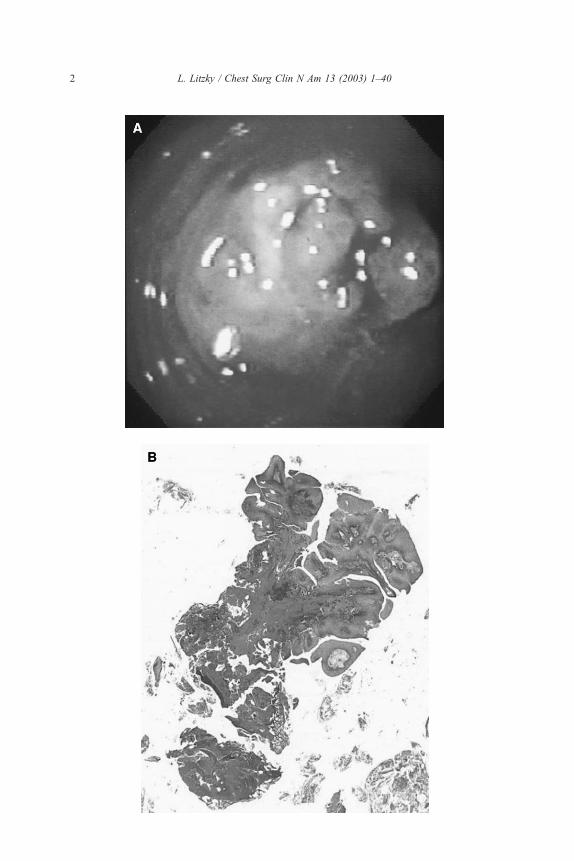
Fig. 1. (A) Endoscopic photograph of a bronchial papilloma. The exophytic polypoid excrescences
are typical. Microscopically, this proved to be a mixed squamous cell and glandular papilloma.
(Courtesy of Michael A. Grippi, Pulmonary, Allergy, and Critical Care Division, University of
Pennsylvania Medical Center.) (B) Squamous papilloma of the trachea. This low-power photomicro-
graph highlights the branching central fibrovascular cores with a surface lined by benign squa-
mous epithelium.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 3

Glandular papilloma (columnar cell papilloma)/ mixed squamous cell and
glandular papilloma
Glandular papilloma is a papillary tumor lined by glandular epithelium. Less
than 20 of these tumors have been reported in the literature [2,12–14]. The
glandular cell types include ciliated columnar cells, nonciliated columnar cells,
cuboidal cells, and goblet cells. These tumors are solitary and central. Equal
numbers of men and women have been reported with a somewhat older median
age than the median age for solitary squamous papillomas (68 years versus
54 years, respectively) [2]. There does not appear to be a significant correlation
with tobacco use. Recurrence has been reported in one incompletely resected
lesion [13]. Cytologic atypia or malignant transformation has not been reported to
date in glandular papillomas. As the name implies, mixed squamous cell and
glandular papilloma contains a mixture of squamous and glandular epithelium.
These papillomas are usually solitary. The squamous epithelium can be dys-
plastic, therefore progression to squamous carcinoma can occur [2,14].
Adenomas of salivary gland-type tumors
Mixed seromucinous glands and ducts are found in the tracheal and large
bronchial submucosa. These glands are believed to give rise to a variety of
salivary gland-like tumors that are histologically indistinguishable from their
major salivary gland counterparts. The category of salivary gland-like tumors
includes benign and malignant lesions. Although rare, salivary gland-type tumors
of the lung are an important subgroup of tracheobronchial tumors because of their
distinctive morphology, growth pattern, and clinical presentation. In general,
patients tend to present with symptoms such as wheezing or hemoptysis, which
are caused by tumor growth within the larger airways. Although the adenomas
described in this article are benign, complete excision is required to prevent
recurrence. The location might therefore necessitate bronchoplastic surgery.
Mucous gland adenoma
Mucous gland adenoma is rare, with less than 30 cases reported to date
[15–17]. In the largest series (by England and Hotelier), about half of the tumors
presented as solitary endobronchial lesions and half presented as coin lesions. The
majority of mucous gland adenomas arise in the main lobar and segmental
bronchi. The tumors range in size from a little less than 1 cm to 7 cm. They are
well circumscribed with a predominantly helophytic growth pattern. Grossly, they
are soft, partially cystic, and mucinous. Histologically, the tumor is composed of
benign-appearing, mucus-filled cysts, tubules, glands, and papillae. The glandular
epithelium consists of tall columnar, flat, cuboidal, goblet, oncocytic, or clear
cells, but there should not be a squamous cell or intermediate cell component
admixed within the tumor. Heard et al, however, reported seven cases of mucous
gland adenoma, four of which contained a small amount of squamous differenti-
ation [18]. Neither invasion of the pulmonary parenchyma nor hilar lymph nodes
metastases has been reported.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–404

Pleomorphic adenoma (benign mixed tumor)
Mixed tumors (pleomorphic adenomas) of the salivary gland are the most
common histologic type of salivary gland tumor, but they are seen rarely in the
lung. Like its salivary gland counterpart, pleomorphic adenomas consist of
varying proportions of epithelial and connective tissue elements. Myoepithelial
cells are prominent within the epithelial component. The associated stroma has a
myxoid or focally chondroid matrix. There is a wide spectrum of histologic
features and a range of clinical behavior. Malignant salivary gland-type mixed
tumors are discussed later in this article. Primary mixed tumors of the lung are
histologically indistinguishable from mixed tumors that occur in other sites;
clinical correlates are therefore essential to exclude a metastasis from another site
such as the salivary gland or breast.
Pleomorphic adenomas occur primarily in middle-aged adults, although they
have occurred in pediatric patients and younger adults [19–22]. The incidence in
men and women has been equal. Moran et al has reported the largest series and
review of the literature to date [19,23]. In their series, there were eight patients, six
of which had what were characterized as histologically benign lesions. Follow-up
information was limited. Three of the patients with benign lesions were lost to
follow-up and two patients died of other diseases. All of the histologically benign
lesions were 2.0 cm to 2.5 cm in size and well circumscribed, but only half were
associated with a bronchus. In their summary of the previously published eight
reports [24–28], the size range was slightly wider (1.5–4.5 cm), but the
distribution of polypoid endobronchial tumors to peripheral parenchymal nodules
was roughly equivalent. Grossly, the tumors were soft to rubbery masses with a
gray–white, myxoid, cut surface. Histologically, most of the tumors resembled the
‘‘cellular’’ variant of mixed tumors of the salivary gland in the relative paucity of
duct-like structures and the predominance of myxoid or hyalinized stroma rather
than a cartilaginous matrix. Small foci of necrosis were present in two tumors.
Mitoses were increased in three tumors on the order of 1 per 10 high power fields.
Nevertheless, the authors concluded that small size and circumscription tended to
correlate best with benign behavior, assuming adequate excision.
Monomorphic adenoma/myoepithelioma/epithelial–myoepithelial tumor
The terms monomorphic adenoma, myoepithelioma, and epithelial–myoepi-
thelial tumor are applied to tracheobronchial gland tumors that are composed
exclusively of epithelial elements (monomorphic adenoma), myoepithelial ele-
ments (myoepithelioma), or a mixture of the two (epithelial/myoepithelial tumor).
They are extremely rare, but their behavior is similar to the mixed tumors of
salivary gland type described previously [29–38].
Oncocytoma
An oncocytoma, whether within the lung or in a far more common site such as
the kidney, is composed of tumor cells with abundant granular eosinophilic
cytoplasm. Other tracheobronchial tumors such as carcinoids, pleomorphic
adenomas, acinic cell tumors, or mucoepidermoid carcinomas can have a
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 5

similar-appearing granular, eosinophilic cytoplasm; therefore, the diagnosis of
oncocytoma cannot be definitively rendered without supporting immunohisto-
chemical or ultrastructural data [39]. By definition, the granular eosinophilic
cytoplasm in an oncocytoma ultrastructurally corresponds to mitochondrial
hyperplasia. Neurosecretory granules should be absent. Immunohistochemistry
reveals that oncocytic carcinoid tumors are positive for neuroendocrine markers
such as chromogranin and synaptophysin. Oncocytomas are negative for these
markers. In retrospect, the earliest case report of a pulmonary oncocytoma by
Black in 1969 [40] probably represents an acinic cell tumor [39], and other
reports in the literature would be more precisely classified as oncocytic carcinoids
[41,42]. Given these ultrastructural criteria, the first example of a pulmonary
oncocytoma was reported by Fechner and Bentinck in 1973 [43]. Other reported
cases within the English literature that conform to these criteria include Santos-
Briz et al, Warter et al, and Nielsen [44–46]. With the exception of the case
reported by Nielsen, the tumors were well circumscribed and in the size range of
3.0 cm to 3.5 cm. The tumor cells ranged from small ovoid cells to larger
polyhedral cells. The tumor cells were arranged in perivascular arrays, ribbon-
like configurations, or cord-like structures. Necrosis was absent and mitoses were
rare or absent. Aggressive behavior—even in the presence of a partially confluent
growth pattern or peribronchial lymph nodes metastasis—has not been reported
to date in these extremely rare tumors.
Epithelial tumors of the tracheobronchial tree—malignant
Endobronchial/tracheal squamous cell carcinoma
Most squamous cell carcinomas occur in central cartilaginous bronchi,
although up to as many as one third develop as peripheral subpleural tumors
[47]. This central location makes it more likely that the tumor will reach an
appreciable size before detection, exfoliate cells into sputum, and to be accessible
to bronchoscopic biopsy (Fig. 2) [48]. In contrast to squamous papillomas, which
tend to occur in the trachea and mainstem bronchi, squamous cell carcinoma is
seen more frequently in lobar and segmental bronchi [48]. Nevertheless, it should
be noted that squamous cell carcinoma occurs with far greater frequency in the
trachea than any other type of tumor [49,50]. Squamous cell carcinomas are
graded by the extent of keratinization, intercellular bridges, or squamous pearl
formation. The presence of squamous differentiation has some prognostic
significance. Data from Gail et al’s study of prognostic factors in patients with
resected stage I non–small-cell carcinoma highlighted the decreased risk for
recurrence in squamous cell carcinoma in comparison with adenocarcinoma and
large-cell carcinoma [51]. Moreover, the risk for recurrence was correlated with
increasing size (T1 versus T2) rather than nodal disease (N0 versus N1). A
similar survival advantage with squamous differentiation was shown using the
1986 staging classification system and was reported by Mountain et al [52].
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–406

Within the subsets of T1N0 and T1N1, patients with squamous cell carcinoma
had a significantly better 5-year survival rate than patients with adenocarci-
noma—83% versus 69% for T1N0 and 75% versus 52% for T1N1, respectively.
In this study, the significance of histologic type as a prognostic factor was
variable in other stages and stage subsets. There was no statistically significant
difference for cell type in T2N0 tumors, suggesting the stronger influence of large
tumor size, pleural involvement, or both on prognosis. Histologic type was
significant in the survival of patients with T2N1 disease (53% for patients with
squamous cell carcinoma versus 25% for patients with adenocarcinoma),
although adenocarcinoma is infrequently diagnosed within this stage subset.
There was no significant difference in survival by histologic cell type in stage III
disease, but this patient subset was more heterogeneous. In general, survival rates
of patients with large-cell, undifferentiated carcinoma and adenosquamous
carcinoma are similar to those of patients with adenocarcinoma.
Papillary variant of squamous cell carcinoma
These tumors are characterized by an extensive or pure exophytic pattern with
delicate fibrovascular papillae projecting into the bronchial lumen. The invasive
component of the tumor can be quite focal; therefore, separation of this tumor
from a squamous papilloma might be difficult on a small biopsy specimen.
Severe cytologic atypia, if present, is a supportive feature of squamous cell
carcinoma; however, many of these tumors are characteristically well differ-
entiated and have histologic features that overlap with dysplastic squamous
papillomas. Dulmet-Brender et al reported a favorable 5-year survival rate in
their series of 34 tumors collected over 35 years [53]. There was a preponder-
ance of male patients with a mean age of 58 years. These tumors were nearly
Fig. 2. Squamous cell carcinoma, right mainstem bronchus. The central location allowed this tumor to
reach an appreciable size before detection.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 7

always T1N0 lesions, but their prognosis did not differ significantly from other
T1N0 lesions.
Basaloid variant of squamous cell carcinoma/basaloid carcinoma
Primary carcinomas of the lung with basaloid features have been reported.
These basaloid histologic features are similar to those seen in other extrapulmo-
nary sites such as the head and neck or the cervix. Most of these tumors develop
within proximal bronchi, and they often have an endobronchial component. A
high percentage of cases will have carcinoma in situ. The key histologic features
are a lobular, trabecular, or palisading growth pattern. The tumor cells are
relatively small with moderately hyperchromatic nuclei, scant cytoplasm, a high
mitotic rate, and absent or focal nucleoli. By WHO/IASLC definition, immuno-
histochemical stains for neuroendocrine markers should be negative [1]. Basaloid
carcinoma can occur as a pure form or it can be mixed with other histologic
subtypes of non–small-cell carcinoma such as squamous cell carcinoma or
adenocarcinoma. In Brambilla et al’s initial series, 21 patients with stage I or
stage II basaloid carcinomas (pure and mixed) were combined in an actuarial
analysis that showed a median survival of 22 months and a 5-year survival
probability of 10% [54]. This study and subsequent reports seem to indicate that
these patients have a significantly shorter survival than those with poorly
differentiated squamous cell carcinoma [55–58].
Endobronchial adenocarcinoma (adenocarcinoma invading into bronchi)
Most pulmonary adenocarcinomas are peripheral and tend to involve the larger
cartilaginous airways secondarily as a consequence of increasing tumor size.
Nevertheless, a small number of endobronchial tumors will prove to be
adenocarcinomas with a histologic growth pattern distinct from the salivary
gland type tumors. As a group, these tumors tend to be grossly polypoid.
Extension along smaller airways or into the parenchyma is variable. A broad
spectrum of histologic patterns has been described. Some tumors have been
papillary with tall columnar cells lining fibrovascular cores [59]. Hirata et al
reported a series of 23 moderately differentiated endobronchial adenocarcinomas
with columnar cells that resembled those seen in the bronchial submucosal gland
[60]. Other unusual histologic patterns have included endometrioid type of cell,
stratified columnar cells with signet ring cells, and clear cells [61–63]. There
does not to appear to be any appreciable difference in prognosis between these
tumors and their more common peripheral counterparts from the limited follow-
up information that is available.
Carcinoid tumor—typical
Carcinoid tumor is one of the most frequently encountered and best charac-
terized of the unusual tumors that occur in the tracheobronchial tree. The reported
incidence generally varies between 1% and 4% of all primary lung tumors [64,65],
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–408

but much attention has been directed towards this entity because of its overall
better prognosis in comparison with other types of pulmonary epithelial malig-
nancy. It should be emphasized from the outset that carcinoid tumors are not
‘‘benign’’ tumors (or ‘‘adenomas’’ as they were termed in older literature); they are
best regarded as a low-grade malignant neuroendocrine carcinoma with a capacity
for invasion and metastasis. On average, the tumor occurs in a younger age group
(< 50 years) than those with the much more common lung cancers, but carcinoid
tumors have been reported in the young and elderly [66–73]. In the previously
cited reports, the incidence is approximately equal between men and women. The
tumor is not associated with cigarette smoking or any other known toxic exposure
[48]. Davila et al reported that 75% of bronchial carcinoid tumors arose in the
lobar bronchi, with 10% originating in the main stem bronchi and 15% arising in
the periphery of the lung [74]. Given their relatively slow growth within a large
bronchus, it is common for patients to present with symptoms or radiographic
evidence of bronchial obstruction [75,76]. Patients with pulmonary carcinoids
usually do not have symptoms of carcinoid syndrome, which is most frequently
reported in patients with liver metastases [72]. Similarly, Cushing’s syndrome is a
rare but well-documented presentation [77–79]. Carcinoid tumors have been
associated with multiple endocrine neoplasia syndrome-type 1 (MEN 1) [80–82].
Grossly, central carcinoids are fleshy, polypoid masses that often have a
cherry-red color that reflects its vascularity. This gross polypoid configuration
can present a problem in accurately determining the margin of resection in a
lobectomy specimen [48]. Attention to the tumor’s characteristic growth pattern,
by surgeons and pathologists alike, can prevent additional unnecessary surgery.
The standard transverse section of the proximal bronchial margin, which is often
appropriate in other types of lung cancer, can create a false-positive appearance to
the margin if a carcinoid is protruding in polypoid configuration proximally in the
lumen, as is often the case. Careful dissection of the true proximal bronchial
margin in a circumferential manner usually proves that the margin is free and that
the base of tumor attachment is really centimeters away. Another characteristic
gross configuration in which there is an obstructing central endobronchial lesion
along with a deeply invasive parenchymal component has been termed ‘‘iceberg’’
by Ishida et al [83].
The histologic appearance of central carcinoids is characteristic of a neuro-
endocrine tumor and recognizable by light microscopy alone. The tumor cells are
arranged in organoid, trabecular, insular, palisading, or ribbon- or rosette-like
patterns. The cells might be round or spindle shaped, but the cell size is generally
uniform and they have moderate amount of eosinophilic, finely granular
cytoplasm and the finely granular nuclear chromatin pattern typical of a neuro-
endocrine tumor. A number of unusual patterns (papillary, sclerosing, follicular,
or glandular), cytologic features (oxphilia, mucin or melanin production), or
stromal changes (bone, cartilage, dense fibrosis, amyloid) have been documented
[1], but their significance is mainly as potential pitfalls in pathologic recognition.
By WHO/IASLC criteria, the tumor must have fewer than two mitoses per 2 mm2
and no necrosis [1]. Although it is well recognized that some carcinoid tumors
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 9

show cytologic atypia, increased cellularity, or lymphatic invasion, these histo-
logic features are not invariably associated with a more aggressive clinical course.
Overall, carcinoid tumors have an excellent prognosis, with 5-year survival rates
in the range of 90% to 100% consistently reported over several decades and from
various institutions [66,68,72,84,85]. There are currently no histologic character-
istics that reliably predict which tumors will behave aggressively, although some
correlation with large tumor size has been noted. In Thunnissen et al’s series,
tumor size predicted for regional lymph node metastasis in 80% of the cases [86].
Hilar and mediastinal lymph node metastases occur in 5% to 10% of cases, but
they do not necessarily indicate a poor prognosis [73]. Recurrences and
metastases tend to occur considerably later than they do in atypical carcinoids
or other types of lung cancer [73]. Some of the earlier analyses were performed
before the 1999 WHO/IASLC revisions. MEN 1-associated genetic alterations
have been reported in sporadic lung carcinoids [87–95]. The group of tumors in
Debelenko et al’s original series [87] was re-reviewed following the 1999 WHO/
IASLC revisions and reclassified as atypical carcinoids (see definition in
following section) [96]. Further investigations are needed to elucidate potential
risk factors.
Carcinoid tumor—atypical
Arrigoni et al were the first to refine the criteria for ‘‘atypical carcinoid’’ in
1972 [97]. Since that time there has been some controversy regarding what
pathologic characteristics define this tumor, how reproducible the diagnosis is,
what the overall prognosis is, and what nomenclature is appropriate for this tumor
given its clinical behavior. Arrigoni et al’s original series described 23 tumors
(out of a total of 215 carcinoid tumors) that appeared to have a general
resemblance to carcinoid tumors but that also had a focally disorganized growth
pattern, areas of tumor necrosis, increased mitoses (5–10/2 mm2), hypercellu-
larity and cellular pleomorphism. Criticism of the use of the word ‘‘atypical’’
soon followed, given the aggressive biologic behavior described in this initial
series, with 30% of patients dead of disease at 3 years, a 70% incidence of
metastases, and an average survival time of 27 months. It was for this reason that
the alternate designation ‘‘well-differentiated neuroendocrine carcinoma’’ [98]
was proposed for the same entity to avoid any confusion with the considerably
better prognosis of carcinoid tumors. Some authors objected to the term ‘‘well-
differentiated’’ given the ultimately fatal outcome for many patients within a
relatively short span of time. Other terms were introduced into the literature in
1980s as more published reports detailing this entity appeared. These terms
included peripheral small-cell carcinoma of lung resembling carcinoid tumor [99]
and Kulchitsky cell carcinoma II [100]. The tumors described in this older
literature are a heterogeneous group in regard to staging and pathological
classification. This was (in part) because of the subjective interpretation of
features such as ‘‘architectural distortion’’ and the lack of generally accepted
criteria for increased mitotic rate and necrosis.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4010

The reproducibility of the diagnosis will (hopefully) in the future be less of an
issue, not just among pathologists. The 1999 WHO/IASLC classification retained
the original nomenclature and defines an atypical carcinoid tumor as a carcinoid
tumor with between two and ten mitoses per 2 mm2 or foci of coagulative necrosis
[1]. The necrosis typically is punctate. Other features such as cytologic atypia,
disorganized architecture, increased cellularity, lymphatic invasion, and nucleoli
have been described in these tumors, but these features are not required for the
diagnosis. It should be noted that the defining characteristics of necrosis or an
increased mitotic rate can be focal; therefore, it might be difficult to distinguish an
atypical carcinoid tumor from a typical carcinoid tumor on a cytology specimen,
small biopsy, or grossly at the time of resection. From a practical point of view,
however, it is not necessary to make the distinction preoperatively because
complete resection remains the treatment of choice for both tumors. A potential
role for chemotherapy or radiation therapy remains to be proven.
The prevalence rate of atypical carcinoid tumors, as estimated from various
series in the literature, ranges from about 10% to 20% of all pulmonary carcinoid
tumors [68,101–103]. The clinical presentation is essentially identical to that of
typical carcinoid tumors. In one recent series, about one third were asymptomatic;
the remainder had the usual common symptoms of cough, hemoptysis, or
evidence of bronchial obstruction [68]. In published reports that conform to this
entity, there appears to be slight male predominance and an overlap of age
incidence with other neuroendocrine tumors. Smoking has proved to be a
consistent association in this tumor in most cases. Central and peripheral
locations have been reported, with patients presenting at all tumor stages. The
incidence of nodal metastases ranges from 30% to 75%. Beasley et al have
published a recent series that examines prognostic indicators for 106 atypical
carcinoid tumors using the 1999 WHO/IASLC criteria [104]. They reported
overall 5- and 10-year survival rates as 61% and 35%, respectively. Five-year
survival rates were significantly better for stage I (71%) than for stage II (46%)
and stages III/IV (37%). Of the clinical features, higher stage and a tumor size of
3.5 cm or greater were associated with a worse prognosis. For unclear reasons,
female gender was also associated with a worse prognosis. Patients were further
subdivided in subgroups of low (2–5 mitoses/2 mm2) versus high (6–10 mitoses/
2 mm2) mitotic rates. When patients were stratified by stage, patients with a
higher mitotic rate had a significantly worse survival. As referenced in the
previous discussion of genetic alterations associated with carcinoid tumors, the
results from studies of atypical carcinoids have been variable, but they tend to
show a higher rate of allelic losses within chromosomal region 11q13 when
compared with carcinoid tumors [92]. Some overlap does exist, and further
investigation will be needed to elucidate these differences and their significance.
Endobronchial small-cell carcinoma/combined small-cell carcinoma
The 1999 WHO/IASLC classification defines small-cell carcinoma as a
malignant epithelial tumor consisting of small cells with scant cytoplasm, ill-
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 11

defined cell borders, finely granular nuclear chromatin, and absent or incon-
spicuous nucleoli [1]. The previous 1981 WHO categories of oat cell carcinoma/
small-cell carcinoma, intermediate cell type/combined oat cell carcinoma, and the
1988 IASLC category of mixed small-cell/large-cell carcinoma have been
dropped because subsequent studies did not confirm their clinical significance
or interobserver reproducibility. The 1999 WHO/IASLC definition of combined
small-cell carcinoma defines the tumor as a small-cell carcinoma with an
additional component that consists of any of the histological types of non–
small-cell carcinoma. The classic common and advanced presentation of small-
cell carcinoma is familiar to all thoracic surgeons and will not be reviewed here.
The diagnosis is more challenging when the occasional small peripheral or
endobronchial nodule is encountered. Small-cell carcinoma remains a diagnosis
by light microscopy. Immunohistochemical demonstration of neuroendocrine
differentiation is of limited utility and can be variable, with about 25% of cases
negative for markers such as chromogranin, synaptophysin, and Leu-7 [1];
however, immunohistochemical stains are reasonable, particularly in a smaller
sample, when the histology suggests a broader differential diagnosis that might
include lymphoma or other small, blue-cell tumors. The tumor cells can be round,
oval, or spindle-shaped. The mitotic rate is high and nuclear molding is
prominent. A few histologic pitfalls can make the diagnosis difficult, especially
in small biopsy specimens obtained by fiberoptic bronchoscopy [71]. There is no
absolute size criterion for the tumor cells, though it is often stated that the general
size should be less than the size of three small resting lymphocytes. Cell size can
be larger in well-fixed specimens, resections, and frozen sections. Similarly,
necrosis is common and often extensive, but it is not an invariable feature,
particularly in small specimens. It might be more difficult to appreciate a high
mitotic rate in small specimens, and this feature is critical in distinguishing small-
cell carcinoma from typical and atypical carcinoid tumors. Finally, although crush
artifact might well preclude a definitive diagnosis (and this is often frustrating
from a pathological and clinical perspective), it is not pathognomic of small-cell
carcinoma or even of malignancy. It is therefore essential given the therapeutic
and prognostic implications of the diagnosis that the complete clinical and
radiographic presentation be taken into consideration and that suboptimal speci-
mens be accepted as nondiagnostic.
Endobronchial large-cell carcinoma/large-cell neuroendocrine carcinoma
Large-cell carcinoma is a diagnosis of exclusion that is based on light
microscopy. By definition, large-cell carcinoma is a malignant epithelial tumor
that lacks squamous or glandular differentiation and lacks the cytologic features of
small-cell carcinoma. Lung tumors are histologically heterogenous, and this
heterogeneity is also expressed in to what degree they show neuroendocrine
differentiation [96]. Neuroendocrine differentiation is usually suggested by
morphologic features such as organoid nesting, trabeculae, rosette-like structures,
and palisading patterns. It can be confirmed by electron microscopy or immuno-
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4012

histochemistry, the latter technique being more widely employed in routine
diagnostic work. There is a general acknowledgment that there are some lung
tumors that are not classic small-cell carcinomas, yet they do suggest neuro-
endocrine differentiation that might or might not be confirmed by special stains. In
1991 Travis et al set forth criteria for what was termed large-cell neuroendocrine
carcinoma (LCNEC) [105]. These tumor cells are generally large with moderate to
abundant cytoplasm and the nuclear chromatin ranges from vesicular to finely
granular. Nucleoli are frequent and often prominent, and they remain the definitive
feature for the separation from small-cell carcinoma. Large areas of necrosis are
also common. By 1999 WHO/IASLC criteria, mitotic counts must be at least
11 per 2 mm2, but in actual practice, the mitotic counts are usually much higher,
averaging in the range of 75 per 2 mm2. LCNEC is classified as a variant of large-
cell carcinoma. The nomenclature is extremely complicated in the 1999 WHO/
IASLC classification because of the current uncertainty as to what effect, if any,
neuroendocrine features have on differences in survival or therapeutic response.
Large-cell neuroendocrine carcinoma has neuroendocrine morphology and
neuroendocrine markers by electron microscopy or immunohistochemistry.
Large-cell carcinoma with neuroendocrine morphology is applied to tumors that
have neuroendocrine morphology but lack neuroendocrine markers by electron
microscopy or immunohistochemistry. Large-cell carcinomas with neuroendo-
crine differentiation lack neuroendocrine morphology but have neuroendocrine
markers by electron microscopy or immunohistochemistry. Large-cell carcinoma
lacks neuroendocrine morphology or neuroendocrine differentiation by special
studies. Complete resection remains the treatment of choice for all of these
tumors. Though there are conflicting reports in the literature, a potential role for
adjuvant or neoadjuvant therapy has yet to be determined. The reproducibility of
the histologic criteria for neuroendocrine tumor subclassification continues to be
an issue [106].
Carcinomas of salivary gland-type
Adenoid cystic carcinoma
Adenoid cystic carcinoma is the most common of the tracheobronchial
salivary gland-type tumors [107,108]. The majority of these tumors arise in large
bronchi, but peripheral primary adenoid cystic carcinomas have been reported
[109–111]. The discovery of a peripheral lesion should prompt clinical consid-
eration of metastasis from a previous or occult head and neck primary. Most
patients are adults, but there are pediatric cases [112,113]. The reported age of
adults at presentation is wide (18–82 years). The tumor occurs in both women
and men, but there is a slight male predominance in some series [23]. There is no
correlation with cigarette smoking. By comparison with other lesions such as
carcinoid tumors and mucoepidermoid carcinomas (which usually present as
intralumenal masses), adenoid cystic carcinomas have a more variable growth
pattern. Typical presenting symptoms such as wheezing, progressive dyspnea,
cough, and hemoptysis tend to reflect tumor size and the extent of intralumenal
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 13

tumor growth [23]. The size of the tumors has generally ranged from about 1 cm
to 4 cm. Some tumors are grossly nodular with minimal invasion of the bronchus,
whereas others have a mixed nodular/infiltrative or predominantly infiltrative
growth pattern [114]. More infiltrative tumors appear as small nodules within the
airway or cause a generalized constriction of the airway. Some tumors have a
tendency to radially spread into the adjacent parenchyma rather than along the
airways. The microscopic level of invasion nearly always exceeds that which is
grossly apparent, and negative resection margins often are difficult to achieve.
Complete resection can be difficult and can require multiple frozen sections to
confirm clear surgical margins. Long-term disease control has been reported with
positive surgical margins [115].
Adenoid cystic carcinoma tumor cells are small with a relatively high nuclear/
cytoplasmic ratio, but nuclear pleomorphism and mitoses are rare. The histologic
pattern varies, with some tumors entirely composed of cells arranged in tubular
and cribriform (cylindromatous) patterns. Some tumors have a significant
component of solid tumor nests [116]. In a large series by Moran et al [117],
prognosis correlated with stage rather than with these variations of cell type. As is
characteristic of their salivary gland counterparts, adenoid cystic carcinomas are
notorious for perineural spread. There can be lymph node involvement, usually
by direct extension. Long-term survival can be achieved with adequate resection,
but local recurrence can occur even late (> 10 years) following resection [50,118].
Mucoepidermoid carcinoma
Mucoepidermoid carcinoma is characterized by an admixture of mucus-filled
cysts and solid areas of transitional (non–mucin-secreting, nonkeratinizing cells)
or squamoid (nonkeratinizing but epidermoid) cells. Mucoepidermoid carcinoma
is distinguished from the bronchial mucous gland adenoma previously described
by the finding of these transitional or intermediate cells in the mucoepidermoid
carcinoma. These tumors are rare, with the reported incidence varying between
0.2% and 1.2% of all lung tumors [119,120]. Unlike its salivary gland counterpart,
mucoepidermoid carcinomas of the lung are divided into low or high-grade
lesions. An intermediate grade variant has not been characterized. This simple
two-grade separation correlates well with clinical presentation, general pathologic
features, and prognosis. The distinction between low-grade and high-grade tumors
is based on nuclear pleomorphism, mitotic activity, the presence of necrosis, and
the degree to which undifferentiated or transitional cells predominate.
Low-grade mucoepidermoid tumors are more common; they occur both in
children and adults. Yousem and Hochholzer have reported the largest series of
adult mucoepidermoid carcinomas of the lung to date [121]. In their series of
58 tumors, 45 tumors were low-grade. There were 18 men and 27 women. The
ages ranged from 9 to 78 years, but 25 of the patients were 30 years of age or
younger. Smoking history was available for only 22 patients, but fewer than half
reported cigarette use. Most of the patients were symptomatic, often with cough
and hemoptysis. A solitary mass was the most common radiographic finding
(29 patients) followed by postobstructive pneumonia (14 patients) and lobar
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4014
atelectasis (seven patients). Although peripheral mucoepidermoid carcinomas
have been reported, they are extremely rare [122]. Low-grade mucoepidermoid
carcinomas characteristically are exophytic and polypoid. The tumors are pre-
dominantly endobronchial with limited involvement of the bronchial submucosa
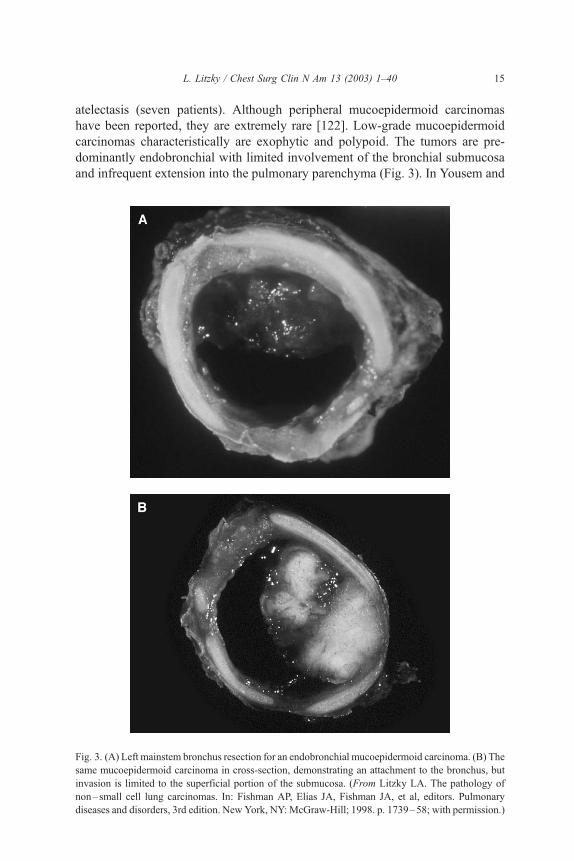
and infrequent extension into the pulmonary parenchyma (Fig. 3). In Yousem and
Fig. 3. (A) Left mainstem bronchus resection for an endobronchial mucoepidermoid carcinoma. (B) The
same mucoepidermoid carcinoma in cross-section, demonstrating an attachment to the bronchus, but
invasion is limited to the superficial portion of the submucosa. (From Litzky LA. The pathology of
non–small cell lung carcinomas. In: Fishman AP, Elias JA, Fishman JA, et al, editors. Pulmonary
diseases and disorders, 3rd edition. New York, NY: McGraw-Hill; 1998. p. 1739–58; with permission.)
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 15

Hochholzer’s series, four tumors were located in the main stem bronchi, but the
rest were in lobar bronchi. The size range was 0.8 cm to 0.6 cm. On cut section,
these tumors are variably cystic and solid lesions. By comparison, the tumors
might appear to be more mucoid or cystic than the more common carcinomas of
the lung. Histologically, there are numerous dilated glandular spaces within the
tumor interspersed with solid areas. Columnar and goblet cells line the cysts, and
these cells also might be scattered through the solid areas. By definition, the solid
areas are composed of transitional and squamoid cells without significant
cytologic atypia, increased mitotic activity, or necrosis. Intermediate cells have
a polygonal shape and eosinophilic cytoplasm, but they lack obvious squamous
or glandular differentiation. Forty-four of the 45 patients in this series had no
nodal involvement and were without evidence of disease at 7 years of follow-up.
The single patient with a low-grade mucoepidermoid carcinoma and a lymph
node metastasis was lost to follow-up. Heitmiller et al’s smaller series of
12 patients with low-grade mucoepidermoid carcinoma showed similar results
following complete resection [119].
By contrast, patients with high-grade mucoepidermoid carcinomas are at
higher risk for recurrent disease and death from disseminated disease. There is
some variation in the reported risk for recurrence and death, but the prognosis
clearly differs from the better outcome in low-grade mucoepidermoid carcinoma
and from the generally worse prognosis in most reported series of adenosqua-
mous carcinoma of the lung if histologic criteria are strictly applied. In Yousem’s
and Hochholzer’s series, there were six men and seven women with an age range
of 13 to 67 years (mean age 45 years). Ten of the 13 patients were symptomatic
with cough, hemoptysis, weight loss, or fatigue. Smoking history was available
for only five patients, but four of the five were cigarette smokers. Radiographic
findings included atelectasis, postobstructive pneumonia, and nodules with a
distal pneumonic infiltrate. All of the tumors were within lobar bronchi and they
ranged in size from 1.5 cm to 4.0 cm. There was an exophytic component to the
tumors, but six of the 13 patients also had invasion of the pulmonary
parenchyma. Histologically, areas of low-grade mucoepidermoid carcinoma were
identified, but solid areas were more conspicuous than in the low-grade tumors.
The tumor cells had high-grade cytologic features with nuclear enlargement,
anaplasia, mitoses, and necrosis. Frank keratinization of cells is not seen in
mucoepidermoid carcinomas; this is a significant feature used in distinguishing
high-grade mucoepidermoid carcinoma from adenosquamous carcinoma [123]. It
is clear from the literature that high-grade mucoepidermoid carcinoma has a
higher incidence of nodal involvement, unresectabilty, and inoperability because
of disseminated disease at the time of presentation [119,121,124,125]. Never-
theless, the magnitude of risk varies from study to study. Three of the 13 patients
(including the two with hilar lymph node metastases) in Yousem and Hochholz-
er’s series died of their disease [121]. A fourth patient had a lobectomy for
recurrent disease 4 years after the initial operation. In Heitmiller et al’s series,
the three patients with high-grade mucoepidermoid carcinoma that was unresect-
able at the time of presentation were all dead of their disease within 16 months
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4016

[119]. All 12 of the patients reported by Turnbull et all were dead in less than
1 year, but eight of these patients presented with disseminated disease or
unresectable lesions [124].
Acinic cell carcinoma
Fechner first described an acinic cell carcinoma of the lung in 1972. These
tumors are extremely rare, with 15 cases reported in the literature to date
[23,126–135]. Most of the tumors have been reported in adults with an
approximately equal male/female incidence and an age range of 27 to 75 years.
There was one pediatric case in a 12-year old girl [128]. The nodules ranged in
size from 1.2 cm to 4.0 cm with some propensity for the right middle lobe.
Tracheal, endobronchial, and intraparenchymal locations have been noted. The
lesions lack sharp circumscription, but proximity or connection to a major
bronchus is usually present. This tumor has the diverse histologic growth patterns
of its salivary gland counterpart, and special attention must be paid to excluding a
salivary gland metastasis by clinical history. The tumor can also be confused with
other tumors such as oncocytic carcinoids or granular cell tumors. Microscop-
ically, the tumors have been predominantly solid with focal acinar or microcystic
areas. Organoid or lobular patterns have also been described. The tumor cells are
cytologically bland with round hyperchromatic nuclei and granular eosinophilic
or clear cell cytoplasm. This cytoplasm is periodic acid Schiff (PAS)-positive in
most cases and ultrastructural studies have confirmed the large, membrane-bound
granules that are typical of serous secretory cells. The tumor cells should be
positive for cytokeratin but negative for chromogranin, synaptophysin, or S-100.
All of the patients reported have been alive and without evidence of recurrence
following surgical resection. One patient with interlobar lymph node metastasis
who underwent radiation therapy was without evidence of disease at 20 months
of follow-up care [130].
Malignant salivary gland-type mixed tumors
In Moran et al’s series of benign and malignant salivary gland-type tumors of
the lung, there were two patients who had what were characterized as histolog-
ically atypical lesions [19]. Both tumors were considerably larger (6 cm and
16 cm) than the benign lesions (2.0–2.5 cm). Both tumors were poorly circum-
scribed and infiltrative, and one patient had satellite nodules within the lobe. The
hilar and regional lymph nodes at the time of initial resection were negative in
both patients. In these two histologically malignant lesions, the tumors grew
predominantly as slender cords and trabeculae of cells within an abundant
myxoid matrix. Angioinvasion was prominent in one patient, and both tumors
had conspicuous areas of hemorrhage and necrosis. Mitotic counts averaged 5 to
10 per 2 mm2. Both patients had recurrent disease within the thorax within 2 to
3 years and one had died of the disease. In their summary of the previous
literature, the authors noted that a high mitotic rate was present in all of the
patients with aggressive disease, although clinical follow-up information was
available for only four of the eight previous reports. Three out of four of these
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 17

patients had late recurrences at 4 years [27], 8 years [136], and 9 years [26].
These data suggest the potential for late recurrence or metastasis, although some
patients have lived for an extended period of time with recurrent disease. It
should also be re-emphasized that clinical history and examination are essential to
exclude a metastasis from another site such as the salivary gland or breast.
Carcinomas with pleomorphic, sarcomatoid, or sarcomatous elements (including
carcinosarcoma and pulmonary blastoma)
The classification of these poorly differentiated, primary, non–small-cell lung
tumors that contain a component of sarcoma or sarcoma-like elements has been a
matter of some controversy, and these tumors have been described under a
variety of names. Designations include spindle cell carcinoma, carcinosarcoma,
sarcomatoid carcinoma (monophasic and biphasic), pleomorphic carcinoma,
giant cell carcinoma, and pulmonary blastoma. Despite the fact that these tumors
might represent a continuum of epithelial and mesenchymal differentiation, the
1999 WHO/IASLC classification set forth criteria for the classification of these
tumors in an attempt to foster uniform classification and potentially provide
more refined prognostic information in the future [1]. Carcinomas with spindle
cells or giant cells are classified as pleomorphic carcinoma, spindle cell
carcinoma, or giant cell carcinoma depending on the predominance of spindle
cells or giant cells. Tumors that are composed exclusively of spindle cells or
giant cells are termed spindle cell carcinoma and giant cell carcinoma, respec-
tively. These pure forms are extremely rare. The more frequently encountered
tumor, which consists of a poorly differentiated squamous cell carcinoma,
adenocarcinoma, or large-cell carcinoma admixed with spindle cells or giant
cells (comprising at least 10% of the tumor) is now termed a pleomorphic
carcinoma. Although the separation is acknowledged as arbitrary, carcinosar-
coma is defined as a malignant tumor having a mixture of carcinoma and
sarcoma containing heterologous elements (ie, differentiated mesenchymal
elements) such as malignant cartilage, bone, or skeletal muscle. In addition to
the histologic recognition of these malignant heterologous elements, immuno-
histochemistry with epithelial markers is often used to confirm epithelial
differentiation. It should be noted that the WHO/IASLC classification relies
on histologic criteria using routine light microscopy and does not require the
demonstration of positive immunohistochemical staining for cytokeratin. When
keratin stains are negative, however, it might be difficult to separate these tumors
from primary or metastatic sarcoma. Pulmonary blastoma is defined as a
biphasic tumor containing a primitive epithelial component that can resemble
well-differentiated fetal adenocarcinoma and a primitive mesenchymal stroma
that occasionally has foci of osteosarcoma, chondrosarcoma, or rhabdomyosar-
coma. This tumor presents mainly in adults [137] and should be distinguished
from pleuropulmonary blastoma, a tumor that occurs almost exclusively in
children of 6 years old or younger at diagnosis [138,139].
In light of this review of changing nomenclature and given that the routine use
of immunohistochemistry has been employed for barely two decades, it is
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4018

somewhat difficult to sort through the literature on these rare tumors. The lack of
adequate staging information that conforms to the current TNM classification
system in older literature adds to this confusion. The most recent series of
carcinosarcomas that utilizes the 1999 WHO/IASLC criteria and provides staging
information is that of Koss et al [140]. In this same series, the 66 carcinosarcomas
were compared with the largest previously published series of pleomorphic
carcinomas [141]. Several generalizations appear to be true from their series.
Pulmonary carcinosarcomas affect men more frequently than women and tend to
present in more elderly patients. The median age in Koss et al’s series was
65 years, but there was a wide patient age range (from 38 to 81 years). When
smoking history is obtained, all are smokers. Frequent symptoms include cough,
chest pain, hemoptysis, and dyspnea, but a substantial number of patients (about
one third in Koss et al’s series) are asymptomatic or have their tumors identified
on a routine chest radiograph. Although the tumors range in size from 2 cm to
16 cm, most are between 3 cm and 11 cm. The tumors are usually gray–white,
and areas of hemorrhage or necrosis are frequent. In an appreciable number of
instances, the large size of the tumor or the lack of information prevented an
accurate assessment of endobronchial or peripheral location. Some carcinosarco-
mas present with an endobronchial component or largely as central, polypoid,
endobronchial masses. Koss et al report a 34% incidence in their series and a 46%
incidence in the literature of tumors that contain an endobronchial component.
They report a 12% incidence and a 17% incidence in the literature of carcino-
sarcomas that were purely polypoid or largely polypoid endobronchial tumors.
Previously published reports and their histologic analysis of these tumors
underscore the need for careful sampling. The malignant stroma is often the
predominant component in the carcinosarcoma, and the foci of carcinoma are
small. Similarly, the poorly differentiated spindle cell component can predom-
inate, and a careful search is required to identify the heterologous elements. The
overall 5-year survival rate was 21%. There was no statistical significance in
5-year survival rates between the patients with carcinosarcomas and patients with
pleomorphic carcinomas (15%). Carcinosarcoma tumor size of greater than 6 cm
correlated with reduced survival. Although stage did not appear to be related to
outcome in this series, nearly one fourth of the cases did not have staging
information. It is still unclear whether or not the distinctly primitive histologic
appearance of pulmonary blastomas makes a prognostic difference. Although
about two thirds of patients have died of their disease within 2 years of diagnosis,
staging information has not always been complete and has lacked statistical
power. Overall, a larger number of cases with more complete staging and follow-
up information will be required to determine whether or not these histologic
differences correlate with clinical behavior.
Metastatic carcinoma
Most tumors metastatic to the bronchus represent intrathoracic spread from
primary pulmonary adenocarcinomas; however, metastases from extrathoracic
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 19

organ sites and intrathoracic tumors like malignant pleural mesothelioma do
occur. Endobronchial metastases from extrapulmonary solid malignant tumors
are rare. Many types of primary tumor have been reported, but breast, colon, and
renal cell adenocarcinomas are the most common [142,143]. Epithelial tumors
predominate, but melanoma and a wide variety of mesenchymal tumors have also
been documented. This latter group of tumors in particular can lead to diagnostic
difficulties because of a long latency period between the initial primary tumor
diagnosis and the development of pulmonary metastases [144]. Symptoms such
as wheezing or cough or radiological findings such as atelectasis can be
indistinguishable from those of primary lung cancer [145,146]. Invasion of the
bronchial wall, the more common pattern of bronchial metastatic spread,
manifests itself as submucosal irregularity or nodularity. Actual polypoid endo-
bronchial masses are quite rare, but they closely mimic a primary bronchogenic
tumor. The histologic diagnosis will be straightforward in most circumstances,
but a history of previous primaries helps to avoid pathologic misinterpretation.
Soft tissue tumors of the tracheobronchial tree—benign and malignant
The supporting fibroconnective tissues of the trachea and bronchi include
cartilage, smooth muscle cells, fibroblasts, adipocytes, nerves, lymphatics, and
blood vessels. These fibroconnective tissues can give rise to a variety of benign
and malignant soft tissue tumors. In comparison with the epithelial tumors
previously described, most of these tumors are exceedingly rare. From a clinical
and pathological perspective, it is essential to recognize that metastases and
epithelial carcinomas far exceed the incidence of primary soft tissue tumors.
Tumors that are initially thought to represent a true primary sarcoma of the lung
often prove upon further inquiry into patient history to represent late metastasis
from a primary soft tissue tumor or to be a carcinosarcoma upon further sampling.
Chondroma/chondrosarcoma
It is common diagnostic error to use the term chondroma for hamartomas that
have a dominant cartilaginous component. True chondromas are quite rare, with
an incidence of three cases in 40,000 autopsies [48]. Chondromas arise in the
cartilaginous rings of the large bronchi or trachea and, by definition, they have a
connection to the rings or with their perichondrium [48]. Chondromas have been
reported in a broad age range of patients, including pediatric patients [147].
Patients typically present with obstructive symptoms. There continues to be a
debate regarding whether true chondromas occur in the setting of Carney’s triad
(pulmonary chondromas, gastric epithelioid smooth muscle tumors, and extra-
adrenal paragangliomas) [148]. Some authors support the notion that the
‘‘chondromas’’ described in Carney’s triad are chondroid hamartomas [48],
whereas others include chondromas within the epidemiologic pathology associ-
ated with Carney’s triad [1]. Grossly, the tumors are 1 cm to 2 cm in size and have
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4020

irregular polypoid projections along a broad base [48]. On cut surface they are
firm and white, often with focal areas of calcification or ossification. Micro-
scopically, the tumor is composed entirely of mature cartilage without cytologic
atypia. Given the intimate connection with the cartilaginous rings, complete
resection might require bronchoplastic surgery.
The distinction between chondroma and chondrosarcoma is based upon the
cytologic atypia of the cartilaginous proliferation. Chondrosarcomas are
extremely rare, but endobronchial and peripheral locations have been described
[149–153]. The central tumors tend to be discovered at an earlier stage and
appear to have a relatively good prognosis until tumor size precludes complete
resection [150,151,153].
Glomus tumor
Glomus tumors are of presumed glomus cell origin or differentiation and have
features of modified smooth muscle by immunohistochemistry or electron
microscopy. Tracheal tumors [132,154–162] and lung tumors with endobronchial
or parenchymal growth have both been reported [154,158,159,162,163]. In
instances in which a detailed description of the tracheal location was reported,
the tumors were described as polypoid lesions arising from the posterior
membranous portion of the trachea [156,157]. Gaertner et al reported four cases
of primary pulmonary glomus tumors and reviewed the literature on four
additional previously reported cases [163]. The average age at presentation was
45 years. The majority of the tumors measured less than 2.5 cm in size. The
behavior of these tumors has been indolent, without evidence of recurrence
following resection. Gaertner et al also reported one patient with a malignant
glomus tumor (glomangiosarcoma) who developed widespread metastases and
died within 10 months after initial therapy.
Vascular tumors—hemangioma/angiosarcoma/Kaposi’s sarcoma
Capillary hemangioma, a benign vascular tumor, is much more frequently
reported in the pediatric literature. The tumor is typically discovered in early
infancy when patients present with respiratory distress and airflow obstruction
[164–167]. There are a few reports of this tumor in adults, who presented with a
chief complaint of hemoptysis or cough [168,169]. Bronchoscopic examination
revealed circumscribed submucosal lesions with an overlying vascularized surface
protruding into the airway. Complete removal has been achieved by bronchoscopy.
Primary pulmonary angiosarcomas are exceeding rare, even within the select
group of primary pulmonary sarcomas or otherwise rare tumors [120,170]. These
patients typically present with late-stage disease. Angiosarcoma in the lung is far
more likely to represent metastatic disease [171].
Kaposi’s sarcoma can present as purple–red, raised nodules along the
tracheobronchial tree or as diffuse bronchial wall thickening, but the most
common patterns of lung involvement are nodular and interstitial infiltrates and
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 21

pleural effusion [172,173]. In most instances, thoracic involvement presents late
in the course of the disease, but the lung might be the primary disease site in 10%
to 15% of AIDS patients [172,174]. Predominant pulmonary involvement
without skin lesions is also seen in rare instances in the sporadic European form
of the disease [175,176] or in the transplant population [177].
Granular cell tumor
Granular cell tumors (GCTs) that are histologically identical to those found in
other anatomic sites have been reported in the lung [178–181]. Deavers et al
reported the largest series on pulmonary granular cell tumors [178]. The patients
were adults with an average age of 45 years and an equal proportion of men to
women. In this study of 23 lesions from 20 patients, two tumors were peripheral,
but the remainder involved large, cartilaginous bronchi. Ten cases presented as
obstructing endobronchial lesions and three others had hemoptysis. Most of the
lesions were solitary, but two patients had multiple lung masses. Pulmonary
GCTs are typically small, with a mean size of 1 cm. The endobronchial lesions
are unencapsulated and polypoid with a tan–white to yellow color. Microscop-
ically, the tumor cells are large, round, or somewhat fusiform cells that have
abundant granular eosinophilic cytoplasm. The nuclei are small, with minimal
atypia and rare—if any—mitoses. The tumor cells can infiltrate peribronchial
structures including nerves and lymph nodes, but this extension is limited. The
overlying bronchial epithelium might show squamous metaplasia or ulceration,
but the characteristic pseudoepitheliomatous hyperplasia that is seen in other sites
has not been reported. The tumor cells are strongly positive for S-100 protein and
negative for cytokeratins. Malignant behavior has not been reported in pulmonary
granular cell tumors, even for those with multicentric involvement. Incompletely
resected lesions appear to be stable over time.
Hamartoma
Hamartoma is a benign, slow-growing mesenchymal tumor consisting of
varying proportions of adipose tissue, loose myxoid fibrous tissue, and cartilage.
Within the tumor are intersecting clefts lined by respiratory epithelium, but these
epithelial elements are not neoplastic and represent passive glandular entrapment
[182,183]. Hamartomas might be more accurately termed fibrolipochondromas
[48], but the historical designation of hamartoma has persisted despite its
misleading implication that these tumors are somehow a congenital malforma-
tion. Hamartoma is the most common benign tumor of the lung. The estimated
incidence of pulmonary hamartomas in the general population is 0.25% [64].
Hamartomas are more frequently intraparenchymal, but roughly 10% will present
in an endobronchial location, often with obstructive symptoms [183–185].
Multiple hamartomas or endotracheal hamartomas have been reported infre-
quently [186–189]. There is some association of pulmonary chondroid hamarto-
mas with Carney’s syndrome [148], hamartomas, and Cowden’s syndrome [190].
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4022

Endobronchial hamartomas are substantially represented (24%) in Shah et al’s
series of 185 benign tumors identified endoscopically and treated with laser
resection [7]. Initial endoscopic biopsies of these lesions are sometimes non-
diagnostic. The overlying benign surface epithelium, the mesenchymal elements
(especially the cartilage), and the frequent surrounding obstructive changes can
make bronchoscopic biopsy difficult. The gross appearance, however, is quite
characteristic. Hamartomas are well circumscribed and often lobulated or
papillary when they project into the airway lumen (Fig. 4). On average,
endobronchial hamartomas are smaller in size than their peripheral counterparts,
which can range from a few millimeters up to 20 cm; this size difference is no
doubt related to obstructive symptoms that lead to earlier detection [48].
Hamartomas are often firm on palpation—even hard—depending on the amount
of cartilage or the extent of calcification or ossification. They range from white to
yellow in color, depending on the proportion of adipose tissue or cartilage. There
is some potential for histologic confusion between this tumor, true chondromas
(as discussed previously), and mixed tumors of salivary gland-type, particularly
with limited sampling. In a cartilagneous hamartoma, the chondroid elements are
Fig. 4. Endobronchial hamartoma. This low-power photomicrograph demonstrates the characteristic
circumscription and lobulated architecture. There is a predominance of cartilaginous elements in this
tumor that might lead to a misdiagnosis of chondroma.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 23

well developed and are usually the predominant component. Moran et al have
observed that this is not typically the case with mixed tumors. In addition, the
epithelial elements of a mixed tumor appear to be an integral part of the lesion
rather than entrapped glandular elements, as in a hamartoma [19]. Chromosomal
abnormalities in pulmonary hamartomas have been reported [191–194]. The
most recent reports center on high-mobility group proteins and show interesting
specific genetic alterations shared by pulmonary hamartomas and other benign,
slow-growing tumors such as lipomas, endometrial polyps, and uterine myomas
[195–198].
Leiomyoma/leiomyosarcoma/rhabdomyosarcoma
Primary pulmonary leiomyomas are solitary tumors that occur as endotra-
cheal/endobronchial or intraparenchymal lesions with approximately equal fre-
quency [107]. Leiomyomas comprise approximately 2% of benign lung tumors
[199]. Although a wide age range that includes pediatric cases has been reported,
most of the tumors tend to occur in individuals in the third and fourth decades of
life [200,201]. Females appear to be affected more commonly than males.
Pulmonary leiomyomas are similar in gross and histologic appearance to
leiomyomas of other common sites such as the uterus or gastrointestinal tract.
Tumors within the tracheobronchial tree appear as fleshy, polypoid masses that
protrude intralumenally and are attached by a wide base [48]. By definition,
leiomyomas do not have significant cytologic atypia, a high mitotic rate, or
necrosis. The prognosis is excellent with complete resection. The location of the
tumor and secondary lung destruction might limit options such as laser therapy or
bronchoplastic surgery.
Excluding those that arise within the pulmonary artery, primary pulmonary
solitary leiomyosarcomas are about as rare as pulmonary leiomyomas. They tend to
be larger tumors and they occur, on average, about a decade later [200,202–205]. It
should be emphasized that the possibility of metastasis from the genital tract should
be rigorously excluded in women who present with what appears to be a primary
pulmonary leiomyosarcoma. Most of these tumors are intraparenchymal, but they
often have an endobronchial component by direct extension. In the small number of
reports that have examined pathologic attributes and prognosis, histologic grade
(ie, lower versus higher mitotic rates and absence or presence of necrosis) in
addition to size (3 cm) seemed to correlate with prognosis [204–206]. Yellin et al
reported an overall 45% 5-year survival rate in patients who underwent complete
surgical resection.
Primary pulmonary rhabdomyosarcomas are extremely rare, but they have
been reported occasionally in children and adults [207–219]. By definition, the
tumor must show evidence of striated muscle differentiation by light microscopy
(ie, cytoplasmic cross-striations), immunohistochemistry, or electron microscopy.
In addition, there are now molecular assays for specific chromosomal trans-
locations in alveolar rhabdomyosarcoma. It is well documented that rhabdomyo-
sarcomas can arise in locations devoid of striated muscle. Endobronchial,
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4024

intraparenchymal, and pulmonary trunk sites have been reported. Most of the
adult patients died within 2 years of diagnosis. Many of the patients presented
late in the course of their disease or at autopsy. As with other rare types of
pulmonary sarcoma, it is important to consider the possibility of metastasis from
another primary site or the more common carcinosarcoma.
Lipoma/liposarcoma
Most lipomas of the lung that have been reported are endobronchial, but it is
not clear to what extent some of these reported cases actually represent
hamartomas with a predominant component of adipose tissue [220–226]. Never-
theless, from a clinical perspective, these tumors are benign and a conservative
approach to excision is indicated. Primary liposarcomas are among the most rare
of the subtypes of pulmonary sarcoma [227–229].
Fibrosarcoma/hemangiopericytoma/malignant fibrous histiocytoma
As Suster asserts in his review of primary sarcomas of the lung, the majority of
the previously reported cases of fibrosarcoma or hemangiopericytoma are
probably intrapulmonary, localized, fibrous tumors or, in the instance of heman-
giopericytoma, primary synovial sarcomas [229]. The most common of the
spindle cell sarcomas without specific differentiation is therefore malignant
fibrous histiocytoma. Most of these tumors have been located in the lung
periphery. Histologically, malignant fibrous histiocytomas are identical in appear-
ance to their soft tissue counterparts. There is a broad differential diagnosis for
this pleomorphic spindled cell tumor that includes more common entities such as
intrathoracic extension from a chest wall primary, extrathoracic metastasis from a
soft tissue primary, pleomorphic carcinoma, and inflammatory pseudotumors.
Benign and malignant nerve sheath tumors—neurofibroma/schwannoma
(neurilemoma)/neurogenic sarcoma
It is far more common to encounter nerve sheath tumors within the posterior
mediastinum or paravertebral region, but a few endotracheal/endobronchial or
intraparenchymal cases have been reported [202,230–239]. Neurofibromas and
schwannomas are more common than malignant tumors. The incidence is equal
among women and men. Although some of these cases involve patients with
neurofibromatosis, other patients do not appear to have any evidence of the
disease. Obstructive symptoms have been noted with intraluminal tumor growth.
Endoscopically, the tumors have a polypoid appearance similar to other types of
tumor. These nerve sheath tumors are histologically identical to similar tumors in
other anatomic sites and have a similar clinical behavior.
Osteosarcoma
Primary osteosarcoma is a rare tumor, with only a handful of cases reported in
the literature [206,240,241]. As Suster cautions in his summary of primary
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 25

pulmonary sarcomas, occasional tumors that were initially felt to be primary lung
osteosarcomas proved to be carcinosarcomas upon additional sampling
[229,240]. The prognosis for these tumors is poor.
Synovial sarcoma
Recent investigations using molecular analysis have confirmed that synovial
sarcomas arise in the lung and are more common among the primary pulmonary
sarcomas than previously thought [170,242–247]. Tissue assays for the char-
acteristic t(X;18) translocation and SYT/SSX fusion genes are available in
specialized laboratories; therefore sufficient tissue should be obtained at the time
of biopsy or resection if sarcoma is a consideration. The largest series to date has
been reported by Zeren et al [247]. In their series of 25 cases, there were an
approximately equal number of men and women, with an age range from 16 to
77 years. The most common symptoms were chest pain, cough, dyspnea, and
hemoptysis. The tumors were solitary and varied in size from 0.6 cm to 20.0 cm.
Two lesions involved the bronchial wall. Follow-up information was available for
18 patients. Six died of the disease, eight were alive with the disease, and four
had no evidence of the disease. The authors emphasize that because of its
distinctive biologic behavior, synovial sarcoma should be distinguished from a
variety of malignant and metastatic lesions.
Miscellaneous tumors
Inflammatory pseudotumor/inflammatory myofibroblastic tumor
Historically, a wide variety of names have been applied to inflammatory
pseudotumor/inflammatory myofibroblastic tumors (or, perhaps, entities). In
addition to inflammatory myofibroblastic tumor [248] or inflammatory pseudo-
tumor [249], these names include plasma cell granuloma [250,251], xanthoma
[252], xanthomatous pseudotumor [253], fibroxanthoma, histiocytoma [254],
fibrous histiocytoma, pseudosarcomatous myofibroblastic tumor, invasive fibrous
tumor of the tracheobronchial tree [255], mast cell tumor [256], and mast cell
granuloma [257]. It is easier to make sense of this bewildering proliferation of
names if one understands that the lesion itself is composed of a variety of cellular
components, and the nature of this lesion (reactive versus neoplastic) is still a
matter of some controversy. These lesions frequently occur in the lung, but many
other extrapulmonary sites have been reported. The 1999 WHO/IASLC criteria
define the entity as ‘‘a spectrum of fibroblastic or myofibroblastic proliferations
with a varying infiltrate of inflammatory cells, typically plasma cells, lympho-
cytes, or foamy histiocytes’’ [1]. The definition encompasses lesions that have a
predominant myofibroblastic or fibroxanthomatous appearance and lesions with a
heavy infiltrate of plasma cells. This lesion is commonly seen in the pediatric age
group, and is encountered more frequently in patients under 40 years old, but
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4026

there is a wide reported age range that includes infants and the elderly
[250,251,258–260]. The incidence in men and women is equal. About 50% of
patients will be asymptomatic; the other half present with cough, hemoptysis, and
dyspnea, and some will have systemic symptoms. The lesions are usually solitary,
but multiple lesions have been reported. The lesion can be entirely intraparen-
chymal or form a polypoid endobronchial/endotracheal mass [250,253,254,261].
Some lesions have mediastinal or chest wall extension. Grossly, the mass is
usually well circumscribed but unencapsulated with variations in color and
consistency that depend upon the degree of inflammation, fibroblastic prolifera-
tion, and extent of collagenization (Fig. 5). The proliferation of names corre-
sponds to microscopic intra- and interlesional variability. Some tumors might
have a predominant spindle cell component; these spindle cells are often high-
lighted by positive vimentin and actin immunohistochemical stains [248].
Cytokeratin stains are positive in about one third of cases. Inflammatory areas
might be alternate with the fibrohistiocytic areas or they might predominate. The
plasma cell areas have been demonstrated to be polyclonal [262]. Although it is
not entirely clear that some authors have excluded other tumors such as malignant
fibrous histiocytoma or deceptively bland sarcomatoid carcinomas, atypical
features such as mitoses, necrosis, vascular invasion, and invasion into adjacent
thoracic structures have been reported. There have been rare instances of
spontaneous regression [259]. Complete excision is generally associated with
an excellent prognosis [259,263,264]. Partially resected lesions might show
slowly infiltrative growth that results in death caused by compromise of adjacent
vital structures [262,265]. In terms of etiology, some authors have suggested a
Fig. 5. Inflammatory myofibroblastic tumor. The lesion is well circumscribed with variegations in
color and consistency. This lesion might easily be mistaken for a more common lung carcinoma on
radiographs and initial gross examination.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 27

reactive process [266,267]. Other authors regard these lesions as benign but
locally invasive tumors and have suggested a similarity to fibromatoses
[248,268]. Recent studies have demonstrated cytogenetic evidence for a clonal
origin [269] in addition to anaplastic lymphoma kinase expression [270,271]. In
the end, one might question whether this is truly one lesion with a diverse
histologic appearance or several distinct reactive or neoplastic entities with
overlapping histologic features [272,273].
Malignant lymphoma
The immunologically specialized bronchial associated lymphoid tissue is an
important component of the larger mucosal associated lymphoid tissue and, as
such, it can give rise to a variety of benign and malignant lymphoproliferative
disorders. Although it is more common to see secondary airway involvement or
disseminated disease from a nodal lymphoma, primary pulmonary lymphomas
have been well characterized. Low-grade lymphomas of the bronchial-associated
lymphoid tissue represent the vast majority of all primary pulmonary lymphomas
[274], but nearly every type of lymphoma has been reported, including primary
Hodgkin’s disease [275] and anaplastic large-cell lymphoma [276]. From a
practical point of view, lymphoid disorders must be considered within the
differential diagnosis of crushed endobronchial biopsies that consist of ‘‘small
blue cells’’ or undifferentiated large cells. By definition, there should be no
histologic evidence of nodal or systemic involvement for the diagnosis of primary
pulmonary lymphoma to be rendered.
Primary pulmonary melanoma
Metastatic melanoma is far more common than primary pulmonary melanoma,
but both lesions can present in an endobronchial location and patients tend to
be symptomatic on the basis of this endobronchial involvement [120,142,
144,277,278]. The tumor can be confused with other more common lung tumors
such as carcinoids or non–small-cell lung cancer, especially when the evaluation
is made on small biopsy samples [48,279]. The diagnosis of primary pulmonary
melanoma is based upon the absence of a melanoma of the skin, eye, or other site
and the finding of a solitary tumor that has at least some endobronchial
component. As Wilson and Moran point out in their review of eight cases, these
criteria for diagnosis—in particular the demonstration of an in situ component—
can be difficult to fulfill [279]. Age of patients in these series has ranged from the
30s to the 80s, and there has been an equal incidence in men and women. Some
of the lesions had a large, central, endobronchial component [278,279]. The
overall prognosis for primary pulmonary melanoma is poor in most circum-
stances, but there have been some reported exceptions. In Wilson and Moran’s
series, one out of eight patients was alive with no evidence of disease at
108 months of follow-up care. Jennings et al reported a 30% survival rate after
complete resection.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4028

Summary
This article provides a broad overview of tumors that can involve the tra-
cheobronchial tree. For the most part, the clinical, radiographic, and endoscopic
presentation of these rare tumors does not differ significantly from the more
common tumors of the lung. Appropriate classification of many tracheobronchial
tumors ultimately requires complete sampling and a thorough microscopic
evaluation. The introduction of ancillary diagnostic techniques such as immuno-
histochemistry and molecular analysis will continue to refine tumor classification.
References
[1] Travis WD, Colby TV, Corrin B, et al. Histological typing of lung and pleural tumours,
3rd edition. Berlin, Germany: Springer; 1999.
[2] Flieder DB, Koss MN, Nicholson A, et al. Solitary pulmonary papillomas in adults: a clinico-
pathologic and in situ hybridization study of 14 cases combined with 27 cases in the literature.
Am J Surg Pathol 1998;22:1328–42.
[3] Laubscher FA. Solitary squamous cell papilloma of bronchial origin. Am J Clin Pathol 1969;
52:599–603.
[4] Zimmermann A, Lang HR, Muhlberger F, et al. Papilloma of the bronchus. Respiration 1980;
39:286–90.
[5] Kramer SS, Wehunt WD, Stocker JT, et al. Pulmonary manifestations of juvenile laryngotra-
cheal papillomatosis. AJR Am J Roentgenol 1985;144:687–94.
[6] Magid MS, Chen YT, Soslow RA, et al. Juvenile-onset recurrent respiratory papillomatosis
involving the lung: a case report and review of the literature. Pediatr Dev Pathol 1998;1:
157–63.
[7] Shah H, Garbe L, Nussbaum E, et al. Benign tumors of the tracheobronchial tree. Endoscopic
characteristics and role of laser resection. Chest 1995;107:1744–51.
[8] Smith PS, McClure J. A papillary endobronchial tumor with a transitional cell pattern. Arch
Pathol Lab Med 1982;106:503–6.
[9] Popper HH, el-Shabrawi Y, Wockel W, et al. Prognostic importance of human papilloma virus
typing in squamous cell papilloma of the bronchus: comparison of in situ hybridization and the
polymerase chain reaction. Hum Pathol 1994;25:1191–7.
[10] Popper HH, Wirnsberger G, Juttner-Smolle FM, et al. The predictive value of human papilloma
virus (HPV) typing in the prognosis of bronchial squamous cell papillomas. Histopathology
1992;21:323–30.
[11] Cook JR, Hill DA, Humphrey PA, et al. Squamous cell carcinoma arising in recurrent respi-
ratory papillomatosis with pulmonary involvement: emerging common pattern of clinical fea-
tures and human papillomavirus serotype association. Mod Pathol 2000;13:914–8.
[12] Ashmore PG. Papilloma of the bronchus. J Thorac Surg 1954;27:293–4.
[13] Basheda S, Gephardt GN, Stoller JK. Columnar papilloma of the bronchus. Case report and
literature review. Am Rev Respir Dis 1991;144:1400–2.
[14] Spencer H, Dail DH, Arneaud J. Non-invasive bronchial epithelial papillary tumors. Cancer
1980;45:1486–97.
[15] Archer RL, Grogg SE, Sanders SP. Mucoepidermoid bronchial adenoma in a 6-year-old girl:
a case report and review of the literature. J Thorac Cardiovasc Surg 1987;94:452–4.
[16] England DM, Hochholzer L. Truly benign ‘‘bronchial adenoma.’’ Report of 10 cases of mucous
gland adenoma with immunohistochemical and ultrastructural findings. Am J Surg Pathol 1995;
19:887–9.
[17] Ferguson CJ, Cleeland JA. Mucous gland adenoma of the trachea: case report and literature
review. J Thorac Cardiovasc Surg 1988;95:347–50.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 29

[18] Heard BE, Corrin B, Dewar A. Pathology of seven mucous cell adenomas of the bronchial
glands with particular reference to ultrastructure. Histopathology 1985;9:687–701.
[19] Moran CA, Suster S, Askin FB, et al. Benign and malignant salivary gland-type mixed tumors
of the lung. Clinicopathologic and immunohistochemical study of eight cases. Cancer 1994;
73:2481–90.
[20] Sweeney EC, McDermott M. Pleomorphic adenoma of the bronchus. J Clin Pathol 1996;49:
87–9.
[21] Basaklar AC, Sonmez K, Memis L, et al. Pleomorphic adenoma of the main bronchus. J Pediatr
Surg 1994;29:1550–2.
[22] Heifetz SA, Collins B, Matt BH. Pleomorphic adenoma (benign mixed tumor) of the trachea.
Pediatr Pathol 1992;12:563–74.
[23] Moran CA. Primary salivary gland-type tumors of the lung. Semin Diagn Pathol 1995;12:
106–22.
[24] Davis PW, Briggs JC, Seal RM, et al. Benign and malignant mixed tumours of the lung. Thorax
1972;27:657–73.
[25] Hayes MM, van der Westhuizen NG, Forgie R. Malignant mixed tumor of bronchus: a biphasic
neoplasm of epithelial and myoepithelial cells. Mod Pathol 1993;6:85–8.
[26] Sakamoto H, Uda H, Tanaka T, et al. Pleomorphic adenoma in the periphery of the lung. Report
of a case and review of the literature. Arch Pathol Lab Med 1991;115:393–6.
[27] Spencer H. Bronchial mucous gland tumours. Virchows Arch A Pathol Anat Histol 1979;383:
101–15.
[28] Wright ES, Pike E, Couves CM. Unusual tumours of the lung. J Surg Oncol 1983;24:23–9.
[29] Higashiyama M, Kodama K, Yokouchi H, et al. Myoepithelioma of the lung: report of two
cases and review of the literature. Lung Cancer 1998;20:47–57.
[30] Horinouchi H, Ishihara T, Kawamura M, et al. Epithelial myoepithelial tumour of the tracheal
gland. J Clin Pathol 1993;46:185–7.
[31] Nistal M, Garcia-Viera M, Martinez-Garcia C, et al. Epithelial–myoepithelial tumor of the
bronchus. Am J Surg Pathol 1994;18:421–5.
[32] Pelosi G, Fraggetta F, Maffini F, et al. Pulmonary epithelial–myoepithelial tumor of un-
proven malignant potential: report of a case and review of the literature. Mod Pathol 2001;14:
521–6.
[33] Ryska A, Kerekes Z, Hovorkova E, et al. Epithelial–myoepithelial carcinoma of the bronchus.
Pathol Res Pract 1998;194(6):431–5.
[34] Shanks JH, Hasleton PS, Curry A, et al. Bronchial epithelial–myoepithelial carcinoma. His-
topathology 1998;33:90–1.
[35] Shirasaki A, Hirakata Y, Kitamura S, et al. A salivary gland-type monomorphic adenoma with
trabecular proliferation in the lung. Intern Med 1995;34:1086–8.
[36] Strickler JG, Hegstrom J, Thomas MJ, et al. Myoepithelioma of the lung. Arch Pathol Lab Med
1987;111:1082–5.
[37] Tsuji N, Tateishi R, Ishiguro S, et al. Adenomyoepithelioma of the lung. Am J Surg Pathol
1995;19:956–62.
[38] Wilson RW, Moran CA. Epithelial–myoepithelial carcinoma of the lung: immunohistochemical
and ultrastructural observations and review of the literature. Hum Pathol 1997;28:631–5.
[39] Ritter JH, Nappi O. Oxyphilic proliferations of the respiratory tract and paranasal sinuses.
Semin Diagn Pathol 1999;16:105–16.
[40] Black III WC. Pulmonary oncocytoma. Cancer 1969;23:1347–57.
[41] Scharifker D, Marchevsky A. Oncocytic carcinoid of lung: an ultrastructural analysis. Cancer
1981;47:530–2.
[42] Sklar JL, Churg A, Bensch KG. Oncocytic carcinoid tumor of the lung. Am J Surg Pathol
1980;4:287–92.
[43] Fechner RE, Bentinck BR. Ultrastructure of bronchial oncocytoma. Cancer 1973;31:1451–7.
[44] Nielsen AL. Malignant bronchial oncocytoma: case report and review of the literature. Hum
Pathol 1985;16:852–4.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4030

[45] Santos-Briz A, Terron J, Sastre R, et al. Oncocytoma of the lung. Cancer 1977;40:1330–6.
[46] Warter A, Walter P, Sabountchi M, et al. Oncocytic bronchial adenoma. Histological, histo-
chemical and ultrastructural study. Virchows Arch A Pathol Anat Histol 1981;392:231–9.
[47] Walter JB, Pryce DM. The site and origin of lung cancer and its relationship to histologic type.
Thorax 1955;10:117–26.
[48] Carter D, Patchefsky AS. Tumors and tumor-like lesions of the lung. 1st edition, Vol. 36.
Philadelphia, PA: WB Saunders; 1998.
[49] Li W, Ellerbroek NA, Libshitz HI. Primary malignant tumors of the trachea. A radiologic and
clinical study. Cancer 1990;66:894–9.
[50] Regnard JF, Fourquier P, Levasseur P. Results and prognostic factors in resections of primary
tracheal tumors: a multicenter retrospective study. The French Society of Cardiovascular Sur-
gery. J Thorac Cardiovasc Surg 1996;111:808–13.
[51] Gail MH, Eagan RT, Feld R, et al. Prognostic factors in patients with resected stage I non-small
cell lung cancer. A report from the Lung Cancer Study Group. Cancer 1984;54:1802–13.
[52] Mountain CF, Lukeman JM, Hammar SP, et al. Lung cancer classification: the relationship of dis-
ease extent and cell type to survival in a clinical trials population. J Surg Oncol 1987;35:147–56.
[53] Dulmet-Brender E, Jaubert F, Huchon G. Exophytic endobronchial epidermoid carcinoma.
Cancer 1986;57:1358–64.
[54] Brambilla E, Moro D, Veale D, et al. Basal cell (basaloid) carcinoma of the lung: a new
morphologic and phenotypic entity with separate prognostic significance. Hum Pathol 1992;
23:993–1003.
[55] Foroulis CN, Iliadis KH, Mauroudis PM, et al. Basaloid carcinoma, a rare primary lung neo-
plasm: report of a case and review of the literature. Lung Cancer 2002;35:335–58.
[56] Lin O, Harkin TJ, Jagirdar J. Basaloid-squamous cell carcinoma of the bronchus. Report of a
case with review of the literature. Arch Pathol Lab Med 1995;119:1167–70.
[57] Moro D, Brichon PY, Brambilla E, et al. Basaloid bronchial carcinoma. A histologic group with
a poor prognosis. Cancer 1994;73:2734–9.
[58] Saltarelli MG, Fleming MV, Wenig BM, et al. Primary basaloid squamous cell carcinoma of the
trachea. Am J Clin Pathol 1995;104:594–8.
[59] Kodama T, Shimosato Y, Koide T, et al. Endobronchial polypoid adenocarcinoma of the lung.
Histological and ultrastructural studies of five cases. Am J Surg Pathol 1984;8:845–54.
[60] Hirata H, NoguchiM, Shimosato Y, et al. Clinicopathologic and immunohistochemical character-
istics of bronchial gland cell type adenocarcinoma of the lung. Am J Clin Pathol 1990;93:20–5.
[61] Iyoda A, Hiroshima K, Toyozaki T, et al. Clear cell adenocarcinoma with endobronchial
polypoid growth. Pathol Int 2000;50:979–83.
[62] Kawabuchi B, Ishikawa Y, Tsuchiya S, et al. Mucosal spreading adenocarcinoma at the hilar
portion of the lung. Acta Pathol Jpn 1993;43:690–5.
[63] Mardini G, Pai U, Chavez AM, et al. Endobronchial adenocarcinoma with endometrioid fea-
tures and prominent neuroendocrine differentiation. A variant of fetal adenocarcinoma. Cancer
1994;73:1383–9.
[64] Carter D, Eggleston JC. Tumors of the lower respiratory tract. In: Hartman WH, editor. Atlas
of Tumor Pathology. 2nd edition, Vol. 17. Washington, DC: Armed Forces Institute of Pathol-
ogy; 1980.
[65] Ribet M, Gosselin B, Gambiez L, et al. Bronchial carcinoids. Eur J Cardiothorac Surg 1993;
7:347–50.
[66] Akiba T, Naruke T, Kondo H, et al. Carcinoid tumor of the lung: clinicopathological study of 32
cases. Jpn J Clin Oncol 1992;22:92–5.
[67] Brandt III B, Heintz SE, Rose EF, et al. Bronchial carcinoid tumors. Ann Thorac Surg 1984;38:
63–5.
[68] Fink G, Krelbaum T, Yellin A, et al. Pulmonary carcinoid: presentation, diagnosis, and outcome
in 142 cases in Israel and review of 640 cases from the literature. Chest 2001;119:1647–51.
[69] Hancock BJ, Di Lorenzo M, Youssef S, et al. Childhood primary pulmonary neoplasms.
J Pediatr Surg 1993;28:1133–6.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 31

[70] Hause DW, Harvey JC. Endobronchial carcinoid and mucoepidermoid carcinoma in children.
J Surg Oncol 1991;46:270–2.
[71] Hurt R, Bates M. Carcinoid tumours of the bronchus: a 33 year experience. Thorax 1984;
39:617–23.
[72] McCaughan BC, Martini N, Bains MS. Bronchial carcinoids. Review of 124 cases. J Thorac
Cardiovasc Surg 1985;89:8–17.
[73] Schreurs AJ, Westermann CJ, van den Bosch JM, et al. A twenty-five-year follow-up of
ninety-three resected typical carcinoid tumors of the lung. J Thorac Cardiovasc Surg 1992;
104:1470–5.
[74] Davila DG, Dunn WF, Tazelaar HD, et al. Bronchial carcinoid tumors. Mayo Clin Proc 1993;
68:795–803.
[75] Jeung MY, Gasser B, Gangi A, et al. Bronchial carcinoid tumors of the thorax: spectrum of
radiologic findings. Radiographics 2002;22:351–65.
[76] Rosado de Christenson ML, Abbott GF, Kirejczyk WM, et al. Thoracic carcinoids: radiologic-
pathologic correlation. Radiographics 1999;19:707–36.
[77] Limper AH, Carpenter PC, Scheithauer B, et al. The Cushing syndrome induced by bronchial
carcinoid tumors. Ann Intern Med 1992;117:209–14.
[78] Salminen US, Halttunen PE, Mattila SP, et al. Bronchial carcinoid: a clinical follow-up study of
33 cases. Scand J Thorac Cardiovasc Surg 1991;25:189–94.
[79] Zhang Z, Li S, Ge F, et al. Surgical treatment and prognosis of bronchial carcinoid tumor. Chin
Med Sci J 1996;11:248–51.
[80] Snyder III N, Scurry MT, Deiss WP. Five families with multiple endocrine adenomatosis. Ann
Intern Med 1972;76:53–81.
[81] Underdahl L, Woolner L, Black B. Multiple endocrine adenopathy: report of eight cases in
which the parathyroids, pituitray, and pancreas were involved. J Clin Endocrinol 1953;13:
20–47.
[82] Williams E, Celestin L. The association of bronchial carcinoid and pluriglandular adenomato-
sis. Thorax 1962;17:120.
[83] Ishida T, Yokoyama H, Sugio K, et al. Carcinoid tumor of the lung: clinicopathological and
immunohistochemical studies. Eur J Surg Oncol 1992;18:180–7.
[84] Okike N, Bernatz PE, Woolner LB. Carcinoid tumors of the lung. Ann Thorac Surg 1976;
22:270–7.
[85] Warren WH, Gould VE. Long-term follow-up of classical bronchial carcinoid tumors. Clin-
icopathologic observations. Scand J Thorac Cardiovasc Surg 1990;24:125–30.
[86] Thunnissen FB, Van Eijk J, Baak JP, et al. Bronchopulmonary carcinoids and regional lymph
node metastases. A quantitative pathologic investigation. Am J Pathol 1988;132:119–22.
[87] Debelenko LV, Brambilla E, Agarwal SK, et al. Identification of MEN1 gene mutations in
sporadic carcinoid tumors of the lung. Hum Mol Genet 1997;6:2285–90.
[88] Finkelstein SD, Hasegawa T, Colby T, et al. 11q13 allelic imbalance discriminates pulmonary
carcinoids from tumorlets. A microdissection-based genotyping approach useful in clinical
practice. Am J Pathol 1999;155:633–40.
[89] Jakobovitz O, Nass D, DeMarco L, et al. Carcinoid tumors frequently display genetic abnor-
malities involving chromosome 11. J Clin Endocrinol Metab 1996;81:3164–7.
[90] Johansson M, Heim S, Mandahl N, et al. Cytogenetic analysis of six bronchial carcinoids.
Cancer Genet Cytogenet 1993;66:33–8.
[91] Onuki N, Wistuba II, Travis WD, et al. Genetic changes in the spectrum of neuroendocrine lung
tumors. Cancer 1999;85:600–7.
[92] Petzmann S, Ullmann R, Klemen H, et al. Loss of heterozygosity on chromosome arm 11q in
lung carcinoids. Hum Pathol 2001;32:333–8.
[93] Ullmann R, Schwendel A, Klemen H, et al. Unbalanced chromosomal aberrations in neuro-
endocrine lung tumors as detected by comparative genomic hybridization. Hum Pathol 1998;
29:1145–9.
[94] Walch AK, Zitzelsberger HF, Aubele MM, et al. Typical and atypical carcinoid tumors of the
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4032

lung are characterized by 11q deletions as detected by comparative genomic hybridization. Am
J Pathol 1998;153:1089–98.
[95] Whang-Peng J, Knutsen T, Gazdar A, et al. Nonrandom structural and numerical chromosome
changes in non–small-cell lung cancer. Genes Chromosomes Cancer 1991;3:168–88.
[96] Brambilla E, Lantuejoul S, Sturm N. Divergent differentiation in neuroendocrine lung tumors.
Semin Diagn Pathol 2000;17:138–48.
[97] Arrigoni MG, Woolner LB, Bernatz PE. Atypical carcinoid tumors of the lung. J Thorac
Cardiovasc Surg 1972;64:413–21.
[98] Warren WH, Memoli VA, Gould VE. Immunohistochemical and ultrastructural analysis of
bronchopulmonary neuroendocrine neoplasms. II. Well-differentiated neuroendocrine carcino-
mas. Ultrastruct Pathol 1984;7:185–99.
[99] Mark EJ, Ramirez JF. Peripheral small-cell carcinoma of the lung resembling carcinoid tumor.
A clinical and pathologic study of 14 cases. Arch Pathol Lab Med 1985;109:263–9.
[100] Paladugu RR, Benfield JR, Pak HY, et al. Bronchopulmonary Kulchitzky cell carcinomas.
A new classification scheme for typical and atypical carcinoids. Cancer 1985;55:1303–11.
[101] Beltrami V, Gallinaro LS, Bezzi M, et al. [Pulmonary carcinoids. Analysis of 53 cases]. Chir
Ital 1999;51:109–12.
[102] Bonikos DS, Bensch KG, Jamplis RW. Peripheral pulmonary carcinoid tumors. Cancer 1976;
37(4):1997–98.
[103] Gould PM, Bonner JA, Sawyer TE, et al. Bronchial carcinoid tumors: importance of prog-
nostic factors that influence patterns of recurrence and overall survival. Radiology 1998;208:
181–5.
[104] Beasley MB, Thunnissen FB, Brambilla E, et al. Pulmonary atypical carcinoid: predictors of
survival in 106 cases. Hum Pathol 2000;31:1255–65.
[105] Travis WD, Linnoila RI, Tsokos MG, et al. Neuroendocrine tumors of the lung with proposed
criteria for large-cell neuroendocrine carcinoma. An ultrastructural, immunohistochemical, and
flow cytometric study of 35 cases. Am J Surg Pathol 1991;15:529–53.
[106] Travis WD, Gal AA, Colby TV, et al. Reproducibility of neuroendocrine lung tumor classi-
fication. Hum Pathol 1998;29:272–9.
[107] Colby TV, Koss MK, Travis WD. Tumors of the lower respiratory tract, Fascicle 13, 3rd
edition. Washington, DC: Armed Forces Institute of Pathology; 1995.
[108] de Lima R. Bronchial adenoma. Clinicopathologic study and results of treatment. Chest 1980;
77:81–4.
[109] Dalton ML, Gatling RR. Peripheral adenoid cystic carcinoma of the lung. South Med J 1990;
83:577–9.
[110] Gallagher CG, Stark R, Teskey J, et al. Atypical manifestations of pulmonary adenoid cystic
carcinoma. Br J Dis Chest 1986;80:396–9.
[111] Inoue H, Iwashita A, Kanegae H, et al. Peripheral pulmonary adenoid cystic carcinoma with
substantial submucosal extension to the proximal bronchus. Thorax 1991;46:147–8.
[112] Ahel V, Zubovic I, Rozmanic V. Bronchial adenoid cystic carcinoma with saccular bronchiec-
tasis as a cause of recurrent pneumonia in children. Pediatr Pulmonol 1992;12:260–2.
[113] Augustin N, Hofmann V, Kap-herr S, et al. Endotracheal and endobronchial tumours in child-
hood. Prog Pediatr Surg 1987;21:136–44.
[114] Nomori H, Kaseda S, Kobayashi K, et al. Adenoid cystic carcinoma of the trachea and main-
stem bronchus. A clinical, histopathologic, and immunohistochemical study. J Thorac Cardio-
vasc Surg 1988;96:271–7.
[115] Grillo HC, Mathisen DJ. Primary tracheal tumors: treatment and results. Ann Thorac Surg
1990;49:69–77.
[116] Ishida T, Nishino T, Oka T, et al. Adenoid cystic carcinoma of the tracheobronchial tree:
clinicopathology and immunohistochemistry. J Surg Oncol 1989;41:52–9.
[117] Moran CA, Suster S, Koss MN. Primary adenoid cystic carcinoma of the lung. A clinicopatho-
logic and immunohistochemical study of 16 cases. Cancer 1994;73:1390–7.
[118] Conlan AA, Payne WS, Woolner LB, et al. Adenoid cystic carcinoma (cylindroma) and mu-
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 33

coepidermoid carcinoma of the bronchus. Factors affecting survival. J Thorac Cardiovasc Surg
1978;76:369–77.
[119] Heitmiller RF, Mathisen DJ, Ferry JA, et al. Mucoepidermoid lung tumors. Ann Thorac Surg
1989;47:394–9.
[120] Miller DL, Allen MS. Rare pulmonary neoplasms. Mayo Clin Proc 1993;68:492–8.
[121] Yousem SA, Hochholzer L. Mucoepidermoid tumors of the lung. Cancer 1987;60:1346–52.
[122] Green LK, Gallion TL, Gyorkey F. Peripheral mucoepidermoid tumour of the lung. Thorax
1991;46:65–6.
[123] Klacsmann PG, Olson JL, Eggleston JC. Mucoepidermoid carcinoma of the bronchus: an elec-
tron microscopic study of the low grade and the high grade variants. Cancer 1979;43:1720–33.
[124] Turnbull AD, Huvos AG, Goodner JT, et al. Mucoepidermoid tumors of bronchial glands.
Cancer 1971;28:539–44.
[125] Wolf KM, Mehta D, Claypool WD. Mucoepidermoid carcinoma of the lung with intracranial
metastases. Chest 1988;94:435–8.
[126] Fechner RE, Bentinck BR, Askew Jr JB. Acinic cell tumor of the lung. A histologic and
ultrastructural study. Cancer 1972;29:501–8.
[127] Gharpure KJ, Deshpande RK, Vlshweshvara RN, et al. Acinic cell tumour of the bronchus
(a case report). Indian J Cancer 1985;22:152–6.
[128] Katz DR, Bubis JJ. Acinic cell tumor of the bronchus. Cancer 1976;38:830–2.
[129] Moran CA, Suster S, Koss MN. Acinic cell carcinoma of the lung (‘‘Fechner tumor’’). A
clinicopathologic, immunohistochemical, and ultrastructural study of five cases. Am J Surg
Pathol 1992;16:1039–50.
[130] Ukoha OO, Quartararo P, Carter D, et al. Acinic cell carcinoma of the lung with metastasis to
lymph nodes. Chest 1999;115:591–5.
[131] Ansari MA, Marchevsky A, Strick L, et al. Upper airway obstruction secondary to acinic cell
carcinoma of the trachea. Use of Nd:YAG laser. Chest 1996;110:1120–2.
[132] Heard BE, Dewar A, Firmin RK, et al. One very rare and one new tracheal tumour found by
electron microscopy: glomus tumour and acinic cell tumour resembling carcinoid tumours by
light microscopy. Thorax 1982;37:97–103.
[133] Horowitz Z, Kronenberg J. Acinic cell carcinoma of the trachea. Auris Nasus Larynx 1994;
21:193–5.
[134] Yoshida K, Koyama I, Matsui T, et al. [Acinic cell tumor of the bronchial gland]. Nippon Geka
Gakkai Zasshi 1989;90:1810–3.
[135] Martin H, Abboud A, Nawka T. [Predominantly mucous-differentiated acinic cell adenocarci-
noma of the trachea]. Zentralbl Allg Pathol 1989;135:751–6.
[136] Payne WS, Schier J, Woolner LB. Mixed tumors of the bronchus (salivary gland type). J Thorac
Cardiovasc Surg 1965;49:663–8.
[137] Koss MN, Hochholzer L, O’Leary T. Pulmonary blastomas. Cancer 1991;67:2368.
[138] Dehner LP. Pleuropulmonary blastoma is THE pulmonary blastoma of childhood. Semin Diagn
Pathol 1994;11:144–51.
[139] Manivel JC, Priest JR, Watterson J, et al. Pleuropulmonary blastoma. The so-called pulmonary
blastoma of childhood. Cancer 1988;62:1516–26.
[140] Koss MN, Hochholzer L, Frommelt RA. Carcinosarcomas of the lung: a clinicopathologic
study of 66 patients. Am J Surg Pathol 1999;23:1514–26.
[141] Fishback NF, Travis WD, Moran CA, et al. Pleomorphic (spindle/giant cell) carcinoma of the
lung. A clinicopathologic correlation of 78 cases. Cancer 1994;73:2936–45.
[142] Braman SS, Whitcomb ME. Endobronchial metastasis. Arch Intern Med 1975;135:543–7.
[143] Heitmiller RF, Marasco WJ, Hruban RH, et al. Endobronchial metastasis. J Thorac Cardiovasc
Surg 1993;106:537–42.
[144] Suster S, Moran CA. Unusual manifestations of metastatic tumors to the lungs. Semin Diagn
Pathol 1995;12:193–206.
[145] Katsimbri PP, Bamias AT, Froudarakis ME, et al. Endobronchial metastases secondary to solid
tumors: report of eight cases and review of the literature. Lung Cancer 2000;28:163–70.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4034

[146] Seo JB, Im JG, Goo JM, et al. Atypical pulmonary metastases: spectrum of radiologic findings.
Radiographics 2001;21:403–17.
[147] Shermeta DW, Carter D, Haller Jr JA. Chondroma of the bronchus in childhood: a case report
illustrating problems in diagnosis and management. J Pediatr Surg 1975;10:545–8.
[148] Carney JA, Sheps SG, Go VL, et al. The triad of gastric leiomyosarcoma, functioning extra-
adrenal paraganglioma and pulmonary chondroma. N Engl J Med 1977;296:1517–8.
[149] Azimullah PC, Teengs JP, Boerma EJ. Chondrosarcoma of the lobar bronchus with prolonged
postoperative survival. Eur Respir J 1994;7:1537–8.
[150] Hayashi T, Tsuda N, Iseki M, et al. Primary chondrosarcoma of the lung. A clinicopathologic
study. Cancer 1993;72:69–74.
[151] Morgenroth A, Pfeuffer HP, Viereck HJ, et al. Primary chondrosarcoma of the left inferior lobar
bronchus. Respiration 1989;56:241–4.
[152] Sun CC, Kroll M, Miller JE. Primary chondrosarcoma of the lung. Cancer 1982;50:1864–6.
[153] Yellin A, Schwartz L, Hersho E, et al. Chondrosarcoma of the bronchus. Chest 1983;84:
224–6.
[154] Alt B, Huffer WE, Belchis DA. A vascular lesion with smooth muscle differentiation presenting
as a coin lesion in the lung: glomus tumor versus hemangiopericytoma. Am J Clin Pathol 1983;
80:765–71.
[155] Fabich DR, Hafez GR. Glomangioma of the trachea. Cancer 1980;45:2337–41.
[156] Ito H, Motohiro K, Nomura S, et al. Glomus tumor of the trachea. Immunohistochemical and
electron microscopic studies. Pathol Res Pract 1988;183:778–84.
[157] Kim YI, Kim JH, Suh JS, et al. Glomus tumor of the trachea. Report of a case with ultra-
structural observation. Cancer 1989;64:881–6.
[158] Koss MN, Hochholzer L, Moran CA. Primary pulmonary glomus tumor: a clinicopathologic
and immunohistochemical study of two cases. Mod Pathol 1998;11:253–8.
[159] Mackay B, Legha SS, Pickler GM. Coin lesion of the lung in a 19-year-old male. Ultrastruct
Pathol 1981;2:289–94.
[160] Sheffield E, Dewar A, Corrin B, et al. Glomus tumour of the trachea. Histopathology 1988;
13:234–6.
[161] Shin DH, Park SS, Lee JH, et al. Oncocytic glomus tumor of the trachea. Chest 1990;98:
1021–3.
[162] Tang CK, Toker C, Foris NP, et al. Glomangioma of the lung. Am J Surg Pathol 1978;2:103–9.
[163] Gaertner EM, Steinberg DM, Huber M, et al. Pulmonary and mediastinal glomus tumors—
report of five cases including a pulmonary glomangiosarcoma: a clinicopathologic study with
literature review. Am J Surg Pathol 2000;24:1105–14.
[164] Franks R, Rothera M. Cardiopulmonary bypass for resection of low tracheal haemangioma.
Arch Dis Child 1990;65:630–2.
[165] Harding JR, Williams J, Seal RM. Pedunculated capillary haemangioma of the bronchus. Br J
Dis Chest 1978;72:336–42.
[166] Kayser K, Zink S, Link B, et al. Endobronchial juvenile hemangioma—a case report of a
neonate including immunohistochemical monitoring and nuclear, cellular, and vascular morph-
ometry. Virchows Arch 2001;438:192–7.
[167] Paul KP, Borner C, Muller KM, et al. Capillary hemangioma of the right main bronchus treated
by sleeve resection in infancy. Am Rev Respir Dis 1991;143:876–9.
[168] Strausz J, Soltesz I. Bronchial capillary hemangioma in adults. Pathol Oncol Res 1999;5:
233–4.
[169] Wigton RB, Rohatgi PK. Isolated bronchial capillary hemangioma: a rare benign cause of
hemoptysis. South Med J 1979;72:1339–40.
[170] Keel SB, Bacha E, Mark EJ, et al. Primary pulmonary sarcoma: a clinicopathologic study of
26 cases. Mod Pathol 1999;12:1124–31.
[171] Patel AM, Ryu JH. Angiosarcoma in the lung. Chest 1993;103:1531–5.
[172] Huang L, Schnapp LM, Gruden JF, et al. Presentation of AIDS-related pulmonary Kaposi’s
sarcoma diagnosed by bronchoscopy. Am J Respir Crit Care Med 1996;153:1385–90.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 35

[173] Purdy LJ, Colby TV, Yousem SA, et al. Pulmonary Kaposi’s sarcoma. Premortem histologic
diagnosis. Am J Surg Pathol 1986;10:301–11.
[174] White DA, Matthay RA. Noninfectious pulmonary complications of infection with the human
immunodeficiency virus. Am Rev Respir Dis 1989;140:1763–87.
[175] Misra DP, Sunderrajan EV, Hurst DJ, et al. Kaposi’s sarcoma of the lung: radiography and
pathology. Thorax 1982;37:155–6.
[176] Chin Jr R, Jones DF, Pegram PS, et al. Complete endobronchial occlusion by Kaposi’s sarcoma
in the absence of cutaneous involvement. Chest 1994;105:1581–2.
[177] Khan GA, Klapper P. Pulmonary haemorrhage following renal transplantation. Thorax 1995;
50:98–9.
[178] Deavers M, Guinee D, Koss MN, et al. Granular cell tumors of the lung. Clinicopathologic
study of 20 cases. Am J Surg Pathol 1995;19:627–35.
[179] Lack EE, Harris GB, Eraklis AJ, et al. Primary bronchial tumors in childhood. A clinicopatho-
logic study of six cases. Cancer 1983;51:492–7.
[180] Thomas de Montpreville V, Dulmet EM. Granular cell tumours of the lower respiratory tract.
Histopathology 1995;27:257–62.
[181] Young CD, Gay RM. Multiple endobronchial granular cell myoblastomas discovered at bron-
choscopy. Hum Pathol 1984;15:193–4.
[182] Bateson EM. So-called hamartoma of the lung—a true neoplasm of fibrous connective tissue of
the bronchi. Cancer 1973;31:1458–67.
[183] van den Bosch JM, Wagenaar SS, Corrin B, et al. Mesenchymoma of the lung (so called
hamartoma): a review of 154 parenchymal and endobronchial cases. Thorax 1987;42:790–3.
[184] Prohm P, Winter J, Schmucker P. Pulmonary hamartoma. Thorac Cardiovasc Surg 1982;30:
302–5.
[185] Ribet M, Jaillard-Thery S, Nuttens MC. Pulmonary hamartoma and malignancy. J Thorac
Cardiovasc Surg 1994;107:611–4.
[186] Dominguez H, Hariri J, Pless S. Multiple pulmonary chondrohamartomas in trachea, bronchi
and lung parenchyma. Review of the literature. Respir Med 1996;90:111–4.
[187] King Jr TE, Christopher KL, Schwarz MI. Multiple pulmonary chondromatous hamartomas.
Hum Pathol 1982;13:496–7.
[188] Laroche CM, Stewart S, Wells F, et al. Multiple recurrent intrapulmonary and endobronchial
mesenchymomas (hamartomas). Thorax 1993;48:572–3.
[189] Suzuki N, Ohno S, Ishii Y, et al. Peripheral intrapulmonary hamartoma accompanied by a
similar endotracheal lesion. Chest 1994;106:1291–3.
[190] Gabrail NY, Zara BY. Pulmonary hamartoma syndrome. Chest 1990;97:962–5.
[191] Fletcher JA, Pinkus GS, Donovan K, et al. Clonal rearrangement of chromosome band 6p21 in
the mesenchymal component of pulmonary chondroid hamartoma. Cancer Res 1992;52:6224–8.
[192] Johansson M, Dietrich C, Mandahl N, et al. Recombinations of chromosomal bands 6p21 and
14q24 characterise pulmonary hamartomas. Br J Cancer 1993;67:1236–41.
[193] Kazmierczak B, Meyer-Bolte K, Tran KH, et al. A high frequency of tumors with rearrange-
ments of genes of the HMGI(Y) family in a series of 191 pulmonary chondroid hamartomas.
Genes Chromosomes Cancer 1999;26:125–33.
[194] Xiao S, Lux ML, Reeves R, et al. HMGI(Y) activation by chromosome 6p21 rearrangements in
multilineage mesenchymal cells from pulmonary hamartoma. Am J Pathol 1997;150:901–10.
[195] Hess JL. Chromosomal translocations in benign tumors: the HMGI proteins. Am J Clin Pathol
1998;109:251–61.
[196] Kazmierczak B, Dal Cin P, Wanschura S, et al. Cloning and molecular characterization of part
of a new gene fused to HMGIC in mesenchymal tumors. Am J Pathol 1998;152:431–5.
[197] Fletcher JA, Longtine J, Wallace K, et al. Cytogenetic and histologic findings in 17 pulmonary
chondroid hamartomas: evidence for a pathogenetic relationship with lipomas and leiomyomas.
Genes Chromosomes Cancer 1995;12:220–3.
[198] Tallini G, Vanni R, Manfioletti G, et al. HMGI-C and HMGI(Y) immunoreactivity correlates
with cytogenetic abnormalities in lipomas, pulmonary chondroid hamartomas, endometrial
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4036

polyps, and uterine leiomyomas and is compatible with rearrangement of the HMGI-C and
HMGI(Y) genes. Lab Invest 2000;80:359–69.
[199] White SH, Ibrahim NB, Forrester-Wood CP, et al. Leiomyomas of the lower respiratory tract.
Thorax 1985;40:306–11.
[200] Yellin A, Rosenman Y, Lieberman Y. Review of smooth muscle tumours of the lower respira-
tory tract. Br J Dis Chest 1984;78:337–51.
[201] Karnak I, Akcoren Z, Senocak ME. Endobronchial leiomyoma in children. Eur J Pediatr Surg
2000;10:136–9.
[202] Attanoos RL, Appleton MA, Gibbs AR. Primary sarcomas of the lung: a clinicopathological
and immunohistochemical study of 14 cases. Histopathology 1996;29:29–36.
[203] Guccion JG, Rosen SH. Bronchopulmonary leiomyosarcoma and fibrosarcoma. A study of
32 cases and review of the literature. Cancer 1972;30:836–47.
[204] Janssen JP, Mulder JJ, Wagenaar SS, et al. Primary sarcoma of the lung: a clinical study with
long-term follow-up. Ann Thorac Surg 1994;58:1151–5.
[205] Moran CA, Suster S, Abbondanzo SL, et al. Primary leiomyosarcomas of the lung: a clinico-
pathologic and immunohistochemical study of 18 cases. Mod Pathol 1997;10:121–8.
[206] Nascimento AG, Unni KK, Bernatz PE. Sarcomas of the lung. Mayo Clin Proc 1982;57:
355–9.
[207] Andrassy RJ, Wiener ES, Raney RB, et al. Thoracic sarcomas in children. Ann Surg 1998;227:
170–3.
[208] Avagnina A, Elsner B, De Marco, L, et al. Pulmonary rhabdomyosarcoma with isolated small
bowel metastasis. A report of a case with immunohistochemical and ultrastructural studies.
Cancer 1984;53:1948–51.
[209] Bleisch VR, Kraus FT. Polypoid sarcoma of the pulmonary trunk: analysis of the literature and
report of a case with leptomeric organelles and ultrastructural features of rhabdomyosarcoma.
Cancer 1980;46:314–24.
[210] Comin CE, Santucci M, Novelli L, et al. Primary pulmonary rhabdomyosarcoma: report of a
case in an adult and review of the literature. Ultrastruct Pathol 2001;25:269–73.
[211] Conquest HF, Thornton JL, Massie JR, et al. Primary pulmonary rhabdomyosarcoma. Ann Surg
1965;161:688–92.
[212] Drennan JM, McCormack RJM. Primary rhabdomyosarcoma of the lung. J Pathol 1960;79:
147–9.
[213] Fallon G, Schiller M, Kilman JW. Primary rhabdomyosarcoma of the bronchus. Ann Thorac
Surg 1971;12:650–4.
[214] Forbes GB. Rhabdomyosarcoma of bronchus. J Pathol 1955;70:427–31.
[215] Gordon LZ, Boss H. Primary rhabdomyosarcoma of the lung. Cancer 1955;8:588–91.
[216] Lee SH, Rengachary SS, Paramesh J. Primary pulmonary rhabdomyosarcoma: a case report and
review of the literature. Hum Pathol 1981;12:92–5.
[217] Micallef-Eynaud PD, Goulden NT, Langdale-Brown B, et al. Intracerebral recurrence of pri-
mary intrathoracic rhabdomyosarcoma. Med Pediatr Oncol 1993;21:132–6.
[218] Przygodzki RM, Moran CA, Suster S, et al. Primary pulmonary rhabdomyosarcomas: a clin-
icopathologic and immunohistochemical study of three cases. Mod Pathol 1995;8:658–61.
[219] Shariff S, Thomas JA, Shetty N, et al. Primary pulmonary rhabdomyosarcoma in a child, with a
review of literature. J Surg Oncol 1988;38:261–4.
[220] Dogan R, Unlu M, Gungen Y, et al. Endobronchial lipoma. Thorac Cardiovasc Surg 1988;
36:241–3.
[221] Hurt R. Benign tumours of the bronchus and trachea, 1951–1981. Ann R Coll Surg Engl 1984;
66:22–6.
[222] Iannicello CM, Shoenut JP, Sharma GP, et al. Endobronchial lipoma: report of three cases. Can
J Surg 1987;30:430–1.
[223] Matsuba K, Saito T, Ando K, et al. Atypical lipoma of the lung. Thorax 1991;46:685.
[224] Moran CA, Suster S, Koss MN. Endobronchial lipomas: a clinicopathologic study of four cases.
Mod Pathol 1994;7(2):212–4.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 37

[225] Nomori H, Horio H, Suemasu K. Two-stage operation for endobronchial lipoma and lung
cancer using bronchoscopy and thoracoscopy in an elderly patient with chronic obstructive
pulmonary disease. Jpn J Thorac Cardiovasc Surg 1999;47:567–9.
[226] Tomashefski Jr JF. Benign endobronchial mesenchymal tumors: their relationship to parenchy-
mal pulmonary hamartomas. Am J Surg Pathol 1982;6:531–40.
[227] Ruiz-Palomo F, Calleja JL, Fogue L. Primary liposarcoma of the lung in a young woman.
Thorax 1990;45:298–9.
[228] Sawamura K, Hashimoto T, Nanjo S, et al. Primary liposarcoma of the lung: report of a case.
J Surg Oncol 1982;19:243–6.
[229] Suster S. Primary sarcomas of the lung. Semin Diagn Pathol 1995;12:140–57.
[230] Davies MJ, Hall DR, Ross BA. Rare tracheal tumours: two case reports of primary neurogenic
tumours occurring in the trachea. Respir Med 1993;87:145–6.
[231] Feldhaus RJ, Anene C, Bogard P. A rare endobronchial neurilemmoma (schwannoma). Chest
1989;95:461–2.
[232] Inoue H, Tsuneyoshi M, Enjoji M, et al. Endotracheal neurilemoma with a lymphoid cuff. An
ultrastructural and immunohistochemical study. Acta Pathol Jpn 1989;39:407–12.
[233] Kitamura H. Primary epithelioid malignant schwannoma of the lung. Pathol Int 1994;44:
317–24.
[234] McCluggage WG, Bharucha H. Primary pulmonary tumours of nerve sheath origin. Histopa-
thology 1995;26:247–54.
[235] Miura H, Kato H, Hayata Y, et al. Solitary bronchial mucosal neuroma. Chest 1989;95:
245–7.
[236] Muhrer KH, Fischer HP. Primary pulmonary neurilemoma. Thorac Cardiovasc Surg 1983;
31:313–6.
[237] Nesbitt JC, Vega DM, Burke T, et al. Cellular schwannoma of the bronchus. Ultrastruct Pathol
1996;20:349–54.
[238] Oosterwijk WM, Swierenga J. Neurogenic tumours with an intrathoracic localization. Thorax
1968;23:374–84.
[239] Silverman JF, Leffers BR, Kay S. Primary pulmonary neurilemoma. Report of a case with
ultrastructural examination. Arch Pathol Lab Med 1976;100:644–8.
[240] Colby TV, Bilbao JE, Battifora H, et al. Primary osteosarcoma of the lung. A reappraisal
following immunohistologic study. Arch Pathol Lab Med 1989;113:1147–50.
[241] Loose JH, el-Naggar AK, Ro JY, et al. Primary osteosarcoma of the lung. Report of two cases
and review of the literature. J Thorac Cardiovasc Surg 1990;100:867–73.
[242] Argani P, Askin FB, Colombani P, et al. Occult pulmonary synovial sarcoma confirmed by
molecular techniques. Pediatr Dev Pathol 2000;3:87–90.
[243] Hisaoka M, Hashimoto H, Iwamasa T, et al. Primary synovial sarcoma of the lung: report of
two cases confirmed by molecular detection of SYT–SSX fusion gene transcripts. Histopathol-
ogy 1999;34:205–10.
[244] Kaplan MA, Goodman MD, Satish J, et al. Primary pulmonary sarcoma with morphologic
features of monophasic synovial sarcoma and chromosome translocation t(X;18). Am J Clin
Pathol 1996;105:195–9.
[245] Roberts CA, Seemayer TA, Neff JR, et al. Translocation (X;18) in primary synovial sarcoma of
the lung. Cancer Genet Cytogenet 1996;88:49–52.
[246] Terasaki H, Niki T, Hasegawa T, et al. Primary synovial sarcoma of the lung: a case report
confirmed by molecular detection of SYT–SSX fusion gene transcripts. Jpn J Clin Oncol
2001;31:212–6.
[247] Zeren H, Moran CA, Suster S, et al. Primary pulmonary sarcomas with features of monophasic
synovial sarcoma: a clinicopathological, immunohistochemical, and ultrastructural study of
25 cases. Hum Pathol 1995;26:474–80.
[248] Coffin CM, Watterson J, Priest JR, et al. Extrapulmonary inflammatory myofibroblastic tumor
(inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases.
Am J Surg Pathol 1995;19:859–72.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4038

[249] Umiker WO, Iverson L. Postinflammatory ‘tumors’ of the lung. Report of four cases simulating
xanthoma, fibroma or plasma cell tumor. J Thoracic Surg 1954;28:55–63.
[250] Bahadori M, Liebow AA. Plasma cell granulomas of the lung. Cancer 1973;31:191–208.
[251] Spencer H. The pulmonary plasma cell/histiocytoma complex. Histopathology 1984;8:903–6.
[252] Grossman RE, Bemis EL, Pemberton AH, et al. Fibrous histiocytoma or xanthoma of the lung
with bronchial involvement. J Thorac Cardiovasc Surg 1973;65:653–7.
[253] Buell R, Wang NS, Seemayer TA, et al. Endobronchial plasma cell granuloma (xanthomatous
pseudotumor): a light and electron microscopic study. Hum Pathol 1976;7:411–26.
[254] Bates HR, Buis LJ, Johns TN. Endobronchial histiocytoma. Chest 1976;69:705–6.
[255] Tan-Liu NS, Matsubara O, Grillo HC, et al. Invasive fibrous tumor of the tracheobronchial tree:
clinical and pathologic study of seven cases. Hum Pathol 1989;20:180–4.
[256] Charrette EE, Mariano AV, Laforet EG. Solitary mast cell ‘‘tumor’’ of lung. Its place in the
spectrum of mast cell disease. Arch Intern Med 1966;118:358–62.
[257] Sherwin RP, Kern WH, Jones JC. Solitary mast cell granuloma (histiocytoma) of the lung.
Cancer 1965;18:634–41.
[258] Hartman GE, Shochat SJ. Primary pulmonary neoplasms of childhood: a review. Ann Thorac
Surg 1983;36:108–19.
[259] Mandelbaum I, Brashear RE, Hull MT. Surgical treatment and course of pulmonary pseudo-
tumor (plasma cell granuloma). J Thorac Cardiovasc Surg 1981;82:77–82.
[260] Souid AK, Ziemba MC, Dubansky AS, et al. Inflammatory myofibroblastic tumor in children.
Cancer 1993;72:2042–8.
[261] Altman H, Pietra GG, LiVolsi VA, et al. Tracheobronchial inflammatory myofibroblastoma. Int
J Surg Pathol 1994;2:93–8.
[262] Pettinato G, Manivel JC, De Rosa N, et al. Inflammatory myofibroblastic tumor (plasma cell
granuloma). Clinicopathologic study of 20 cases with immunohistochemical and ultrastructural
observations. Am J Clin Pathol 1990;94:538–46.
[263] Cerfolio RJ, Allen MS, Nascimento AG, et al. Inflammatory pseudotumors of the lung. Ann
Thorac Surg 1999;67:933–6.
[264] Ishida T, Oka T, Nishino T, et al. Inflammatory pseudotumor of the lung in adults: radiographic
and clinicopathological analysis. Ann Thorac Surg 1989;48:90–5.
[265] Warter A, Satge D, Roeslin N. Angioinvasive plasma cell granulomas of the lung. Cancer 1987;
59:435–43.
[266] Barbareschi M, Ferrero S, Aldovini D, et al. Inflammatory pseudotumour of the lung. Immu-
nohistochemical analysis on four new cases. Histol Histopathol 1990;5:205–11.
[267] Matsubara O, Tan-Liu NS, Kenney RM, et al. Inflammatory pseudotumors of the lung: pro-
gression from organizing pneumonia to fibrous histiocytoma or to plasma cell granuloma in 32
cases. Hum Pathol 1988;19:807–14.
[268] Coffin CM, Dehner LP, Meis-Kindblom JM. Inflammatory myofibroblastic tumor, inflamma-
tory fibrosarcoma, and related lesions: an historical review with differential diagnostic consid-
erations. Semin Diagn Pathol 1998;15:102–10.
[269] Su LD, Atayde-Perez A, Sheldon S, et al. Inflammatory myofibroblastic tumor: cytogenetic
evidence supporting clonal origin. Mod Pathol 1998;11:364–8.
[270] Cook JR, Dehner LP, Collins MH, et al. Anaplastic lymphoma kinase (ALK) expression in the
inflammatory myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg
Pathol 2001;25:1364–71.
[271] Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3–ALK and TPM4–ALK oncogenes in
inflammatory myofibroblastic tumors. Am J Pathol 2000;157:377–84.
[272] Corrin B. Pathology of the lungs. London, UK: Churchill Livingstone; 2000.
[273] Dehner LP. The enigmatic inflammatory pseudotumours: the current state of our understanding,
or misunderstanding. J Pathol 2000;192:277–9.
[274] Koss MN. Pulmonary lymphoid disorders. Semin Diagn Pathol 1995;12:158–71.
[275] Yousem SA, Weiss LM, Colby TV. Primary pulmonary Hodgkin’s disease. A clinicopathologic
study of 15 cases. Cancer 1986;57:1217–24.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–40 39

[276] Rush WL, Andriko JA, Taubenberger JK, et al. Primary anaplastic large cell lymphoma of the
lung: a clinicopathologic study of five patients. Mod Pathol 2000;13:1285–92.
[277] Cagle P, Mace ML, Judge DM, et al. Pulmonary melanoma. Primary vs metastatic. Chest 1984;
85:125–6.
[278] Jennings TA, Axiotis CA, Kress Y, et al. Primary malignant melanoma of the lower respiratory
tract. Report of a case and literature review. Am J Clin Pathol 1990;94:649–55.
[279] Wilson RW, Moran CA. Primary melanoma of the lung: a clinicopathologic and immunohis-
tochemical study of eight cases. Am J Surg Pathol 1997;21:1196–202.
L. Litzky / Chest Surg Clin N Am 13 (2003) 1–4040














![Primary tracheal schwannoma a review of a rare entity ......include benign, intermediate, and malignant tumors [1]. Neurogenic tumors of the tracheobronchial tree are ex-tremely rare](https://img.pdfslide.us/doc/110x75/60f7b75799a3976448468c79/primary-tracheal-schwannoma-a-review-of-a-rare-entity-include-benign-intermediate.jpg)














