Embed Size (px)
Citation preview

EVIEWS
M a m m a l i a n genetics is not only important for its relevance to medicine and agriculture, but is also of considerable fundamental interest because certain epi- genetic phenomena that occur in mammals have not yet been found in any other vertebrates. These epigenetic phenomena include genetic imprinting and X-chromo- some inactivation. Imprinting is the phenomenon in which genes or chromosomes retain some memory or 'imprint' of their gametic origin, and behave differently according to whether they are inherited from the mother or from the father ~-3. In X inactivation, one of the two X chromosomes in somatic cells of females becomes inactive early in embryonic development; this results in dosage compensation for X-linked genes, so that chromosomally XX females and XY males both have effectively a single dose of these genes ¢-s.
The phenomena of X inactivation and imprinting are associated, in that in some cases imprinting is involved in the choice of X chromosome for inacti- vation, with the paternally derived X chromosome being preferentially inactivated. Another feature in common is that DNA methylation is known or believed to be involved in the mechanism in both cases 9. However, these processes also differ from each other in some ways. X inactivation involves coordinated regulation of a whole chromosome. By contrast, imprinting, while appearing at one level to apply to a whole genome set (the haploid complement of chromosomes derived from one parent), when exam- ined in more detail seems to affect individual genes in different ways.
Evidence for imprinting Imprinting is found in a wide range of organisms,
including yeast, some plants, and various animals, particularly insects, including mealy bugs and scale insects3. It may be manifested either by differential expression of genes, or by differential inactivation or elimination of chromosomes derived from one or other parent. In mammals, both these types of imprinting are seen: differential expression in the au tosomes and preferential inactivation in the X chromosomes. Evidence for imprinting of autosomal genes comes from the phenotypic abnormalities seen in animals that have genome sets, single chromosomes or chromo- some regions of inappropriate parental origin. By nuclear transfer in fertilized eggs, animals can be pro- duced that have two maternal or two paternal genome sets la0. Neither type can survive. In those with two matemal sets (gynogenotes or pa~-thenogenotes), the embryonic tissues begin development fairly normally, but the extraembryonic tissues required for placental development fail to grow. Converseb', in andro- genotes, which have two paternal genome sets, the extraemb~onic tissues develop but embryonic tissues fail.
Moore and Haig n have suggested that in a viviparous organism such as a mammal there may be a conflict between maternally and paternally inherited genes that affect growth. It may be selectively advan- tageous for the father's genes to promote the growth of the fetus, and thus give it a better chance of sur- vival, and advantageous for the mother's genes for the
"riG APRIL 1 9 9 3
Epigenetic inheritance in mammals MARY F. LYON
The epigenettc phenomena of genome imprinting and X-chromosome inactivation, found in mammals, both entail homologous genes or chromosomes behaving differently within the same cell Although both have consequences for genie balance in the whole genome, in imprinting the control seems mainly at the single gene leve~ whereas in X-chromosome tmgffvatio~ there is coordinated regulation of the whole chromosome, and single gene effects are relatively minor.
fetus to remain relatively small, to promote the mother's chances of having further offspring. Thus, they suggest there will be maternal and patemal imprints affecting genes involved in fetal growth. Clearly, the results from parthenogenotes, gynogenotes and androgenotes are in line with these ideas.
However, more detailed studies reveal a much more complex picture. By making use of Robertsonian or reciprocal chromosome translocations, mice can be bred that have single chromosomes, or chromosome segments, of inappropriate parental origin (uniparental disomy). By this means Cattanach 2 and others have shown, firsdy, that only for some chromosome regions does uniparental disomy have any phenotypic effect, and thus presumably only certain regions are imprinted. Secondly, the phenotypic effects are varied. In one case paternal disomics are abnormally large and ma- ternal disomics are small, broadly in line with the phenotypes of androgenotes and gynogenotes, and with the ideas of Moore and Haig. However, in other cases these effects on growth are reversed. In yet others there is prenatal lethality and in others again there are behavioural effects accompanied by post- natal lethality z.
Some human genetic phenomena have also been attributed to imprinting and again the phenotypic effects are diverse. A particularly clear-cut example concerns the Prader-Willi and Angelman syndromes 12, both of which are associated with deletions of a region of chromosome 15q11-13. Failure to in]~erit the pa- ternal region results in Prader-Willi syndrome (PWS), whereas Angelman syndrome (AS) occurs in indi- viduals who lack a matemai homologue. The two syn- dromes have contrasting symptoms; AS patients have ataxia, suffer seizures and are hyperactive, while PWS is characterized by retarded motor development, severe hypotonia, feeding difficulties and, later in life, excessive appetite.
Imprinting has also been invoked as an expla- nation for the chromosomal findings in certain tumours. The Philadelphia chromosome characteristic of chronic myelogenous leukaemia results from a translocation between chromosome 9 and chromosome 22. Recent work has shown that the chromosome 9 component is preferentially of paternal origin and the chromosome 22 component is preferentially maternal 1-~. Similar parental origin effects have been seen in other
rOt. 9 NO. 4
©;.993 Elsevier Science Publishers Ltd (L:K) 0168 - 9525/93/$06.00 t23

EVIEWS
(a)
C I I
I
F7 I
Initiation
I I
I I
Spreading I ~ I
[////~/////A
(b)
I
(.~No methylation _ Early replication " ~
, / - - Transcription - -
gene, SNRPN (small nuclear riboprotein polypeptide N), that is deleted in patients with Prader-Willi syndrome 17. The mouse equivalent, Snrpn, maps to a region known to be homologous with the Prader-Willi region, and is imprinted in that only the paternal allele is expressed mA9. Cattanach et al} 9 made a mouse model of PWS by breed- ing mice with two maternal copies of the rel- evant chromosome 7 region. The animals did not express Snrpn and, although their pre- natal viability was apparently unaffected, they failed to thrive and died soon after birth. These effects mimic some of the symptoms that are characteristically found in infants who have PWS. Thus, Snrpn is a candidate I gene for some, but not all, effects of PWS 17-19. As with the other imprinted mouse genes, a nearby locus, in this case Gabrb-3, was not imprinted 19. Thus, on both mouse chromosomes 7 and 17 closely linked genes are differently affected by imprinting.
Imprinting is shown not only by endogen- ous genes but also by transgenes. Some transgenes are expressed only when they have been inherited from one particular parent, usually the male 20,21. Yet there is further complexity; in some cases, the effect depends on the background strain into which the transgene is introduced. This implies that one or more tram-acting genes affect the imprinting process and one such gene, Ssm-1 (strain-specific modifier), has been mapped 22. Evidence on imprinting obtained from studies on transgenes must be viewed with caution, since it is possible that the cell may treat a transgene as 'foreign DNA', and indeed the frequency of imprint-
ing among transgenes seems high compared with that of endogenous genesZ0, zl.
There is, however, evidence of a genetic influ- ence on imprinting of endogenous genes. Forejt and Gregorova 23 crossed laboratory mice carrying the chromosome 17 deletion TbP, which covers the Igf2r locus, to a different mouse subspecies, Mus muscu- lus musculus. In contrast to previous results, men- tioned above, animals that inherited the deletion from the mother survived, had no malformations, and were somewhat bigger than their littermates. As in the transgene case, the authors postulated an unlinked gene affecting the imprinting process. The paternal Igf2r gene remained imprinted (not expressed) in these animals, and the authors suggest that some other, as yet unidentified, gene in the deleted region was responsible for the lethal effect seen in comparable crosses with laboratory mice. However, another possible explanation, that there is subtle change in expression of Igf2r in a specific tissue or stage of development, seems not to be excluded. De Chiara et al. 15 found that the maternal allele of Igf2, although silent in most tissues, was expressed in the choroid plexus and the leptomeninges. Possibly the paternal allele of Igf2r is also expressed in some tissues.
Methylation I Late replication ~
v / / / ~ / / / / 1 ~
No transcription
[/// /~/////A
FIG[I[ The initiation and maintenance of X-chromosome inactivation.
The X-inactivation centre is shown as an open box. (a) At initiation, a trans.acting factor (stippled arrow) acts on one inactivation centre and
blocks its activity (stippled box). A signal for the spreading of inactivation (hatching) is then initiated in both directions from the unblocked centre. (b) In maintenance, the active or inactive state of each X chromosome is maintained through successive cell divisions by a feedback loop, thought
to involve both late replication and DNA methylation. Reproduced, with permission, from Ref. 45.
chromosome anomalies associated with tumours. Thus, it appears that only some genes are imprinted, and that the effects are developmentally complex.
A similar picture comes from molecular studies on individual imprinted genes. On mouse chromosome 17, there are two small deletions (TbP and #ub2) which show imprinting, in that animals receiving the deletion from the father survive normally, whereas those that receive the deletion from the mother and a normal chromosome 17 from the father die before birth. Barlow et al. 14 identified four genes that mapped within these deletions. One of these, the gene that encodes the insulin-like growth factor 2 receptor (Igf2r), was expressed only when inherited from the mother whereas the others, the genes for mitochondrial superoxide dismutase 2 (Sod-2), t complex peptide 1 (Tcp-1) and plasminogen (Pig) were expressed nor- mally from both parental alleles. Similar differential behaviour of individual genes occurs on chromosome 7, where the Igf2 gene coding for insulin-like growth factor 2 is expressed only if derived from the male parent ]5, while only the maternal allele of the closely linked gene 1-119 is expressed 1o.
The most recently identified imprinted mouse gene, also on chromosome 7 but remote from lgf2 and H19, is of particular interest because of its homology with a
"riG APRIL 1993 VOL. 9 NO. 4
]REVIEWS
On the basis of the correlation between methylation of imprinted transgenes and their lack of ex- pression it has been suggested that imprinting and transcriptional inac- tivation may be achieved through a mechanism that involves DNA methylation20, zl. It will be very interesting to see if this suggestion is borne out as more imprinted endogenous genes are investigated in detail.
X-chromosome inactivation X inactivation contrasts with
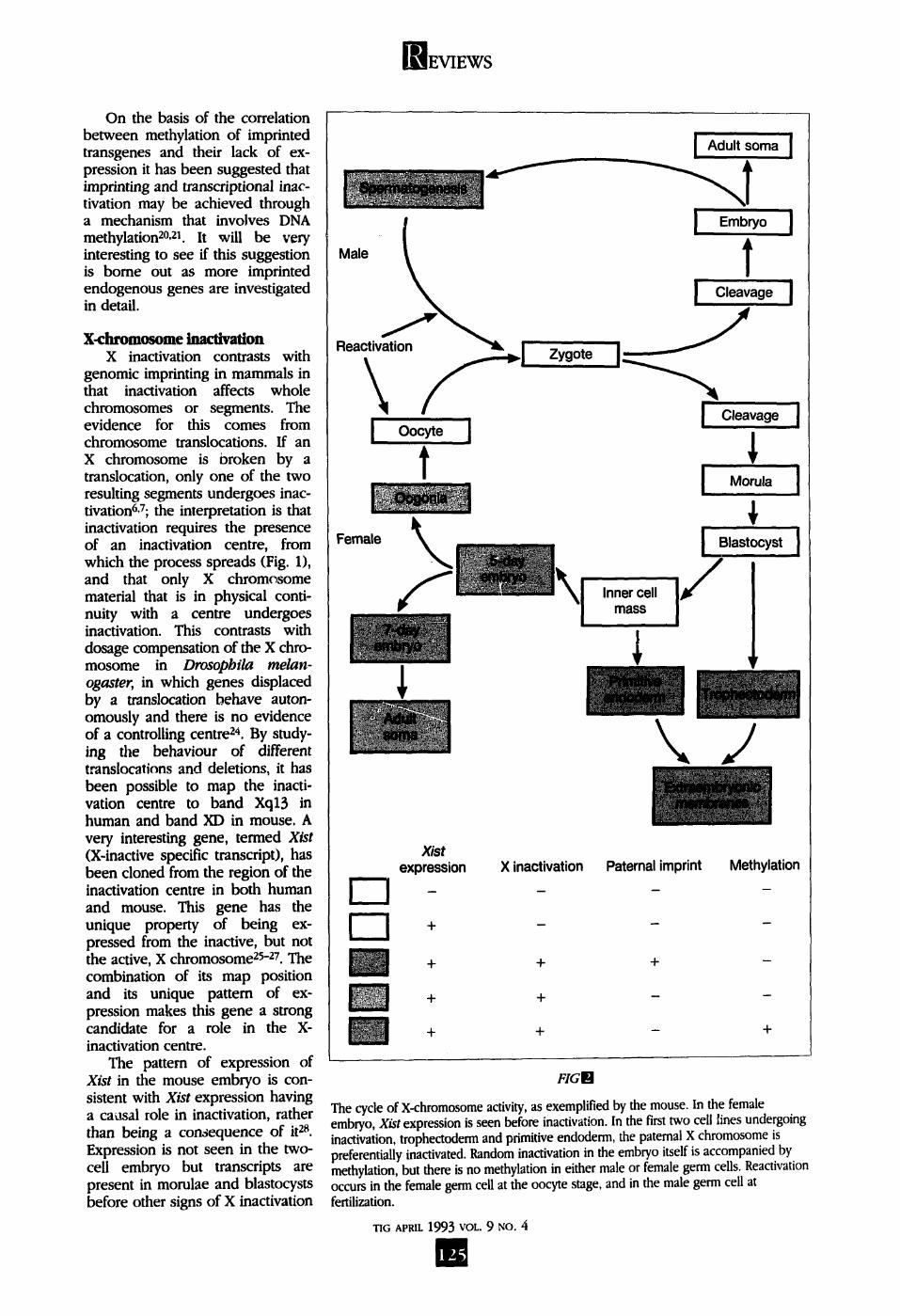
genomic imprinting in mammals in that inactivation affects whole chromosomes or segments. The evidence for this comes from chromosome translocations. If an X chromosome is broken by a translocation, only one of the two resulting segments undergoes inac- tivation6,7; the interpretation is that inactivation requires the presence of an inactivation centre, from which the process spreads (Fig. 1), and that only X chromosome material that is in physical conti- nuity with a centre undergoes inactivation. This contrasts with dosage compensation of the X chro- mosome in Drosophila melan- ogaster, in which genes displaced by a translocation behave auton- omously and there is no evidence of a controlling centre 24. By study- ing the behaviour of different translocations and deletions, it has been possible to map the inacti- vation centre to band Xql3 in human and band XD in mouse. A very interesting gene, termed Xist (X-inactive specific transcript), has been cloned from the region of the inactivation centre in both human and mouse. This gene has the unique property of being ex- pressed from the inactive, but not the active, X chromosome 25-27. The combination of its map position and its unique pattern of ex- pression makes this gene a strong candidate for a role in the X- inactivation centre.
The pattern of expression of Xist in the mouse embryo is con- sistent with Xist expression having a causal role in inactivation, rather than being a consequence of it 28. Expression is not seen in the two- cell embryo but transcripts are present in morulae and blastocysts before other signs of X inactivation
Adult soma I i
J Embryo J
Male 1 L
l Cleavage 1
Rea I Zygote
Female
Oocyte J I Cleavage
f /
i
\ /
Xist expression
m +
m +
X inactivation Paternal imprint Methylation
+
+
+
+
+
F/GB
The cycle of X-chromosome activity, as exemplified by the mouse. In the female embryo, Xist expression is seen before inactivation. In the first two cell lines undergoing inactivation, trophectoderm and primitive endoderm, the paternal X chromosome is preferentially inactivated. Random inactivation in the embryo itself is accompanied by methylation, but there is no methylation in either male or female germ cells. Reactivation occurs in the female germ cell at the oocyte stage, and in the male germ cell at fertilization.
"FIG APRIL 1993 VOL. 9 NO. 4
125

EVIEWS
can be recognized (Fig. 2). Similarly, expression of Xist is not seen in embryonic stem (ES) cell lines in which both X chromosomes are active, but appears when these cells are allowed to differentiate 2s, a process accompanied by X inactivation. Changes in X chromosome activity occur in germ cells, with reacti- vation of the inactive X chromosome in oocytes and inactivation of the single X chromosome in male germ cells. Expression of Xist has now been found in testis at an appropriate stage 29"-31 (Fig. 2), giving a first indi- cation that the mechanism of inactivation in male germ cells may be the same as, or at least similar to, that in female somatic cells.
An important link between imprinting and X inacti- vation is seen in marsupials, in the choice of X chromo- some to remain active. In eut|,erians the choice is ran- dom. By contrast, in marsupials there is preferential inactivation of the paternally derived X chromosome (reviewed in Refs 4--8). This imprinted type of X inacti- vation is also seen in the trophectoderm and primitive endoderm of rats and mice (Fig. 2). However, studies in the mouse indicate that the expression of one or other parental X chromosome is not fixed as in autosomal imprinting. In diploid parthenogenotes, in which both X chromosomes are of maternal origin, inactivation of one X chromosome can still occur; con- versely, the paternal X chromosome can remain active when no maternal X chromosomes are present. It is not yet clear whether the X chromosome imprint involves differential gene expression, as in autosomal imprinting, or whether there is some other change in the chromatin, such as accessibility to binding of some factors, or a change in timing of replication.
Differential susceptibility of two homologues to inactivation would reduce the risk that both will simul- taneously be inactivated or activated (a presumably lethal error): this may be the selective advantage of X chromosome imprinting32. Eutherians have developed an inactivation mechanism without imprinting, perhaps by making inactivation occur later in development, when the imprint may already have been lost. Presum- ably the risk of error still remains low. The selective advantage of this system would be more stable inacti- vation and thus better dosage compensation. In eu- therians, X inactivation is very stable, whereas in mar- supials reactivation of the inactive X chromosome can occur both in cultured cells and in some tissues in vivo.
Function of the X-inactivation centre Studies on aneuploids that have abnormal numbers
of X chromosomes show that only one X chromosome remains active in diploid cells, no matter how many are present. In triploids either one or two X chromo- somes remain active, and in tetraploids two remain active33. Thus, a single X chromosome remains active for every two sets of autosomes, and the X-inactivation centre must in some way be involved in the counting mechanism that brings this about. In Drosophila, where both sex determination and dosage compen- sation depend on the ratio of X chromosomes to sets of autosomes (X:A), specific genes that have numer- ator and denominator function have been identified 24. In mammals no such genes have yet been found, and over the years speculation has ranged widely. Some
models suggest that there is a limited supply of a particular molecule, or even a single nuclear site, to which the active X chromosome can attach (reviewed in Ref. 34). Other ideas involve cooperative binding of some factor to the inactivation centre 4,5. In either case, one would expect the inactivation centre to contain a factor-specific binding site. An essential requirement for inactivation is the presence of an active X chromo- some in the cell, and hence it is possible that the active X chromosome itself provides a trigger for inacti- vation. In that case some other explanation would be needed for X inactivation in male germ cells, in which only a single X chromosome is present. An interesting recent observation was made in a boy who had a small duplication of the X chromosome35, apparently involving the inactivation centre and the XIST gene. The XIST gene was not expressed, suggesting that the XIST locus can only be transcribed when it is physi- cally separate from the inactivation centre on the active X chromosome.
In addition to receiving a signal that determines its activation state, the inactivation centre must disseminate the signal to spread inactivation along the chromo- some (Fig. 1). Although there has been much conjec- ture, the mechanism of spreading is not yet known. At one time, DNA methylation seemed an attractive candidate (reviewed in Ref. 8). In somatic cells of eutherians, there are clear methylation differences between the active and inactive X chromosomes. The CpG islands in the 5' promoter regions of genes on the inactive X chromosome are heavily methylated, in con- trast to the unmethylated state of these islands in the rest of the genome s. However, differential methylation is not found in marsupials, nor is there any apparent modification of the DNA of the inactive paternal X chromosome in tile extraembryonic tissue of eu- therians, or of the inactive X chromosome in sperm and oocytes36,.¢7 (Fig. 2). Thus, differential methylation is not a primary factor in spreading inactivation; rather, ~ it appears to have evolved in eutherians as a means of stabilizing inactivation by forming a feedback loop s, in which hemimethylated sites are methylated by a maintenance methylase.
Other properties of the inactive X that could poten- tially be involved in spreading are its late replication and its condensation to form the sex chromatin body, which is attached to the nuclear membrane. Late repli- cation and condensation are also properties of other types of inactive chromatin, such as centromeric hetero- chromatin and other heterochromatic blocks, and these two phenomena are clearly linked. Riggs s and Grant and Chapman 4 have suggested that, like methyl- ation, late replication could be involved in maintaining the inactive state of the X chromosome via a feedback loop, in which late replication prevents transcription, and transcription is required for early replication. In embryogenesis, condensation is visible only after late replication is evident, and is clearly a secondary event. However, it presumably results from a change in chromatin structure at the level of the DNA fibre. Riggs s3s has suggested that condensation could be brought about by type I DNA reeling, with an enzyme at a scaffold attachment site reeling DNA towards itself from both directions. However, the causal connections
WIG APRIL 1993 VOL. 9 NO. 4
126

WEVIEWS
between the various properties of the inactive X remain to be eluci- dated, as does the role, if any, of the Xist gene in spreading. Sequence analysis of the Xist gene in mouse and human has revealed no substantial open reading frame, although there is general conser- vation of the transcriptional and structural organization of the locus, including various blocks of repeats39.'io. Thus, the gene may act through a functional RNA.
Although the inactivation cen- tre has a key part in X inactivation there is evidence that other se- quences and individual genes on the chromosome also have a role. A remarkable feature of spreading
) ) )
FIGSFt
Suggested involvement of chromosome booster elements and individual genes in X inactivation. (a) Inactivation spreads along the chromosome (arrow), promoted by booster elements (hatched boxes) at intervals of megabases. Stably inactivated loci are indicated by filled circles. (b) The inactivation signal passes through genes (open circles), one of which has regulatory elements (open box) that protect the gene either from the inactivating signal or from the signal for stabilization of inactivation.
is the length of spread. There is evidence from X-autosome translocations that inactivation can spread over several G-bands and many megabases into the autosome 41. However, spreading is apparently less effective in autosomal material than in the X chromo- some itself 42, and this has led Riggs 43 to propose that the X chromosome has 'booster elements' which pro- mote the spread (Fig. 3). Evidence that genes may have individual responses to the spreading signal comes from the human X chromosome, on which cer- tain genes escape inactivation. Some of these genes are in inactivated regions, with genes that are known to be inactivated on both sides of them (reviewed in Ref. 7). This implies that the spreading signal must run through these genes, and they must in some way either resist it or undergo reactivation. In the mouse, the homologous genes undergo typical inactivation 44. This suggests that in the course of mammalian evol- ution there has been both selection for sequences (pre- sumably interspersed repetitive DNA) that promote the spread of inactivation, and selection against sequences that cause X-linked genes to resist inactivation (Fig. 3).
Thus the autonomous response of some individual genes to signals for X-chromosome inactivation rep- resents another similarity between X inactivation and imprinting. A difference, according to present knowl- edge, lies in the relative importance of these individual responses. In imprinting, phenotypic effects are seen at the level of whole genome sets, as well as at the chromosome level. However detailed investigations have revealed evidence that individual genes behave autonomously, and chromosome-wide controls have yet to be discovered. In X-chromosome inactivation, the predominant control is at the chromosomal level, probably in determining the state of the chromatin fibre. Individual genes can respond autonomously to these chromosomal controls, but the effect seems of relatively minor importance. It is interesting to specu- late on whether imprinting and X-chromosome inacti- vation have a common evolutionary origin, and whether further similarities will be unveiled in future. Cattanach and Beechey 2 have speculated that control genes analogous to the X-inactivation centre will be
it is expressed from the opposite chromosome to the neighbouring imprinted Igf2 gene.
Acknowledgements I am grateful to many colleagues for stimulating dis-
cussions, particularly to Dr B.M. Cattanach. I acknowledge the many important primary papers that could not be cited owing to space limitations.
References 1 Solter, D. (1988) Annu. Rev. Genet. 22, 127-146 2 Cattanach, B.M. and Beechey, C.V. (1990) in Genomic
Imprinting (Development Suppl.) (Monk, M. and Surani, A., eds), pp. 63--72, Company of Biologists
3 Monk, M. and Surani, A., eds (1990) Genomic Imprinting (Development Suppl.), Company of Biologists
4 Grant, S.G. and Chapman, V.M. (1988) Annu. Rev. Genet. 22, 199-233
5 McBumey, M.W. (1988) BioEssays 9, 85.-88 6 Lyon, M.F. (1989) Prog. Nucleic Acid Res. Mol. Biol. 36,
119-130 7 Lyon, M.F. (1992) Annu. Rev. Genet. 26, 15-27 8 Riggs, A.D. (1990) Philos. Trans. R. Soc. London, Ser. B
326, 285-297 9 Bird, A.P. (1992) Cell70, 5-8
I0 Surani, M.A. et al. (1990) in Genomic Imprinting (Development Suppl.) (Monk, M. and Surani, A., eds), pp. 89-98, Company of Biologists
11 Moore, T. and Haig, D. (1991) Trends Genet. 7, 45--49 12 Hall, J.G. (1990) in Genomic Imprinting (Development
Suppl.) (Monk, M. and Surani, A., eds), pp. 141-148, Company of Biologists
13 Haas, O.A., Argyriou, A. and Lion, T. (1992) Nature 359, 414--416
14 Badow, D.P. et al. (1991) Nature 349, 84--87 /5 DeChiara, T.M., Robertson, E.J. and Efstratiadis, A. (1991)
Cell 64, 849--859 16 Bartolomei, M.S., Zemel, S. and Tilghman, S.M. (1991)
Nature 351,153-155 17 Ozcelik, T. et ai. (1992) Nature Genet. 2, 265-269 18 Left. S.E. et ai. (1992) Nature Genet. 2, 259-264 19 Cattanach, B.M. et al. (1992) Nature Genet. 2, 271-274 20 Reik, W., HowleR, S.K. and Surani, M.A. (1990) in
Genomic Imprinting (Development Suppl.) (Monk, M. and Surani, A., eds), pp. 99-106, Company of Biologists
21 Surani, M.A. et al. (1990) Philos. Trans. R. Soc. London, Ser. B 326, 313-327
found near imprinted genes; in this context, the H19 22 Engler, P. et ai. (1991) Cell 65, 939-947 gene has provocative similarities to Xist (Ref. 39), since 23 Foreijt, J. and Gregorova, S. (1992) Cell 70, 443-450
TIG APRIL 1993 VOL. 9 NO. 4
12"7

EVIEWS
24 Lucchesi, J.C. and Manning, J.E. (1987) Adv. Genet. 24, 371--429
25 Brown, C.J. et ai. (1991) Nature 349, 38--44 26 Borsani, G. etal. (1991) Nature 351, 32%329 27 Brockdorff, N. et al. (1991) Nature 351,329--331 28 Kay, G.F. etai. (1993) Cell72, 171-172 29 Richler, C., Soreq, H. and Wahrman, J. (1992) Nature
Genet. 2, 192-195 .fro Salido, E.C., Yen, P.H., Mohandas, T.K. and Shapiro, L.J.
(1992) Nature Genet. 2, 196-199 31 McCarrey, J.R. and Dilworth, D.D. (1992) Nature Genet. 2,
200-203 32 Lyon, M.F. (1988) Am.J. Hum. Genet. 42, 8-16 33 Webb, S., DeVries, T.J. and Kaufman, M.H. (1992) Genet.
Res. 59, 20%214 34 Gartler, S.M. and Riggs, A.D. (1983) Annu. Rev. Genet. 17,
15%190 35 Muscatelli, F., Lena, D., Mattei, M.G. and Fontes, M.
(1992) Hum. Mol. Genet. 1, 115--119 36 Kafri, T. et al. (1992) Genes Dev. 6, 705-714
37 Grant, M., Zuccotti, M. and Monk, M. (1992) Nature Genet. 2, 161-166
38 Riggs, A.D. and Pfeifer, G.P. (1992) Trends Genet. 8, 169--174
39 Brockdorff, N. et al. (1992) Cell 71, 51%526 40 Brown, C.J. et al. (1992) Cell71, 527-542 41 Russell, L.B. (1983)in Cytogenetics o f the Mammalian X
Chromosome (Part A) (Sandberg, A.A., ed.), pp. 205-250, Alan R. Liss
42 Rastan, S. (1983) J. Embryol. Exp. Morpbol. 78, 1-22 43 Riggs, A.D. (1990) Aust.J. Zool. 37, 419-441 44 Ashworth, A., Rastan, S., LoveU-Badge, R. and Kay, G.
(1991) Nature 351,406-408 45 Short, R.V. (1993) The Differences Between the Sexes,
Cambridge University Press
M.F. LYON IS IN THE MRC RxmOmOLOGr UNn; CHILTON, DmcoT, OXON, UK OXI I ORD.
A variety of chronic degenerative diseases that involve the brain, heart, muscle, kidney and endocrine glands have been shown to result from mutations in mitochondrial DNA (mtDNA) (Refs 1, 2; Fig. 1). The first pathogenic mtDNA mutations identified were associated with rare syndromes such as Leber's her- editary optic neuropathy3 (LHON), myoclonic epilepsy and ragged-red fiber disease 4 (MERRF), and the Keams--Sayre syndrome s (KSS). However, the unortho- dox genetics, late onset, and progression of these dis- eases suggest that a variety of more common degener- ative disorders might also be caused by mitochondrial mutations. These include heart failure 6-~, adult-onset diabetes'~, and even ageing t0-t3,
While the genetics of mtDNA reveals important insights into a variety of chronic degenerative diseases, it also presents several enigmas. How can a pathologi- cal mtDNA mutation be present at birth, yet only be clinically manifest in adults? How can the same mtDNA mutation be present in all cells of the body, yet only affect certain tissues? Studies on the accumulation of somatic mtDNA mutations with age and on the control of mitochondrial gene expression during development are beginning to answer these questions 1.
The genetics of mitochondrtal DNA The mitochondrial genome is a 16 569 base pair
(bp) circular molecule. It encodes 13 polypeptide sub- units of the mitochondrial ATP-generating pathway, oxidative phosphorylation (OXPHOS), together with the 12S and lOS ribosomal RNAs and the 22 mitochon- drial transfer RNAs necessary for mRNA expression (Fig. 1). Each human cell has hundreds of mitochon- dria and thousands of mtDNAs, and the high copy- number and cytoplasmic location of these DNAs means that their genetic behavior is surprisingly unorthodox I.
Mitochondrial DNA is maternally inherited, since it is predominantly transmitted through the egg cytoplasm. When a new mtDNA mutation arises in a
1'~03 Elsevier Science Puhlishers Ltd (I tK) 0168 - 9525/93/$06.00
"FIG APRIL 1 9 9 3
Mitochondrial diseases: genotype versus phenotype DOUGLAS C. WALLACE
Recently, a variety of degenerative diseases have been attributed to mutations in mitochoodrlal DNA. Even though these mutations are inherited and present throughout the body, they frequently cause late.onset, ttssue.specO~ disease, This may be explained by a combi~tatioa of the tissue.spec ic accumulation of somatic mtDNA mutations with age and the variation between tissues in the expression of nuclear genes that encode mttochondrial frictions.
cell, an intracellular mixture of mutant and normal mtDNAs is created; this is known as heteroplasmy. As heteroplasmic cells divide, the mtDNA genotype undergoes replicative segregation and the proportion of mutant mtDNAs drifts, such that cells tend towards having either all mutant or all normal mtDNAs (homo- plasmy) I.
Different tissues and organs rely on mitochondrial energy to various extents, with the central nervous system, heart, muscle, kidney and endocrine glands being particularly oxidative, and therefore especially dependent on energy produced by the mitochondrion. As the proportion of mutant mtDNAs increases among maternal relatives, mitochondrial energy output de- clines until the ATP-generating capacity of the cell falls below the energy threshold necessary for normal tissue function, and disease ensue#.
Mitochondrial DNA has a very high mutation rate, involving both nucleotide substitution and insertion- deletion mutations. Mutations that occur in the germ line result in familial disease, while those that occur
VOL. 9 NO. 4





























