Embed Size (px)
Citation preview

Entirely plasmid-based reverse genetics systemfor rotavirusesYuta Kanaia, Satoshi Komotob, Takahiro Kawagishia,c, Ryotaro Noudaa,c, Naoko Nagasawaa, Misa Onishia,Yoshiharu Matsuurac, Koki Taniguchib, and Takeshi Kobayashia,1
aLaboratory of Viral Replication, International Research Center for Infectious Diseases, Research Institute for Microbial Diseases, Osaka University, Suita,Osaka, 565-0871 Japan; bDepartment of Virology and Parasitology, Fujita Health University School of Medicine, Toyoake, Aichi, 470-1192 Japan;and cDepartment of Molecular Virology, Research Institute for Microbial Diseases, Osaka University, Suita, Osaka, 565-0871 Japan
Edited by Peter Palese, Icahn School of Medicine at Mount Sinai, New York, NY, and approved December 28, 2016 (received for review November 7, 2016)
Rotaviruses (RVs) are highly important pathogens that cause severediarrhea among infants and young children worldwide. The under-standing of the molecular mechanisms underlying RV replication andpathogenesis has been hampered by the lack of an entirely plasmid-based reverse genetics system. In this study, we describe the recoveryof recombinant RVs entirely from cloned cDNAs. The strategy requirescoexpression of a small transmembrane protein that accelerates cell-to-cell fusion and vaccinia virus capping enzyme. We used this systemto obtain insights into the process by which RV nonstructural proteinNSP1 subverts host innate immune responses. By insertion into theNSP1 gene segment, we recovered recombinant viruses that encodesplit-green fluorescent protein–tagged NSP1 and NanoLuc luciferase.This technology will provide opportunities for studying RV biologyand foster development of RV vaccines and therapeutics.
rotavirus | reverse genetics | vaccine | reporter virus
Group A rotaviruses (RVs), members of the family Reoviridae,are a highly prevalent cause of severe diarrhea in infants and
young children worldwide and are responsible for ∼215,000 deathsannually, mostly in developing countries (1). RVs are nonenvelopedicosahedral viruses containing a genome of 11 gene segmentscomposed of double-stranded (ds) RNA.Reverse genetics systems for manipulating viral genomes
provide key critical insights into viral replication and pathogen-esis and facilitate development of novel vaccines and viral vec-tors through direct gene modification and attenuation. Entirelyplasmid- or RNA transcript-based reverse genetics systems havenow been established for several genera of Reoviridae, includingmammalian orthoreovirus (MRV), Nelson Bay orthoreovirus(NBV) (Orthoreovirus genus), and bluetongue virus, Africanhorse sickness virus, and epizootic hemorrhagic disease virus(Orbivirus genus) (2–9). The development of the plasmid-basedreverse genetics system for MRV (2) raised expectations that thistechnology could be readily applied to genus Rotavirus. Partialplasmid-based reverse genetics systems that are dependent onhelper viruses have been developed for RV, and these strategieshave been used to generate recombinant RVs containing a singlerecombinant gene segment derived from cloned cDNAs (10–13).The breakthrough for generating recombinant RVs was de-veloped to manipulate the gene segment that encodes outer-capsid protein VP4 (13). In this system, a plasmid cDNA con-taining the VP4 gene segment was transfected into monkeykidney epithelial COS-7 cells expressing T7 RNA polymerase(T7pol) from attenuated vaccinia virus (VV) recombinant-strainrDIs-T7pol. The cells were then infected with human RV strainKU as a helper virus. Distinct recombinant VP4 monoreassortantviruses were isolated using neutralizing monoclonal antibodiesspecific for helper virus VP4. Subsequently, other helper-virus–dependent techniques were developed by modification of the firstsystem. Troupin et al. reported a reverse genetics method for RVsbased on preferential packaging of rearranged gene segments. Inthis system, a recombinant monoreassortant virus containing thenonstructural protein NSP3 gene segment was engineered by
extensive serial selective passage at a high multiplicity of infection(MOI) (11). A method for generating a recombinant virus containingthe NSP2 gene segment uses independent selection mechanisms: atemperature-sensitive (ts) mutant in which NSP2 is defective atnonpermissive temperature as a helper virus, and siRNA-mediatedgene silencing against the NSP2 message-sense ssRNA of the tsmutant (12). However, despite extensive efforts in many laboratories,no entirely plasmid-based reverse genetics system that does not re-quire a selection method against helper virus and is applicable to allgene segments of RV strains has been developed (14).Here, we demonstrate that recombinant RV can be recovered
following transfection of baby hamster kidney cells constitutivelyexpressing T7pol (BHK-T7) with 11 RV cDNA plasmids andexpression plasmids encoding NBV fusion-associated smalltransmembrane (FAST) protein and VV capping enzyme. Wetested the plasmid-based reverse genetics system by generatinga recombinant virus lacking the C-terminal region of NSP1 andused it to investigate the function of this protein as an antag-onist of the innate immune response in infected cells. In ad-dition, we established efficient gene transfer systems for use inlive-cell imaging, trafficking, and antiviral screening.
ResultsDevelopment of a Reverse Genetics System for RV. In the efforts todevelop improved reverse genetics systems for Reoviridae viruses,we discovered two important modifications that significantlyincrease nonfusogenic MRV and RV replication and enhance
Significance
Rotaviruses (RVs) are a group of viruses that cause severe gas-troenteritis in infants and young children. Until now, no strategyhas been developed to generate infectious RVs entirely fromcloned cDNAs. The absence of a reliable reverse genetics plat-form has been a major roadblock in the RV field, precludingnumerous studies of RV replication and pathogenesis and ham-pering efforts to develop the next generation of RV vaccines.Here, we developed a plasmid-based reverse genetics systemthat is free from helper viruses and independent of any selectionfor RV. This technology will accelerate studies of RV pathobiol-ogy, allow rational design of RV vaccines, and yield RVs suitablefor screening small molecules as potential antivirals.
Author contributions: Y.K., Y.M., K.T., and T. Kobayashi designed research; Y.K., S.K.,T. Kawagishi, R.N., N.N., and M.O. performed research; Y.K. and T. Kobayashi analyzeddata; and Y.K., Y.M., K.T., and T. Kobayashi wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The complete genome sequences of strain SA11 reported in this paperhave been deposited in the GenBank database (accession nos. LC178564–LC178574).
See Commentary on page 2106.1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1618424114/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1618424114 PNAS | February 28, 2017 | vol. 114 | no. 9 | 2349–2354
MICRO
BIOLO
GY
SEECO
MMEN
TARY
Dow
nloa
ded
by g
uest
on
Mar
ch 2
6, 2
020
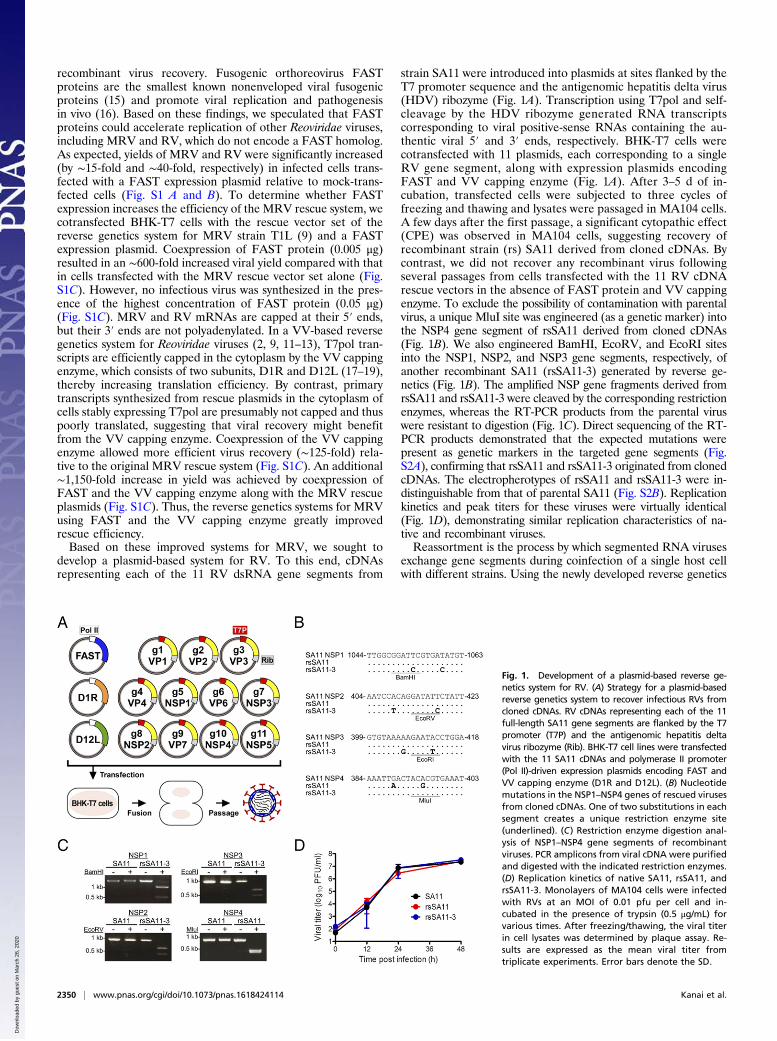
recombinant virus recovery. Fusogenic orthoreovirus FASTproteins are the smallest known nonenveloped viral fusogenicproteins (15) and promote viral replication and pathogenesisin vivo (16). Based on these findings, we speculated that FASTproteins could accelerate replication of other Reoviridae viruses,including MRV and RV, which do not encode a FAST homolog.As expected, yields of MRV and RV were significantly increased(by ∼15-fold and ∼40-fold, respectively) in infected cells trans-fected with a FAST expression plasmid relative to mock-trans-fected cells (Fig. S1 A and B). To determine whether FASTexpression increases the efficiency of the MRV rescue system, wecotransfected BHK-T7 cells with the rescue vector set of thereverse genetics system for MRV strain T1L (9) and a FASTexpression plasmid. Coexpression of FAST protein (0.005 μg)resulted in an ∼600-fold increased viral yield compared with thatin cells transfected with the MRV rescue vector set alone (Fig.S1C). However, no infectious virus was synthesized in the pres-ence of the highest concentration of FAST protein (0.05 μg)(Fig. S1C). MRV and RV mRNAs are capped at their 5′ ends,but their 3′ ends are not polyadenylated. In a VV-based reversegenetics system for Reoviridae viruses (2, 9, 11–13), T7pol tran-scripts are efficiently capped in the cytoplasm by the VV cappingenzyme, which consists of two subunits, D1R and D12L (17–19),thereby increasing translation efficiency. By contrast, primarytranscripts synthesized from rescue plasmids in the cytoplasm ofcells stably expressing T7pol are presumably not capped and thuspoorly translated, suggesting that viral recovery might benefitfrom the VV capping enzyme. Coexpression of the VV cappingenzyme allowed more efficient virus recovery (∼125-fold) rela-tive to the original MRV rescue system (Fig. S1C). An additional∼1,150-fold increase in yield was achieved by coexpression ofFAST and the VV capping enzyme along with the MRV rescueplasmids (Fig. S1C). Thus, the reverse genetics systems for MRVusing FAST and the VV capping enzyme greatly improvedrescue efficiency.Based on these improved systems for MRV, we sought to
develop a plasmid-based system for RV. To this end, cDNAsrepresenting each of the 11 RV dsRNA gene segments from
strain SA11 were introduced into plasmids at sites flanked by theT7 promoter sequence and the antigenomic hepatitis delta virus(HDV) ribozyme (Fig. 1A). Transcription using T7pol and self-cleavage by the HDV ribozyme generated RNA transcriptscorresponding to viral positive-sense RNAs containing the au-thentic viral 5′ and 3′ ends, respectively. BHK-T7 cells werecotransfected with 11 plasmids, each corresponding to a singleRV gene segment, along with expression plasmids encodingFAST and VV capping enzyme (Fig. 1A). After 3–5 d of in-cubation, transfected cells were subjected to three cycles offreezing and thawing and lysates were passaged in MA104 cells.A few days after the first passage, a significant cytopathic effect(CPE) was observed in MA104 cells, suggesting recovery ofrecombinant strain (rs) SA11 derived from cloned cDNAs. Bycontrast, we did not recover any recombinant virus followingseveral passages from cells transfected with the 11 RV cDNArescue vectors in the absence of FAST protein and VV cappingenzyme. To exclude the possibility of contamination with parentalvirus, a unique MluI site was engineered (as a genetic marker) intothe NSP4 gene segment of rsSA11 derived from cloned cDNAs(Fig. 1B). We also engineered BamHI, EcoRV, and EcoRI sitesinto the NSP1, NSP2, and NSP3 gene segments, respectively, ofanother recombinant SA11 (rsSA11-3) generated by reverse ge-netics (Fig. 1B). The amplified NSP gene fragments derived fromrsSA11 and rsSA11-3 were cleaved by the corresponding restrictionenzymes, whereas the RT-PCR products from the parental viruswere resistant to digestion (Fig. 1C). Direct sequencing of the RT-PCR products demonstrated that the expected mutations werepresent as genetic markers in the targeted gene segments (Fig.S2A), confirming that rsSA11 and rsSA11-3 originated from clonedcDNAs. The electropherotypes of rsSA11 and rsSA11-3 were in-distinguishable from that of parental SA11 (Fig. S2B). Replicationkinetics and peak titers for these viruses were virtually identical(Fig. 1D), demonstrating similar replication characteristics of na-tive and recombinant viruses.Reassortment is the process by which segmented RNA viruses
exchange gene segments during coinfection of a single host cellwith different strains. Using the newly developed reverse genetics
Fig. 1. Development of a plasmid-based reverse ge-netics system for RV. (A) Strategy for a plasmid-basedreverse genetics system to recover infectious RVs fromcloned cDNAs. RV cDNAs representing each of the 11full-length SA11 gene segments are flanked by the T7promoter (T7P) and the antigenomic hepatitis deltavirus ribozyme (Rib). BHK-T7 cell lines were transfectedwith the 11 SA11 cDNAs and polymerase II promoter(Pol II)-driven expression plasmids encoding FAST andVV capping enzyme (D1R and D12L). (B) Nucleotidemutations in the NSP1–NSP4 genes of rescued virusesfrom cloned cDNAs. One of two substitutions in eachsegment creates a unique restriction enzyme site(underlined). (C) Restriction enzyme digestion anal-ysis of NSP1–NSP4 gene segments of recombinantviruses. PCR amplicons from viral cDNA were purifiedand digested with the indicated restriction enzymes.(D) Replication kinetics of native SA11, rsSA11, andrsSA11-3. Monolayers of MA104 cells were infectedwith RVs at an MOI of 0.01 pfu per cell and in-cubated in the presence of trypsin (0.5 μg/mL) forvarious times. After freezing/thawing, the viral titerin cell lysates was determined by plaque assay. Re-sults are expressed as the mean viral titer fromtriplicate experiments. Error bars denote the SD.
2350 | www.pnas.org/cgi/doi/10.1073/pnas.1618424114 Kanai et al.
Dow
nloa
ded
by g
uest
on
Mar
ch 2
6, 2
020
system, we recovered a monoreassortant virus, rsSA11/KUVP6,containing the human strain KU VP6 gene segment in an other-wise simian strain SA11 genetic background (Fig. 2A). The elec-trophoretic pattern of rsSA11/KUVP6 revealed comigration of theVP6 RNA with that of strain KU (Fig. 2B), and the mono-reassortant virus exhibited replication kinetics similar to those ofnative SA11 (Fig. 2C). The recovery of monoreassortant virusesdemonstrated the utility of the reverse genetics system for rescueof reassortant viruses with unique genetic combinations that hadnot been reported previously.
The C-Terminal Region of NSP1 Is Essential for Targeting of InterferonRegulatory Factor 3 and Evasion of Innate Immune Surveillance.RV NSP1 antagonizes interferon (IFN) signaling by promotingdegradation of IFN regulatory factor 3 (IRF3) (20). The im-portance of the C-terminal region of NSP1, including the pLxISmotif, for interaction with IRF3, followed by degradation, wasdemonstrated using recombinant NSP1 proteins and spontane-ous mutants encoding C-terminal truncations in duplicate se-quences of NSP1 (20–22). To determine whether the C-terminalregion of NSP1 is essential for inhibiting IFN signaling by an-tagonizing the function of IRF3, we engineered isogenic rsSA11-dC103, which differs from wild-type rsSA11 only by harboring aC-terminal 103-residue truncation of NSP1 (Fig. 3A). Genotypesof recombinant viruses were confirmed by electrophoretic anal-ysis of viral dsRNA. The truncated NSP1 gene segment wassmaller than the wild-type NSP1 gene segment (Fig. 3B). Ex-pression of NSP1 in cells infected with rsSA11 or rsSA11-dC103was confirmed by immunoblotting with NSP1-specific antiserum(Fig. S3A). A comparison of the truncated NSP1 mutant andwild-type viruses revealed that replication of rsSA11-dC103 washampered in simian and human cell lines (Fig. 3C). We observedsignificant degradation of IRF3 in HT29 cells infected with
rsSA11 (Fig. 3D). By contrast, rsSA11-dC103 did not induceIRF3 degradation (Fig. 3D). Luciferase assays revealed that theIFN-β promoter element was activated in cells infected withrsSA11-dC103, but not in cells infected with rsSA11 (Fig. S3B).In addition, we assessed the replication of rsSA11 and rsSA11-dC103 in mouse embryonic fibroblasts (MEFs) deficient inTANK-binding kinase 1 (TBK1), which is required for the nu-clear translocation of IRF3 (23). Replication of rsSA11-dC103was significantly greater in TBK1-deficient (TBK1−/−) MEFsthan in TBK1+/− MEFs, even though the titers of rsSA11-dC103were lower than those of rsSA11 in TBK−/− cells (Fig. 3E).Taken together, these results indicate that the C-terminal regionof NSP1 is required to antagonize the innate immune responseby inducing the degradation of IRF3, a transcription factor re-quired for IFN expression.
Fig. 2. Generation of a monoreassortant virus between strains SA11 andKU. (A) Representative monoreassortant, rsSA11/KUVP6, which contains thestrain KU VP6 gene segment on an otherwise SA11 genetic background. (B)Coelectrophoretic pattern of rsSA11/KUVP6. Viral genomic dsRNAs extractedfrom native SA11, KU, and rsSA11/KUVP6 virions were separated in 10%polyacrylamide gels and visualized by silver staining. Numbers indicate theorder of the SA11 and KU gene segments. (C) Monolayers of MA104 cells wereinfected with native SA11, KU, and rsSA11/KUVP6 at an MOI of 0.01 pfu percell for various intervals. Results are expressed as the mean viral titer fromtriplicate experiments. Error bars denote the SD. *P < 0.05 (t test).
Fig. 3. Analysis of the role of the C-terminally truncated NSP1 mutant virusin the innate immune response. (A) Construction of the C-terminally trun-cated NSP1 mutant. The NSP1-dC103 cDNA construct was generated by de-leting 103 residues (nucleotides 1,192–1,490) from the C-terminal region ofNSP1. (B) Electrophoretic pattern of rsSA11-dC103. Viral genomic dsRNAsextracted from recombinant RV virions were separated in 10% poly-acrylamide gels. Numbers indicate the order of the SA11 gene segments.(C) Replication of rsSA11 and rsSA11-dC103 in simian and human cell lines.Monolayers of cells were infected with rsSA11 or rsSA11-dC103 at an MOI of0.001 pfu per cell. Cells were harvested 48 h postinfection, and the titer ofinfectious virus in the cell lysate was determined by plaque-forming assay inCV-1 cells. Data are expressed as the means ± SD n = 4, *P < 0.05 and **P <0.01 (t test). (D) Impaired degradation of IRF3 in cells infected by rsSA11-dC103. HT29 cells were infected with rsSA11 or rsSA11-dC103 at an MOI of10 pfu per cell. Six hours after infection, cells were harvested and primaryantibodies raised against IRF3 and NSP5 were used to detect the intrinsicIRF3 and SA11 NSP5 proteins, respectively. An actin-specific antibody wasused as a loading control. Molecular masses were determined by coelec-trophoresis of prestained protein markers. (E) Replication of rsSA11 andrsSA11-dC103 in IFN signaling-deficient cells. MEFs from TBK1+/− or TBK1−/−
mice were infected with rsSA11 or rsSA11-dC103 at an MOI of 0.001 pfu percell and incubated with 0.25 μg/mL trypsin. Cells were harvested, and thetiter of infectious virus was determined by plaque assay. Fold increases in thepfu in TBK1−/− MEFs versus TBK1+/− MEFs are shown. Data are expressed asthe mean ± SD, n = 3. *P < 0.05 (t test).
Kanai et al. PNAS | February 28, 2017 | vol. 114 | no. 9 | 2351
MICRO
BIOLO
GY
SEECO
MMEN
TARY
Dow
nloa
ded
by g
uest
on
Mar
ch 2
6, 2
020
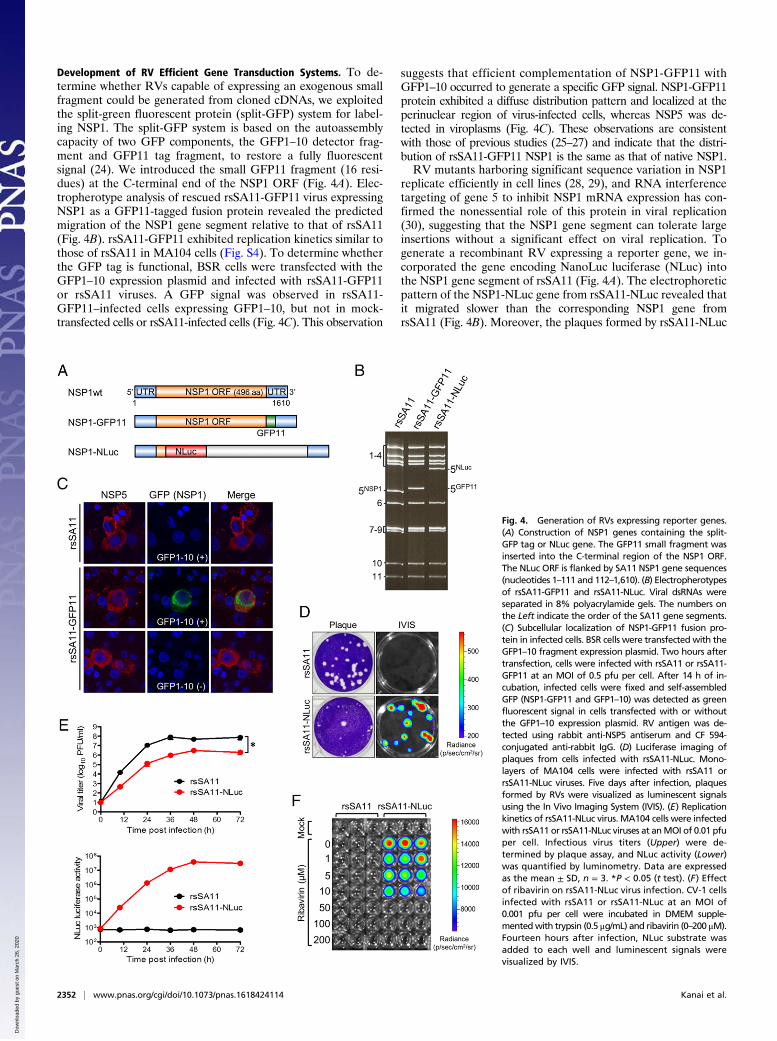
Development of RV Efficient Gene Transduction Systems. To de-termine whether RVs capable of expressing an exogenous smallfragment could be generated from cloned cDNAs, we exploitedthe split-green fluorescent protein (split-GFP) system for label-ing NSP1. The split-GFP system is based on the autoassemblycapacity of two GFP components, the GFP1–10 detector frag-ment and GFP11 tag fragment, to restore a fully fluorescentsignal (24). We introduced the small GFP11 fragment (16 resi-dues) at the C-terminal end of the NSP1 ORF (Fig. 4A). Elec-tropherotype analysis of rescued rsSA11-GFP11 virus expressingNSP1 as a GFP11-tagged fusion protein revealed the predictedmigration of the NSP1 gene segment relative to that of rsSA11(Fig. 4B). rsSA11-GFP11 exhibited replication kinetics similar tothose of rsSA11 in MA104 cells (Fig. S4). To determine whetherthe GFP tag is functional, BSR cells were transfected with theGFP1–10 expression plasmid and infected with rsSA11-GFP11or rsSA11 viruses. A GFP signal was observed in rsSA11-GFP11–infected cells expressing GFP1–10, but not in mock-transfected cells or rsSA11-infected cells (Fig. 4C). This observation
suggests that efficient complementation of NSP1-GFP11 withGFP1–10 occurred to generate a specific GFP signal. NSP1-GFP11protein exhibited a diffuse distribution pattern and localized at theperinuclear region of virus-infected cells, whereas NSP5 was de-tected in viroplasms (Fig. 4C). These observations are consistentwith those of previous studies (25–27) and indicate that the distri-bution of rsSA11-GFP11 NSP1 is the same as that of native NSP1.RV mutants harboring significant sequence variation in NSP1
replicate efficiently in cell lines (28, 29), and RNA interferencetargeting of gene 5 to inhibit NSP1 mRNA expression has con-firmed the nonessential role of this protein in viral replication(30), suggesting that the NSP1 gene segment can tolerate largeinsertions without a significant effect on viral replication. Togenerate a recombinant RV expressing a reporter gene, we in-corporated the gene encoding NanoLuc luciferase (NLuc) intothe NSP1 gene segment of rsSA11 (Fig. 4A). The electrophoreticpattern of the NSP1-NLuc gene from rsSA11-NLuc revealed thatit migrated slower than the corresponding NSP1 gene fromrsSA11 (Fig. 4B). Moreover, the plaques formed by rsSA11-NLuc
Fig. 4. Generation of RVs expressing reporter genes.(A) Construction of NSP1 genes containing the split-GFP tag or NLuc gene. The GFP11 small fragment wasinserted into the C-terminal region of the NSP1 ORF.The NLuc ORF is flanked by SA11 NSP1 gene sequences(nucleotides 1–111 and 112–1,610). (B) Electropherotypesof rsSA11-GFP11 and rsSA11-NLuc. Viral dsRNAs wereseparated in 8% polyacrylamide gels. The numbers onthe Left indicate the order of the SA11 gene segments.(C) Subcellular localization of NSP1-GFP11 fusion pro-tein in infected cells. BSR cells were transfected with theGFP1–10 fragment expression plasmid. Two hours aftertransfection, cells were infected with rsSA11 or rsSA11-GFP11 at an MOI of 0.5 pfu per cell. After 14 h of in-cubation, infected cells were fixed and self-assembledGFP (NSP1-GFP11 and GFP1–10) was detected as greenfluorescent signal in cells transfected with or withoutthe GFP1–10 expression plasmid. RV antigen was de-tected using rabbit anti-NSP5 antiserum and CF 594-conjugated anti-rabbit IgG. (D) Luciferase imaging ofplaques from cells infected with rsSA11-NLuc. Mono-layers of MA104 cells were infected with rsSA11 orrsSA11-NLuc viruses. Five days after infection, plaquesformed by RVs were visualized as luminescent signalsusing the In Vivo Imaging System (IVIS). (E) Replicationkinetics of rsSA11-NLuc virus. MA104 cells were infectedwith rsSA11 or rsSA11-NLuc viruses at anMOI of 0.01 pfuper cell. Infectious virus titers (Upper) were de-termined by plaque assay, and NLuc activity (Lower)was quantified by luminometry. Data are expressedas the mean ± SD, n = 3. *P < 0.05 (t test). (F) Effectof ribavirin on rsSA11-NLuc virus infection. CV-1 cellsinfected with rsSA11 or rsSA11-NLuc at an MOI of0.001 pfu per cell were incubated in DMEM supple-mentedwith trypsin (0.5 μg/mL) and ribavirin (0–200 μM).Fourteen hours after infection, NLuc substrate wasadded to each well and luminescent signals werevisualized by IVIS.
2352 | www.pnas.org/cgi/doi/10.1073/pnas.1618424114 Kanai et al.
Dow
nloa
ded
by g
uest
on
Mar
ch 2
6, 2
020

on CV-1 monolayer cells were smaller than those of rsSA11.However, cells infected with rsSA11-NLuc were positivefor bioluminescent signals (Fig. 4D). rsSA11-NLuc replicatedefficiently in MA104 cells (over 106 pfu/mL at 48 h post-infection), although its replication kinetics were slightly impairedcompared with rsSA11 (Fig. 4E). The activities of NLuc in celllysates infected with rsSA11-NLuc could be detected as early as12 h postinfection and increased over time (Fig. 4E). Finally, wedemonstrated the utility of rsSA11-NLuc for antiviral screeningusing a known RV inhibitor, ribavirin (31). CV-1 cells infectedwith rsSA11 or rsSA11-NLuc were cultured with ribavirin atincreasing concentrations; the cells did not exhibit significantcytotoxicity in the range of concentrations used in the experiment(Fig. S5A). Strong bioluminescence signals were observed in cellsinfected with rsSA11-NLuc at low concentrations (0–10 μM)of ribavirin and decreased in a dose-dependent manner (Fig. 4F).Similarly, viral titers in infected cell lysates also exhibited dose-dependent inhibition (Fig. S5B), supporting the validity of screeningwith rsSA11-NLuc. The genetic stability of rsSA11-NLuc to expressNLuc was unchanged through five passages (Fig. S6 A and B).These results demonstrate that replication-competent RVs encod-ing a reporter gene can be recovered by plasmid rescue and used forantiviral screening.
DiscussionWe succeeded in developing an entirely plasmid-based RV re-verse genetics system in which FAST protein and capping en-zyme were coexpressed along with rescue plasmids in BHK-T7cells (Fig. 1). The mechanism underlying the remarkable effectsof FAST protein, which induce cell-to-cell fusion and syncytiumformation in nonfusogenic RV and MRV replication in infectedand transfected cells, remains unclear. However, previous studies(15, 16, 32) suggest that the cell fusion activity of FAST proteinsaccelerates cell-to-cell transmission of virus infection and en-sures rapid release of progeny virions from apoptotic syncytia,thereby promoting systematic infection. FAST proteins also mayincrease cotransfection efficiency, resulting in increased virusproduction in reverse genetic systems via fusion of transfectedcells with neighboring cells, leading to formation of syncytiacarrying all 14 transfected plasmids (11 RV cDNA plasmidsand expression plasmids encoding FAST and VV capping en-zyme). The currently available reverse genetics systems for RVsare based on vaccinia-driven T7pol expression (11–13). Al-though it is possible to rescue Reoviridae viruses that havecapped and nonpolyadenylated mRNA using VV expressingT7pol, VV infection has negative effects on RV replication andrescue efficiency in reverse genetics systems. Accordingly, theimproved FAST- and VV capping enzyme-based reverse ge-netics system free of any helper virus for RV and MRV de-scribed herein is applicable to the recovery of any member ofthe Reoviridae family, particularly attenuated recombinantviruses that replicate poorly.Two licensed RV vaccines, Rotarix (GlaxoSmithKline) and
RotaTeq (Merck), are currently available. Rotarix is based on asingle human strain, and RotaTeq is a combination of five bovine ×human strain monoreassortants. In addition, a new RV vac-cine, Rotavac (Bharat Biotech International), was licensed inIndia in 2014 (33). Although these vaccines are effective againstRV-associated severe gastroenteritis, concerns about their effi-cacy, safety, and cost have inspired the development of newvaccines. We generated a recombinant RV containing silentmutations in three gene segments (NSP1, NSP2, and NSP3) anda monoreassortant virus harboring the human RV strain KUVP6 gene on the strain SA11 genetic background (Figs. 1 and 2).Thus, in contrast to earlier helper virus-based reverse geneticssystems, the RV rescue system described here can be easily usedfor rapid generation of infectious RVs containing multiple mu-tations in several different gene segments simultaneously, as well
as reassortants with any desired gene segment combination andfeatures that could serve as vaccine candidates.We confirmed that the C-terminal 103 residues of NSP1 are
required to inhibit IFN signaling by inducing proteasome-dependent degradation of IRFs (Fig. 3). According to previousstudies, NSP1 also inhibits NF-κB activation by inducing degra-dation of β-TrCP and down-regulating p53, which induces apo-ptosis and transactivates several genes involved in antiviralresponses (34, 35). Additionally, NSP1 interacts with the p85subunit of the phosphoinositide 3-kinase (PI3K)-mediated anti-apoptotic PI3K/Akt pathway (36). Taken together, these obser-vations indicate that NSP1 can interfere with multiple antiviralpathways, including IFN and apoptosis signaling, to promoteefficient viral replication and infection. NSP1 mutants, includinga C-terminal truncation incapable of blocking IFN signaling andapoptosis pathways, may be attractive candidates for the devel-opment of new attenuated RV vaccines.We used the reverse genetics system to modify the NSP1
gene segment to engineer RVs expressing reporter genes. Arecombinant RV harboring the split-GFP system was generatedby inserting a small GFP11 tag into the C terminus of the NSP1ORF (Fig. 4). Thus, the split-GFP–based recombinant NSP1mutants will be useful tools for understanding NSP1 traffickingand interactions with host proteins, including IFN signalingcomponents, in infected cells. Furthermore, a similar approachusing the split-GFP system could be used to study other RVproteins in living cells. We also applied the reverse geneticssystem to generate a replication-competent recombinant RVexpressing the NLuc gene fused to the N-terminal 27 residues ofNSP1 (Fig. 4). The results confirm that NSP1 is not required forviral replication, a finding consistent with a previous study thatused siRNA gene silencing (30), and that the NSP1 gene segmentis suitable for insertion of a heterologous sequence. The attenu-ated replication kinetics of rsSA11-NLuc could be explained bythe defect in the IFN suppressor activity of NSP1, or by the in-fluence of reduced packaging efficiency. In addition, the replica-tion and luciferase activity of rsSA11-NLuc were inhibited byribavirin, a known anti-RV inhibitor, suggesting that the reportervirus would be a useful tool for high-throughput screening forantiviral therapeutics (Fig. 4). Furthermore, the replication-com-petent RV carrying the NLuc gene makes it possible to track RVinfection in vivo and develop an oral RV vector.In this study, we developed a plasmid-only–based reverse ge-
netics system for RV. This technique opens new horizons for thestudy of RV replication and pathogenesis, as well as for thedevelopment of antiviral drugs and new vaccines that protectagainst this important gastrointestinal pathogen.
Materials and MethodsCells and Viruses. Monkey kidney epithelial MA104, CV-1, Vero, murine fi-broblast L929, and human colon epithelial Caco-2 cells were cultured inDMEM (Nacalai Tesque) supplemented to contain 5% (vol/vol) fetal bovineserum (FBS) (Gibco), 100 units/mL penicillin, and 100 μg/mL streptomycin(Nacalai Tesque). BHK/T7-9 cells were grown in DMEM supplemented tocontain 5% FBS, 10% (wt/vol) tryptose phosphate broth, 100 units/mL pen-icillin, and 100 μg/mL streptomycin (37). To establish another BHK-T7 cell line(BHK-T7/P5), BSR cells (3), a derivative of BHK cells, were selected by trans-fection with a eukaryotic expression plasmid encoding T7pol under thecontrol of a strong CMV early enhancer/chicken β-actin (CAG) promoter (38),followed by incubation in the presence of 4 μg/mL puromycin (Sigma-Aldrich). Immortalized MEFs derived from TBK1+/− IKKi−/− (TBK1+/−) andTBK1−/− IKKi−/− (TBK1−/−) mice were prepared as previously described (39, 40)and grown in DMEM supplemented to contain 10% FBS, 100 units/mLpenicillin, and 100 μg/mL streptomycin. Human cancer cell lines, HT29, PC3,HCC-2998, and OVCAR-4, were cultured in RPMI1640 (Nacalai Tesque) sup-plemented to contain 10% FBS, 100 units/mL penicillin, and 100 μg/mLstreptomycin. Simian RV strain SA11 (SA11-L2) (G3P[2]) (41) and human RVstrain KU (G1P[8]) (42) were propagated in MA104 cells cultured in DMEMsupplemented with 0.5 μg/mL trypsin (Sigma-Aldrich). MRV strain T1L, alaboratory stock originally obtained from Bernard Fields, Harvard Medical
Kanai et al. PNAS | February 28, 2017 | vol. 114 | no. 9 | 2353
MICRO
BIOLO
GY
SEECO
MMEN
TARY
Dow
nloa
ded
by g
uest
on
Mar
ch 2
6, 2
020

School, Boston, was propagated in L929 cells. Infectious titers of RV and MRVwere determined by plaque assay using CV-1 cells and L929 cells, re-spectively, as previously described (43, 44).
Recovery of Recombinant RVs from Cloned cDNAs. Monolayers of BHK-T7 cells(8 × 105) in six-well plates were cotransfected with plasmids using 2 μL of TransIT-LT1 transfection reagent per microgram of plasmid DNA, as follows: 0.8 μg ofeach strain SA11 rescue plasmid [pT7-VP1SA11, pT7-VP2SA11, pT7-VP3SA11, pT7-VP4SA11, pT7-VP6SA11 (or pT7-VP6KU), pT7-VP7SA11, pT7-NSP1SA11 (pT7-NSP1-BamHI, pT7-NSP1-dC103, or pT7-NSP1-GFP11), pT7-NSP2SA11 (or pT7-NSP2-EcoRV), pT7-NSP3SA11 (or pT7-NSP3-EcoRI), pT7-NSP4SA11 (or pT7-NSP4-MluI),and pT7-NSP5SA11], 0.015 μg of pCAG-FAST, and 0.8 μg of each capping enzymeexpression plasmid. After 2 d of incubation in FBS-free medium, MA104 cells (105
cells) were added to the transfected cells and cocultured for 3 d in FBS-freemedium supplemented with trypsin (0.5 μg/mL). After incubation, transfectedcells were lysed by freeze/thaw, trypsin was added to cell lysates at a final
concentration of 10 μg/mL, and the samples were incubated at 37 °C for 30 minto activate infectious RVs. The lysates were then transferred to freshMA104 cells.After adsorption at 37 °C for 1 h, the lysate-adsorbed MA104 cells were washedand cultured in FBS-free DMEM supplemented with 0.5 μg/mL trypsin and in-cubated at 37 °C for 7 d. When CPE was observed following RV infection ofmonolayer cells, recombinant viruses were isolated from passaged cells by pla-que purification using CV-1 cells.
ACKNOWLEDGMENTS. We thank Terence S. Dermody for reviewing themanuscript, Kaede Yukawa for secretarial work, Toru Okamoto for technicaladvice, Saori Fukuda for technical assistance, Polly Roy for providing BSRcells, and Naoto Ito for providing BHK/T7-9 cells. This work was supportedin part by grants-in-aid for the Research Program on Emerging and Re-Emerging Infectious Diseases from the Japan Agency for Medical Researchand Development and Japanese Society for the Promotion of Science(JSPS) KAKENHI Grants JP16K19138, JP15J04209, and JP26292149.
1. Tate JE, Burton AH, Boschi-Pinto C, Parashar UD;World Health Organization–CoordinatedGlobal Rotavirus Surveillance Network (2016) Global, regional, and national estimates ofrotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis 62(Suppl 2):S96–S105.
2. Kobayashi T, et al. (2007) A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1(2):147–157.
3. Boyce M, Celma CC, Roy P (2008) Development of reverse genetics systems for blue-tongue virus: Recovery of infectious virus from synthetic RNA transcripts. J Virol82(17):8339–8348.
4. Kaname Y, Celma CC, Kanai Y, Roy P (2013) Recovery of African horse sickness virusfrom synthetic RNA. J Gen Virol 94(Pt 10):2259–2265.
5. Matsuo E, Saeki K, Roy P, Kawano J (2015) Development of reverse genetics forIbaraki virus to produce viable VP6-tagged IBAV. FEBS Open Bio 5:445–453.
6. Yang T, et al. (2015) Development of a reverse genetics system for epizootic hem-orrhagic disease virus and evaluation of novel strains containing duplicative generearrangements. J Gen Virol 96(9):2714–2720.
7. Kawagishi T, et al. (2016) Reverse genetics for fusogenic bat-borne orthoreovirusassociated with acute respiratory tract infections in humans: Role of outer capsidprotein σC in viral replication and pathogenesis. PLoS Pathog 12(2):e1005455.
8. Pretorius JM, Huismans H, Theron J (2015) Establishment of an entirely plasmid-basedreverse genetics system for bluetongue virus. Virology 486:71–77.
9. Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS (2010) An improved reversegenetics system for mammalian orthoreoviruses. Virology 398(2):194–200.
10. Johne R, Reetz J, Kaufer BB, Trojnar E (2015) Generation of an avian-mammalianrotavirus reassortant by using a helper virus-dependent reverse genetics system.J Virol 90(3):1439–1443.
11. Troupin C, et al. (2010) Rearranged genomic RNA segments offer a new approach tothe reverse genetics of rotaviruses. J Virol 84(13):6711–6719.
12. Trask SD, Taraporewala ZF, Boehme KW, Dermody TS, Patton JT (2010) Dual selectionmechanisms drive efficient single-gene reverse genetics for rotavirus. Proc Natl AcadSci USA 107(43):18652–18657.
13. Komoto S, Sasaki J, Taniguchi K (2006) Reverse genetics system for introduction ofsite-specific mutations into the double-stranded RNA genome of infectious rotavirus.Proc Natl Acad Sci USA 103(12):4646–4651.
14. Richards JE, Desselberger U, Lever AM (2013) Experimental pathways towards de-veloping a rotavirus reverse genetics system: synthetic full length rotavirus ssRNAs areneither infectious nor translated in permissive cells. PLoS One 8(9):e74328.
15. Ciechonska M, Duncan R (2014) Reovirus FAST proteins: Virus-encoded cellular fus-ogens. Trends Microbiol 22(12):715–724.
16. Brown CW, et al. (2009) The p14 FAST protein of reptilian reovirus increases vesicularstomatitis virus neuropathogenesis. J Virol 83(2):552–561.
17. Morgan JR, Cohen LK, Roberts BE (1984) Identification of the DNA sequences en-coding the large subunit of the mRNA-capping enzyme of vaccinia virus. J Virol 52(1):206–214.
18. Niles EG, Lee-Chen GJ, Shuman S, Moss B, Broyles SS (1989) Vaccinia virus gene D12Lencodes the small subunit of the viral mRNA capping enzyme. Virology 172(2):513–522.
19. Ensinger MJ, Martin SA, Paoletti E, Moss B (1975) Modification of the 5′-terminus ofmRNA by soluble guanylyl and methyl transferases from vaccinia virus. Proc Natl AcadSci USA 72(7):2525–2529.
20. Barro M, Patton JT (2005) Rotavirus nonstructural protein 1 subverts innate immuneresponse by inducing degradation of IFN regulatory factor 3. Proc Natl Acad Sci USA102(11):4114–4119.
21. Graff JW, Ewen J, Ettayebi K, Hardy ME (2007) Zinc-binding domain of rotavirus NSP1is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1stability. J Gen Virol 88(Pt 2):613–620.
22. Zhao B, et al. (2016) Structural basis for concerted recruitment and activation of IRF-3by innate immune adaptor proteins. Proc Natl Acad Sci USA 113(24):E3403–E3412.
23. Fitzgerald KA, et al. (2003) IKKepsilon and TBK1 are essential components of the IRF3signaling pathway. Nat Immunol 4(5):491–496.
24. Cabantous S, Terwilliger TC, Waldo GS (2005) Protein tagging and detection withengineered self-assembling fragments of green fluorescent protein. Nat Biotechnol23(1):102–107.
25. Welch SK, Crawford SE, Estes MK (1989) Rotavirus SA11 genome segment 11 proteinis a nonstructural phosphoprotein. J Virol 63(9):3974–3982.
26. Hua J, Patton JT (1994) The carboxyl-half of the rotavirus nonstructural protein NS53(NSP1) is not required for virus replication. Virology 198(2):567–576.
27. Martínez-Álvarez L, Piña-Vázquez C, Zarco W, Padilla-Noriega L (2013) The shift fromlow to high non-structural protein 1 expression in rotavirus-infected MA-104 cells.Mem Inst Oswaldo Cruz 108(4):421–428.
28. Patton JT, et al. (2001) Effect of intragenic rearrangement and changes in the 3′consensus sequence on NSP1 expression and rotavirus replication. J Virol 75(5):2076–2086.
29. Taniguchi K, Kojima K, Urasawa S (1996) Nondefective rotavirus mutants with anNSP1 gene which has a deletion of 500 nucleotides, including a cysteine-rich zincfinger motif-encoding region (nucleotides 156 to 248), or which has a nonsense codonat nucleotides 153-155. J Virol 70(6):4125–4130.
30. Silvestri LS, Taraporewala ZF, Patton JT (2004) Rotavirus replication: Plus-sense tem-plates for double-stranded RNA synthesis are made in viroplasms. J Virol 78(14):7763–7774.
31. Smee DF, Sidwell RW, Clark SM, Barnett BB, Spendlove RS (1982) Inhibition of rota-viruses by selected antiviral substances: mechanisms of viral inhibition and in vivoactivity. Antimicrob Agents Chemother 21(1):66–73.
32. Salsman J, Top D, Boutilier J, Duncan R (2005) Extensive syncytium formation medi-ated by the reovirus FAST proteins triggers apoptosis-induced membrane instability.J Virol 79(13):8090–8100.
33. Bhandari N, et al.; India Rotavirus Vaccine Group (2014) Efficacy of a monovalenthuman-bovine (116E) rotavirus vaccine in Indian infants: A randomised, double-blind,placebo-controlled trial. Vaccine 383(9935):2136–2143.
34. Nandi S, et al. (2014) MAVS protein is attenuated by rotavirus nonstructural protein 1.PLoS One 9(3):e92126.
35. Graff JW, Ettayebi K, Hardy ME (2009) Rotavirus NSP1 inhibits NFkappaB activation byinducing proteasome-dependent degradation of beta-TrCP: A novel mechanism ofIFN antagonism. PLoS Pathog 5(1):e1000280.
36. Bagchi P, et al. (2010) Rotavirus nonstructural protein 1 suppresses virus-inducedcellular apoptosis to facilitate viral growth by activating the cell survival pathwaysduring early stages of infection. J Virol 84(13):6834–6845.
37. Ito N, et al. (2003) Improved recovery of rabies virus from cloned cDNA using a vac-cinia virus-free reverse genetics system. Microbiol Immunol 47(8):613–617.
38. Niwa H, Yamamura K, Miyazaki J (1991) Efficient selection for high-expressiontransfectants with a novel eukaryotic vector. Gene 108(2):193–199.
39. Ono C, et al. (2014) Innate immune response induced by baculovirus attenuatestransgene expression in mammalian cells. J Virol 88(4):2157–2167.
40. Hemmi H, et al. (2004) The roles of two IkappaB kinase-related kinases in lipopoly-saccharide and double stranded RNA signaling and viral infection. J Exp Med 199(12):1641–1650.
41. Taniguchi K, et al. (1994) Differences in plaque size and VP4 sequence found in SA11virus clones having simian authentic VP4. Virology 198(1):325–330.
42. Urasawa S, Urasawa T, Taniguchi K, Chiba S (1984) Serotype determination of humanrotavirus isolates and antibody prevalence in pediatric population in Hokkaido, Ja-pan. Arch Virol 81(1-2):1–12.
43. Arnold M, Patton JT, McDonald SM (2009) Culturing, storage, and quantification ofrotaviruses. Curr Protoc Microbiol Chapter 15:Unit 15C.3.
44. Virgin HW, 4th, Bassel-Duby R, Fields BN, Tyler KL (1988) Antibody protects againstlethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62(12):4594–4604.
45. Schnell MJ, Mebatsion T, Conzelmann KK (1994) Infectious rabies viruses from clonedcDNA. EMBO J 13(18):4195–4203.
46. Maan S, et al. (2007) Rapid cDNA synthesis and sequencing techniques for the geneticstudy of bluetongue and other dsRNA viruses. J Virol Methods 143(2):132–139.
47. Shiokawa M, et al. (2014) Novel permissive cell lines for complete propagation ofhepatitis C virus. J Virol 88(10):5578–5594.
2354 | www.pnas.org/cgi/doi/10.1073/pnas.1618424114 Kanai et al.
Dow
nloa
ded
by g
uest
on
Mar
ch 2
6, 2
020







![Emergence of Unusual G6P[6] Rotaviruses in Children ...Emergence of Unusual G6P[6] Rotaviruses in Children, Burkina Faso, 2009–2010 Technical Appendix Technical Appendix Table 1](https://img.pdfslide.us/doc/110x75/5f0ac8c87e708231d42d537a/emergence-of-unusual-g6p6-rotaviruses-in-children-emergence-of-unusual-g6p6.jpg)





















