Embed Size (px)
Citation preview

Published Ahead of Print 27 July 2011. 2011, 85(19):10319. DOI: 10.1128/JVI.00787-11. J. Virol.
Jinghua Yan and George F. GaoGao, Daizong Lin, Wangke Qian, Hong Liu, Hualiang Jiang, Guangwen Lu, Jianxun Qi, Zhujun Chen, Xiang Xu, Feng Disease Virus Drug DesignSubstrates and Anti-Hand, Foot, and MouthProteases: Binding to Rupintrivir and Their Enterovirus 71 and Coxsackievirus A16 3C
http://jvi.asm.org/content/85/19/10319Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/85/19/10319#ref-list-1at:
This article cites 44 articles, 18 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://jvi.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from

JOURNAL OF VIROLOGY, Oct. 2011, p. 10319–10331 Vol. 85, No. 190022-538X/11/$12.00 doi:10.1128/JVI.00787-11Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Enterovirus 71 and Coxsackievirus A16 3C Proteases: Binding toRupintrivir and Their Substrates and Anti-Hand, Foot, and
Mouth Disease Virus Drug Design�
Guangwen Lu,1,2 Jianxun Qi,1 Zhujun Chen,1,3 Xiang Xu,1,3 Feng Gao,4,5 Daizong Lin,6
Wangke Qian,6 Hong Liu,6 Hualiang Jiang,6 Jinghua Yan,1* and George F. Gao1,2,7,8*Chinese Academy of Sciences Key Laboratory of Pathogenic Microbiology and Immunology (CASPMI), Institute of Microbiology,Chinese Academy of Sciences, Beijing 100101, China1; Graduate University, Chinese Academy of Sciences, Beijing 100049, China2;
College of Life Science, Anhui Agricultural University, Anhui 230036, China3; National Laboratory of Macromolecules, Institute ofBiophysics, Chinese Academy of Sciences, Beijing 100101, China4; School of Life Sciences, Sichuan University,
Chengdu 610064, Sichuan, China5; Shanghai Institute of Materia Medica, Chinese Academy of Sciences,Shanghai 201203, China6; China-Japan Joint Laboratory of Molecular Immunology and
Molecular Microbiology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101,China7; and Beijing Institutes of Life Science, Chinese Academy of
Sciences, Beijing 100101, China8
Received 19 April 2011/Accepted 16 July 2011
Enterovirus 71 (EV71) and coxsackievirus A16 (CVA16) are the major causative agents of hand, foot, andmouth disease (HFMD), which is prevalent in Asia. Thus far, there are no prophylactic or therapeuticmeasures against HFMD. The 3C proteases from EV71 and CVA16 play important roles in viral replicationand are therefore ideal drug targets. By using biochemical, mutational, and structural approaches, we broadlycharacterized both proteases. A series of high-resolution structures of the free or substrate-bound enzymeswere solved. These structures, together with our cleavage specificity assay, well explain the marked substratepreferences of both proteases for particular P4, P1, and P1� residue types, as well as the relative malleabilityof the P2 amino acid. More importantly, the complex structures of EV71 and CVA16 3Cs with rupintrivir, aspecific human rhinovirus (HRV) 3C protease inhibitor, were solved. These structures reveal a half-closed S2subsite and a size-reduced S1� subsite that limit the access of the P1� group of rupintrivir to both enzymes,explaining the reported low inhibition activity of the compound toward EV71 and CVA16. In conclusion, thedetailed characterization of both proteases in this study could direct us to a proposal for rational design ofEV71/CVA16 3C inhibitors.
Hand, foot, and mouth disease (HFMD) is a common viralillness among infants and young children, with clinical charac-terizations of prodromal fever followed by pharyngitis, mouthulcers, and a rash on the hands and feet (7, 8). Human entero-virus 71 (EV71) and coxsackievirus A16 (CVA16) are the twomajor causative agents of HFMD. Clinically, infections withthe two viruses manifesting as HFMD are indistinguishable.However, EV71 can also result in severe neurological diseases,such as aseptic meningitis and acute flaccid paralysis (AFP),and even death (7, 8, 38). Since the first reported case ofHFMD in New Zealand in 1957 (31), it has continued tospread globally and is a continuing threat to global publichealth (2, 5, 14, 16, 17, 29). In the last decade, regularly reoc-curring outbreaks of HFMD have been relatively centralized inthe Asia-Pacific region (9, 10, 33, 40). In 2008, an unexpectedHFMD outbreak hit mainland China, resulting in �480,000cases nationwide, �120 fatal cases, and great economic losses(41). Thus far, no prophylactic or therapeutic method is avail-able to treat HFMD (43). These urgent issues and the poten-tial for an HFMD pandemic in the future prompted us to
exploit a more effective approach to combat these highlypathogenic viruses.
Both EV71 and CVA16 belong to the genus Enterovirus inthe family Picornaviridae (30). Like other members of the fam-ily, both viruses contain a genome of single-stranded, positive-sense RNA with a single open reading frame (ORF) encodinga large polyprotein precursor. In infected cells, this polyproteinis further cleaved into four structural (Vp1 to Vp4) and sevennonstructural (2A to 3D) proteins via the virus-encoded 2Aand 3C proteases. Upon translation of the polyprotein, the 2Aprotease automatically cleaves the joining sequence betweenVp1 and 2A. However, 3C is the main protease, because it isresponsible for the cleavage of the other eight junction siteswithin the remainder of the polyprotein (30). In addition, the3C protease also acts as a constituent of the replication com-plex via its binding to the 5� untranslated region (UTR) of theviral genomic RNA (32). There are also reports demonstratingthat the EV71 3C facilitates progeny virus production andhelps the virus evade host antiviral immunity by interactionwith or cleavage of host factors (22, 37). The pivotal roles ofthe 3C protease in the life cycles of EV71 and CVA16 make itan ideal target for anti-HFMD drug design.
Rupintrivir (also referred to as AG7088) is a drug that wasinitially designed as a specific inhibitor of the human rhinovirus(HRV) 3C protease but was later found to exhibit broad-
* Corresponding author. Mailing address: No. 1 West Beichen Road,Chaoyang District, Beijing 100101, China. Phone: 86-10-64807688. Fax:86-10-64807882. E-mail: [email protected] or [email protected].
� Published ahead of print on 27 July 2011.
10319
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from

spectrum antiviral activity against other members of the familyPicornaviridae (6, 23, 27). Compared to its extremely highpotency against HRV 3C, the compound exhibits nearly 2orders of magnitude lower inhibition activity toward 3C pro-teases from EV71 and CVA16 (21, 36). Therefore, structure-based modifications of rupintrivir are urgently required to gen-erate more specific and effective inhibitors of EV71/CVA163C, which necessitates assistance from high-resolution struc-tures of the free and/or the substrate-bound and/or the inhib-itor-bound enzymes. However, with the atomic structures ofEV71 proteins (such as 3C and 3D RdRp) starting to beunveiled recently (12, 39), to date, only a 3-Å structure ofEV71 3C is available (12).
Here, we thoroughly characterize the 3C proteases fromEV71 and CVA16 by defining their substrate specificities andreporting a series of high-resolution structures of both en-zymes in free-, peptide-bound, or inhibitor-bound form. Thesedata enabled us to explain the substrate preferences of EV71and CVA16 3Cs for particular P4, P1, and P1� residue typesand their relative malleability for P2 amino acids. Further-more, a half-closed S2 subsite and a size-reduced S1� subsiteare revealed by the enzyme-rupintrivir complexes, which webelieve are unique to the 3C proteases of members of humanenterovirus group A. These structural features cause rupintri-vir to tilt the P1� ester group away from the enzyme afterbinding to EV71/CVA16 3C, demonstrating that the inhibitoris not as well accommodated as in HRV 3C. This explains whyrupintrivir exhibits dramatically lower efficacy for EV71 andCVA16 than for HRV. We also propose a strategy for rupin-trivir modification to more effectively inhibit EV71 andCVA16.
MATERIALS AND METHODS
Expression and purification of the 3C proteases from EV71 and CVA16. Theoriginal viral strains selected in this study are Anhui1-09-China (GenBank ac-cession no. GQ994988) for EV71 and Beijing0907 (GenBank accession no.,ACV33372) for CVA16. The DNA fragment encoding the full-length 3C pro-tease from either virus was synthesized such that the protease gene was imme-diately followed by a hexahistidine tag coding sequence (Takara Corporation).The gene was then subcloned into pET-21a via NdeI and XhoI restriction sites.
For site-directed mutagenesis, the plasmid encoding the wild-type EV71(or CVA16) 3C protease was used as a template to generate the constructcoding for the C147A mutant enzyme. The following primer pairs were used:CVA16-3C-C147A-mutant-forward, 5�-GCAGGACAGGCTGGAGGTGTG-3�; CVA16-3C-C147A-mutant-reverse, 5�-CACACCTCCAGCCTGTCCTGC-3�; EV71-3C-C147A-mutant-forward, 5�-GCAGGACAGGCTGGGGGAGTG-3�; EV71-3C-C147A-mutant-reverse, 5�-CACTCCCCCAGCCTGTCCTGC-3�.
Both mutant constructs were successfully obtained using the Phusion SiteMutagenesis Kit (NEB) according to the manufacturer’s instructions. All theconstructs made in this study were verified by direct DNA sequencing.
Each of the four proteases was expressed and purified using the followingprotocol. One microliter of the plasmid was transformed into Escherichia coliBL-21(DE3) competent cells, and cultures were grown to an optical density at600 nm (OD600) of 0.8 in LB medium (supplemented with ampicillin) andinduced with 0.1 mM IPTG (isopropyl-�-D-thiogalactopyranoside) for 5 h at37°C. The cells were harvested and resuspended in cold lysis buffer (50 mMHEPES [pH 6.5] and 150 mM NaCl) and homogenized by sonication. Cell debriswas removed by centrifugation at 16,000 rpm for 30 min. The resultant super-natant was added to Ni-nitrilotriacetic acid (NTA) resin (GE) and gently mixedat 4°C for 1 h. The resin was then collected and treated with 2.5 M NaCl in 50mM HEPES (pH 6.5) for another 5 h. This extra step disrupted the nonspecificinteraction between the 3C protease and RNA contaminants. The nonspecificcontaminants were then removed by washing the resin with 20 column volumesof lysis buffer. The 3C protease was subsequently eluted with 200 mM imidazole
in lysis buffer and concentrated to an appropriate volume. The eluate was furtherpurified by gel filtration chromatography using a Superdex 75 Hiload 16/60column (GE). The enzyme fractions were pooled and concentrated to approxi-mately 10 to 12 mg/ml in buffer containing 50 mM HEPES (pH 6.5), 150 mMNaCl, 1 mM EDTA, and 5 mM dithiothreitol (DTT).
Preparation of the peptide-enzyme, as well as rupintrivir-enzyme, complexes.The peptides used in this study were purchased from ChinaPeptides Co., Ltd.,with purity of over 95%. Each peptide was dissolved in 100% dimethyl sulfoxide(DMSO) to a final concentration of 50 mM and stored in small aliquots at �80°Cbefore use in the cleavage assay or the cocrystallization trials. Rupintrivir waspurchased from Toronto Research Chemicals Inc. and dissolved at 30 mM inDMSO.
The noncovalent peptide-enzyme complex was prepared at 1:5 molar ratio ofenzyme to peptide, since this has been shown to work well for related proteases(28, 45). Typically 150 �l of the C147A mutant 3C protease at 12 mg/ml wasmixed with 30 �l of the 50 mM peptide and incubated with rotation at 4°Covernight. This yields a complex with a 10-mg/ml protein concentration that isready for subsequent crystallization screenings.
A similar method was used to obtain the complex of rupintrivir bound toEV71/CVA16 3C, except that the enzyme: inhibitor molar ratio was set to 1:3.After an overnight incubation of 150 �l wild-type enzyme (12 mg/ml) with 30 �lrupintrivir solution (30 mM), the complex preparation still has a protein con-centration of 10 mg/ml and is immediately screened for crystallizing conditions.
Crystallization, data collection, and structure determination. All the crystalswere obtained by the hanging-drop vapor diffusion method at 18°C. For thepeptide-enzyme complexes, we screened a series of 16-mer peptides (corre-sponding to the substrate-peptides in this study) or 10-mer peptides (correspond-ing to the “P” portion of the respective substrate-peptide) to cocrystallize withthe C147A mutant proteases from either EV71 or CVA16. Finally, only peptideFAGLRQAVTQ with the CVA16 enzyme and peptide KPVLRTATVQGPSLDF with the EV71 enzyme yielded crystals. The final conditions used tocrystallize each protein preparation were as follows: (i) CVA16 3C, 0.1 MBis-Tris (pH 5.5), 0.1 M ammonium acetate, and 17% (wt/vol) polyethyleneglycol (PEG) 10000; (ii) FAGLRQAVTQ plus CVA16-C147A 3C, 0.1 MHEPES, pH 7.5, 0.2 M lithium sulfate, and 25% (wt/vol) PEG 3350; (iii) rupin-trivir plus CVA16 3C, 0.1 M sodium acetate (pH 4.6), 0.1 M magnesium chloride,and 25% (wt/vol) PEG 4000; (iv) KPVLRTATVQGPSLDF plus EV71-C147A3C, 0.1 M Tris-HCl (pH 8.5), 20 mM lithium sulfate, and 25% (wt/vol) PEG 5000monomethyl ether; and (v) rupintrivir plus EV71 3C, 0.1 M sodium acetate (pH4.6), 0.2 M ammonium sulfate, and 25% (wt/vol) PEG 4000.
Seventeen percent (vol/vol) glycerol in mother liquor worked well as a cryo-protectant for all of the crystal species in this study. For data collection, a singlecrystal was mounted on a nylon loop and flash cooled with a nitrogen gas streamat 100 K. Diffraction data for the rupintrivir plus EV71 3C crystal were collectedat beam line NE3A (wavelength, 1.0000 Å) of KEK, Japan, while data sets for therest of the crystal species were obtained with an in-house Rigaku MicroMax007rotating-anode X-ray generator equipped with an image plate detector. Raw datawere processed and scaled using HKL2000 (26).
All crystal structures were determined by the molecular-replacement method.We first solved the structure of CVA16 3C using the CVB3 3C structure (ProteinData Bank [PDB] code 2ZTY) as the search model. Then, with the newlyobtained CVA16 3C structure as the input model, the remaining structures weresolved. In each case, the initial model was obtained by MOLREP and subse-quently refined in Refmac5 (CCP4 suite) (11) using rigid-body refinement andmaximum-likelihood procedures. Then, a series of iterative cycles of manualrebuilding were performed in COOT (13) and followed by refinement withPhenix.refine (1). During the course of model building and refinement, thestereochemistry of the structure was monitored by PROCHECK (19). The de-tailed statistics are summarized in Table 1. All figures were generated usingPyMOL (http://pymol.sourceforge.net) and ESPript (15).
In vitro cleavage assay. For each peptide (natural or mutated), the cleavageassay was performed in a reaction volume of 200 �l, using 2.1 �l enzyme (10mg/ml) and 2 �l peptide (50 mM) in the reaction buffer (50 mM HEPES, pH 6.5,150 mM NaCl, 1 mM EDTA, 2 mM DTT, 10% glycerol) to yield a final con-centration of 5 �M enzyme and 500 �M peptide. Cleavage reactions wereroutinely incubated at 25°C for 5 min and then terminated by the addition of anequal volume of 2% trifluoroacetic acid. The samples were analyzed by reverse-phase high-performance liquid chromatography (HPLC) on a C18 column (4.6 by250 mm) using a 20-min, 5 to 45% linear gradient of acetonitrile in 0.1%trifluoroacetic acid. The absorbance was monitored at 215 nm. The area of theproduct peak was calculated and transformed into a product-peptide concentra-tion, based on which the efficiency of each peptide being processed by EV71/CVA16 3C could be determined. The cleavage efficiency was defined as the
10320 LU ET AL. J. VIROL.
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from
average rate within 5 min of a selected peptide being cleaved by 5 �M eitherprotease with an initial peptide concentration of 500 �M.
Enzyme inhibition assay. A fluorescent peptide with the sequence Dabcyl-KIGNTIEALFQGPPKFRE-Edans (purchased from GL BioChem [Shanghai]Ltd., with a purity of 90%) was used as the substrate for the inhibition assay. Thispeptide contains the 2C/3A junction site. In our in vitro cleavage assay, the sitewas demonstrated to be most efficiently processed by both 3C proteases. Withexcitation at 340 nm, enhanced fluorescence could be observed at 490 nm afterthe cleavage of the peptide. This enabled us to monitor the peptide cleavage inreal time.
To determine the 50% inhibitory concentrations (IC50s) of rupintrivir againstEV71/CVA16 3C, 1 �M protease, 20 �M fluorescent peptide, and gradientconcentrations of inhibitor were mixed in a buffer containing 50 mM HEPES(pH 6.5), 150 mM NaCl, 1 mM EDTA, 2 mM DTT, and 10% glycerol. The initialvelocities of the enzymatic reactions (within the first 5 min) were determined andfitted to a sigmoidal dose-response equation with nonlinear regression analysisusing the program GraphPad Prism. The data from three independent assayswere used as input for Prism to calculate the IC50 and 95% confidence intervalvalues.
Protein structure accession numbers. The atomic coordinates and structurefactors of all the structures solved in this study have been deposited in theProtein Data Bank (Table 1).
RESULTS
Cleavage specificities of EV71 and CVA16 3C proteases. Wefirst determined the cleavage efficiencies of EV71 and CVA163C proteases for the junctions derived from both viruses. Thetwo 3Cs we characterized in this study were from strainAnhui1-09-China (for EV71) and Beijing0907 (for CVA16).The whole-genome sequence for isolate Beijing0907 is notavailable; therefore, the junction sequence information for thecoxsackievirus was obtained from strain shzh01. The twoCVA16 isolates encode 3C proteases that differ from eachother at only one position (V103I).
As with other picornaviruses, the 3C proteases from bothEV71 and CVA16 are responsible for the cleavage of eightjunction sites within the respective viral polyproteins. All thesesites contain a Q/G or Q/S scissor bond (Fig. 1). According tothe nomenclature of Berger and Schechter (4), the amino acidswithin each junction site are designated “P” or “P�” residues.The newly generated C terminus after the cleavage of the
TABLE 1. Data collection and refinement statistics
Parameter
Value
CVA16 3C EV71-C147A 3C CVA16-C147A 3C �FAGLRQAVTQ
CVA16 3C �rupintrivir EV71 3C � rupintrivir
Data collectionSpace group P43 R3 P43 P43 P1Cell dimensions
a, b, c (Å) 40.6, 40.6, 99.3 74.3, 74.3, 102.6 62.5, 62.5, 110.1 40.4, 40.4, 98.7 41.3, 82.2, 98.0�, �, � (°) 90.0, 90.0, 90.0 90.0, 90.0, 120.0 90.0, 90.0, 90.0 90.0, 90.0, 90.0 89.96, 89.99, 90.13
Wavelength (Å) 1.5418 1.5418 1.5418 1.5418 1.0000Resolution (Å)a 50.0–2.2 (2.28–2.20) 50.0–2.1 (2.18–2.10) 50.0–2.4 (2.49–2.40) 50.0–1.8 (1.86–1.80) 50.0–1.7 (1.76–1.70)Rsys or Rmerge
a,b 0.073 (0.510) 0.087 (0.477) 0.157 (0.576) 0.073 (0.416) 0.052 (0.312)I/Ia 21.07 (2.36) 17.87 (2.18) 20.16 (5.07) 20.50 (2.93) 20.64 (3.18)Completeness (%)a 99.7 (99.0) 98.6 (92.7) 99.9 (100) 96.0 (86.5) 96.8 (95.6)Redundancya 4.5 (3.7) 4.7 (3.5) 13.7 (13.9) 5.2 (4.3) 3.0 (2.8)Total no. of reflections 37,064 57,833 124,527 74,110 403,163No. of unique reflections 8,199 12,238 9,106 14,147 136,411
RefinementResolution (Å) 17.3–2.2 17.6–2.1 27.5–2.4 28.6–1.8 34.5–1.7Rwork/Rfree
c 0.192/0.226 0.207/0.238 0.195/0.241 0.179/0.218 0.205/0.222No. of atoms
Protein 1,387 1,445 1,387 1,394 11,128Ligand/ion 61 44 344Water 75 104 111 179 919
B factorsProtein 38.9 42.4 26.1 27.9 30.7Ligand/ion 33.1 26.6 26.3Water 37.5 42.1 28.5 38.0 26.6
RMSDBond length (Å) 0.014 0.006 0.008 0.004 0.008Angle (°) 1.358 0.903 0.758 0.869 1.060
Ramachandran plotd
Most favored region 84.8% 84.1% 86.6 84.8% 85.3%Additionally allowed region 14.6% 14.6% 12.7 13.9% 13.2%Generously allowed region 0.7% 1.3% 0.6 1.3% 1.5%Disallowed region 0 0 0 0 0
PDB accession code 3SJ8 3SJK 3SJ9 3SJI 3SJO
a Values for the outermost resolution shell are given in parentheses.b Rmerge �i�hkl � Ii � �I� �/�i�hklIi, where Ii is the observed intensity and �I� is the average intensity from multiple measurements.c Rwork � � � Fo � � � Fc � �/� � Fo �, where Fo and Fc are the structure-factor amplitudes from the data and the model, respectively. Rfree is the R factor for a subset
(5%) of reflections that was selected prior to refinement calculations and was not included in the refinement.d Ramachandran plots were generated by using the program PROCHECK.
VOL. 85, 2011 COMPLEX STRUCTURES OF EV71 AND CVA16 3Cs 10321
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from

scissor bond is denoted P1, preceded by the P2, P3, etc., resi-dues, and the N terminus yielded by cleavage is denoted P1�,followed by the P2�, P3�, etc., residues. Accordingly, those siteswithin the 3C protease that accommodate substrate “P” or“P�” residues are designated “S” or “S�” subsites. Overall,conservation in sequence at each site between the EV71 andCVA16 viruses was observed for the junctions within 2A to 2Cand 3A to 3C of the viral polyprotein, whereas the joiningsequences are relatively discrepant for the Vp2/Vp3, Vp3/Vp1,and 3C/3D junctions. Therefore, 11 peptides (P10 to P6�) cov-ering all eight cleavage sites within the polyprotein precursorsof both viruses were synthesized and investigated for theirsusceptibilities to 3C cleavage (Table 2).
On the whole, for each substrate peptide (SP) tested, theproteases exhibit similar cleavage efficiencies, which is consis-tent with the high sequence identity between the two enzymes(94%). Of the 11 SPs, the one that could be most efficientlyprocessed was SP-7, which corresponds to the 2C/3A junctionsite, indicating fast and effective separation of the P3 regionfrom the P2 region of the viral polyprotein. Both enzymes alsoshow high enzymatic activities toward peptides SP-1, -9, -10,and -11, corresponding to Vp2/Vp3 (EV71), 3B/3C (CVA16),3C/3D (EV71), and 3C/3D (CVA16) junctions, respectively.Moderate proteolytic efficiency was observed for peptide SP-8,representing the sequence joining coxsackievirus 3A and 3Bsubunits. Finally, the substrates to be least processed are thosepeptides representing the junction sites within the P2 region ofthe polyprotein precursor, including SP-5 (2A/2B of CVA16)and SP-6 (2B/2C of CVA16), as well as SP-2; the sequencelinks the Vp2 and Vp3 subunits in CVA16 (Table 2). We failedto determine the cleavage efficiencies of both proteases towardpeptides SP-3 and SP-4 due to severe dissolving problems forthe two peptides. Still, it was quite unexpected that the Vp2/
Vp3 site of EV71 was much more (about 17-fold) efficientlycleaved than that of CVA16.
To further elucidate the substrate specificities of the EV71and CVA16 3Cs, a series of mutational peptides (MPs) withsubstitutions for amino acids in the P6-P1� region of SP-1 weretested in vitro. The “P-side” portion of SP-1 was also success-fully cocrystallized with the CVA16 enzyme; therefore, thecleavage result for these MPs is presented alongside a descrip-tion of the substrate-enzyme complex structure. Overall, it isnotable that both EV71 and CVA16 3Cs exhibit marked pref-erence for P4, P1, and P1� residue types, whereas there is littlesequence conservation at the rest of the P and P� positions inthe natural cleavage sites of both proteases (Table 2). Accord-ingly, within the context of SP-1 sequence, mutations at the P4and P1� positions could dramatically reduce the rate of peptidecleavage by EV71/CVA16 3C. Variations at the P6, P5, and P2positions only moderately affect its ability to be processed byboth proteases (Table 2).
Overall structures of CVA16 3C and EV71-C147A 3C. Asexpected, the CVA16 3C protease also forms a chymotrypsin-like fold, which is a typical feature of all reported structures of3C/3C-like proteases (3, 20, 23, 24, 35, 42). The CVA16 3Cstructure contains 179 amino acids from residues L4 to E182,forming two domains. The first domain is largely composed ofa 7-stranded �-barrel structure (aI to gI), surrounded by sur-face loops. The second domain also contains a compact barrelcore, which is composed of seven �-strands (aII to cII and fIIto iII) arranged in an antiparallel manner. This core structureis further flanked by the most N-terminal �-helix (helix A) anda strand-loop-strand �-ribbon structure (dII-eII), which (ac-cording to the previous reports on other 3C proteases) shouldplay an important role in substrate recognition by the enzyme(34, 45). Overall, the two domains are connected via a long
FIG. 1. Overview of the domain organization within the viral polyproteins of EV71 and CVA16. The single ORF of EV71 or CVA16 could befurther divided into three regions (P1, P2, and P3). The four structural subunits encoded by the P1 region and the seven nonstructural proteinsencoded by the P2 and P3 regions are colored cyan, light pink, and gray, respectively. The eight cleavage junction sites that are to be processedby the 3C protease are indicated, with each joining sequence spanning from the P10 to P6� residues listed above or below the schematic boxesrepresenting viral protein subunits Vp4 to 3D. The scissor bonds are highlighted in red.
10322 LU ET AL. J. VIROL.
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from
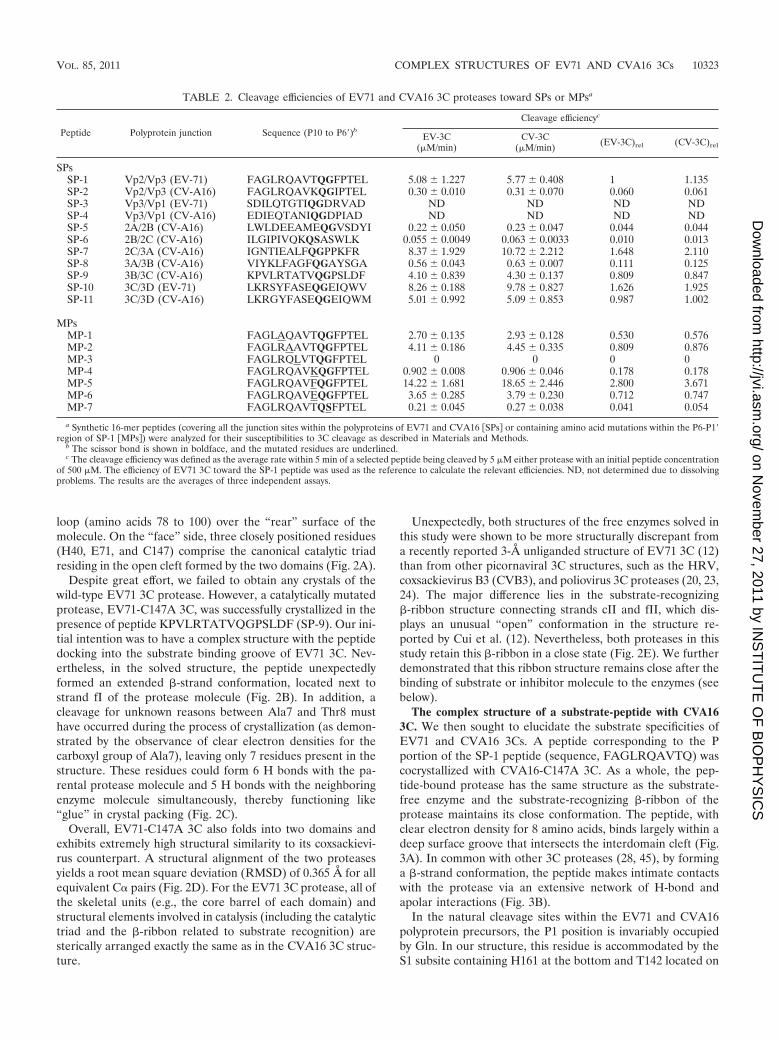
loop (amino acids 78 to 100) over the “rear” surface of themolecule. On the “face” side, three closely positioned residues(H40, E71, and C147) comprise the canonical catalytic triadresiding in the open cleft formed by the two domains (Fig. 2A).
Despite great effort, we failed to obtain any crystals of thewild-type EV71 3C protease. However, a catalytically mutatedprotease, EV71-C147A 3C, was successfully crystallized in thepresence of peptide KPVLRTATVQGPSLDF (SP-9). Our ini-tial intention was to have a complex structure with the peptidedocking into the substrate binding groove of EV71 3C. Nev-ertheless, in the solved structure, the peptide unexpectedlyformed an extended �-strand conformation, located next tostrand fI of the protease molecule (Fig. 2B). In addition, acleavage for unknown reasons between Ala7 and Thr8 musthave occurred during the process of crystallization (as demon-strated by the observance of clear electron densities for thecarboxyl group of Ala7), leaving only 7 residues present in thestructure. These residues could form 6 H bonds with the pa-rental protease molecule and 5 H bonds with the neighboringenzyme molecule simultaneously, thereby functioning like“glue” in crystal packing (Fig. 2C).
Overall, EV71-C147A 3C also folds into two domains andexhibits extremely high structural similarity to its coxsackievi-rus counterpart. A structural alignment of the two proteasesyields a root mean square deviation (RMSD) of 0.365 Å for allequivalent C� pairs (Fig. 2D). For the EV71 3C protease, all ofthe skeletal units (e.g., the core barrel of each domain) andstructural elements involved in catalysis (including the catalytictriad and the �-ribbon related to substrate recognition) aresterically arranged exactly the same as in the CVA16 3C struc-ture.
Unexpectedly, both structures of the free enzymes solved inthis study were shown to be more structurally discrepant froma recently reported 3-Å unliganded structure of EV71 3C (12)than from other picornaviral 3C structures, such as the HRV,coxsackievirus B3 (CVB3), and poliovirus 3C proteases (20, 23,24). The major difference lies in the substrate-recognizing�-ribbon structure connecting strands cII and fII, which dis-plays an unusual “open” conformation in the structure re-ported by Cui et al. (12). Nevertheless, both proteases in thisstudy retain this �-ribbon in a close state (Fig. 2E). We furtherdemonstrated that this ribbon structure remains close after thebinding of substrate or inhibitor molecule to the enzymes (seebelow).
The complex structure of a substrate-peptide with CVA163C. We then sought to elucidate the substrate specificities ofEV71 and CVA16 3Cs. A peptide corresponding to the Pportion of the SP-1 peptide (sequence, FAGLRQAVTQ) wascocrystallized with CVA16-C147A 3C. As a whole, the pep-tide-bound protease has the same structure as the substrate-free enzyme and the substrate-recognizing �-ribbon of theprotease maintains its close conformation. The peptide, withclear electron density for 8 amino acids, binds largely within adeep surface groove that intersects the interdomain cleft (Fig.3A). In common with other 3C proteases (28, 45), by forminga �-strand conformation, the peptide makes intimate contactswith the protease via an extensive network of H-bond andapolar interactions (Fig. 3B).
In the natural cleavage sites within the EV71 and CVA16polyprotein precursors, the P1 position is invariably occupiedby Gln. In our structure, this residue is accommodated by theS1 subsite containing H161 at the bottom and T142 located on
TABLE 2. Cleavage efficiencies of EV71 and CVA16 3C proteases toward SPs or MPsa
Peptide Polyprotein junction Sequence (P10 to P6�)b
Cleavage efficiencyc
EV-3C(�M/min)
CV-3C(�M/min) (EV-3C)rel (CV-3C)rel
SPsSP-1 Vp2/Vp3 (EV-71) FAGLRQAVTQGFPTEL 5.08 1.227 5.77 0.408 1 1.135SP-2 Vp2/Vp3 (CV-A16) FAGLRQAVKQGIPTEL 0.30 0.010 0.31 0.070 0.060 0.061SP-3 Vp3/Vp1 (EV-71) SDILQTGTIQGDRVAD ND ND ND NDSP-4 Vp3/Vp1 (CV-A16) EDIEQTANIQGDPIAD ND ND ND NDSP-5 2A/2B (CV-A16) LWLDEEAMEQGVSDYI 0.22 0.050 0.23 0.047 0.044 0.044SP-6 2B/2C (CV-A16) ILGIPIVQKQSASWLK 0.055 0.0049 0.063 0.0033 0.010 0.013SP-7 2C/3A (CV-A16) IGNTIEALFQGPPKFR 8.37 1.929 10.72 2.212 1.648 2.110SP-8 3A/3B (CV-A16) VIYKLFAGFQGAYSGA 0.56 0.043 0.63 0.007 0.111 0.125SP-9 3B/3C (CV-A16) KPVLRTATVQGPSLDF 4.10 0.839 4.30 0.137 0.809 0.847SP-10 3C/3D (EV-71) LKRSYFASEQGEIQWV 8.26 0.188 9.78 0.827 1.626 1.925SP-11 3C/3D (CV-A16) LKRGYFASEQGEIQWM 5.01 0.992 5.09 0.853 0.987 1.002
MPsMP-1 FAGLAQAVTQGFPTEL 2.70 0.135 2.93 0.128 0.530 0.576MP-2 FAGLRAAVTQGFPTEL 4.11 0.186 4.45 0.335 0.809 0.876MP-3 FAGLRQLVTQGFPTEL 0 0 0 0MP-4 FAGLRQAVKQGFPTEL 0.902 0.008 0.906 0.046 0.178 0.178MP-5 FAGLRQAVFQGFPTEL 14.22 1.681 18.65 2.446 2.800 3.671MP-6 FAGLRQAVEQGFPTEL 3.65 0.285 3.79 0.230 0.712 0.747MP-7 FAGLRQAVTQSFPTEL 0.21 0.045 0.27 0.038 0.041 0.054
a Synthetic 16-mer peptides (covering all the junction sites within the polyproteins of EV71 and CVA16 �SPs� or containing amino acid mutations within the P6-P1�region of SP-1 �MPs�) were analyzed for their susceptibilities to 3C cleavage as described in Materials and Methods.
b The scissor bond is shown in boldface, and the mutated residues are underlined.c The cleavage efficiency was defined as the average rate within 5 min of a selected peptide being cleaved by 5 �M either protease with an initial peptide concentration
of 500 �M. The efficiency of EV71 3C toward the SP-1 peptide was used as the reference to calculate the relevant efficiencies. ND, not determined due to dissolvingproblems. The results are the averages of three independent assays.
VOL. 85, 2011 COMPLEX STRUCTURES OF EV71 AND CVA16 3Cs 10323
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from
one side wall of the pocket. These 2 residues in total formthree strong hydrogen bonds (2.8 to 2.9Å) with the side chainof P1-Gln in the substrate (Fig. 3B). Therefore, the S1 pocketdisplays a character of extremely good chemical complemen-tarity to the glutamate residue.
In contrast to the absolute conservatism for P1-Gln, the P2residues of the 3C substrates from both viruses are rathervariable (Table 2). Our cleavage assay with peptides containingmutations at P2 showed that amino acids with various sidechains (acidic, basic, aliphatic, and polar) are acceptable. Con-
sistent with this, a pocket with enough size to accommodatevarious residue side chains is observed in our structure (Fig.3A). The P2-Thr, located at the entry of the S2 subsite, leavesmost of the specificity pocket unoccupied and mainly hydro-phobically interacts with L127 and I162 of the enzyme (Fig.3B). Despite the malleability of the P2 residue, both proteasesfavor aliphatic or aromatic side chains at this specific position.Replacement of P2-Thr by Phe can increase the rate of cleav-age by about 3-fold, whereas introduction of Glu or Lys at P2results in a reduction of the cleavage rate. It is notable that a
FIG. 2. Overall structures of the 3C proteases from CVA16 and EV71. (A) Cartoon representation of the structure of CVA16 3C. Domain I(strands aI to gI) and domain II (strands aII to iII) are colored green and magenta, respectively. The �-helices are marked A to D according totheir occurrence along the primary structure. The long loop over the “rear” surface connecting domains I and II is highlighted in blue. The catalytictriad, which is composed of H40, E71, and C147, is shown as cyan sticks. The N and C termini are indicated. (B) Structure of the C147A mutantenzyme from EV71 (EV71-C147A 3C). The structural elements involved are colored and labeled as in the CVA16 3C protease. The extra strandthat is located next to strand fI of the protease molecule is indicated. (C) In the solved structure of EV71-C147A 3C, residues KPVLRTA (orange)of the extra strand could form 6 hydrogen bonds with E61, V63, and R134 of the parental (green) protease molecule and could form 5 H-bondswith D99, S111, M112, and F113 of the neighboring (magenta) enzyme molecule. The dashed lines indicate H bonds, and the residues involvedare labeled. (D) Superimposition of the 3Cs from CVA16 (cyan) and EV71 (orange). Alignment of the two proteases yields an RMSD of 0.365Å for all equivalent C� pairs, demonstrating strong similarities between the enzymes. The well-aligned catalytic triads are highlighted. (E) Su-perimposition of the free-enzyme structures (cyan for CVA16 3C, orange for EV71 3C) in this study on a recently reported 3-Å unligandedstructure of EV71 3C (green). The substrate-recognizing �-ribbon, which exhibits major differences, and the well-aligned cII and fII strands arehighlighted.
10324 LU ET AL. J. VIROL.
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from
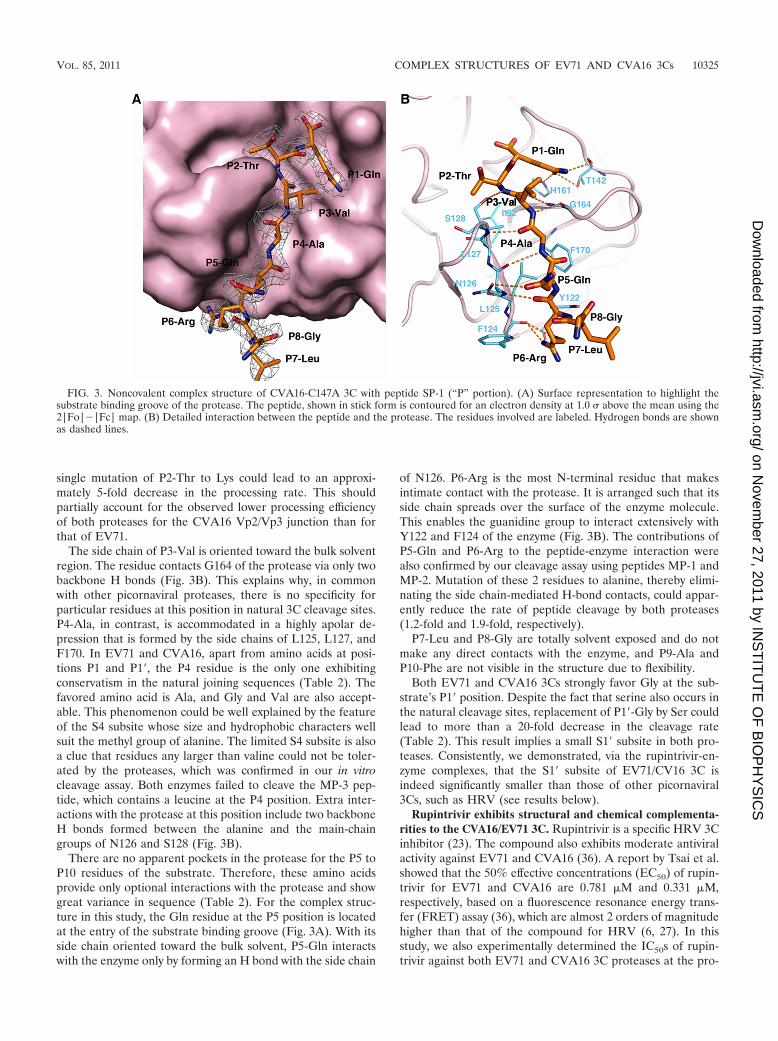
single mutation of P2-Thr to Lys could lead to an approxi-mately 5-fold decrease in the processing rate. This shouldpartially account for the observed lower processing efficiencyof both proteases for the CVA16 Vp2/Vp3 junction than forthat of EV71.
The side chain of P3-Val is oriented toward the bulk solventregion. The residue contacts G164 of the protease via only twobackbone H bonds (Fig. 3B). This explains why, in commonwith other picornaviral proteases, there is no specificity forparticular residues at this position in natural 3C cleavage sites.P4-Ala, in contrast, is accommodated in a highly apolar de-pression that is formed by the side chains of L125, L127, andF170. In EV71 and CVA16, apart from amino acids at posi-tions P1 and P1�, the P4 residue is the only one exhibitingconservatism in the natural joining sequences (Table 2). Thefavored amino acid is Ala, and Gly and Val are also accept-able. This phenomenon could be well explained by the featureof the S4 subsite whose size and hydrophobic characters wellsuit the methyl group of alanine. The limited S4 subsite is alsoa clue that residues any larger than valine could not be toler-ated by the proteases, which was confirmed in our in vitrocleavage assay. Both enzymes failed to cleave the MP-3 pep-tide, which contains a leucine at the P4 position. Extra inter-actions with the protease at this position include two backboneH bonds formed between the alanine and the main-chaingroups of N126 and S128 (Fig. 3B).
There are no apparent pockets in the protease for the P5 toP10 residues of the substrate. Therefore, these amino acidsprovide only optional interactions with the protease and showgreat variance in sequence (Table 2). For the complex struc-ture in this study, the Gln residue at the P5 position is locatedat the entry of the substrate binding groove (Fig. 3A). With itsside chain oriented toward the bulk solvent, P5-Gln interactswith the enzyme only by forming an H bond with the side chain
of N126. P6-Arg is the most N-terminal residue that makesintimate contact with the protease. It is arranged such that itsside chain spreads over the surface of the enzyme molecule.This enables the guanidine group to interact extensively withY122 and F124 of the enzyme (Fig. 3B). The contributions ofP5-Gln and P6-Arg to the peptide-enzyme interaction werealso confirmed by our cleavage assay using peptides MP-1 andMP-2. Mutation of these 2 residues to alanine, thereby elimi-nating the side chain-mediated H-bond contacts, could appar-ently reduce the rate of peptide cleavage by both proteases(1.2-fold and 1.9-fold, respectively).
P7-Leu and P8-Gly are totally solvent exposed and do notmake any direct contacts with the enzyme, and P9-Ala andP10-Phe are not visible in the structure due to flexibility.
Both EV71 and CVA16 3Cs strongly favor Gly at the sub-strate’s P1� position. Despite the fact that serine also occurs inthe natural cleavage sites, replacement of P1�-Gly by Ser couldlead to more than a 20-fold decrease in the cleavage rate(Table 2). This result implies a small S1� subsite in both pro-teases. Consistently, we demonstrated, via the rupintrivir-en-zyme complexes, that the S1� subsite of EV71/CV16 3C isindeed significantly smaller than those of other picornaviral3Cs, such as HRV (see results below).
Rupintrivir exhibits structural and chemical complementa-rities to the CVA16/EV71 3C. Rupintrivir is a specific HRV 3Cinhibitor (23). The compound also exhibits moderate antiviralactivity against EV71 and CVA16 (36). A report by Tsai et al.showed that the 50% effective concentrations (EC50) of rupin-trivir for EV71 and CVA16 are 0.781 �M and 0.331 �M,respectively, based on a fluorescence resonance energy trans-fer (FRET) assay (36), which are almost 2 orders of magnitudehigher than that of the compound for HRV (6, 27). In thisstudy, we also experimentally determined the IC50s of rupin-trivir against both EV71 and CVA16 3C proteases at the pro-
FIG. 3. Noncovalent complex structure of CVA16-C147A 3C with peptide SP-1 (“P” portion). (A) Surface representation to highlight thesubstrate binding groove of the protease. The peptide, shown in stick form is contoured for an electron density at 1.0 above the mean using the2|Fo|�|Fc| map. (B) Detailed interaction between the peptide and the protease. The residues involved are labeled. Hydrogen bonds are shownas dashed lines.
VOL. 85, 2011 COMPLEX STRUCTURES OF EV71 AND CVA16 3Cs 10325
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from

tein level using a fluorescent substrate peptide (for details, seeMaterials and Methods). These enzyme-based values weremeasured at 1.65 and 2.06 �M for EV71 3C and CVA16 3C,respectively (Table 3). The enzyme-based data obtained in thisstudy and the cell-based results of Tsai et al. all indicate potentinhibition by rupintrivir of EV71/CVA16 3C, although not asefficient as the compound for HRV 3C. To explore the specificbinding mode of rupintrivir to EV71/CVA16 3C and, thereby,to provide a structural basis for the development of EV71/CVA16 3C-specific inhibitors, we also determined the struc-tures of both proteases in complex with rupintrivir.
Overall, rupintrivir can be described as a peptide mimicinhibitor containing a lactam at P1, a fluorophenylalanine atP2, a Val at P3, a 5-methyl-3-isoxazole at P4, and an �,�-unsaturated ester at P1� (Fig. 4) (23). In both proteases, thecompound is accommodated in the substrate binding groovewith a covalent linkage to the nucleophilic Cys147 (Fig. 5A andB). Consistent with the extremely high structural similaritiesbetween the EV71 and CVA16 3Cs, rupintrivir exhibits thesame binding mode in both structures. Superimposition ofthe two complex structures yields an RMSD of 0.221 Å for theequivalent C� atoms of the protein molecules and 0.236 Å forall atoms of the inhibitor moiety (Fig. 5C). In light of theseobserved similarities, the structure of rupintrivir bound toCVA16 3C was used as a representative for subsequent ana-lyses.
As shown in Fig. 6, rupintrivir binds to CVA16 3C via anetwork of extensive H bonds and hydrophobic interactions.These include three hydrogen bonds provided by the P4 isoxa-zole group, two by the P3-Ala, four at the P1 position, and oneby the terminal P1� ester group. Conversely, the P2 group andthe aliphatic portion of P4 mainly contribute to the enzyme-inhibitor interaction by contacting the hydrophobic S2 and S4subsites of the protease. In addition, the fluoride at the tip ofP2 fluorophenylalanine could also have polar interaction withR39 of the protein (see results below).
The detailed interactions, including H bonds and hydropho-bic contacts, between rupintrivir and EV71 3C are exactly thesame as those observed in the CVA16 3C with compoundstructure. Therefore, overall, rupintrivir exhibits structural andchemical complementarities to both CVA16 and EV71 3Cs.
Rupintrivir is not as well accommodated in CVA16/EV71 3Cas in HRV 3C. Compared to a previously reported structure ofrupintrivir in HRV 3C (23), the compound binds to CVA16and HRV 3Cs with similar modes. Superimposition of the twocomplex structures reveals well-aligned enzyme entities, as wellas inhibitor molecules at the P4, P3, and P1 positions, wheremost (9 out of 10) of the H-bond contacts with the enzyme arelocated. However, great discrepancies were observed for theother two groups of the compound in the CVA16 3C structure:(i) the P2 group makes a rotary movement (around atom C-5as the rotating axis) of �1.7 Å out of the S2 pocket and (ii) the
P1� group adopts a tilted conformation, exposing its ester chainto the solvent (Fig. 7A). The latter difference is extremelyunexpected, because the P1� group could be well accommo-dated in the S1� subsite of HRV 3C. By “lying down,” rupin-trivir’s P1� group forms two H bonds with the oxyanion hole(main-chain amide of G145 and C147) of HRV 3C. However,in the context of the 3C protease from CVA16, the tilted P1�group of rupintrivir supports only one H-bond interaction withthe enzyme (Fig. 7B). Therefore, despite the complementarityof rupintrivir to CVA16/EV71 3C, the compound binds the 3Cprotease of HRV more stably, well explaining the observedlower efficacy of rupintrivir for EV71/CVA16 than for HRV(23). Next, we set out to explore the structural basis for theseobserved differences by comparison of the S2 and S1� subsitesin the HRV and CVA16/EV71 3C proteases.
Structural basis for the modification of rupintrivir. Withinthe CVA16-3C-rupintrivir structure, it is noteworthy that aresidue with a long side chain (K130) is located at the distalend of the S2 subsite. With its side chain atoms half closing thepocket, the P2 group of rupintrivir could not be deeply buriedin the S2 subsite of CVA16 3C and thus adopted a relativelytilted conformation. This tilted P2-fluorophenylalanine wasfurther stabilized via extensive polar interactions between thegroup and R39 of CVA16 3C, a residue located at the top rightcorner of the S2 subsite. These include a direct polar attractionof the P2 group from the R39 residue (�3.6 Å) and an indirectinteraction between the two entities via a water-mediated H-bond network (Fig. 8A). However, in the rhinoviral protease,the R39 and K130 residues are replaced by T39 and N130,respectively. Not only do these substitutions leave the S2 sub-site fully open to the solvent, but they also disrupt the potentialcontact of T39 with the rupintrivir fluoride. With a pocket fullyopen to the solvent, the P2 group of rupintrivir is easily burieddeep in the S2 subsite of HRV 3C. This lying-down conforma-tion for the inhibitor’s P2 group was further stabilized via aweak H bond (3.5 Å) contributed by a bottom-residing glu-tamic acid (E71) (Fig. 8B).
For the S1� subsite in both CVA16 and HRV 3Cs, thespecificity pocket is formed by the backbone atoms of theoxyanion hole residues (G145 to C147), the main-chain atomsof H24/K24, and the side chain of F25. Superimposition of thesubsites reveals that these residues are well aligned, except forthe H24/K24 pair, where the carbonyl group of H24 in CVA163C is moved �0.9 Å into the pocket relative to that of K24 inHRV 3C (Fig. 8C). This results in a smaller S1� pocket inCVA16 3C than in the rhinoviral enzyme. Further analysis
FIG. 4. Chemical structure of rupintrivir. Overall, rupintrivir couldbe described as a peptide-mimic inhibitor containing a lactam at P1, afluorophenylalanine at P2, a Val at P3, a 5-methyl-3-isoxazole at P4,and an �,�-unsaturated ester at P1�. The five groups (P4 to P1�) of theinhibitor are indicated.
TABLE 3. IC50s of rupintrivir against EV71 3C and CVA16 3C
Protease IC50 (�M) 95% Confidenceinterval (�M)
EV71 3C 1.65 1.56–1.75CVA16 3C 2.06 1.93–2.20
10326 LU ET AL. J. VIROL.
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from

shows that residues Q19, E107, and H108 are positioned insteric proximity to H24 in CVA16 3C. By surrounding the sidechain of H24 like a cap, these residues confine the histidineresidue within a limited space. They also form three hydrogenbonds with H24 and thereby help stabilize its observed confor-mation. However, in HRV 3C, these 3 residues are replaced byT19, N107, and Q108 (respectively), and due to a differentorientation of the aII-bII loop (the loop connecting the aII andbII strands), residue K24 is easily exposed to solvent (Fig. 8D).
The confinement arising from residues Q19, E107, and H108 inCVA16 3C might cause a 0.9-Å shift for the main-chain car-bonyl group of residue 24 relative to that of the HRV enzyme.
It should be noted that the inhibitor’s P2 group is uplifted �1.7Å compared to that in HRV 3C. Taking the rigidity of the druginto account, this shift should be transferred to the opposite sideof the inhibitor, moving the P1 and P1� groups toward the body ofthe enzyme. In support of this, the compound indeed inserts its P1lactam deeper into the S1 subsite of CVA16 3C. For the S1�subsite, which is smaller and lacks malleability, the ester chain atthe P1� position would have conflicted with CVA16 3C if it wasretained in the conformation observed in the HRV 3C-rupintrivircomplex. Together, these factors force the P1� chain of the inhib-itor to adopt an alternative (in this case tilted) conformation whenit binds to CVA16 3C.
Identical rupintrivir-enzyme interaction modes at the S2 and
FIG. 5. Rupintrivir exhibits the same mode of binding to the EV71 and CVA16 3C proteases. (A and B) Overall structure of the enzyme-inhibitor complex. The structures of rupintrivir bound to the 3C proteases from CVA16 and EV71 are shown in panels A and B, respectively.Rupintrivir is in orange, and Cys147 is cyan. The covalent bond formed between the inhibitor and the protease is highlighted with an arrow. Theinhibitor is shown in stick representation with a 1.0- contoured map. There are 8 molecules per asymmetric unit in the structure of rupintrivirbound to EV71 3C. These molecules have essentially the same structure, and molecule A was selected for depiction or comparison. (C) Super-imposition of the two complex structures reveals the same binding mode of rupintrivir to the EV71 (green) and CVA16 (cyan) 3C proteases. Theinhibitor moieties in the EV71 and CVA16 enzymes are shown as orange and magenta sticks, respectively.
FIG. 6. Detailed interactions between rupintrivir and CVA16 3C.H-bond interactions are shown as dashed lines. The five groups of theinhibitor are labeled P1� to P4 and colored orange. The residues in theenzyme that interact with rupintrivir are shown as thin cyan sticks andare labeled.
FIG. 7. Rupintrivir is not as well accommodated in CVA16 3C as inHRV 3C. (A) Comparison of rupintrivir in CVA16 (magenta) andHRV (cyan) 3C proteases. The 1.7-Å uplift of the P2 group and thetilted P1� ester chain of the inhibitor in CVA16 3C relative to those ofthe compound in HRV 3C are indicated. (B) Contributions of the P1�group to the enzyme-inhibitor interaction. Two H bonds are formedbetween the ester group and the protein when the compound binds toHRV 3C (magenta), whereas only one is observed in the case ofCVA16 3C (cyan). The inhibitor moiety in the CVA16 and HRVenzymes is shown as green and orange sticks, respectively.
VOL. 85, 2011 COMPLEX STRUCTURES OF EV71 AND CVA16 3Cs 10327
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from

S1� subsites were observed in the structure of the inhibitor inEV71 3C. Therefore, the half-closed S2 and size-reduced S1�subsites are also typical features of the EV71 enzyme. Webelieve that the special characters of the S2 and S1� subsiteswithin the EV71/CVA16 enzyme together cause rupintrivirto tilt both its P2 and P1� groups and that the less avidbinding of rupintrivir to EV71/CVA16 3C is the result of thesubpar fit of the compound to the protease at both the S2and S1� subsites.
DISCUSSION
EV71 and CVA16 are phylogenetically closely related (25).Overall, the 3C proteases from different EV71 and CVA16strains display 90 to 98% sequence identity. Accordingly, the
two proteases in this study exhibited similar in vitro cleavagecapabilities and did not show a preference for peptides derivedfrom their respective viruses. It was unexpected that the Vp2/Vp3 junction of EV71 is much more efficiently processed thanthat of CVA16, especially in the context where the nonstruc-tural junctions (2A-2C and 3A-3D) of both viruses are eachcleaved in parallel by EV71/CVA16 3C (Table 2). Despite thefact that we failed to determine the cleavage rates of the twoproteases for peptides representing Vp3/Vp1 junctions ofEV71 and CVA16, a more effective in vivo processing of thecapsid region for EV71 than for CVA16 could still be ex-pected. This could lead to fast assembly and production ofEV71 progeny virus in infected individuals. Supportive evi-dence comes from a report by Zhang et al. (44), who demon-strated that CVA16 might experience a lower evolution rate
FIG. 8. The half-closed S2 subsite and an S1� pocket with reduced size in CVA16 3C. (A and B) Surface representation of the S2 subsites inthe CVA16 (A) and HRV (B) 3C proteases. The residues referred to in the text are presented as thin sticks and are labeled. The P2 groups ofrupintrivir in the CVA16 and HRV enzymes are shown as green and orange sticks, respectively. The dashed lines indicate H-bond or polarinteractions. The red balls represent water molecules. (C) Superimposition of the 3C proteases from CVA16 (magenta) and HRV (cyan) at theS1� subsite. The residues constituting the specificity pocket in the respective proteases are indicated. The 0.9-Å shift observed for the main-chaincarbonyl of His24 in CVA16 3C relative to that of the corresponding residue (Lys24) in the rhinoviral protease is highlighted. The P1� group ofrupintrivir, which exhibits great conformational differences in the respective proteases, is shown in stick representation (green for rupintrivir inCVA16-3C and orange for the compound in HRV-3C). (D) Structural features leading to the reduced size of the CVA16 3C S1� subsite. Theresidues involved and the aII-bII loop (the loop connecting the aII and bII strands) are indicated.
10328 LU ET AL. J. VIROL.
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from
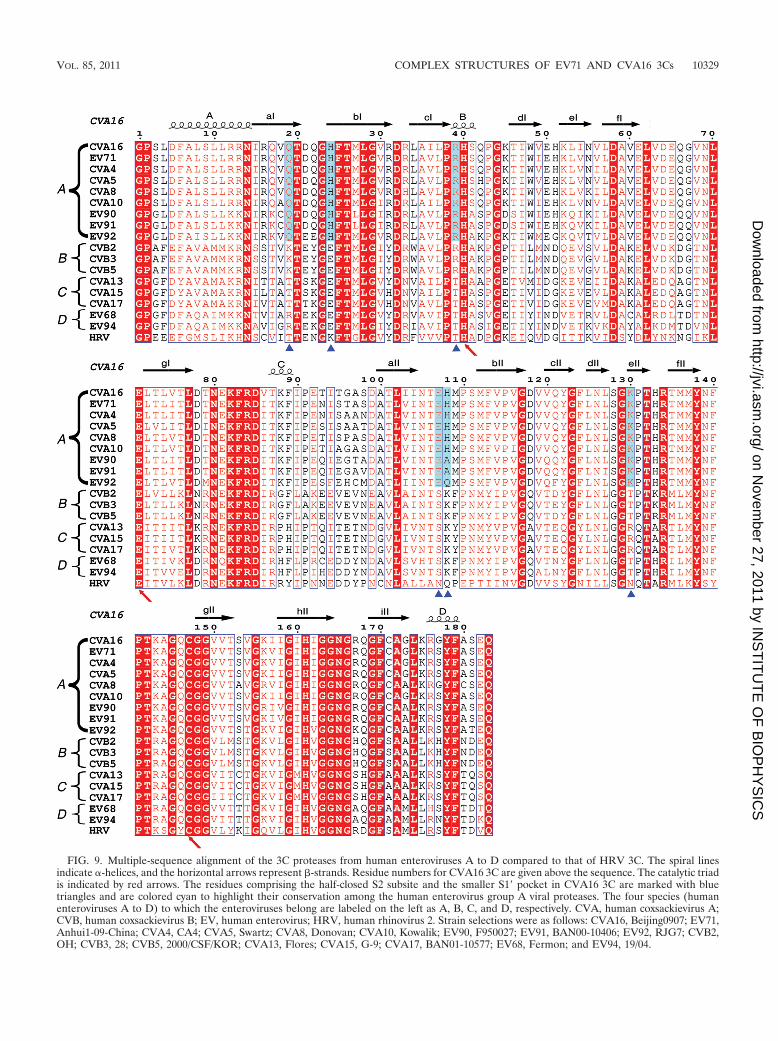
FIG. 9. Multiple-sequence alignment of the 3C proteases from human enteroviruses A to D compared to that of HRV 3C. The spiral linesindicate �-helices, and the horizontal arrows represent �-strands. Residue numbers for CVA16 3C are given above the sequence. The catalytic triadis indicated by red arrows. The residues comprising the half-closed S2 subsite and the smaller S1� pocket in CVA16 3C are marked with bluetriangles and are colored cyan to highlight their conservation among the human enterovirus group A viral proteases. The four species (humanenteroviruses A to D) to which the enteroviruses belong are labeled on the left as A, B, C, and D, respectively. CVA, human coxsackievirus A;CVB, human coxsackievirus B; EV, human enterovirus; HRV, human rhinovirus 2. Strain selections were as follows: CVA16, Beijing0907; EV71,Anhui1-09-China; CVA4, CA4; CVA5, Swartz; CVA8, Donovan; CVA10, Kowalik; EV90, F950027; EV91, BAN00-10406; EV92, RJG7; CVB2,OH; CVB3, 28; CVB5, 2000/CSF/KOR; CVA13, Flores; CVA15, G-9; CVA17, BAN01-10577; EV68, Fermon; and EV94, 19/04.
VOL. 85, 2011 COMPLEX STRUCTURES OF EV71 AND CVA16 3Cs 10329
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from

than EV71. Further experiments are needed to confirm thisinference.
It is also noteworthy that a single mutation of P2-Thr toP2-Lys within the SP-1 peptide could lead to a decrease incleavage efficiency of about 6-fold (Table 2). In terms of the17-fold difference in the 3C processing rates between peptidesSP-1 and SP-2, which differ from each other at only two posi-tions (P2 and P2�), it is a natural inference that an F-to-Imutation at the P2� position should also affect the cleavagerate of the peptide by about 3-fold.
We also reported the rupintrivir-enzyme complex structuresin this study. Compared to rupintrivir in HRV 3C, the P1�group of the inhibitor is oriented toward the bulk solvent afterit is bound to CVA16/EV71 3C. We hypothesize that this tiltedester group orientation will decrease the binding affinity ofrupintrivir to CVA16/EV71 3C. Although the ester group caninteract with the enzyme molecule by forming one H bond, theswitch from a Z conformation (as observed in the HRV 3C-rupintrivir complex) to the tilted conformation (as observed inour structures) requires energy. In support of this, a recentreport compared the abilities of two compounds (differing onlyat the P1� position) to inhibit the EV71 3C protease anddemonstrated that substitution of the ester chain by aldehydeimproves the potency of the compound by �100-fold (18).
Based on the findings in this study, we propose that modi-fications of rupintrivir at the P2 and P1� positions will allow thecompound to bind more tightly to EV71/CVA16 3C. An optionfor P1� modification is to replace the entire ester chain withaldehyde. For the P2 group, the fluorophenylalanine chain istoo large to be accommodated by the half-closed S2 subsite.Therefore, an unmodified benzyl ring would likely fit the S2pocket better.
It is also noteworthy that the residues forming the featuredS2 and S1� subsites in our enzyme structures are highly con-served (with either no variations or one variation at H108)among the main proteases of viruses from human enterovirusgroup A but not those of other groups (e.g., human enterovirusgroups B, C, and D) (Fig. 9). A reasonable inference is that thehalf-closed S2 subsite and an S1� pocket with a reduced size arecommon characteristics of group A enteroviral 3C proteases.Moreover, apart from EV71 and CVA16, many other group Aenteroviruses, such as coxsackieviruses A4, A5, A8, and A10,also clinically cause HFMD. Consequently, we believe that thestructures presented in this study provide a solid basis for thedevelopment of 3C-based anti-HFMD virus drugs.
ACKNOWLEDGMENT
G.F.G. is a leading principal investigator of the Innovative ResearchGroup of the National Natural Science Foundation of China (NSFC)(grant no. 81021003).
REFERENCES
1. Adams, P. D., et al. 2010. PHENIX: a comprehensive Python-based systemfor macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr.66:213–221.
2. Alexander, J. P., Jr., L. Baden, M. A. Pallansch, and L. J. Anderson. 1994.Enterovirus 71 infections and neurologic disease—United States, 1977–1991.J. Infect. Dis. 169:905–908.
3. Anand, K., et al. 2002. Structure of coronavirus main proteinase revealscombination of a chymotrypsin fold with an extra alpha-helical domain.EMBO J. 21:3213–3224.
4. Berger, A., and I. Schechter. 1970. Mapping the active site of papain with theaid of peptide substrates and inhibitors. Phil. Trans. R. Soc. Lond. B Biol.Sci. 257:249–264.
5. Bible, J. M., et al. 2008. Molecular epidemiology of human enterovirus 71 inthe United Kingdom from 1998 to 2006. J. Clin. Microbiol. 46:3192–3200.
6. Binford, S. L., et al. 2005. Conservation of amino acids in human rhinovirus3C protease correlates with broad-spectrum antiviral activity of rupintrivir, anovel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Che-mother. 49:619–626.
7. Chan, K. P., et al. 2003. Epidemic hand, foot and mouth disease caused byhuman enterovirus 71, Singapore. Emerg. Infect. Dis. 9:78–85.
8. Chen, K. T., H. L. Chang, S. T. Wang, Y. T. Cheng, and J. Y. Yang. 2007.Epidemiologic features of hand-foot-mouth disease and herpangina causedby enterovirus 71 in Taiwan, 1998–2005. Pediatrics 120:e244–e252.
9. Chen, S. C., H. L. Chang, T. R. Yan, Y. T. Cheng, and K. T. Chen. 2007. Aneight-year study of epidemiologic features of enterovirus 71 infection inTaiwan. Am. J. Trop. Med. Hyg. 77:188–191.
10. Chua, K. B., et al. 2007. Genetic diversity of enterovirus 71 isolated fromcases of hand, foot and mouth disease in the 1997, 2000 and 2005 outbreaks,Peninsular Malaysia. Malays. J. Pathol. 29:69–78.
11. Collaborative Computing Project Number 4. 1994. The CCP4 suite: pro-grams for protein crystallography. Acta Crystallogr. D Biol. Crystallogr.50:760–763.
12. Cui, S., et al. 2011. Crystal structure of human enterovirus 71 3C protease.J. Mol. Biol. 408:449–461.
13. Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for moleculargraphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132.
14. Gilbert, G. L., et al. 1988. Outbreak of enterovirus 71 infection in Victoria,Australia, with a high incidence of neurologic involvement. Pediatr. Infect.Dis. J. 7:484–488.
15. Gouet, P., E. Courcelle, D. I. Stuart, and F. Metoz. 1999. ESPript: analysis ofmultiple sequence alignments in PostScript. Bioinformatics 15:305–308.
16. Ho, M., et al. 1999. An epidemic of enterovirus 71 infection in Taiwan.Taiwan Enterovirus Epidemic Working Group. N. Engl. J. Med. 341:929–935.
17. Ishimaru, Y., S. Nakano, K. Yamaoka, and S. Takami. 1980. Outbreaks ofhand, foot, and mouth disease by enterovirus 71. High incidence of compli-cation disorders of central nervous system. Arch. Dis. Child. 55:583–588.
18. Kuo, C. J., et al. 2008. Design, synthesis, and evaluation of 3C proteaseinhibitors as anti-enterovirus 71 agents. Bioorg. Med. Chem. 16:7388–7398.
19. Laskowski, R. A., M. W. Macarthur, D. S. Moss, and J. M. Thornton. 1993.PROCHECK: a program to check the stereochemical quality of proteinstructures. J. Appl. Crystallogr. 26:283–291.
20. Lee, C. C., et al. 2009. Structural basis of inhibition specificities of 3C and3C-like proteases by zinc-coordinating and peptidomimetic compounds.J. Biol. Chem. 284:7646–7655.
21. Lee, J. C., et al. 2008. A mammalian cell-based reverse two-hybrid system forfunctional analysis of 3C viral protease of human enterovirus 71. Anal.Biochem. 375:115–123.
22. Lei, X., et al. 2010. The 3C protein of enterovirus 71 inhibits retinoidacid-inducible gene I-mediated interferon regulatory factor 3 activation andtype I interferon responses. J. Virol. 84:8051–8061.
23. Matthews, D. A., et al. 1999. Structure-assisted design of mechanism-basedirreversible inhibitors of human rhinovirus 3C protease with potent antiviralactivity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U. S. A.96:11000–11007.
24. Mosimann, S. C., M. M. Cherney, S. Sia, S. Plotch, and M. N. James. 1997.Refined X-ray crystallographic structure of the poliovirus 3C gene product.J. Mol. Biol. 273:1032–1047.
25. Oberste, M. S., S. Penaranda, K. Maher, and M. A. Pallansch. 2004. Com-plete genome sequences of all members of the species Human enterovirus A.J. Gen. Virol. 85:1597–1607.
26. Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction datacollected in oscillation mode. Methods Enzymol. 276:307–326.
27. Patick, A. K., et al. 1999. In vitro antiviral activity of AG7088, a potentinhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother.43:2444–2450.
28. Phan, J., et al. 2002. Structural basis for the substrate specificity of tobaccoetch virus protease. J. Biol. Chem. 277:50564–50572.
29. Qiu, J. 2008. Enterovirus 71 infection: a new threat to global public health?Lancet Neurol. 7:868–869.
30. Racaniello, V. R. 2007. Picornaviridae: the viruses and their replication,796–839. In D. M. Knipe et al. (ed.), Fields virology, 5th ed. LippincottWilliams & Wilkins, Philadelphia, PA.
31. Robinson, C. R., F. W. Doane, and A. J. Rhodes. 1958. Report of an outbreakof febrile illness with pharyngeal lesions and exanthem: Toronto, summer1957; isolation of group A Coxsackie virus. Can. Med. Assoc. J. 79:615–621.
32. Shih, S. R., et al. 2004. Mutations at KFRDI and VGK domains of entero-virus 71 3C protease affect its RNA binding and proteolytic activities.J. Biomed. Sci. 11:239–248.
33. Shimizu, H., et al. 2004. Molecular epidemiology of enterovirus 71 infectionin the Western Pacific Region. Pediatr. Int. 46:231–235.
34. Sweeney, T. R., N. Roque-Rosell, J. R. Birtley, R. J. Leatherbarrow, and S.Curry. 2007. Structural and mutagenic analysis of foot-and-mouth disease
10330 LU ET AL. J. VIROL.
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from

virus 3C protease reveals the role of the beta-ribbon in proteolysis. J. Virol.81:115–124.
35. Tian, X., et al. 2009. Structure and cleavage specificity of the chymotrypsin-like serine protease (3CLSP/nsp4) of Porcine Reproductive and RespiratorySyndrome Virus (PRRSV). J. Mol. Biol. 392:977–993.
36. Tsai, M. T., et al. 2009. Real-time monitoring of human enterovirus (HEV)-infected cells and anti-HEV 3C protease potency by fluorescence resonanceenergy transfer. Antimicrob. Agents Chemother. 53:748–755.
37. Weng, K. F., M. L. Li, C. T. Hung, and S. R. Shih. 2009. Enterovirus 71 3Cprotease cleaves a novel target CstF-64 and inhibits cellular polyadenylation.PLoS Pathog. 5:e1000593.
38. Wong, K. T., et al. 2008. The distribution of inflammation and virus in humanenterovirus 71 encephalomyelitis suggests possible viral spread by neuralpathways. J. Neuropathol. Exp. Neurol. 67:162–169.
39. Wu, Y., et al. 2010. Structures of EV71 RNA-dependent RNA polymerase incomplex with substrate and analogue provide a drug target against thehand-foot-and-mouth disease pandemic in China. Protein Cell 1:491–500.
40. Wu, Y., et al. 2010. The largest outbreak of hand, foot and mouth disease inSingapore in 2008: the role of enterovirus 71 and coxsackievirus A strains.Int. J. Infect. Dis. 14:e1076–e1081.
41. Yang, F., et al. 2009. Enterovirus 71 outbreak in the People’s Republic ofChina in 2008. J. Clin. Microbiol. 47:2351–2352.
42. Yang, H., et al. 2003. The crystal structures of severe acute respiratorysyndrome virus main protease and its complex with an inhibitor. Proc. Natl.Acad. Sci. U. S. A. 100:13190–13195.
43. Zhang, D., and J. Lu. 2010. Enterovirus 71 vaccine: close but still far. Int.J. Infect. Dis. 14:e739–e743.
44. Zhang, Y., et al. 2010. Molecular evidence of persistent epidemic and evo-lution of subgenotype B1 coxsackievirus A16-associated hand, foot, andmouth disease in China. J. Clin. Microbiol. 48:619–622.
45. Zunszain, P. A., et al. 2010. Insights into cleavage specificity from the crystalstructure of foot-and-mouth disease virus 3C protease complexed with apeptide substrate. J. Mol. Biol. 395:375–389.
VOL. 85, 2011 COMPLEX STRUCTURES OF EV71 AND CVA16 3Cs 10331
on Novem
ber 27, 2011 by INS
TIT
UT
E O
F B
IOP
HY
SIC
Shttp://jvi.asm
.org/D
ownloaded from





























