Embed Size (px)
Citation preview

Energy Storage in Polymer Chains Under Stress
ROBERT COOK, Lawrence Livermore National Laboratory, Livermore, California 94550
Synopsis
The local conformation and storage of energy in individual polymer chains during a deforma- tion of a bulk polymer sample are examined by the computer simulation of a relatively simple model. It is shown that as the interaction between the chain atoms and surrounding medium increases, rotational angle motion is suppressed during the deformation, and large amounts of energy are stored in backbone bond angle and bond length distortions. The relationship of this phenomena to Tg and the implications for chain relaxation are discussed.
INTRODUCTION
In recent years our understanding of polymer chain conformational transi- tions for polymers in dilute solution has increased markedly through a combination of theoretical and computer simulation It is a fact, however, that modern technology is more interested in bulk polymers than dilute solution systems, and although it has long been realized that conforma- tional transitions, and chain motion in general, are key to understanding many properties of bulk polymer systems,' progress in understanding these motions in bulk systemss-12 has lagged far behind dilute solution systems. The reasons hardly need to be expanded upon; we only note that a fundamen- tal difference is that in dilute solution the Brownian action of the solvent promotes and mediates conformational motion, whereas in the bulk state, particularly below T', the surrounding chains inhibit or prevent significant conformational motion. For glassy bulk polymers significant conformational motion can be expected only when the sample is subjected to deformation, and, in fact, the ease of executing these deformations is intimately related to the freedom individual chains have in executing conformational motions.
To further clarify this point and to set the stage for the calculations described in this paper, consider a portion of a single chain imbedded in a bulk polymeric solid which is elongationally deformed by a factor of 2. The chain begins in some conformation, i.e., sequence of rotational angles, appropriate for its end-to-end length, but after the deformation the end-to-end length has doubled, and the original conformation is no longer appropriate. If the constraining forces are not too great, a situation one might encounter in a material significantly above T', the chain will relax by changing conformation to a sequence of rotational angles consistent with its new end-to-end length. These kinds of motion require the translation of chain segments and thus significant local free volume and mobility of the neighboring chains. In the new extended and relaxed conformation we would find the various rotational
Journal of Polymer Science: Part B: Polymer Physics, Vol. 26, 1349-1359 (1988) 0 1988 John Wiley & Sons, Inc. CCC 0098-1273/88/071349-11$04.00

1350 COOK
angles oscillating about local minima as will also be the case with the bond lengths and backbone bond angles. In this respect the chain appears locally much like it did before deformation; that is, the internal energy of the chain has changed little, and, in fact, may have decreased due to a higher population of lower energy rotational states. Stability, of course, is dictated by the free energy, not the internal energy, and the unfavorable entropy of the elongated chain will produce a strong restoring force, or if the chain ends are free to move, a recoiling of the chain to a more entropically favored situation.
If we are sufficiently below Tg, however, the situation will be markedly different. During the deformation the chain conformational motion will be partially suppressed by neighboring chains, and those motions necessary to relax the chain will be blocked. Since the chain is still being elongated, a good deal of the deformation energy may be taken up in the backbone bond angles and bond lengths. In the work that follows we explore this area of energy storage during deformation by modeling a polyethylene-like chain imbedded in a bulk medium which is mechanically strained.
MODEL
Our model is based on a polyethylene-like chain similar to that used by Helfand et aL3 The potential function can be expressed as
1)T = Vb + 0, + 04 + 0, (1)
where the total energy, uT, is expressed as a sum of contributions from the bond lengths, vb, backbone bond angles, v,, bond rotational angles, v+, and the constraints of neighboring chains, v,. No intrachain excluded volume terms are included, since the segments of chain we will be examining are relatively short. The first three terms of eq. (l), which represent the intrinsic chain potential, can be modeled with the following functional forms:
I n - 2
and
The number of atoms in the chain is n; Yb and ye are the force constants for the vibrations in the bond lengths, bi, around the equilibrium value, bo, and for vibrations in the backbone bond angles, Bi, around the equilibrium value of do, where 8 is measured as the supplement of the skeletal bond angle. The expression for the rotational angle potential, v,, is a fifth-order polynomial series which, following Helfar~d,~ allows one to fit six independent constants of the potential: E,, the energy of the trans state; ECi,, the energy of the cis state; EB, the energy of the gauche state; E*, the energy at the top of the
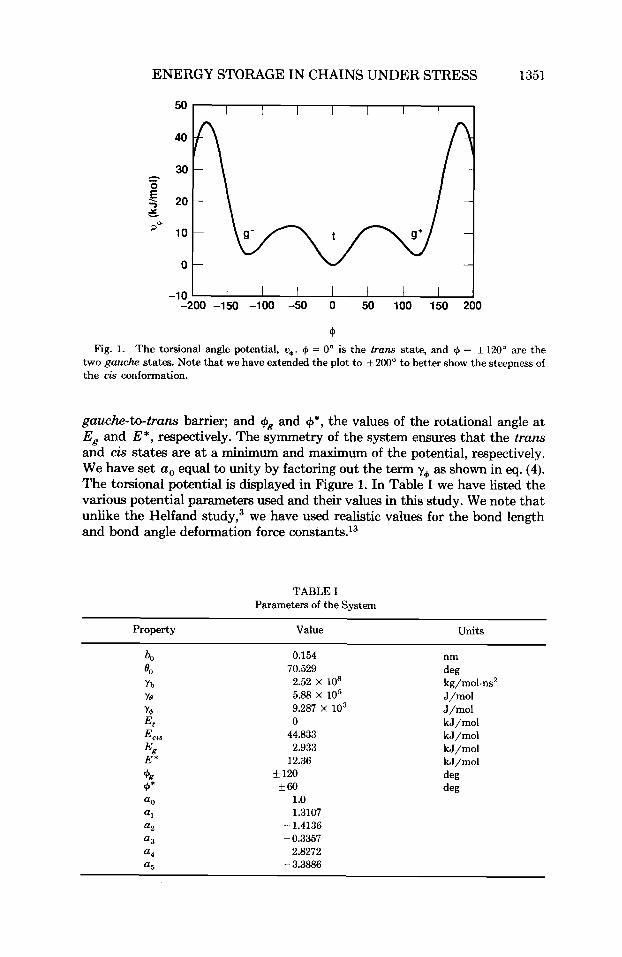
ENERGY STORAGE IN CHAINS UNDER STRESS 1351
50 I I I I I I I
-1 0 I I I I I I -200 -150 -100 -50 0 50 100 150 200
cp Fig. 1. The torsional angle potential, u+. + = 0" is the trans state, and + = f120" are the
two gauche states. Note that we have extended the plot to * 200" to better show the steepness of the cis conformation.
gauche-to-trans barrier; and +g and $I*, the values of the rotational angle at Eg and E*, respectively. The symmetry of the system ensures that the trans and cis states are at a minimum and maximum of the potential, respectively. We have set a , equal to unity by factoring out the term y, as shown in eq. (4). The torsional potential is displayed in Figure 1. In Table I we have listed the various potential parameters used and their values in this study. We note that unlike the Helfand study,3 we have used realistic values for the bond length and bond angle deformation force con~tants.'~
TABLE I Parameters of the System
Property Value Units
Ecis
E E.,
h +* a0
a1
a 2
a3
a4
a5
0.154 70.529 2.52 X 10'
9.287 X lo3 0
44.833 2.933
12.36
5.88 x lo5
f 120 *60
1.0 1.3107
- 1.4136 - 0.3357
2.8272 - 3.3886
nm
kg/mol-ns2 J/mol J/mol kJ/mol kJ/mol kJ/mol kJ/mol
deg
deg deg

1352 COOK
Let us digress for a moment to discuss the inclusion of bond angle and bond length terms in our potential. It has become common practice to suppress bond angle and bond length distortions with rigid links, the motivation being a combination of computational time savings and the expectation that the relatively small oscillations in these degrees of freedom do not contribute much to the overall chain dynamics. Although this may be the case for certain equilibrium measurements, particularly for chains in dilute solution, we feel the contribution of these modes, particularly that of bond angle bending, cannot be ignored when one is concerned with imposed deformations on polymeric materials. To support this view it can be noted that the actual curvature at the minimum of the v, potential well is only a factor of about 7 greater than that for the v+ well, a fact partially obscured by the functional forms used in eqs. (3) and (4). Thus for a unit angle deformation the restoring force due to the bond angle potential is only seven times greater than that from the rotational angle potential, or, alternatively, if the potentials were linked in series, the energy storage in the softer rotational angle potential would be only seven times greater than that in the bond angle potential. The importance of the bond angle (and bond length) potential in the rates of conformational transitions was also pointed out in a series of papers by Helfand and collaborators,'~'4~'5 in which they study the details of the trans-to-gauche barrier passage using both analytic methods based on Kramers' theory and Brownian dynamics simulations. They find that the rate of barrier passage is slowed by stiffer bond angle and length potentials due to the coupling of these potentials into the local mode describing the transition. In effect, the flexibility in these degrees of freedom lowers the barrier to the transition. As we will show shortly, this coupling becomes more important for bulk materials during deformation.
The modeling of the constraining potential due to neighboring chains was accomplished as follows. To simplify matters we will consider only the case where a section of chain is situated such that the line between the ends of this section of chain is aligned with the direction of an elongational deformation. As the medium is elongated the neighboring constraining medium is also elongated. Thus chain motion in the direction of the deformation is likely to be less constrained than motion in a direction perpendicular to the elongation direction. In order to model this situation we attach to each atom in the undeformed chain segment Hookean constraining springs, which act only in directions normal to the deformation direction. Thus if we take the deforma- tion to be along the z direction, the potential, v,, can be expressed as
where y, and ycy are the Hookean force constants for displacements in the x and y directions, and xi ,o and yi ,o are the x and y coordinates of particle i in the undeformed chain. A similar term could be added for displacements in the z direction, presumably with a smaller value of ycz, but for the reasons outlined above we have set ycr equal to zero. We have also assumed isotropy normal to the deformation direction and have thus set yCx = ycy = yc. The magnitude of y, clearly controls the strength of the constraining forces and

ENERGY STORAGE IN CHAINS UNDER STRESS 1353
must be related to the interaction potential of elements on neighboring chains, as well as the local free volume. This will certainly vary from atom to atom, and thus we are modeling an average effect.
The deformation is executed as follows. The undeformed, relaxed chain segment, with bond lengths, bond angle, and torsional angles at their minima, is first aligned so that the first atom is at the origin and the last atom on the positive z axis. The chain is then stretched by an amount A, typically taken to be 0.001 nm, by multiplying the z coordinate of each particle by (1 + A/z,) where z, is the z coordinate of the end particle in the n-particle chain. This afine deformation method places most of the initial strain energy in the bond lengths. We then let the system relax to its lowest energy conformation consistent with the current end-to-end separation. The conformational and energetic variables for this "relaxed" configuration are then recorded. The process is repeated until the chain has been strained the desired amount.
We note that our method of relaxation is consistent with a zero tempera- ture simulation. Our goal at this point is to understand the basic mechanics of the model and forego explicit thermal effects. Furthermore, we believe this treatment is consistent with the mean field treatment of the surrounding chains through yc. In this context we note that the concept of thermal motion is implicitly contained in the magnitude of yc, since it controls the degree of translational freedom the atoms of the chain enjoy. A proper treatment of thermal effects would require an explicit modeling of the chain environment. Although such studies are computationally within reach," our goal at this point is to focus on simpler models.
In the work reported here, we have examined the conformational geometry and energetics associated with the gauche-to-trans transition by examining the extension of a four-atom chain originally in a gauche configuration, as well as six and eight atom chains in tgt and ttgtt configurations, respectively. We have examined these systems with constraining force constant, yc, that varies from zero to values high enough to suppress almost all translational motion normal to the chain direction, effectively eliminating the torsional trans-to-gauche motion. Our particular interest is in energy storage; thus we have followed the energy stored in the bond lengths, backbone bond angles and torsional angles as a function of chain extension and the magnitude of the constraining forces. Our results follow.
RESULTS Before proceeding to an examination of chain conformations and energy
storage for a chain constrained by its environment, let us first look at a chain without any external constraints, except those on the first and last atoms which control the degree of chain extension. In our discussions here and later we will focus on the four-atom chain, initially in a gauche configuration. The results for the six- and eight-atom chains are qualitatively the same, except as noted. In Figure 2, using the left ordinate, we plot the value of the torsional angle and a backbone bond angle with heavy dotted and dashed lines, respectively, as a function of d14, the distance between the chain ends. We see a decrease in the torsional angle from the gauche state at 120" to the trans state a t 0". The relatively stiff backbone bond angles open slightly (which is
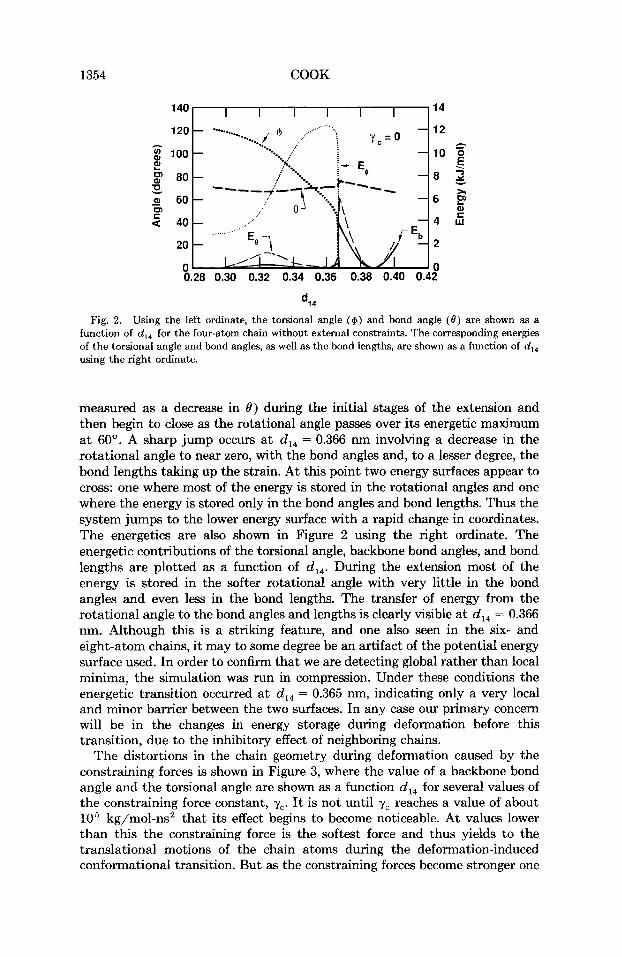
1354 COOK
120
l 4 O C l 4
0.28 0.30 0.32 0.34 0.36 0.38 0.40 0.42
d14
Fig. 2. Using the left ordinate, the torsional angle ($) and bond angle (0) are shown as a function of d,, for the four-atom chain without external constraints. The corresponding energies of the torsional angle and bond angles, as well as the bond lengths, are shown as a function of d,, using the right ordinate.
140 14 I I I I I I
A
Q) 60 - cn
0.28 0.30 0.32 0.34 0.36 0.38 0.40 0.42
d14
Fig. 2. Using the left ordinate, the torsional angle ($) and bond angle (0) are shown as a function of d,, for the four-atom chain without external constraints. The corresponding energies of the torsional angle and bond angles, as well as the bond lengths, are shown as a function of d,, using the right ordinate.
measured as a decrease in 0) during the initial stages of the extension and then begin to close as the rotational angle passes over its energetic maximum a t 60". A sharp jump occurs a t d,, = 0.366 nm involving a decrease in the rotational angle to near zero, with the bond angles and, to a lesser degree, the bond lengths taking up the strain. At this point two energy surfaces appear to cross: one where most of the energy is stored in the rotational angles and one where the energy is stored only in the bond angles and bond lengths. Thus the system jumps to the lower energy surface with a rapid change in coordinates. The energetics are also shown in Figure 2 using the right ordinate. The energetic contributions of the torsional angle, backbone bond angles, and bond lengths are plotted as a function of d14. During the extension most of the energy is stored in the softer rotational angle with very little in the bond angles and even less in the bond lengths. The transfer of energy from the rotational angle to the bond angles and lengths is clearly visible a t d,, = 0.366 nm. Although this is a striking feature, and one also seen in the six- and eight-atom chains, i t may to some degree be an artifact of the potential energy surface used. In order to confirm that we are detecting global rather than local minima, the simulation was run in compression. Under these conditions the energetic transition occurred a t d,, = 0.365 nm, indicating only a very local and minor barrier between the two surfaces. In any case our primary concern will be in the changes in energy storage during deformation before this transition, due to the inhibitory effect of neighboring chains.
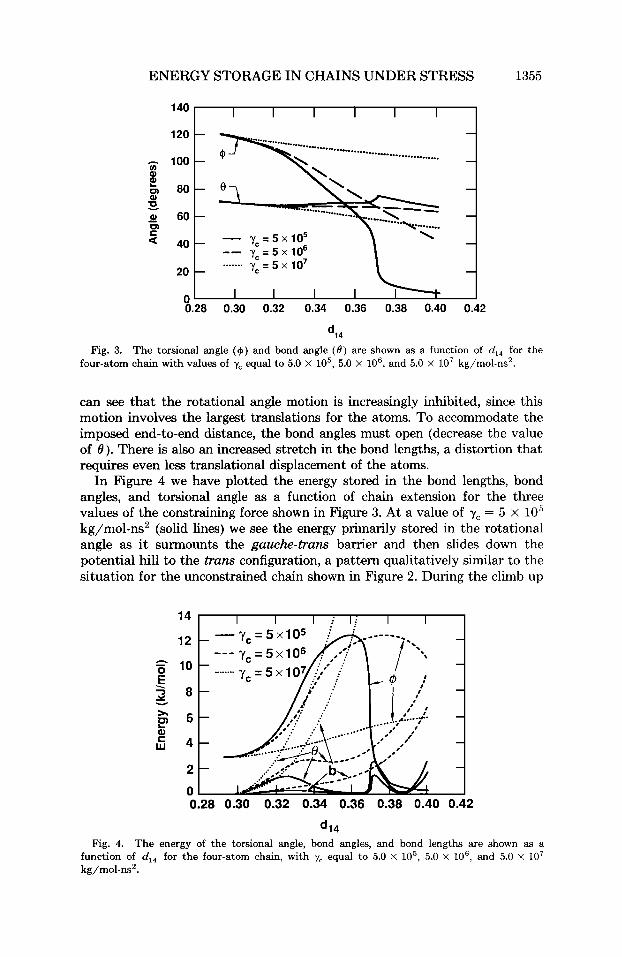
The distortions in the chain geometry during deformation caused by the constraining forces is shown in Figure 3, where the value of a backbone bond angle and the torsional angle are shown as a function d,, for several values of the constraining force constant, y,. It is not until y, reaches a value of about lo5 kg/mol-ns* that its effect begins to become noticeable. At values lower than this the constraining force is the softest force and thus yields to the translational motions of the chain atoms during the deformation-induced conformational transition. But as the constraining forces become stronger one
ENERGY STORAGE IN CHAINS UNDER STRESS 1355
d,, Fig. 3. The torsional angle (+) and bond angle (0) are shown as a function of d,, for the
four-atom chain with values of yc equal t o 5.0 X lo5, 5.0 X lo6, and 5.0 X lo7 kg/mol-ns2.
can see that the rotational angle motion is increasingly inhibited, since this motion involves the largest translations for the atoms. To accommodate the imposed end-to-end distance, the bond angles must open (decrease the value of 0). There is also an increased stretch in the bond lengths, a distortion that requires even less translational displacement of the atoms.
In Figure 4 we have plotted the energy stored in the bond lengths, bond angles, and torsional angle as a function of chain extension for the three values of the constraining force shown in Figure 3. At a value of y, = 5 x lo5 kg/mol-ns2 (solid lines) we see the energy primarily stored in the rotational angle as i t surmounts the gauche-trans barrier and then slides down the potential hill to the trans configuration, a pattern qualitatively similar to the situation for the unconstrained chain shown in Figure 2. During the climb up
14
12
g 10 E ' 5 8 25
6 h
Q) F 5 4
2
0
I I I i I; I I . .
0.28 0.30 0.32 0.34 0.36 0.38 0.40 0.42
d14 Fig. 4. The energy of the torsional angle, bond angles, and bond lengths are shown as a
function of d,, for the four-atom chain, with y, equal t o 5.0 X lo5, 5.0 X lo6, and 5.0 X lo7 kg/mol-ns2.

1356 COOK
the barrier some energy is stored in the bond angles as they open slightly a t about d,, = 0.32 nm and again during the slide down to the trans configura- tion as described during the discussion of Figure 3. Note that very little energy is distributed to the stiffer bond angles during the initial deformation. When the constraining potential is increased to a value of 5 X lo6 kg/mol-ns2 (dashed lines) the torsional angle motion is significantly inhibited, and the peak in the torsional angle energy curve shifts to longer d14. This delay is compensated for by increased deformation in the bond angles and lengths as can be seen by the increased energy storage in these modes. At a value of y, = 5.0 x lo7 kg/mol-ns2 (dotted lines) the rotational angle motion is nearly eliminated, and the energy of the deformation is stored largely in the bond angles and lengths. Although not shown, a t higher values of yc more energy is stored in the bond lengths than angles, since an increase in end-to-end length can be accommodated with less atom motion normal to the extension direc- tion by bond length deformation than by bond angle deformation.
The situation for the longer chains of six and eight atoms is qualitatively similar to what has been described above. In principal, the extra degrees of freedom allow for a distribution of the strain energy, but in fact it remains fairly concentrated in the neighborhood of the gauche bond. For example, in the six-atom chain in a tgt conformation, when y, < lo6 kg/mol-ns2 more than 80% of the energy stored in the bond angles was stored in angles 2 and 3. At higher values of y, the storage becomes progressively more uniform as the rotational angle transition becomes suppressed. A qualitative difference occurs during the extension of the eight-atom chain. It turns out that the tttgt conformation has an end-to-end distance between that of the ttgtt and ttttt conformations, and if the constraining forces are not too great there is a transition between these two conformers during the extension. The ttttg conformation has an end-to-end length between the ttgtt and tttgt conformers; however, this low energy state is not sensed during the simulation, probably due to the separation of the two gauche locations and the large energy barrier between them. Even the transition from ttgtt to tttgt occurs significantly past the tttgt minimum energy end-to-end distance due to the local barriers which are independent of the constraining forces. Although these qualitative differ- ences for the eight-atom chain are of some interest, particularly with regard to the dynamics of energy transmission between the two states, it is important to note that the basic pattern of conformational motion suppression by the constraining forces remains remarkably the same, with similar shifts in energy to the backbone bond angles and bond lengths a t about the same magnitude of Y,.
DISCUSSION
Let us now turn our attention to an interpretation of y,, particularly to the question of what numerical values might be appropriate under certain condi- tions. Based on the discussion above, we would expect to find that values in excess of 106 kg/mol-ns2 would be appropriate for materials below 2''. What we seek is a method of checking this prediction by calculating an approximate value from molecular parameters. To do this we will consider the repulsive interaction as largely a steric one due to the van der Waals repulsions of a pair

ENERGY STORAGE IN CHAINS UNDER STRESS 1357
of chain elements. To the extent that this picture applies, we can calculate an approximate yc by expanding the van der Waals pair interaction energy,
U ( r ) = 4Rr[ ( :)I2 - ( :)6] to quadratic terms. In eq. (6), R = 8.314 J/deg-mol, and r and u are the appropriate van der Waals interaction parameters for polyethylene-like chain elements. Expanding eq. (6) around 2'I6u, the position of minimum energy, gives
By comparing with eq. (5) we can identify y, as
72 Rr y c = 21/3a2'
In order to evaluate this expression we need values of c and 0. We can roughly approximate (c , a ) by examining the yalues for Ne, CH,, 05 n-C4Hlo, which are (35.7 K, 2.789 A), (137 K, 3.822 A), and (410 K, 4.997 A), respectively.16 Certainly the true value falls somewhere in this range. Plugging these values into eq. (8) and converting to appropriate units gives values of yc equal to 2.2 x lo5, 4.5 x lo5, and 7.8 x lo5 kg/mol-ns2, respectively. Note that these values are at the level which we identified with the onset of configurational motion inhibition. It might be argued that the chain elements are, on average, separated by more than the minimum energy distance, particularly above Tg, and thus the actual y, would be less than these calculated values. However, below Tg we expect the average distance between chain elements to be closer to the position of minimum van der Waals energy. Furthermore, in glassy polymers we expect the effective value of y, to be significantly larger than the roughly 5 X lo5 kg/mol-ns2 calculated above because the actual repulsive part of the van der Waals potential is much steeper than the quadratic fit of eq. (7). For example, the actual repulsive force at r = a from the van der Waals expression in eq. (6) is more than seven times that calculated from the harmonic approximation of eq. (7). Since conformational motion involves relatively large translations, it is easy to see how a value of y, several orders of magnitude greater than 5 X lo5 would be appropriate for a glassy material. Let us emphasize again the approximate nature of the calculation, since yc is at best an average model parameter. Our objective is simply to show that the range of values considered is consistent with what one might calculate from molecular data.
Let us now continue the discussion, begun in the introduction, of the deformation of a glassy polymer. Certainly if the deformation is sufficiently small the sample will behave elastically, and the modulus will depend on the local chain chemical structure as well as the magnitude of the constraining forces. As the magnitude of the deformation grows, however, the fate of the individual chains, as well as the sample as a whole, will depend upon the

1358 COOK
relative strength of the constraining forces and bond-length restoring forces. If the constraining forces are sufficiently strong, the deformation energy cannot be dissipated through conformational motions and will instead be loaded into the bond angles and lengths, ultimately causing bond breakage, crack propa- gation, and material failure. If, on the other hand, bond failure does not occur, the imposed deformation must cause some combination of conformational motion and bond angle and length deformation, in many cases allowing the material to yield. Clearly, one must also consider the time scale and method of deformation. One can obtain significant strains even with brittle substances using various techniques, for example testing under hydrostatic pressure. The glassy material which has yielded must be in a very complex and high energy molecular state. Much of the energy of the deformation will be stored not only in the distribution of partially opened rotational angles but also to a signifi- cant degree in the deformed bond angles and lengths. This is to be contrasted with a polymeric material above Tg, where most of the energy of deformation is stored entropically. The energy stored in a deformed material below T, can generally be quantitatively recovered by heating the sample above Tg, which allows for conformational relaxations by effectively reducing the constraining forces. It seems reasonable, however, particularly in these high energy de- formed glassy samples, that independent partial relaxation of the bond lsngth and bond angle deformations are possible, and further that the onset of these relaxations should occur below T,, since full scale conformational motion may not be necessary. Such an observation has been made in some preliminary differential scanning calorimetry work on highly strained epoxy systems by LeMay.17 He sees small exotherms attended by negligible dimensional changes significantly below Tg on the first heating cycle, possible indicating partial relaxation of the bond lengths and angles in the sample. I t is to be expected that these relaxations would involve only very small dimensional changes in the sample and would be much smaller than the dimensional and entropic relaxation that occurs a t T,. More detailed experimental work on these systems is in pr0gre~s.l~
The author thanks Dr. Jim LeMay for several useful discussions. The programming contribu- tions of David Turner, a summer visitor from Drexel University, and Jonathan Mohr and Deborah Weiss, Associated Western University Summer Fellowship participants, is gratefully acknowledged. This work was performed under the auspices of the U.S. Department of Energy by the Lawrence Livermore National Laboratory under contract no. W-7405-ENG-48.
References 1. E. Helfand, J . Chem. Phys., 64, 4651 (1971). 2. M. Fixman, J . Chem. Phys., 69, 1527 (1978); Zbid., 69, 1538 (1978). 3. E. Helfand, Z. R. Wasserman, and T. A. Weber, Macromolecules, 13, 526 (1980). 4. C. K. Hall and E. Helfand, J . Chem. Phys., 77, 3275 (1982). 5. R. Cook and L. Livornese, Macromokcules, 16, 920 (1983). 6. R. Cook and E. Helfand, J. Chem. Phys., 82, 1599 (1985). 7. See, for example, R. T. Bailey, A. M. North, and R. A. Pethrick, Molecular Motions in
8. R. Cook and M. B. Mercer, Mat. Chem. Phys., 12, 571 (1985). 9. Y. Termonia, P. Meakin, and P. Smith, Macromolecules, 18, 2246 (1985); Ibid., 19, 154
High Polymers, Clarendon Press, Oxford, 1981.
(1986). 10. D. Brown and J. H. R. Clarke, J . Chem. Phys., 84, 2858 (1986).

ENERGY STORAGE IN CHAINS UNDER STRESS 1359
11. D. N. Theodorou and U. W. Suter, Macromolecules, 19, 139, 379 (1986). 12. R. Cook, J . Polym. Sci. Polym. Phys. Ed. 26, 1337 (1988). 13. R. Zbinden, Znfrared Spectroscopy of High Polymers, Academic Press, New York, 1964. 14. J. Skolnick and E. Helfand, J . Chem. Phys., 72, 5489 (1980). 15. E. Helfand, Z. R. Waseman, T. A. Weber, J. Skolnick, and J. H. Runnels, J . Phys. Chem.,
16. J. 0. Hirschfelder, C. F. Curtis, and R. B. Bud, Molecular Theory of Gases and Liqulds,
17. J. LeMay (private communication).
75, 4441 (1981).
John Wiley & Sons, New York, 1964.
Received August 11, 1987 Accepted November 23, 1987





























