Embed Size (px)
Citation preview

ENDOCRINE SYSTEM
PATHOLOGY
Mousa Al-Abbadi, MD, FCAP,CPE, CPHQ,FIAC,ABMQ
Professor of Pathology & Cytopathology
University of Jordan
College of Medicine

MY DUTIES• 5 lectures; 90 minutes each
• Simplify
• Understand the concepts
• Help U all Understand…understand…
understand X 10…only then memorize and
recall
• Answer questions & inquiries
• Respect

YOURDUTIES
This Book is your
main source of
knowledge &
EXAMQUESTIONS

YOURDUTIES• ON TIME ATTENDANCE
• Attendance will be taken at the beginning of lecture this system.
• No entrance after 8 am or 9:30 am
• Plz…plz…plz…NO CHATTING during lecture
• Understand first then memorize and recall
• Respect to the process
• NO MOBILE
• No inquiries about the nature
of the exam…I tellyou

PLEASE DON’T ASK THESE
QUESTIONS ATALL
• How many questions on my material?
• What should we concentrate on?
• Are the slides enough?
• Should we memorize this or that?
• Is this or that required?

[YOU SHOULD NOT ONLY STUDY
FOR THEEXAM]
[YOU ARE NOT STUDYING FOR ME
EITHER]
[YOU ARE LEARNING SO THAT YOU
WILL BE A GOOD CARING &
THOROUGH PHYSICIAN WHO WILL
APPLY THE STNADRAD OFCARE]

REMEMBER“Success in life is
90% hard work and
10% talent, luck and
high IQ”
“ALSOREMEMBER”IF YOU ARE LATE….YOU
MAY LOSE YOUR PATINET

INTENDED LEARNINGOBJECTIVES
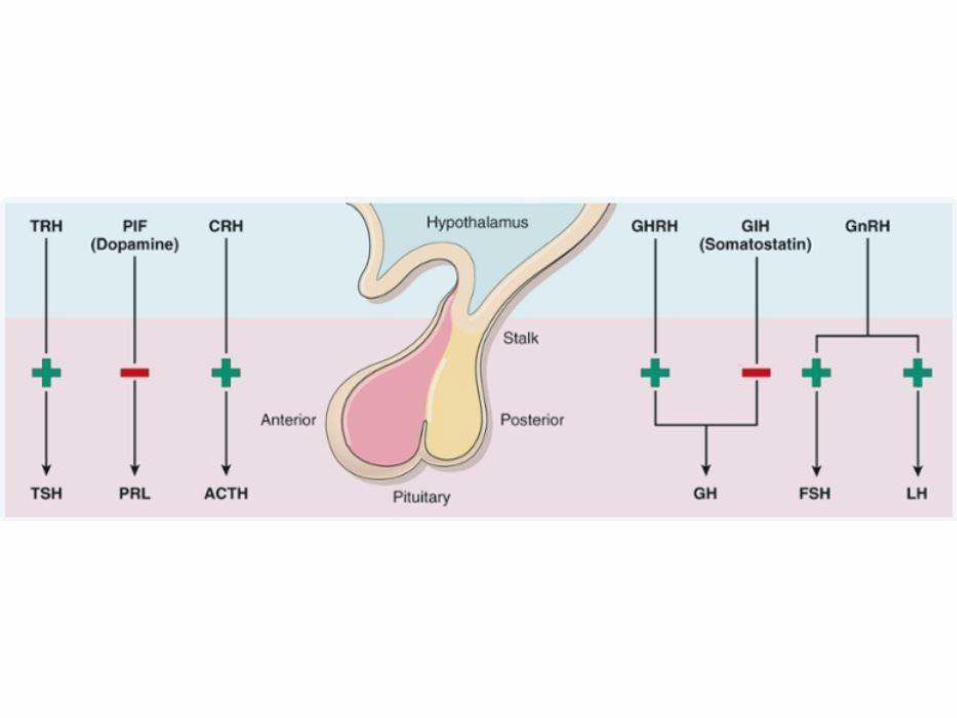
• Brief review of the anatomy, physiology and biochemistry of human endocrine system.
• Clear understanding of hyper-function and hypo-function of different glands and grasp the concept of positive and negative feedback control.
• Understand the pathogenesis of pituitary adenomas and its hyper-function associated neoplasms.
• Comprehend common causes and features of hypopituitarism.
• Grasp the general features and causes of hyperthyroidism and hypothyroidism.
• Recognize and understand common autoimmune thyroid diseases including Hashimoto thyroiditis.

ILOs….continue
• Absorb the pathology of common benign thyroid diseases including subacute granulomatous thyroiditis (de Quervain thyroiditis), subacute lymphocytic thyroiditis, and Gravesdisease.
• Recognize all the features of the common diffuse and multi-nodular goiter.
• Recognize the pathology of common thyroid neoplasms, adenomas and carcinomas and understand the concept of evaluation by fine needle aspiration for pre-management diagnosis.
• Understand the clinicopathological characteristics of parathyroid gland diseases including hyper-parathyroidism (primary and secondary) and hypo-parathyroidism.
• Brief review of normal insulin physiology and glucosehomeostasis.

ILOs….continue
• Grasp the detailed understanding of the pathogenesis of type 1 & type 2 diabetes mellitus and the differences between these two.
• Recognize other types of diabetes mellitus.
• Recognize and understand the acute and chronic complications of diabetes mellitus.
• Understand and recognize common neuroendocrine pancreatic tumors such as insulinomas andgastrinomas.
• Grasp the features of adrenocortical hyperfunction syndromes including Cushing syndrome, hyperaldosteronism, and adrenogenital syndrome.
• Recognize the pathology of benign breast epithelial tumors
• Understand the pathophysiology of primary and secondary adrenal insufficiency; acute and chronic (Addison disease).

ILOs….continue
• Grasp the detailed understanding of the pathogenesis of type 1 & type 2 diabetes mellitus and the differences between thesetwo.
• Recognize other types of diabetes mellitus.
• Recognize and understand the acute and chronic complications of diabetes mellitus.
• Understand and recognize common neuroendocrine pancreatic tumors such as insulinomas and gastrinomas.
• Grasp the features of adrenocortical hyperfunction syndromes including Cushingsyndrome, hyperaldosteronism, and adrenogenital syndrome.
• Recognize the pathology of benign breast epithelial tumors
• Understand the pathophysiology of primary and secondary adrenal insufficiency; acute and chronic (Addison disease).
• Recognize common adrenocortical tumors and its general features.• Understand the pathology of common adrenal medulla tumor including
Pheochromocytoma, Neuroblastoma and other neuronalneoplasms.
• Understand the concept of multiple endocrine neoplasia type 1 and 2 and its pathogenesis

GENERAL CONCEPTS OF ENDOCRINESYSTEM:
• Basics of Hormonephysiology– Cell surface receptor binding hormones (GH)
– Intracellular receptor binding hormones (ER)
• Feedback inhibition
• Laboratory chemical testing
• Pathology:
1. Hyper or hypo production
2. End organresistance
3. Neoplasms (functional or non functional)
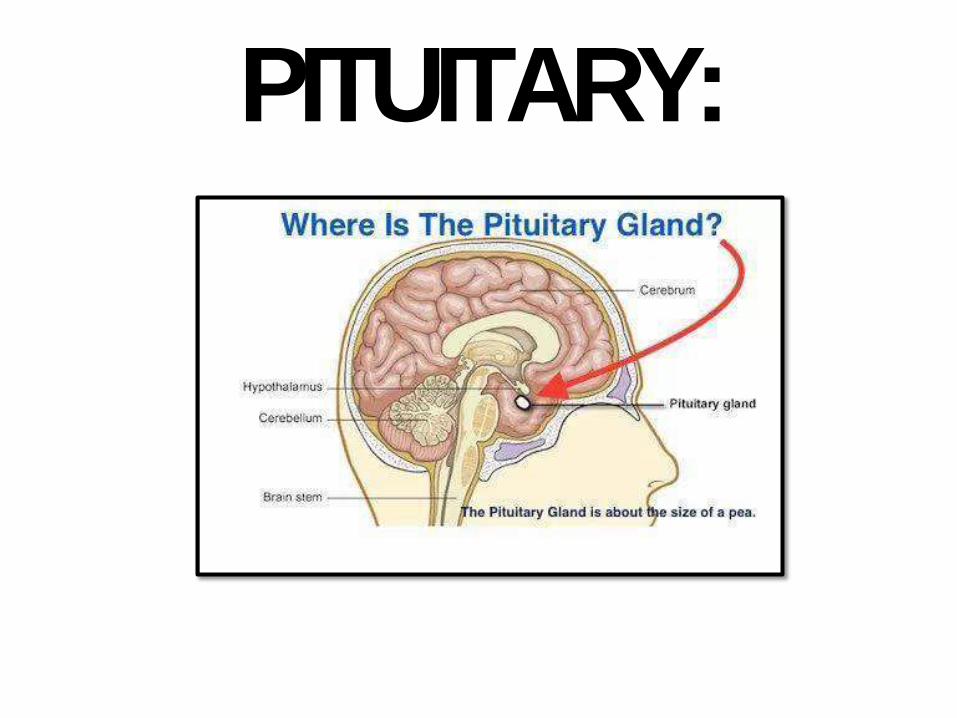
PITUITARY:

PITUITARYDISEASES:• Hyperpituitarism: hormones, usually anterior
pituit. adenomas, primary morecommon
• Hypopituitarism: hormones (ischemia, surgery, radiation or inflammation). Or non functional adenoma with pressureeffects
• Local mass effects: sella turcica changes by radiology, visual field defects (chiasm, bi-temporal hemianopsia). ICP symptoms (headache, nausea,vomiting).
• Sometimes: they cause seizures, obstructive hydrocephalus, and cranial nerve palsies
PITIUITARYAPOPLEXY:• Sudden acute hemorrhage
into pituitary tissue or
neoplasm; causing rapid
enlargement
• Sudden severe headache,
diplopia and hypopituitarism
• Acute neurosurgical
emergency/maybe fatal
(specially ACTH sudden
deficiency

ANTERIOR PITUITARYTUMORS:
• The most common are benign adenomas
• Can be functional, non functional or silent; all may cause mass effect
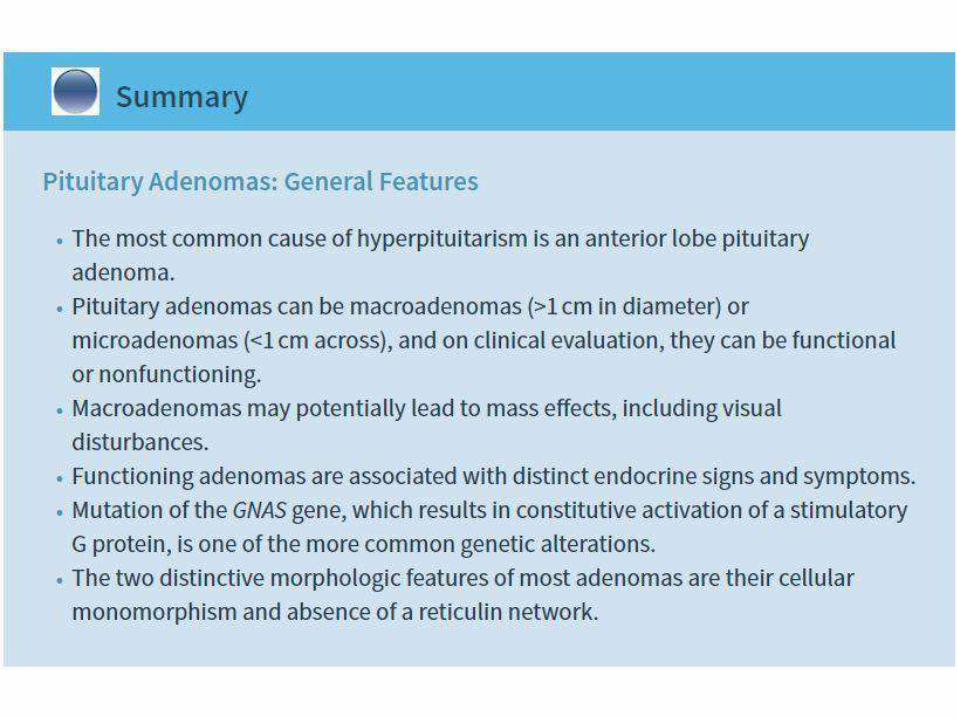
• P. Adenoma of anterior lobe is the most common cause of hyperpituitarism
• Microadenoma (< 1cm) or Macroadenoma (> than 1 cm)
• Non functional ones: late presentation and may causehypopituitarism

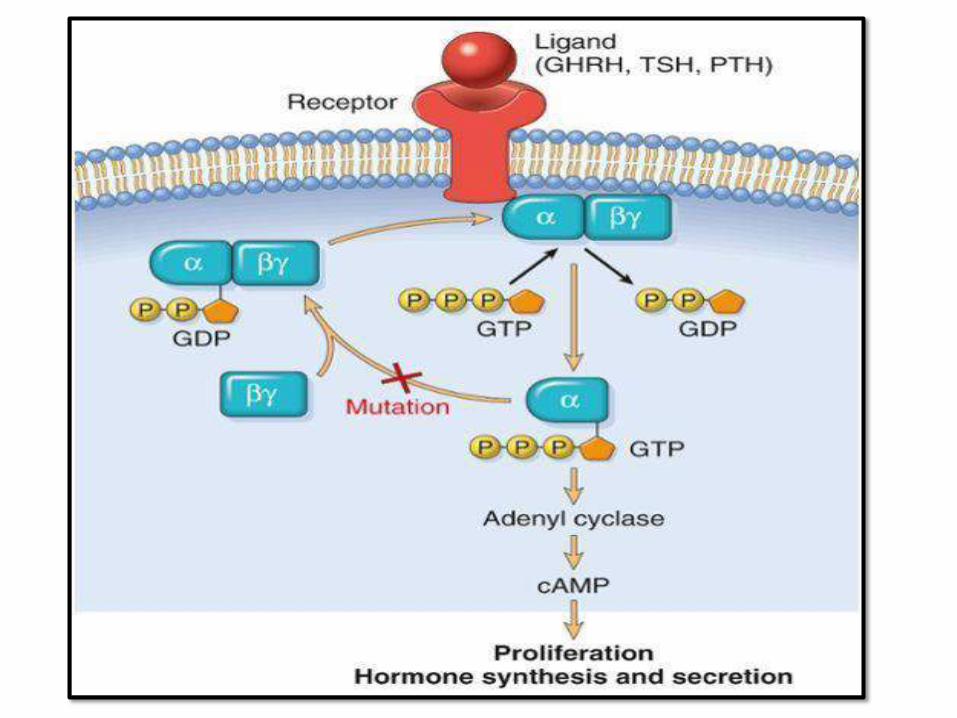
PATHOGENESIS:1. G-protein mutations: most common
2. Familial gene mutations: MEN1,
CDKN1B,…
3. Molecular defects: cyclin-D1, TP53,
RB, RAS oncogene (aggressive tumors
and pituitary carcinomas)
MORPHOLOGY:

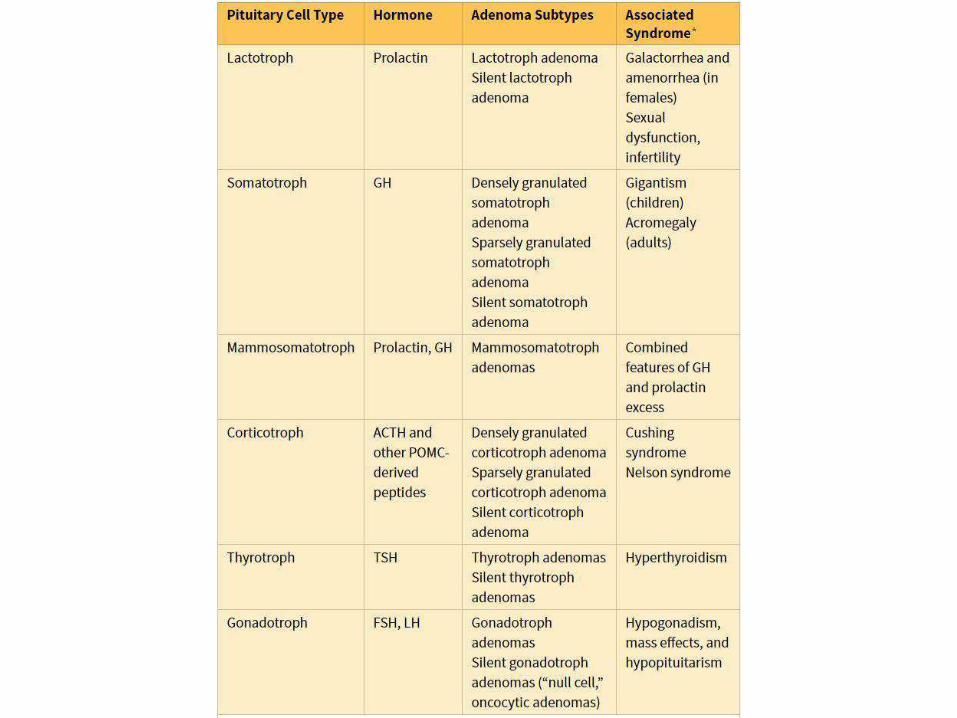
LACTOTROPH ADENOMAS(PROLACTINOMAS):
• Most frequent hyperfunctioning (30%)
• IHC of tumor cells +ve for PRL
• Clinically: amenorrhea, galactorrhea, loss of
lipido, infertility
• Earlier and easier to diagnose in younger
females )in contrast to older F andmales)
• Serum PRL level usually >200 μg

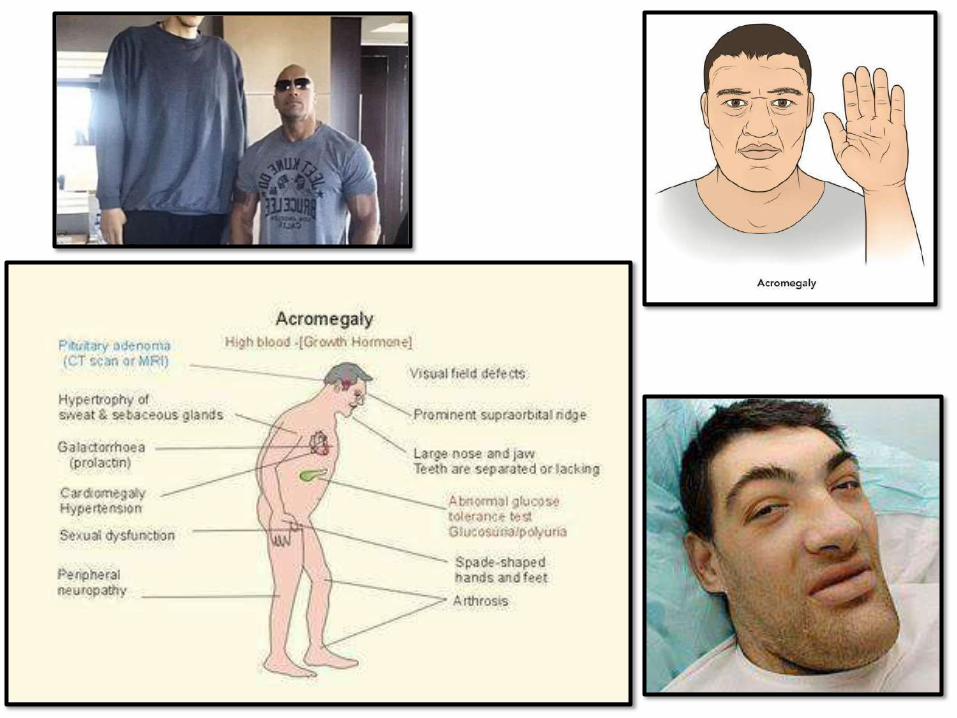
SOMATOTROPHADENOMAS:
• 2nd mostcommon
• Gigantism in children and acromegaly in
adults
peripheral• Diabetes mellitus due to
resistance to insulin
• Slow growing and usually macroadenomas
• Gonadal dysfunction, muscle weakness, HT,
arthritis, CHF, risk of GITcancers

CORTICOTROPHADENOMAS:
• Hypercortisolism “Cushingsyndrome”
• When the cause is ACTH producing adenoma “Cushingdisease”
• Usually microadenoma
• S.Times large one develop after surgical removal of adrenal glands to treat CS (Nelson syndrome) mass effect no hypercortisolism
• Hyperpigmentation due to MSH

OTHER ANT. PITUIT.NEOPLASMS
• Non functioning Gonadotroph adenomas
(FSH and LH), mainlyFSH
• Thyrotroph adenomas: 1%, rare cause of
hyperthyroidism
• Most are usually non functioning and may
cause hypopituit due to mass effect and
apoplexy
• Carcinomas are rare and aggressive

HYPOPITUITARISM:• Occurs when >75%tissue loss
• Congenital are rare; moreacquired
• S.times associated with posteriorpit dysfunction (Diabetes insipidus)
• Causes:
– Non funct adenomas (masseffect)
– Sheehan syndrome (ischemic necrosis): pregnancy, shock, DIC, Sickle CD,trauma, ICP,iatrogenic causes and other less commcauses

HYPOPITUITARISM:CLINICALLY
• Depends on the deficient hormone
• Pituitary dwarfism in children
• GnRH: amenorrhea and infertility inwomen
• Hypothyroidism and hypoadrenalism
• Prl: no lactation postpartum
• Hypopigmentation ( MSH)

POSTERIOR PIYUITARYSYNDROMES:
• ADH deficiency (Diabetes inspidus): polyuria.
• Causes: head trauma, surgery and
inflammatory conditions. Maybe idiopathic.
• Central (see above) or nephrogenic DI
(unresponsive renal tubules)
• Thirst and polydipsia; severe dehydration if
untreated. Serumsodiumand urine
specific gravity (dilute urine)

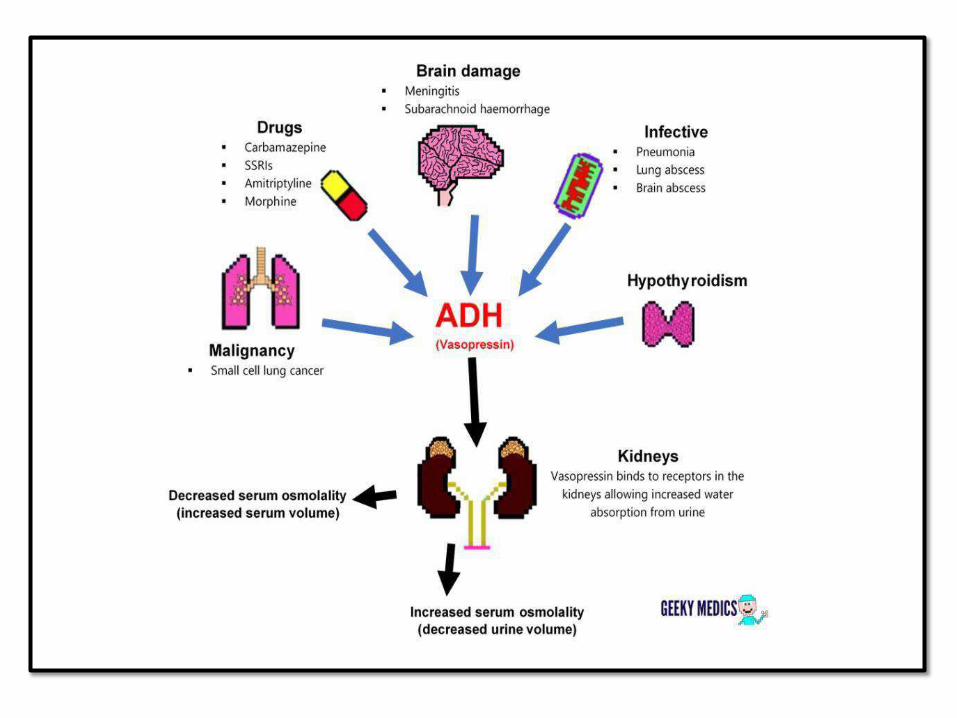
SYNDROME OF INAPPROPRIATE ADH
SECRETION(SIADH):
• Ectopic ADH (small cell carcinoma of lung and
other causes)
• Hyponatremia, cerebral edema, neurologic
dysfunction
• No peripheral edema
THYROID

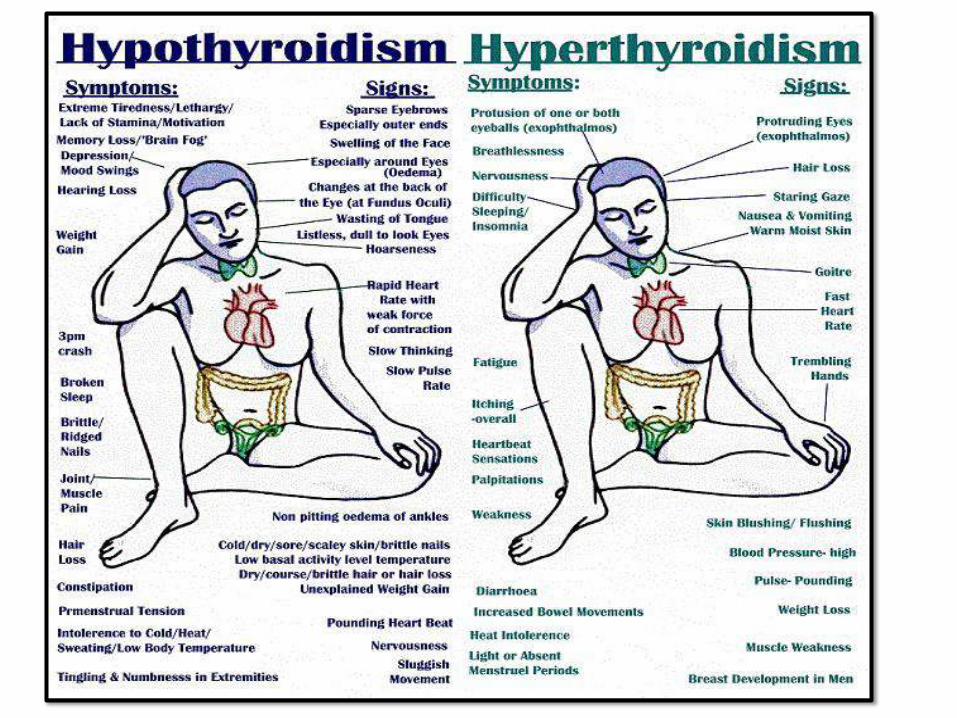
THYROIDDISORDERS:
• Very common diseases
• Hypo and hyperthyroidism
• Thyroiditis (autoimmune and others)
• Enlargement (Diffuse and Multinodular
Goiter MNG)
• Neoplasms
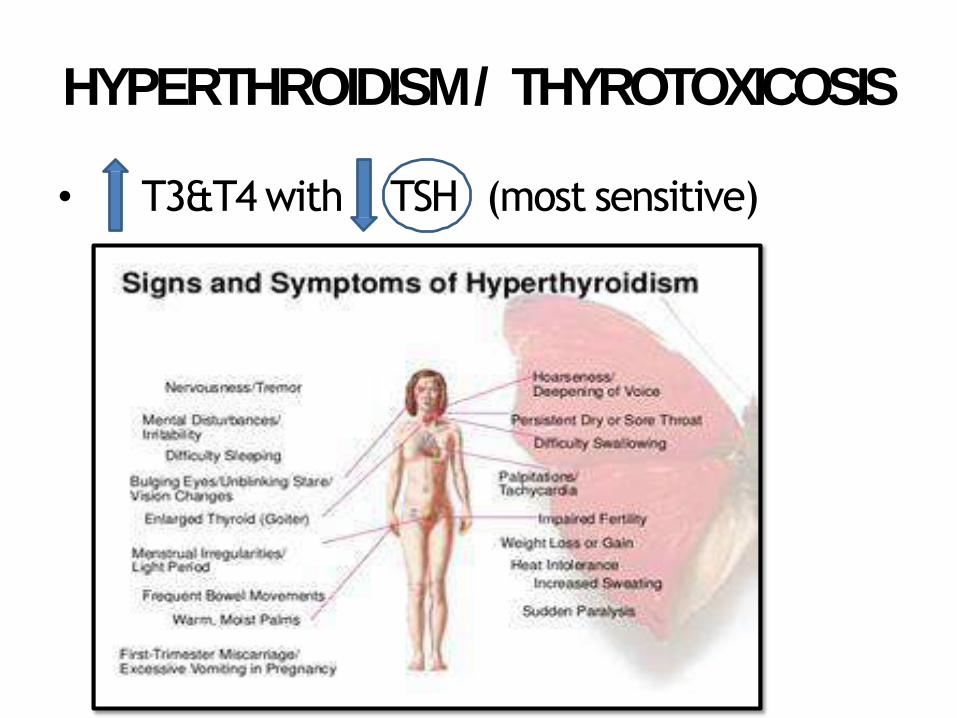
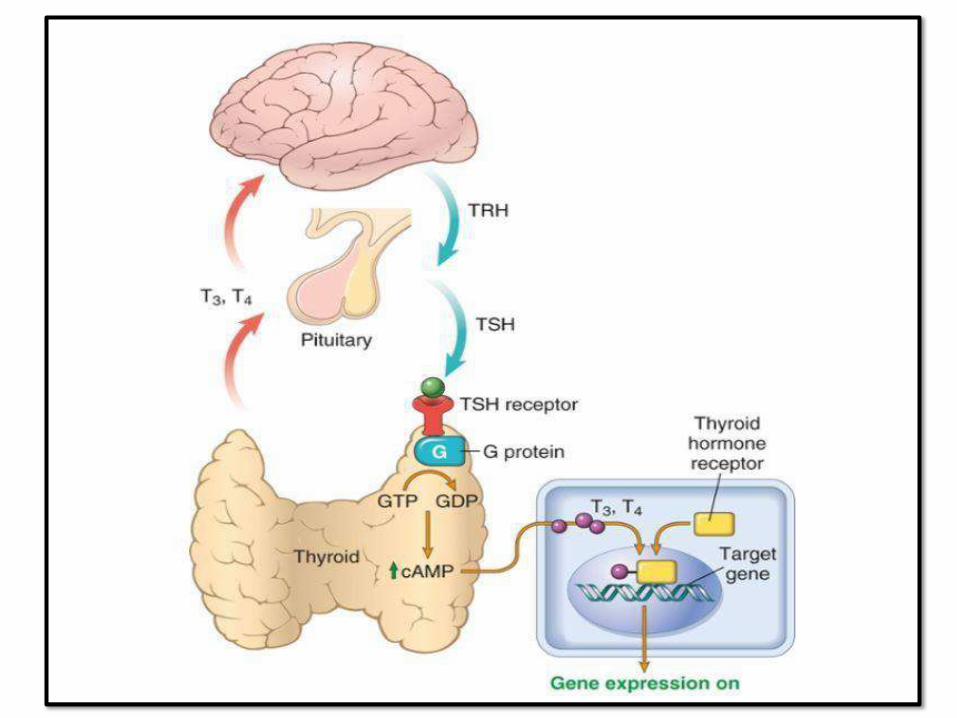
HYPERTHROIDISM / THYROTOXICOSIS
• T3&T4 with TSH (most sensitive)
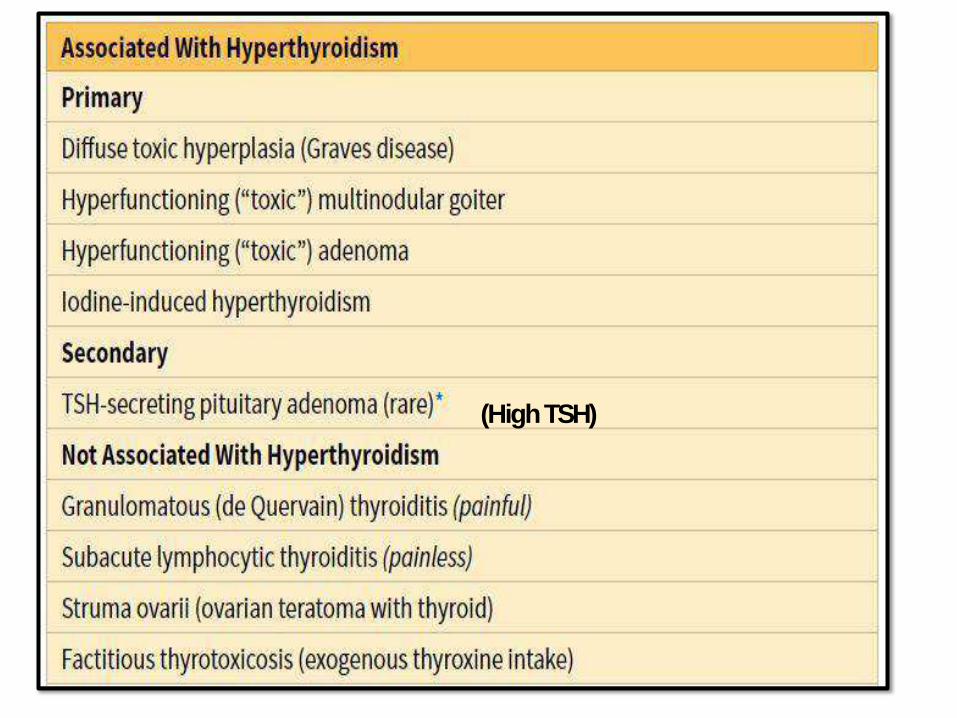
(HighTSH)
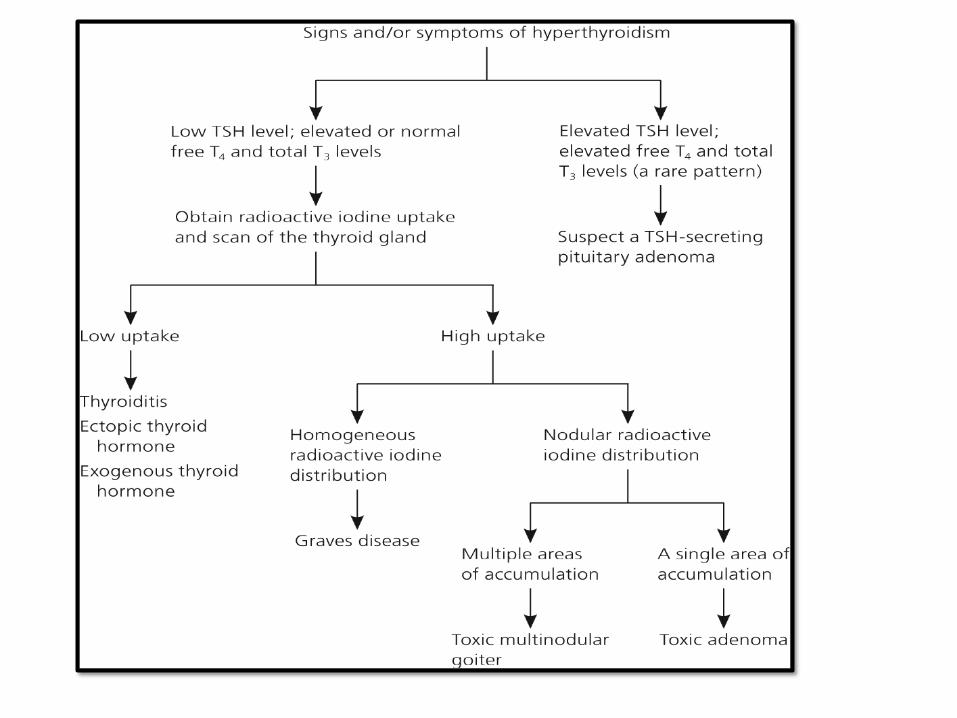
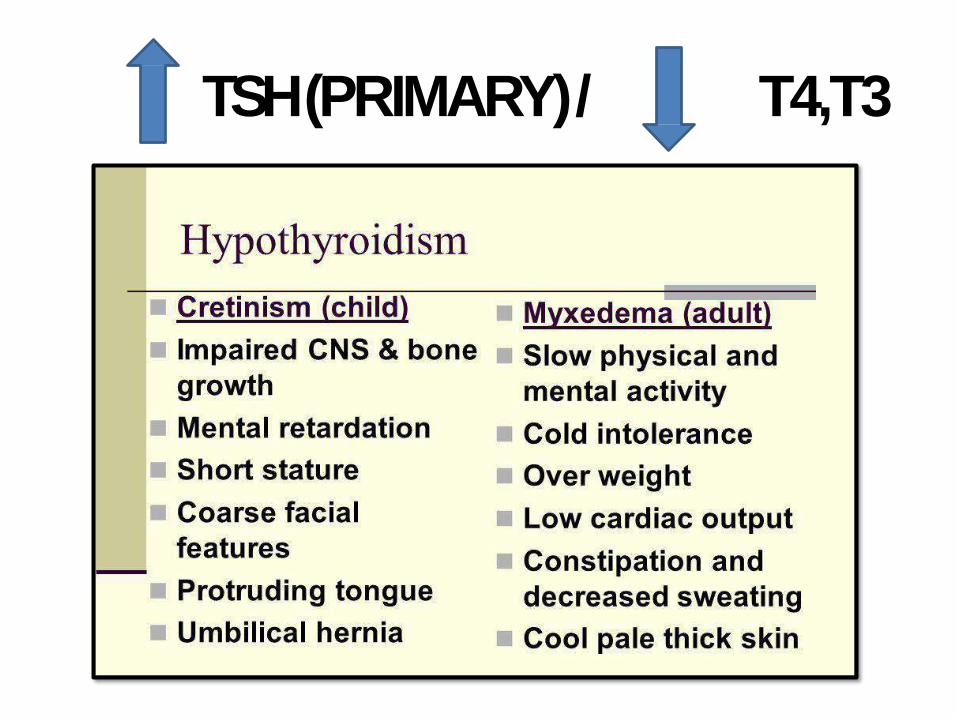
TSH (PRIMARY)/ T4,T3
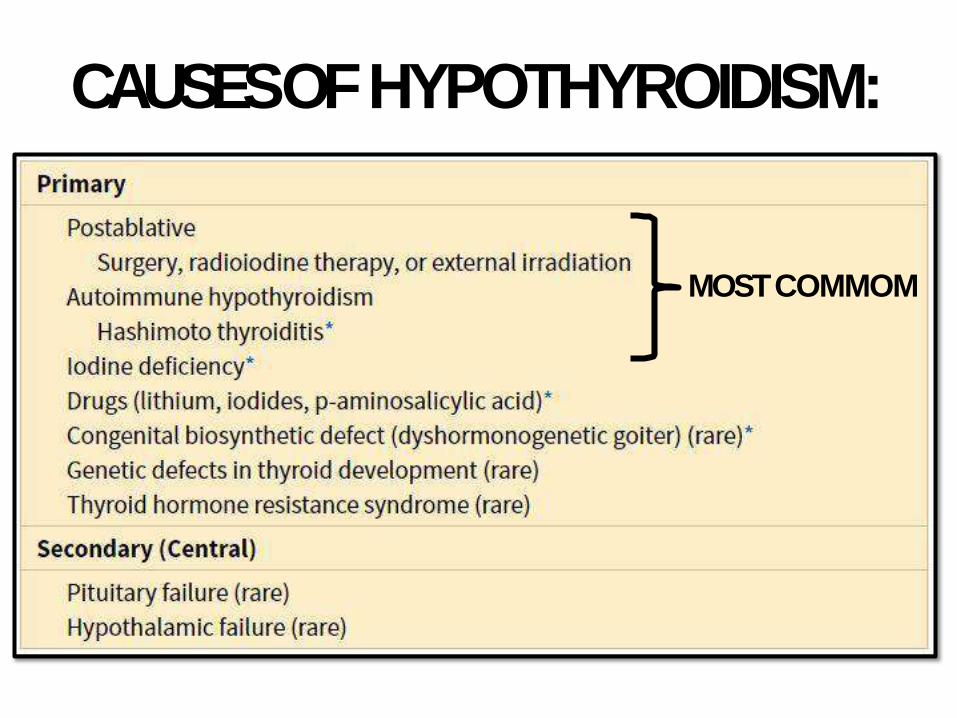
CAUSES OFHYPOTHYROIDISM:
MOSTCOMMOM
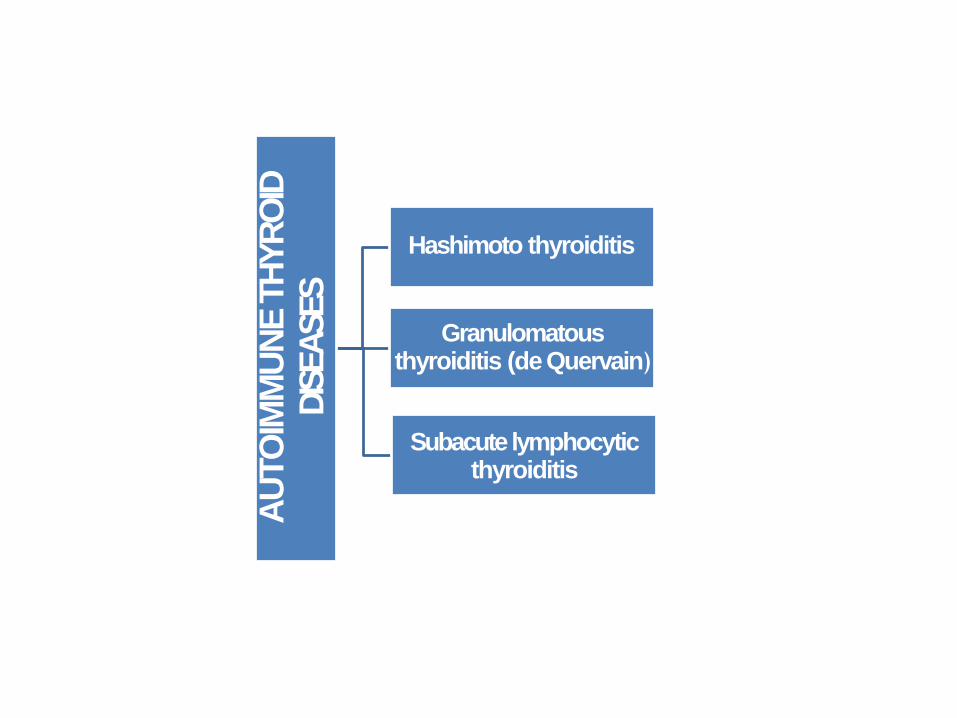
AU
TO
IMM
UN
ETH
YR
OID
DIS
EA
SES
Hashimoto thyroiditis
Granulomatous thyroiditis (de Quervain)
Subacute lymphocytic thyroiditis

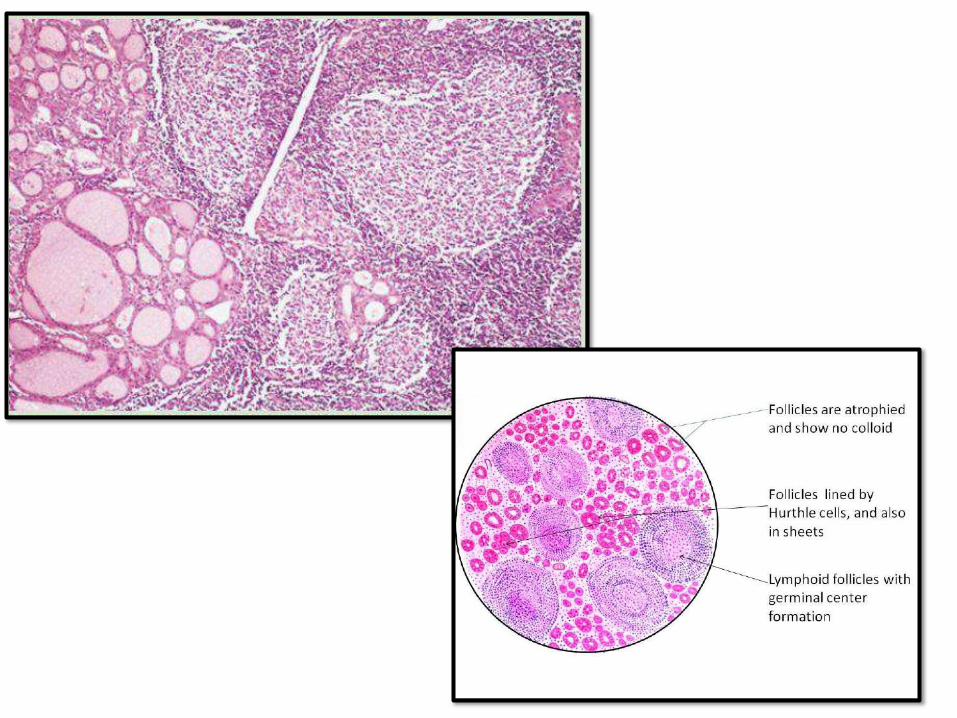
HASHIMOTO THYROIDITIS (CHRONIC
LYMPHOCYTICTHYROIDITIS)
• Most common cause of hypothyroidism in areas with no iodine deficiency
• Gradual hypothyroidism (rarely initial transient Hashitoxicosis)
• Middle aged females (45-60years)
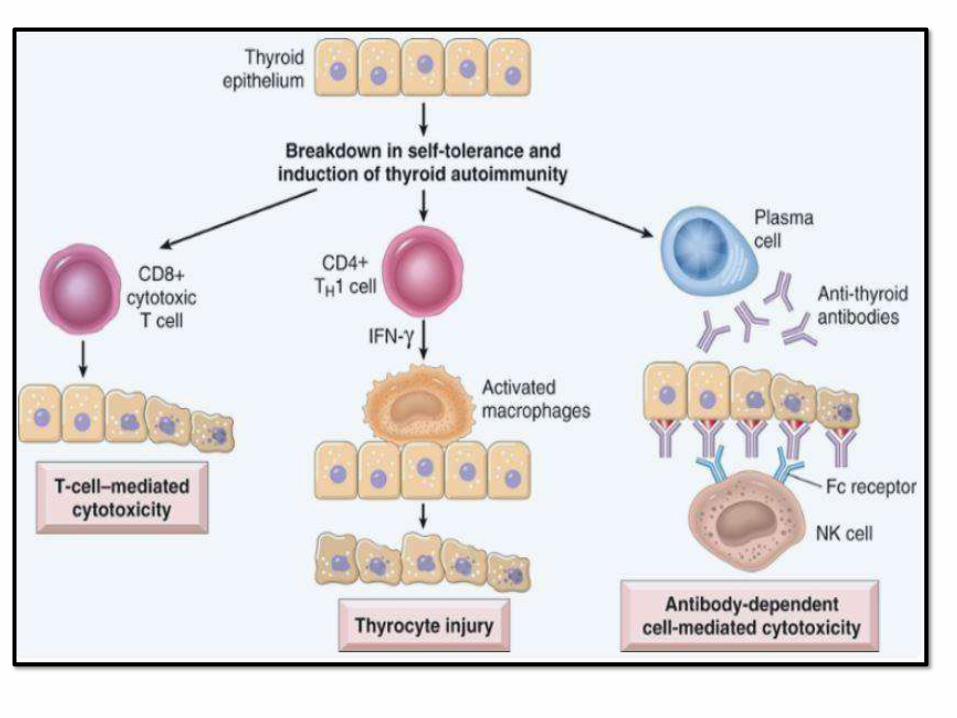
• Autoimmune destruction of thyroid epithelial cells, high anti-thyroid antibodies
• Increase risk for papillary thyroid carcinoma and B-cell NH lymphoma

SUBACUTE GRANULOMATOUS(DE
Quervain)THYROIDITIS
• Granulomatous thyroiditis, more acute
with neck pain, firm thyroid
• ? Virally associated or induced
• Females, 30-50 years
• Maybe initial transient thyrotoxicosis
followed by hypothyroidism
• Self limiting disease (6-8 weeks)

OTHER LESSCOMMONTHYROIDITIS:
• Subacute lymphocytic thyroiditis: middle
aged women, post partum, initial transient
thyrotoxicosis then gradual hypothyroidism.
Autoimmune with circulating antibodies.
Gland is usually normal size. Lymphocytic
thyroiditis
• Riedel thyroiditis: IgG4 associated disease,
stony-hard thyroid due to severe fibrosis

GRAVES DISEASE (TOXIC DIFFUSEGOITER)
• Described by Robert Graves in 1835
• Most common cause of endogenous hyperthyroidism
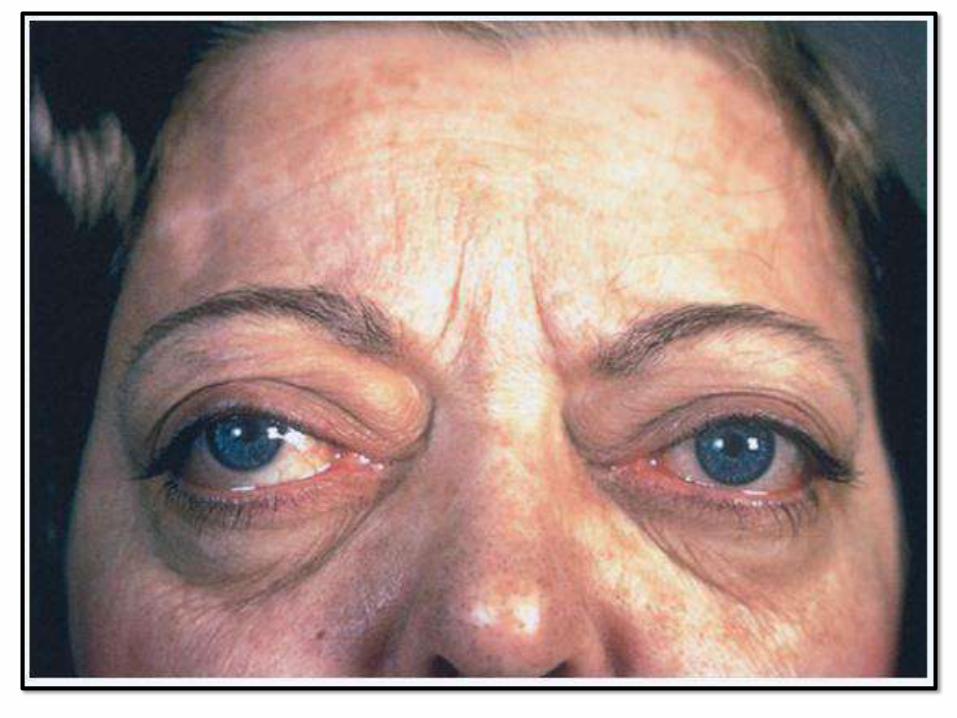
• Triad: thyrotoxicosis + opthalmopathy (exopthalmos) + dermopathy (pretibial myxedema)
• Autoimmune, HLA-DR3 andCTLA-4
• Women, 20-40 years

GRAVES DISEASE (TOXIC DIFFUSE
GOITER)
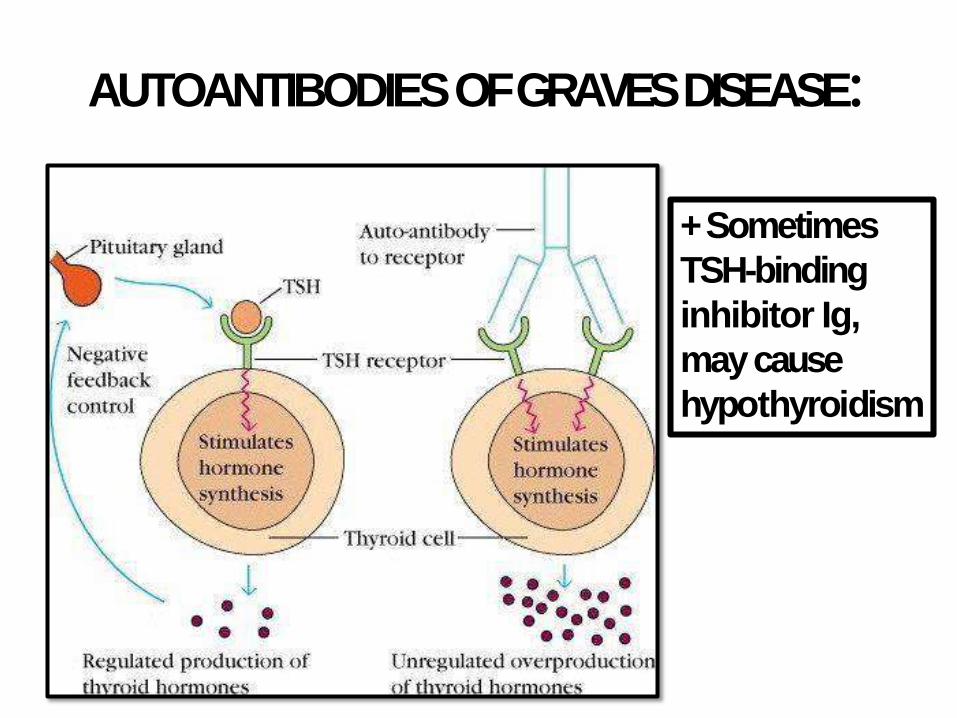
AUTOANTIBODIES OF GRAVESDISEASE:
+ Sometimes
TSH-binding
inhibitor Ig,
may cause
hypothyroidism
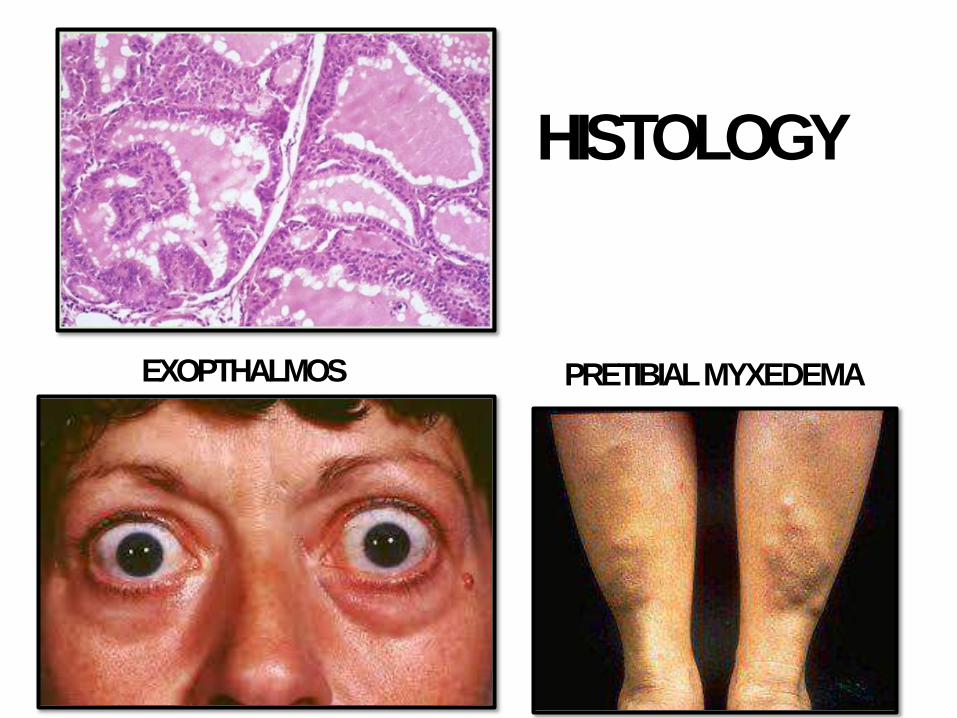
HISTOLOGY
EXOPTHALMOS PRETIBIALMYXEDEMA

DIFFUSE AND MULTINODULARGOITER:
• Very common; most common thyr disease
• Impaired hormone synthesis, iodine deficiency
• TSH, hyperplasia & hypertrophy
• In most cases; euthyroid; rarely goitrous hypothyroidism
• Endemic or sporadic. Females
• Initially diffuse then multinodular
• Clinically: mass effects andcosmetic
• Rare: toxic MNG (Plummer syndrome)
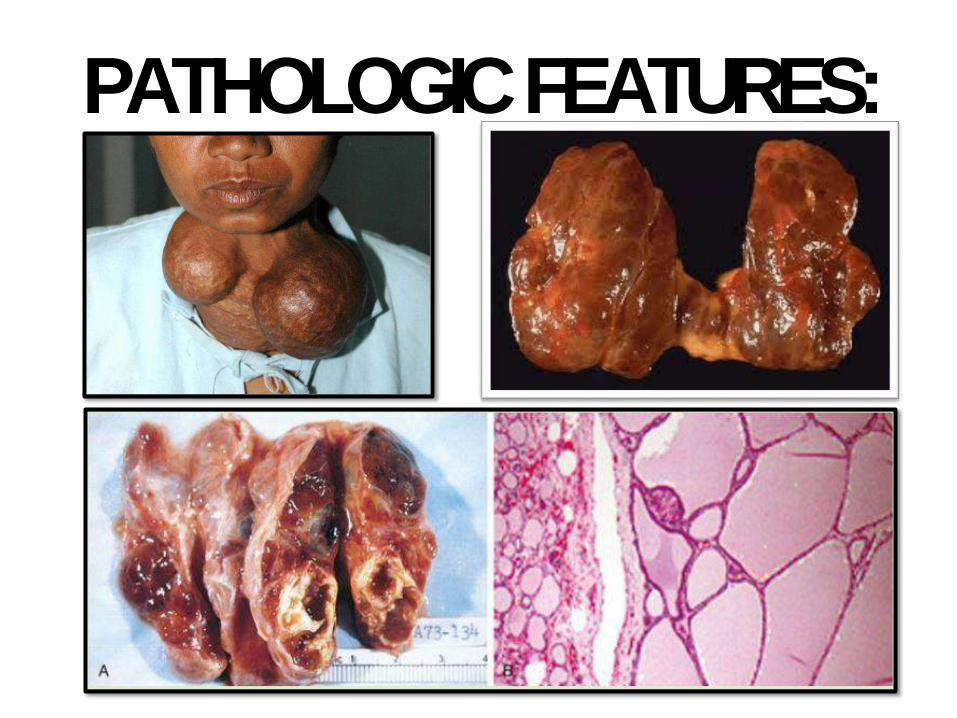
PATHOLOGICFEATURES:

THYROIDNEOPLASMS:
• Benign >>>>>>> malignant.
• Most are adenomas
• Risk increases when:
– Solitary nodule > than multiple ones
– Male nodules > than female ones
– Age < than 20 or > than 70 year
– Family Hx. And hx, of radiation
– Cold nodule >>>>>Hot nodules
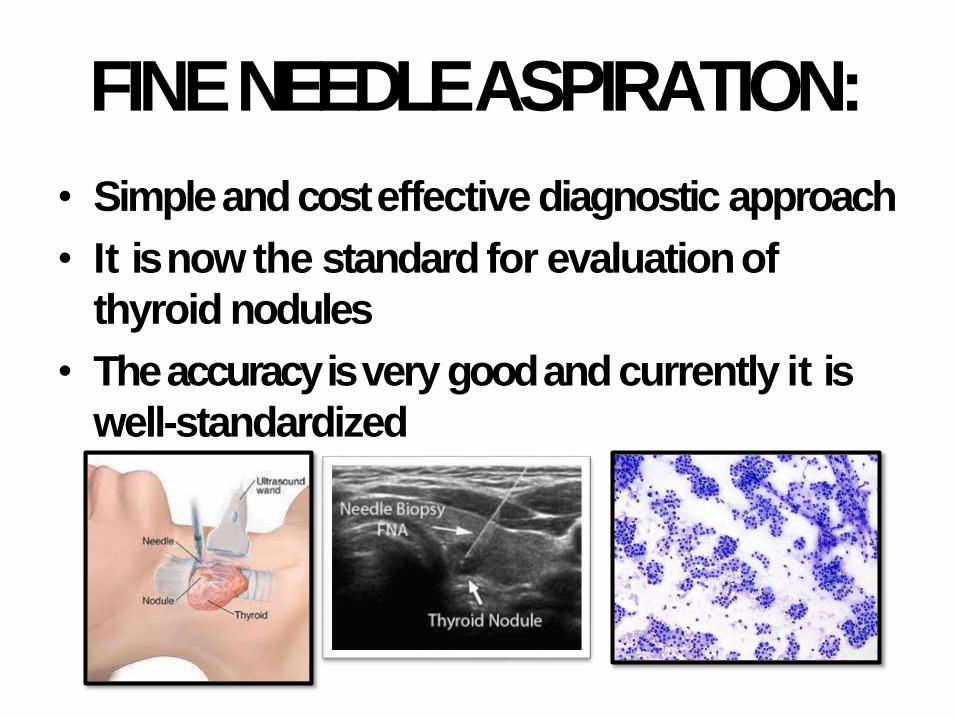
FINE NEEDLEASPIRATION:
• Simple and cost effective diagnostic approach
• It is now the standard for evaluationof
thyroid nodules
• The accuracy is very good and currently it is
well-standardized

FOLLICULARADENOMAS:
• Almost all adenomas are follicular
• Autonomous adenoma; driver mutations in
TSH stimulation; rarely RASmutations
• Solitary, well-circumscribed with intact thick
capsule. Bland cells or Hurthle cell (Hurthle
cell adenoma). Occasional atypia can be seen
• Intact capsule is the main distinguishing
feature from follicular carcinoma
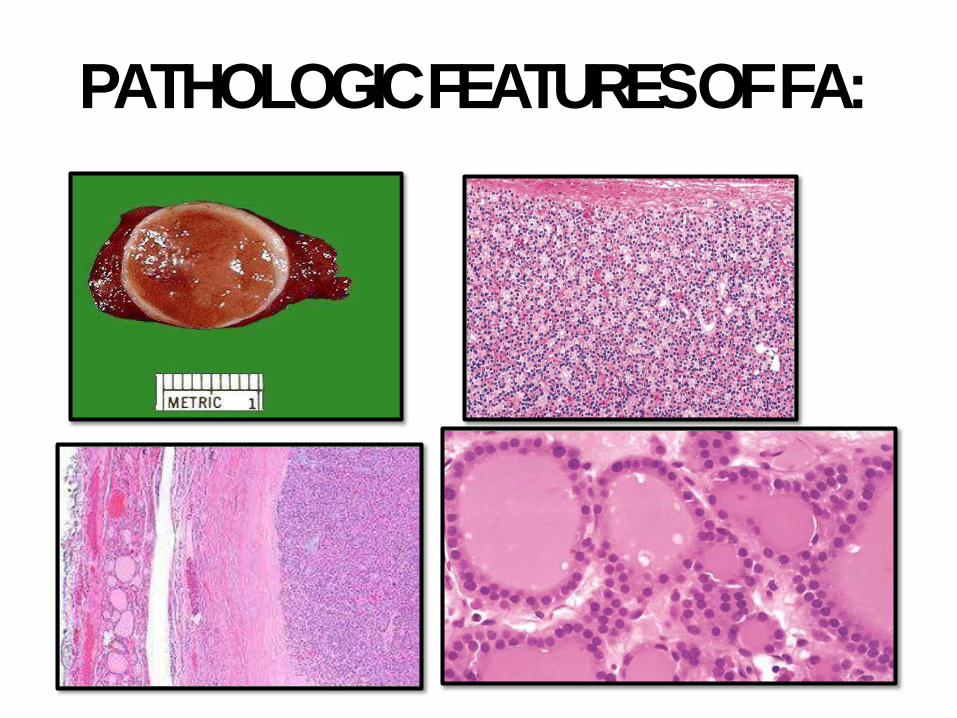
PATHOLOGIC FEATURES OFFA:
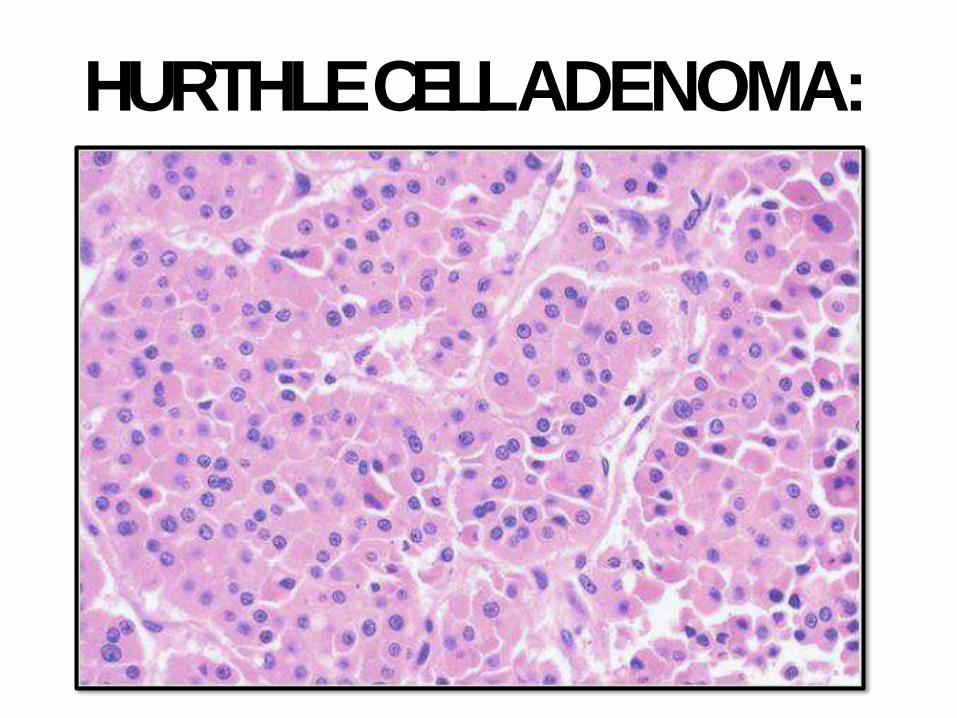
HURTHLE CELLADENOMA:

THYROIDMALIGNANCIES:
• Relatively common but not aggressive
• More common in females
• Risk factors: ionizing radiation (Chernobyl
1986) and iodine deficiency
PAPILLARYCARCINOMA 85% LYMPH NODEMETASTASIS
FOLLICULARCARCINOMA 5-15% HEMATOGENOUSSPREAD
ANAPLASTICCARCINOMA < 5%; VERYAGGRESSIVE
MEDULLARY CARCINOMA (C-CELLS) 5%, MAYBE PART OF MEN2SYNDROMES
LYMPHOMA 1% B CELL NONHODGKIN
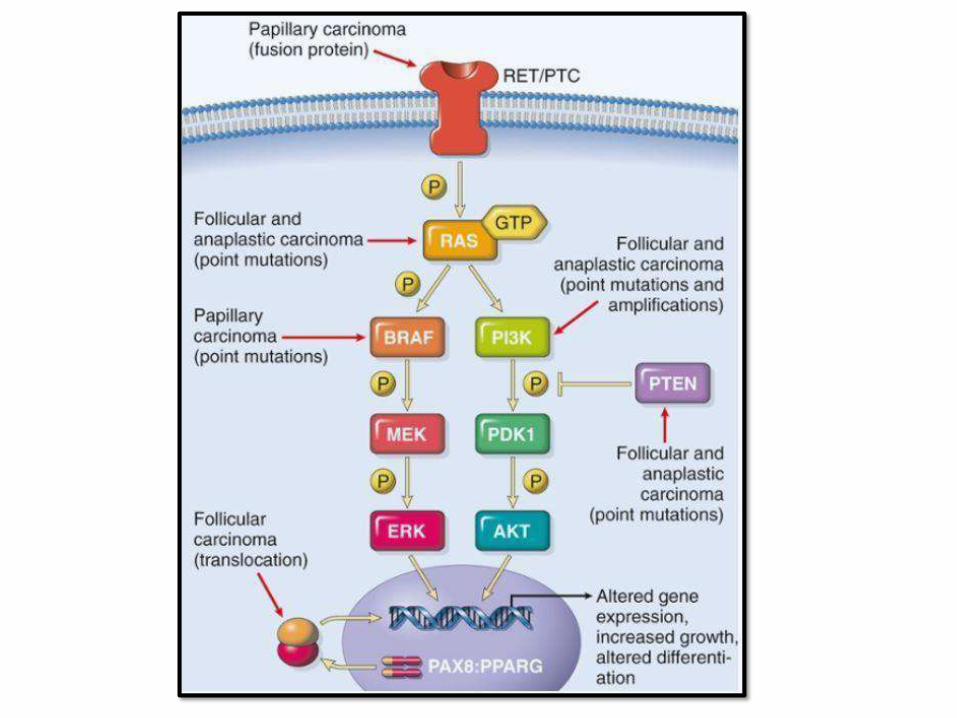

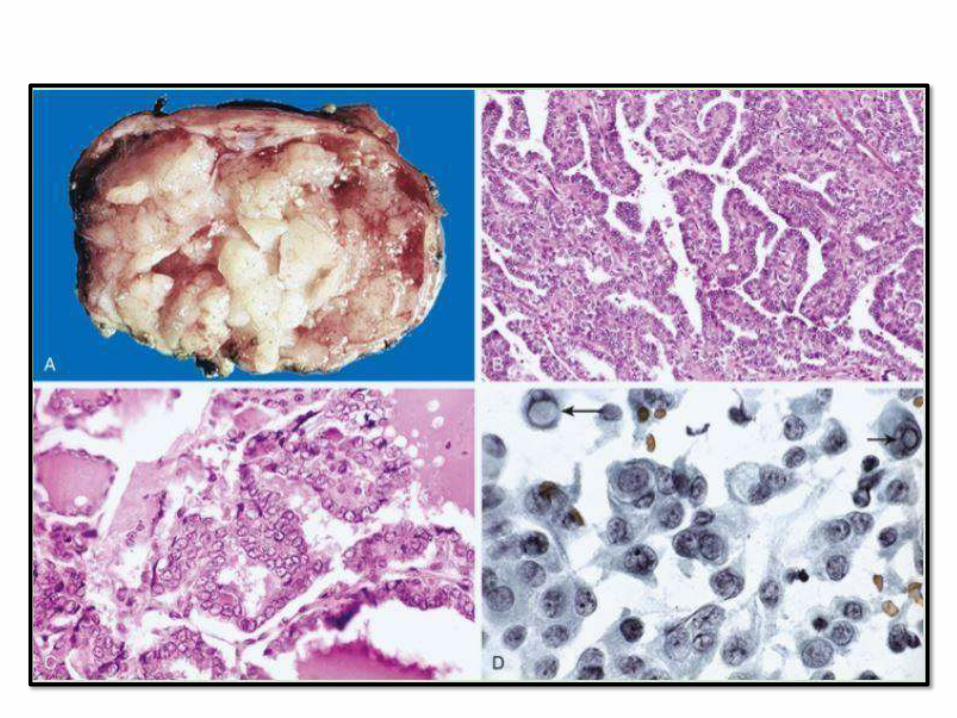
PAPILLARY THYROID CARCINOMA
• Most common
• Relatively indolent, 10 year survival > than 95%, even with lymph node metastasis
• Uni and multifocal
• Preoperative dx by FNA isaccurate
• Nuclear features most important
• Features: papilae, nuclear grooves, pseudonuclear inclusions, psammoma bodies, Orphan Annie eye nuclei
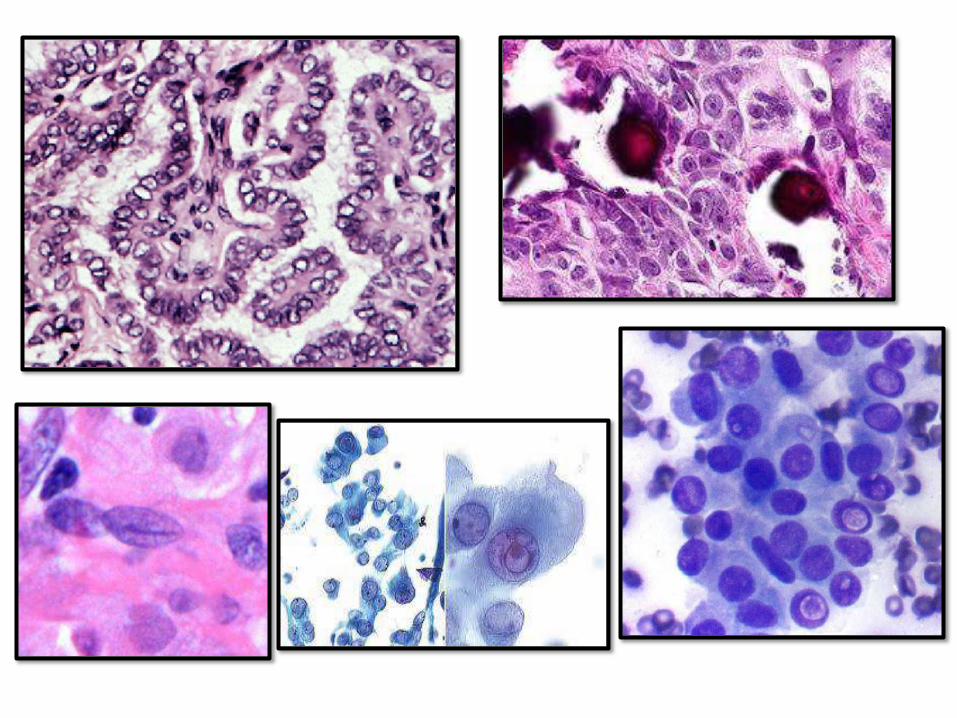

FOLLICULARCARCINOMA
• Women, 40-60 years
• > common in iodine deficient regions
• Solitary coldnodule
• Hematogenous spread to bone, lung and liver
• 50% die within 10 years
• Capsular and vascular invasion is the
distinguishing feature from F.adenoma
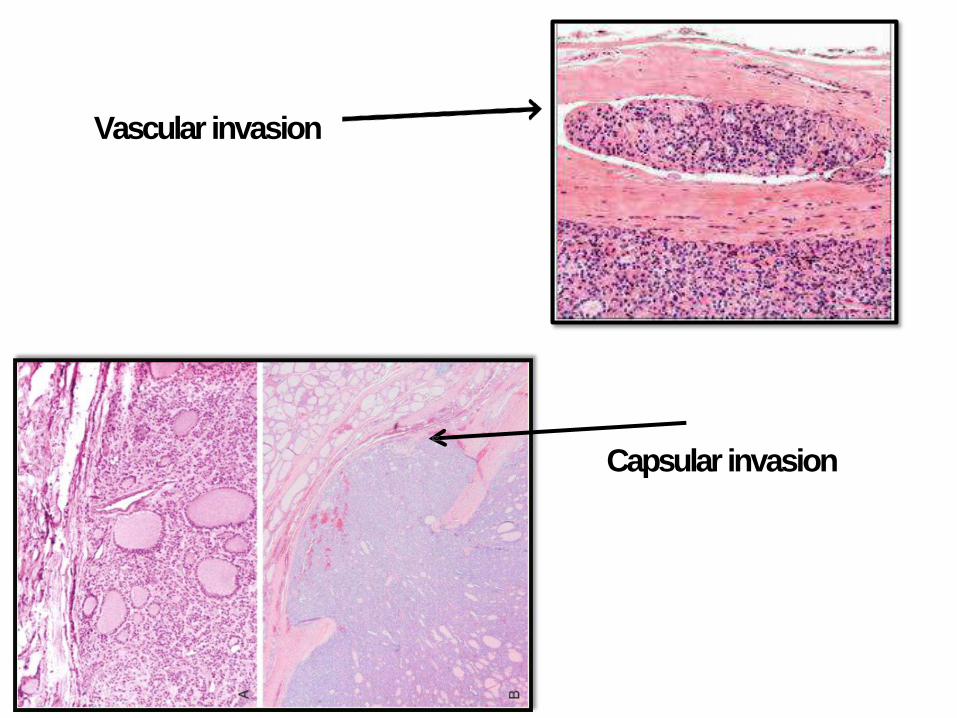
Capsular invasion
Vascular invasion
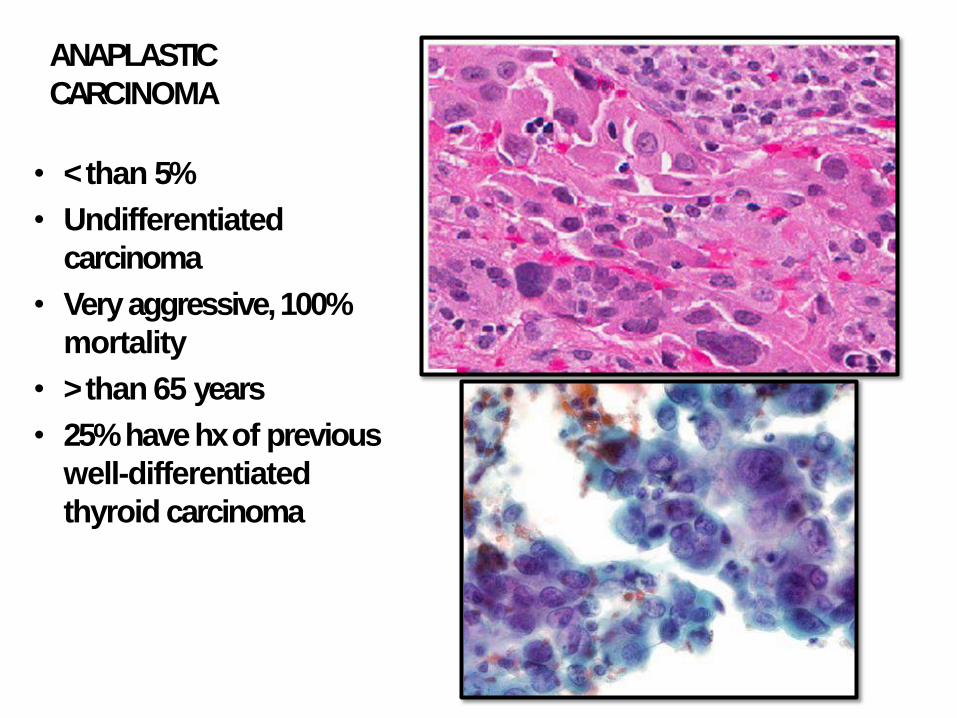
ANAPLASTIC
CARCINOMA
• < than 5%
• Undifferentiated
carcinoma
• Very aggressive, 100%
mortality
• > than 65 years
• 25% have hx of previous
well-differentiated
thyroid carcinoma

MEDULLARYCARCINOMA:
• Arise from C cells (parafollicular cells) that secretes Calcitonin (increase level and hypercalcemia)
• 70% sporadic, 30% familial (MEN 2A&B)
• RET receptor tyrosine kinasemutations
• Sporadic 50-60 years; familial younger
• Multicentric, contain amyloid
• RET +ve family members require prophylactic thyroidectomy
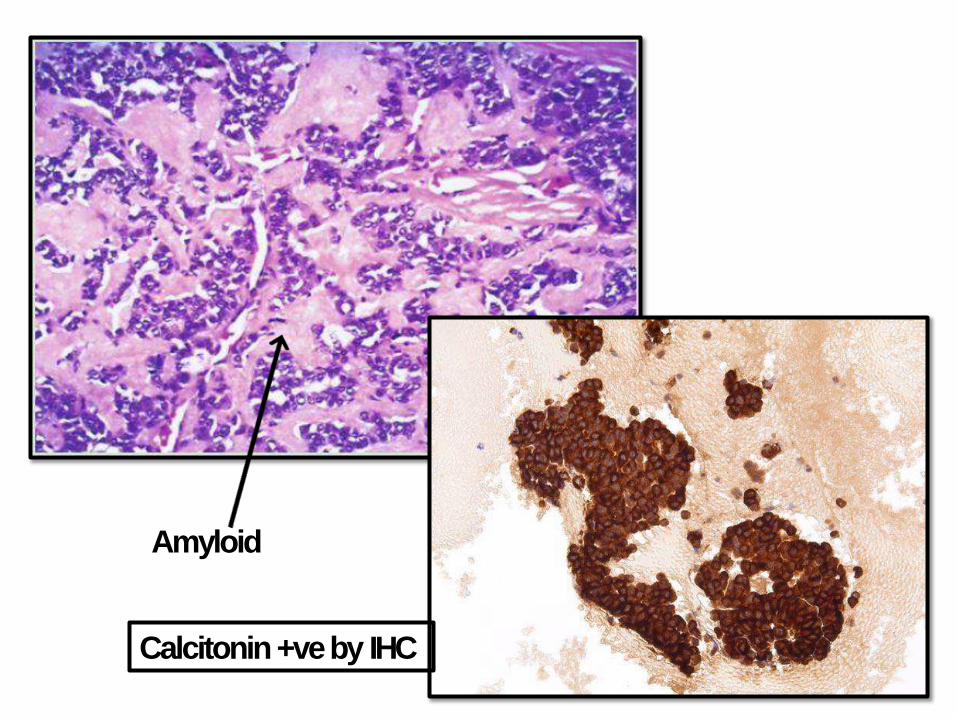
Amyloid
Calcitonin +ve by IHC

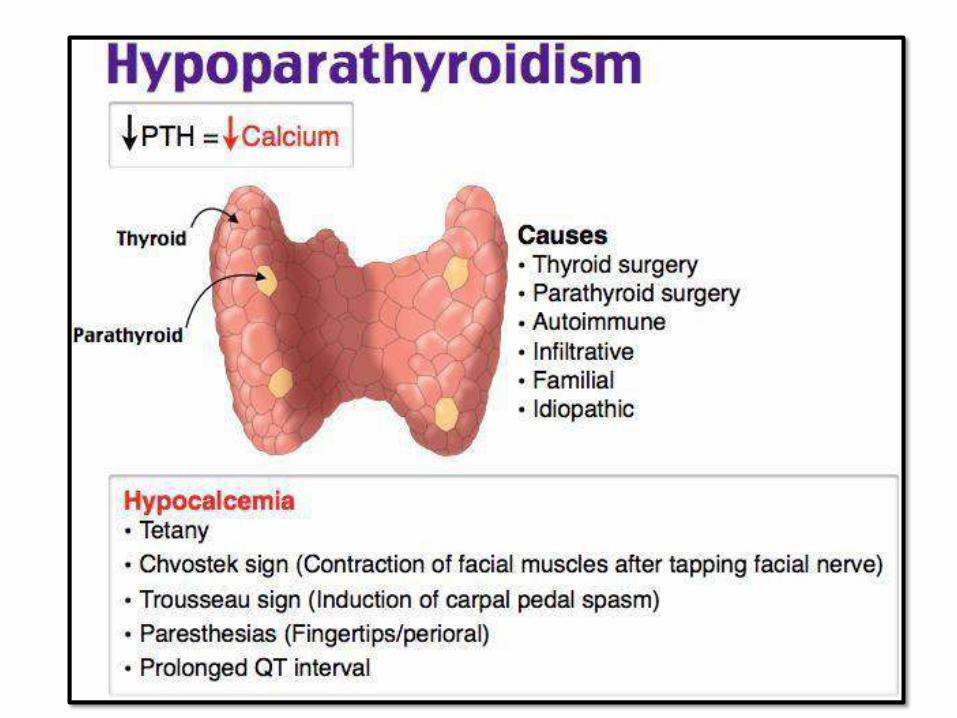
PARATHYROIDGLAND:• PTH secreted by Chief cells.
• Controlled mainly by free C+2 level in serum less than trophic hormones
• Hyper, Hypo and tumors (rare mass effects)
• Functions of PTH:
– Reabsorption of Ca from renal tubules
– Excretion of PO4 into urine
– Vit D conversion to active form
– Stimulates osteoclast activity on bone resorption

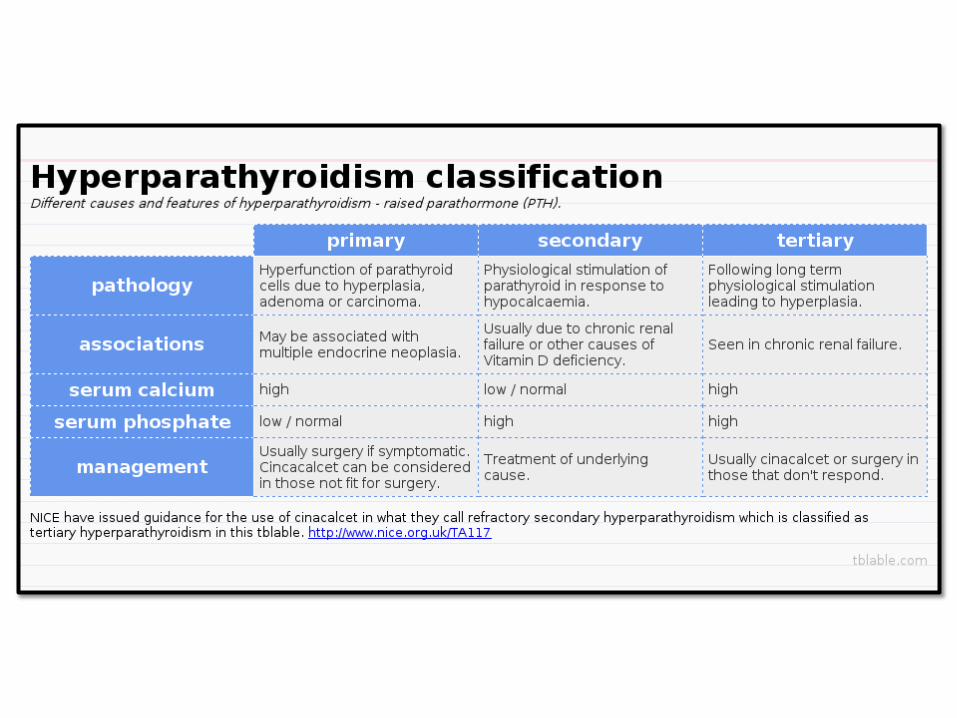
HYPERPARATHYROIDISM:
• Primary, secondary and tertiary
• Osteitis fibrosa cystica, Brown tumor of bone, nephrolithiasis, nephrocalcinosis and metastatic calcifications
• Primary HPT:
–Adenomas (85-95%, Hyperplasia (5-10%), carcinoma(1%)
–Mutations: Cyclin D1 gene on chromosome 1 or MEN1 mutations
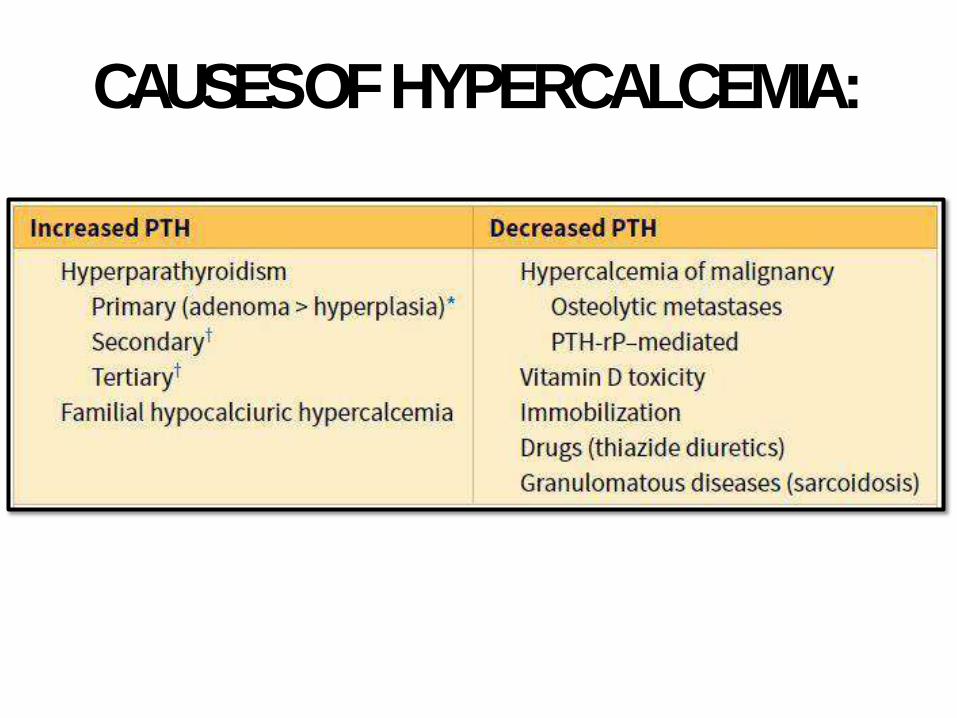
CAUSES OFHYPERCALCEMIA:
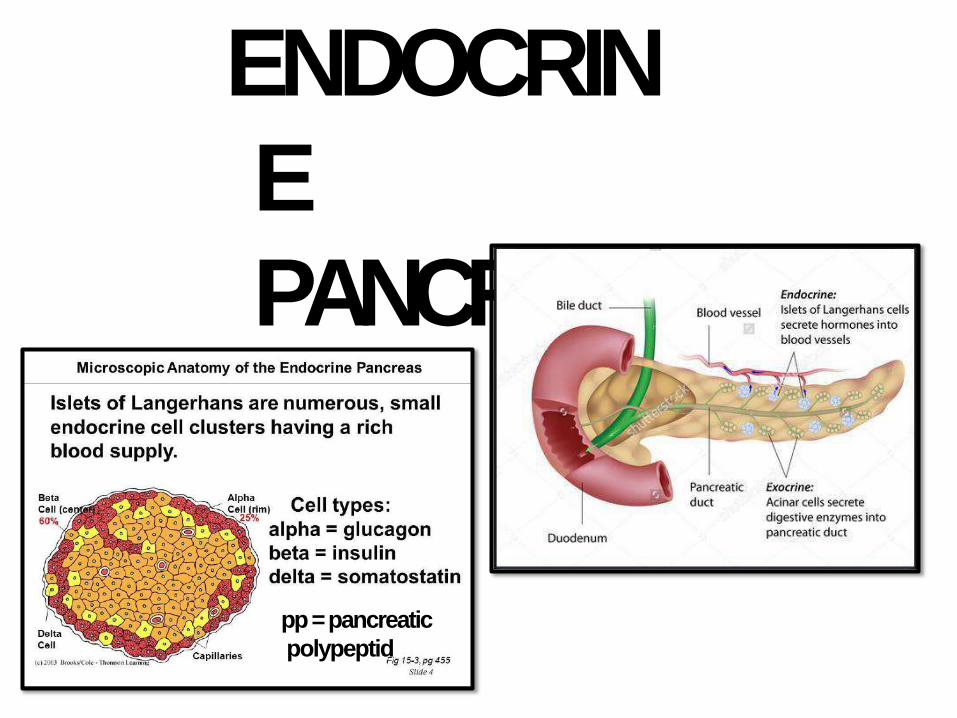
ENDOCRIN
E
PANCREAS
pp = pancreatic
polypeptid

DIABETESMELLITUS:• A group of systemic metabolic disorders
characterized by chronic hyperglycemia
• Chronic hyperglycemia = multiple organ
damage
• Leading cause of ESRD, adult blindness, non-
traumatic amputations (USAdata)
• Prevalence: very common (see graphs)
• Huge health cost worldwide
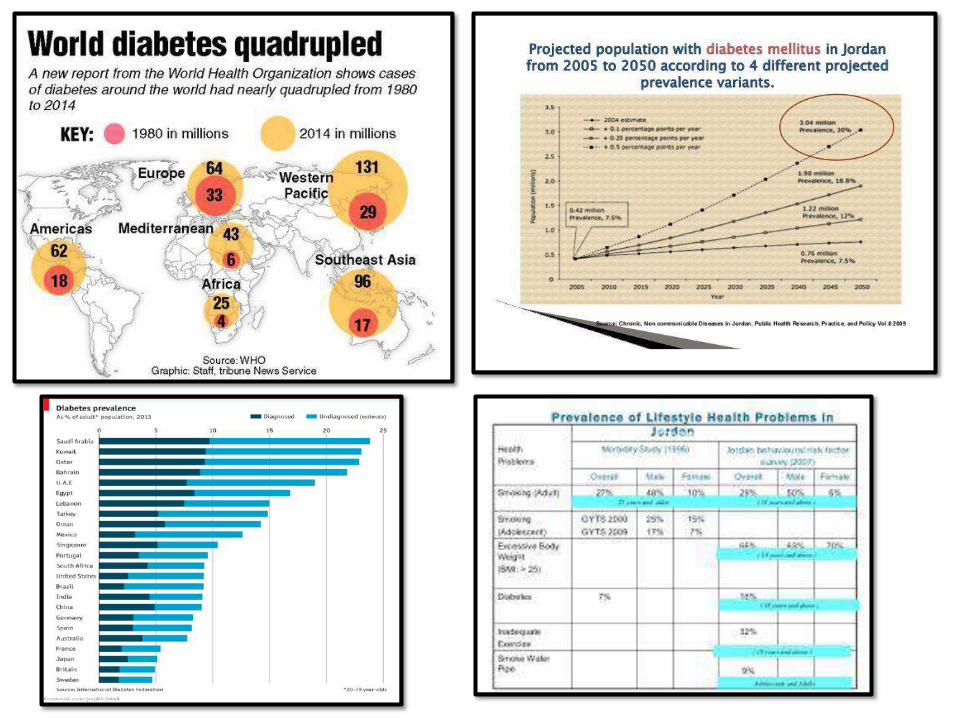

Diabetes: A global
emergency
IDF 2016 : The 7th
Atlas
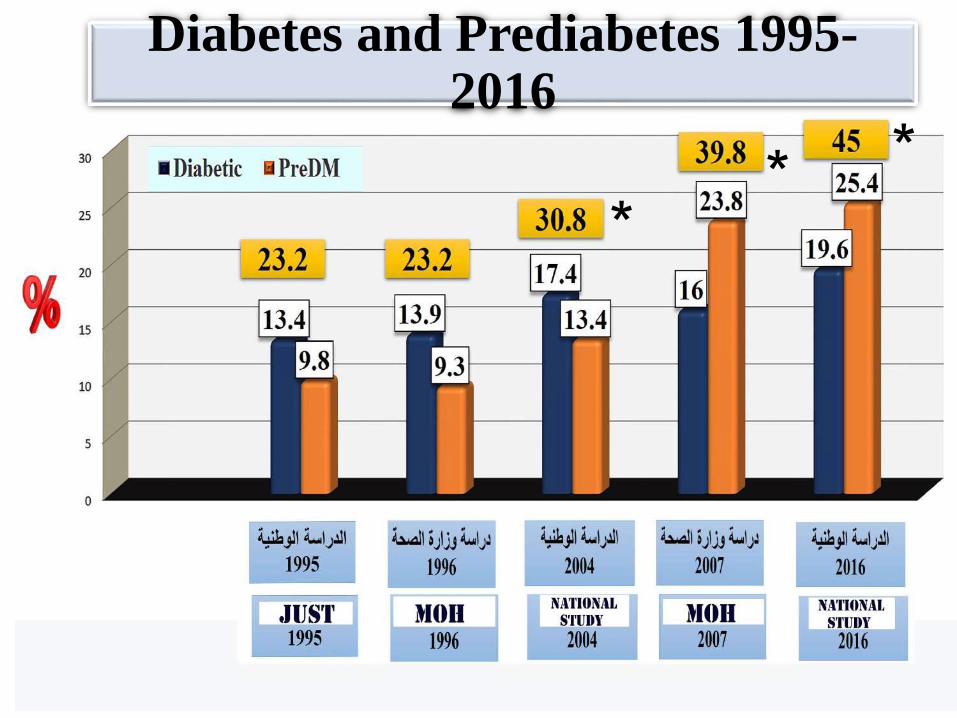
Diabetes and Prediabetes 1995-2016
** *

DIAGNOSIS:

CLASSIFICATION:5-10%
90-95%

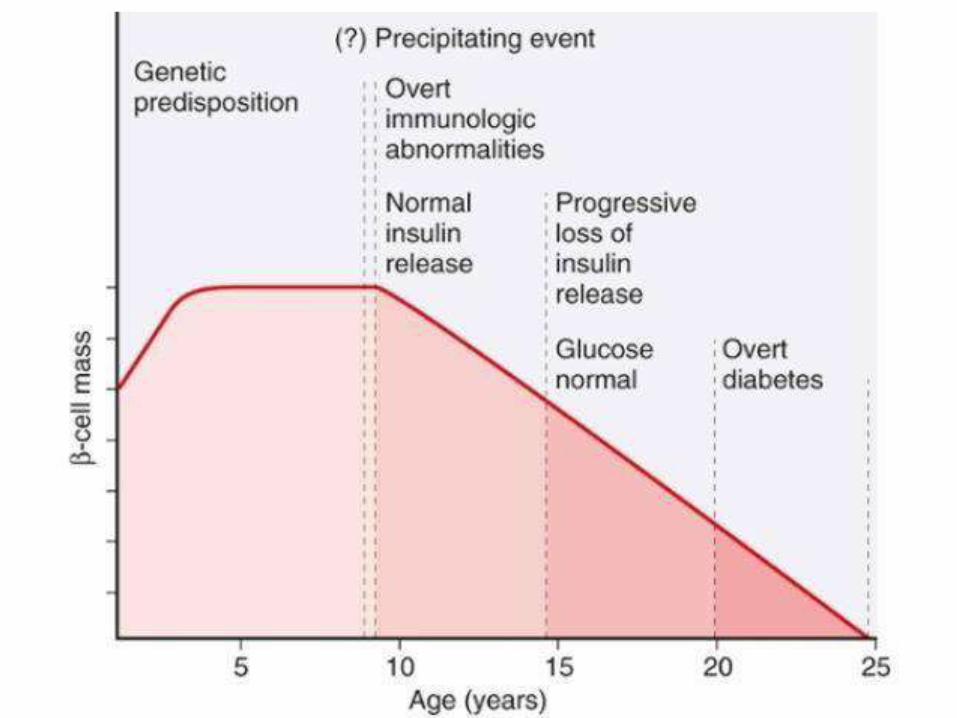
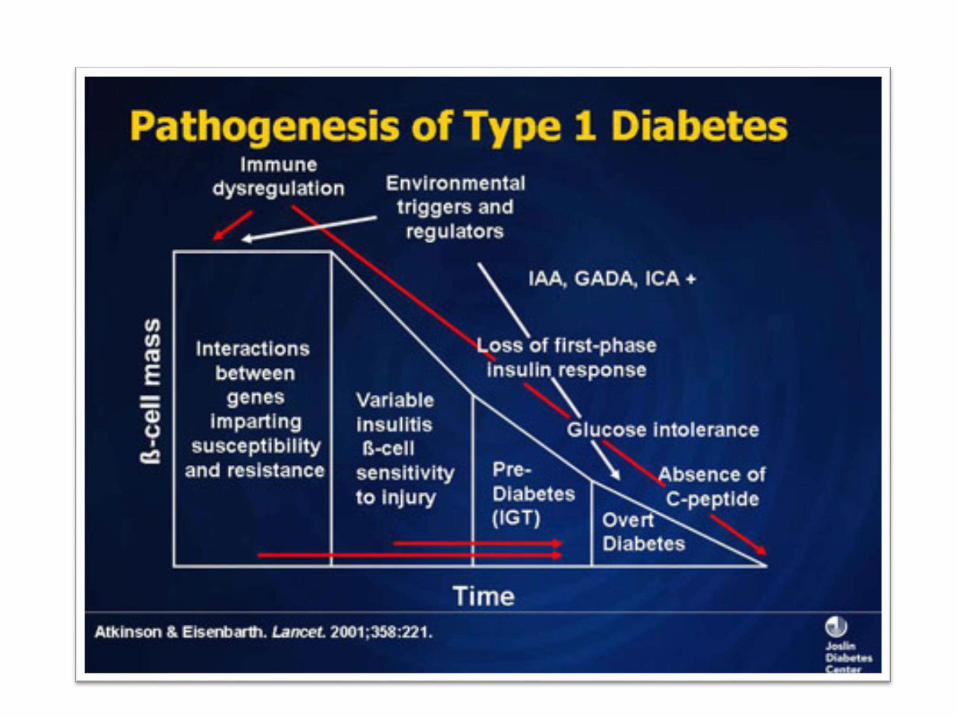
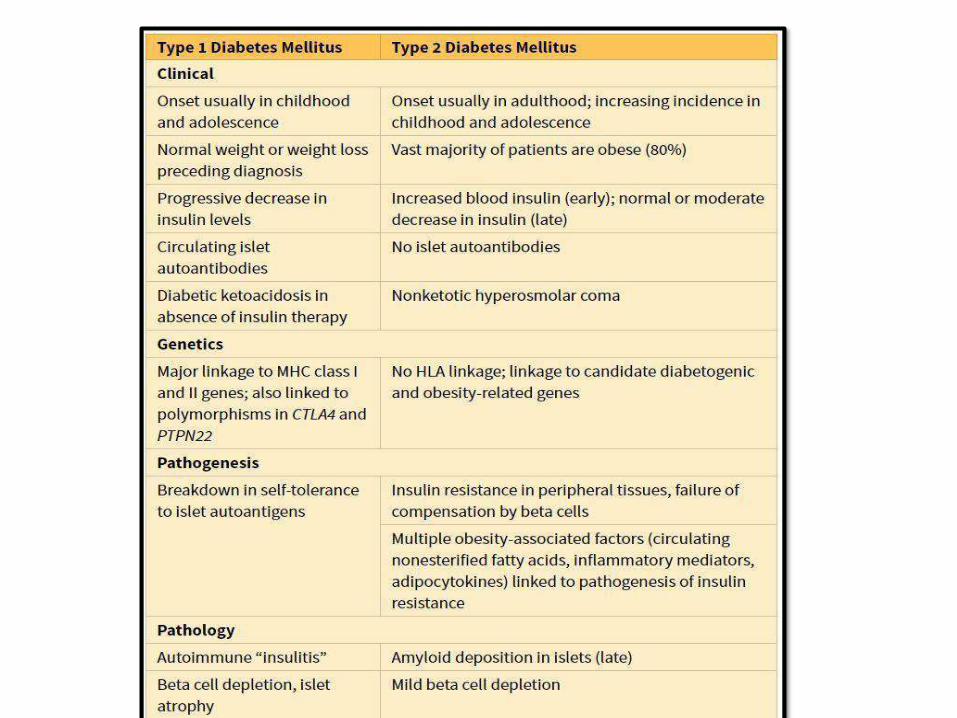
PATHOGENEISIS OF TYPE I DM:
• Autoimmune disease; destruction of beta
cells, “insulitis” (ultimately insulindeficiency)
• Genetic susceptibility: HLA-DR3/4 and other
non-HLA genes CTLA4 & PTPN22
• Environmental factors: viruses (mumps,
rubella & coxackie B); intestinal dysbiosis
• Failure of self tolerance (regulatory T-cells:
Tregs) auto AB against beta cellantigens

PATHOGENEISIS OF TYPE 2DM:
• Multifactorial: genetics, environmental
and inflammation (NO
AUTOIMMUNITY)
• Insulin resistance + beta cell
dysfunction
• Insulin resistance is increased with
obesity; specially central
obesity
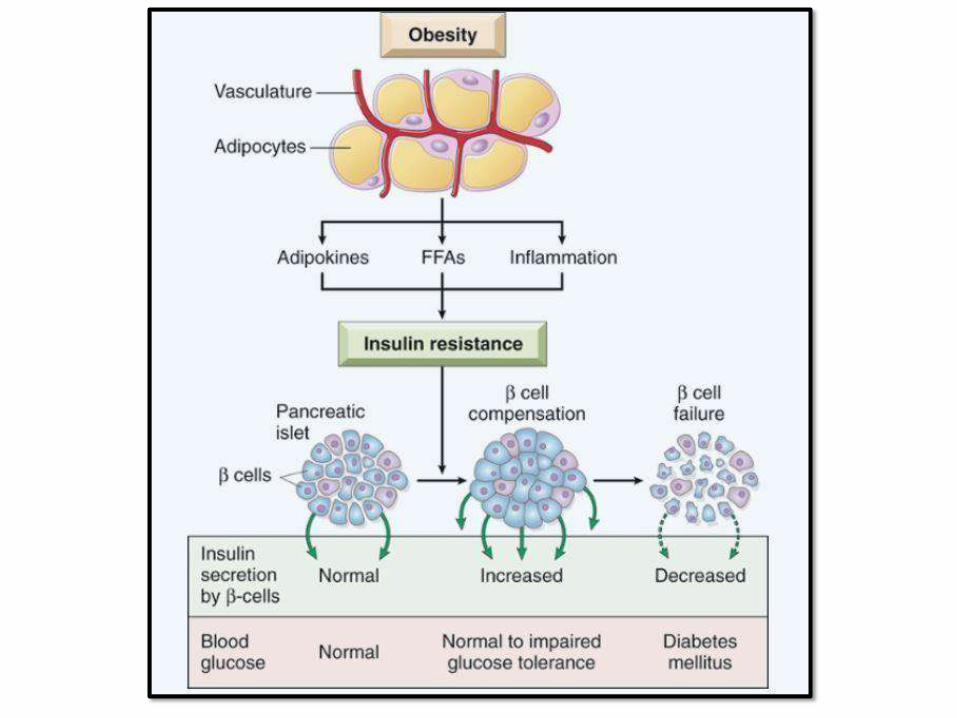
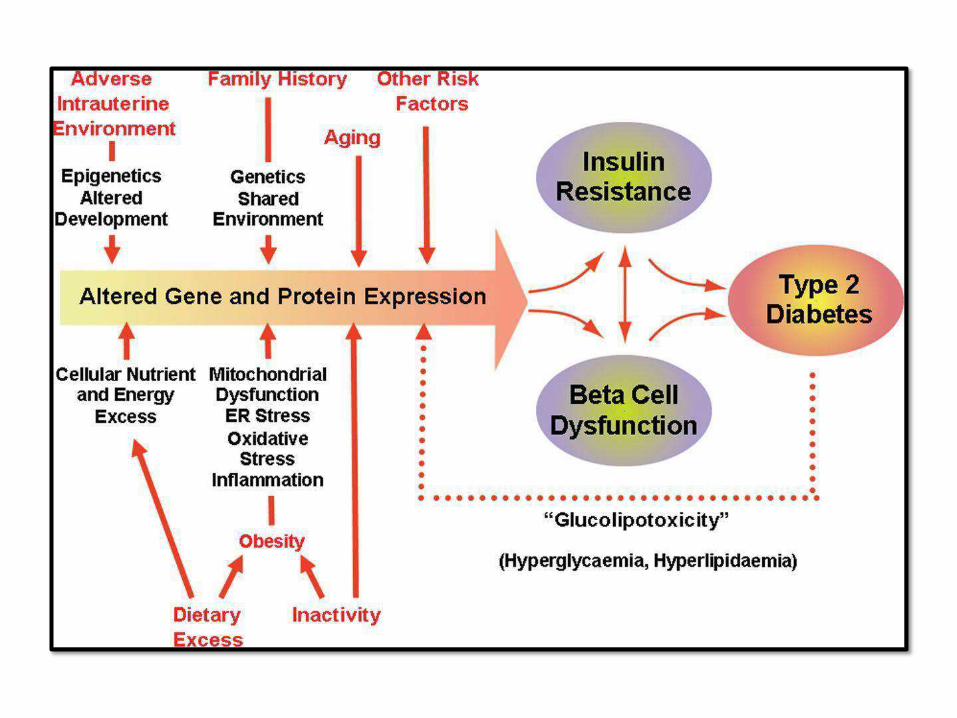

BETA CELLDYSFUNCTION:
1. Excess FFA(lipotoxicity)
2. Chronic hyperglycemia (Glucotoxicity)
3. Abnormal incretin effect
4. Amyloid deposition
5. Polymorphisms in genes that control
insulin

MATURITY ONSET DIABETES OF THE YOUNG(MODY):
• Resemble type 2 but occurs in young
patients, < than 25 years of age
• Gene mutations of beta cell function
CONGENITAL EARLY ONSETDIABETES
• Neonatal period diabetes
• Insulin receptor signaling mutations (insulin
resistance and hyperinsulinemia)
PANCREATICOGENICDIABETES:
• Hyperglycemia due to exocrine pancreas disease
• Heterogeneous: CF, Ch. Pancreatitis and Ca pancreas

GESTATIONALDIABETES:
• 5% of pregnancies (diabetogenicstate)
• Mainly insulin resistance due to increased
steroid hormones of pregnancy
• High risk of stillbirth if no early control
• Later in pregnancy: baby with macrosomia
• May need insulin to control sugarlevel
• Long term: higher risk ofDM
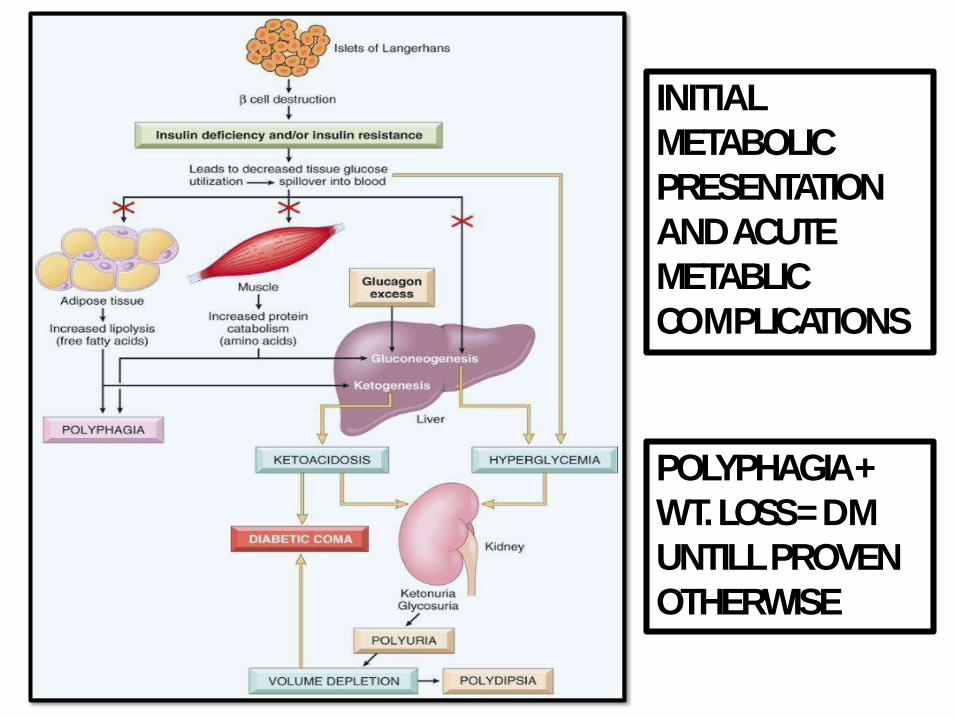
INITIAL
METABOLIC
PRESENTATION
AND ACUTE
METABLIC
COMPLICATIONS
POLYPHAGIA +
WT. LOSS = DM
UNTILL PROVEN
OTHERWISE

CHRONIC COMPLICATIONS OFDM:
• Result from chronic hyperglycemia
• Macrovascular and microvascular
• Macrovascular: MI, Stroke, LLischemia
• Microvascular: retinopathy, nephropathy and
neuropathy
• Varies among patients
• Tighter control of sugar delays almost all
these complications
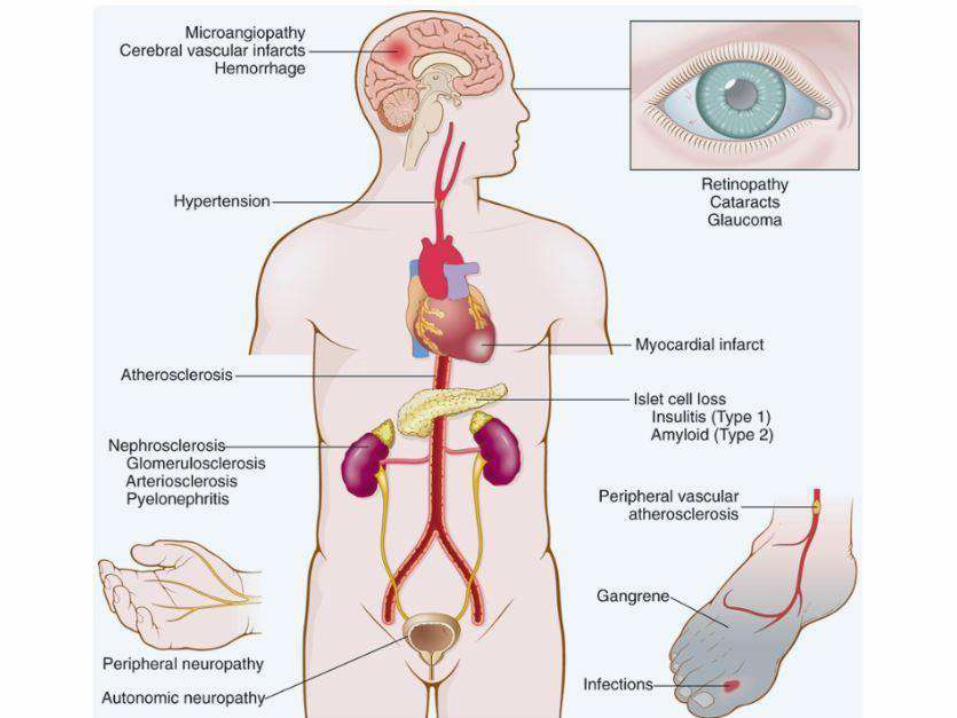

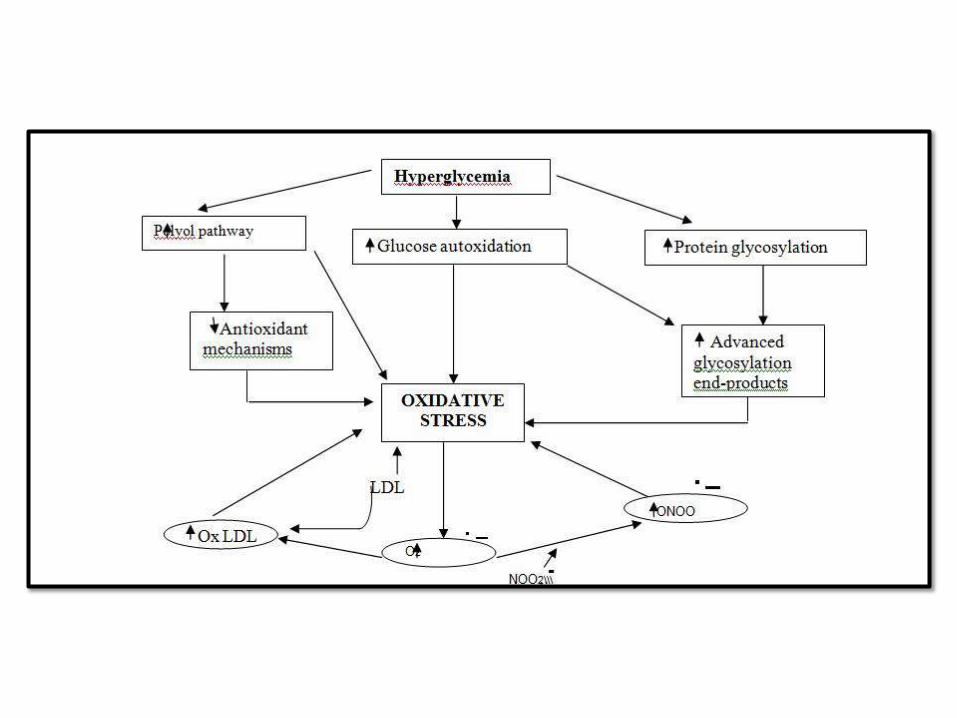
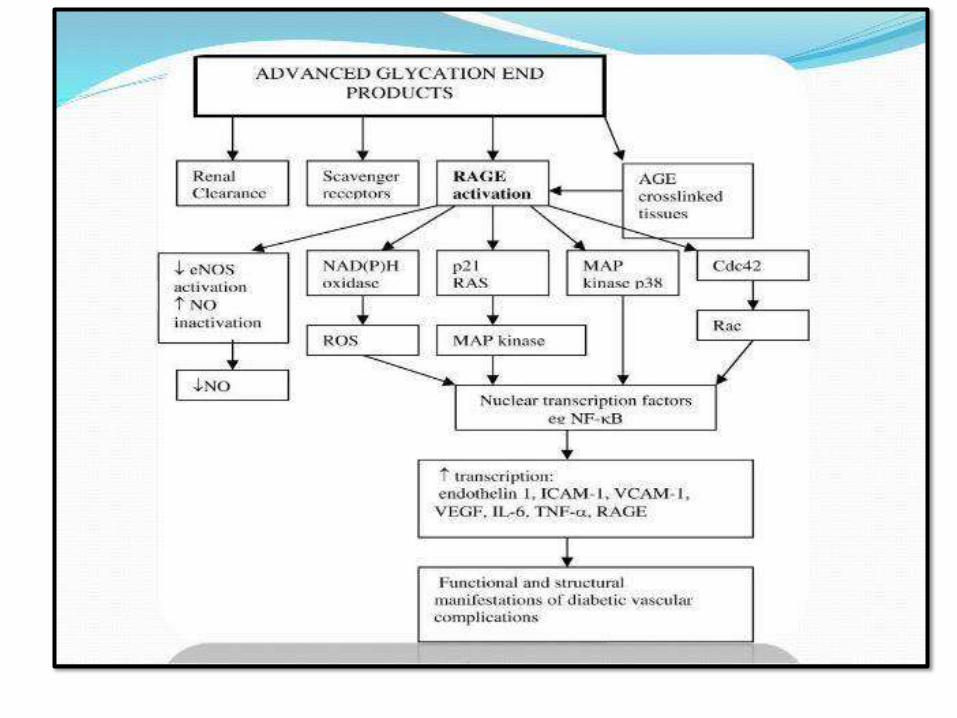
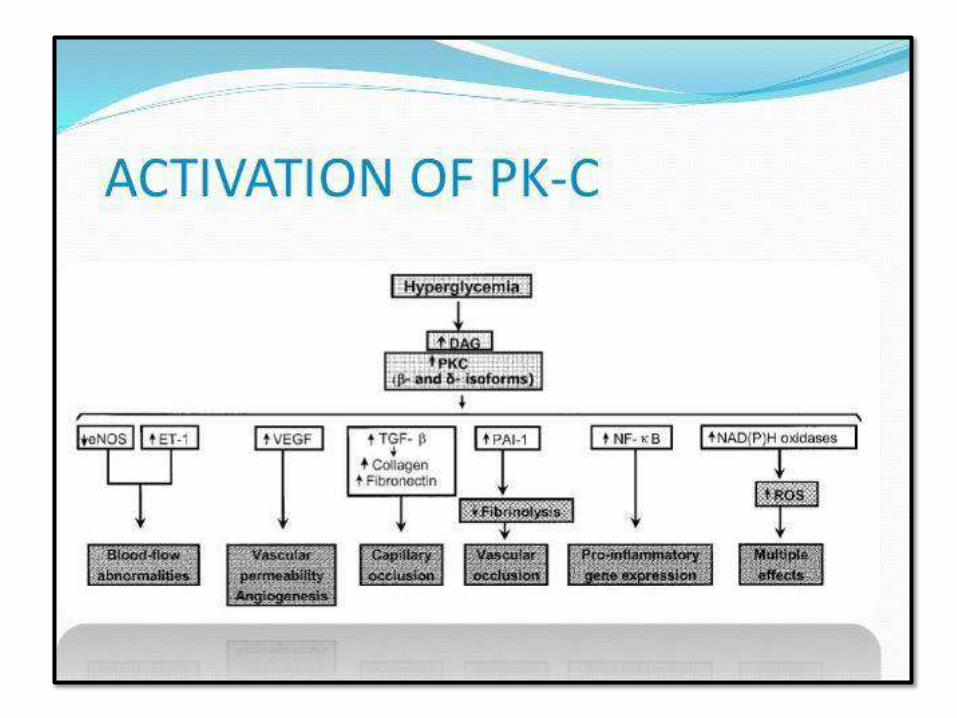
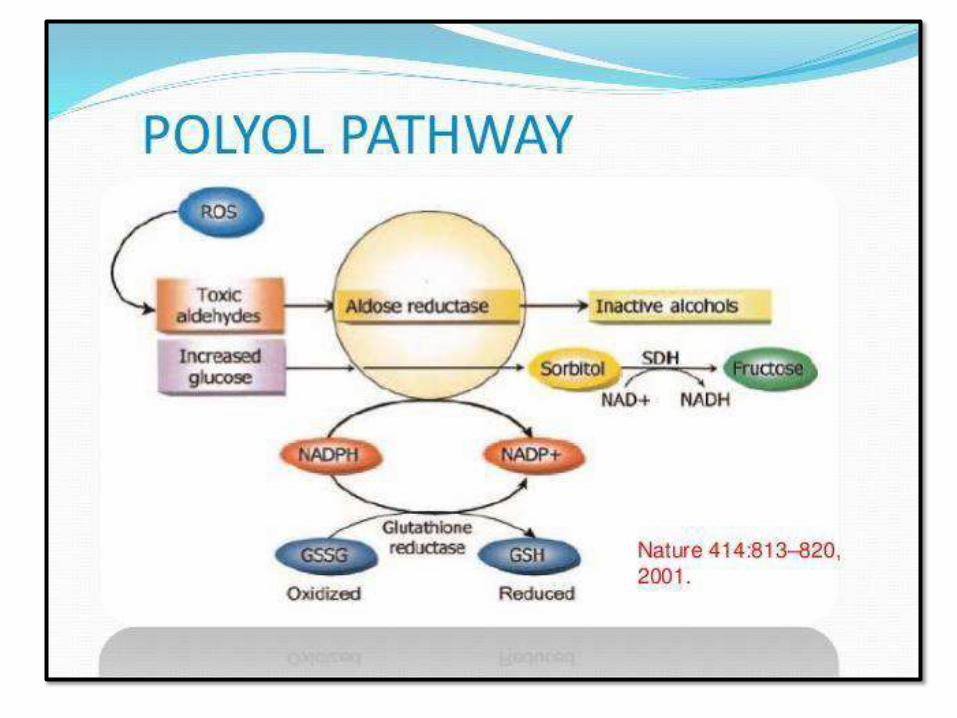
PATHOGENESIS OF CHRONIC COMPLICATIONS
(GLUCOTOXICITY):
1. Advanced glycation end products (AGEs):
AGE-RAGEsignaling
– CK andTGFβ
– Procoagulant activity
– ROS
– Smooth muscle proliferation and matrix
2. Activation of protein kinase C:DAG
3. Disturbance of polyol pathways: GSH
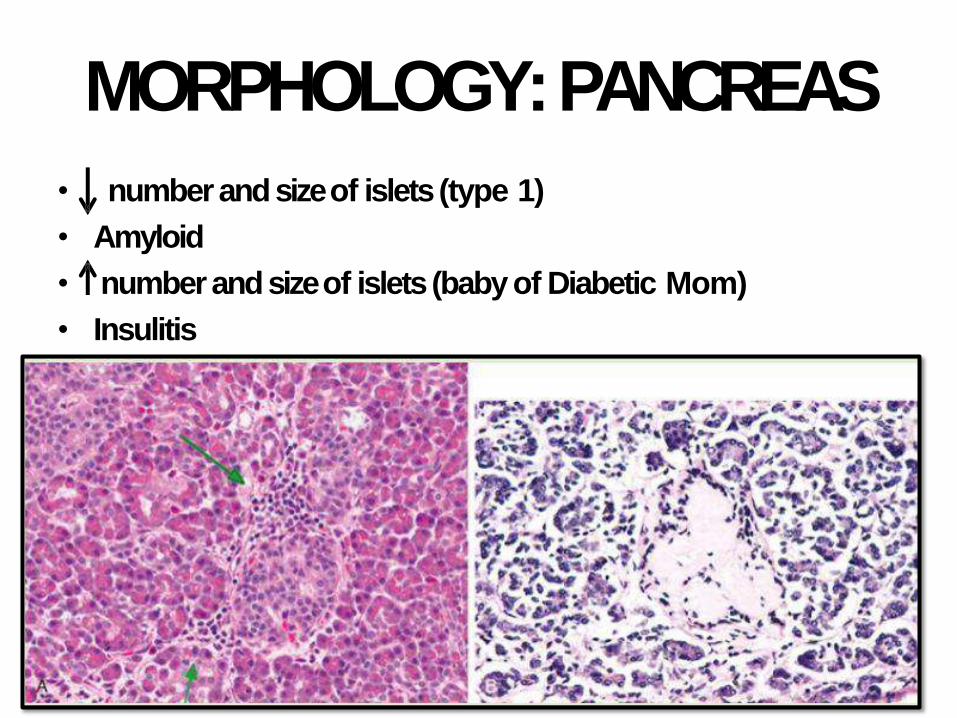
MORPHOLOGY:PANCREAS
• number and size of islets (type 1)
• Amyloid
• number and size of islets (baby of Diabetic Mom)
• Insulitis

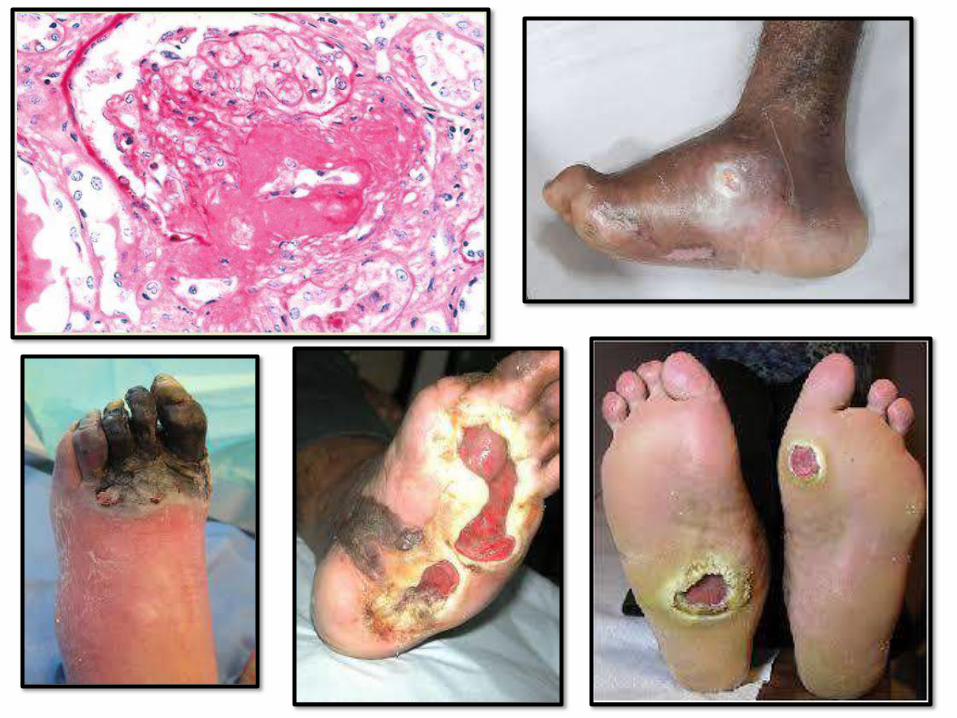
VASCULARCHANGES:
• Myocardial infarction: most common cause
of death in diabetics (accelerated
atherosclerosis)
• Gangrene and ischemia of lower extremities
• Hyaline arteriosclerosis (increase risk of HT)
• Diabetic microangiopathy: diffuse BM
thickening. Underlines diabetic retinopathy,
nephropathy and neuropathy

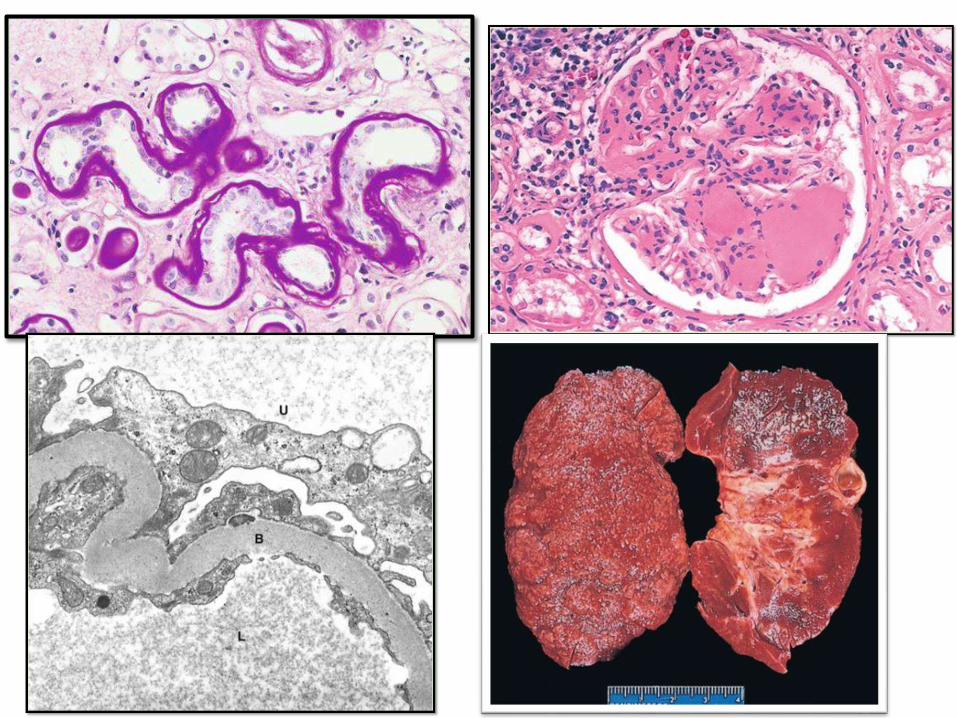
D. NEPHROPATHY: second to MI as a
cause of mortality
• Glomeruli: BM thickening, diffuse
mesangial sclerosis & nodular
glomerulosclerosis (Kimmelstiel-Wilson
lesion)
• Vessels: atherosclerosis and
arteriolosclerosis
• Pyelonephritis: acute and chronic and
occasionally necrotizing papillitis
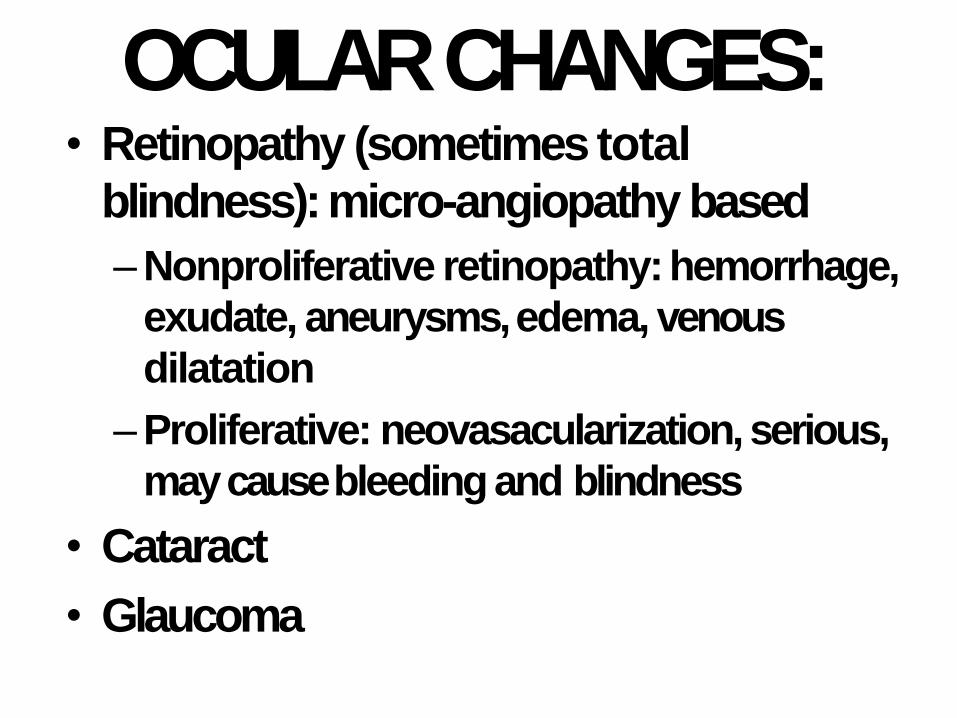
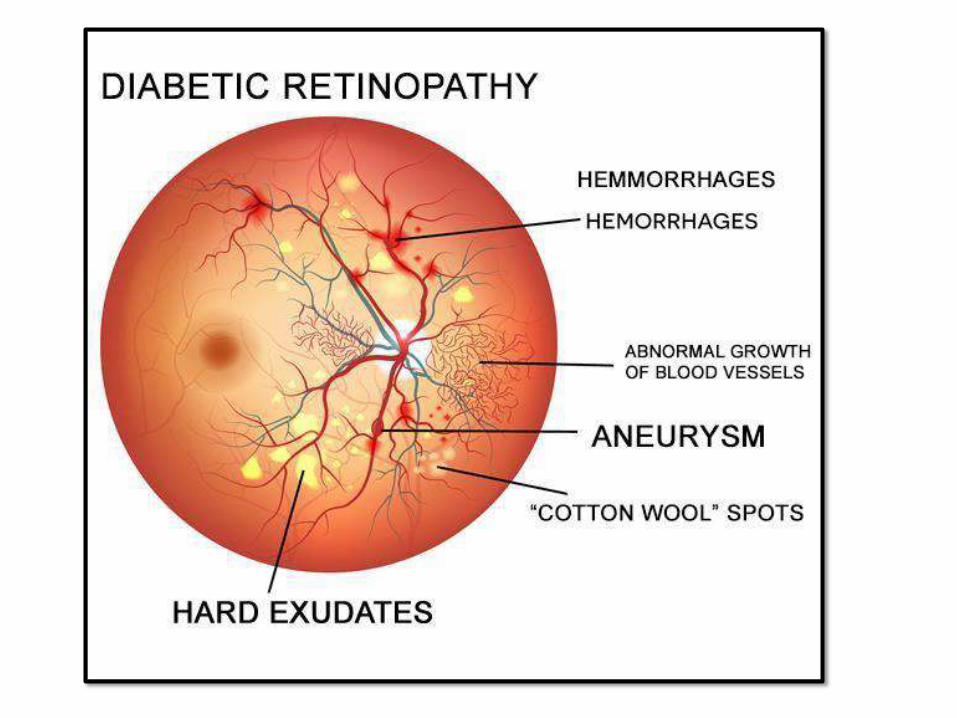
OCULARCHANGES:• Retinopathy (sometimes total
blindness): micro-angiopathy based
–Nonproliferative retinopathy:hemorrhage,
exudate, aneurysms, edema, venous
dilatation
–Proliferative: neovasacularization, serious,
may cause bleeding and blindness
• Cataract
• Glaucoma

DIABETICNEUROPATHY:
• Mechanism: microangiopathy
• Peripheral > central
• Symmetric neuropathy, motor and
sensory (more common)
• Autonomous neuropathy: impotence, U.
bladder and GI tract

MANAGEMENT:• Strict glycemic control is key
• Type 1: insulin replacement therapy
• Type 2: diet, exercise, medications,
ultimately insulin therapy will be
needed
• HbA1c: long term monitoring level,
recommendation is to keep it below 7%
• LDL and HDL cholesterolcontrol

PANCREATIC NEUROENDOCINE TUMORS
(Pan NETs):
• Rare compared to exocrine tumors, 2%
of all pancreatic tumors
• Single or multifocal; functional or not
• Almost all have malignant potential
except insulinomas
• Mutations in tumor suppressor genes:
MEN1, PTEN or inactivating gene
mutations (ATRX)

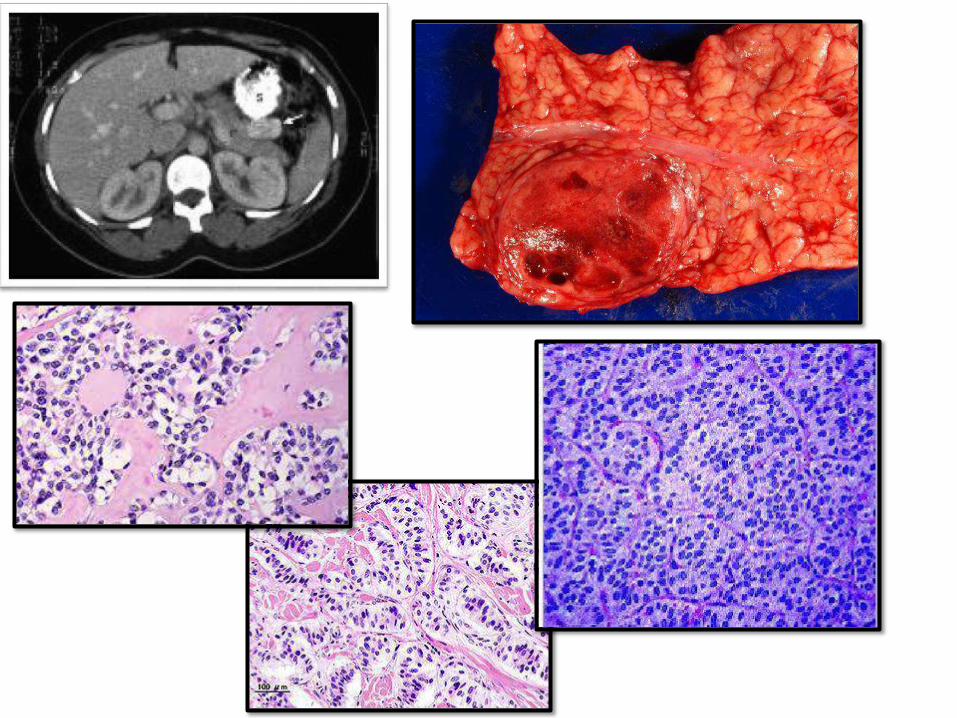
INSULINOMAS:• Most commonPanNETs
• DX: Whipple triad (hypoglycemia + Symptoms
of hypoglycemia + relief of symptoms with
glu
• Symptoms: stupor, confusion, LOC
• 10% malignant potential (invasion and mets)
• Histology: giant islets + amyloid deposition
• Cured by surgical removal

GASTRINOMAS:• PanNET secretinggastrin
• Location: duodenum, peripancreatic tissue and
pancreas
• Zollinger-Ellison syndrome: gastrinoma +
increase gastric acid + severe peptic ulceration
• Ulcers: severe, multifocal and unusual location
• >50% of gatrinomas are malignant at dx
• 25% part of MEN-1 syndrome
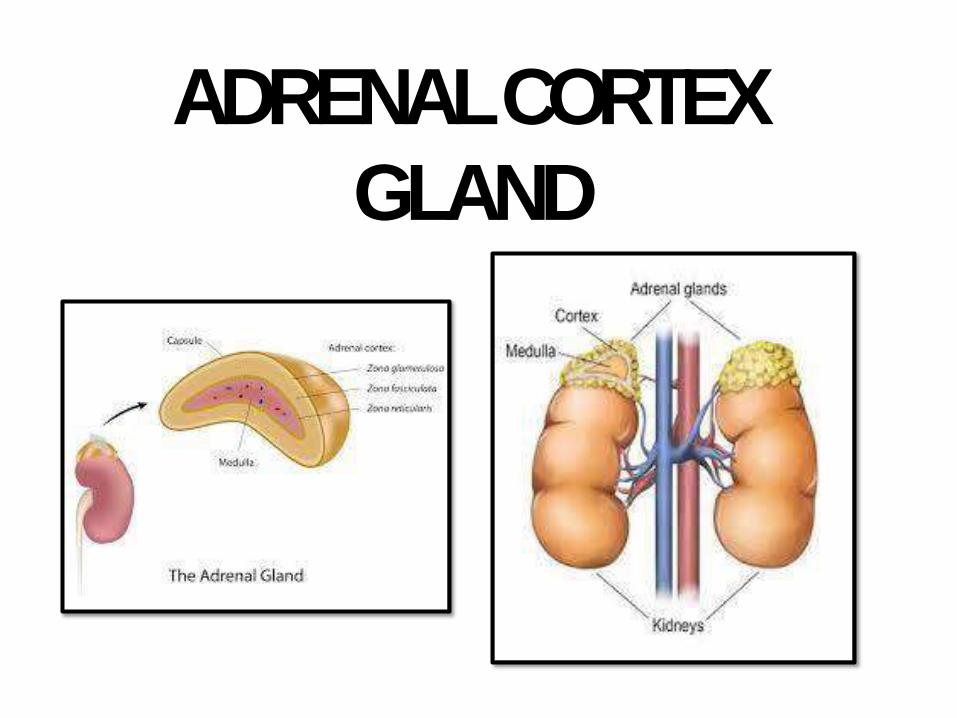
ADRENAL CORTEX
GLAND
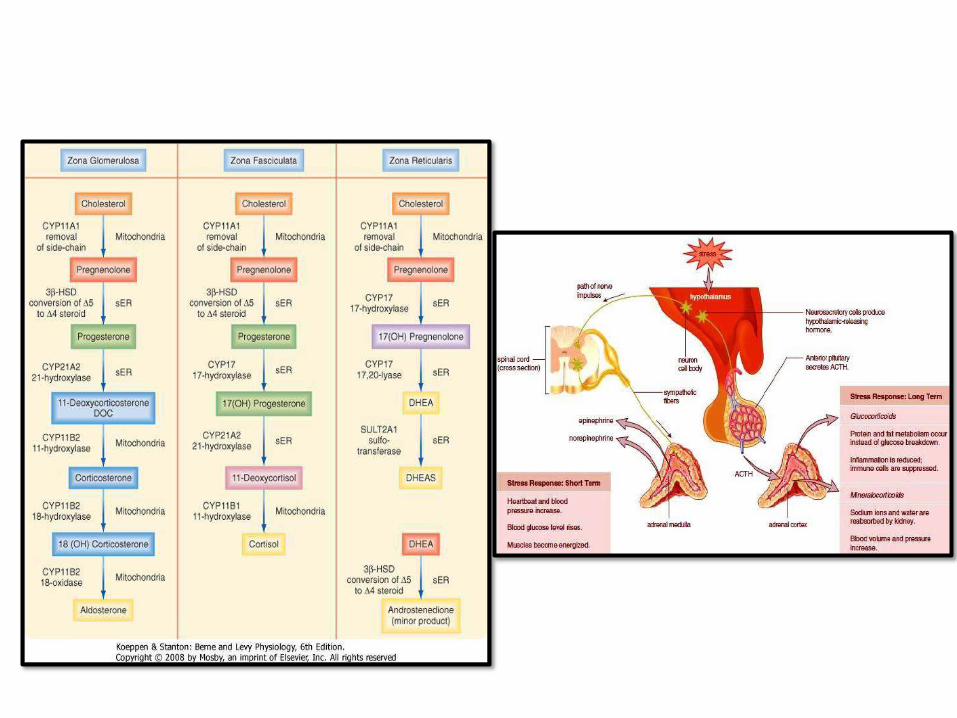
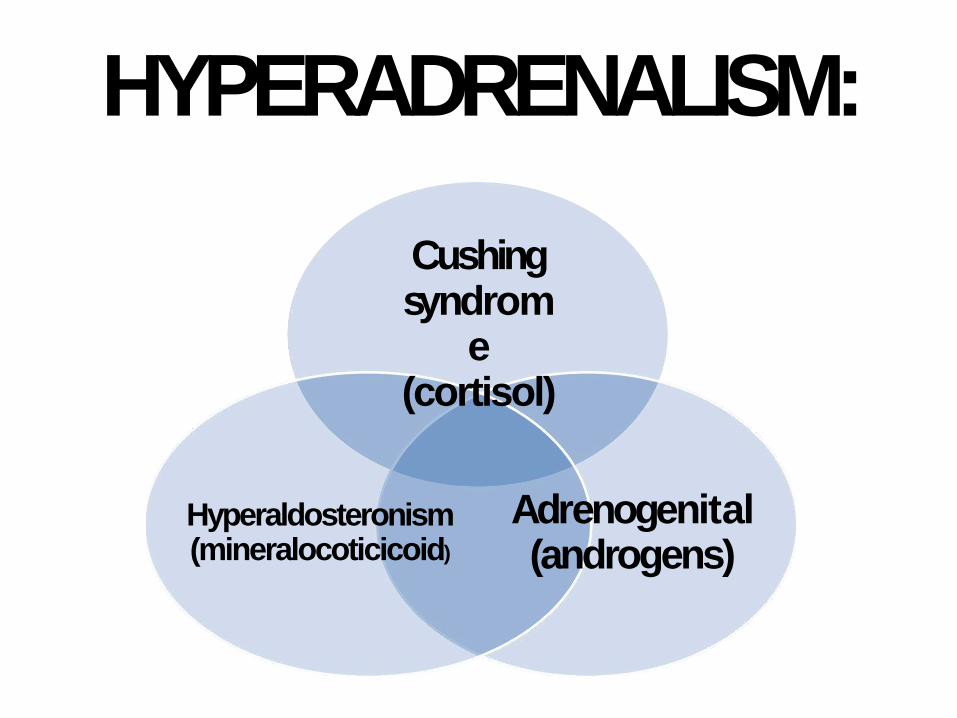
HYPERADRENALISM:
Cushing syndrom
e (cortisol)
Adrenogenital (androgens)
Hyperaldosteronism (mineralocoticicoid)
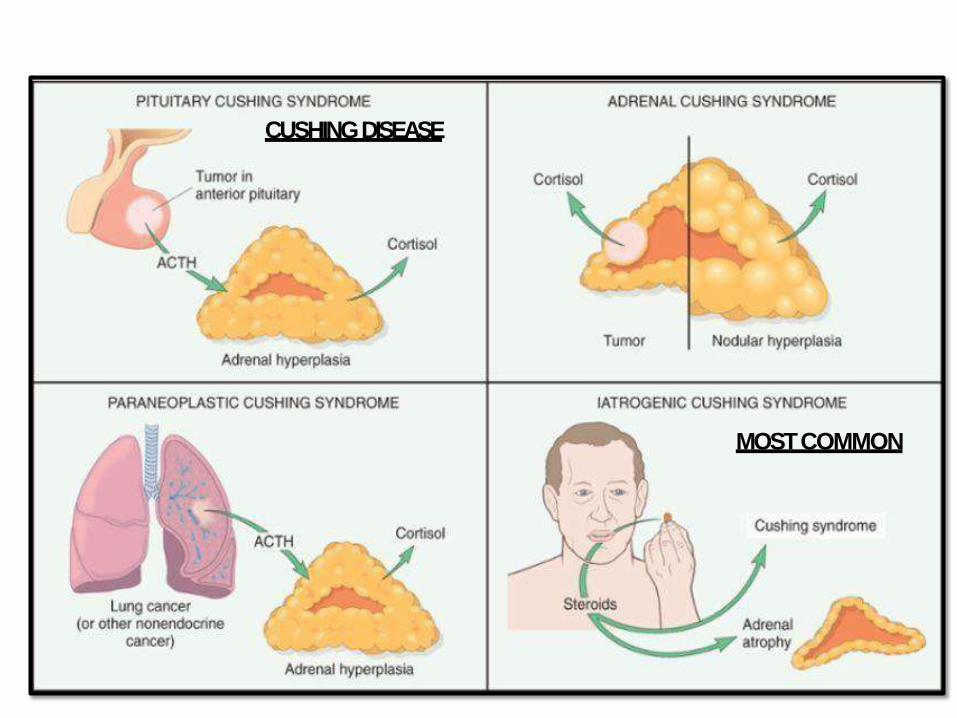
MOSTCOMMON
CUSHINGDISEASE
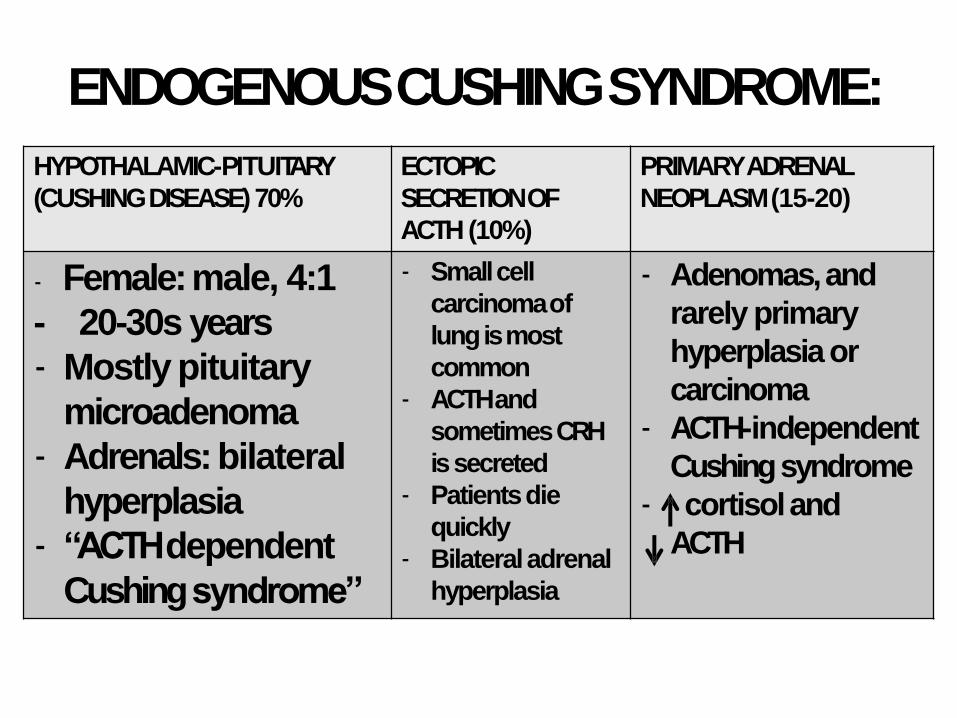
ENDOGENOUS CUSHINGSYNDROME:
HYPOTHALAMIC-PITUITARY
(CUSHING DISEASE) 70%
ECTOPIC
SECRETION OF
ACTH (10%)
PRIMARY ADRENAL
NEOPLASM(15-20)
- Female: male, 4:1
- 20-30s years
- Mostly pituitary
microadenoma
- Adrenals: bilateral
hyperplasia
- “ACTH dependent
Cushingsyndrome”
- Small cell
carcinomaof
lung is most
common
- ACTH and
sometimesCRH
issecreted
- Patientsdie
quickly
- Bilateraladrenal
hyperplasia
- Adenomas, and
rarely primary
hyperplasia or
carcinoma
- ACTH-independent
Cushingsyndrome
- cortisoland
ACTH
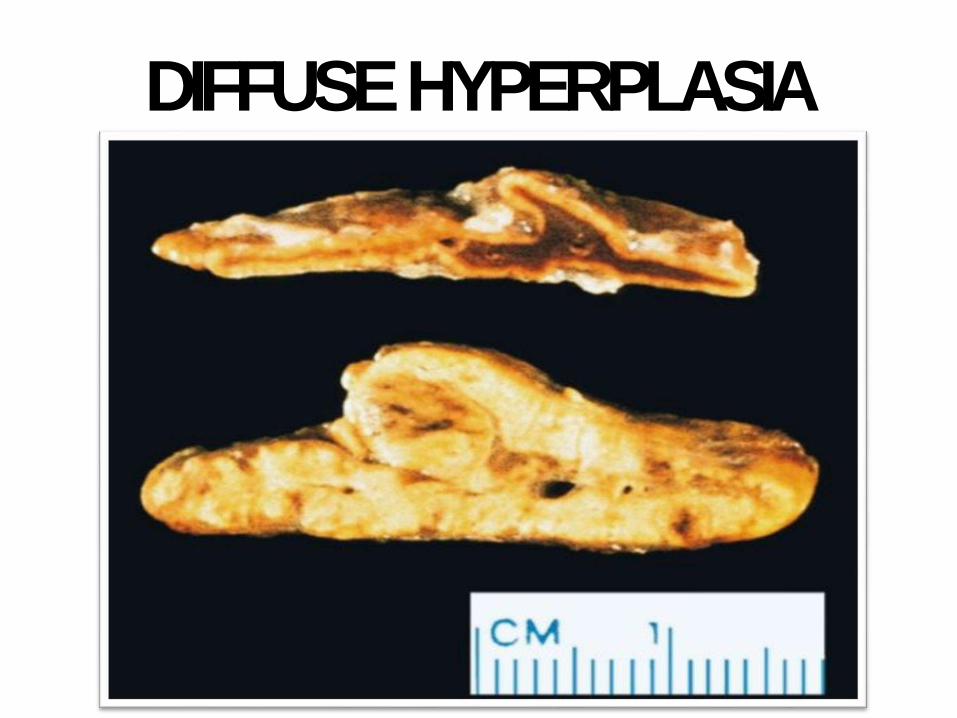
DIFFUSEHYPERPLASIA
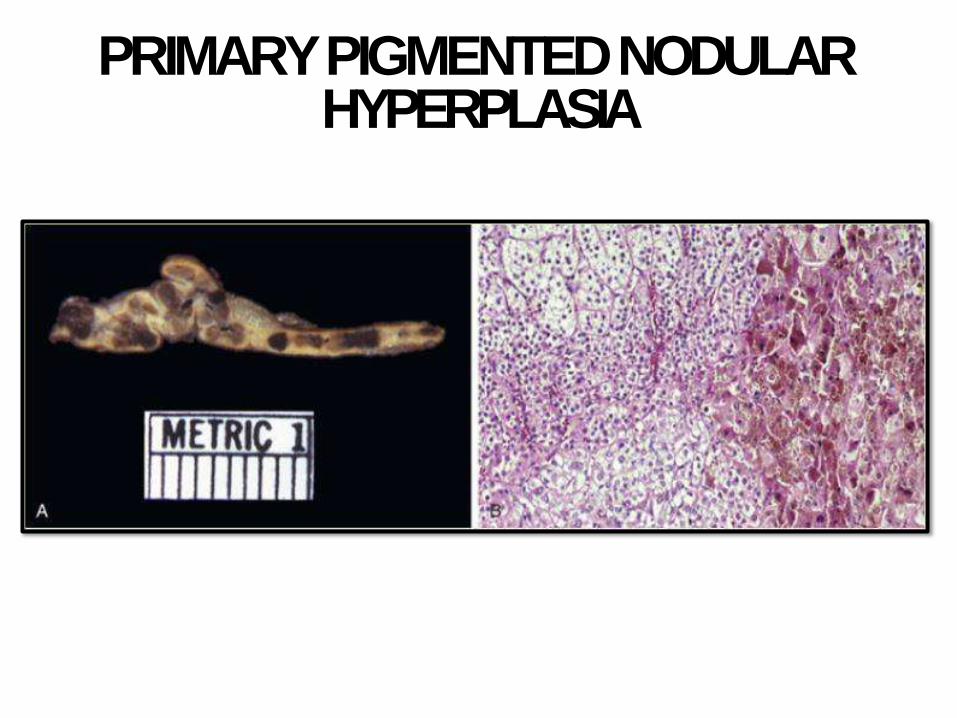
PRIMARY PIGMENTED NODULAR HYPERPLASIA
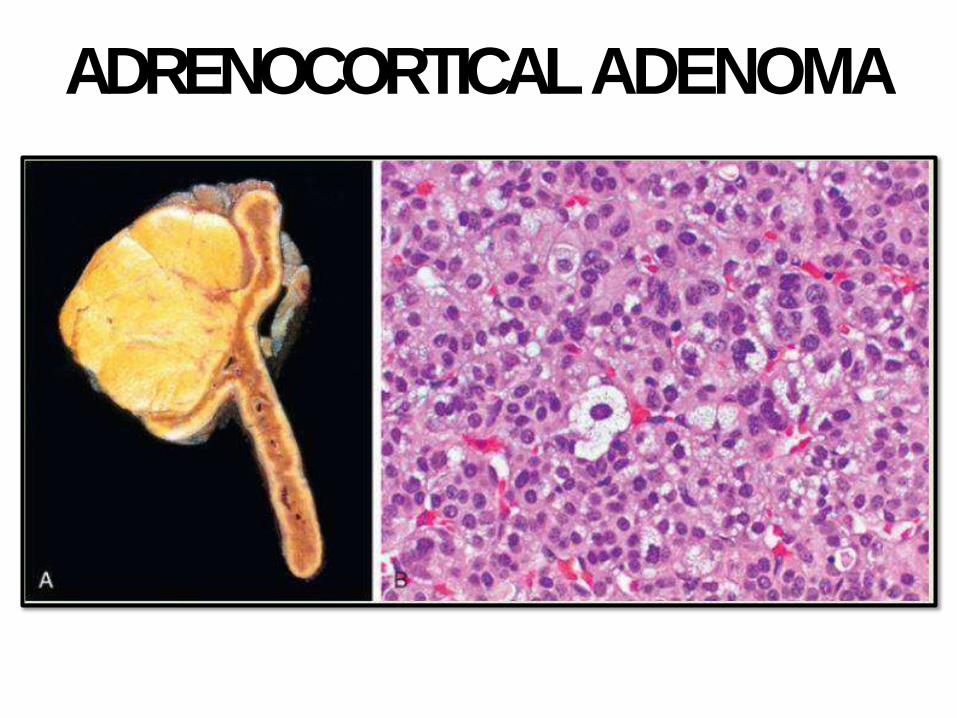
ADRENOCORTICALADENOMA
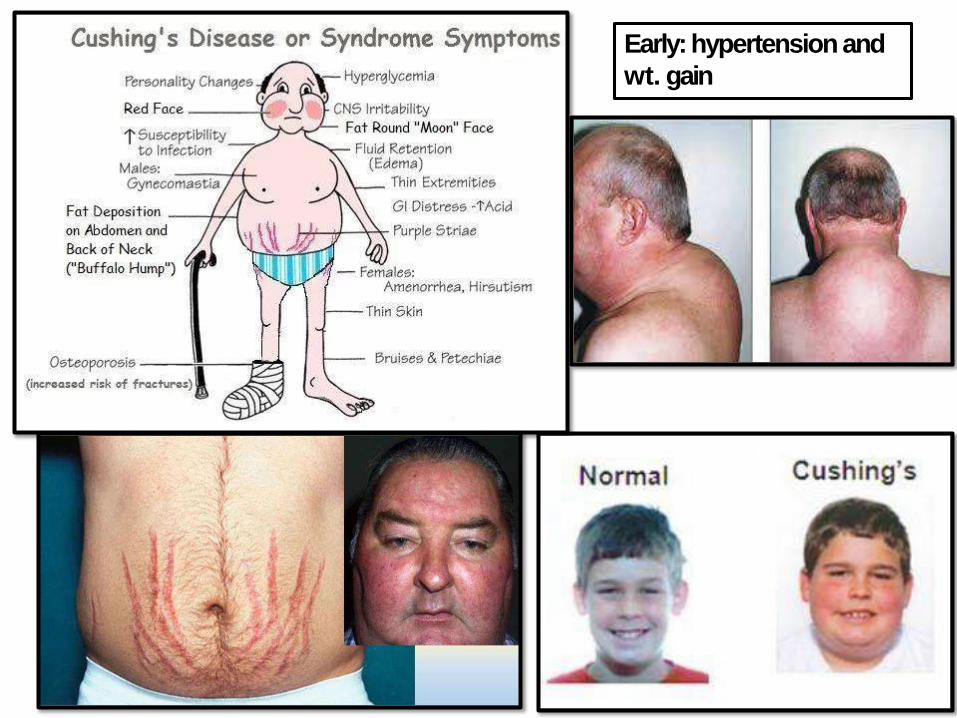
Early: hypertension and
wt. gain

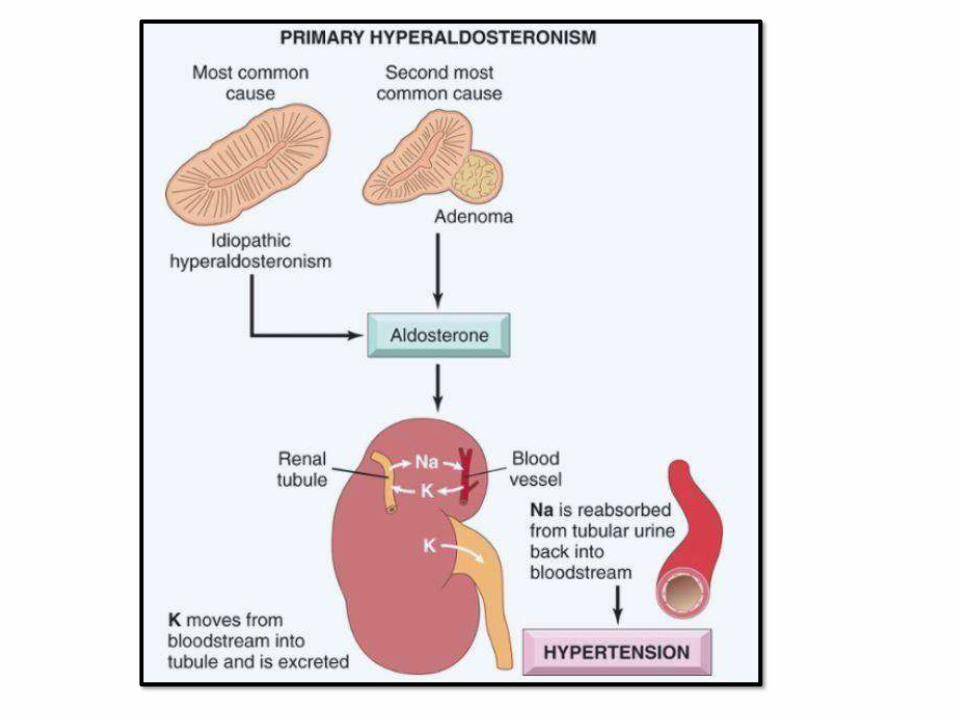
HYPERALDOSTERONISM:• Chronic excess aldosteronesecretion
• Primary or secondary
• Primary: autonomous aldosterone + suppression of renin-angiotensin systemand
serum renin activity. Causes:
➢Bilateral idiopathic hyperaldosteronism (most common)
➢ Neoplasm (adenoma: Conn syndrome) orcarcinoma
➢ Familial: Aldosterone synthase gene (CYP11B2)

SECONDARYHYPERALDOSTERNISM:
• aldosterone release due to activation of
renin-angiotensinsystem.
• Associatedwith:
– renal perfusion (arteriolar nephrosclerosis,
renal artery stenosis)
– Hypovolemia and edema (CHF, Cirrhosis,
Nephrotic syndrome)
– Pregnancy

HYPERALDOSERONISMCLINICALLY:
• HT. Most common cause of secondaryHT
• LVH, strokes,MI
• K (50% of cases): weakness, paresthesia,
visual disturbances and sometimes tetany
• Treatment: surgery for adenomas and anti-
aldosterone agents for others
(spironolactone)

ADRENOGENITALSYNDROME:
• Group of disorders: excessandrogens
• Primary gonadal or primary adrenal
• Adrenal ones maybe associated with Cushing
• Adrenal neoplasm(carcinoma>adenoma)
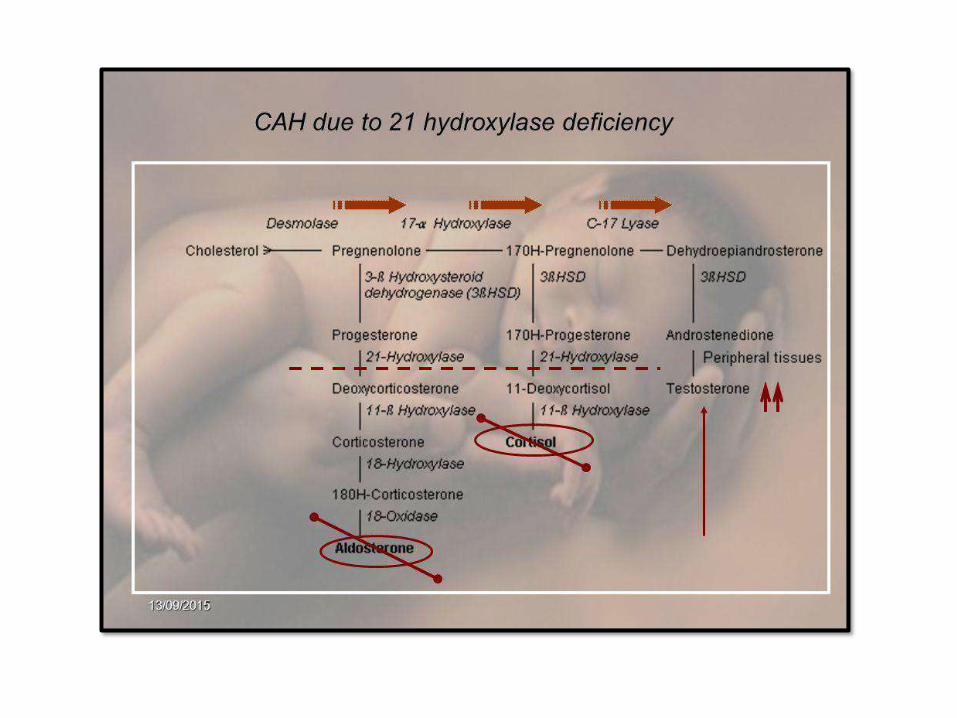
• Congenital adrenal hyperplasia (CAH): AR,
enzymatic deficiency, most common is 21-
hydroxylasedeficiency
• Histology: bilateral adrenalhyperplasia
ADRENALINSUFFICIENCY:

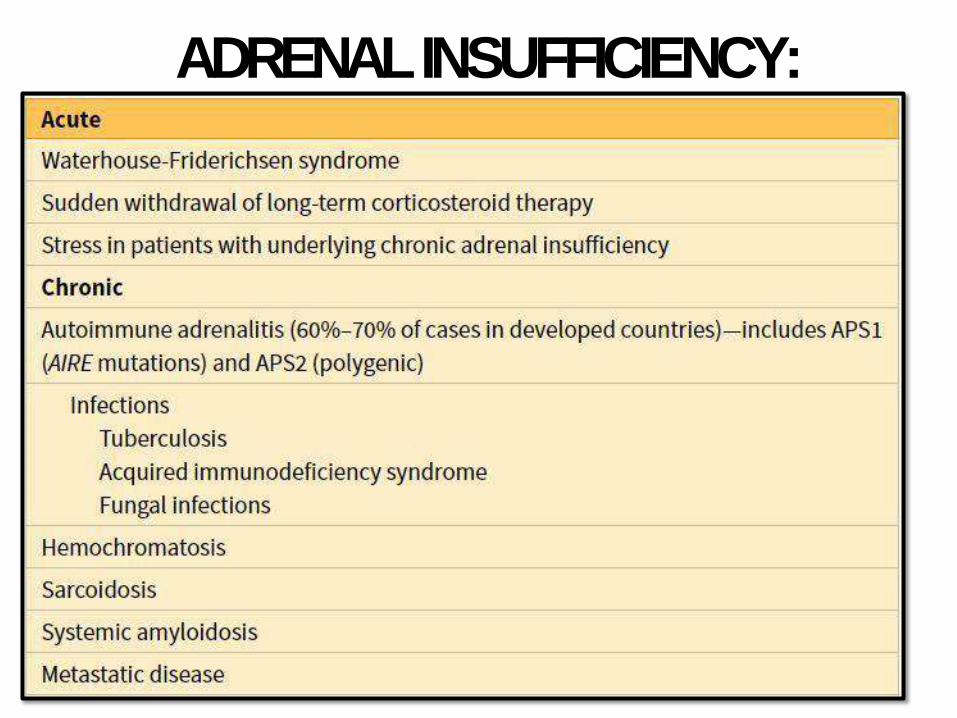
ACUTE ADRENOCORTICALINSUFFICIENCY
• Acute on top of Chronic or sudden
withdrawal of long term steroid therapy
• Massive adrenal hemorrhage:
anticoagulant tx, DIC, pregnancy or
severesepsis.
• In sepsis: Waterhouse-Friderichsen
syndrome specially Neisseria
meningitidis

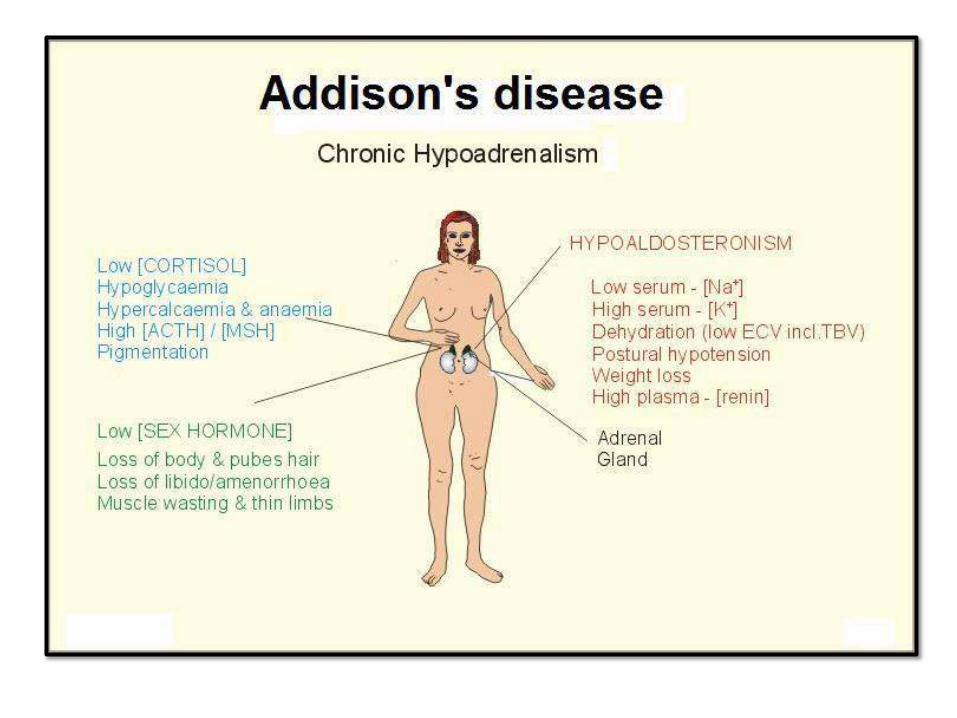
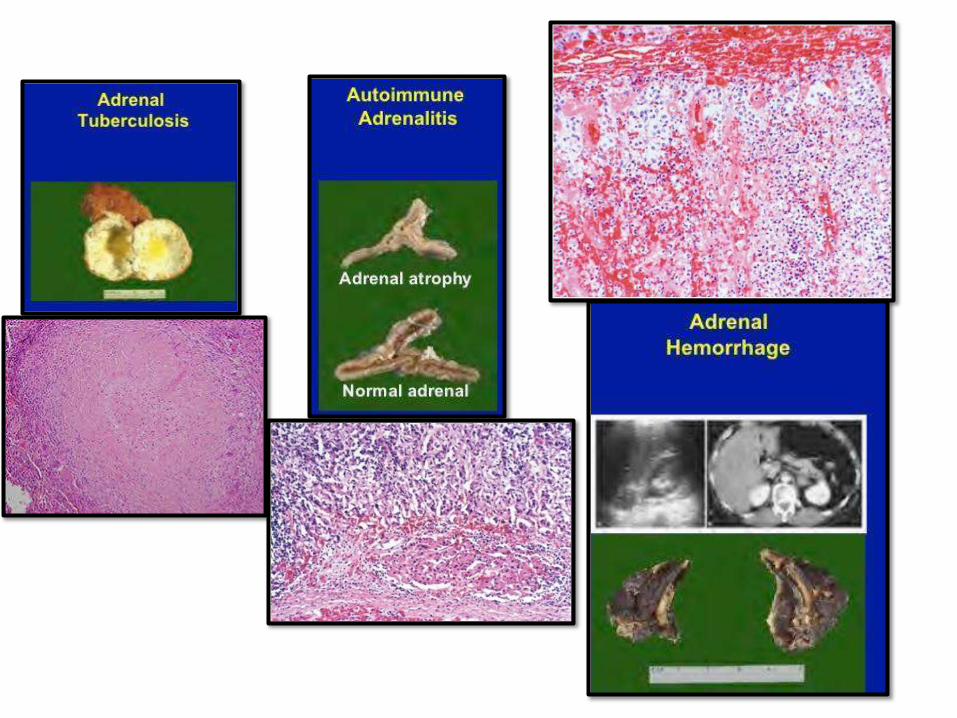
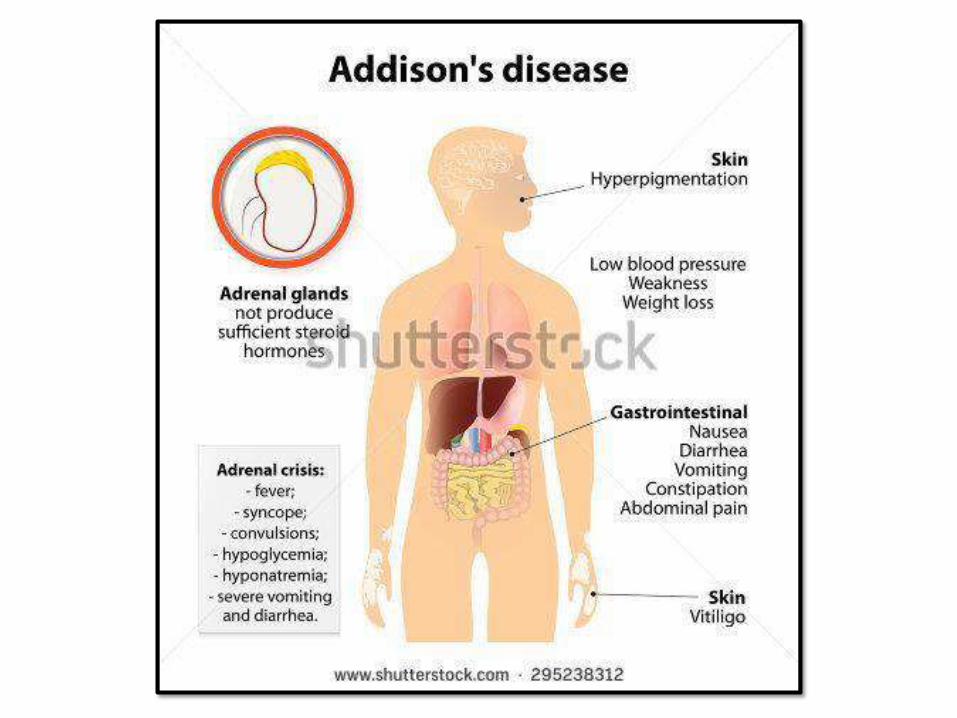
CHRONID ADRENOCORTICAL
INSUFFICIENCY: ADDISSONDISEASEAutoimmune adrenalitis 60-70% - Antibodies againstenzymes.
- Part of autoimmune
polyendocrine syndromes (APS1),
mutation of autoimmune
regulator gene (AIRE),
chromosome21.
- Mucocutaneous candida, dental,
skin and nail abnormalities
Infections - TB andfungi
AIDS - Infectious inorigin
Metastatic carcinoma - Lung andbreast
Secondary adrenocortical insufficiency:
hyopathalamic or pituitary in origin

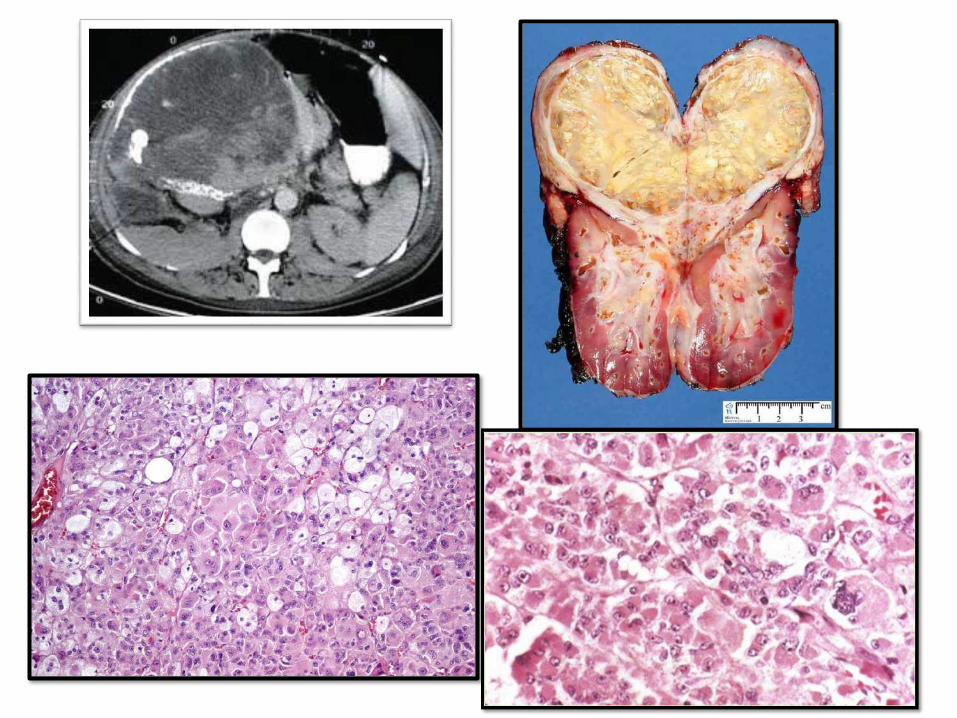
ADRENOCORTICALTUMORS:• Functional adenomas: hyperaldosteronism and
Cushing syndrome. Virilizing neoplasms are more commonly carcinomas
• Adrenocortical adenomas: small, sometimes incidentally found at autopsy. 1-2 cm.
• Carcinomas: rare, large, infiltrative, at any age. Li-Fraumeni and Beckwith-Weidemann syndromes are inherited causes
• Mets to adrenal is >>>> than primary carcinomas

ADRENAL MEDULLA:
PHEOCHROMOCYTOMA
• The “10%” tumor
• 10% extraadrenal (paraganglioma)
• 10% bilateral (50% in familal cases)
• 10% malignant (> in extraadrenal sites)
• 10% not associated with HT
• 25% have germline mutations (RET in
MEN-2, NF1, VHL, and others
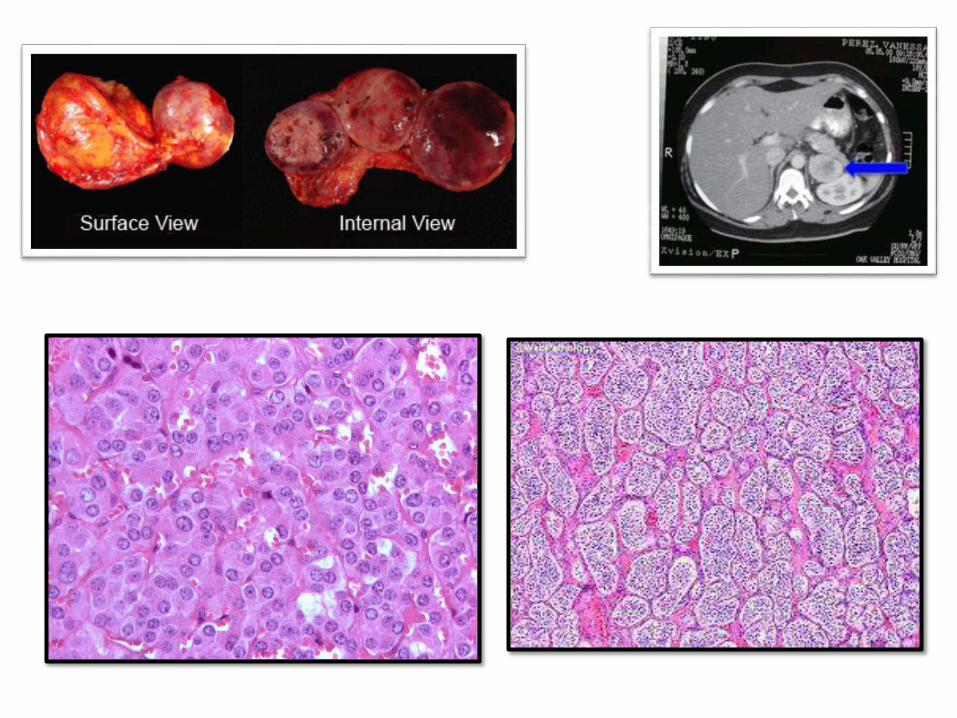
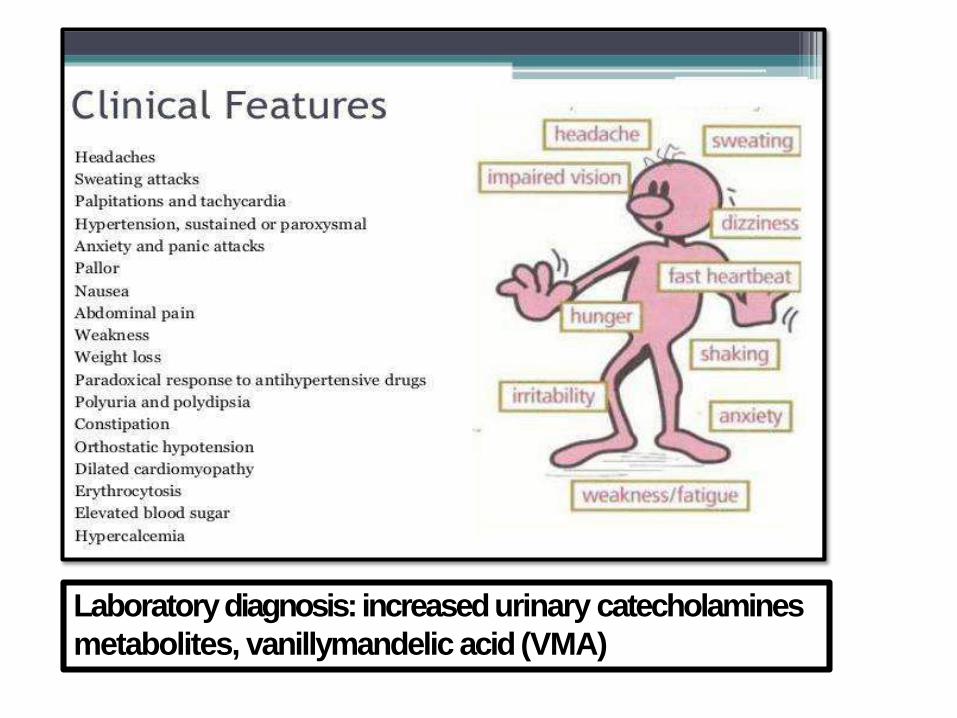
Laboratory diagnosis: increased urinary catecholamines
metabolites, vanillymandelic acid (VMA)

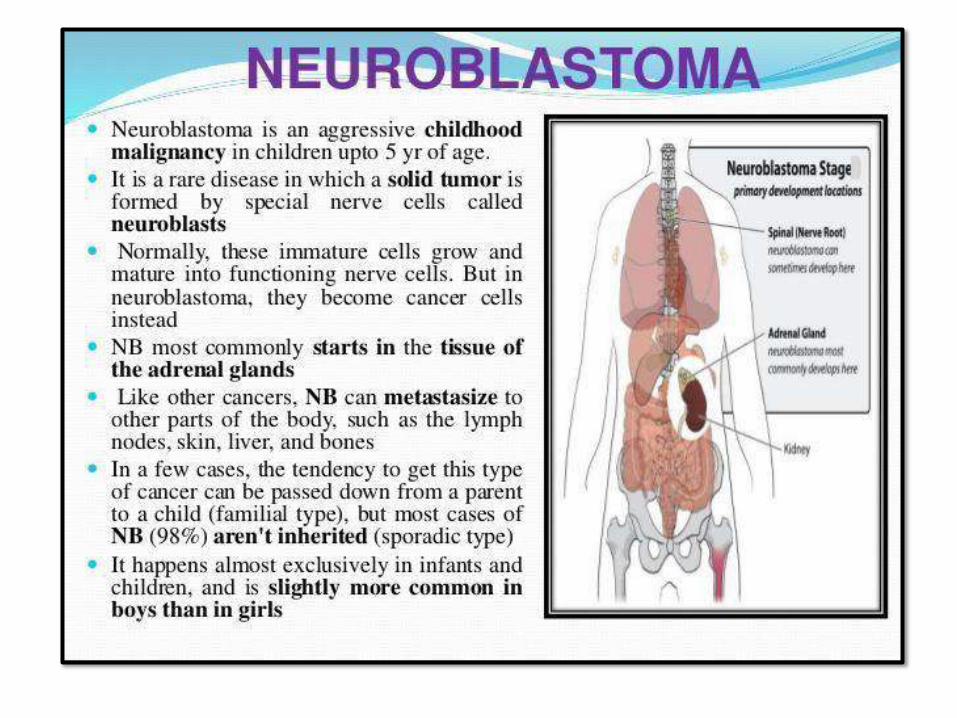
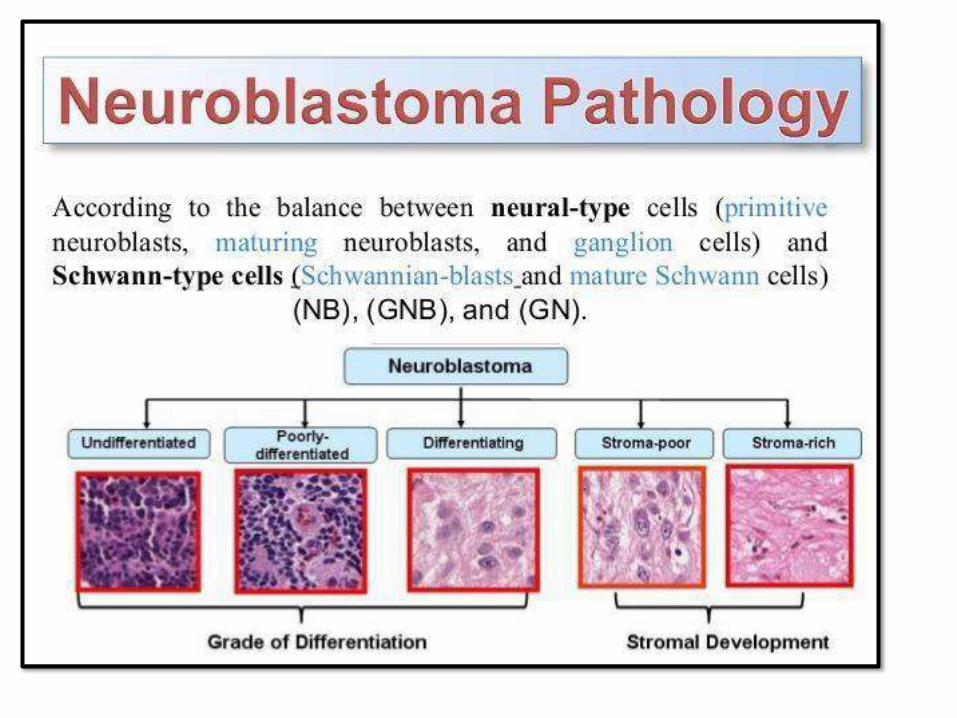
NEUROBLASTOMA:• Most common extracranial solid tumor
of childhood
• < than 5 years, sometimes infants
• Can occur anywhere, but abdominal is
the mostcommon
• Most are sporadic

MULTIPLE ENDOCRINE NEOPLASIA
SYNDROMES(MEN):• Inherited disorders, proliferative of multiple
endocrine organs
• Younger age groups
• Synchronous or meta-chronous in multiple
organs
• Often muti-focal in the sameorgan
• Often preceded by asymptomatic hyperplasia
• More aggressive than their sporadic
counterparts
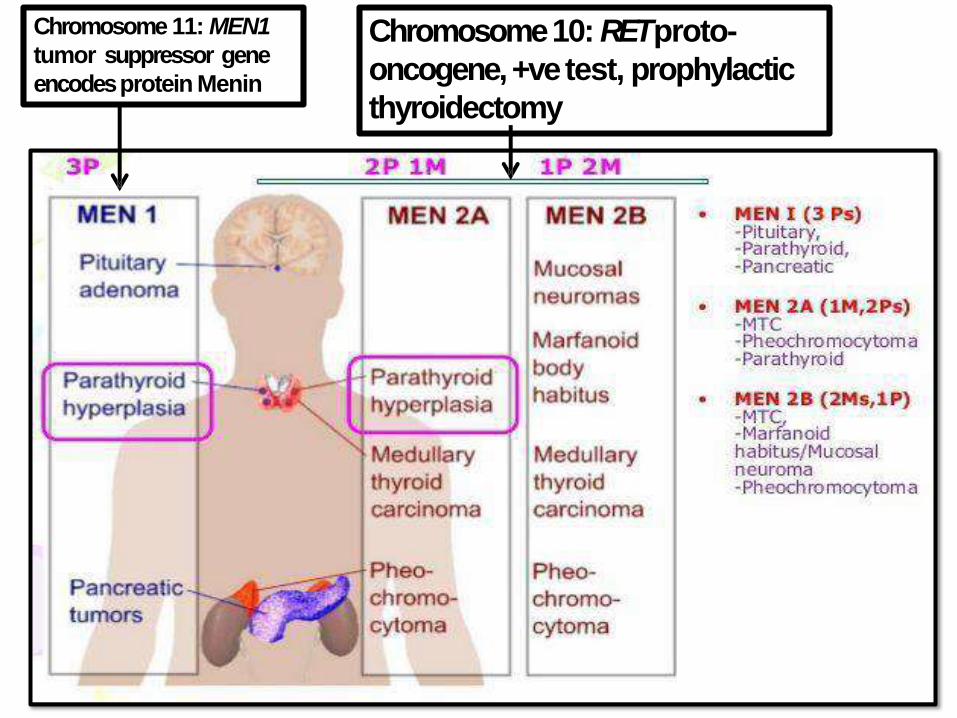
Chromosome 11: MEN1
tumor suppressor gene
encodesprotein Menin
Chromosome 10: RET proto-
oncogene, +ve test, prophylactic
thyroidectomy
Goo
d
luck































![Endocrine Pathology, 4E (2014) [UnitedVRG]](https://img.pdfslide.us/doc/110x75/577c7de31a28abe054a00b57/endocrine-pathology-4e-2014-pdf-unitedvrg.jpg)





