Embed Size (px)
Citation preview

Emission of Occluded Volatiles During Deformation of Polycarbonate Due to Strain-Enhanced Diffusion
J. T. DICKINSON,‘ 1. C. JENSEN,’ S. C. LANGFORD,’ and R. P. DION’
‘Physics Department, Washington State University, Pullman, Washington 991 64-281 4; and ’The Dow Chemical Company, Designed Thermoplastics Research, Midland, Michigan 48667
SYNOPSIS
Measurements of the emission of purposely entrained volatiles (Ar and D 2 0 ) during the loading and unloading of a bisphenol-A polycarbonate in vacuum are made by quadrupole mass spectrometry. Transient loading events are accompanied by dramatic increases in emission, reflecting a similar rise in the diffusion constant of the measured species. We attribute this change to an increase in size of molecular voids in the polymer network, which accompany the increase in sample volume under load. The results are interpreted in terms of the Dolittle relation in which the diffusion constant depends exponentially upon u * / u f o , the ratio between an activation volume for diffusion and the average size of the relevant voids in the polymer network. Our data suggests that u * / u f o is unusually low in the D20-polycarbonate system, which we attribute to a relatively large value of u f o ; this would be consistent with the relatively long distance between flexible links in the polycar- bonate structure. 0 1994 John Wiley & Sons, Inc. Keywords: deformation polycarbonate diffusion strain-enhanced diffusion free volume
INTRODUCTION
The diffusion of volatile gases through polymers is significantly affected by the ambient temperature and pressure, as well as the mechanical/thermal history of the polymer.’ These effects are commonly attributed to changes in the size and density of nanometer-scale voids, which together constitute the “free volume” of the polymer structure. Strain- induced changes in the permeability of thin films have previously been demonstrated in polystyrene, polyethylene and polypropylene, and polycarbonate and p~lyimide.~ Permeability is a function of both the rate of diffusion and the solubility of the mobile species, both of which are affected by pressure and temperature. Strain-induced changes in solubility have been studied in terms of the “swelling” behav- ior of rubbers and other polymer^.^^^ The experi- ments reported here involve monitoring the emission of volatile species from a polymer strained in vac- uum. Although our lack of knowledge of the con-
Journal of Polymer Science: Part B Polymer Physics, Vol. 32, 993-999 (1994) 0 1994 John Wiley & Sons, Inc. CCC 0sS7-6266/94/060993-07
centration gradients in the near surface region makes it difficult to determine absolute values of the diffusion constant, relative changes are readily measured. In addition, emission measurements do not require attaining saturation and allow measure- ments of transient effects over a wide range of time scales.
Polymeric materials typically contain substantial amounts of entrained volatiles, including water, at- mospheric gases, and products of incomplete poly- merization.6-8 Significant quantities of these vola- tiles can remain in the interior of reasonably thick samples even after days in vacuum conditions, as may be verified by mass spectroscopy of the volatiles diffusing to the surface of the sample and desorbing into vacuum. Any transient changes in the diffusion constant of these volatiles will make a corresponding change in the signal detected by the mass spectrom- eter. Volatile species are readily introduced into most polymers by storing them in the appropriate at- mosphere or liquid. This work focuses on Ar and D20, whose masses are well removed from common vacuum system contaminants.
We argue that the diffusion of Ar and D20 from
993

994 DICKINSON ET AL.
1 J
- c l 01,
the near-surface region of bulk polycarbonate sam- ples in vacuum is strongly affected by the applied uniaxial stress. We report measurements of the emission of these species during loading and frac- ture, as well as under transient loading (on the order of 1 s ) prior to yield. The enhanced diffusion of D20 under transient loading is analyzed according the theory of Cohen and Turnbull.2 In the traditional interpretation, the results suggest that a relatively small number of polymer segments are involved in the molecular motion responsible for diffusion. In the present work, this low value more likely reflects an unusually large average void size, consistent with the unusually long, stiff segments that make up this material.
(b) Load
*
EXPERIMENT
The polycarbonate used in this work was Calibre 300-10 provided by The Dow Chemical Company. Dogbone-shaped samples were milled from 3.2 mm injection molded sheet to produce a gauge section 2.5 cm long and 3.2 X 3.2 mm2 in cross section. Ar- loaded samples were produced by storing samples for periods ranging from 24 h to several weeks in an Ar atmosphere. Similarly, D20-loaded samples were prepared by storing samples in heavy water for a few days. The diffusion constant for Ar in polycar- bonate is roughly DAr = 1.5 X cm2/s, while that
Ar Signal and Load during Continuous Loading
i Time (s)
i3
of D20 should be about DH~O = 6.8 X lo-' cm2/s.11 The characteristic decay time for the removal of these volatiles from 3.2 mm thick samples should be about two days for water and eight days for Ar. Experiments were carried out in vacuum within 12 h of mounting the sample, a period significantly less than the characteristic outgassing decay time; thus quite intense emission intensities can be obtained. The high solubility of water in polycarbonate (about two orders of magnitude higher than that of Ar)' results in particularly strong signals from the D20- loaded samples, despite the relatively fast pumpout of this material.
The samples were mounted in a tensile loading apparatus in an ultrahigh vacuum system. Typical background pressures were Pa. The neutral emission was monitored with a UTI lOOC quadru- pole mass spectrometer (QMS) tuned to 40 amu ( Ar ) or 20 amu ( D,O ) . The applied load was mon- itored with a Scaime force transducer. Tensile de- formation was introduced from the outside of the vacuum through a stainless steel bellows.
RESULTS
A typical mass 40 signal during loading and fracture of Ar-soaked polycarbonate appears in Figure 1, where the sample had been stored in Ar for three days prior to loading in the vacuum system, and remained in the vacuum system for 12 h prior to testing. A transient increase in emission at the onset of yield is barely observable at this level of sensitiv- ity. (When desired, measurements can be made with 2-3 orders of magnitude greater sensitivity.) The signal then rises slowly as the sample draws, in- creasing roughly linearly with time. At the onset of strain hardening, the Ar emission increases more rapidly. An intense peak is observed at fracture, fol- lowed by a long, slow decay. Stronger emission in- tensities can be obtained by increasing the time the sample is stored in Ar prior to mounting in vacuum. (Three days is insufficient to establish an equilib- rium concentration of Ar in the interior of the sample.)
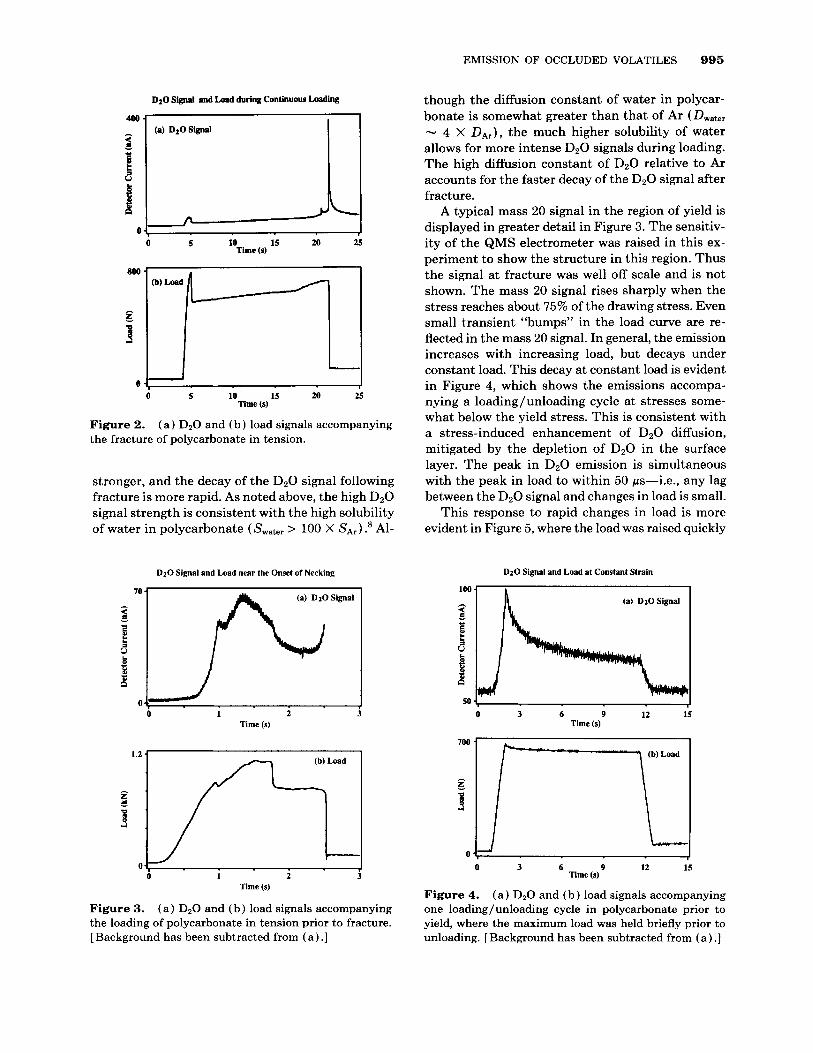
A comparable mass 20 signal from D20-soaked polycarbonate appears in Figure 2. This sample was soaked in D20 at room temperature for three days prior to mounting in vacuum. Note the different time scale employed due to the lower strain rate. The general form of the emission is the same, although several times stronger. The principle differences be- tween the Ar and D20 signals are that the transient increase in the D20 signal at yield is relatively much
EMISSION OF OCCLUDED VOLATILES 995
4ooJ
i3 *
E 5
0 %
(a) D ~ O S i g n d
/r 0 5 10 15 20
Time (s)
8ooJ
!i 4 4
(b) Load
I_
stronger, and the decay of the DzO signal following fracture is more rapid. As noted above, the high D20 signal strength is consistent with the high solubility of water in polycarbonate ( Swater > 100 X SA,) ? Al-
I ” - (a) DzOSignal
z v - e: e
E g
DzO Signal and Load near the Onset of Necking
(a) DzOSignal
3 - - E ‘;I P a’
B
s
though the diffusion constant of water in polycar- bonate is somewhat greater than that of Ar (Dwater - 4 X DA,), the much higher solubility of water allows for more intense D20 signals during loading. The high diffusion constant of DzO relative to Ar accounts for the faster decay of the D20 signal after fracture.
A typical mass 20 signal in the region of yield is displayed in greater detail in Figure 3. The sensitiv- ity of the QMS electrometer was raised in this ex- periment to show the structure in this region. Thus the signal at fracture was well off scale and is not shown. The mass 20 signal rises sharply when the stress reaches about 75% of the drawing stress. Even small transient “bumps” in the load curve are re- flected in the mass 20 signal. In general, the emission increases with increasing load, but decays under constant load. This decay at constant load is evident in Figure 4, which shows the emissions accompa- nying a loading/unloading cycle at stresses some- what below the yield stress. This is consistent with a stress-induced enhancement of D20 diffusion, mitigated by the depletion of D 2 0 in the surface layer. The peak in DzO emission is simultaneous with the peak in load to within 50 ps-i.e., any lag between the DzO signal and changes in load is small.
This response to rapid changes in load is more evident in Figure 5, where the load was raised quickly
D z 0 Signal and Load at Constant Strain
I
Time (s)
Time (s)
Figure 3. ( a ) D20 and ( b ) load signals accompanying the loading of polycarbonate in tension prior to fracture. [Background has been subtracted from ( a ) .]
0 3 6 9 12 15 Time (s)
700 J
E I
0 3 6 9 12 I Time (s)
Figure 4. ( a ) D20 and ( b ) load signals accompanying one loading/unloading cycle in polycarbonate prior to yield, where the maximum load was held briefly prior to unloading. [Background has been subtracted from ( a ) .]

996 DICKINSON ET AL.
and quickly relaxed. The nominal strain a t the peak load is about 7.5%. Note that the strain acts like a valve presumably due to reversible void opening and closing, with essentially no near surface damage (which would be expected to result in erratic and perhaps sustained emission upon stress release ) . Assuming a linear response, the fractional change in diffusion constant per unit strain ( 1/D) ( d D / d e ) x 8.5, which is reasonably consistent with a pub- lished value measured under more steady-state strain on another commercial polycarbonate [ ( 1 / D) ( d D / d e ) = 7.81 .5 Emission responses at even faster loading and unloading were measured showing relaxation times of less than 10 ms.
Similar reversible, rapidly responding Ar emission has been observed from Ar-loaded polystyrene. Strain-enhanced permeability has been observed in polystyrene, but the enhancement at high pressures can be attributed to enhanced transport through crazes. In a separate work,12 we have shown that a large increase in volatile emission accompanies the growth of crazes in polystyrene in vacuum. In this work, the increases observed are at low loads, where crazing is negligible, and can be turned off imme- diately by releasing the strain. Thus, strain-en- hanced diffusion is observed in polystyrene as well.
DISCUSSION
We attribute the enhanced diffusion prior to yield to stress-induced increases in “free volume.” The principle alternative to a free volume mechanism is thermally enhanced diffusion due to small amounts of plastic deformation. Although a certain amount of anelastic behavior is expected prior to yield, Mat- suoka and Bair report that the surface temperature actually drops slightly (about 0.5 K ) during the ini- tial stages of loading, reaching a minimum near the yield point.13 Although they employed much lower loading rates than those of this work (8.6 X s-l versus typically 5 X lop2 s-l here), the basic relationship between mechanical and thermal be- havior should be unchanged. Any changes in the mechanical behavior (e.g., a higher yield stress) should actually result in more cooling prior to yield. Thus we expect that the temperature of the poly- carbonate during the peak in D20 emission prior tc yield is actually somewhat below ambient. In con- trast, the free volume should peak at the point of maximum stress, immediately prior to yield. The rapid drop in emission accompanying yield (where the temperature rises and the free volume falls) rules
D20 Signal and Load during LoaWnload Cycle
100.- - (a) D,OSignal a J
0 1 2 3 4 5 Time (s)
0 1 2 3 4 Time (s)
Figure 5. ( a ) D20 and ( b ) load signals accompany- ing another loading/unloading cycle in polycarbonate. [Background has been subtracted from (a) .]
out thermal effects as the cause of our observed in- crease in emission.
Free volume effects also account for the large, nearly reversible change in emission intensities during the transient loading of Figure 5 (prior to macroscopic yield), as well as the relatively slow decay of the D20 signal a t constant load. Any tem- perature rise and subsequent cooling (by thermal conduction) would be comparable in these two in- stances, but the D20 signals contrast markedly. The D20 signal follows changes in the load when deple- tion of D20 in the near surface region is taken into account. The rapid falloff of emission when the load is quickly released is counter to a thermal mecha- nism. Thus, prior to yield, free volume effects dom- inate any thermal effects on the diffusion of D20 in the near surface region.
The increase in emission intensities during the early stages of cold drawing are readily explained on the basis of the increasing surface area of the drawn portion, as well as the increasing concentra- tion gradients that accompany the decrease in the thickness. These effects are consistent with a roughly linear increase in volatile emission during cold drawing. The free volume of the drawn material is expected to be about the same as that in the un- drawn material, suggesting that diffusion in the

EMISSION OF OCCLUDED VOLATILES 997
drawn material is not enhanced by free volume ef- fects. Diffusion would be enhanced by any heating that accompanies cold drawing. With the onset of strain hardening, the free volume rises again, ac- counting for the nonlinear rise in emission between the onset of strain hardening and fracture.
To quantitatively account for the observed sig- nals, one must account for the both the diffusion of gas in the strained polycarbonate and the response of the vacuum system to transient releases of volatile material. The output of the QMS is proportional to the density of the measured species in the electron impact ionizer. Since volatile gases released by the sample are eventually pumped away by the vacuum pumps, the net density in the system is determined by the competition between these two processes. The rate of change of the density may be expressed as
1 d N 1 . 1 v dt v V _ _ = -Nd - - N p
where N is the number of the molecules of interest in the vacuum system, V is the volume of the vacuum system, Nd is the rate at which molecules are emitted from the sample, and NP is the rate at which mol- ecules are removed from the system by pumping.
Assuming that the rate of emission from the sample is Fickian, the flux of particles leaving the sample surface is proportion to the product of the diffusion constant D and the concentration gradient in the near surface region, V C . The total number of particles leaving the sample is just the product of this flux and the surface area of the sample A . Therefore,
Nd = DAVC ( 2 )
Similarly, the rate at which molecules are pumped out of the system is related to the pumping speed of the vacuum pumps S (units of volume per unit time) for the species of interest. This rate is given by
NS V
I\j = - - ( 3 )
The particle density in the system as a function of time is then given by
( 4 ) 1 dN - DAVC NS V dt V V 2
Although eq. ( 4 ) is difficult to solve in the general case, the difference between the two terms on the
right-hand side is generally much smaller than the magnitude of either term. In this case, the particle density is well approximated by equating N d and N, and solving for N / V .
This approximation will be valid as long as the rate of change in the measured particle density is much smaller than the rate at which particles are removed from the vacuum system, i.e.,
(6 ) 1 dN NS 1 dN S
4- or -- 4- V dt V 2 N dt V _ _
The quantity S / V is characteristic of the vacuum system and the species of interest. The pumping speed of our system is typically in excess of 4000 1/ s for Ar and D20, and the system volume is about 40 1, so that S / V > 100. As long as the fractional change in the particle density per second is much less than 100, the approximation leading to eq. ( 5 ) is valid. In the work above, the fractional change in particle density per unit time was seldom more than 1 s-l, so this condition is well satisfied.
The dependence of the diffusion constant D on the unknown concentration gradient V C does not allow for an absolute measurement of D from eq. ( 5 ) . However, transient increases in D will have only small effects on VC, so that to first order VC will remain constant during a particular transient strain event. Thus the fractional change in the measured signal (proportional to N ) will be equal to the frac- tional changes in D to a good approximation. From our measurements, typical stresses can readily in- crease D by a factor of two.
The dependence of the diffusion constant on the free volume may be rather simply modeled in terms of a Dolittle relation,
D = A exp(-yu*/uf) ( 7 )
where A and y are constants, u* is the minimum void size for molecular diffusion, and uf is the average volume of the voids making up the free volume. In the theory of Cohen and Turnbull,' the voids in- volved in the diffusion process are created by the cooperative motion of polymer segments. They in- terpret y as accounting for overlap of voids asso- ciated with adjacent polymer segments, so that 0.5 > y > 1. The change in D with changing free volume may then be expressed as4

998 DICKINSON ET AL.
= exp( - % ($ - 1)) ( 8 )
where ufo is the initial average void size and ufi is the final average void size. Assuming that the num- ber of voids is constant during straining, the average void size is proportional to the sum of the volumes of all the voids Vf . Then writing Vf l = Vfo + SV, eq. ( 8 ) becomes
- = e x p ( - z ( D1 -6V ) ) DO v, + 6V
where Vo is the initial volume of the sample. The change in total free volume under uniaxial
strain 6V is equal to the volume change of the Sam- ple. In the limit of small strains, 6V/Vo = ( 1 - 2p)q where p is Poisson’s ration ( -0.4 for this material) and c is the strain. Thus 6V is readily computed from experimental measurements of stress or strain. Similarly, the ratio between Dl and Do equals the ratio of the mass spectrometer outputs, denoted by Il and Io , respectively [ eq. (5) 1. These experimen- tally determined quantities allow us to estimate y u * / u f o . At the emission peak, c - 0.075, so that 6V/Vo - 0.015; comparing the DzO signal at the peak with that prior to loading, D1/Do = I l / Io - 1.9. We assume that Vfo/Vo is about 0.025 on the basis of experimental measurements on similar materiaLg Substituting these values into eq. (9) and solving for yv*/ u f o yields a value of 1.7. Identifying u fo with the total free volume in the sample divided by the total number of segments, i.e., the free volume per segment, Cohen and Turnbull take yu*/ufo as an indication of the minimum number of polymer seg- ments involved the cooperative motion responsible for diffusion.
Molecular dynamics models of segmental motion in polycarbonate (and other glassy polymers) show clearly that any major segment motion involves the coordinated motions of a large number of segments.14 The value of yv*/ ufo for the DzO-polycarbonate system is quite small relative to most other polymer- solute systems analyzed in this formalism, where
yu*/ ufo is typically between 7 and 30. Thus for most polymers, the formalism of Cohen and Turnbull is consistent with the cooperative motion of many polymer segments. This suggests that it is inappro- priate to approximate the average void size ufo by the total free volume divided by the number of seg- ments, and that (relative to many other polymers), the polycarbonate is characterized by a small num- ber of large voids. Such a void size distribution is consistent with the unusually great distance between flexible links along the polycarbonate backbone, where small misorientations between segments would yield unusually large voids.
If ufo is known and assuming that 0.5 I y I 1.0, u * may be estimated. The average void size u f o , can be determined from positron annihilation studies. Void size measurements in another commercial polycarbonate yielded ufo = 45 A3,15 which would suggest that yu* is about 75 A3. With 0.5 I y I 1.0, the activation volume u * is bracketed by the values 75 A3 I u* I 150 A 3 , which is reasonable for a small molecule like water. (The volume per molecule in the liquid state of water is about 110 A3.)
Although it is speculative at this point, we suggest that the principle molecular motion associated with this diffusion involves the rotation of phenylene groups in the polycarbonate backbone, probably in concert with an adjacent carbonyl group. (We can- not rule out the possibility, for instance, that the molecule motions involved are so complex that no particular segment motions correlate with diffusion.) The hydrogen atoms on a rotating phenylene group interact strongly with the adjacent carbonyl group, l6
so that water molecules in the “pocket” between the phenylene and carbonyl groups would be swept along with the rotation. (Water would prefer sites next to the polar carbonyl group.) From the pressure de- pendence of proton NMR line widths, the activation free volume for ring rotation has been determined to be about 40 A’.’’ This activation volume is rea- sonable in view of the volume swept out by a rotating phenylene group (about 70 A 3 ) , which must be re- duced by the volume of the phenylene group at rest. Since the volume swept out by a rotating phenylene group is significantly less than the volume of a water molecule, we expect that a t least two phenylene groups on adjacent chains, probably with at least one adjacent carbonyl group, must rotate to accom- modate the motion of water. Phenylene group ro- tation plays an important role in the unusual me- chanical properties of polycarbonate (e.g., high im- pact toughness).

EMISSION OF OCCLUDED VOLATILES 999
Finally, we point out that oscillating the load at about 1 Hz on a DzO-loaded sample for a few tens of cycles provided sustained pulses of DzO, suggest- ing that perhaps a form of fatigue testing could be done where deviations from well-formed, periodic emissions might be detected and related to very early onset of irreversible damage.
CONCLUSION
Measurements of Ar and DzO emission accompa- nying the loading of polycarbonate indicate that dif- fusion is strongly enhanced under mechanical de- formation. We attribute this enhancement to the increase in free volume that accompanies increasing strain. Interpreting these results for D20 according to the formalism of Cohen and Turnbull, we find unusually low values of yu* / ufo N 1.7 for diffusion in this material. In the traditional interpretation, this would suggest that a relatively small number of polymer segments are involved in the molecular motion responsible for diffusion. In the present work, this low value more likely reflects an unusually large average void size. Although the identification of any particular molecular motions responsible for the diffusion of DzO is speculative, a cooperative motion involving phenylene group rotations is con- sistent with the observed diffusion kinetics.
Diffusion under low concentration gradients and/ or stress, such as those studied here, can have im- portant practical applications, e.g., in packaging and membranes. Our qethods also serve as a dynamic probe of stress-induced void growth, which may play an important role in the mechanical properties of polycarbonate, e.g., facilitating the molecular mo- tions responsible for the onset of yield. These mea- surements can probe transient changes on small time scales (-milliseconds), which are not acces- sible to most permeability measurements, and thus may prove useful in the study of fast relaxation pro- cesses and their effect on free volume. Outgasing under vacuum conditions also allows for solute con- centrations well below saturation in many polymer- solute systems, which may simplify the interpreta- tion of results.
This work was supported by The Air Force Office of Sci- entific Research Contract AFOSR-F49620-91-C-0093, The Dow Chemical Company, and the Washington Technology Center.
REFERENCES AND NOTES
1. V. Stannett, in Diffusion in Polymers, J. Crank and G. S. Park (eds.) , Academic Press, London, 1968, pp.
2. C. A. Kumins and T. K. Kwei, in Diffuswnin Polymers, J. Crank and G. S. Park (eds.), Academic Press, Lon- don, 1968, pp. 107-140.
41-73.
3. B. Rosen, J. Polym. Sci., 47, 19 (1960). 4. H. Yasuda, V. Stannett, H. L. Frisch, and A. Peterlin,
5. T. L. Smith and R. E. Adam, Polymer, 22,299 (1981). 6. L. R. G. Treloar, The Physics of Rubber Elasticity, 3rd
ed., Claredon, Oxford, 1975, pp. 150-159. 7. C. J. Wolf and Holly Fu, Proc. Am. Chem. SOC. Div.
Polym. Muter., Sci. Engin. ( 1993), to appear. 8. M. A. Grayson, C. J. Wolf, R. L. Levy, and D. B.
Miller, J . Polym. Sci., Polym. Phys., 14,1601 (1976). 9. N. S. Enikolopian, L. S. Zarkhin, and E. V. Prut, J .
Appl. Polym. Sci., 30, 2991 (1985). 10. V. R. Regel, T. M. Muinov, and 0. F. Pozdnyakov, in
Physical Basis of Yield and Fracture, A. C. Stickland (ed.) , Institute of Physics, London, 1966, pp. 194- 199.
Macromol. Chem., 73, 188 (1964).
11. F. J. Norton, J. Appl. Polym. Sci., 7 , 1649 (1963). 12. J. T. Dickinson, L. C. Jensen, S. C. Langford, and
R. P. Dion, J. Polym. Sci. B, Polym. Phys. Ed., 31, 1441 ( 1993).
13. S. Matsuoka and H. E. Bair, Appl. Phys., 48, 4058 (1977).
14. M. Hutnik, A. S. Argon, and U. W. Suter, Macro- molecules, 24,5970 (1991).
15. B. D. Malhotra and R. A. Pethrick, Eur. Polym. J., 19, 457 (1983).
16. J. T. Bendler, Ann. NYAcad. Sci., 371,299 (1981). 17. J. H. Walton, M. J. Lizak, M. S. Conradi, Terry Gul-
lion, and Jacob Schaefer, Macromolecules, 23, 416 (1990).
Received June 22, 1993 Revised September 21, 1993 Accepted September 27, 1993





























