Embed Size (px)
Citation preview

1
Electronic Supporting Information
Experimental-Theoretical Approach to the Adsorption Mechanisms for Anionic,
Cationic and Zwitterionic Surfactants at the Calcite-Water Interface.
Agustín Durán-Álvarez†,Ѧ, Mauricio Maldonado-Domínguez§, Oscar González-Antonio§,
Cecilia Durán-Valencia†, Margarita Romero-Ávila§, Fernando Barragán-Aroche†, Simón
López-Ramírez†*.
†Universidad Nacional Autónoma de México, Facultad de Química, Departamento de
Ingeniería Química/USIP, Ciudad Universitaria, México, D. F., C.P. 04510, México.
§Universidad Nacional Autónoma de México, Facultad de Química, Departamento de
Química Orgánica, Ciudad Universitaria, México, D. F., C.P. 04510, México.
ѦUniversidad Nacional Autónoma de México, Facultad de Ingeniería, Departamento de
Ingeniería Petrolera, Ciudad Universitaria, México, D. F., C.P. 04510, México.
*) Corresponding author’s e-mail. [email protected]
Contents Page
Characterization of the CaCO3 Substrate 2
Characterization of Water for Streaming Potential Analyses 3
Determination of Critical Micelle Concentration (CMC) 4
DFT Study of Electron Density and Adsorption Modes for Surfactant Molecules 6
Energy Profiles for MD Simulations 13
2
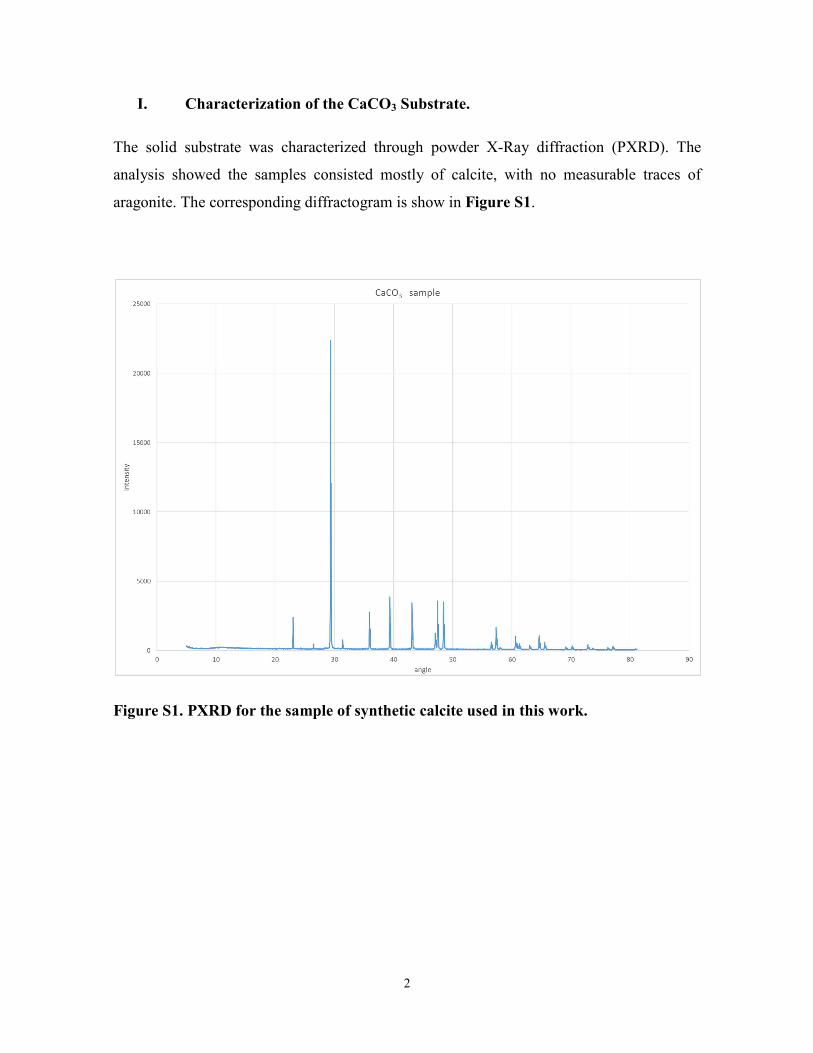
I. Characterization of the CaCO3 Substrate.
The solid substrate was characterized through powder X-Ray diffraction (PXRD). The
analysis showed the samples consisted mostly of calcite, with no measurable traces of
aragonite. The corresponding diffractogram is show in Figure S1.
Figure S1. PXRD for the sample of synthetic calcite used in this work.

3
II. Characterization of Water for Streaming Potential Analyses.
For streaming zeta potential experiments, deionized water was equilibrated with an excess
amount of calcite, previous to measurement. This process was monitored through pH and
conductivity, until a constant behavior was observed. In figure S2, this can be seen as
plateau behavior in both parameters, which persisted once saturation was reached.
Figure S2. Conductivity (k) and pH values, during CaCO3 equilibration with deionized
water.
4
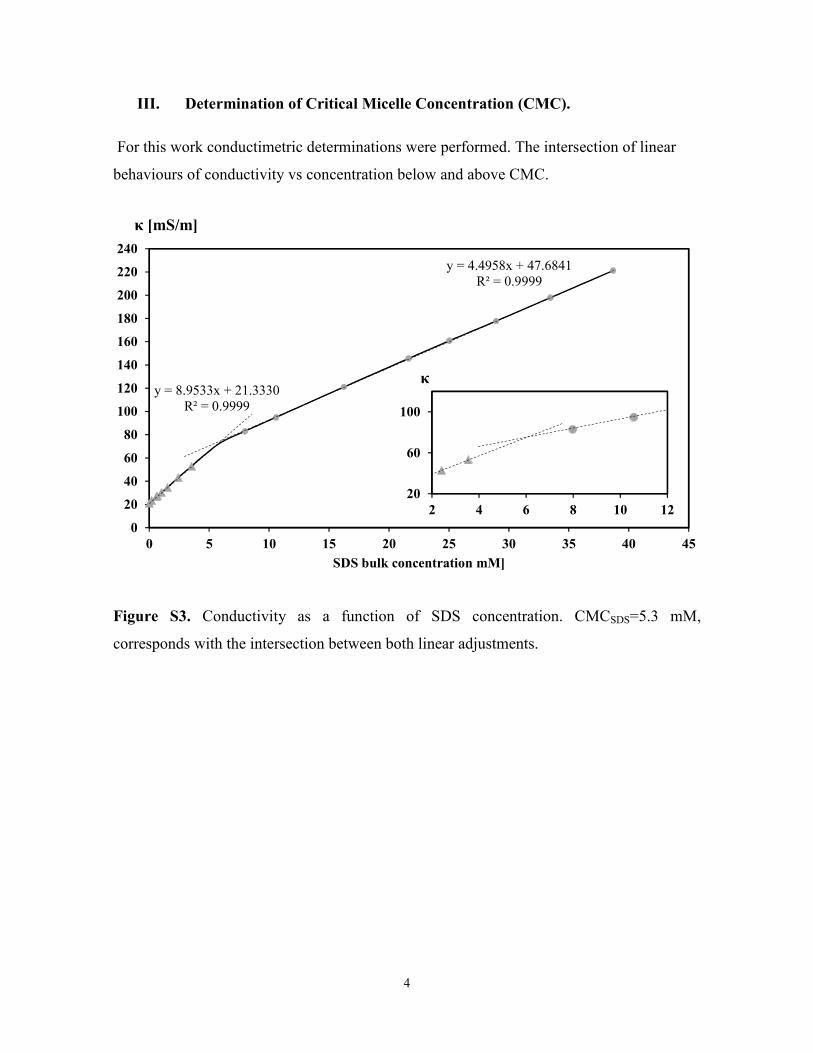
III. Determination of Critical Micelle Concentration (CMC).
For this work conductimetric determinations were performed. The intersection of linear
behaviours of conductivity vs concentration below and above CMC.
Figure S3. Conductivity as a function of SDS concentration. CMCSDS=5.3 mM,
corresponds with the intersection between both linear adjustments.
y = 8.9533x + 21.3330R² = 0.9999
y = 4.4958x + 47.6841R² = 0.9999
0 5 10 15 20 25 30 35 40 45
0
20
40
60
80
100
120
140
160
180
200
220
240
SDS bulk concentration mM]
κ [mS/m]
2 4 6 8 10 12
20
60
100
κ
5
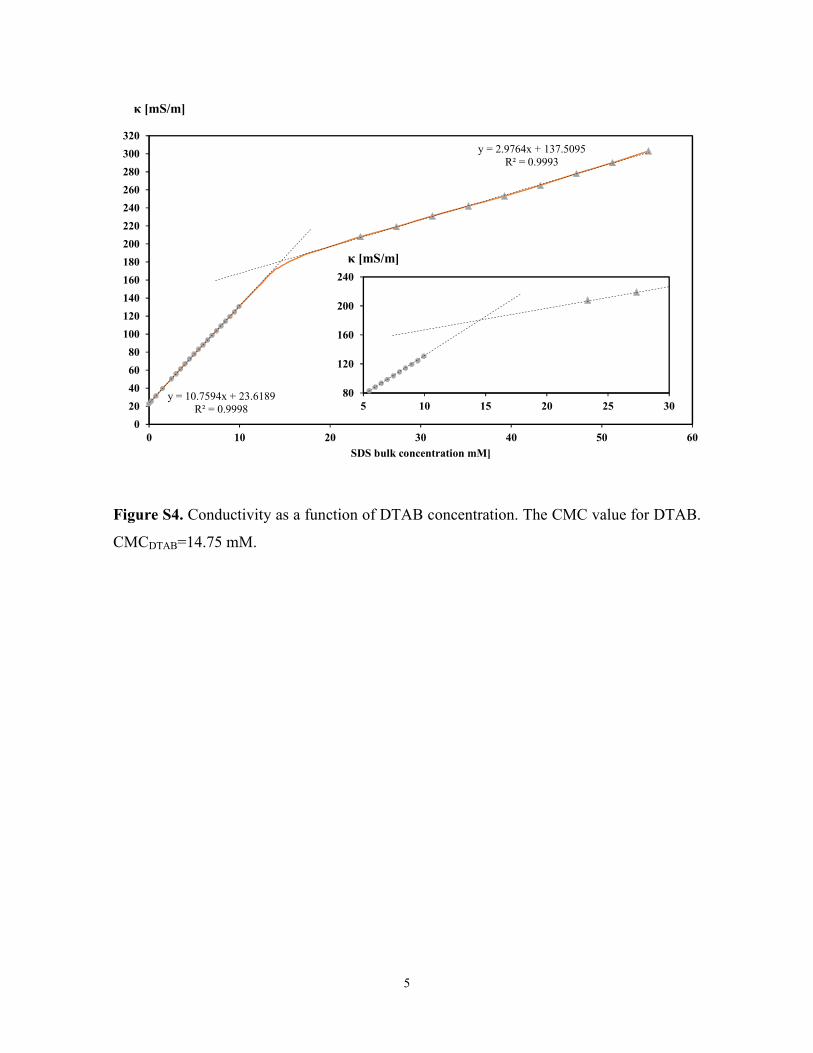
Figure S4. Conductivity as a function of DTAB concentration. The CMC value for DTAB.
CMCDTAB=14.75 mM.
y = 2.9764x + 137.5095R² = 0.9993
y = 10.7594x + 23.6189R² = 0.9998
0 10 20 30 40 50 60
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
SDS bulk concentration mM]
κ [mS/m]
5 10 15 20 25 30
80
120
160
200
240
κ [mS/m]

6
IV. DFT Analysis of Surfactant Molecules and MD-derived Modes of
Adsorption.
The studied surfactant molecules SDS, CAPB and DTAC were built and subject to
molecular dynamics simulations using the Materials Studio 8 suite for all calculations. The
M06-L functional, with DNP numerical basis set, was employed throughout this work.
Population analyses were performed with the Hirshfeld scheme.
The binding of these three surfactants on the (1 0 4) surface of calcite was simulated
through molecular dynamics in vacuo. Our goal was to elucidate the supramolecular factors
involved in the adsorption of each surfactant on the mineral surface.
Since the changed in zeta potential followed the charge of the lipophilic ion in both SAS
and DTAB, the counterions can be safely assumed to have greater mobility, plus enthalpic
solvation-contacts as hardness of the ion increases. Thus, Na+ and Br- were not considered
during the docking and annealing processes in the following studies in vacuo. The effect of
ions and solvent was also computed, to compare both experiments.
Sodium dodecylsulfate anion: This anionic surfactant features bidentate binding modes
when adsorbed onto neutral calcite in the absence of water (Figure S5). These coulombic
contacts are within ionic-bonding distance, so chemisorption is intrinsically preferred in
this model system.
Figure S5. Electrostatic potential maps for the dodecylsulfate anion. The yellow surface
encloses negative charge values which, as can be seen, concentrate on the anionic head.

7
Figure S6. Geometry and energies computed at the MMCOMPASS level for the adsorption of
dodecylsulfate anion on the (104) face of calcite, in vacuo. This strong electrostatic
interaction arises due to the hard ionic nature of the species involved and the spacing of
calcium cations, adequate for bidentate binding.
ECOMPASS (kcal/mol)
Average energy -47737.4
Standard deviation 1.2
Sum of isolated components -47650.3
Energy of unimolecular adsorption -87.1

8
Dodecyltrimethylammonium cation (DTA): Adsorption of the DTA cation on calcite
displays no characteristic directionality. This suggests a soft electrostatic behavior in this
molecule (Figure S7).
Figure S7. Electrostatic potential map for the N,N,N,-trimethyldodecylammonium cation.
The blue volume indicates positive charge values, which, as can be seen, delocalize towards
the hydrophobic chain. Computed at the DFT M06-L/DNP+ level, with implicit solvation
(water) using the COSMO scheme.

9
ECOMPASS (kcal/mol)
Average energy -47661.2
Standard deviation 2.1
Sum of isolated components -47625.9
Energy of unimolecular adsorption -35.3
Figure S8. Geometry and energies computed at the MMCOMPASS level for the adsorption of
dodecyltrimethylammonium cation on the (104) face of calcite, in vacuo. The diffuse, non-
directional electrostatic interaction of the species involved can be observed.

10
Cocoamidopropyl betaine: The electrostatic potential surface for CAPB shows its dual
ionic nature and soft-hard contrasting features for negative loci and positive zones.
Figure S9. Electrostatic potential map for the cocoamidopropyl betaine zwitterion (CAPB). The blue volume indicates positive charge values, which permeate the hydrophobic chain. Computed at the DFT M06-L/DNP level, with implicit solvation (water) using the COSMO scheme.
The prevailing adsorption mode of this amphoteric surfactant on calcite consists on a
tridentate binding, with the carboxylate head chemisorbed on two calcium ions. The
carboxylate-calcite interaction displays an average O-Ca internuclear distance of 2.1 Å,
smaller than the sum of ionic radii of the involved atoms.
Figure S10. In the bidentate chemisorbed interaction, the Ca-O distances are 2.1 Å, shorter than the sum of ionic radii, which is 2.4 Å. In the physisorption interaction, the Ca-O distance is 2.7 Å.

11
The dipolar carbonyl group of the amide functionality is also found to establish a short
contact with the mineral surface. This ion-dipole interaction between the signaled atoms in
figure S10 displays a Ca-O internuclear distance of 2.7 Å.
Figure S11. Bidentate mode observed during MD simulation of the adsorption of CAPB on calcite. As concentration increases, chemisorption contacts displace physisorbed ones.
A second bidentate mode appears upon increasing concentration. In this
configuration, an interaction between O and Ca is sacrificed in favor of ionic contacts with
neighboring molecules adsorbed in the vicinity (see Figure S11). Another bidentate mode
featuring a carboxylate-calcium chemical contact and a N-H----O3C hydrogen bond is
consistently observed (Figure S12).

12
Figure S12. An additional contribution to total adsorption observed during multiscale dynamics is a tridentate mode, where the NH group within the amide portion of CAPB establishes hydrogen bonding (ca. 1.8 Å).with a carbonate group at the interface. Bidentate modes become more important as concentration increases (12mpc), allowing for
a larger adsorption density, thus a larger total mineral/surfactant contacts. At 16 molecules
per cell, tridentate modes was virtually absent. In particular, physisorption of the amidic
carbonyl group is displaced by more exothermic, ionic interactions between carboxylate
polar heads and calcium ions. Upon reaching a concentration of 20 molecules per cell,
dynamic trajectories point towards a structural shift from mostly anionic-head adsorption to
a mixed distribution. This way, coverage is maximized and, at the same time, accumulation
of negative charge on the surface is alleviated.
ECOMPASS (kcal/mol)
Average energy -47803.4
Standard deviation 1.9
Sum of isolated components -47747.6
Energy of unimolecular adsorption -55.8
13
V. Energy Profiles for MD Simulations
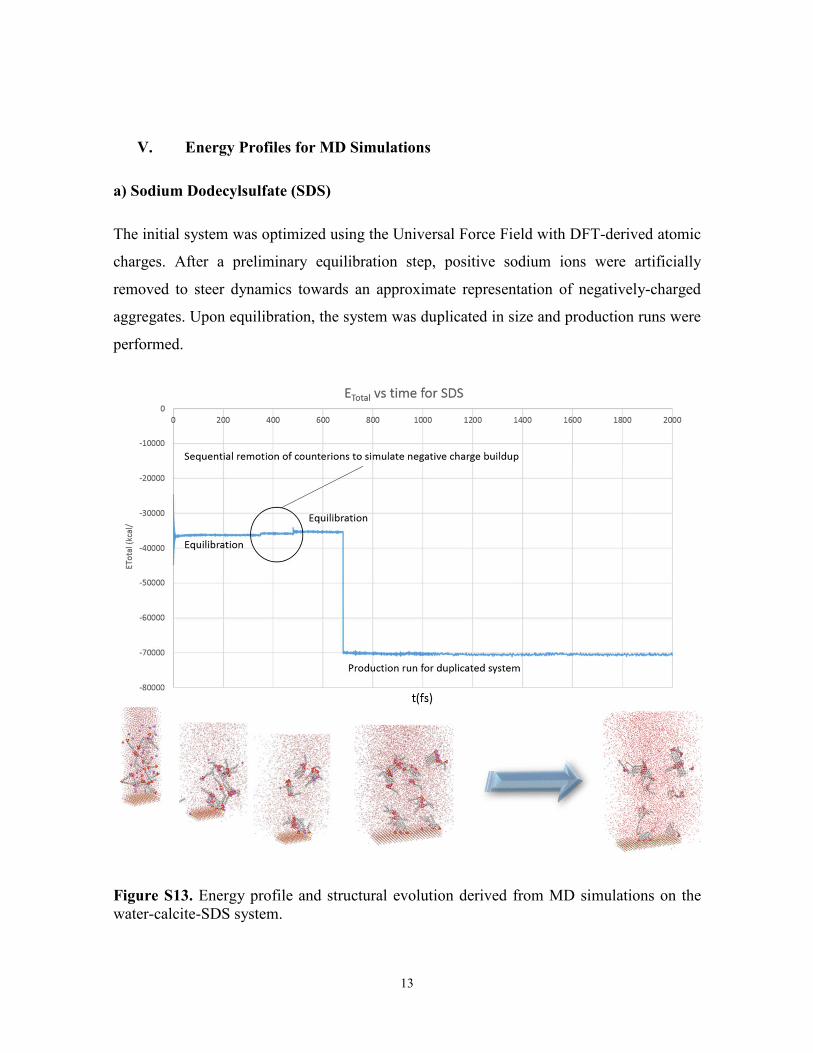
a) Sodium Dodecylsulfate (SDS)
The initial system was optimized using the Universal Force Field with DFT-derived atomic
charges. After a preliminary equilibration step, positive sodium ions were artificially
removed to steer dynamics towards an approximate representation of negatively-charged
aggregates. Upon equilibration, the system was duplicated in size and production runs were
performed.
Figure S13. Energy profile and structural evolution derived from MD simulations on the water-calcite-SDS system.
14
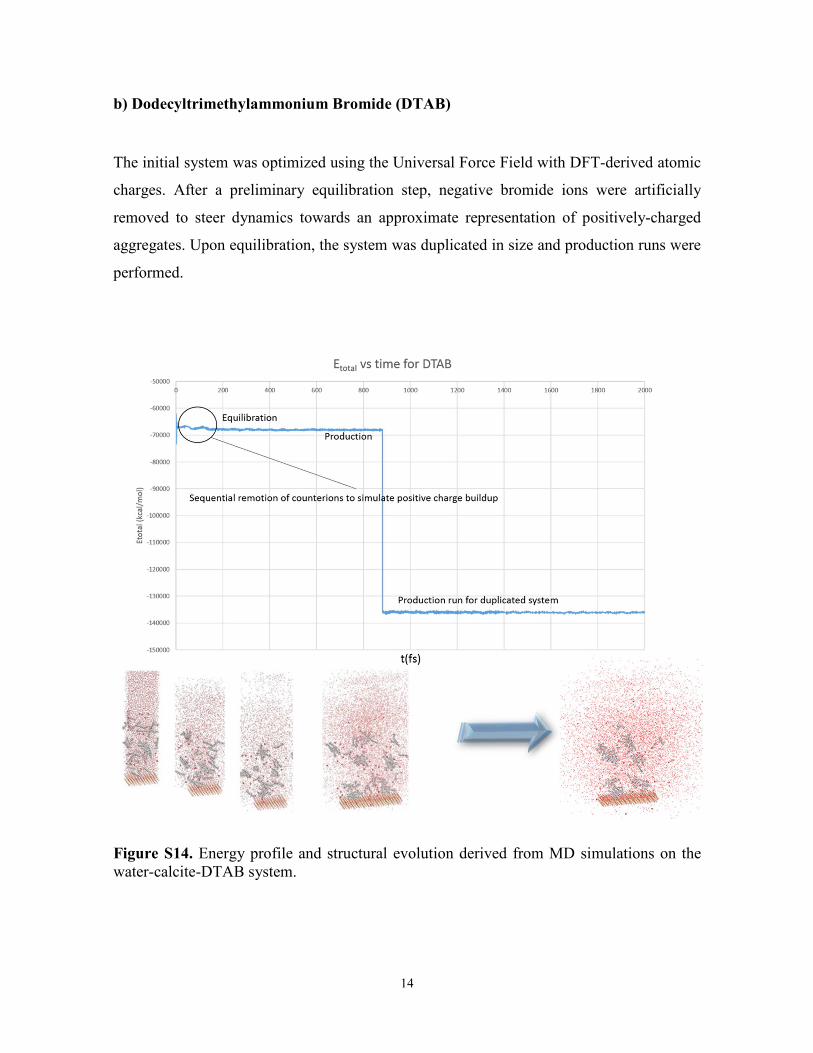
b) Dodecyltrimethylammonium Bromide (DTAB)
The initial system was optimized using the Universal Force Field with DFT-derived atomic
charges. After a preliminary equilibration step, negative bromide ions were artificially
removed to steer dynamics towards an approximate representation of positively-charged
aggregates. Upon equilibration, the system was duplicated in size and production runs were
performed.
Figure S14. Energy profile and structural evolution derived from MD simulations on the water-calcite-DTAB system.
15
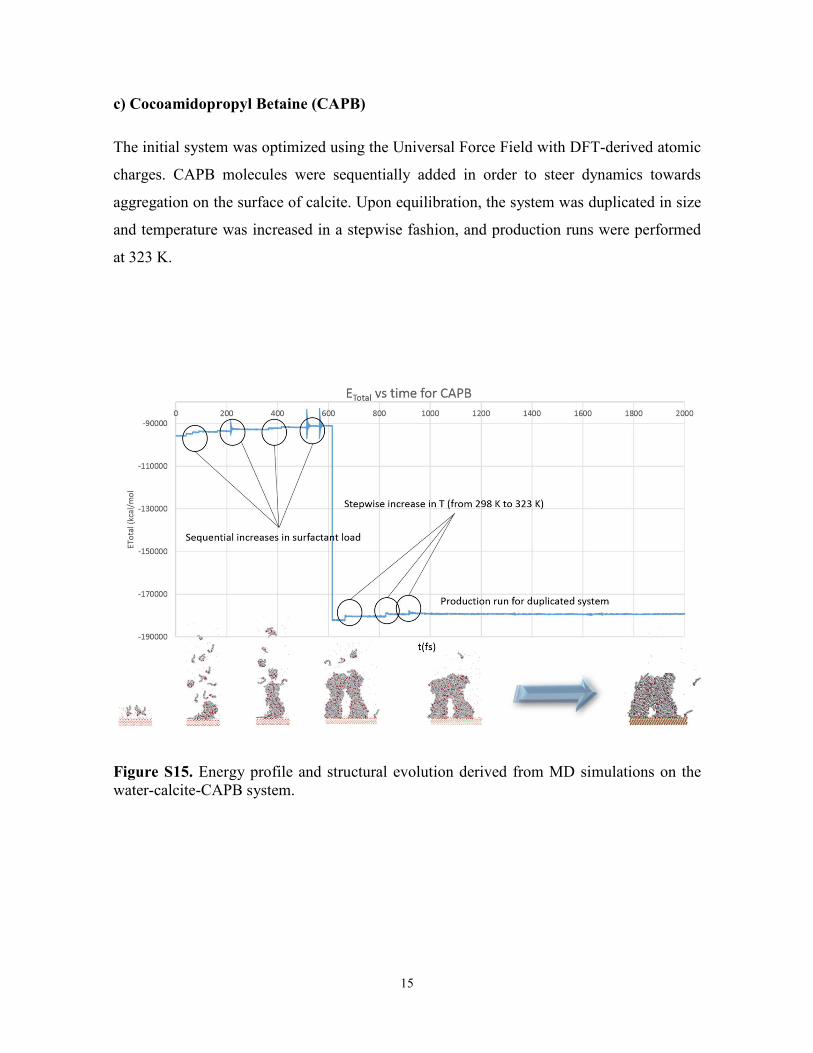
c) Cocoamidopropyl Betaine (CAPB)
The initial system was optimized using the Universal Force Field with DFT-derived atomic
charges. CAPB molecules were sequentially added in order to steer dynamics towards
aggregation on the surface of calcite. Upon equilibration, the system was duplicated in size
and temperature was increased in a stepwise fashion, and production runs were performed
at 323 K.
Figure S15. Energy profile and structural evolution derived from MD simulations on the water-calcite-CAPB system.











![isoprenylazides. Experimental and theoretical studies of ... · 1 Supporting Information for Experimental and theoretical studies of the [3,3]-sigmatropic rearrangement of isoprenylazides](https://img.pdfslide.us/doc/110x75/5f07b75d7e708231d41e6170/isoprenylazides-experimental-and-theoretical-studies-of-1-supporting-information.jpg)

















