Embed Size (px)
Citation preview

~ " ~ .-, ,~Ii "' ! U
ELSEVIER Journal of Electroanalytical Chemistry 422 (1997) 7-12
~ O J :
Prelinii'aa~-y note
Electrocatalytic oxidation of hydrogen peroxide by nitroxyl radicals
Beno~t Limoges, Chantal Degrand Equipe Electrochimie Organique. UMR "Synth~se, Eiectrosynti~se et Etude de Systkmes ~ lntdr~t Biologique", Unk, ersit~ Blaise-Pascal de
Ciermont-Ferrand, 24 Avenue des Landais. F-63177 Aubi~re, France
Received 8 October 1996: revised 19 November 1996
Abstract
A series of nitroxyi radicals have been used as efficient organic catalysts for the mediated oxidation of hydrogen peroxide at a carbon paste electrode immersed in aqueous media. Second order catalytic rate constants were measured and indicate an efficient inner sphere catalytic process. A nitmxide-loaded carbon paste electrode was prepared and used as a sensitive reagentless hydrogen peroxide sensor. The sensitivity of the resulting H202 anode was 0.23 Acre -2 M - t at 0.6V and a detection limit of 5 x 10 -s M was obtained.
Kev~ords: Nitroxyl radicals: Hydrogen peroxide: Electrocatalysis: Nitroxide-loaded carbon paste
1. Introduction
The selective and sensitive determination of hydrogen peroxide is of practical importance in biological and clini- cal fields, and in chemical, pharmaceutical and food indus- tries. Hydrogen peroxide is enzymatically generated or consumed by a large variety of oxidase enzymes which are the base of numerous biosensors [l], or which are com- monly used as labels in enzyme immunoassays [2]. Spec- trophotometric methods are widely used for the determina- tion of H202 [3], but the need for more simple, inexpen- sive, rapid and reliable detection systems has resulted in extensive research in the development of sensors and biosensors, especially those associated with electrochemi- cal detection [4-19]. Although amperometric techniques provide an attractive route to the quantification of H202, its oxidation or reduction at carbon electrodes requires high overpotentials and is thus susceptible to interference from electroactive species in sample matrices. To circum- vent these shortcomings several strategies have been devel- oped. Permselective coatings can be used to minimize the interference of electroactive constituents [4,5,8-11,16]. The detection potential of H202 can be lowered in the presence of metallized carbon surfaces [6-10] or by using organometallic mediators [11,12] such as metallophthalo- cyanines [12] which selectively electrocatalyse the oxida- tion or reduction of H202, probably through an inner- sphere process. Another strategy involves the use of horseradish peroxidase (HRP) which catalyses the reduc- tion of H202, and so HRP-based biosensors have been developed, which proceed by direct [7,13] or indirect
[14-19] electron transfer between the enzyme and the electrode. These biosensors are highly selective towards H202 and they can offer very high sensitivities [7,16,18,19]. However, they possess severals drawbacks, i.e. the carbon surface must have a proper conditioning so that facile direct electron transfer between the electrode and the enzyme can take place. Also the HRP tends to react with a variety of reducing substrates such as ascot- bate, and the instability of the enzyme limits the life of the biosensor. Conversely, metallized surfaces [6-10] or met- allophthalocyanine-modified electrodes [ 12] provide stable H202 sensors.
This note reports on the use of nitroxyl radicals as selective and efficient organic catalysts for the mediated anodic oxidation of H202, in view of their future applica- tions in H202 sensors, biosensors, or affinity binding assays.
2. Experimental
A 30% hydrogen peroxide solution was purchased from Aldrich and dilute solutions of H 202 were prepared daily. TEMPO (2,2,6,6-tetramethylpiperydinyloxy), 4.-hydroxy- TEMPO, 4-amino-TEMPO and 3-carboxy-PROXYL (PROXYL -- 2,2,5,5-tetramethylpyrrolidinyloxy) were used as received from Sigma. Graphite powder was sup- plied by Alfa Johnson Matthey (UCP-IM).
The unmodified carbon paste was prepared by mixing thoroughly 250mg of graphite powder with 200rag of
0022-0728/97/$17.00 Copyright © 1997 Elsevier Science S.A. All rights reserved. I'll S0022-0728(96)05052-8

8 B. Limoges, C. Degrand/Journal of Electroanalytical Chemistry 422 (1997) 7-12
silicon oil in mortar and pestle. Nitroxide-modificd carbon paste was obtained by adding the desired amount of TEMPO (0.2 wt%). Portions of the resulting paste were then packed into the end of a plastic tip with an internal diameter of 3 nun, and a stainless steel wire was inserted through the opposite extremity to establisb electrical con- tact, as described previously [20]. New surfaces were smoothed on a paper. Between the experiments the nitrox- ide-modified paste was stored at 4°C.
An EG&G PAR model 273 potentiostat interfaced to an IBM XT 286 computer system with a PAR M270 software was used for cyclic voltammetry (CV). Ampero- metric measurements were performed with a Tacussel Polaropulse type PRG5. The output response was dis- played on a SEFRAM TGV 164 x - t chart recorder. The electrochemical measurements were performed at room temperature in a standard three-electrode cell, equipped with a platinum wire counter electrode and an AglAgCII3 M NaCI reference electrode. The volume of the cell was 50 ml, but it contained generally 20 ml of the test solution which was stirred magnetically when necessary. Non- deaerated 0.1 M phosphate buffer (pH 7.4) or 0.1 M citrate buffer (pH 5.0) was used as electrolyte.
For the amperometric measurements the electrochemi- cal cell was placed inside a Faraday cage and a potential of 0.65 V was applied to the carbon paste electrode immersed in the buffer solution, and the transient background current was allowed to decay to a constant value before spiking the first aliquot of the stock H202 solution.
3. Results and discussion
The nitroxide derivatives are known as stable radicals and they undergo reversible one-electron oxidation in a large range of pHs, to form oxoammonium salts [21].
- + c ( 1 ) "N-O" o
They are able to mediate the oxidation of primary and secondary alcohols [22,23] and the mediation can be car- ried out advantageously at a nitroxide-modified electrode [24,25]. Two decades ago, Sen et al. showed that the chemically generated oxoammonium salt of 4-hydroxy- TEMPO can oxidize H20 2 [26], and the authors suggested the mechanism corresponding to Eqs. (2)-(4) and to the global reaction (5). According to these authors, the mecha- nism would be an inner-sphere process and would be analogous to the general mechanism for HeO 2 oxidation by polyvalent metal ions [27].
H202 ~ HO2 + H + (2)
~ + k) ,,N:O ÷ I-IO2- ~ , ~N-O" ÷ H02" (3)
"+ HO2" ~ "N-O" ÷ 02 ÷ H + (4) ,,N'O ÷ /
\+ 2"N-O" * O~ + 2H + 2 N:O * H20, ~ - (5)
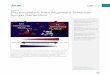
Taking into account this pioneering work by Sen et al. [26], we have presumed that the reversible anodic oxida- tion of nitroxide derivatives would provide an electrocat- alytic way for the oxidation of H202. TO validate this hypothesis, cyclic voltammetry of 4-hydroxy-TEMPO in the presence or the absence of H2O 2 was carried out at a carUon paste electrode immersed in a citrate buffer (pH 5.0). The voltammograms are shown in Fig. 1. The re- versible one-electron behavior of the redox couple 4-hy- droxy-TEMPO/4-hydroxy-TEMPO + (E ° = 0.59 V, aver- age of the anodic and cathodic peak potentials, A Ep = 71 m V , / p / V 1/2 -- constant, DRe d ---- 5 . 0 X 10 - 6 cm 2 s- t) in Fig. I(A) is consistent with the literature [28]. Addition of H202 leads to an enhancement of the oxidation peak while its reverse cathodic peak is suppressed (Fig. I(B)), which demonstrates that the electrochemically generated oxoammonium salt oxidizes catalytically H202. The com- parison of the cyclic voltammograms obtained for H202, with and without nitroxide (Fig. l(B)Fig. I(C) respec- tively) shows that the oxidation of H 2 0 2 a t a carbon paste elecarode is much more easily achieved with the mediated process, since the oxidation potential of H202 is lowered by ca. 0.55 V.
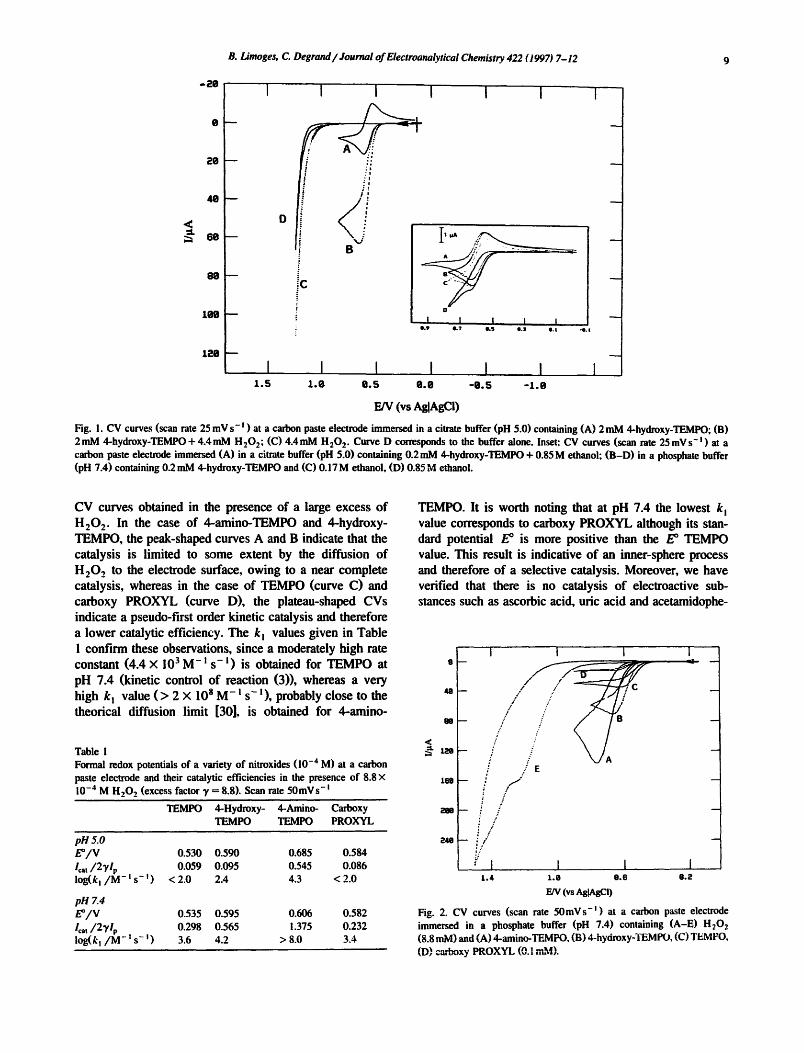
The CV behavior of several nitroxide derivatives was investigated at a carbon paste electrode with and without H202. Their formal redox potentials E °, their catalytic efficiency and their second order catalytic rate constant k~ are given in Table 1 at two pHs (5.0 and 7.4). The catalytic parameters were obtained with the help of the theory developed by Savtant and coworkers for homogeneous redox catalysis in the context of cyclic voltammetry [29]. The catalytic efficiency is expressed by the ratio Icat/2~/l p, where lea , is the catalytic peak current of the nitroxide derivative in the presence of H202, lp is the diffusion-cow trolled peak current of the nitroxide alone and the excess factor 7 is the ratio of the H202 to nitroxide bulk concen- trations. The additional factor 2 is related to the overall two-electron oxidation of H2O 2. The log(k~) values were derived from the working curve in fig. 3 of Ref. [29]. For each nitroxide, the ratio I~a,/271 p and therefore the rate constants k~ are higher at pH 7.4 than at pH 5.0, which is consistent with the global reaction (5). With the TEMPO series, the catalytic efficiency increases with increasing electron-withdrawing effect of the substituents. Indeed, the highest ratio is obtained for the protonated form of 4- ahfino-TEMPO which possesses the most positive E ° value, while the unsubstituted TEMPO has both the lowest E ° value and catalytic efficiency. Fig. 2 shows the catalytic
B. Limoges, C. Degrand i Journai of Electroanalytical Chemistry 422 (1997) 7-12 9
- 2 0
20
40
< ~k
60
80
100
120
I I I I I I I
i/ /:
i c
T'+ :
c a . --~ :
O
i I I I I l l . l t I kT 0 .5 l l . 1 I . ! " l . l
I I I I I I I 1 . 5 1 . 0 0 . 5 0 . 0 - 0 . 5 - 1 . 0
E/V (vs AglAgCi)
Fig. 1. CV curves (scan rate 25 mV s - I ) at a carbon paste electrode immersed in a citrate buffer (pH 5.0) containing (A) 2 mM 4-hydroxy-TEMPO; (B) 2 mM 4-hydroxy-TEMPO + 4.4 mM H202; (C) 4.4 mM H202. Curve D corresponds to the buffer alone. Inset: CV curves (scan rate 25 mV s -I ) at a carbon paste electrode immersed (A) in a citrate buffer (pH 5.0) containing 0.2 mM 4--hydroxy-TEMIK) + 0.85 M ethanol; (B-D) in a phosphate buffer (pH 7.4) containing 0.2 mM 4-hydroxy-TEMPO and (C) 0.17 M ethanol, (D) 0.85 M ethanol.
CV curves obtained in the presence of a large excess of H 2 0 2. In the case of 4.-amino-TEMPO and 4-hydroxy- TEMPO, the peak-shaped curves A and B indicate that the catalysis is limited to some extent by the diffusion of H 2 0 2 to the electrode surface, owing to a near complete catalysis, whereas in the case of TEMPO (curve C) and carboxy PROXYL (curve D), the plateau-shaped CVs indicate a pseudo-first order kinetic catalysis and therefore a lower catalytic efficiency. The k n values given in Table 1 confirm these observations, since a moderately high rate constant (4.4 × 103 M- l s - i) is obtained for TEMPO at pH 7.4 (kinetic control of reaction (3)), whereas a very high k n value ( > 2 × 108 M-n s - n), probably close to the theorical diffusion limit [30], is obtained for 4-amino-
Table 1 Formal redox potentials of a variety of nitroxides (10 -4 M) at a carbon paste electrode and their catalytic efficiencies in the presence of 8.8 × 10 -4 M H20 2 (excess factor 3'--8.81. Scan rate 50mVs -n
TEMPO 4-Hydroxy- 4-Amino- Carboxy TEMPO TEMPO PROXYL
pH 5.0 E°/V 0.530 0.590 !ca, 123"1 p 0.059 0.095 log(k I / M - I s- I ) < 2.0 2.4
pH 7.4 g'/v 0.535 0.595
lea,/23'/p 0.298 0.565 Iog(k t / M - * s- n ) 3.6 4.2
0.685 0.584 0.545 0.086 4.3 < 2.0
0.606 0.582 1.375 0.232
> 8.0 3.4
TEMPO. It is worth noting that at pH 7.4 the lowest k n value corresponds to carboxy PROXYL although its stan- dard potential E ° is more positive than the E ° TEMPO value. This result is indicative of an inner-sphere process and therefore of a selective catalysis. Moreover, we have verified that there is no catalysis of electroactive sub- stances such as ascorbic acid, uric acid and acetamidophe-
8 8
< :::k
120
168
2eO
2 4 8
B - -
4 8 - -
I I I i
/
/ / C / .:- / _,
i i .: / B f :." .: \
_ - _
- ! ; : /
-- : ...-
i / • . - - / / - -
L ~ I I I i
t .4 x.e e.e e.a
~ , (vs ASlAgCl)
Fig. 2. CV curves (scan rate 50mVs -n ) at a carbon paste electrode immersed in a phosphate buffer (pH 7.4) containing (A-E) H20 2 (8.8 raM) and (A) 4-amino-TEMPO, (B) 4-hyaroxy-TEMPO, (C) TEMFO, (D) carboxy PROXYL (0.1 mM).
10 B. Limoges, C. Degrand / Journal of Eiectroanalytical Chemistry 422 (1997) 7-12
~.0
4 . 8
I I I I i
B
I 8 .1
I I I I 0 . 9 8 . 7 0 . 5 8 . 3
EN (vs Agi^sCi)
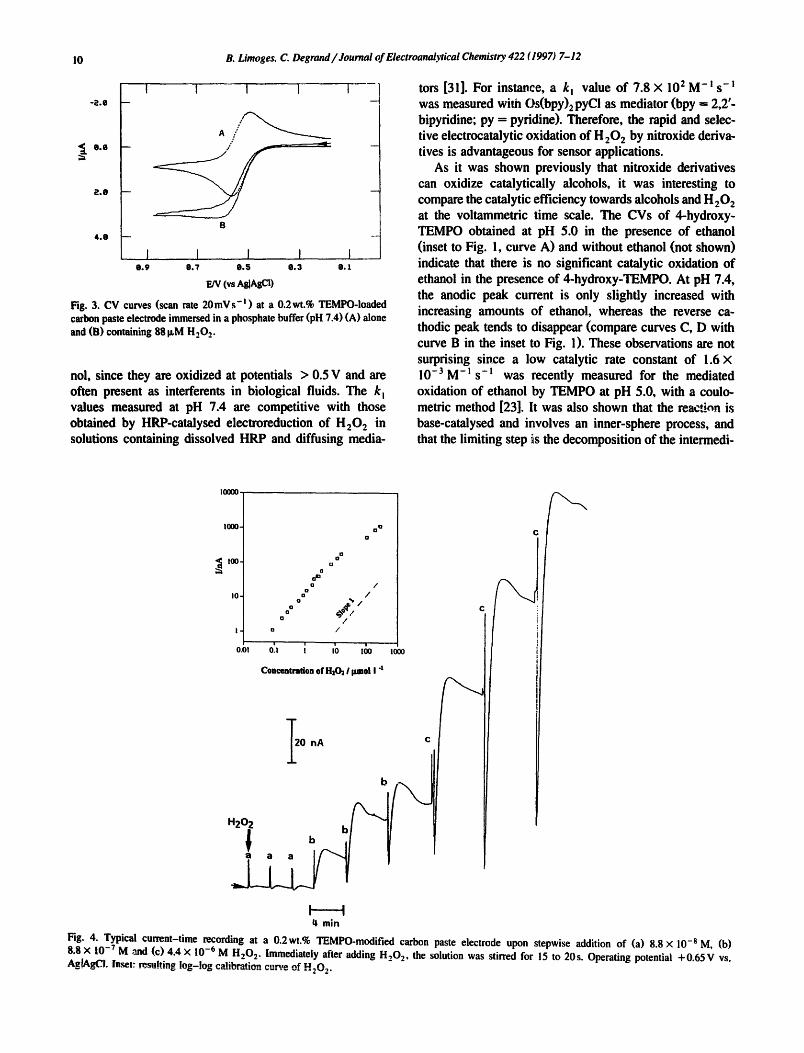
Fig. 3. CV curves (scan rate 20mVs- ' ) at a 0.2wt.% TEMPO-loaded carbon paste electrode immersed in a phosphate buffer (pH "/.4) (A) alone and (B) containing 88 ILM H202.
nol, since they are oxidized at potentials > 0.5 V and are often present as interferents in biological fluids. The k, values measured at pH 7.4 are competitive with those obtained by HRP-catalysed electroreduction of H202 in solutions containing dissolved HRP and diffusing media-
tors [31]. For instance, a k I value of 7.8 X 102 M- ' s - i was measured with Os(bpy)z pyC! as mediator (bpy -- 2,2'- bipyridine; py = pyridine). Therefore, the rapid and selec- tive electrocatalytic oxidation of H 202 by nitroxide deriva- tives is advantageous for sensor applications.
As it was shown previously that nitroxide derivatives can oxidize catalytically alcohols, it was interesting to compare the catalytic efficiency towards alcohols and H 202 at the voltammetric time scale. The CVs of 4-hydroxy- TEMPO obtained at pH 5.0 in the presence of ethanol (inset to Fig. 1, curve A) and without ethanol (not shown) indicate that there is no significant catalytic oxidation of ethanol in the presence of 4-hydroxy-TEMPO. At pH 7.4, the anodic peak current is only slightly increased with increasing amounts of ethanol, whereas the reverse ca- thodic peak tends to disappear (compare curves C, D with curve B in the inset to Fig. 1). These observations are not surprising since a low catalytic rate constant of 1.6 × 10 -~ M -m s-~ was recently measured for the mediated oxidation of ethanol by TEMPO at pH 5.0, with a coulo- metric method [23]. It was also shown that the reaction is base-catalysed and involves an inner-sphere process, and that the limiting step is the decomposition of the intermedi-
I0000-
I000-
!00-
I0-
1
0.01
O
°
o % / o° ,,,,oez
u /
° /
oi, i ,b i~o
Concentnttion of HzO21 limol I .!
1000
20 nA
H202
a a a
min
Fig. 4. Typical current-time recording at a 0.2wt.% TEMPO-modified carbon paste electrode upon stepwise addition of (a) 8.8 × 10 -s M, (b) 8.8 × 10-TM ,and (c)4.4 × 10-6M H202. Immediately after adding H20 z, the solution was stirred for 15 to 20s. Operating potential +0.65V vs. Ag[AgCI. Inset: resulting log-log calibration cu.,',-e of H202.

B. Limoges, C. Degrand / Journai of Electroanalytical Chemistry. 422 (1997) 7-12 11
ate complex, which would explain the loss of reversibility of the redox peak systems in the inset to Fig. 1. Clearly, H202 can be selectively detected in the presence of alipbatic alcohols, even at pH 7.4.
The efficient catalytic oxidation of H202 by nitroxide mediators suggested to us the construction of a nitroxide- loaded carbon paste electrode and its analytical application as a sensitive reagentless hydrogen peroxide sensor.
Although 4-amino-TEMPO is the most efficient cata- lyst, the use of TEMPO for the development of a nitrox- ide-containing carbon paste electrode was preferred, be- cause it is more hydropbobic than the other nitroxides. Hence, it can be more strongly retained within ~ e paste. Fig. 3 shows the cyclic voltammograms obtained at a carbon paste electrode containing 0.2 wt% of TEMPO in the absence and in the presence of 88 IxM of H 202 (curves A and B respectively). The redox behavior of TEMPO contained in the paste is diffusion controlled (Fig. 3(A), A Ep = 70 mV and E ° = 0.526 V), i.e. as in solution (Table 1). This behavior suggests that TEMPO is not adsorbed or immobilized within the paste, but that it freely diffuses through it. In the presence of H202 a well-defined plateau-shaped curve is observed (Fig. 3(B)), which indi- cates that the catalytic oxidation of H 202 proceeds. Fig. 4 shows a typical amperometric response of the 0.2wt% tEMpo-modified carbon paste electrode to the stepwise addition of H202 when the applied potential was +0.65 V. The spikes appearing after injection of H202 result from stirring the solution for 15 to 20 s. Afterwards the signal increases to a maximum in quiet solution. This maximum current was adopted as the output signal and its depen- dence on the concentration of H202 is depicted by the inset to Fig. 4. The time for 90% response ranges between 55 and 80s for low and high adducts of H202 respec- tively. The current increases linearly with the H202 con- centration over 2 orders of magnitude, from ca. 10 -7 to 10 -s M, with a detection limit of ca. 5 × 10-SM ( s i g n a l / n o i s e ratio 3) and a sensit ivity of 0.23 A cm- 2 M- !. At the highest concentrations, deviation from linearity is observed. The sensitivity obtained in this work is ca. 5 times lower than the best sensitivities (ca. 1 Acm 2 M-L) found in the literature for H202 detection [10,18,19]. High sensitivities are generally obtained for a three-dimensional distribution of the catalytic sites [10,16,18,19]. Indeed, when the mediator is covalently attached to a polymer backbone on the electrode surface, the diffusion of the mediator is restricted [ 17]. Our results suggest that the nitroxide mediator would be released from the carbon paste during its oxidation and would form a three-dimensional steady-state diffusion layer adjacent to the electrode surface. Therefore an excellent catalytic effi- ciency is achieved, since the mediator can freely diffuse.
No change in the calibration plot was observed during a three-month utilization of the 0.2wt% TEMPO carbon paste electrode, indicating that TEMPO was stable within the carbon paste matrix.
4. Conclusions
This preliminary study has shown that nitroxyl radicals are efficient selective organic catalysts for the mediated anodic oxidation of H202. Moreover, low current re- sponses of H 202 at a TEMPO-modified carbon paste have been achieved, and it can be envisaged that an electrode of this type can advantageously replace a Pt electrode. How- ever, an important question remains concerning practical applications of this system in biological fluids where the presence of endogenous species electroactive at about 0.6 V can interfere. Further work is under way to study the potential use of the mediated anodic oxidation of H202 as an electrochemical detection method for enzyme im- munoassays involving oxidase labels, and to construct low-cost reagentless biosensors.
References
If]
[2] [31
[4]
Is]
[6] [7]
[8] [9]
[10] [Ill
[121
[13l
[141
[15l
[16]
[17]
[18l [19]
[2o]
[211
[22]
[23]
[24]
[251
A.P.F. Turner, I. Karube and G.S. Wilson (Eds.), Biosensor: Funda- mentals and Applications, Oxford University Press, Oxford, 1987. J.P. Gosling, Clin. Chem., 36 (1990) 1408. W.C. Schumb, C.N. Satterfield and R.L. Wentworth, Hydrogen Peroxide, Reinhold, New York, 1975. D.S. Bindra, Y. Zhang, G.S. Wilson, R. Stemherg, D.R. Th~venot, D. Moaui and G. Reach, Anal Chem., 63 (1991) i692. J.P, Lewry, IC McAreer, S.S. El A~rash, A. Duff and R.D. O'Neill, Anal. Chem., 66 (1994) 1754. L. Gorton, Anal. Chim. Acta, 178 (1985) 247. W.O. Ho, D. Athey and CJ. McNeil, J. Electroanal. Chem., 351 (1993) 185. J. Wang, J. Liu, L. Chen and F. Lu, Anal. Chem., 66 (1994) 3600. I. Wang and H. Wu, J. Electroanal. Chem., 395 (1995) 287. Z. Zhang, H. Liu and J. Deng, Anal. Chem., 68 (1996) 1632. I. Taniguchi, K. Matsushita, M. Okamoto, I,P. Collin and J.P. Sauvage, I. Electroanal. Chem., 280 (1990) 221. M.A.T. Gilmanin, RJ. Ewen and J.P. Hart, ]. Electroanal. Chem., 401 (1996) 127. U. Wollenherger, J. Wang, M. Ozsoz and E.G. Romero, Bioelect. Bioenerg., 26 (1991) 287. T. Tatsuma, Y. Okawa and T. Watanahe, Anal. Chem., 61 (1989)
2352. J. Kulys, U. Bilitewski and R.D. Schmid, Bioelect. Bioenerg., 26 (1991) 277. P. Dominguez-Sanchez, AJ. Miranda-Ordieres, A. Costa-Garcia and P. Tunon-Blanco, Electroanalysis, 3 (1991) 281. A. Mulchandani, C.L. Wang and H.H. Weetall, Anal. Chem., 67
0995) 94. M. Vreeke, R. Maidan and A. Heller, Anal. Chem., 64 (1992) 3084. T. Lumley-Woodyear, P. Rocca, J. Lindsay, Y. Dror, A. Freeman and A. Heller, Anal. Chem., 67 (1995) 1332. S. Rapicault, B. Limoge.~ and C. Degrand, Anal. Chem., 68 (1996)
930. J.M. Bobbitt and C.L. Flores, Heterocycles, 27 0988) 509 and
references cited therein. M.F. Semmelhack, C.S. Chou and D.A. Cones, J. Am. Chem. Soc.,
105 (1983) 4492. B. Chmielewska, P. Krzyczmonik and H. Scholl, J. Electroanal.
Chem., 395 (1995) 167. A. Dcronzier, D. Limosiu and J.C. Moutet, Electrochim. Acta, 32
(1987) 1643. A. Mel-z and H. Bachmann, J. Am. Chem. Soc., 117 (1995) 901

12 B. Limoges, C. Degrand / Journal of Electroanalytical Chemistry 422 (1997) 7-12
[26] V.D. Sen, V.A. Golubev, I.V. Kulyk and E.G. Rosantsev, Izv. Akad. Nauk SSSR, Ser. Khim., 8 (1976) 1745.
[27] T.E. Jones and R.E. Hamm, inorg. Chem., 13 (1974) 1940. [28] G. Thomas and J.G. Mohanty, Indian J. Chem., A21 (1982) 451. [29] C.P. Andrieux, C. Blockman, J.M. Dumas-Bouchat, F. M'Halla and
J.M. Say,ant, J. Electroanal. Chem., 113 (1980) 19.
[30] C.P. Andrieux, C. Blockman, J.M. Dumas-Bouchat, F. M'Halla and J.M. Say,ant, J. Am. Chem. Soc., 101 (1979) 3431.
[31] M.G. Garguilo, N. Huynh, A. Proctor and A.C. Michael, Anal. Chem., 65 (1993) 523.