Embed Size (px)
Citation preview

溶液の分子論
京都大学化学研究所
松林 伸幸

第0章 はじめに
講義日程:水曜日午後 4 時半~6 時 C345 号室 講義の目的:溶液の分子論 講義内容:
1) 熱力学の復習 流体の相について 化学ポテンシャルの導入 化学ポテンシャルが統計熱力学で最も重要な物理量である
2) 統計力学の基礎的な部分 化学ポテンシャルの統計力学的表現 自由エネルギーの最小化と化学平衡との関連
3) 分布関数の考え方 構造の不規則な系では、構成分子の配置は確率的である 情報の縮約の遂行=さまざまな「便利」な分布関数 相互作用ポテンシャルと分布関数に関する密度汎関数理論 溶液系の分子配置の実現法=分子シミュレーションと積分方程式
4) 時間相関関数の考え方 Liouville 演算子の使用による、古典力学と量子力学の「同型」な取り扱い 確率的時間発展に対する Schrödinger 描像と Heisenberg 描像 久保の線形応答理論と Redfield の緩和理論 応用例としての NMR 分光法
各自の名前、所属研究室、学年、e-mail アドレスを教えてください 連絡先:電話 内線 3071 (吉田地区からかけるときは 17-3071)
e-mail [email protected]

第 1 章 流体の相
1.1 はじめに
流体とは、読んで字のごとく、流れる物質状態を意味し、気体・液体をそこに含む。
流体の特性は、温度と密度(圧力)によって左右される。温度については、流体内に
おかれた物質の物性・反応性を制御する重要な変数であるとの認識は広く行き渡って
いる。例えば、温度の効果はアレニウス式に見られるように大変大きく、日常生活で
溶媒の温度特性を(知らない間に)利用していることも多い。これに対して、密度を
日常生活で変数として利用することはあまり無いが、その効果も重要なものである。
実際、気体は、密度の低い流体であり、拡散能は大きいが、溶媒和能は小さい。逆に、
液体は、密度の高い流体であり、溶媒和によって、様々な影響を溶質に与えることが
できる。密度を連続的に変化させるためには、気体とも液体ともつかない超臨界流体
を使うのがよい。超臨界流体では、密度の変化を伴う相転移は存在しない。それゆえ、
密度軸に焦点を当てて、気体のような低い密度から液体のような高い密度までを連続
的に走査して、溶媒和された分子の示す物性・反応性を系統的に探索・制御すること
が可能になる。
本講義では、流体を分子レベルで記述する方法論を紹介する。気体・液体のみなら
ず超臨界流体までを含めて流体を捉えるとき、広い温度・密度領域が連続的にカバー
されるので、方法論もまた温度・密度を変化させても共通に使用することができるも
のでなくてはならない。本講義は、「溶液の分子論」と題して、分布関数や相関関数
の理論、および、構造とダイナミクスを実験的に探る NMR 分光法を紹介する。これ
らの方法は、液体への適用を主目的として発展してきたものの、特定の温度・密度領
域への適用に偏らない一般的な原理を身につけることが、流体の物理化学的研究のた
めに重要である。
化学における流体系の研究では、近年の環境関連問題への超臨界流体の適用は、注
目に値するものである。しかし、本講義の目的は、気体・液体およびそれをつなぐ媒
質としての超臨界流体を記述する基礎的な方法論(の一端)を紹介することである。
超臨界流体の応用については、成書があるので、そちらを参考にしていただきたい
[1,2]。
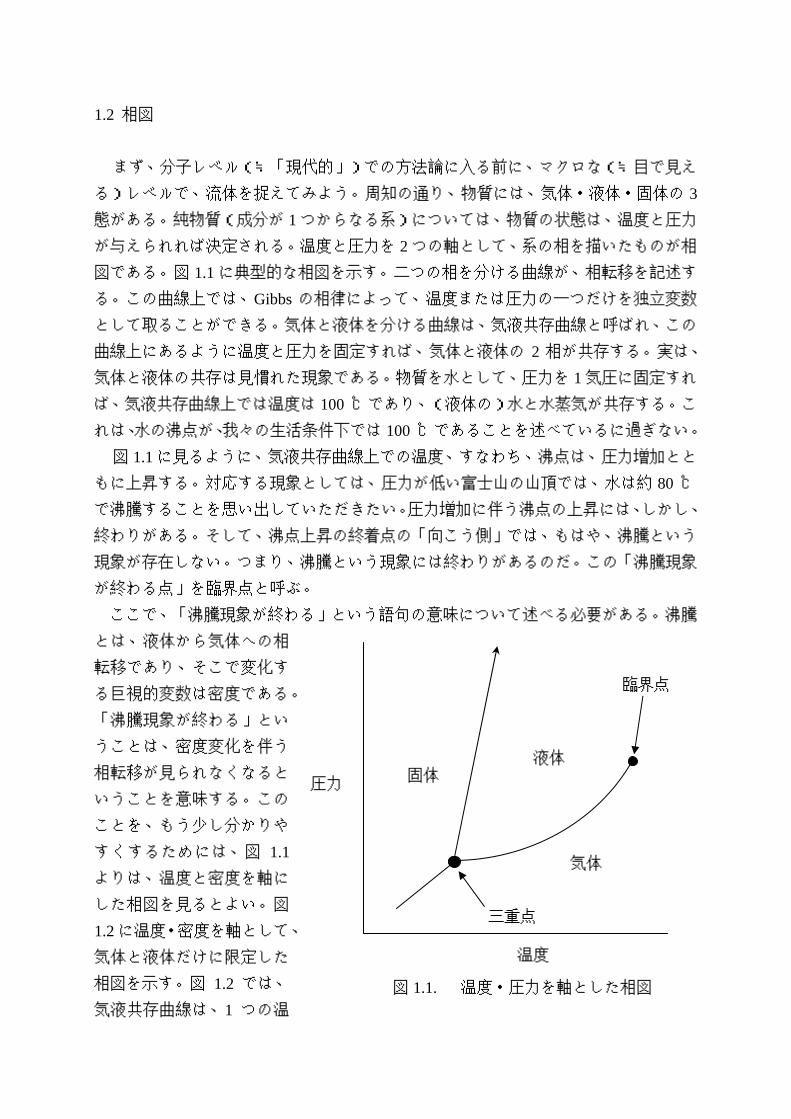
1.2 相図
まず、分子レベル(≒「現代的」)での方法論に入る前に、マクロな(≒目で見え
る)レベルで、流体を捉えてみよう。周知の通り、物質には、気体・液体・固体の 3
態がある。純物質(成分が 1 つからなる系)については、物質の状態は、温度と圧力
が与えられれば決定される。温度と圧力を 2 つの軸として、系の相を描いたものが相
図である。図 1.1 に典型的な相図を示す。二つの相を分ける曲線が、相転移を記述す
る。この曲線上では、Gibbs の相律によって、温度または圧力の一つだけを独立変数
として取ることができる。気体と液体を分ける曲線は、気液共存曲線と呼ばれ、この
曲線上にあるように温度と圧力を固定すれば、気体と液体の 2 相が共存する。実は、
気体と液体の共存は見慣れた現象である。物質を水として、圧力を 1 気圧に固定すれ
ば、気液共存曲線上では温度は 100 ℃であり、(液体の)水と水蒸気が共存する。こ
れは、水の沸点が、我々の生活条件下では 100 ℃であることを述べているに過ぎない。
図 1.1 に見るように、気液共存曲線上での温度、すなわち、沸点は、圧力増加とと
もに上昇する。対応する現象としては、圧力が低い富士山の山頂では、水は約 80 ℃
で沸騰することを思い出していただきたい。圧力増加に伴う沸点の上昇には、しかし、
終わりがある。そして、沸点上昇の終着点の「向こう側」では、もはや、沸騰という
現象が存在しない。つまり、沸騰という現象には終わりがあるのだ。この「沸騰現象
が終わる点」を臨界点と呼ぶ。
ここで、「沸騰現象が終わる」という語句の意味について述べる必要がある。沸騰
とは、液体から気体への相
転移であり、そこで変化す
る巨視的変数は密度である。
「沸騰現象が終わる」とい
うことは、密度変化を伴う
相転移が見られなくなると
いうことを意味する。この
ことを、もう少し分かりや
すくするためには、図 1.1
よりは、温度と密度を軸に
した相図を見るとよい。図
1.2 に温度・密度を軸として、
気体と液体だけに限定した
相図を示す。図 1.2 では、
気液共存曲線は、1 つの温
温度
圧力
気体
液体 固体
三重点
臨界点
図 1.1. 温度・圧力を軸とした相図

度について 2 値の曲線で表わされている。これは、気体および液体という 2 つの密度
の異なる系が、沸点で共存していることを表わす。曲線の内部は、沸騰によって密度
がジャンプするために、現実には存在し得ない領域である。そして、温度上昇にとも
なって、気液共存曲線上の気体と液体の密度の差は狭まり、臨界点で、気液共存曲線
上の密度差が 0,つまり、共存する気体と液体の密度が一致する。これ以上の温度で
は、密度のジャンプは存在せず、「沸騰」は起きない。
臨界点における温度・圧力・密度を、それぞれ、臨界温度・臨界圧力・臨界密度と
呼び、まとめて、臨界定数と呼ばれる。臨界定数は、物質固有の量であり、理科年表
などにもまとめられている。そして、温度が臨界温度以上になった状態を「超臨界流
体」と呼ぶ(圧力が臨界圧力以上ということも「超臨界流体」の定義に加えることも
あるが、ここでは、自由に密度を変えることが可能な媒質という立場に立ち、「超臨
界流体」を温度だけで定義する)。図 1.2 に見るように、臨界温度を超えると、密度
の自由な変化が可能になる。水を例に取れば、0.3 g/cm3 や 0.7 g/cm3 の水というものは、
通常の条件では存在しないが、臨界温度以上では存在する。それゆえ、超臨界流体で
は、これまで可能でなかった密度領域の研究が可能になる。つまり、密度効果の系統
的な研究が可能になるわけである。
余談ではあるが、図 1.1 で液体と固体を分ける共存曲線には、終わりがないと信じ
られている。つまり、液体と固体の間の相転移に関する臨界点は存在しないと考えら
密度
温度
気液共存曲線
液体 気体 この領域には
入れない
超臨界流体
臨界密度
臨界温度 臨界点
図 1.2. 温度・密度を軸とした相図

れている。また、気体・液体・固体の全てが共存できる点を三重点と呼ぶ。三重点で
は、Gibbs の相律によって、温度と圧力はただ一通りに決まる。三重点も、臨界点と
同様に、物質固有の量である。
ここでは、1 成分系の相図について述べたが、2 成分系以上では、相図は大変複雑
なものになる。2 成分系以上の相図については、成書を参照されたい[3]。
問1.1 Gibbs の相律を調べる。
問1.2 水・メタノール・二酸化炭素の臨界定数を調べる。
問1.3 上記の物質について、1 気圧の沸騰での密度変化を調べる。
問1.4 液体と固体の間の相転移に関する臨界
点が存在しない理由を考える。
問1.5 共 存 曲 線 を 記 述 す る
Clausius-Clapeyron の式を調べる。
問1.6 図 1.3 のように、体積一定の容器の中
に室温で液体を密閉した系が、温
度上昇とともにどのように変化し
ていくかを、図 1.2 に即して述べる。
また、超臨界状態における密度が
どのように与えられるかを考える。
第 1 章の参考文献
[1] 超臨界流体の環境利用技術 (エヌ・ティー・エス) 1999 年
[2] 水熱科学ハンドブック (技報堂出版) 1997 年
注:上記 2 つの本は大冊で大変高価であり、本屋にはおそらく無いので、図書
館を探された方がよいだろう。
[3] 化学便覧 基礎編 (丸善)
図 1.3 体積が一定の容器に 液体を密閉した状態

第 2 章 溶液の統計熱力学の基礎
溶液を取り扱うためには、統計熱力学の素養が必要である。本章では、溶液の統計
熱力学の基礎的事項について述べる。
2.1 溶けるということと溶けないということ
砂糖は水によく溶ける。また、油は水には溶けない。これらのことは、日常生活に
おける常識である。溶液の統計熱力学とは、「溶ける」「溶けない」という日常的感
覚を発展させて、数量的な関係をきちんと押さえて精密化し体系化した学問体系であ
る。
今、精密化という言葉を述べたが、実際に様々な概念を曖昧さのない明瞭な形であ
らわすためには、溶液のかかわる現象についての用語の定義が必要である。まず、溶
液(solution)という言葉であるが、これは一般に砂糖水などから連想されるように、流
体状態にある均一な混合物(mixture)をさす。均一な混合物が固体状態を取るときは固
溶体(solid solution)と呼ばれるが、ここでは流体状態にある溶液だけを対象にする。溶
液はある流体に他の物質を溶かす(溶解させる)ことでつくられる。そして、もとに
なる流体を溶媒(solvent)と呼び、溶解した物質を溶質(solute)と呼ぶ。冒頭にあげた砂
糖水の例では、水が溶媒であり、砂糖が溶質である。エタノールを水に溶かす場合な
どは、エタノールと水が任意の組成比で混合するため、溶媒と溶質の区別は明らかで
はないが、便宜上多量に存在する成分を溶媒と見なすことが多い。
次に、「よく溶ける」「あまり溶けない」というのは日常的感覚であり、その意味
するところは人によって若干異なることがある。そこで、明瞭に定義された物理量で
もって、「どれだけ溶けるか」を数量的にきちんと表すと、すべての人が共通の尺度
を用いることが出来て大変便利である。溶解度(solubility)とは、「どれだけ溶けるか」
を表す物理量である。それ以上は溶けないという限界まで溶質を溶かした溶液を飽和
溶液(saturated solution)と呼ぶが、溶解度とは飽和溶液中の溶質の濃度を表す量である。
実用的には、溶液を 1 リットルにしたときの溶質の量や、溶媒 1 kg に溶ける溶質の
量でもって、溶解度をあらわすことが多い。
しばしば使われる濃度の単位が、体積モル濃度(molarity)と重量モル濃度(molality)
である。体積モル濃度は、溶液 1 リットルあたりに溶けた溶質のモル数を表し、重量
モル濃度は、溶媒 1 kg あたりに溶けた溶質のモル数を表す。ここで、1 リットルや 1 kg
を単位にするのには、慣用上以上の理由はないが、単位になる量を決めて、その中の

溶質量を測ることが重要である。同じ体積モル濃度の溶液でも、5 リットルの溶液は
1 リットルの溶液に比べて 5 倍の量の溶質を含む。
溶解度を分子のレベルで論じるためには、統計熱力学の助けを借り、化学ポテンシ
ャル(chemical potential)の概念を用いなくてはならない。そこで、以下では、簡単な熱
力学のおさらいをして化学ポテンシャルを導入し、溶解現象を化学ポテンシャルの言
葉で論じる。
2.2 熱力学の簡単なおさらい
一つの系が与えられたときに(この場合は溶液と考えてよい)、dQ だけの熱量を
吸収し、圧力 P を媒介にして系の体積 V が dV だけ変化したときに、系の内部エネル
ギーU は
dU=dQ-PdV (2.2.1)
で表される量だけ変化する。また、系の変化が非常に遅いときには熱量 dQ は、温度
T とエントロピーS を用いて
dQ=TdS (2.2.2)
と書くことができる。すると、式(2.1)と式(2.2)をまとめると、
dU=TdS-PdV (2.2.3)
となる。式(2.2.3)が、熱力学の基本方程式であり、熱力学の体系は式(2.2.3)の上に構
築されている。ここで注意すべきことは、式(2.2.3)において、エントロピーS と体積 V
の変化量が内部エネルギーU の変化量と結び付つけられていることである。このこと
を、式(2.2.3)ではエントロピーS と体積 V が独立変数になっているという。例えば、
エントロピーS を一定にして体積を変化させると、内部エネルギーの変化は単純に系
にかかっている圧力で決まる。一方、式(2.2.3)によると、温度 T を一定にして体積を
変化させると、内部エネルギーの変化率は圧力に一致しない。しかしながら、実際の
実験をエントロピー一定の条件下で行うことは、稀である。むしろ、温度を制御しな
がら実験を行うことが一般的であり、それにしたがって熱力学の関係式も、温度 T の
変化量 dT があらわに出てくる形で書かれることが望ましい。つまり、温度を独立変
数とする式(2.2.3)に類似した熱力学の関係式が欲しいのである。
温度 T と体積 V を独立変数にする際に用いられるのが
A=U-TS (2.2.4)
で定義されるヘルムホルツ自由エネルギー(Helmholtz free energy) A である。ヘルムホ
ルツ自由エネルギーについては、式(2.2.3)に類似した関係式として
dA=-SdT-PdV (2.2.5)

が成り立つ。ここでの独立変数は温度 T と体積 V であり、例えば、温度を一定にして
体積を変化させたときのヘルムホルツ自由エネルギーの変化率が圧力そのものであ
る。また、体積を一定にしてヘルムホルツ自由エネルギーの変化量を求めると、エン
トロピーを求めることができる。式(2.2.5)から注意すべきことは、温度一定で行われ
た仕事量はヘルムホルツ自由エネルギーに等しいということである。これに対して、
式(2.2.3)に見るように、エントロピー一定の条件の下では、仕事量は内部エネルギー
の変化に等しい。同様にして、温度 T と圧力 P を独立変数にする際に用いられるのが
G=U+PV-TS=A+PV (2.2.6)
で定義されるギブス自由エネルギー(Gibbs free energy) G であり、
dG=-SdT+VdP (2.2.7)
が成り立つ。ここでは、独立変数は温度 T と圧力 P であり、例えば、温度を一定にし
て圧力を変化させたときのギブス自由エネルギーの変化率が系の体積である。
内部エネルギーや自由エネルギーは、総称して熱力学ポテンシャルと呼ばれる。こ
れは、系の状態は熱力学ポテンシャルが小さいほど安定であることに由来する。例え
ば、温度・圧力が一定の状態では、ギブス自由エネルギーが最小の状態が最も安定で
あり、自然界の自発的変化はギブス自由エネルギーを小さくする方向に起こる。一方、
温度・体積が一定の状態では、ヘルムホルツ自由エネルギーが最小の状態が最も安定
である。さらに、内部エネルギーはエントロピー・体積が一定の時の熱力学ポテンシ
ャルとして働くが、このことからしても、ヘルムホルツおよびギブス自由エネルギー
が、実用上は自然を記述するうえで便利であることがわかる。自由エネルギーを実験
的に決定することも可能である。例えば、式(2.2.7)に見るように、定温条件下で体積
を圧力の関数として測り、圧力上で積分すると、ギブス自由エネルギーを算出するこ
とができる。このことは、定積比熱の温度上の積分によって内部エネルギーが求まる
ことと同様である。
これまでの議論では、系の粒子数の増減は扱ってこなかった。つまり、粒子数 N
を変数としてあらわに扱ってこなかったということである。そこで、温度・圧力が一
定の条件下で系の粒子数 N が単位量(例えば、1 粒子)変化したときのギブス自由エ
ネルギーの変化量を化学ポテンシャルµと呼ぶ。化学ポテンシャルµも含めると、粒子
数 N の変化量を dN として、式(2.2.7)は
dG=-SdT+VdP+ µdN (2.2.8) と一般化される。式(2.2.8)では温度 T と圧力 P に加えて、粒子数 N も独立変数である。
式(2.2.4) 、(2.2.6) 、(2.2.8)から
VSVTPT NU
NA
NG
,,,
∂∂
=
∂∂
=
∂∂
=µ (2.2.9)
となることに注意されたい。つまり、µは、粒子数を単位変化させたときの、定温定

圧下における系の Gibbs 自由エネルギー変化であると同時に、定温定積下における系
の Helmholtz 自由エネルギー変化であり、また、定エントロピー定積下における系の
内部エネルギー変化である。この性質があるがゆえに、化学ポテンシャルの議論では、
温度圧力の対応が正しく取られている限り、定圧下での粒子数変化を対象にしている
のか定積下での粒子数変化を対象にしているのかを問題にする必要が無い。これに対
して、系の粒子数変化に対するエネルギーやエントロピーの変化を問題にするときは、
常に、定圧下での議論であるのか定積下での議論であるのかを明らかにする必要があ
る。
系に含まれる成分(component)の種類が複数の時も、それぞれの成分に対して、化学
ポテンシャルを導入することができる。例えば、系が二つの成分からなっているとき
には、それぞれの成分の粒子数を N1、N2 とし、化学ポテンシャルをµ1、µ2 とすると、
式(2.8)は
dG=-SdT+VdP+µ1dN1+µ2dN2 (2.2.10) の形に拡張される。
温度・圧力が一定下での粒子の移動に対しても、自発的変化はギブス自由エネルギ
ーを小さくする方向に起こる。ここから、相平衡・相変化によって二つの相があると
きに粒子の移動する方向を知ることができる。例えば、定温定圧下で粒子が dN だけ、
化学ポテンシャル がµAである相 A から化学ポテンシャル がµB である相 B に移動す
ると、ギブス自由エネルギーの変化は(µB-µA)dN で与えられる。このため、µA > µBで
あれば、相 A から相 B への粒子の移動によってギブス自由エネルギーは減少し、粒
子の移動が起こる。逆に、µA < µBであれば、相 A から相 B への粒子の移動によって
ギブス自由エネルギーは増加し、粒子移動は相 B から相 A へ起こる。µA = µBであれ
ば、相 A と相 B の間の粒子移動でギブス自由エネルギーは変化せず、平衡状態にあ
る。よって、粒子はその化学ポテンシャルが高いところから低いところへ移動する傾
向があり、そこに、化学ポテンシャルの名の由来がある。
また、化学反応は、粒子の変換と考えられ、相の間での粒子の移動と同様に化学ポ
テンシャルで扱うことができる。反応物(reactant)が生成物(product)になる過程を考え
たとき、反応物の化学ポテンシャルが生成物の化学ポテンシャルよりも高いときに、
反応は自発的に進行する(もちろん、反応には活性化障壁があるのが普通であり、「自
発的」進行は充分長い間待った場合の意味である)。逆に、反応物の化学ポテンシャ
ルが生成物の化学ポテンシャルよりも低いときには、反応物は生成物に転換しない。
圧力と体積が状態方程式でもって、互いに関係している場合には、化学平衡にかか
わる平衡定数は、その平衡過程が、定温・定圧で行われるか定温・定積で行われるか
には依存しない。これに対応して、化学ポテンシャルは、実は、温度・体積が一定の
条件下で系の粒子数が単位量変化したときのヘルムホルツ自由エネルギーの変化量

にも等しい。つまり、定温・定圧条件下で粒子数が単位量変化したときのギブス自由
エネルギーの変化量と、定温・定積条件下で粒子数が単位量変化したときのヘルムホ
ルツ自由エネルギーの変化量とは等しく、その変化量が化学ポテンシャルと呼ばれる
のである(定温・定圧条件下または定温・定積条件下でのギブス自由エネルギーの変
化量とヘルムホルツ自由エネルギーの変化量とは一致しないことに注意する必要が
ある)。
2.3 化学ポテンシャルと溶解過程
ある溶媒(例えば、水)の中に一種類の溶質(例えば、蔗糖)が溶けている溶液を
考えよう。このとき、溶質のモル数の溶液中の全成分のモル数に対する比を、その溶
質のモル分率と呼ぶ。すると、次節で説明するが、モル分率 x があまり大きくないと
きには、気体定数 R を用いて、溶質の化学ポテンシャルµは
µ = RTlnx +µ0 (2.3.1) と表される。µ0 は仮想的に x=1 と取ったとき(これを標準状態と呼ぶ)の化学ポテン
シャルであり、x に依存しない。式(2.3.1)によると、溶質のモル分率 x が 0 に近いと
きには、溶質・溶媒の組合せにかかわり無く、RTlnx は大きな絶対値を持つ負の量で
あり、µは非常に低い。このことは、極めてわずかの量であれば、どのような溶質で
も溶媒に溶けうることを表している。モル分率 x が大きくなってくると、µはもはや
低くなくなってきて、µ0 の効果が重要になってくる。µ0 は溶質・溶媒の組合せに依存
する量なので、溶質と溶媒の相互作用を知りたければµ0 に着目する必要がある。
蔗糖が水に溶ける過程を考えてみよう。はじめ、蔗糖は結晶相にあり、結晶相での
蔗糖の化学ポテンシャル µ (結晶)は、水溶液相での蔗糖の化学ポテンシャルµ (溶液)
よりも高い。そこで、蔗糖は水に溶け出していく。ある程度溶けると、µ (結晶)とµ (溶
液)の差は小さくなり、その差が 0 になったところで、蔗糖はもう溶け出さなくなる。
これは、µ (溶液)がµ (結晶)より高くなると、溶液から結晶相への蔗糖の移動、すなわ
ち、析出が起こるからである。
さらに、物質 A から B への反応を考えよう。最初に、系には A のみがあり、B は
無いものとする。すると、式(2.3.1)によって、最初の状態では、B の化学ポテンシャ
ルは-∞である。そこで、A は B に転換し始め(kinetics がとても遅いとしても、充
分待てば、という留保付きで)、反応の進行にともなって、RTlnx 項は有限になり、
A と B の化学ポテンシャルが等しくなるところで反応は止まる。注意しなくてはなら
ないのは、B の標準状態における化学ポテンシャルµ0 がどれだけ高くても、A と B が
平衡に達するまでは反応が起きるという点である。自由エネルギー差と標準自由エネ

ルギー差は、厳密に区別されねばならない概念であり、自由エネルギー差が反応の進
行の向きを決定し、標準自由エネルギー差が平衡の場所を決める。
2.4 化学ポテンシャルの統計力学的表現
これまでに、熱力学に基づいて、化学ポテンシャルを導入した。熱力学は、分子間
の相互作用にかかわらずに成立する理論で、それゆえに、強力であり一般的であるが、
同時に、分子のレベルで自然を語る際には適していない。分子間相互作用から、溶液
に関わる現象を記述し熱力学を基礎づけるのが、統計力学である。ここでの目的は、
化学ポテンシャルの統計力学的な表式を与え、溶質―溶媒相互作用との関わりについ
て述べることである。
粒子数が N で、体積が V、温度が T のカノニカル集団(canonical ensemble)を考える。
このとき、系のハミルトニアン (Hamiltonian)を H とすると、分配関数 (partition
function)Q は、P および X を、それぞれ、系の分子の運動量および位置を集合的に表
した座標として
∫
−=
TkHdd
hNQ
BfN exp
!1 XP (2.4.1)
で表される。ここで、kB はボルツマン定数(Boltzmann constant)であり、h はプランク
定数(Planck constant)である。1/N!の因子は N 個の粒子が同じもので区別できないこと
によって生じている。また、h のベキである f は 1 分子当りの自由度の個数である。x、
y、z 方向の並進の 3 つの自由度を持つ場合は f=3 であり、回転の自由度を持つ場合な
どは、それに応じて、自由度の個数が h のベキとして表われる。体積と温度が与えら
れたときには、分配関数 Q から、ヘルムホルツ自由エネルギーA が
A = -kBTlnQ (2.4.2)
によって与えられる。式(2.4.1)と式(2.4.2)は、分子の運動や相互作用を表すハミルト
ニアンと、系の熱力学を記述するヘルムホルツ自由エネルギーを結び付けるもので、
統計力学の出発点となる式である。一般に、ハミルトニアンは運動エネルギーT とポ
テンシャルエネルギーU の和で表される。そのとき、式(2.4.1)における運動量に関す
る部分の積分(P 上の積分)は簡単に実行できて、式(2.4.1)は系の分子の位置だけを
集合的に表した座標 X を用いて
∫
−
Λ=
TkUd
NQ
BN exp
!1 X (2.4.3)

と書かれる。ここで、1/Λは粒子一個あたりの運動量部分からの寄与であり、並進の
一自由度に対するものは熱ドブロイ波長(thermal de Broglie wavelength)と呼ばれ、長さ
の次元を持つ。剛体分子のΛは分子の質量と慣性モーメントのみによって決まる量で
あり、分子間の相互作用にはよらない。よって、運動エネルギーに由来するΛの関わ
る項だけでは、溶液系(より一般的には多粒子系)のもつ自然現象の多様性を理解す
ることはできない。統計力学は、理想気体などのごく単純なケースを除けば、微視的
な分子間相互作用の効果がいかに巨視的な現象として現れてくるかを研究する学問
である。分子間相互作用はポテンシャルエネルギーU の中に含まれているため、式
(2.4.3)の形を、溶液・流体の統計力学の出発点と取る。量子流体を扱う場合は、式(2.4.3)
の形を出発点にすることはできない。
式(2.4.1)および式(2.4.3)では、1 成分系を考えてきた。これは、現在の文脈では、純
溶媒を取り扱っていることに相当する。そこで次に、溶液のモデルとして、溶質分子
を n 個と溶媒分子を N 個含む溶液を考える。この場合、溶媒分子同士のポテンシャル
エネルギーの和を U と書き、溶質分子と溶媒分子との間のポテンシャルエネルギーの
和を un と書き、また、溶質分子同士のポテンシャルエネルギーの和を Wn と書くと、
書くと、溶媒分子および溶質分子の運動量部分からの寄与をそれぞれΛ、λとして、溶
液の分配関数 Q(n,N)は n 個の溶質分子の位置座標を集合的にあらわした yn 上での積
分でもって
( ) ∫
++−
Λ=
TkuWU
ddNn
NnQB
nnnNn exp
!!1, yXλ
(2.4.4)
と書かれる。
溶質の化学ポテンシャルとは、溶質数の単位変化に対する系の自由エネルギー変化
である。系が統計熱力学の対象である場合、n(および N)は、アボガドロ数程度に
大きく、それゆえに、n での微分を n→n+1 への変化と同一視してもよい。つまり、
溶質の化学ポテンシャルµは、Q(n+1,N)と Q(n,N)および式(2.4.2)によって、
( )( )
++−
++−
−=
+−=
∫
∫ +++
TkuWU
dd
TkuWU
dd
nTk
NnQNnQTk
B
nnn
B
nnn
B
B
exp
exp1log
,,1log
111
yX
yX
λ
µ
(2.4.5)
と書くことができる。さらに、V は系全体の体積であり、ρsは溶質の濃度であるとす
ると、式(2.4.5)は、

( )
++−
++−
−=
++−
++−
−
=
∫
∫
∫
∫
+
+++
+++
TkuWU
dd
TkuWU
ddTkTk
TkuWU
ddV
TkuWU
ddTk
VnTk
B
nnn
B
nnn
BsB
B
nnn
B
nnn
BB
exp
exploglog
exp
exploglog
1
111
111
yX
yX
yX
yX
λρ
λµ
(2.4.6)
となる。ここで、式(2.4.6)の第 2 行の第 2 項の log 内の分母では、n+1 番目の溶質分子
は、系の他の分子とまったく相互作用しないという(仮想的な)条件での積分が実行
されていることに注意されたい。つまり、式(2.4.6)の第 2 行の第 2 項は、n+1 番目の
溶質分子と系の他の分子との間の相互作用を入れていく可逆仕事に相当する。
ここで、簡単のために、溶質分子の濃度が溶媒分子に比べて充分薄く、溶質分子間
の相互作用が無視できるものとする。また、分子間の相互作用が分子間の相対的な配
置のみに依存するものとすると、溶媒座標を溶質の場所を原点とするように取り直し
た上で、式(2.4.6)は
( )
−
+−
−=
∫
∫
TkUd
TkuUd
TkTk
B
BBsB
exp
exploglog
1
X
Xλρµ (2.4.7)
と変形される。式(2.4.7)においては、溶質は原点に固定されており、積分は溶媒分子
の座標 X 上でのみ行われる。すると、u1 は溶媒分子に対する外場と考えることができ、
式(2.4.67)の第 2 項は、その外場を系に入れていく可逆仕事に相当する。すると、 L
で純溶媒系での平均を表し、ある量 A に対して、その純溶媒系での平均が
∫
∫
−
−
=
TkUd
TkUAd
A
B
B
exp
exp
X
X (2.4.8)
であらわされることを用いると、式(2.4.7)は、
−−=
TkuTkTkB
BsB explnln λρµ (2.4.9)
となることに注意されたい。
式(2.4.6)の第 1 項は、溶質粒子が系内のどの場所にいることも可能であることによ

る寄与であり、系内の分子間相互作用には関係しない。第 2 項は、逆に、分子間相互
作用によって決定される項であり、溶質―溶媒の組合せによる溶液の多様な振る舞い
はこの項によって決定される。式(2.4.6)では、溶媒の運動エネルギーから来る寄与で
あるΛが相殺されていることに注意されたい。
もし、系が希薄な気体であり、溶質―溶媒相互作用の影響が無視できるときには、
式(2.4.9)の第 2 項は無視できて、 λρµ sBTk ln= (2.4.10)
となる。つまり、式(2.4.10)は理想気体中での化学ポテンシャルの表現である。そこで、
式(2.4.6)の第 1 項は理想気体的な寄与であると言ってよい。第 2 項は、過剰化学ポテ
ンシャルと呼ばれる。
希薄気体中から溶液中への溶質の溶解を考える。ただし、溶液内においても溶質は
希薄であるものとする。このとき、希薄気体中での溶質の密度をρsg とすると、希薄
気体での溶質の化学ポテンシャルは式(2.4.10)でρs をρsg に置き換えたものに等しい。
一方、溶液中での溶質の密度をρsl とすると、溶液内の溶質の化学ポテンシャルは式
(2.4.9)でρsをρslに置き換えたもので与えられる。すると、溶解平衡に達しているとき
には、希薄気体中と溶液中とで溶質の化学ポテンシャルが等しいので、希薄気体から
の溶解によって飽和に達している溶液について
−=
Tku
Bgs
ls exp
ρρ
(2.4.11)
が成り立つ。但し、式(2.4.11)での L は溶液側での平均である。ここで、溶解平衡が
温度一定で達成されているので、溶質の運動エネルギーから来るλが相殺されている
ことに注意されたい。式(2.4.11)にみるように、溶質―溶媒相互作用が引力的で u が負
の時はρslが大きくなり、溶質がよく溶けることを表す。逆に、溶質―溶媒相互作用が
斥力的で u が正の時はρsg が大きくなり、溶質があまり溶けないことを表す。
式(2.4.9)において、ρsが小さいときには、ρsは溶質のモル分率 x に比例するため、
式(2.3.1)の形に変形できる。式(2.3.1)でも式(2.4.9)でも、化学ポテンシャルが溶質濃度
0 の極限で-∞になることは、第 1 項で表されているが、両式の第 1 項(および第 2
項)は等しくない。つまり、化学ポテンシャルの値そのものは系が決まれば決定され
るが、その成分への分割の仕方は一意的ではない。このことは、平衡を論じる際に重
要となる「標準状態」の同定が一意的ではないことを意味する。よって、文献で「標
準の熱力学量」を挙げるときには、問題としている熱力学量がどのように成分分割さ
れているかをはっきりさせることが重要である。溶質―溶媒相互作用の効果を取り出
してやるためには、式(2.3.1)の形よりも式(2.4.9)の形が便利である。そこで、式(2.4.9)

の形に化学ポテンシャルを整理して、その第 2 項、すなわち、過剰化学ポテンシャル
だけを取り出して溶質―溶媒相互作用を議論することが、溶液化学では推奨される。
問2.1 式(2.3.1)のµ0 を過剰化学ポテンシャルで表わす
問2.2 定温定圧下における粒子数の単位変化に対するヘルムホルツ自由エネルギー
および内部エネルギーの変化の表式をしめす。
問2.3 溶質が A と B の 2 種類存在し、それぞれの個数が nAおよび nBであるとき、
系の自由エネルギーの統計力学的表式を書き下す
問2.4 A と B の間の化学平衡について、系の自由エネルギーを最小にするように、
nAおよび nBを決定することによって、平衡定数を、化学種 A および B の
の過剰化学ポテンシャルで表わす。
問2.5 A+C ⇔ B なる化学平衡についても同様の事を行う。
問2.6 さらに、化学種 C が溶媒分子 S そのものであるときを考察する。 問2.7 粒子の運動エネルギーは mp 22 (p は運動量、m は質量)のとき、熱ドブロ
イ波長が ( ) 212 2 Tmkh Bπ であり、長さの次元を持つことを見る。また、分
子量が M である分子について、25 ℃における熱ドブロイ波長を計算する。
問2.8 慣性モーメントが IA、IB、ICである剛体分子を考えよう。その配向を表すオイ
ラー角をθ、φ、ψとしたとき、分子の配向に関わる部分の運動エネルギー
Trotは
( ){ }
( ){ } 222
22rot
21cossinsincos
sin21
sinsincoscossin21
ψθψφ
θψφ
ψθψθθ
ψθψθθ
pI
pppI
pppI
T
CB
A
++−+
−−=
で与えられる。このとき、
θππππψφθ sin
888exp12
2
2
2
2
2rot
3 hTkI
hTkI
hTkI
TkT
dpdpdph
BCBBBA
B
=
−∫
であることを見る。この式から配向θ、φ、ψのみの関数 f(θ,φ,ψ)について
( )
( )ψφθψφθθπππ
ψφθψφθψφθ
,,sin222
,,exp1
222
rot3
fdddh
TkIh
TkIh
TkI
fTk
Tddddpdpdph
BCBBBA
B
∫
∫
=
−
となる。dxdydz が並進(原点の取り方)によって不変であるように、
sinθdθdφdψは回転によって不変である(座標を回転させたときの Jacobian

が 1)。つまり、座標軸の方向を変えて、オイラー角θ、φ、ψの定義を変
えても、sinθdθdφdψは変化しない(dθdφdψは変化する)。この意味で、
dxdydz や sinθdθdφdψは「不変測度」と呼ばれる。式(2.4.3)の形に分配関数
を書き換えたときに不変測度が表われることは、分配関数が座標の取り
方によらないことに対応している。
問2.9 2.4 節での議論は、カノニカル集団(温度・体積・粒子数一定)で行ったが、
これを温度・圧力・粒子数一定の等温等圧集団に拡張する。また、グラ
ンドカノニカル集団(温度・体積・化学ポテンシャル一定)ではどうな
るだろうか。
問2.10 溶質に分子内自由度(例えば振動)がある場合の化学ポテンシャルの表式を
与える
問2.11 Boltzmann の原理によると、エネルギー(E)、体積(V)、粒子数(N)を与えたとき
に、系のエントロピーS(E,V,N)は kBlog(E,V,N を持つ微視的状態の数)であ
る。もし、S(E,V,N)が対数ではなく、別のある関数 f でもって S(E,V,N) =
f(E,V,N を持つ微視的状態の数)と表されているものとするときに、カノニ
カル、定温定圧、およびグランドカノニカル分布の表式を与える
問2.12 前問の設定の条件下での化学ポテンシャルの表式を与える
問2.13 前々問の設定の条件下で f が対数形でなければ、物理的に不合理なことが起
きることを見る。このことは、Boltzmann の原理が妥当であることについ
て、数学的証明ではないが、支持を与えるものである。

第 3 章 溶液の分布関数
気体・液体を含む流体は、巨視的に密度が一様の系である。しかし、分子レベルで
見ると、ある分子同士は近づく傾向があり、別のあるものは遠ざけ合う傾向があると
いうように、完全に均一ではない。分子レベルでの不均一性を表わす関数が分布関数
である。本章では、分布関数の理論への導入を行う。
3.1 分布関数の表示
溶液として、「溶質」分子を一つ含み、残りの分子が全て「溶媒」分子である系を
考えよう。実は、この規約は、とくに意味があるわけではない。例えば、純溶媒を考
えるときは、溶媒分子の 1 つを「溶質」分子と考え、他の分子を「溶媒」分子と見な
せばよいし、無限希釈に無い系(溶質の濃度が有限である系)では、溶質と溶媒から
なる分子集団をまとめて混合溶媒系と見ればよい。つまり、ある特定の分子を「溶質」
と呼び、他を「溶媒」と呼んでいるだけのことである(以下では、溶質と溶媒にいち
いち「」をつけないこととする)。
溶質分子は、系の原点にその配向も含めて固定されているものとしよう。すると、
溶質と溶媒の相対配置は、溶媒分子の位置と配向を与えることで決定される(これは、
分子振動を無視しているときの話である)。つまり、溶媒分子の位置と配向という 6
次元の変数によって、溶質に対する溶媒の配置を完全に記述することができる。6 次
元の位置と配向をまとめて x と書こう。また、i 番目の溶媒分子の位置と配向を xiと
する。そうすると、溶液のある瞬間的な配置において、x における溶媒の密度 ( )xρ̂ は
( ) ( ) の和は全溶媒分子について∑∑=ii
ix-xx δρ̂ (3.1)
で与えられる。ここで、密度という言葉を使ったが、x という位置・配向の周りの微
小領域 dx にある溶媒分子の個数が ( ) xx dρ̂ であるという意味である。 ( )xρ̂ は、溶液の瞬間的な配置が決まれば決定されるものであり、統計力学的な平
均操作は入っていない。 ( )xρ̂ を、平均したものが 1 体の分布関数と呼ばれる。つまり、
1 体の分布関数 ( )xρ は
( ) ( )xx ρρ ˆ= (3.2)
で与えられる。ここで、<・・・>が統計力学的な平均を表わし、温度・圧力(密度)
の関数である。溶媒の配置 x が溶質分子から遠く離れているとき、 ( )xρ は単に溶媒の
(バルク)密度ρに帰する。つまり、 ( )xρ は溶媒の配置が、溶質の存在によってどの
ような影響を受けるかを記述している。この意味で、 ( )xρ は溶質分子の配置と 1 つの
溶媒分子の配置との間の相関を表わす関数であるといえる。同様に、2 体の分布関数( )( )yx,2ρ は
( )( ) ( ) ( ) についての和つの溶媒分子の全ペアは2,2 ∑∑≠≠
−−=jiji
ji xyxxyx δδρ
で与えられ、溶質分子の配置と溶媒分子のペアの配置との間の相関を表わす。より多
くの溶媒分子を含む多体の分布関数の定義も同様である。 ここでは、 ( )xρ を 1 体の分布関数と呼んだ。しかし、実際には、1 つの溶媒分子
に対する溶質分子の影響を記述しているので、溶質-溶媒の 2 体の相関を記述する関
数と考えてもよい。 ( )xρ を 1 体と呼ぶか 2 体と呼ぶかは便宜的なものである。溶質を
溶媒と「似たように」扱いたいときは、2 体と呼ぶのが自然であろうし、「溶質」を
外場の源と考える場合には1体と呼んだ方が理論的な構造がスッキリする。ここでは、
後者の立場を取り、 ( )xρ を 1 体と呼ぶ。 ( )( )yx,2ρ についても、溶質-溶媒-溶媒の 3
体の相関を記述しており、同様の注意が当てはまる。 溶質および溶媒が球形であるとき、 ( )xρ はとりわけ簡単になる。このとき、 ( )xρ は、
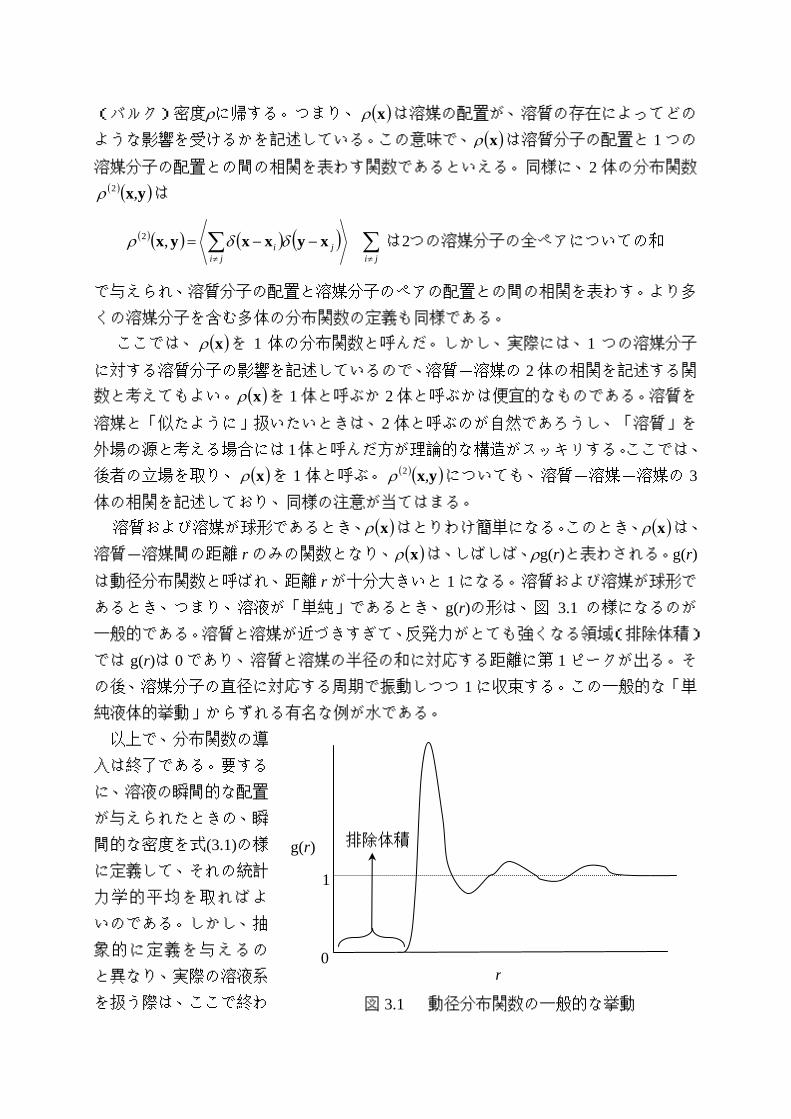
溶質-溶媒間の距離 r のみの関数となり、 ( )xρ は、しばしば、ρg(r)と表わされる。g(r)
は動径分布関数と呼ばれ、距離 r が十分大きいと 1 になる。溶質および溶媒が球形で
あるとき、つまり、溶液が「単純」であるとき、g(r)の形は、図 3.1 の様になるのが
一般的である。溶質と溶媒が近づきすぎて、反発力がとても強くなる領域(排除体積)
では g(r)は 0 であり、溶質と溶媒の半径の和に対応する距離に第 1 ピークが出る。そ
の後、溶媒分子の直径に対応する周期で振動しつつ 1 に収束する。この一般的な「単
純液体的挙動」からずれる有名な例が水である。
以上で、分布関数の導
入は終了である。要する
に、溶液の瞬間的な配置
が与えられたときの、瞬
間的な密度を式(3.1)の様
に定義して、それの統計
力学的平均を取ればよ
いのである。しかし、抽
象的に定義を与えるの
と異なり、実際の溶液系
を扱う際は、ここで終わ
g(r)
r
排除体積
図 3.1 動径分布関数の一般的な挙動
1
0

るわけにいかない。溶媒の位置と配向を表わす完全な変数の組 x は 6 次元であり、6
次元の独立変数を持つ関数を現実に扱うわけにいかないからだ。それゆえ、独立変数
の数を減らし、溶媒の配置を指定する座標を低次元化しなくてはならない。
溶媒座標の低次元化のために最もよく使われるのが、相互作用点表示である。ここ
では、溶質分子および溶媒分子を、原子または原子団を表わす球形の相互作用点(サ
イト)の集合と見なし、その相互作用点の距離を独立変数として動径分布関数を考え
る。例えば、水(H2O)を考えよう(溶質も溶媒も H2O)。この場合、O-O、O-H、H-H
の 3 種類の相互作用点距離があり、それに対応して、3 種類の動径分布関数が構成さ
れる。もう少し一般的に述べると、溶質のサイトαと溶媒のサイトγとの間の動径分布
関数 gαγ(r)を
( ) ( )( ) ( )xxx ρδπ
ρ αγαγ rrdr
r −= ∫241g (3.3)
と定義する。ただし、式(3.3)において、rαγ(x)とは溶媒の位置と配向が x にある時の溶
質のサイトαと溶媒のサイトγとの間の距離である。式(3.3)に見るように、6 次元だっ
た独立変数が「射影」されて 1 次元化されていることが分かる。当然、 ( )xρ と比べる
と、情報量は落ちており、 ( )xρ から gαγ(r)を構成することは式(3.3)によって可能であ
るが、逆は一般に可能ではない。また、溶質のサイト数が n、溶媒のサイト数が m で
ある場合、nm(または、対称性によってそれより少ない数)種類のサイト-サイト動
径分布関数がある。
溶媒座標を低次元化する別の方法として、筆者によって導入されたエネルギー表示
の方法がある。これは、溶質分子と溶媒分子の間の相互作用ポテンシャルエネルギー
関数が v(x)のとき、分布関数 ( )ερ e を
( ) ( )( ) ( )xxx ρεδερ ∫ −= vde (3.4)
として、溶質と溶媒の相関を記述するものである。エネルギー表示の方法でも、分布
関数の独立変数は 1 次元である。また、これまで、溶質と溶媒分子は剛体(分子内振
動のないもの)として扱ってきたが、たとえ、分子内振動があっても、溶質-溶媒相
互作用エネルギーの分布を定義するのにとくに困ることはない。gαγ(r)の時と同様に、
( )xρ から ( )ερ e を構成することは式(3.4)によって可能であるが、逆は可能ではない。
さらに、gαγ(r)から ( )ερ e を構成することおよびその逆は一般に可能ではない。
上に述べたもの以外に、球関数展開の方法がある。球関数展開の方法は、分布関数
を、等方的な(角度依存しない)分布関数からずれたものとして無限級数で表したも
のである。このような級数展開は、等方的な場合からの摂動展開と見なすことができ
る。しかし、現実に興味のある分子は球状から少しずれたものと扱うことはできず、
また、球関数展開の方法は級数展開であるため、数学的にややこしく(難しくはない)、

直感的にも分かりにくいので、ここでは紹介しない。興味のある方は、参考文献を見
ていただきたい[1,2]。
相互作用点表示やエネルギー表示を採用するメリットは、溶媒座標の低次元化であ
る。溶媒座標が1次元になると、直観的に分かり易くなり(≒見やすいグラフが書け
る)、実用的に取り扱いやすくなる(≒計算機上での取り扱いが簡単になる)。情報
量については、多ければよいというものではないので、その減少を低次元化に伴う「デ
メリット」と取るかどうかは、使う人の観点によるであろう。むしろ、様々な表示の
メリット・デメリットは、次節で述べる近似手法の構成に関わって表われてくるもの
である。
問3.1 水の動径分布関数を文献で調べる。
問3.2 次の場合、サイトーサイト動径分布関数は何種類あるか?
1. 異核 3 原子分子の溶質と異核 2 原子分子の溶媒
2. 異核 3 原子分子の溶質と等核 2 原子分子の溶媒
3. 異核 2 原子分子の純溶媒
(=異核 2 原子分子の溶質と異核 2 原子分子の溶媒)
4. 等核 2 原子分子の純溶媒
問3.3 溶質分子と溶媒分子の間の相互作用ポテンシャルエネルギー関数が v(x)のと
き、溶質-溶媒相互作用エネルギーの和の平均値を v(x)と ( )xρ を用いて表
わす。
問3.4 溶質分子と溶媒分子の間の相互作用ポテンシャルエネルギー関数 v(x)が
( ) ( )∑=αγ
αγ rvv x という形で書けるとき、溶質-溶媒相互作用エネルギーの
和の平均値を vαγ(r)と gαγ(r)を用いて表わす。 問3.5 溶質-溶媒相互作用エネルギーの和の平均値を ( )ερ e で表わす。
3.2 溶質-溶媒相互作用ポテンシャルと分布関数の関係
溶液内過程を記述する最も重要な量は、その過程に対応する自由エネルギー(変化)
である。特に、ある過程の始状態と終状態にある化学種の化学ポテンシャルが分かれ
ば、その過程の自由エネルギー変化は分かるので、溶液内にある溶質の溶媒和自由エ
ネルギー(化学ポテンシャル)を評価する方法の確立が、溶液の統計力学の最も重要
な課題である。ここでは、化学ポテンシャルを軸にして、溶質-溶媒相互作用と分布

関数との間の関係を述べる。
問題としている溶液の溶質-溶媒相互作用ポテンシャルを v(x)としよう。また、溶
質-溶媒相互作用が 0 のときを「純溶媒」と呼ぼう。すると、溶質の位置と配向を固
定したままで、溶質-溶媒相互作用を 0 から v(x)まで変えたときの自由エネルギー変
化が、溶質の過剰化学ポテンシャル∆µである。∆µは、溶質の化学ポテンシャルから
理想気体的な寄与を除いたものである。古典統計力学の枠内では、
( )0
expln
−−=∆ ∑ TkvTk B
iiB xµ (3.5)
が成立する。式(3.5)で、kBはボルツマン定数、T は(絶対)温度、0
L は純溶媒系で
取った統計力学平均である。
そこで、溶質-溶媒相互作用ポテンシャルを 0 から v(x)まで変化させる過程を考え
よう。溶媒の座標 x 以外に、結合パラメーター(coupling parameter)とよばれる変化の
度合いを記述する変数λ(0≦ λ ≦1)に依存する溶質-溶媒相互作用ポテンシャル
( )xλu をもってくるわけである。もちろん、変化の始状態および終状態での溶質-溶
媒相互作用エネルギーは与えられているので、 ( )
( ) ( )xxx
vuu
==
1
0 0 (3.6)
が満たされている必要がある。また、溶媒―溶媒相互作用や温度・体積といった外部
変数は変化しないものとする。すると、式(3.5)は
( ) ( )λλ ρλ
λµ uudd ;1
0
xxx∫∫ ∂∂
=∆ (3.7)
と変形することができる。ただし、 ( )λρ u;x は、溶質-溶媒相互作用を ( )xλu としたと
きに式(3.2)によって与えられる分布関数である。式(3.7)では結合パラメーターλの上
の積分が含まれているにもかかわらず、式(3.6)を満たす限り、 ( )xλu をどのように取
っても、∆µの値は変らない。∆µは自由エネルギー変化であり、自由エネルギー変化
は始状態と終状態のみによって決まるという統計熱力学の大原則に沿うものである。
式(3.7)を(Kirkwood の)charging 式とよぶ。 ( )xλu の取り方を限定することで、相互作用
点表示やエネルギー表示で charging 式を構成することも可能である。
これまでは、対象とする溶質-溶媒相互作用ポテンシャル v(x)を固定して考えてき

たが、v(x)を変化させて、v(x)とそれに対応する分布関数 ( )v;xρ との関係について考え
よう。2 つの異なる v(x)、w(x)を取ってきたとき(v(x)-w(x)=定数のときは v(x)と w(x)
は同じであると見なす),
( ) ( )( ) ( ) ( )( ) ( )vvwdTkvwTkv
Bi
iiB ;expln xxxxxx ρ−<
−−− ∫∑ (3.8)
が成り立つ。ただし、v
L は溶質-溶媒相互作用が v(x)である溶液系で取った統計力
学平均である。また、v と w を入れ替えることによって
( ) ( )( ) ( ) ( )( ) ( )wwvdTkwvTkw
Bi
iiB ;expln xxxxxx ρ−<
−−− ∫∑ (3.9)
が成り立つ。それゆえ、異なる v(x)と w(x)に対して ( ) ( )wv ;; xx ρρ = (3.10)
とすると、式(3.8)と式(3.9)の間に矛盾が生じる。よって、2 つの異なる v(x)、w(x)を取
ってきたとき、対応する分布関数 ( )v;xρ 、 ( )w;xρ は異なる。つまり、
式(3.2)によって引き起こされる、溶質-溶媒相互作用ポテンシャル v(x)の 集合から、分布関数 ( )v;xρ の集合への写像は 1 対 1 である
という定理が証明できた。この定理によって、系の変化を、溶質-溶媒相互作用 v(x)の変化と見る視点から、分布関数 ( )xρ の変化と見る視点に移行することができる。
溶質-溶媒相互作用が v(x)から v(x)+δv(x)へ変化をしたとき、分布関数が応答して、
ρ(x)からρ(x)+δρ(x)に変化したものとする。このとき、変化δv(x)が微小であれば、変
化の 1 次までを残して、
( ) ( ) ( )yyxyx v;vd δχδρ ∫= , (3.11)
と書くことができる。ここで、χ(x,y;v)は相関行列と呼ばれ、 ( ) ( ) ( ) ( )( ) ( ) ( )vvv,vv, ;;;;; 2 yxyxxyxyx ρρρρδχ −+−= (3.12)
で与えられる。上の定理によって、χ(x,y;v)には逆行列が存在することが分かる。そし
て、
( ) ( ) ( ) ( )( )0;;0;,; 1 xxyxyx ρρχ −= −∫ vdvc (3.13)
と定義される c(x;v)は直接相関関数と呼ばれ、式(3.13)は Ornstein-Zernike 方程式と呼
ばれる。ただし、 ( )0;xρ は、溶質-溶媒相互作用が存在しない状況(v=0)での分布関数
である。

溶質と溶媒の間に相互作用は存在するが、溶媒-溶媒間の相互作用が無視できる状
況では、
( ) ( ) ( )0;exp; xxx ρρ
−=
Tkvv
B
(3.14)
が成り立つ。言い換えると、溶質-溶媒間の相互作用のみを考え、多体の絡み合い(相
関)の効果を無視したものが式(3.14)である。そこで、溶媒-溶媒間の相互作用が無
視できない普通の状況でも、多体の相関の効果を補正するために、溶質-溶媒間の平
均力ポテンシャルの間接部分 ( )v;xω を
( ) ( )( ) ( )xxxx vvTkv B −−=
0;;ln;
ρρω (3.15)
の形で定義するとよい。この関数を使うと、式(3.7)は式(3.6)の下で
( ) ( )( ) ( ) ( ) ( ) ( )
∂
∂−+−−=∆ ∫∫
1
0
;;;;0;;λ
ρρωλωρρρµ λλ uTk
dTkvvvdTk
BBB
xxxxxxx (3.16)
と変形される。ここで、 ( )λρω ;x は ( )λω u;x と同じものであるが、式(3.16)では溶質-
溶媒相互作用ではなく、分布関数が変化するものとして積分が捉え直されているので、
溶質-溶媒間の平均力ポテンシャルの間接部分の引数を分布関数とした。これは、v
を引数として持つ関数は、1 対 1 対応の定理によって、ρを引数にすることが可能で
あることによる。溶媒-溶媒間の相互作用が存在しないとき(低い密度の極限と考え
てよい)、 ( )λρω ;x は 0 であるので、式(3.16)のλ上の積分は、溶質-溶媒および溶媒
-溶媒の多体間相関の過剰化学ポテンシャルへの寄与を記述する。 式(3.14)や式(3.15)では、c(x;v)や ( )v;xω を溶質-溶媒相互作用 v によって決まるもの
と見て、v を引数にもってきたが、1 対 1 対応の定理によって、式(3.16)ではρを引数
とした。c(x;v)や ( )v;xω を v で摂動展開してうまくいくことは物理的・化学的に興味の
ある系の場合ほとんど無いが、 ( )ρω ;x を ( ) ( )( )0;; xx ρρ −v で摂動展開してうまくいくこ
とがしばしばある。溶液論・液体論で最もよく使われてきた Percus-Yevick 近似(PY近似)および超網状鎖(hypernetted-chain)近似(HNC 近似)は、 ( )ρω ;x またはその
類似物の ( ) ( )( )0;; xx ρρ −v による摂動展開で得られる[3]。 ( )ρω ;x の近似が得られれば、
式(3.16)によって、過剰化学ポテンシャルの近似が構成できる。PY 近似は斥力的な相
互作用、HNC 近似は引力的な相互作用の場合に良い結果を与えることが経験的に(つ
まり、そうなることの物理的な意味が分からずに)知られている。
相互作用点表示やエネルギー表示の場合には、溶質-溶媒相互作用の集合と分布関
数の集合との間の 1 対 1 対応が成立するためには、溶質-溶媒相互作用の集合を制限

する必要がある。相互作用点表示に関する定理については参考文献[4]を、エネルギー
表示に関する定理については参考文献[5]を読んでいただきたい。1 対 1 対応の定理と
類似のものが成立し、PY近似やHNC近似に対応する近似を定式化することができる。
近似の定式化についても、参考文献[4,5]を参照されたい。
問3.6 式(3.5)を導く。
問3.7 式(3.5)に類似した
( )
=∆ ∑ TkvTk B
iiB xexplnµ
を導く。ただし、L は問題とする溶液系で取った統計力学平均である(式
(3.5)では純溶媒での平均であることに注意)。
問3.8 溶媒分子をある配置 x から別の配置 y に移動させたときの自由エネルギー変
化が ( ) ( )( )yx ρρlnTkB− であることを証明する。
問3.9 式(3.7)を導く。
問3.10 溶質分子と溶媒分子の間の相互作用ポテンシャルエネルギー関数 v(x)が
( ) ( )∑=αγ
αγ rvv x という形で書けるとき、相互作用点表示での charging 式を
書く。このとき、 ( )xλu の取り方にどのような限定が加わるか?
問3.11 エネルギー表示での charging 式を書く。このとき、 ( )xλu の取り方にどのよう
な限定が加わるか?
問3.12 式(3.8)を次の手順で導こう。
(i) F(0)=F(1)=0 であり、F(2)(x)<0 を満たす関数 F(x)について、F(x)>0 が
0<x<1 で成り立つ。
(ii) 2 つの異なる値 x1、x2 について、0<λ<1 なるλを取ったとき、f(2) (x)>0
である関数 f(x)に対して、 ( ) ( ) ( ) ( )( )2121 11 xxfxfxf λλλλ −+>−+
を証明する。
(iii) (ii)の結果を n 個の異なる値がある場合に拡張する。
(iv) 2 値以上を持つ確率変数 X に対して、f(2) (x)>0 である関数 f(x)をとった
とき、 ( ) ( )XfXf > が成り立つ( L は平均を表わす)。

(v) 式(3.8)を導く。
問3.13 式(3.10)を仮定すると、式(3.8)と式(3.9)の間に矛盾が生じることを示す。
問3.14 溶液系における溶質-溶媒相互作用エネルギーの和の平均値が、必ず、過剰
化学ポテンシャルより小さいことを証明する。
問3.15 式(3.11)を証明する。
問3.16 実は、式(3.13)は Ornstein-Zernike 方程式としての、通常用いられる形ではな
い。「通常の」Ornstein-Zernike 方程式の形を調べ、それが式(3.13)に等価で
あることを証明する。
問3.17 溶媒―溶媒間の相互作用がないとき、式(3.14)を導く。
問3.18 相互作用点表示での、式(3.14)の類似物を導く。
問3.19 エネルギー表示での、式(3.14)の類似物を導く。 問3.20 ( )v;xω は「溶質-溶媒間の平均力ポテンシャルの間接部分」の名で呼ばれる
が、式(3.15)の x での微分を考えることでその名の由来を考える。
問3.21 式(3.16)を導く。 問3.22 c(x;v)や ( )v;xω を v の 1 次まで展開するとどのような表式が得られるか?
問3.23 PY 近似は ( )( )Tkv B;exp xω− を ( ) ( )( )0;; xx ρρ −v の 1 次まで展開したものである。
PY 近似が
( ) ( ) ( )( )
( )( )
( )
−−=
−=
Tkvvv
Tkvvc
BB
;exp0;
;0;
;exp1; xxx
xxxx ω
ρρ
ρρ
と表されることを示す。 問3.24 HNC 近似は ( )v;xω を ( ) ( )( )0;; xx ρρ −v の 1 次まで展開したものである。HNC
近似が
( ) ( )( )
( )Tkvvvc
B
;10;
;; xxxx ω
ρρ
+−=
と表されることを示す。
問3.25 溶質と溶媒が同じであるとき、PY 近似や HNC 近似を用いると、溶媒の分布
関数を解として与える方程式を定式化することができる。位置と配向を完
全に指定した表示において、そのような方程式を書き下す。
問3.26 単純液体系について、PY 近似や HNC 近似を用いた結果の成否について、参
考文献[6]の 5.7 および 5.8 節を参照して調べる。
問3.27 相互作用点表示で、溶質-溶媒相互作用と分布関数との間の 1 対 1 対応が成
立するためには、溶質-溶媒相互作用の集合をどのように制限する必要が
あるかを、参考文献[4]を読んで考える。
問3.28 相互作用点表示では、「溶質-溶媒間の平均力ポテンシャルの間接部分」の
構成が難しく、式(3.16)の類似物が構成しにくい。この点について、参考文
献[4]を読んで考える。
問3.29 相互作用点表示において、溶質と溶媒が同じであるときの、溶媒の分布関数
を解として与える方程式を書き下す。
問3.30 エネルギー表示で、溶質-溶媒相互作用と分布関数との間の 1 対 1 対応が成
立するためには、溶質-溶媒相互作用の集合をどのように制限する必要が
あるかを、参考文献[5]を読んで考える。
問3.31 エネルギー表示で、式(3.16)の類似物を構成する。
問3.32 エネルギー表示で、溶媒の分布関数に関わる方程式を PY 近似または HNC 近
似で書き下し、その問題点を考える。
第 3 章の参考文献
[1] W. A. Steele, J. Chem. Phys., 39, 3197 (1963).
[2] L. Blum and A. J. Torruella, J. Chem. Phys., 56, 303 (1972).
[3] J. K. Percus, Phys. Rev. Lett., 8, 462 (1962).
[4] D. Chandler, J. D. McCoy, and S. J. Singer, J. Chem. Phys., 85, 5971 (1986).
[5] N. Matubayasi and M. Nakahara, J. Chem. Phys., 113, 6070 (2000). [6] J. P. Hansen and I. R. McDonald, Theory of Simple Liquids, 2nd Ed. Academic Press
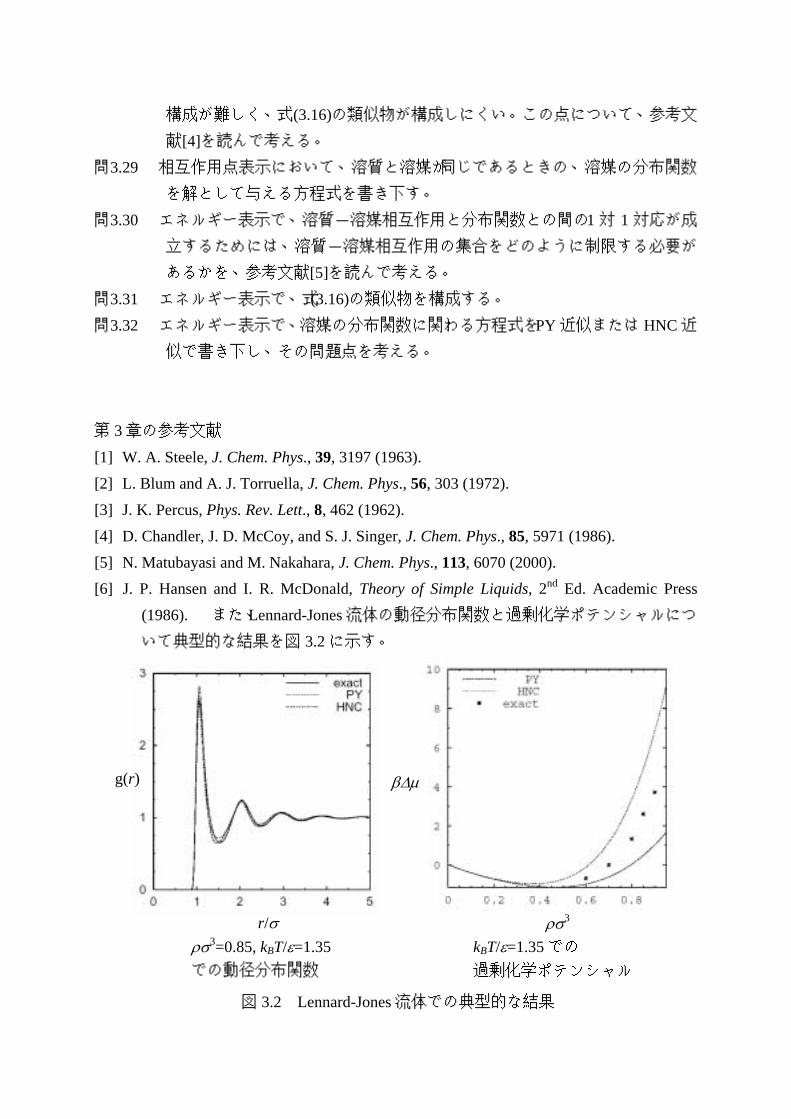
(1986). また、Lennard-Jones 流体の動径分布関数と過剰化学ポテンシャルにつ
いて典型的な結果を図 3.2 に示す。
図 3.2 Lennard-Jones 流体での典型的な結果
β∆µ
ρσ3 kBT/ε=1.35 での 過剰化学ポテンシャル
r/σ ρσ3=0.85, kBT/ε=1.35 での動径分布関数
g(r)

第 4 章 時間相関関数の理論
前章では、溶液の静的な(時間依存する項がない)構造を記述する方法論として分
布関数の理論を紹介した。本章では、系に時間依存する外場がかかったときの物理量
の応答を記述する方法論を紹介する。
4.1 時間発展の方程式
f 個の自由度を持つ N 個の粒子があるとき、その状態は fN 個の座標(q1,…qfN)と fN
個の運動量(p1,…pfN)の組で特定される。fN 個の座標および fN 個の運動量からなる 2fN
次元の空間を位相空間と呼び、位相空間内の特定の点を位相点と呼ぶ。以下では、
(q1,…qfN)を総称して q、(p1,…pfN) を総称して p と書く。この表示によって、あたかも
(q,p)を 1 自由度系の座標と運動量の組と見たときと同様の定式化ができ便利である。
系のハミルトニアン(Hamiltonian)を H(q,p)としよう。このとき、系の時間発展はハミ
ルトン(Hamilton)の(正準)運動方程式
qH
dtdp
pH
dtdq
∂∂
−=
∂∂
= (4.1)
で決定される。そこで、(q,p)の関数である任意の物理量 A(q,p)を考えると、その時間
発展は、
iLAqH
pA
pH
qA
dtdA
=∂∂
∂∂
−∂∂
∂∂
= (4.2)
と書くことができる。ここで、L はリウビル(Liouville)演算子と呼ばれ、式(4.2)で定義
される。式(4.2)は ( ) ( ) ( )0exp AiLttA = (4.3)
と形式的に変形できるので、L は時間発展を「推進する」演算子である。また、(q(0),p(0))
から(q(t),p(t))への時間発展を「変換」と捉えると、その変換の行列式は ( ) ( )( )( ) ( )( ) 1
0,0,det =
∂∂
pqtptq (4.4)
を満たし、変換は「体積保存的」であるといえる。
次に、位相空間上での確率分布ρ(q,p;t)を考えよう。ρ(q,p;t)の引数としての(q,p)は位
相空間内の(6N 次元の)座標であり、上のような時間発展の対象ではないことに注
意する必要がある。そのとき、任意の位相空間上で定義された物理量 A(q,p)の時刻 t

における平均<A(t)>は
( ) ( ) ( )∫= 0;,, pqpqdqdpAtA tt ρ (4.5)
である。ここで、(qt, pt)は、時刻 0 のとき(q,p)にあった状態が、時刻 t まで時間発展し
たときの状態である。つまり、(qt, pt)は、(q,p)(および t)の関数である。式(4.5)では、
(q,p)は積分が取られる位相空間内の座標であることに注意されたい。そこで、(qt, pt)
を積分座標に採用し、(q,p)から変換すると、式(4.4)によって
( ) ( ) ( )( ) ( )∫
∫−−=
=
0;,,
0;,,
tt
tttt
pqpqdqdpA
pqpqAdpdqtA
ρ
ρ (4.6)
となる。ここで、(qt, pt)が単なる積分座標となったことを使った。(q-t, p-t)は、時刻 0
のとき(q,p)にあった状態を時刻-t まで時間を戻したときの状態である。ところで、定
義によって
( ) ( ) ( )∫= tpqpqdqdpAtA ;,, ρ (4.7)
である。A は任意であるので、 ( ) ( )0;,;, tt pqtpq −−= ρρ (4.8)
が成り立つ。式(4.8)は
( ) ( ) ( ) ( )
( ) ( ) ( )0;,exp;,
;,;,;,;,
pqiLttpq
tpqiLqH
ptpq
pH
qtpq
ttpq
ρρ
ρρρρ
−=
−=∂∂
∂∂
+∂∂
∂∂
−=∂
∂ (4.9)
に等価である。式(4.2)または式(4.3)に対して、式(4.9)は時間 t の向きが逆になってい
ることに注意されたい。式(4.2)または式(4.3)における(q,p)は運動方程式に従って時間
発展する状態点を表しているのに対して、式(4.9) における(q,p)は位相空間の座標で
あり、時間発展の対象ではない(式(4.9)の第 1 式の最左辺は t に対する偏微分であっ
て(q,p)は一定に保たれていることに注意)。式(4.5)と式(4.7)の等価性が、式(4.9)にお
ける時間 t の向きの逆転を要求する。式(4.9)が、時間依存する系の統計的取り扱いの
基本式(の古典力学版)である。
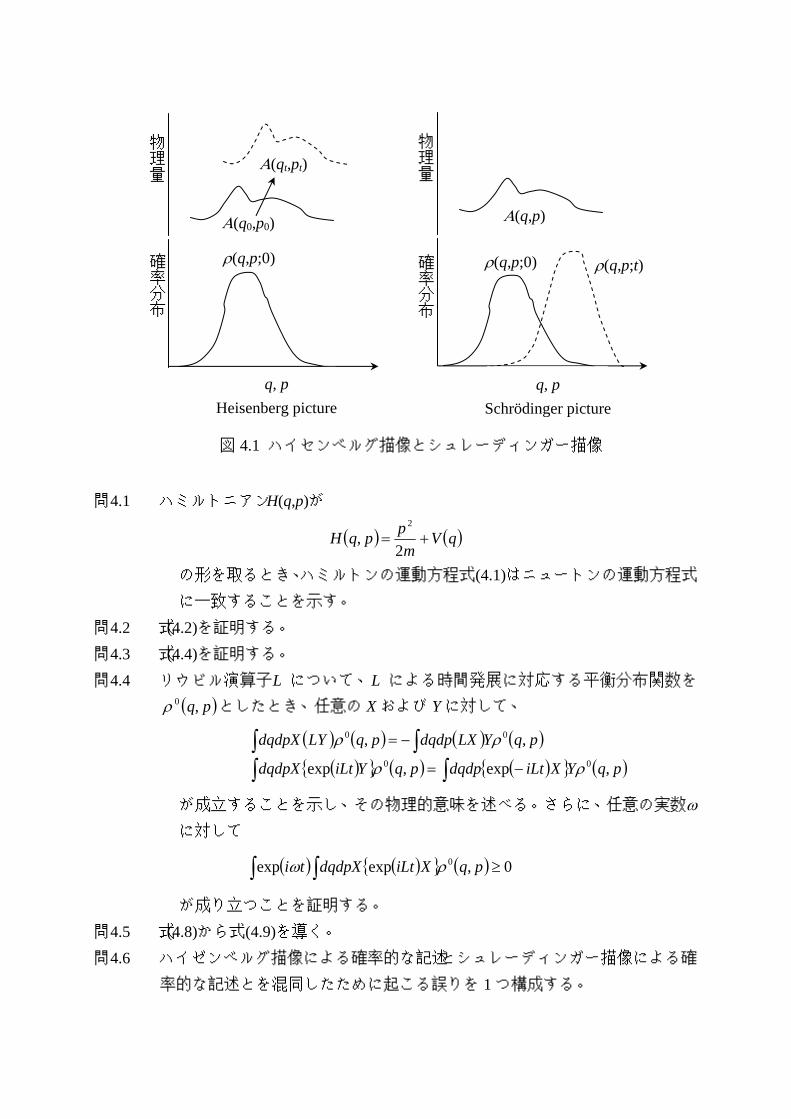
式(4.5)では、<A(t)>の時間発展を担うのは A(qt, pt)であり、確率分布として、時刻 0
でのρ(q,p;0)が使われている。このような<A(t)>の時間発展に対する見方をハイゼンベ
ルグ描像と呼ぶ。逆に、式(4.7)では、A(q,p)は単に位相空間上で定義された関数であ
り、<A(t)>の時間発展を担うのは、確率分布であるρ(q,p;t)である。このような<A(t)>
の時間発展に対する見方をシュレーディンガー描像と呼ぶ。ハイゼンベルグ描像とシ
ュレーディンガー描像の違いを図 4.1 に模式的に表す。
問4.1 ハミルトニアン H(q,p)が
( ) ( )qVm
ppqH +=2
,2
の形を取るとき、ハミルトンの運動方程式(4.1)はニュートンの運動方程式
に一致することを示す。
問4.2 式(4.2)を証明する。
問4.3 式(4.4)を証明する。
問4.4 リウビル演算子 L について、L による時間発展に対応する平衡分布関数を
( )pq,0ρ としたとき、任意の X および Y に対して、
( ) ( ) ( ) ( )( ){ } ( ) ( ){ } ( )∫∫
∫ ∫−=
−=
pqYXiLtdqdppqYiLtdqdpX
pqYLXdqdppqLYdqdpX
,exp,exp
,,00
00
ρρ
ρρ
が成立することを示し、その物理的意味を述べる。さらに、任意の実数ω
に対して
( ) ( ){ } ( ) 0,expexp 0 ≥∫∫ pqXiLtdqdpXti ρω
が成り立つことを証明する。
問4.5 式(4.8)から式(4.9)を導く。
問4.6 ハイゼンベルグ描像による確率的な記述とシュレーディンガー描像による確
率的な記述とを混同したために起こる誤りを 1 つ構成する。
図 4.1 ハイセンベルグ描像とシュレーディンガー描像
物理量
Α(q0,p0)
Α(qt,pt) 物理量
Α(q,p)
確率分布
ρ(q,p;0)
q, p Heisenberg picture
確率分布
q, p Schrödinger picture
ρ(q,p;t) ρ(q,p;0)

4.2 線形応答理論
摂動として扱うことのできる外場がかかったときの系の応答を、式(4.9)に基づいて
定式化することが、線形応答理論の目的である。線形応答理論は、多くの分光法の基
礎をなしている。
時間依存する外場 F(t)が、系の変数 A と結合して、時間に依存する摂動ハミルトニ
アン H’が
H’=-AF(t) (4.10)
で与えられるものとしよう。このとき、リウビル演算子 L も対応して、無摂動項 L0
と摂動項 L’の和として書かれる。すると、「時間発展の演算子」U(t)を形式的に導入
すると ( ) ( ) ( )
( ) ( ) ( )( ) ( )∫ ′−−−−=
−=t
sULstiLdsitiLtU
tUtiLdt
tdU
0
00 expexp (4.11)
が厳密になりたつ。ここでの取り扱いでは、全リウビル演算子 L が陽に時間 t に依存
するので、 ( ) ( )iLttU −= exp とできないことに注意されたい。1 次摂動論を行うには、
最後に出てくる U(t)を無摂動リウビル演算子 L0(L0 は時間 t に陽に依存しない)の下
での時間発展の演算子に置き換えて、
( ) ( ) ( )( ) ( )∫ −′−−−−=−t
siLLstiLdsitiLiLt0
000 expexpexpexp (4.12)
を得る。この式が、小さな外場がかかったときの系の時間発展を記述する。系に外場
が加わるのは t>0 であって、それ以前の系は無摂動ハミルトニアンで決まる平衡状態
であったとすると、無摂動平衡状態での分布関数をρ0(q,p)として(時間に依存しない
ことに注意)、式(4.9)と式(4.12)から、
( ) ( ) ( )( ) ( )∫ ′−−−=t
pqLstiLdsipqtpq0
000 ,exp,;, ρρρ (4.13)
という形で摂動を受けた分布関数ρ(q,p;t)が表される。
式(4.13)から、任意の物理量 B(q,p)について、その平均が
( ) ( )( ) ( )∫ ∫ ′−−−=t
pqLstiLBdqdpdsiBtB0
000 ,exp ρ (4.14)
と表される。式(4.14)で、<B(t)>は摂動を受けた系での平均であり、0
L は無摂動平衡

状態で取った平均を意味する。ここで、記号の簡略化の一つとして、
{ }qY
pX
pY
qXYX
∂∂
∂∂
−∂∂
∂∂
=, (4.15)
でもって、ポアソン(Poisson)括弧を導入する。すると、式(4.14)は
( ) ( ){ } 0, AtBtBA −=Φ (但し、B(t)は無摂動系で
時間発展したときの B の値である) (4.16)
で定義される応答関数ΦBA(t)を用いて、式(4.10)によって、
( ) ( ) ( )∫ −Φ=−t
BA stsdsFBtB0
0 (4.17)
と書き直すことができる。また、式(4.16)は
( )( ) ( )
Tk
AtB
Tk
AtBt
BBBA
00 &&−==Φ (上の黒点は無摂動系での時間微分を表す)(4.18)
と変形することができる。
一般に、n 個の物理量 A1、・・・、An が与えられたとき n 点の時間相関関数は
( ) ( ) 011 nn tAtA L で定義される。上の取り扱いで見たように、外場の 1 次の摂動に対す
る応答は 2 点の時間相関関数によって与えられる。平衡状態は、時間に依存しない状
態なので、2 点の時間相関関数は、2 つの物理量を考える時間の差にのみ依存する。
つまり、平衡状態での 2 点時間相関関数は、1 つの時間の引数を持つ。また、上の取
り扱いは、外場の 1 次の応答に関するものであり、線形応答の取り扱いと呼ばれる。
外場の 2 次以上の応答を、非線形応答と呼び、そこでは、3 点以上の時間相関関数に
よって、系の応答を記述することができる。
次に、系が外部から吸収するエネルギー(系内エネルギーの増分)を考えよう。時
刻 0 に外場がスイッチオンされて、T の時刻でのエネルギー吸収 U(T)は
( ) ( ) ( )∫ ∂
∂−=
T
ttFtAdtTU
0
(4.19)
である。そこで、F(t)が単色(1 つの周波数成分を持つという意味)で
F(t) = F0cos(ωt+α) (4.20) の形を持つとき、単位時間当りのエネルギー吸収は、T が大きいとき(応答の線形性
が破れるほど大きくてもいけないのだが)
( ) ( ) ( ) ( ) ( )∫∫∞∞
Φ=Φ=0
20
0
20 expIm
2sin
2titdtFttdtF
TTU
AAAA ωωωω (4.21)

と表される。ここで、応答関数ΦBA(t)から
( ) ( ) ( )∫∞
Φ=0
exp titdt BABA ωωχ (4.22)
で定義されるχBA(ω)を一般化感受率と呼ぶ。式(4.21)に見るように、外場による系のエ
ネルギー吸収を測ることで、一般化感受率の虚部を知ることができ、系の応答関数に
対する情報を得ることができる。
問4.7 式(4.11)を導く。
問4.8 リウビル演算子 L が陽に時間 t に依存するとき、式(4.11)の U(t)が
( ) ( ) ( ) ( )
( ) ( ) ( ) ( ) ( ) L+−+−+=
−+=
∫∫∫
∫
210
20
12
10
1
110
1
1
1
1
tLtLdtdtitLdti
tUtLdtitU
ttt
t
を満たすことを見る。また、この式が
( ) ( )
−= ∫
t
sdsLitU0
exp
に等しくなるには、L(t)がどのような条件を満たさなくてはならないかを考
える。
問4.9 式(4.13)は L’の 1 次までの表式である。式(4.11)に基づいて、より高次までの
表式を導く。
問4.10 任意の X、Y、Z に対して、ポアソン括弧が、 { }{ } { }{ } { }{ } 0,,,,,, =++ YXZXZYZYX
を満たすことを示す。
問4.11 任意の X、Y、Z に対して、ポアソン括弧が、
{ } { } { }∫∫ ∫ == YXdqdpZXZdqdpYZYdqdpX ,,,
を満たすことを示す(但し、積分の端で被積分関数は 0 になるとしてよい)。
問4.12 式(4.17)を導く。
問4.13 式(4.16)から式(4.18)を導く。
問4.14 式(4.17)において、外場 F(s)が s によらず一定値 F0 をとるとき、長い時間の経
ったあと、

( ) ( )∫∞
Φ=−∞0
00 tdtFBB BA
となることが分かる。このとき、右辺が時間によらない 1 次摂動論を行っ
たときの結果に一致することを示す。
問4.15 式(4.19)を導く。
問4.16 式(4.21)を導く。
問4.17 式(4.20)では、F(t)を単色としたが、F(t)がいくつかの周波数成分をもつとき、
式(4.21)はどのような形になるだろうか?
問4.18 応答関数ΦBA(t)が t→∞で 0 になるとき、対応する一般化感受率χBA(ω)は
Kramers-Kronig 関係式
( ) ( ) ( )ωχωκ
κχκ
πωχ BA
BABA idP Im
Im1∫∞
∞−
+−
= (P は主値積分)
を満たすことを示す。
問4.19 自己相関に関する一般化感受率χAA(ω)がωIm χAA(ω) > 0 を満たすことを示す。
4.3 Redfield 方程式
前節の線形応答理論では、全粒子を含む系を取って、分布関数を構成した。しかし、
問題にしたい部分系だけをとりだし、残りを熱浴として扱いたいことも多い。本節で
は、全系をまるまる取り扱うのでなく、熱浴に関わる自由度を積分し去って、興味の
ある部分系だけの分布関数を構成することを目的とする。
問題としている部分系の座標と運動量を(qs,ps)と総称し、熱浴の座標と運動量を
(qb,pb)としよう。すると、問題としている部分系の分布関数ρs(qs,ps;t)は、全系の分布
関数ρ(qs,ps,qb,pb;t)から熱浴に関わる自由度を縮約して、
( ) ( )tpqpqdpdqtpq bbssbbsss ;,,,;, ρρ ∫= (4.23)
と与えられる。問題としている系(以下、単に系と呼ぶ)のハミルトニアンは、(qs,ps)
だけの関数であり、熱浴のハミルトニアンは(qb,pb) だけの関数である。それゆえ、系
のハミルトニアンから生成されるリウビル演算子 Ls は(qs,ps)のみに作用し、熱浴から
のリウビル演算子 Lb は(qb,pb)のみに作用する。系と熱浴の相互作用 V は、
( ) ( ) ( )bbi
i
ssi
bbss pqFpqApqpqV ,,,,, ∑−= (4.24)
の形を持つものとする。時刻 0 以前に系と熱浴の間に相互作用は無く、系と熱浴が、

それぞれのハミルトニアンに対応する平衡状態にあったものとする。系と熱浴の平衡
状態での分布関数を、それぞれ、ρs0(qs,ps)、ρb0(qb,pb)とすると、もちろん、 ( ) ( ) 0,0, 00 == bbbbssss pqLpqL ρρ (4.25)
であり、系と熱浴全体の分布関数は積ρs0(qs,ps)ρb0(qb,pb)である。また、熱浴の物理量
Fi(qb,pb)は平衡状態ρb0(qb,pb)で平均すると 0 になるものとする(そのように定義し直
す)。すると、系と熱浴の相互作用に対応するリウビル演算子を Lv として、式(4.11)
から
( ) ( ) ( ) ( ) ( )
( ) ( )( )( ) ( )ypqpqLytLLidyLi
pqpqiLtpqpqLLit
tpqpq
bbssvt
bsv
bbbsssvbbssbsbbss
;,,,exp
,,;,,,;,,,
0
2
00
ρ
ρρρρ
∫ −+−−+
−+−=∂
∂
(4.26)
が厳密に成り立つ。
ここで、熱浴は莫大な自由度を持ち、系との相互作用は熱浴に無視できる程度の影
響しか与えないと仮定する。つまり、系と熱浴の相互作用 V(qs,ps,qb,pb)が常に早く散
逸してしまい、どのような時刻 t においても、
{ } 0, 0 ≈biF ρ (4.27)
が成り立つと仮定するのである。このとき、熱浴の分布関数は、平衡状態のρb0(qb,pb)
のままであり、 ( ) ( ) ( )bbbsssbbss pqtpqtpqpq ,;,;,,, 0ρρρ = (4.28)
が成立する。式(4.27)の仮定および ( ) 0=BtF (B
L は熱浴の平衡分布関数ρb0(qb,pb)
で取った平均を意味する)のために、式(4.26)で熱浴の自由度(qb,pb) を積分し去ると、
系の分布関数ρs(qs,ps;t)だけの方程式として
( ) ( )
( ) ( ) ( )( ) ( ){ }{ }∑∫ −−+
−=∂
∂
ji
tsss
js
i
B
ji
sssssss
ypqAytiLAyFtFdy
tpqiLt
tpq
, 0
;,,exp,
;,;,
ρ
ρρ
(4.29)
を得る。式(4.29)で第 1 項は、熱浴との相互作用がないときの系の時間発展に対応す
る項であり、第 2 項が熱浴との相互作用によって系の分布が変化する寄与を示してい
る。式(4.29)の第 2 項に、熱浴の物理量に関する 2 点の時間相関関数が表われている
ことに注意されたい。この時間相関関数は、熱浴の平衡状態での平均量であり、2 つ
の物理量を考える時間の差にのみ依存する。さらに、系の分布関数ρs(qs,ps;t)の時間変
化が、熱浴の揺らぎよりはるかに遅いとすると、熱浴の時間相関関数 ( ) ( ) B
ji tFF 0 の緩
和時間よりはるかに大きな t について

( ) ( )
( ) ( ) ( ) ( ){ }{ }∑∫∞
−+
−=∂
∂
ji
sssi
sj
B
ji
sssssss
tpqAyiLAyFFdy
tpqiLt
tpq
, 0
;,,exp,0
;,;,
ρ
ρρ
(4.30)
とすることができる。式(4.30)を Redfield 方程式と呼び、分光学における緩和現象の
記述の基礎をなす方程式である。
系内の任意の物理量 B(qs,ps)を考えよう。その平均 ( ) stB は
( ) ( ) ( )tpqpqBdpdqtB ssssssss ;,,∫= ρ (4.31)
で与えられ、時間発展は、式(4.30)によって、
( ) ( ) ( ) ( ){ }{ }∑∫∞
+=ji
s
js
i
B
ji
sss
BAyiLAyFFdyBiLdt
tBd
, 0
,exp,0 (4.32)
を満たす。この式は、両辺を確率分布ρs(qs,ps;t)で平均すると等しいという意味で
( ) ( ) ( ){ }{ }∑∫∞
+
=
jij
si
B
ji BAyiLAyFFdydtdB
dtdB
, 00
,exp,0 (4.33)
と形式的に書ける。式(4.33)で、第 1 項は iLsB の意味であり、熱浴との相互作用がな
いときの時間微分を表す。熱浴からの揺動による効果は第 2 項である。熱浴の揺動場
の 2 点時間相関関数が表われていることに注意されたい。また、 ( )yiLsexp の項がある
が、これが、揺動場の時間相関関数の緩和に影響を及ぼす周波数成分を同定する。例
えば、系内の物理量 Ai(qs,ps)が揺動場の揺らぎよりはるかにゆっくりと時間変化する
ものとすると、式(4.33)の第 2 項にある ( )yiLsexp を 1 とおいて良く(式(4.33)の時間積
分の間、Ai(qs,ps)を「止める」ことになる)、 ( ) ( ) B
ji tFF 0 の全時間積分、つまり、周
波数 0 の成分によって揺動場の影響が表われる。
問4.20 式(4.26)を導く。
問4.21 式(4.27)の仮定の下で、式(4.28)が導かれることを示す。
問4.22 式(4.29)を導く。
問4.23 式(4.30)を導く。
問4.24 式(4.32)を導く。

4.4 密度行列
本章での展開は、古典力学の枠内で行ってきた。量子力学的に理論展開を行うには、
密度行列を使うとよい。状態ベクトルが|Ψi>である i 番目の状態が確率 piで出現する
アンサンブルでは、密度行列ρは
∑ ΨΨ=i
iiipρ
で定義される。量子的な取り扱いでは、密度行列が、古典的な取り扱いにおける分布
関数の役割を果たす。特に、Redfield 理論の展開は、4.3 節では敢えて古典的に行った
が、おそらくどこを見ても量子的に行われているだろう(例えば、参考文献[1,2])。
本章の結果を、密度行列を用いて、量子力学版に書き換えることはよい演習である。
問4.25 ハミルトニアンを H としたとき、密度行列ρの時間発展の方程式が
[ ]ρρ ,Hit h
−=∂∂
であることを示す。 問4.26 式(4.15)で与えたポアソン括弧{ }YX , を
{ } [ ] ( )YXXYi
YXi
YX −=→hh
1,1, と置き換えると、式(4.18)を除くと、本章の結果が全て量子力学的な表式と
して通用することを確かめる。また、式(4.18)の量子力学版が
( ) ( ) ( ) ( ) 01
0
expexp HAHtBdtTk
BA
B
λλλ −=Φ ∫ &
であることを示し、古典論との対応について考える。
問4.27 リウビル演算子を使った定式化が、古典論でも量子論でも通用する原因を特
定する。
第 4 章の参考文献
[1] C. P. スリクター「磁気共鳴の原理」(シュプリンガー・フェアラーク東京) 1998
年、178-192 頁。
[2] J. McConnell, The Theory of Nuclear Magnetic Relaxation in Liquids, Cambridge
University Press (1987), pages 32-46.

第 5 章 NMR 分光法の基礎
NMR とは核磁気共鳴(nuclear magnetic resonance)の略である。原子・分子に埋め込
まれた原子核の磁気的な共鳴現象を用いて、分子構造・分子間相互作用・分子運動を
調べる方法である。原子核を変えること(同位体置換と呼ぶ)で、分子の化学的性質
を変えることなく、分子をラベルすることができる。また、電磁場を様々な方法で掛
けていくこと(パルス系列の構成)によって、分光から得られる情報を選択すること
ができる。これらの理由によって、NMR 法は、現在もっと有力な(もちろんオール
マイティではない)分光法となっており、爆発的な進歩をしている。本章では、FT-NMR
法および緩和現象を線形応答理論と Redfield 理論に基づいてその初歩を紹介する。
5.1 静磁場の中の運動
原子核が M=(Mx, My, Mz)なるベクトルで表される角運動量を持つとき(スピンを持
つと言ってよい)、それは同時に磁気モーメントµをもち、
µ =γM (5.1) の関係式で結ばれている。ここで、γは磁気回転比と呼ばれる原子核(の状態)に固
有の量であり、原子核の「回転」による原子核内および表面の「電流」が誘起する磁
化の強さを決定する。外部から静磁場 H がかかっているとき、磁気エネルギーE は
E = -µ・H = -γM・H (5.2)
と表される。ここで、角運動量の満たすポアソン括弧式は
{Mx, My} = Mz {My, Mz} = Mx {Mz, Mx} = My (5.3)
である。式(5.2)と式(5.3)から、静磁場の中に置かれた原子核の角運動量は
HMMM×== γ},{ E
dtd
(5.4)
という運動方程式によって時間発展することが分かる。外部からの静磁場 H が z 軸に
平行で H= (0, 0, H)と表されるとき、式(5.4)の解を成分ごとに書き下すと、 ( ) ( ) ( ) ( ) ( )( ) ( ) ( ) ( ) ( )( ) ( )0
0cos0sin0sin0cos
zz
yxy
yxx
MtMMHtMHttM
MHtMHttM
=+−=
+=γγ
γγ (5.5)
となる。式(5.5)から、Mzは時間に対して不変であり、Mxと Myは x-y 平面上をγH の角
速度で時計回りに(通常、反時計回りを回転の正の方向に取るので、角速度-γH と
いえる)その大きさを保存したまま回転する。この回転の様子を図 5.1 に模式的に示
す。x-y 平面上の運動は周期運動であり、その周波数で共鳴を起こすことができる。

Mx
My
Mz
図 5.1 角運動量ベクト
ルの運動
このγH をラーモア周波数とよび、NMR の共鳴周波数
に当たる。物質に固有の量は磁気回転比γであり、ラ
ーモア周波数は外部磁場の強さに比例する。それゆえ、
強い磁場をかけるとそれだけ高い周波数で共鳴が起
こることに注意する必要がある。また、スピン量子数
が 0 である原子核は、どれだけ強い磁場をかけてもラ
ーモア周波数は 0 であり、磁気共鳴を示さない。スピ
ンが 0 の原子核としては、12C と 16O(もっともよく見
られる炭素および酸素の原子核)がある。
一般に、原子核の感じる磁場はその置かれている分
子内の環境によって異なる。つまり、同じ原子核でも、
周りの電子雲の違いによって、受ける磁場が微妙に異
なる。i 番目の原子核の感じる磁場を Hiとしたとき、i 番目の原子核の角運動量を(Mxi,
Myi, Mz
i)とし、全角運動量を(Mx, My, Mz)とすると、
( ) ( ) ( ) ( ) ( )[ ]( ) ( ) ( ) ( ) ( )[ ]( ) ( )∑
∑∑
=
+−=
+=
i
izz
i
iy
iix
iy
i
iy
iix
ix
MtM
MtHMtHtM
MtHMtHtM
0
0cos0sin
0sin0cos
γγ
γγ
(5.6)
と表され、いくつかの周波数成分を持つ回転が見出される。基準の磁場を H0 とした
ときに、(Hi-H0)/H0 を化学シフトと呼ぶ。原子核の受ける磁場の違いは、周りの電子
雲の違いによるものなので、化学シフトは、分子構造や分子間相互作用の指標である。
化学シフトを電子構造と結びつける理論については、参考文献[1]を参照していただき
たい。
式(5.4)では、原子核の回転は、静磁場とのみ相互作用をするものとしていた。しか
し、現実には、M は熱浴(NMR の取り扱いでは、熱浴をしばしば格子と呼ぶ)と相
互作用をし、ある平衡値に戻っていく。系が等方的なとき、M の平衡値は静磁場 Hと平行であり、
γM = χH (5.7) として、比例定数χが導入される。このχを磁化率と呼び、平衡状態での系の磁化を記
述する。
そこで、磁化の運動に平衡値への緩和の現象を付け加えると、現実の磁化の運動が
より正確に記述できるものと期待される。静磁場が z 軸に平行で H= (0, 0, H)と表され
るとき、Mzの平衡値 Mz0 は(χ/γ)H であり、平衡値への減衰が時定数 T1 で表されるもの
としよう。また、Mx および My の平衡値は 0 であり、減衰の時定数を T2 とする。T1

と T2 という 2 つの時定数を考慮して、式(5.4)を書き直したものが
( )
( )
( )2
2
1
0
TM
dtdM
TM
dtdM
TMM
dtdM
yy
y
xx
x
zzz
z
−×=
−×=
−−×=
HM
HM
HM
γ
γ
γ
(5.8)
であり、Bloch 方程式と呼ばれる。Bloch 方程式は、やや直感的に導かれたものである
が、磁気共鳴の基本式であり、統計力学的に基礎づけることが可能である。T1 はスピ
ン-格子緩和時間(縦緩和時間)と呼ばれ、エネルギーの緩和にほぼ対応する。T2
はスピン-スピン緩和時間(横緩和時間)と呼ばれ、様々な核の運動がコヒーレンス
を失う緩和にほぼ対応する。両緩和時間とも、分子間相互作用・分子運動に由来する
ものであるので、緩和時間測定によって、分子間相互作用や分子運動に関する情報を
得ることができる。
問5.1 式(5.4)を導く。
問5.2 陽子(水素の原子核)のγの値を調べる。
問5.3 陽子が 100、270、400、600 MHz で共鳴を起こす磁場の強さを求める。
問5.4 100、270、400、600 MHz での 13C、17 O、2H(重陽子、陽子と中性が一つず
つ結合したもの)、14N の共鳴周波数を求める。
問5.5 Bloch 方程式が成り立つという条件の下で、dM/dt = 0 が成り立っているとき
の磁化 M を求める。
5.2 FT-NMR 法
単純な分光法では、周波数を変えながら電磁場をあててやって、共鳴を見るという
方法をとる。これに対して、FT 法(FT がフーリエ変換の意味)では、4.2 節で導入
した応答関数を時間の関数として直接観測し、スペクトルを求めるものである。現在
の NMR の主流は、FT 法であり、これによって観測時間の大幅な短縮化が達成された。
z 軸に平行な静磁場 H= (0, 0, H)がかかっている系を考えよう(i 番目の原子核にか
かる磁場は Hiである)。このとき、摂動として x 軸に平行な磁場 H’(t)をかけ、摂動
エネルギーE’が
( ) ( )tHMtHMEi
ixx ′−=′−=′ ∑ (5.9)

であるとすると、4.2 節で導入した Mxに対する応答関数Φ(t)(添字は省いた)は
( ) ( ) ( )∑∑
−=
−=Φ
i
ii
ii
i
iz
ii HtH
TtMtH
Ttt
γχγγ sinexpsinexp
2
0
2
(5.10)
で与えられる。ただし、χiおよび T2iは、それぞれ、i 番目の原子核の磁化率およびス
ピン-スピン緩和時間である(磁気回転比は一定と取っておいた)。
摂動磁場 H’(t)がパルス的でδ(t)に比例する様に取ると、式(4.17)によって、磁化の観
測がそのまま応答関数Φ(t)の決定であることが分かる。応答関数Φ(t)が決定されると、
そのフーリエ変換をすることでγHi を得ることができる。つまり、化学シフトをΦ(t)
のフーリエ変換によって得ることができるわけである。このように、δ(t)的なパルス
をかけて応答関数Φ(t)を測定し、フーリエ変換をすることでスペクトルを得る方法が、
FT 法の基本型である。
問5.6 式(5.10)を導く。
問5.7 式(5.9)の形の摂動エネルギーの下における My および Mz に対する応答関数を
導く。
問5.8 現実の測定において、摂動磁場 H’(t)をδ(t)に比例する様に取ることは不可能で
ある。そこで、H’(t)をどのように取れば、H’(t)がパルス的であってδ(t)に比
例するものと考えても問題ないか、ということを式(5.10)に基づいて考える。
問5.9 実際の FT-NMR 測定では、応答関数Φ(t)を測定する時間 t の原点が必ずしも
明確でない場合が多い。応答関数の時間 t の原点がαだけずれていたとき、
フーリエ変換で決定される一般化感受率がどのような影響を受けるかを、
式(4.22)に基づいて考える。さらに、αの影響を除去するには、どのような
方法があるかを考える。
5.3 磁気緩和現象
ここでは、4.3 節の Redfield 理論に基づいて、T1 および T2 について考察する。簡単
のために、原子核の種類は 1 種類であるとしよう。無摂動系として、外部からの静磁
場を H= (0, 0, H)として式(5.2)をとる。そして、熱浴との相互作用 V として、
V = -γ (Mxhx + Myhy + Mzhz) (5.11)
とおこう。ここで、(hx, hy, hz)は熱浴によって及ぼされる揺動磁場であって、原子核(4.3
節における「系」に相当)の立場から見ると、時間依存する確率変数である。ただし、
熱浴からの揺動磁場(hx, hy, hz)は外部静磁場 H よりはるかに弱いものとしよう。すると、

式(4.33)によって、
( ) ( ) ( ) ( ) ( ) ( )( )( ) ( ) ( ) ( ) ( ) ( )( )( ) ( ) ( ) ( ) ( ) ( )( )
+−×=
+−×=
+−×=
∫
∫
∫
∞
∞
∞
0
2
0
2
0
2
0cos0
0cos0
00cos
Bzz
Bxxyy
y
Bzz
B
yyxxx
B
yyB
xxzzz
shhHsshhdsMdt
dM
shhHsshhdsMdt
dM
shhshhHsdsMdt
dM
γγγ
γγγ
γγγ
HM
HM
HM
(5.12)
となることが分かる。この式は、x および y 成分の T2 が同じではないものの、まさに
Bloch 方程式である(Mz0 が出てきていないことについては、もう少し込み入った議論
が必要)。つまり、やや直感的に導入した Bloch 方程式は Redfield 理論から導き出す
ことが可能であることが示された。さらに、熱浴が均一な流体であって、原子核との
相互作用が等方的で、揺動磁場の相関関数が
( ) ( ) ( ) ( ) ( ) ( ) ( )( ) ( ) ( ) ( ) ( ) ( ) 0000
000
===
≡==B
xz
B
zy
B
yx
Bzz
B
yyB
xx
thhthhthh
tCthhthhthh (5.13)
となると仮定しよう。すると、式(5.12)によって
( ) ( )
( )( ) ( )
+=
=
∫
∫∞
∞
0
2
2
0
2
1
cos11
cos21
tCHtdtT
tCHtdtT
γγ
γγ (5.14)
であることが分かる。この式から、磁気緩和は、熱浴からの揺動磁場の時間相関関数
によって決定されることが分かる。逆に、磁気緩和の測定から、測定している原子核
に影響を及ぼす相互作用モードの強度と分子運動の情報を得ることができる。
次に、相関関数 C(t)が時定数τによって
( )
−=
τtCtC exp0 (5.15)
の形を持つものとしよう。このとき、NMR 緩和時間は
( )
( )
+
+=
+=
202
2
202
1
1111
1121
τγτγ
τγτγ
HC
T
HC
T (5.16)
と表される。C0 は揺動磁場の強さを表すパラメーターであり、これが大きくなると、
熱浴との相互作用が大きくなり、T1 と T2 はともに小さくなる(緩和が速くなる)。
また、揺動磁場の相関時間τの値にかかわらず、T1≧T2 が常に成り立つ。さらに、T1
はτがラーモア周波数の逆数 1/γH に等しいとき、最小値をとり、T2 はτが大きくなる

と小さくなる。通常の低分子の流体条件では、相関時間τがラーモア周波数の逆数 1/γH
よりはるかに小さいので(τは ps のオーダーで、γH は GHz 以下のオーダー)、式(5.16)
は
τγ 02
21
211 CTT
== (5.17)
となる。γHτ≪1 は極度先鋭化の条件と呼ばれる。極度先鋭化の条件が成り立つとき
T1 と T2 は等しく、τが大きくなると磁気緩和が速くなる。C0 が既知のとき、T1 または
T2 を測定して、分子レベルの運動の緩和時間τを求めることができる。
問5.10 揺動磁場が外部磁場よりはるかに弱いという条件下で、式(5.12)を導く。
問5.11 式(5.16)を導く。
問5.12 C(t)の減衰時間がラーモア周波数の逆数 1/γH よりはるかに小さいとき、式
(5.15)の仮定が成り立たなくても、式(5.17)の形に T1 と T2 を表すことができ
て、T1 = T2 である。このとき、相関時間τはどのように C(t)で表されるだろ
うか?
5.4 補遺
前章を古典的に叙述したのに対応して、本章の解説も古典的に行った(例えば、プ
ランク定数は出てこなかった)。分光学における最も重要な概念である「共鳴」と「緩
和」は、古典論の範囲で扱うことができるのである(もちろん、現実系に量子論的要
素が含まれていないと言っているわけではない)。共鳴が起きるために必要なものは
運動の周期性であり、緩和が起きるのに必要なものは熱浴からのランダムな揺動であ
る。このことを押さえた上で、古典論での式展開を量子論でのものに書き換えていく
のはよい演習である。ある現象を見る・考える際に、その現象が古典的にも理解しう
るものなのか、それとも、量子的にのみ理解しうるものなのかを考えることは、その
現象の本質を理解するための有効なアプローチの一つである。
本章では、化学シフトの微視的理論については述べなかった。分子の電子構造と化
学シフトを結びつける理論については、ぜひ、参考文献[1]を見ていただきたい。ここ
で、化学シフトの微視的理論を述べなかったのは、流体系における化学シフトの理論
の決定版が無い(と筆者は思う)からである。化学シフトは、測定している原子核で
の磁場に対応する量であるが、実は、この値は流体系の形状に依存するのだ。もちろ
ん、分子構造や分子間相互作用を評価する量として化学シフトを捉えるためには、分

子レベルの構造や相互作用に関係のない系の形状(系が球状であるか、細長いかなど)
の影響を取り除かなくてはならない。流体系における化学シフトの理論は、流体系の
形状に対する依存性を正しく把握し、分子構造や分子間相互作用からの寄与だけを取
り出す方策を厳密に基礎づけるものでなくてはならない。そのような理論構成が未だ
なされていないと言う意味で、流体系における化学シフトの理論の決定版が無いと言
った。現状での到達点については、参考文献[2]を参照していただきたい。また、超臨
界流体の構造の実験的研究という立場からは、参考文献[3]が NMR 法の「実際の使わ
れ方」を知る上で役に立つと思う。
第 5 章の参考文献
[1] C. P. スリクター「磁気共鳴の原理」(シュプリンガー・フェアラーク東京) 1998
年、85-98 頁。磁気共鳴をきちんと学びたいなら、この本を読むべきである。
[2] T. Yamazaki, H. Sato, and F. Hirata, J. Chem. Phys., 115, 8949-8957 (2001).
[3] (a) N. Matubayasi, C. Wakai, and M. Nakahara, J. Chem. Phys. 107, 9133-9140 (1997).
NMR 化学シフトの測定による超臨界水の水素結合構造の決定
(b) N. Matubayasi, C. Wakai, and M. Nakahara, J. Chem. Phys. 110, 8000-8011 (1999).
超臨界水の化学シフトと分子間相互作用の関連についての半経験的な解析
(c) N. Matubayasi, N. Nakao, and M. Nakahara, J. Chem. Phys. 114, 4107-4115 (2001).
超臨界水の NMR 緩和測定による分子運動の解析





























