Embed Size (px)
Citation preview

EFFORTS TOWARD THE TOTAL SYNTHESIS OF BIELSCHOWSKYSIN
By
Nina Renee Collins
Thesis
Submitted to the Faculty of the
Graduate School of Vanderbilt University
in partial fulfillment of the requirements
for the degree of
MASTER OF SCIENCE
in
Chemistry
August, 2013
Nashville, Tennessee
Approved:
Professor Gary Sulikowski
Professor Carmelo Rizzo

ii
TABLE OF CONTENTS
Page
LIST OF FIGURES........................................................................................................... iii
LIST OF SCHEMES......................................................................................................... iv
Chapter
I. Background.................................................................................................................... 1
Introduction............................................................................................................ 1
Biosynthesis........................................................................................................... 1
Alternate Routes..................................................................................................... 3
II. Current Progress........................................................................................................... 12
Sulikowski Route.................................................................................................. 12
Model System....................................................................................................... 16
Future Plans.......................................................................................................... 18
Experimental Methods.......................................................................................... 20
Appendix
A. SPECTRA OF RELEVANT COMPOUNDS....................................................... 26
REFERENCES................................................................................................................. 41

iii
LIST OF FIGURES
Figures Page
1 Furanocembranoid Skeletons................................................................................. 1
A1 1H NMR spectra (400 MHz, CDCl3) of compound 103....................................... 27
A2 13C NMR spectra (100 MHz, CDCl3) of compound 103...................................... 28
A3 1H NMR spectra (400 MHz, CDCl3) of compound 106....................................... 29
A4 13C NMR spectra (100 MHz, CDCl3) of compound 106...................................... 30
A5 1H NMR spectra (400 MHz, CDCl3) of compound 108....................................... 31
A6 13C NMR spectra (100 MHz, CDCl3) of compound 108...................................... 32
A7 1H NMR spectra (400 MHz, CDCl3) of compound 109....................................... 33
A8 13C NMR spectra (100 MHz, CDCl3) of compound 109...................................... 34
A9 1H NMR spectra (400 MHz, CDCl3) of compound 110....................................... 35
A10 13C NMR spectra (100 MHz, CDCl3) of compound 110...................................... 36
A11 1H NMR spectra (400 MHz, CDCl3) of compound 111....................................... 37
A12 1H NMR spectra (400 MHz, CDCl3) of compound 100....................................... 38
A13 1H NMR spectra (400 MHz, CDCl3) of compound 84......................................... 39
A14 1H NMR spectra (400 MHz, CDCl3) of compound 113....................................... 40

iv
LIST OF SCHEMES
Scheme...........................................................................................................................Page
1. Conversion of Geranylgeranyl pyrophosphate to rubifolide.................................. 2
2. Proposed Biosynthesis of Bielschowskysin........................................................... 2
3. Lear’s Route........................................................................................................... 3
4. Nicolaou’s Route.................................................................................................... 4
5. Mulzer’s Route....................................................................................................... 6
6. Stoltz’s Synthesis of Enantiomeric Alcohols 47 and 48........................................ 7
7. Stoltz’s Approach to Cyclobutane 56.................................................................... 8
8. Stoltz’s Route to Unexpected Compound 61......................................................... 9
9. Theoretical Approach to Verrillin and Bielschowskysin........................................ 9
10. Theodorakis’ Synthesis of the C7–C12 fragment................................................. 10
11. Theodorakis’ Synthesis of the C1–C13 fragment................................................ 10
12. Theodorakis’ Completion of Route to Intermediate 79........................................ 11
13. Retrosynthetic Analysis of Bielschowskysin........................................................ 12
14. Revised Synthetic Analysis of Bielschowskysin.................................................. 13
15. Synthesis of Lactone 90........................................................................................ 14
16. Synthesis of Bis-Butenolide 96............................................................................. 15
17. Synthesis of Advanced Intermediate 84............................................................... 16
18. Synthesis of Aldehyde 104................................................................................... 17
19. Synthesis of Butenolide 112................................................................................. 17
20. Application of Model System Chemistry to Front End........................................ 18

v
21. Bielschowskysin End-Game Strategy................................................................... 19
22. Proposed conversion to the Tris-Butenolide 119.................................................. 19

1
CHAPTER 1
BACKGROUND
Introduction
Isolated from Pseudopterogorgia kallos by Rodriguez, bielschowskysin is a
unique furanocembranoid natural product.1 In addition to its structural novelty and
complexity, bielschowskysin exhibits antiplasmoidial activity against Plasmodium
falciparum (IC50 = 10 µg/ mL) as well as in vitro cytotoxicity against EKVX nonsmall
cell lung cancer (GI50 < 0.01 µM) and CAKI-1 renal cancer (GI50 < 0.51 µM). Its
structural and biological profile renders the natural product an interesting target for total
synthesis.
Biosynthesis
Bielschowskysin belongs to a unique class of diterpene natural products
assembled by gorgonian corals. These marine species are responsible for the production
of most known furanocembranoids, pseudopteranes and gersolanes. Differing most
markedly in carbocyclic ring size, these natural products are highly oxygenated – often
featuring furan and butenolide moieties – and biosynthetically related.2
66 O 33
O 1212
1111
O(furano)cembrane
skeleton
O
O
O
O
O
Opseudopterane
skeletongersolaneskeleton

2
Figure 1: Furanocembranoid skeletons
The proposed biosynthesis of the furanocembranoid ring system begins with
geranylgeranyl diphosphate 1. Cyclization leads to generation of the cembrane skeleton,
which upon deprotonation, forms neocembrane 3. Subsequent oxidation promotes
formation of rubifolide 4, a supposed precursor for more complex furanocembranoids.2
Scheme 1: Conversion of geranylgeranyl (1) diphosphate to rubifolide (4).
Trauner has postulated that biosynthesis of bielschowskysin occurs through
rubifolide 4. Advancement to intermediate 5 is accomplished through various oxidative
processes. Further oxidation and hydration generates compound 6 which undergoes [2+2]
photocycloaddition to yield the natural product.2
Scheme 2: Proposed biosynthesis of bielschowskysin (7).
OPP
- OPP- H - H+O
OO21 34
[O]
O
OO
4
O
5
O
O
O
HOAc
HOH O
H
HOH
O
OH
HO Me
OO
H
O
OAcOO
HO Me
H
H
H
Me
OOHH
H
6
7
[O] [O]H2O
[2 + 2]hv
OH

3
Alternate Routes
Due to its structural complexity, bielschowskysin has proven an interesting yet
elusive target. Several groups, in addition to ours, have undertaken a total synthesis
program. A noteworthy point of divergence among these routes is the manner in which
the core cyclobutane functionality is installed.
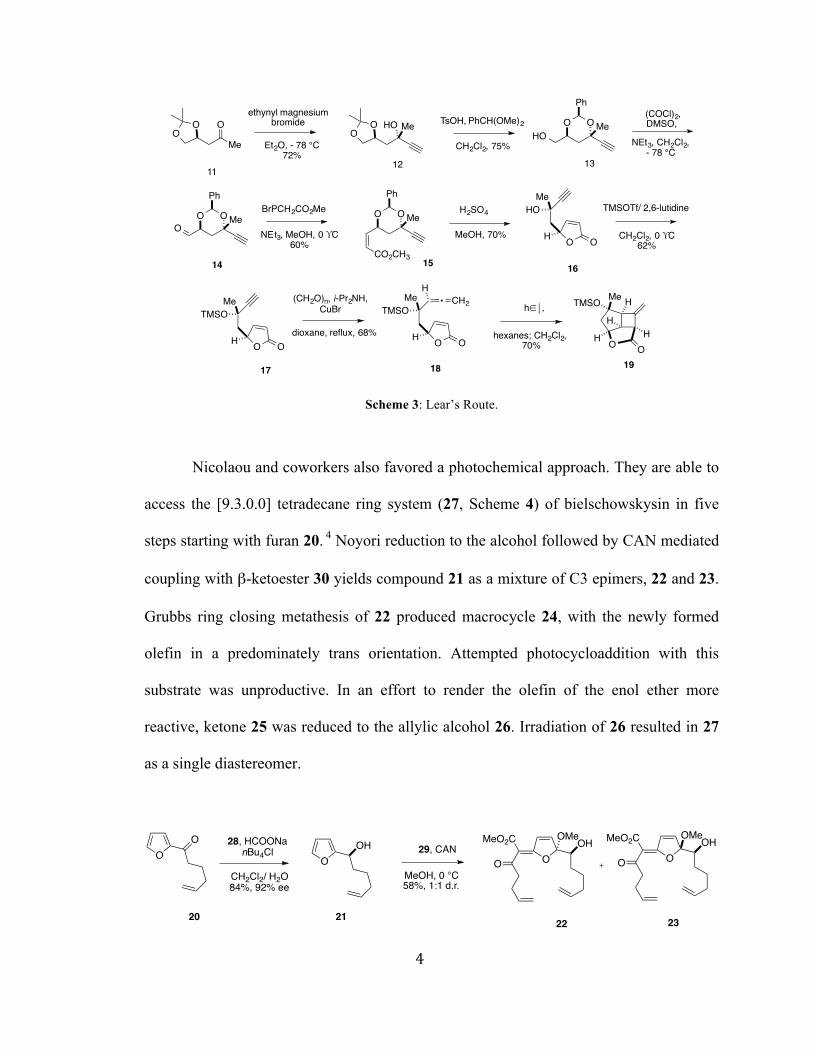
Similar to our route, Lear proposes a transannular [2+2]-photocycloaddition
between an allene and a butenolide.3 In order to investigate the feasibility of this
transformation, the group implemented a model system.
Malic acid was reduced to triol 9 and converted to acetonide 10. Swern oxidation
of the primary alcohol, addition of methyl magnesium bromide, and subsequent oxidation
afforded methyl ketone 11. Ethynyl magnesium bromide addition resulted in tertiary
alcohol 12, which underwent transketalization to the benzylidene acetal. Oxidation of the
primary alcohol to the aldehyde followed by Wittig homologation yielded the
predominately cis α,β-unsaturated ester 15. Upon treatment with sulfuric acid, the diol
was liberated and spontaneously formed butenolide 16. Reaction of silyl ether 17 with
paraformaldehyde gave photocycloaddition precursor 18. Upon irradiation in a solution
of hexanes and dichloromethane, the allene butenolide was converted to cyclobutane 19.
As of yet, there have been no reports of this methodology being employed in a
transannular setting.
HO OH
O
O
OH BH3‚ãÖSMe2
95%HO OH
OH p-TsOH, CuSO4
acetone, 67% OH
OO
i. (COCl)2, DMSO, NEt3, CH2Cl2, - 78 °C
iii. PCC, NaOAcDCM43%
8 9 10
ii. MeMgBr, Et2O- 78 °C
4
Scheme 3: Lear’s Route.
Nicolaou and coworkers also favored a photochemical approach. They are able to
access the [9.3.0.0] tetradecane ring system (27, Scheme 4) of bielschowskysin in five
steps starting with furan 20. 4 Noyori reduction to the alcohol followed by CAN mediated
coupling with β-ketoester 30 yields compound 21 as a mixture of C3 epimers, 22 and 23.
Grubbs ring closing metathesis of 22 produced macrocycle 24, with the newly formed
olefin in a predominately trans orientation. Attempted photocycloaddition with this
substrate was unproductive. In an effort to render the olefin of the enol ether more
reactive, ketone 25 was reduced to the allylic alcohol 26. Irradiation of 26 resulted in 27
as a single diastereomer.
Me
OO
Oethynyl magnesium
bromide
Et2O, - 78 °C 72%
OO
HO Me
CH2Cl2, 75%
TsOH, PhCH(OMe)2 O OHO
Ph
Me (COCl)2, DMSO,
NEt3, CH2Cl2, - 78 °C
1112 13
O OO
Ph
MeNEt3, MeOH, 0 !C
60%
BrPCH2CO2Me O O
Ph
Me
CO2CH3
MeOH, 70%
H2SO4
O OH
HOMe
CH2Cl2, 0 !C 62%
TMSOTf/ 2,6-lutidine
O OH
TMSOMe • CH2
H
O OH
TMSOMe
dioxane, reflux, 68%
(CH2O)n, i-Pr2NH, CuBr
hexanes; CH2Cl2, 70%
h"#,
O OHH
TMSO Me H
H
14 15 16
17 1918
OO
OOH
O
MeO2C
O
OHOMe
O
MeO2C
O
OHOMe
20 2122 23
28, HCOONanBu4Cl
CH2Cl2/ H2O 84%, 92% ee
29, CAN
MeOH, 0 °C58%, 1:1 d.r.

5
Scheme 4: Nicolaou’s Route.
Mulzer opted for a non-photochemical approach and began his synthesis with
known bicyclic alcohol 31.5 Silyl ether 32 was converted to enone 33 via
photooxygenation. Conjugate addition of a methyl cuprate followed by silyl ether
formation and Saegusa oxidation yielded 35. Luche reduction and MOM-protection
afforded compound 36, which underwent Mukaiyama-Isayama reaction to the tertiary
alcohol. Silyl protection, selective deprotection yielded alcohol 38. Oxidation and
alkylation at the α-position with formalin generated the quaternary carbon of the
molecule. TES protection of the primary alcohols and treatment with Petasis’ reagent
gave methylene 40. Exposure to Jones reagent resulted in tandem liberation of the
primary alcohols and oxidation to the dicarboxylic acid. MOM deprotection was
accompanied by lactone formation. Selective reduction of the remaining carboxylic acid
O
MeO2C
O
OHOMe
O
MeO2C
O
OMe
OH
O
MeO2C
HO
OMe
OH
24 25
26
30, CH2Cl2
reflux, 90%trans/ cis 15:1
NaBH4
THF/ H2O, 0 °C83%
O
OHOMeO
OMe
H
HO
H
NTs
Ru
TsNPh
Ph
i-Pr
Me
Ru
P(Cy)3
PhP(Cy)3
ClCl
OCO2Me
28
27
2930
h!
C6H6, 90%

6
and subsequent oxidation to the aldehyde produced 43, establishing the eastern portion of
bielschowskysin.
Scheme 5: Mulzer’s Route.
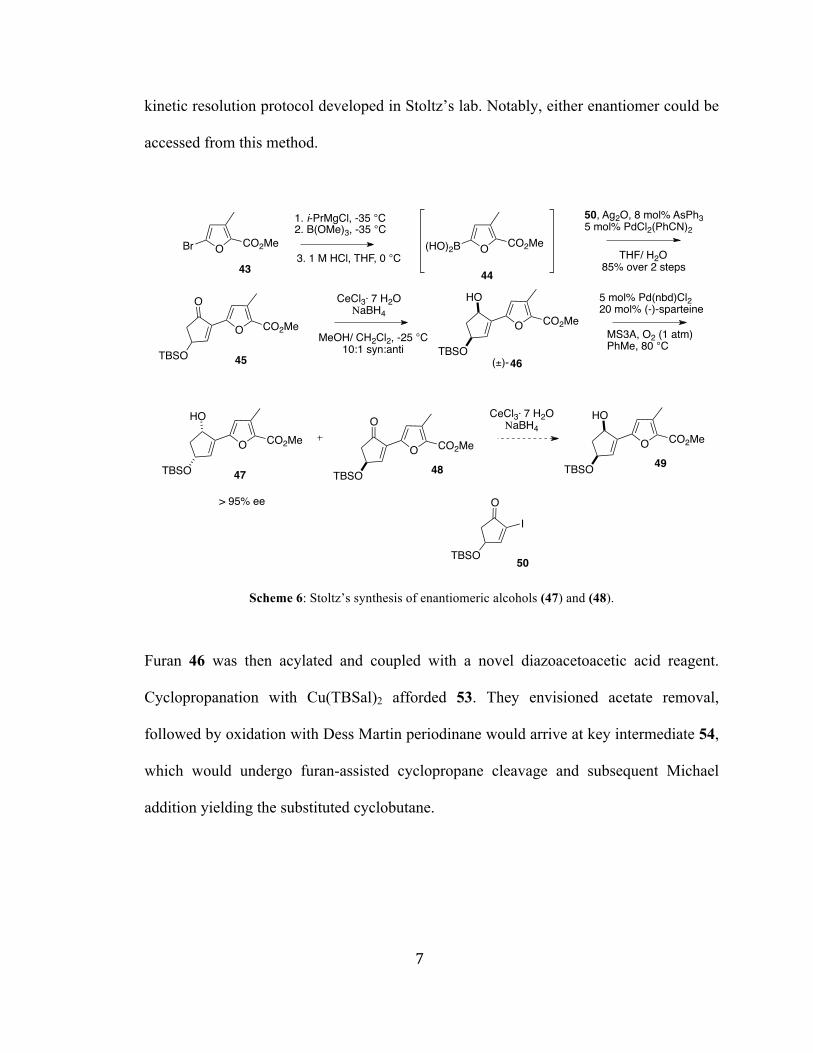
Stoltz and coworkers propose a tandem cyclopropane fragmentation/ Michael
addition of intermediate 54 to arrive at 56. To this end, furan 43 was converted to the
boronic acid and coupled with iodide 50.6 The group later found that the electron
deficient olefin of the enone precluded cyclopropane formation. Thus, ketone 45 was
selectively reduced to afford allylic alcohol 46. This substrate is amenable to an oxidative
H
H
OH TBSCl, NEt3, DMAP
CH2Cl2, 94%
H
H
OTBSAc2O, pyr, DMAPporphyrin, O2, h!
CH2Cl2, 0 °C,
H
H
OTBS
O
i. CuI, MeLi, Et2O -78 °C
ii. TMSOTf, 0 °C
31 32 33
H
H
OTBS
TMSO
Pd(OAc)2, O2
DMSO, 40 °C
H
H
OTBS
O
1. NaBH4, CeCl3MeOH, 0 °C
2. MOMCl, i-Pr2NEt
75% from X
H
H
OTBS
MOMO
Co(acac)2, O2PhSiH3
THF, 64%
H
H
OTBS
MOMO
HO 1. TBSOTf, 2,6-lutidineTHF, 0 °C, 86%
2. 70% HF"pyr
H
H
OH
MOMO
TBSO 1. IBX, EtOAcreflux
2. formalin, DBU90%
H
H
O
MOMO
TBSO
HO
OH
1. TESCl, imi CH2Cl2, 93%
2. Cp2TiMe2, toluene, 94%
H
HMOMO
TBSO
TESO
OTES
HTBSO
OH
1. Jones reagentacetone, 0 °C
2. Et3N, ethyl chloroformatethen NaBH4, 0 °C
54%
O O
H
(COCl)2, DMSOCH2Cl2, - 78 °C
then NEt3, 0 °C94%
HTBSO
OO O
H
34 35 36
37 38
3940
41 42
7
kinetic resolution protocol developed in Stoltz’s lab. Notably, either enantiomer could be
accessed from this method.
Scheme 6: Stoltz’s synthesis of enantiomeric alcohols (47) and (48).
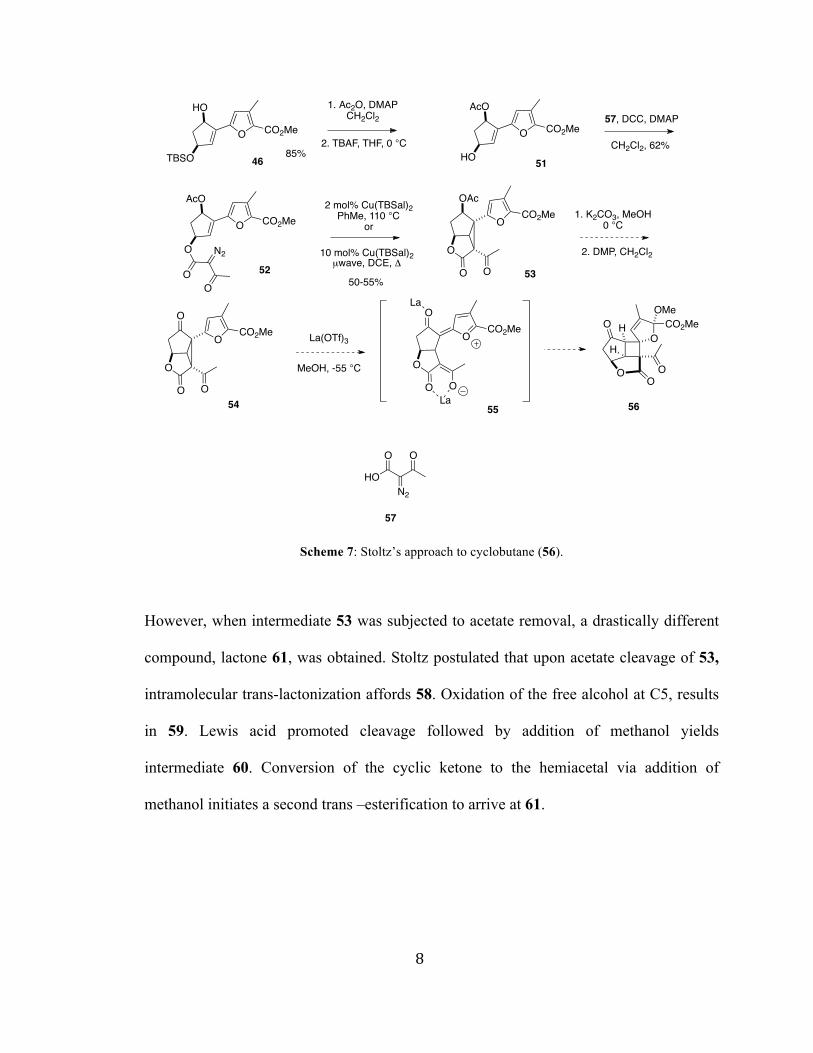
Furan 46 was then acylated and coupled with a novel diazoacetoacetic acid reagent.
Cyclopropanation with Cu(TBSal)2 afforded 53. They envisioned acetate removal,
followed by oxidation with Dess Martin periodinane would arrive at key intermediate 54,
which would undergo furan-assisted cyclopropane cleavage and subsequent Michael
addition yielding the substituted cyclobutane.
OBr CO2Me
1. i-PrMgCl, -35 °C2. B(OMe)3, -35 °C
3. 1 M HCl, THF, 0 °CO(HO)2B CO2Me
50, Ag2O, 8 mol% AsPh35 mol% PdCl2(PhCN)2
THF/ H2O85% over 2 steps43 44
O
O
TBSO
CO2Me
CeCl3! 7 H2O"aBH4
MeOH/ CH2Cl2, -25 °C10:1 syn:anti
O
TBSO
CO2Me
HO 5 mol% Pd(nbd)Cl220 mol% (-)-sparteine
MS3A, O2 (1 atm)PhMe, 80 °C
(±)-
O
TBSO
CO2Me
> 95% ee
HO
O
TBSO
CO2Me
OCeCl3! 7 H2O
"aBH4
O
TBSO
CO2Me
HO
I
TBSO
O
50
45 46
47 48 49
8
Scheme 7: Stoltz’s approach to cyclobutane (56).
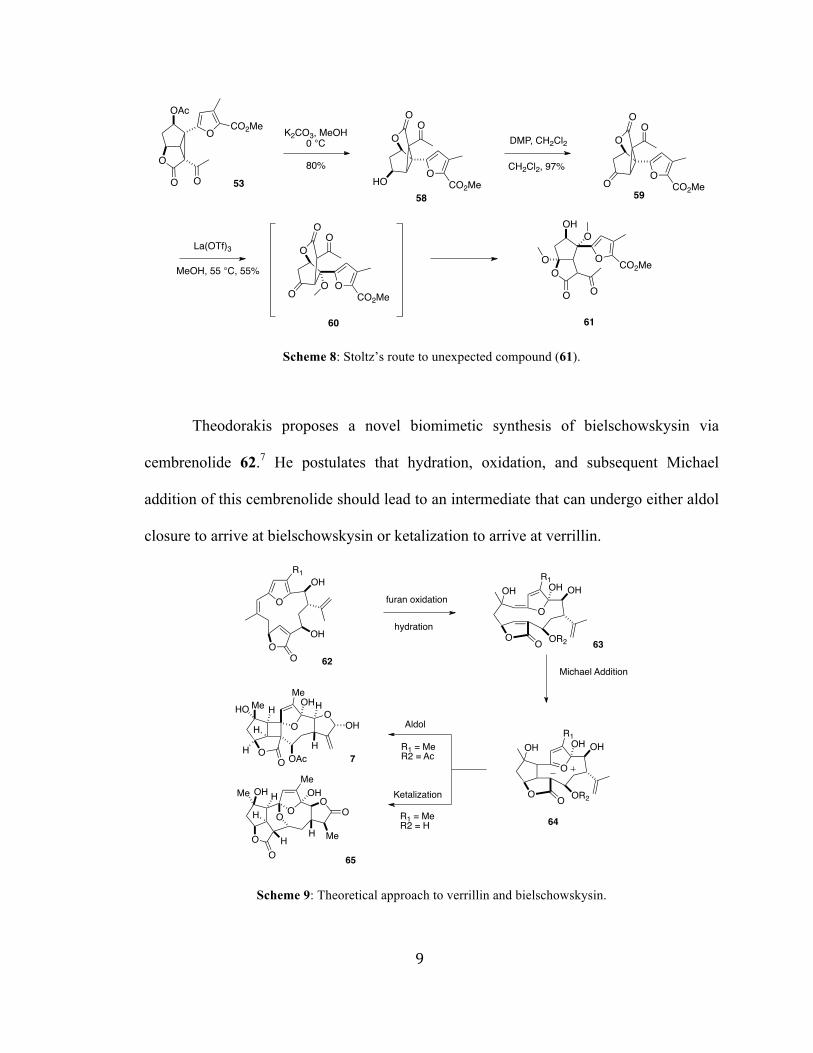
However, when intermediate 53 was subjected to acetate removal, a drastically different
compound, lactone 61, was obtained. Stoltz postulated that upon acetate cleavage of 53,
intramolecular trans-lactonization affords 58. Oxidation of the free alcohol at C5, results
in 59. Lewis acid promoted cleavage followed by addition of methanol yields
intermediate 60. Conversion of the cyclic ketone to the hemiacetal via addition of
methanol initiates a second trans –esterification to arrive at 61.
1. Ac2O, DMAPCH2Cl2
2. TBAF, THF, 0 °CO
TBSO
CO2Me
HO
85%
O
HO
CO2Me
AcO57, DCC, DMAP
CH2Cl2, 62%
O
O
CO2Me
AcO
O
N2
O
2 mol% Cu(TBSal)2PhMe, 110 °C
or
10 mol% Cu(TBSal)2µwave, DCE, !
50-55%
O
OAc
O
OCO2Me
O
1. K2CO3, MeOH0 °C
2. DMP, CH2Cl2
46 51
52 53
HON2
O O
O
O
OCO2Me
O
O
La(OTf)3
MeOH, -55 °C O
O
OCO2Me
O
OLa
La
OO
O
OCO2Me
OMeO H
H
57
54 55 56
9
Scheme 8: Stoltz’s route to unexpected compound (61).
Theodorakis proposes a novel biomimetic synthesis of bielschowskysin via
cembrenolide 62.7 He postulates that hydration, oxidation, and subsequent Michael
addition of this cembrenolide should lead to an intermediate that can undergo either aldol
closure to arrive at bielschowskysin or ketalization to arrive at verrillin.
Scheme 9: Theoretical approach to verrillin and bielschowskysin.
O
OAc
O
OCO2Me
O
K2CO3, MeOH0 °C DMP, CH2Cl2
53
80%HO
OO
O
OCO2Me
CH2Cl2, 97%
OO
O
OCO2MeO
MeOH, 55 °C, 55%
La(OTf)3
O
OH
O
O O
O
O CO2Me
58 59
61
OO
O
OCO2MeO
60
O
O
R1
OO
OH
OH
furan oxidation
hydrationO O
O
OHOHR1
OH
Michael Addition
O O
O
OHOHR1
OH
OR2
OR2
Aldol
Ketalization
R1 = Me R2 = Ac
R1 = Me R2 = H
O
OAcOO
HO Me
H
H
H
Me
OOH
H
OHH
O
O
OO
O
Me OH H
H
H
MeOH
O
MeH
65
62
63
64
7

10
The C7-C12 fragment of the cembrenolide was synthesized from butynol 66.
Conversion to ester 67 was accomplished in three steps. Copper mediated reduction of
the alkyne followed by acid-induced cyclization afforded lactone 69. Treatment with
LHMDS and PhSeBr effected selenation at the alpha position.
Scheme 10: Theodorakis’ synthesis of the C7–C12 fragment.
Methyl zirconation and iodination of propargyl alcohol followed by silylation
yielded vinyl iodide 72. Lithium halogen exchange and addition of oxirane resulted in
homoallylic alcohol 73 which was further oxidized to the aldehyde completing the C1 –
C13 fragment of the cembrenolide intermediate.
Scheme 11: Theodorakis’ synthesis of the C1–C13.
Coupling of the two fragments and oxidation/ elimination of the selenide moiety
proceeded to give butenolide 75 as a mixture of diastereomers. Silyl protection followed
OH
i
HOCO2Et
i
HOEtO
O
OO
I
OO
I
SePh
NaBH4, CuI
- 78 °C
TsOH
benzene79% over 2 steps
LHMDS, PhSeBr
- 78 °C, 76%
1. Me3Al, Cp2ZrCl2 then I22. DMP, NaHCO3
3. CO2EtLDA, THF
6667
68
6970
HO
i. Me3Al, Cp2ZrCl2 then I2
ii. TBSCl, imi50% over 2 steps
TBSOI
t-BuLi
then OTBSO OH
55%71
72 73
DMP, NHCO3
95% TBSO O
74

11
by selective deprotection gave primary alcohol 76. Coupling of fufural 80 and iodide 77
was accomplished via a modified Stille. Appel reaction of the allylic alcohol resulted in
formation of the bromide, which underwent NHK cyclization to macrocycle 79.
However, attempted oxidation of the furan moiety with CAN led to a complex mixture of
products rather than enol ether 63.
Scheme 12: Theodorakis’ completion of route to common intermediate (79)
.
OO
I
SePhLHMDS, - 78 °C
TBSO O
74 70 OO
I
SePhOH
TBSOH2O2
THF/ EtOAc
78% over 2 steps
75
OO
I
SePhOH
TBSO
i. TBSCl, DCM
ii. PPTS, EtOH82% over 2 steps O
O
I
SePhOTBS
HO
7776
80, Pd(PPh3)4Cui
DMF
OO
SePhOTBS
HOO
CHO
1. NBS, PPh3
2. Cr2Cl2NiCl2(DME)
O
OO
OH
79
OTBS
78
OH
OMe3Sn
80
CANX complex
mixture

12
CHAPTER II
CURRENT PROGRESS
Sulikowski Route
Initially, efforts toward the molecule centered on construction of the tetracyclic
core. With this strategy in mind, bis-butenolide 82 was envisioned as a precursor to
cyclobutane 81 via a [2+2] photocycloaddition. The required bis-butenolide could
ultimately be derived from (-)-malic acid.
Scheme 13: Retrosynthetic analysis of bielschowskysin (7).
In a 2006 manuscript, Doroh and Sulikowski disclosed the synthesis of
intermediate 82 and its subsequent photocyclization to cyclobutane 81.8 Irradiation of an
acetone solution of bis-butenolide 9 resulted in a 5:1 mixture of [2+2] photoadducts with
the major product having the desired stereochemistry. Stereochemical assignments were
confirmed by NOE and single-crystal x-ray analysis.
With an established protocol for the [2+2] photocycloddition, the group’s focus
shifted to synthesis of an α–substituted butenolide which would provide a handle for
elaboration of the eastern portion of the molecule. After several failed attempts to install
O
OAcOO
HO Me
H
H
H
Me
OOH
H
OHH
OO
HO Me
H
H
HO
O
Me
7
HO Me OO
MeO
O
81
O
HOO
OH
OH
882

13
the substituted butenolide directly, the group resorted to Grieco’s method of selenation-
oxidation, accessing this moiety through lactone 85.9
In a revised synthetic analysis, it was envisioned that formation of the macrocycle
would arise from an intramolecular vinylogous aldol reaction of 83. Aldehyde 84 would
enable installation of the required butenolide for this key reaction. The
photocycloaddition precursor could be accessed from lactone 85, which could be derived
from epoxide 86. As before (-)-malic acid would serve as the starting point of the
synthesis.
Scheme 14: Revised synthetic analysis of bielschowskysin (7).
Dr. Steve Townsend developed the route to advanced intermediate 84. At the
outset of the synthesis, (-)-malic acid was converted to methyl ester 87 in three steps via
a protocol reported by Saito.8, 10 Formation of the Weinreb amide and subsequent reaction
with methyl Grignard generated ketone 11.11 Chelation-controlled addition of ethynyl
magnesium bromide resulted in a 6:1 mixture of tertiary alcohols 12 and 88, with the
major product having the desired 1,3 anti-oxygenated relationship observed in
bielschowskysin.12, 13 Subsequent benzyl protection of the free alcohol gave benzyl ether
O
OAcOO
HO Me
H
H
H
Me
OOH
H
OHH
OAcOO
HO Me
H
H
H
CHOO
O
Me
HO Me OO
MeO
O
O
HOO
OH
OH
783 84
858
HO MeO
86
OAcOO
BnO Me
H
H
HO
O
Me
O
CO2MeO

14
89. Exposure to acid liberated the diol, which upon addition to a slurry of sodium hydride
and tosyl chloride resulted in concomitant formation of epoxide 8614, 15 The action of
ynamine 91 on the epoxide followed by desilylation gave lactone 90 in 88% yield.16
Scheme 15: Synthesis of lactone (90).
With formation of the lactone complete, the synthesis was directed toward
extension of the carbon chain for construction of the alkylidene butenolide. Sonogashira
coupling of alkyne 90 and vinyl iodide 97 resulted in enyne 92.17, 18 Generation of the
lithium enolate of 92 followed by addition of diphenyl diselenide resulted in 93. In the
same manner, conversion to the disubstitued lactone was achieved upon aldol reaction
with D-glyceraldehyde acetonide (98), producing the aldol adduct as a single
diastereomer. Acylation was followed by TBAF deprotection and allylic oxidation to the
aldehyde. Pinnick oxidation simultaneously resulted in conversion of the aldehyde to the
carboxylic acid and oxidative elimination of the phenyl selenoxide to give the butenolide.
OO
OMe
OO
O
Me
OO
O HO Me OO Me OH
87 11 12 88
HCCMgBri. (Meo)MeNH•HClii. MeMgBr, THF
77-91%CH2Cl286%, 6:1
BnO MeOOO BnO Me BnO MeO
O
O N TMS
91
89 8690
BnBr, NaH 1) 2N HCl, CH3CN2) TsCl, NaH, DMF
i. BF3OEt2, 91CH2Cl2
ii. aq. KHF2, CH3CN
DMF85 - 95% 90%
78-88%

15
Completion of photocycloadditon precursor 96 was realized upon silver catalyzed
cycloisomerization of acid 95.19, 20
Scheme 16: Synthesis of bis-butenolide (96).
Using the previously established protocol for the [2+2] photocycloaddition,
irradiation of bis-butenolide 96 in acetone afforded cyclobutane 99 as a single isomer.8
Subsequent TFA removal of the acetonide and lead tetraacetate cleavage of the resultant
diol provided aldehyde 84. With the aldehyde in place, our focus shifted to
functionalization of the eastern portion of the molecule and pursuit of an end-game
strategy for bielschowskysin.
PhSe
BnO MeOO BnO MeO
O
Me
OTBS
90 92
97, CuI, Pd(PPh3)4
NEt3, CH3CN
i. LDA, THF
1) LHMDS, 98
2) Ac2O, DMAP pyr, CH2Cl2
ii. PhSe2, HMPA
BnO MeOO
Me
OTBS
93
BnO MeOO
Me
OTBS
PhSe
AcO
OO
94
85-91% 80-91%
76-80%
> 95%
BnO MeOO
Me
OAcO
OO
HO BnO Me OO
MeO
O
AcO
O O95 96
I OTBS97
O
HOO
98
1) TBAF, THF, 70%
3) NaOCl2, 85%
AgNO3aq. MeOH
2) MnO2, 90% 92%

16
Scheme 17: Synthesis of advanced intermediate (84).
Model System
To complete our synthetic vision of a vinylogous aldol to install the C2, C3 bond,
a final butenolide must be constructed from aldehyde 12. In the interest of conserving
valuable material, a model system was adopted to develop methodology before applying
it to any advanced intermediates.
Octenol, 101, serves as the starting point of our exploratory synthesis. The alcohol
was smoothly converted to ester 102. Lemieux Johnson oxidation resulted in aldehyde
104 in modest yield.21 The aldehyde was also obtained via a separate dihydroxylation/
sodium periodate cleavage pathway, a route more consistent with the reactions to be
applied to the actual system.22 At this point the model system resembles C13 and C14 of
intermediate 84.
BnO Me OO
MeO
O
AcO
O O96
hvacetone86-95%
OAcOO
BnO Me
H
H
HO
O
Me
OO
99
OAcOO
BnO Me
H
H
HO
O
Me
OO
OAcOO
BnO Me
H
H
HO
O
Me
OHOH
1313OAcO
O
BnO Me
H
H
H
CHO1414OO
Me
99100 84
TFACHCl3
Pb(OAc)4EtOAc
70% 62%

17
Scheme 18: Synthesis of aldehyde (104).
Wittig olefination with triphenyl phosphorane 105 afforded homologated
aldehyde 106. Sodium borohydride reduction resulted in allylic alcohol 107, an
important intermediate allowing for synthetic divergence within the model system.
Transformation of 107 to bromoacetal 108 was accomplished with NBS and neat ethyl
vinyl ether. Employing a protocol established by Stork, radical cyclization of the
bromoacetal was observed upon reflux with Bu3SnH and AIBN in benzene for five
hours.23 Jones’ oxidation of the lactol yielded lactone 110. Subsequent enolization and
reaction with Mander’s reagent provided the substituted lactone. Presumably, selenation
followed by oxidation elimination would provide butenolide 112. Satisfied with the
preliminary findings of the model system, we shifted our efforts toward scale-up of
material and application of the newly explored chemistry to the front end.
OH
1014
Ac2O, DMAPpyr, CH2Cl2
(91%)
OAc
1024
OsO4, NaIO4THF50%
OOAc
1044
OsO4, NMOacetone, H2O
57 ! 81%O
OAc
1044
NaIO4THF/ H2O
82%
OAc
103
4OH
OH
OAc
107
4OH
ONBS78%
OAc
108
4O OEt
Br
Bu3SnH, AIBN
benzene, 80°C75%
OAc
109
4
OOEt
OOAc
1044
Ph3PCHO
105CH2Cl2
87%
OAc
106
4 CHO EtOH78%
NaBH4

18
Scheme 19: Synthesis of butenolide (112).
Aldehyde 84 was converted to α,β-unsaturated aldehyde 113 without incident.
However, attempted reduction to the allylic alcohol resulted in decomposition.
Scheme 20: Application of model system chemistry to front end.
Future Plans
Once intermediate 84 is advanced to butenolide 83, we will turn our attention to
the completion of the molecule. We propose deprotonation at C2 will lead to a
vinylogous aldol reaction resulting in formation of macrocycle 115. DIBAL reduction
and subsequent hydrogenation is expected to provide lactol 116. Tosylation of the free
primary alcohol followed by Mitsunobu reaction should correct the stereochemistry at
C16. Subsequent reaction of 117 with p-toluensulfonyl hydrazide is expected to install
Jones OAc
110
4
OO
LHMDS OAc
111
4
OO
CO2Me
1) LHMDS, Ph2Se2
2) [O]
OAc
112
4
OO
CO2Me67% CNCO2Me61%
OAcOO
BnO Me
H
H
HO
O
Me
OO
OAcOO
BnO Me
H
H
HO
O
Me
OHOH
OAcOO
BnO Me
H
H
H
CHOO
O
Me
TFA
CHCl3
Pb(OAc)4
EtOAc
DCM
OAcOO
BnO Me
H
H
HO
O
Me
CHO
Ph3PCHO
NaBH4
EtOH
OAcOO
BnO Me
H
H
HO
O
Me
OH
70% 62%
58%
x
113
99 100 84
114
19
the exocyclic olefin and trans ring juncture simultaneously via an intermediate diazene.24
Finally benzoate removal will result in bielschowskysin, 7.
Scheme 21: Bielschowskysin end-game strategy.
In an alternate strategy, we envision advancing bis-butenolide 96 to tris-
butenolide 119 via the model system chemistry and then attempting the
photocycloaddition.
Scheme 22: Proposed conversion to tris-butenolide 119.
O OH
83
base O
OAcOO
BnO Me
H
H
H
Me
O
CO2Me
OHH
115
1) DIBAL
2) H2, Pd(OH)2
O
OAcOO
HO Me
H
H
H
Me
OOHH
116
OHOAcOO
BnO Me
H
H
HO
O
Me
O
CO2MeO
1) TsCl, pyr2) PPh3, DEAD PhCOOH
OBzO
OAcOO
HO Me
H
H
H
Me
OOHH
117
OTs
NH2NHTsbase ORO
OAcOO
HO Me
H
H
H
Me
OOHH
H
118 R = Bzbielschowskysin (7, R = H)
BnO Me OO
MeO
O
AcO
O O
BnO Me OO
MeO
O
AcO
OMeO2C
O
OAcOO
BnO Me
H
H
HO
O
Me
O
O
hv
MeO2C
1199683

20
Experimental Methods
General. All non-aqueous reactions were performed under an argon atmosphere in oven-
dried glassware. Reagents were purchased at the highest commercial quality and used
without further purification, unless otherwise stated. Diethyl ether (Et2O), acetonitrile
(CH3CN), dichloromethane (CH2Cl2), and dimethylformamide (DMF) were obtained by
passing commercially available formulations through activated alumina columns
(MBraun MB-SPS solvent system). Tetrahydrofuran (THF) was obtained by distillation
from benzophenone-sodium. Triethylamine (Et3N) and diisopropylamine were distilled
from calcium hydride and stored over potassium hydroxide. Reactions were monitored by
thin-layer chromatography (TLC) using E. Merck precoated silica gel 60 F254 plates.
Visualization was accomplished with UV light and aqueous stain followed by charring on
a hot plate. Flash chromatography was conducted using the indicated solvents and silica
gel (230-400 mesh). Yields refer to chromatographically and spectroscopically
homogenous materials. 1H and 13C NMR spectra were recorded on a Bruker 400 MHz
spectrometer and are reported relative to deuterated solvent signals (7.26 and 77.2).
Preparative Procedures
To a solution of alcohol 101 (1.0 eq, 9.5 g, 74.1 mmol) in CH2Cl2 (120
mL) at ambient temperature was added pyridine (100 mL), acetic
anhydride (60 mL), and DMAP (1 crystal). The reaction was stirred 1 h, washed with 1 N
HCl (3 x 50 mL), brine (1 x50 mL), dried (MgSO4), filtered, and concentrated in vacuo.
OAc
102

21
The crude residue was purified by distillation (72-74 °C) to provide acetate 102 (12.0 g,
70.0 mmol, 95%) as colorless oil. Spectral data was consistent with reported values.25
To a solution of acetate 102 (1.0 eq, 3.0 g, 17.6 mmol) in THF (90 mL)
at ambient temperature was added OsO4 (1 crystal) and sodium
periodate (1.5 eq, 5.7 g, 26.5 mmol) as a slurry in water (25 mL). The reaction was stirred
4 h, diluted with Et2O (100 mL) and washed with saturated sodium bicarbonate (3 x 50
mL), brine (1 x 50 mL), dried (MgSO4), filtered, and concentrated in vacuo. The crude
residue was purified by column chromatography (7:1 hexanes/ ethyl acetate) to provide
aldehyde 104 (1.84 g, 10.7 mmol, 61%) as a colorless oil. Spectral data was consistent
with reported values. 25
To a solution of aldehyde 104 (1.0 eq, 900 mg, 5.2 mmol) in
CH2Cl2 (50 mL) at ambient temperature was added
formylmethylene triphenylphosphorane (1.0 eq, 1.6 g, 5.2 mmol). After warming to
reflux, the reaction was stirred 3 h. Celite (1 g) was added and the reaction was
concentrated in vacuo. The crude residue was purified by column chromatography (4:1
hexanes/ ethyl acetate) to provide aldehyde 106 (680 mg, 3.4 mmol, 66%) as a pale
yellow oil. 1H NMR (400 MHz CDCl3): δ 9.7 (d, J = 7.6 Hz, 1H), 6.75 (dd, J = 15.6, 4.8
Hz, 1 H), 6.21 (dd, J = 15.6, 7.6 Hz, 1H), 5.52 (dd, J = 6.8, 5.2, 1H), 2.13 (s, 3H), 1.75-
1.69 (m, 2H), 1.44-1.27 (m, 6H), 0.93-0.89 (m, 3H); 13C (100 MHz, CDCl3) δ 192.9,
170.0, 153.9, 131.4, 33.5, 31.3, 24.6, 22.3, 20.9, 13.9.
OAcO
103
OAc
106
CHO

22
To a solution of aldehyde 106 (1.0 eq, 198 mg, 1 mmol) in
EtOH (10 mL) at ambient temperature was added sodium
borohydride (1,0 eq, 380 mg, 10 mmol) and the reaction was stirred 1 h. The reaction
mixture was concentrated in vacuo and the crude residue dissolved in ethyl acetate (10
mL). The organics were washed with water (1 x 10 mL), brine (1 x 10 mL), dried
(MgSO4), filtered and concentrated in vacuo. The crude alcohol was lowered to – 20 °C.
Ethyl vinyl ether (10 eq, 740 mg, 10 mmol) was added and the reaction was allowed to
stir 5 minutes. N-bromosuccinimide (1.0 eq, 178 mg, 1 mmol) was added while
maintaining an internal temperature below 0 C. After 30 min, hexanes (10 mmol) was
added and the reaction filtered through a plug of celite. The organics were washed with
1N HCl (3 x 10 mL), dried (MgSO4), filtered, and concentrated in vacuo. The crude
residue was purified by flash column chromatography (6:1 hexanes/ ethyl acetate) to
provide acetal 108 (284 mg, 0.81 mmol, 81%) as a colorless oil. 1H NMR (400 MHz,
CDCl3) δ 5.83-5.68 (m, 2H), 5.27 (dd, J = 6.8, 6.4 Hz, 1 H), 4.71 (t, J = 5.6 Hz, 1H),
4.19-4.07 (m, 2H), 3.74-3.56 (m, 2H), 3.38 (d, J = 5.6 Hz, 1H), 2.06 (s, 3H), 1.66-1.54
(m, 2H), 1.30-1.21 (m, 6H), 0.91-0.88 (m, 3H); 13C (100 MHz, CDCl3) δ 170.3, 131.4,
128.3, 100.9, 73.9, 66.3, 62.4, 34.2, 31.6, 31.4, 24.7, 22.4, 21.2, 15.1, 13.9.
To a solution of bromoacetal 108 (1.0 eq, 40 mg, 0.11 mmol) in
benzene (5 mL) was added tributyl tin hydride (1.1 eq, 30 µL, 0.12
mmol). The solution was degassed and then AIBN (1 crystal) was added. Heated to reflux
(80 °C). After 3 hours, concentrated in vacuo. The residue was dissolved in Et2O (5 ml),
washed 2 x 5 mL 10% KF, dried (MgSO4), filtered through Celite, and concentrated in
OAc
108
O OEt
Br
OAc
109
4
OOEt

23
vacuo. The crude residue was purified by column chromatography (8:1 hexanes/ ethyl
acetate) to provide lactol 109 (23 mg, 0.08 mmol, 75%) as a colorless oil. 1H NMR (400
MHz, CDCl3) δ 5.29 (s, 1H), 5.12-5.08 (m, 1H), 4.91-4.83 (m, 1H), 3.78 (dt, J = 24, 8
Hz, 1H), 3.72 (ddd, J = 8, 7, 1 Hz, 1H), 3.47 (m, 2H), 2.35-2.15 (m, 2H), 2.04 (s, 3H),
1.73-1.60 (m, 2H), 1.49-1.43 (m, 4H), 1.30-1.19 (m, 6H), 0,9-0.86 (m, 3H) 13C (100
MHz, CDCl3) δ 170.7, 104.1, 73.4, 73.2, 71.6, 71.5, 39.3, 39.1, 37.3, 37.2, 35.4, 31.2,
24.8, 22.4, 15.1, 13.9.
To a solution of lactol 109 (1.0 eq, 80 mg, 0.30 mmol) in acetone (3
mL) added 1 mL of a solution of Jones reagent (1:9 Jones reagent:
acetone). The reaction was monitored by TLC. After 30 minutes, the reaction was
quenched with ethanol (1 mL). The reaction was diluted with ether (5 mL) and washed
with H2O (1 x 5 mL), sodium bicarbonate (1 x 5 mL), and brine (1 x 5 mL). The organics
were dried (MgSO4) and filtered. The crude residue was purified by column
chromatography (4:1 hexanes/ ethyl acetate) to provide lactone 110 (50 mg, 0.20 mmol,
71%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.90 (m, 1H), 4,42 (m, 1H), 3.93
(dd, J = 16, 8 Hz, 1H), 2.70-2.55 (m, 2H), 2.21 (dd, J = 16, 8 Hz, 2H), 2.06 (s, 3H), 1.73
(m, 2H), 1.58-1.53 (m, 3H), 1.27 (m, 6H), 0.90-0.87 (m, 3H) 13C (100 MHz, CDCl3) δ
176.7, 170.7, 73.2, 72.9, 72.4, 72.1, 37.4, 32.8, 31.5, 24.8, 22.4, 21.1, 13.9.
To a freshly prepared solution of LDA (1.0 eq, 0.10 mmol) in THF (1
mL) was added lactone 110 (1.0 eq, 25 mg, 0.10 mmol) at 0 °C.
Stirred at 0 °C for 1 hour, then cooled to -78 °C and added HMPA (20 µL) and methyl
OAc
110
4
OO
OAc
111
4
OO
CO2Me

24
cyanoformate (1.0 eq, 10 µL, 0.10 mmol). Stirred 30 min at -78 °C then poured reaction
into cold water (1 mL). Extracted (3 x 5 mL) Et2O, dried (MgSO4), and concentrated with
Celite. The crude residue was purified by column chromatography (4:1 hexanes/ ethyl
acetate) to provide ester 111 (18 mg, 0.07 mmol, 70%) as a colorless oil. 1H NMR (400
MHz, CDCl3) δ 4.69 (m, 1H), 4.46 (t, J = 8 Hz, 1H), 3.94 (t, J = 8 Hz, 1 H), 3.78 (s, 3H),
3.47 (d, J = 8 Hz, IH), 3.15 (dd, J = 16, 8 Hz, 1H), 2.43 (s, 3H), 1.78 – 1.74 (m, 2H),
1.35-1.20 (m, 6H), 0.90-0.88 (m, 3H).
To a solution of acetonide 99 (1.0 eq, 5 mg, 10 µmol) in CDCl3 (600
µL) in an NMR tube was added 1 drop of trifluoroacetic acid.
Reaction progress was monitored by NMR. After 1 hour, the reaction
was concentrated in vacuo and the crude residue was purified by column chromatography
(1:1 hexanes/ ethyl acetate) to provide diol 100 (2 mg, 4.0 µmol, 50%) as a white solid.
1H NMR (400 MHz, CDCl3) δ 7.37-7.31 (m, 5H), 6.95 (d, J = 1.5 Hz, 1H), 5.69 (d, J = 4,
1 Hz, 1H), 5.33 (td, J = 11.4, 4 Hz, 1H), 4.37 (dd, J = 18, 11.2 Hz, 2H), 4.14 (dd, J = 14,
6.7 Hz, 1H), 3.95 (m, 1H), 3.68-3.63 (m, 2H), 3.55 (dd, J = 7.8, 1.5 Hz, 1H), 3.35 (t, J =
8 Hz, 1H), 3.15 (t, J = 6 Hz, 1H), 2.90 (ddd, J = 10, 8, 1.8 Hz, 1H), 2.15 (s, 3H), 2.05 (d,
J = 1.8 Hz, 1H), 1.95 (s, 3H), 1.34 (s, 3H).
To a solution of diol 100 (1.0 eq, 3 mg, 6.0 µmol) in ethyl acetate (500
µL) at ambient temperature was added lead tetraacetate (3 mg). After 30
min, filtered reaction through a plug of celite and obtained aldehyde 84
(2 mg, 4 µmol, 75%) as a pure white solid. 1H NMR (400 MHz, CDCl3) δ 9.43 (s, 1H),
OAcOO
BnO Me
H
H
HO
O
Me
OHOH
100
OAcOO
BnO Me
H
H
H
CHOO
O
Me
84

25
7.38-7.31 (m, 5H), 7.02 (d, J = 1.4 Hz, 1H), 5.76 (s, 1H), 5.35 m, 1H), 4.37 (dd, J = 19,
11.4 Hz, 2H), 4.14 (dd, J = 14, 7 Hz, 1H), 3.59-3.51 (m, 2H), 2.90 (ddd, J = 10, 8, 2 Hz,
1H), 2.23 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H).
To a solution of aldehyde 84 (1.0 eq, 3 mg, 7.0 µmol) in CH2Cl2 (1
mL) was added formylmethylene triphenylphosphorane (1.0 eq, 2 mg,
7.0 µmol). The reaction was heated to reflux (40 °C). After 2 hours,
concentrated reaction with Celite and purified residue by column chromatography (1:1
hexanes/ ethyl acetate) to provide aldehyde 113 (2 mg, 4.0 µmol, 61%) as a white solid.
1H NMR (400 MHz, CDCl3) δ 9.53 (d, J = 7.6 Hz, 1H), 7.38-7.31 (m, 5H), 7.02 (d, J =
1.4 Hz, 1H), 6.73 (dd, J = 15, 6 Hz, 1H), 6.29 (dd J = 15.7, 6.7 Hz, 1H), 6.1 (d, J = 5.1
Hz, 1H), 5.29-5.25 (m, 1H), 4.35 (dd, J = 20, 11.3 Hz, 1H), 2.87 (dd, J = 16, 8 Hz, 1H),
2.16 (s, 3H), 1.91 (s, 3H), 1.5 (s, 3H).
OAcOO
BnO Me
H
H
HO
O
Me
CHO
113

26
Appendix A:
Spectra of Relevant Compounds
27
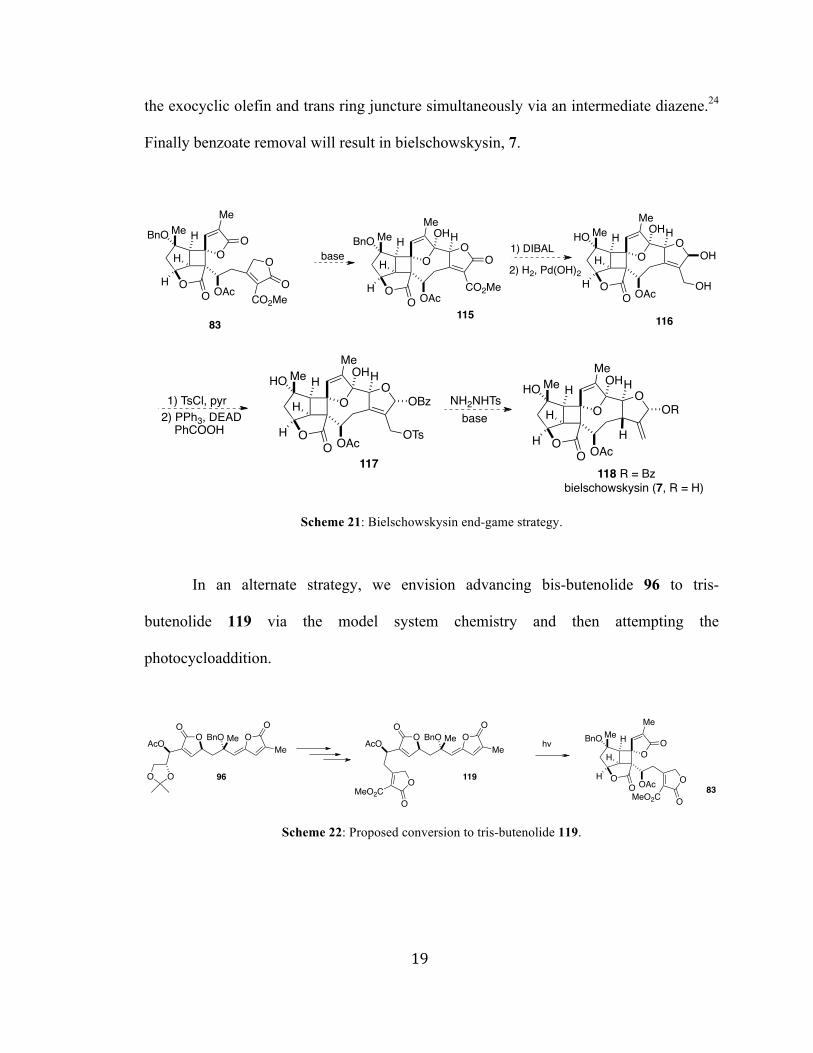
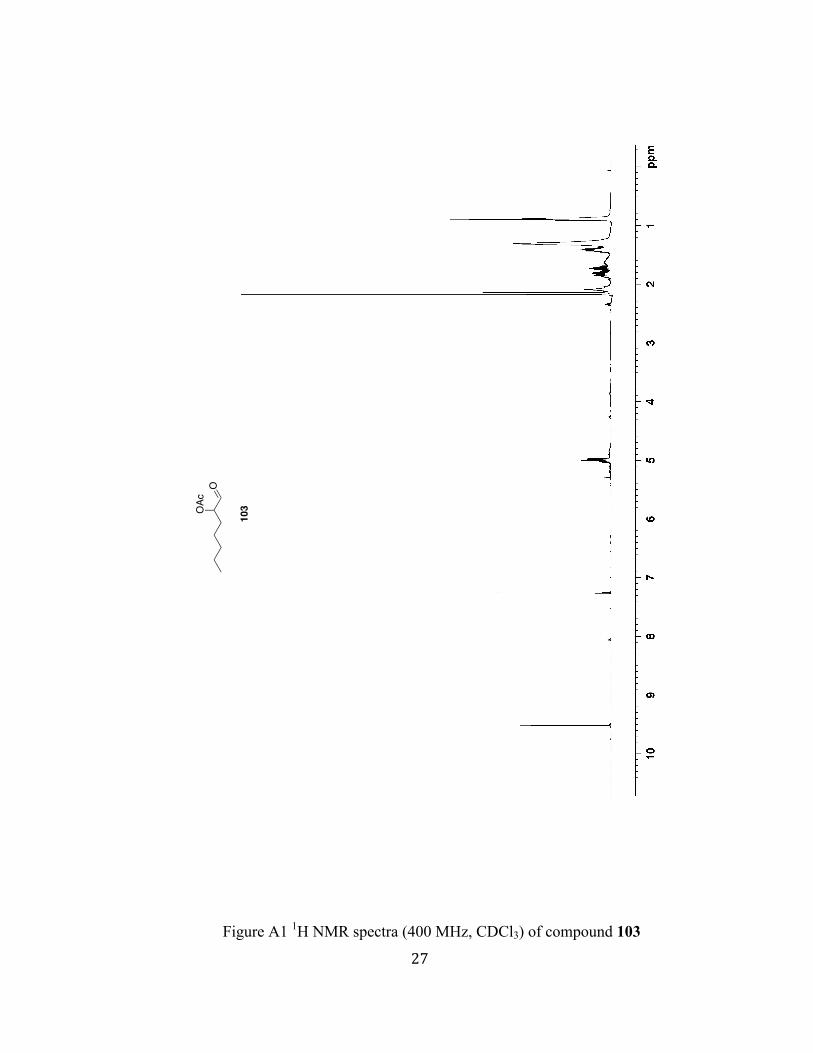
Figure A1 1H NMR spectra (400 MHz, CDCl3) of compound 103
OAc
O
103

28
Figure A2 13C NMR spectra (100 MHz, CDCl3) of compound 103
OAc
O
103
29
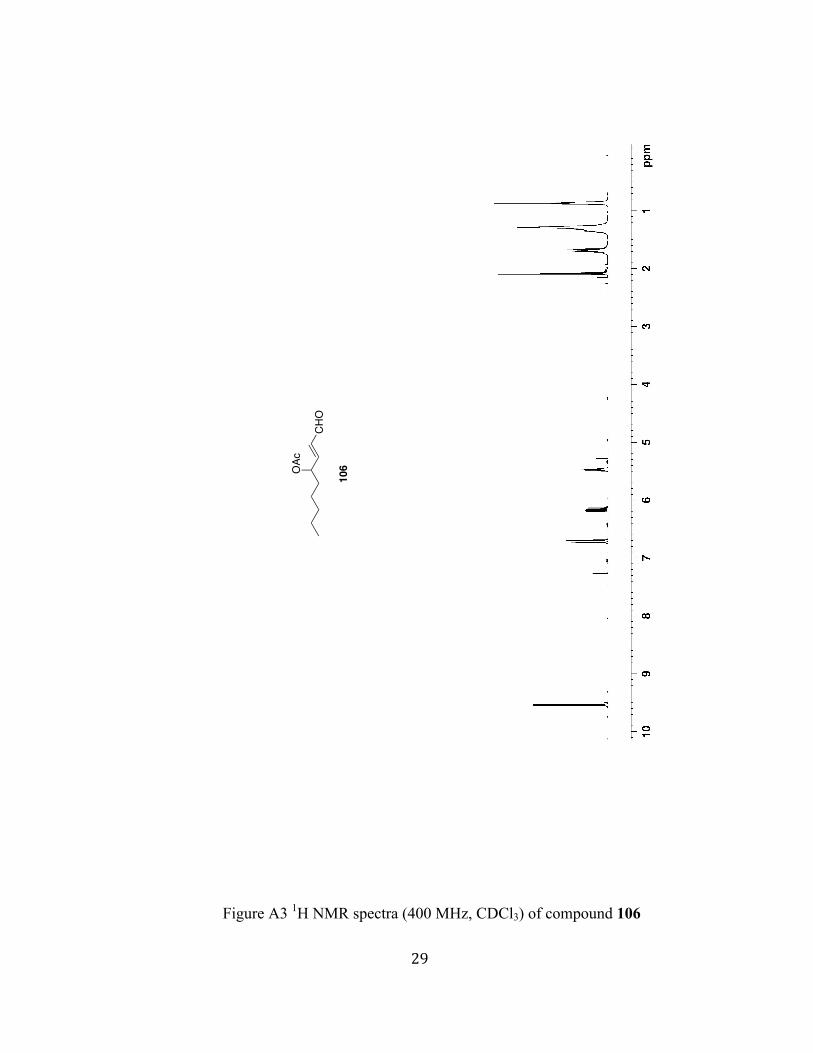
Figure A3 1H NMR spectra (400 MHz, CDCl3) of compound 106
OAc
106
CHO

30
Figure A4 13C NMR spectra (100 MHz, CDCl3) of compound 106
OAc
106
CHO

31
Figure A5 1H NMR spectra (400 MHz, CDCl3) of compound 108
OAc
108
OOEt
Br

32
Figure A6 13C NMR spectra (100 MHz, CDCl3) of compound 108
OAc
108
OOEt
Br

33
Figure A7 1H NMR spectra (400 MHz, CDCl3) of compound 109
OAc
109
4
OOEt

34
Figure A8 13C NMR spectra (100 MHz, CDCl3) of compound 109
OAc
109
4
OOEt
35
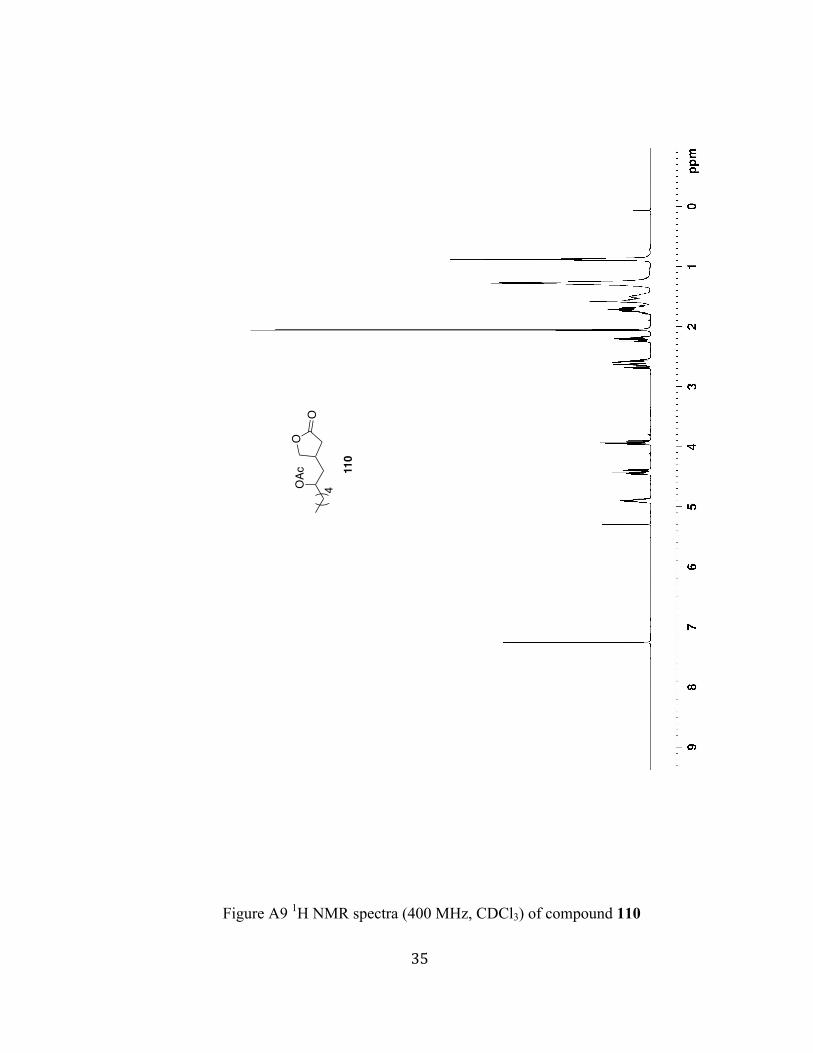
Figure A9 1H NMR spectra (400 MHz, CDCl3) of compound 110
OAc 11
0
4
OO

36
Figure A10 13C NMR spectra (100 MHz, CDCl3) of compound 110
OAc 11
0
4
OO
37
Figure A11 1H NMR spectra (400 MHz, CDCl3) of compound 111
OAc 11
1
4
OO
CO2Me
38
Figure A12 1H NMR spectra (400 MHz, CDCl3) of compound 100
OAc
OO
BnOMe
H
H
HO
O
Me
OH
OH
100
39
Figure A13 1H NMR spectra (400 MHz, CDCl3) of compound 84
OAc
OO
BnOMe
H
H
H
CHO
OO
Me
84
40
Figure A14 1H NMR spectra (400 MHz, CDCl3) of compound 113
OAc
OO
BnOMe
H
H
HO
O
Me
CHO
113

41
REFERENCES
1. Marrero, J.; Rodriguez, A. D.; Baran, P.; Raptis, R. G.; Sanchez, J. A.; Barria, E.;
Capson, T, L. Org. Lett. 2004, 6, 1661–1664.
2. Roethle, P.A.; Trauner, D. Nat. Prod. Rep. 2008, 25, 298– 317.
3. Miao, R.; Gramani, S. G.; Lear, M. J. Tet. Lett. 2009, 50, 1731–1733.
4. Nicolaou, K. C.; Adsool, V. A.; Hale, C. R. Angew. Chem. Int. Ed. 2011, 50, 5149–5142.
5. Farcet, J.; Himmelbauer, M.; Mulzer, J. Org. Lett. 2012, 14, 2195–2197.
6. Meyer, M. E.; Phillips, J. H.; Ferreira, E. M.; Stoltz, B. M. Tetrahedron 2013,
http://dx.doi.org/10.1016/j.tet.2013.02.034
7. Saitman, A.; Sullivan, S. D.; Theodorakis, E. A. Tet. Lett. 2013, 54, 1612–1615.
8. Doroh, B.; Sulikowski, G. A. Org. Lett. 2006, 8, 903–906.
9. Greico, P. A.; Miyashit, M. J. Org. Chem. 1974, 39, 120–122.
10. Saito, S.; Ishikawa, T.; Kuroda, A.; Koga, K.; Moriwake, T. Tetrahedron 1992, 48, 4067–4086.
11. Williams, J. M.; Jobson, R. B.; Yasuda, N.; Marchesini, G.; Dolling, U. H.;
Grabowski, E. J. J. Tetrahedron Lett. 1995, 36, 5461–5464.
12. Weber, B.; Seebach, D. Angew. Chem. Int. Ed. 1992, 31, 84–86.
13. Reetz, M. T.; Jung, A. J. Am. Chem. Soc. 1983, 105, 4833–4835.
14. Ohgi, T.; Kondo, T.; Goto, T. Tetraheron Lett. 1977, 46, 4051–4054.
15. Murthy, V. S.; Gaitonde, A. S.; Rao, S. P. Synthetic Commun. 1993, 23, 285–289 .
16. Movassaghi, M.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 2456–2457.
17. Duboudin, J. G.; Jousseaume, B.; Bonakdar, A. Organomet. Chem. 1979, 168, 227–232.
18. Scarlato, G. R.; DeMattei, J. A.; Chong, L. S.; Ogawa, A. K.; Lin, M. R.;
Armstrong, R. W. J. Org. Chem. 1996, 61, 6139–6152.

42
19. Jong, T. T.; Leu, S. J. J. Chem. Soc. Perk. T1 1990, 2, 423–424.
20. Xu, C. D.; Negishi, E. Tetraheron Lett. 1999, 40, 431–434.
21. Pappo, R.; Allen, D. S.; Lemieux, R. U.; Johnson, W. S. J. Org. Chem. 1956, 21, 478–479.
22. VanRheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett. 1976, 17, 1973–
1976.
23. Stork, G.; Mook, R.; Biller, S. A.; Rychnovsky, S. D. J. Am. Chem. Soc. 1983, 105, 3741–3742.
24. Meyers, A. G.; Kukkola, P. J. J. Am. Chem. Soc. 1990, 112, 8208–8210.
25. Steinfeldt, N.; Abdallah, R.; Dingerdissen, U.; Jahnisch, K. Org. Process Res.
Dev. 2007, 11, 1025–1031.





























