Embed Size (px)
Citation preview

Neuroseience Letters, 87 (1988) 23 28 23 Elsevier Scientific Publishers Ireland Ltd.
NSL 05251
Effects of D-(-)-aminophosphonovalerate on behavioral and histological changes induced by
systemic kainic acid
Wladyslaw Lason*, Jeffrey N. Simpson and Jacqueline F. McGinty
Department of Anatomy, School of Medicine, East Carolina University, Greenville, NC 27858-4354 (U.S.A.)
(Received 5 October 1987; Revised version received 22 December 1987; Accepted 22 December 1987)
Keyword~." Kainic acid; N-methyl-D-aspartate (NMDA) receptor; o-(-)-Aminophosphonovalerate (D(-)APV); Hippocampus; Seizure: Neurotoxicity
In rats, the seizures induced by systemic kainic acid (KA) are followed by extensive neuronal damage, notably in the hippocampal region. We report that the specific N-methyl-o-aspartate (NMDA) receptor antagonist, o-(-)-aminophosphonovalerate (D(-)APV), given i.c.v, prior to or 2 h after i.p. KA injection markedly protected CA 1 but not other hippocampal neurons against degeneration. In contrast, D( - )APV had no effect on KA-induced wet dog shakes or on behavioral seizures. We conclude that NMDA recep- tors participate in the neurotoxic but not in the behavioral effects of systemic KA.
Intracerebral or systemic injection of kainic acid (KA), a conformationally rigid analog of glutamate, evokes seizures which are followed by extensive nerve cell damage in the limbic system [2, 10]. For this reason both routes of KA administra- tion are widely used in experimental models of temporal lobe epilepsy. However, several observations indicate that there are important differences in the pathological manifestations of intracerebral and systemically administered KA. Intracerebral or intraventricular injection of KA (0.14).8/tg) produces neuronal loss selectively in the CA3--4 cell fields of hippocampus [11] which correlates well with the high density of KA receptors in this area [7]. In contrast, systemic KA causes seizures and extensive brain edema and the most consistent neuronal cell loss in the hippocampus is in the CA 1 region [8, 13] where there is a low density of KA receptors [7]. The CA 1 damage caused by systemic KA closely resembles that produced by cerebral ischemia [3]. Excitatory amino acid receptors which are most sensitive to N-methyl-D-aspartic acid (NMDA) are highy concentrated in CAI [9] and their activation contributes to the development of neuronal ischemic damage [14]. Thus, we hypothesized that NMDA
*Permanent address: Institute of Pharmacology, Polish Academy of Sciences, 31-343 Krakow, Poland. Correspondence." J.F. McGinty, Department of Anatomy, School of Medicine, East Carolina University, Greenville, NC 27858-4354, U.S.A.
0304-3940/88/$ 03.50 O 1988 Elsevier Scientific Publishers Ireland Ltd.

24
receptors may contribute to the neuronal loss in CA I which is induced by systemic KA. To test this hypothesis, we investigated the effects of the selective NMDA recep- tor antagonist, ~-(-) -aminophosphonovalerate (D( - )APV) , on the behavioral and histological changes that follow i.p. KA administration.
Twenty-seven male Sprague-Dawley rats (28(~300 g) were injected i.p. with 10 mg/kg of KA (Sigma) dissolved in saline and adjusted to pH 7.2_+ 0.2 with NaOH. Immediately prior to the KA injection, 21 animals of this group received an i.c.v. injection of 20/tg (n = 17) or 40/~g (n---4) of APV in 5/tl phosphate-buffered saline (PBS) (pH 7.2_+0.2) delivered via a chronically implanted cannula. The remaining 6 animals received 20/tg D ( - )APV/5 /A PBS after status epilepticus had developed (120___ 20 min after i.p. KA injection). An additional 4 rats received i.c.v. 20/~g (n = 2) or 40/~g (n = 2) of D ( - ) A P V alone. Nineteen rats which received 5/A PBS i.c.v, prior to or after i.p. KA injection served as controls. The rats were placed in individual plastic cages and were observed continuously for 3 11 after KA injection. The number of wet dog shakes, latency to the first clonic convulsion, and whether or not status epilepticus developed were recorded.
Rats brains were perfused with a buffered 4% paraformaldehyde solution 72 h after the injections. Five 50/~m frozen sections were cut at 100/~m intervals through the extent of the hippocampus and Nissl stained. The extent of neuronal damage in each hippocampal cell field on each section was evaluated under a × 4 objective by an in- vestigator (J.F.M.) who was unaware of the identity of each experimental group. The evaluation of neuronal damage was made using a 4 point scale (4 + = most severe cell loss: 1 + = least severe).
Twenty-five to 35 min after an i.p. KA injection, rats developed wet dog shakes interspersed with short periods of immobility. In all experimental groups the frequen- cy of wet dog shakes was highest between 40 and 60 min after i.p. KA injection (pla- teau period) and became less frequent when episodes of clonic convulsions (facial muscle myoclonus, forelimb clonus, 'praying' position accompanied by hypersaliva- tion) appeared. Gradually (100-140 min after KA injection), the animals developed status epilepticus characterized by constant head nodding and a 'praying' position with occasional rearing and falling. D ( - ) A P V given i.c.v, prior to a systemic KA injection did not significantly change the number of wet dog shakes (control: mean=77.0-+6.75 (S.E.M.); 20 /~g D ( - ) A P V : mean=72 .0+6 .05 (S.E.M.); 40 ~g D ( - ) A P V : mean = 60.5-+ 11.21 (S.E.M.) per 20 min plateau period). D ( - ) A P V also did not affect the latency to clonic seizures in minutes (control: mean = 73.1 + 3.52 (S.E.M.); 20 ~g D ( - ) A P V : mean=80.5-+6.02 (S.E.M.); 40 /~g D ( - ) A P V : mean = 74.8_+ 9.04 (S.E,M.)). After the 40/lg dose of D ( - ) A P V , the animals were more excited than after KA alone and showed pronounced ataxia; they frequently lost the righting reflex during wet dog shakes or during their attempts to adopt a "praying' position. The 20 pg dose of D ( - ) A P V plus KA produced only slight ataxia and no motor excitation. D ( - ) A P V (20/~g) given to animals after status epilepticus had developed did not attenuate behavioral convulsions and falling became more fre- quent. D ( - ) A P V alone (20/~g) did not evoke significant behavioral changes. After the higher dose (40 #g) of D ( - ) A P V alone, motor hyperactivity accompanied by
25
ataxia was seen dur ing the first 30 min following drug injection. Two rats which were
given vehicle i.c.v, and 4 rats which received D ( - ) A P V (20/tg) pr ior to systemic K A
injection did no t develop any of the behavioral syndromes. Five animals given vehicle
i.c.v, and 2 animals which received 20/~g D ( - ) A P V i.c.v, died between 5 and 36 h
after K A adminis t ra t ion .
Histological analysis of the brains of each of the surviving rats 72 h after K A
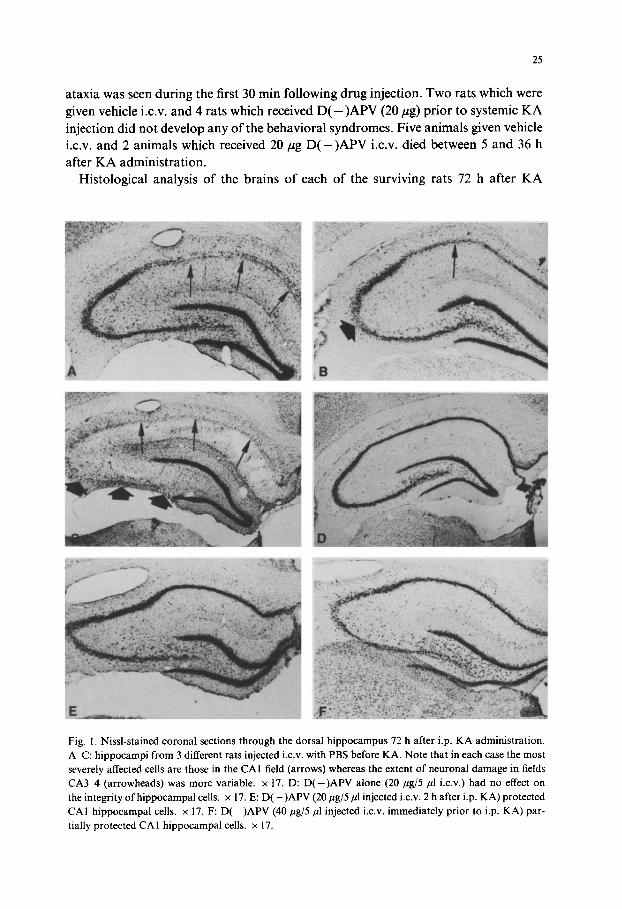
Fig. 1. Nissl-stained coronal sections through the dorsal hippocampus 72 h after i.p. KA administration. A-C: hippocampi from 3 different rats injected i.c.v, with PBS before KA. Note that in each case the most severely affected cells are those in the CA 1 field (arrows) whereas the extent of neuronal damage in fields CA3 ~, (arrowheads) was more variable, x 17. D: D(-)APV alone (20/zg/5/zl i.c.v.) had no effect on the integrity of hippocampal cells, x 17. E: D(-)APV (20/1g/5/zl injected i.c.v. 2 h after i.p. KA) protected CAI hippocampal cells. × 17. F: D(-)APV (40/tg/5/zl injected i.c.v, immediately prior to i.p. KA) par- tially protected CA1 hippocampal cells. × 17.
26
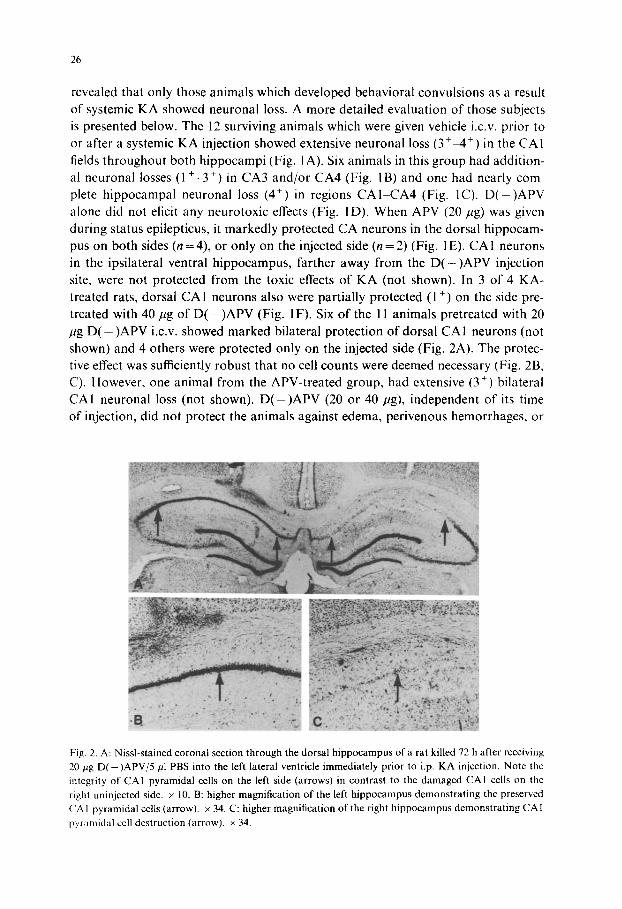
revealed that only those animals which developed behavioral convulsions as a result of systemic KA showed neuronal loss. A more detailed evaluation of those subjects is presented below. The 12 surviving animals which were given vehicle i.c.v, prior to or after a systemic KA injection showed extensive neuronal loss (3 + 4 +) in the CA1 fields throughout both hippocampi (Fig. I A). Six animals in this group had addition- al neuronal losses (1 +-3 +) in CA3 and/or CA4 (Fig. IB) and one had nearly com- plete hippocampal neuronal loss (4 +) in regions CA1 CA4 (Fig. 1C). D ( - ) A P V alone did not elicit any neurotoxic effects (Fig. I D). When APV (20/~g) was given during status epilepticus, it markedly protected CA neurons in the dorsal hippocam- pus on both sides (n = 4), or only on the injected side (n = 2) (Fig. I E). CA1 neurons in the ipsilateral ventral hippocampus, farther away from the D ( - ) A P V injection site, were not protected from the toxic effects of KA (not shown). In 3 of 4 KA- treated rats, dorsal CA1 neurons also were partially protected (1 +) on the side pre- treated with 40/tg of D ( - ) A P V (Fig. 1F). Six of the 11 animals pretreated with 20 pg D ( - ) A P V i.c.v, showed marked bilateral protection of dorsal CAI neurons (not shown) and 4 others were protected only on the injected side (Fig. 2A). The protec- tive effect was sufficiently robust that no cell counts were deemed necessary (Fig. 2B, C). However, one animal from the APV-treated group, had extensive (34) bilateral CA1 neuronal loss (not shown). D ( - ) A P V (20 or 40 pg), independent of its time of injection, did not protect the animals against edema, perivenous hemorrhages, or
Fig. 2. A: Nissl-stained coronal section through the dorsal hippocampus of a rat killed 72 h after receiving 20 pg D ( - ) A P V / 5 / t l PBS into the left lateral ventricle immediately prior to i.p. KA injection. Note the integrity of CAI pyramidal cells on the left side (arrows) in contrast to the damaged CAI cells on the right uninjected side. x 10. B: higher magnification of the left h ippocampus demonstrat ing the preserved ( IA I pyramidal cells (arrow). × 34. C: higher magnification of the right hippocampus demonstrat ing CA 1
pyramidal cell destruction (arrow), × 34.

27
neuronal loss in pyriform/entorhinal cortex resulting from systemic KA administra- tion (not shown).
Our data demonstrate that blockade of N M D A receptors by APV protects CA1 cells from marked destruction by systemic KA. Furthermore, the data indicate that N M D A receptors participate in a subspectrum of the neurotoxic but not the behav- ioral sequelae of systemic KA administration. The latter result is in agreement with a previous observation that N M D A antagonists do not prevent KA induced seizures [4]. Our histological data confirm the view that the neurons most vulnerable to syste- mic K A are those localized in the N M D A receptor-rich CA1 region of the hippocam- pus [2, 8, 13]. Although KA initiates its excitatory effects through an interaction with a KA selective receptor, it may also enhance the activation of N M D A receptors by two possible mechanisms. The first is the removal of a Mg 2+ blockade of the N M D A channel by KA-induced depolarization [5]. Electrophysiological evidence indicates that an i.c.v. KA injection leads to expression of N M D A receptor-mediated depolari- zation in response to suprathreshold orthodromic activation of CA1 pyramidal cells [1]. The second mechanism may involve an increase in the glutamate/aspartate con- centration in the vicinity of CA 1 N M D A receptors evoked by prolonged activation of CA3 Schaffer collaterals [12]. D ( - ) A P V has been reported to inhibit the K ÷-stim- ulated release of glutamate from dentate slices by a presynaptic mechanism which also requires strong depolarization for its functional activation [6]. We conclude that severe KA-induced seizures may provide the necessary conditions for a secondary N M D A activation in CAl . The prolonged seizures after systemic KA are thought to induce hypoxia which is an important factor in the pathogenic mechanism of CA1 neuronal loss [15]. In contrast, i.c.v. KA administration is less likely to produce hypoxia-related neuronal death because the injection is usually performed under long-acting anesthesia and the subsequent status epilepticus is less severe.
Interestingly, it has been demonstrated that ischemic hippocampal neuron loss evoked by bilateral carotid occlusion is effectively prevented by another selective N M D A receptor antagonist, 2-aminophosphonoheptanoic acid [14]. Although it is still unclear whether seizures or ischemia contribute more to neuronal degeneration in CA1 after systemic KA, the present data provide evidence that N M D A receptors are involved in this type of cell damage.
Supported by Grant DA 03982 (J.F.M.).
1 Ashwood, T.Y. and Wheal, H.V., Effects of D-2-amino-5-phosphonovalerate on epileptiform activity recorded from the kainic acid lesioned hippocampus, In T.P. Hicks, D. Lodge and H. McLennan (Eds.), Excitatory Amino Acid Transmission, Liss, New York, 1987, pp. 333-336.
2 Ben-Ari, Y., Limbic seizures and brain damage produced by kainic acid mechanism and relevance to human temporal lobe epilepsy, Neuroscience, 14 (1985) 375-403.
3 Brown, A.W. and Brierly, I.B., Anoxic ischaemic cell change in rat brain: light and microscopic and fine-structural observations, J. Neurol. Sci., 16 (1972) 59-84.
4 Czuczwar, S.J. and Meldrum, B., Protection against chemically induced seizures by 2-amino-7-phos- phonoheptanoic acid, Eur. J. Pharmacol., 83 (1982) 335 338.

28
5 Engberg, J., Flatman, J.A., Lambert, J.D.C. and Lindsay, A., An analysis of bioelectric phenomena evoked by microiontophoretically applied excitoxic amino-acids in the feline spinal cord. In D. Fuxe, P. Roberts and R. Schwarz (Eds.), Excitotoxins, Macmillan, London, 1983, pp. 17(~ 183.
6 Errington, M.L., Lynch, M.A. and Bliss, T.V.P., Long-term potentiation in the dentate gyrus: induc- tion and increased glutamate release are blocked by D(-)aminophosphonovalerate, Neuroscience, 20 (1987) 279-284.
7 Foster, A.C., Mena, E.E., Monaghan, D.T. and Cotman, C.W., Synaptic localization of kainic acid binding sites, Nature (Lond.), 289 (1981) 73--75.
8 Lassman, H., Petsche, V., Kitz, K., Baran, H., Sperk, G., Seitelberger, F. and Hornykiewicz, O., The role of brain edema in epileptic brain damage induced by systemic kainic acid injection, Neurosciencc, 13 (1984) 691-704.
9 Monaghan, D.T. and Cotman, C.W., Distribution of N-methyl-o-aspartate-sensitive L-[3H] glutamate- binding sites in rat brain, J. Neurosci., 5 (1984) 2909-2919.
10 Nadler. J.V., Kainic acid as a tool for the study of temporal lobe of epilepsy, Life Sci., 29 (1981) 2031 2042.
11 Nadler, J.V., Perry, B.W. and Cotman, C.W., Intraventricular kainic acid preferentially destroys hip- pocampal pyramidal ceils. Nature (Lond.), 271 (1978) 676-677.
12 Nadler, J.V., Vaca, K.W., White, W.F., Lynch, G.S. and Cotman, C.W., Aspartate and glutamate as possible transmitters of excitatory hippocampal afferents, Nature (Lond.), 306 (1983) 176 179.
13 O'Shaughnessy, D. and Gerber, G.Y., Damage induced by systemic kainic acid in rats is dependent upon seizure activity a behavioral and morphological study, Neurotoxicology, 7 (1986) 187 202.
14 Simon, R.P., Swan, J.H., Griffiths, T. and Meldrum, B.S., Blockade of N-methyl-l)-aspartate receptors may protect against ischemic damage in the brain, Science, 226 (1984) 85(~-852.
15 Sperk, G., Lassman, H., Baran, H., Kish, S.J., Seitelberger, F. and Hornykiewicz, O., Kainic acid induced seizures neurochemical and histopathological changes, Neuroscience, 10 (1983) 1301 1315.





























