Embed Size (px)
Citation preview

EDQM: 50 YEARS OF LEADERSHIP IN THE QUALITY OF MEDICINES
PAVING THE WAY FOR THE FUTURE
6-8 October 2014Strasbourg, France
WORKSHOP
QUALITY BY DESIGN
1

The Pharmacopoeia and QbD
7 October 2014
Dr. Øyvind Holte
Norwegian Medicines Agency/ EDQM PAT WP
Overview
• Quality by Design (QbD)
– High-level principles
– Role of the pharmacopoeia
• EDQM PAT working party
– Background, efforts and future prospects
• European pharmacopoeia and QbD
– General guidance in the pharmacopoeia
– Recent revisions, interpretation and practice• Ph.Eur. 2.9.47
Uniformity of dosage units using large sample sizes
• NIR, Raman, General notices2
2

Quality by DesignPrinciples
• Demonstration of product quality:
– QbD: Focus shifted from end testing of the productvia product knowledge to process control
3
End testing
Processcontrol
Enhancedproduct
knowledge
Quality assurance/ Control strategy
Quality by DesignRole of the Pharmacopoeia
• European Pharmacopoeia– Quality standards
• General dosage forms, drug substances, excipients, (drug products)
– Harmonised general methods
• QbD often involves on-line analysis during manufacture, rather than off-line end testing– Is this compatible with pharmacopoeial requirements?
• QbD is all about achieving and demonstrating therequired product quality– This is well within the scope of the Pharmacopoeia!
4
Contributing to the quality of
medicines
3

EDQM PAT working party
• Work programme
– Revision of relevant chapters
– Consider need for new chapters
• Key chapters
– Uniformity of dosage units (Ph.Eur. 2.9.40)
– Near infrared spectroscopy (Ph.Eur. 2.2.40)
– Raman spectroscopy (Ph.Eur. 2.2.48)
– Ph.Eur. 1. General notices
• Current and future status– PAT is not a stand-alone area, but should be considered within
the remit of any type of analytic methodology5
European Pharmacopoeia and QbD1. General notices
• An article is not of Pharmacopoeia quality unless it complies with all the requirements stated in the monograph
• The manufacturer may obtain assurance that a product is of Pharmacopoeia quality on the basis of its design, together with its control strategy and data derived, for example, from validation studies of the manufacturing process.
• Real-time release testing […] is not precluded by the need to comply with the Pharmacopoeia
6
4

European Pharmacopoeia and QbD5.15 Functionality-related characteristics
• FRC sections are non-mandatory
• FRCs are not exhaustive, but they are typical for the excipient:
– Particle size distribution
– Powder flow
– Bulk and tapped density
– Viscosity
– Melting point
• Knowledge of FRCs may facilitate the applicationof process analytical technology (PAT)
7
European Pharmacopoeia and QbDQbD-relevant chapter: Uniformity of dosage units
• Specific issues raised on the UDU test (2.9.40):
– Acceptance criteria directly linked to the prescribed sample size N = 30
– Rigid requirement: no single unit outside +/- 25 %.N >> 30: one or few largely deviating units is expected?
– Improved batch knowledge with large sample was not appreciated in the acceptance criteria
“The pharmacopoeia should not represent a barrier/ disincentive to the implementation of PAT/ QbD”
ksXMAV
8
5

European Pharmacopoeia and QbDQbD-relevant chapter: Uniformity of dosage unitsPh.Eur. 2.9.47
• Option 1– Parametric
Depending onsample size:
Limit for number ofunits outside(1 ± L2 x 0.01)T
• Option 2– Non-parametric
Depending onsample size:
Limit for number ofunits outside(1 ± L1 x 0.01)T
(1 ± L2 x 0.01)T
ksXMAV
9~90 % probability to pass Ph.Eur. 2.9.40 on any small sample
European Pharmacopoeia and QbDQbD-relevant chapters: NIR, Raman
• 2.2.40 Near-infrared spectroscopy
– revised in parallel with the EMA NfG on NIRs
• 2.2.48 Raman spectroscopy
• Revised to adequately describe equipmentand procedures suitable for in-line analysis
10
6

European Pharmacopoeia and QbDQbD-relevant chapter: 1. General notices
Demonstration of compliance with the Pharmacopoeia
• (2) An enhanced approach to quality control could utiliseprocess analytical technology (PAT) […] strategies as alternatives to end-product testing alone.
Real-time release testing Parametric release in circumstances deemed appropriate by the competent authority is thus not precluded by the need to comply with the Pharmacopoeia.
Ph.Eur. 8.2
11
European Pharmacopoeia and QbDSummary
Ph.Eur. provides quality standards
The manufacturer must assure his productcomplies with these standards
Ph.Eur. requirements apply regardless ofcontrol strategy
QbD/ PAT is one way of demonstrating quality
This approach is fully in line with Ph.Eur.
1. General notices
5.15 Functionality-related characteristics
12
7

Back-up slides/ References
• High-level overview of QbD and PAT analytical methods as concepts
• Brief overview of ICH Q8, Q9, Q10, Q11
• Details of Ph.Eur. 2.9.47 Uniformity ofdosage units using large sample sizes
13
Quality by DesignOverview
• Quality can be planned
• QbD for pharmaceutics
– US FDA initiative (2004)
– ICH Q8, Q9, Q10, Q11
– European Pharmacopoeia
14
8

Quality by DesignOverview: PAT analytical methods
Traditional analytics
• Off-line testing
– Starting materials
– Isolated intermediates
– End products
• Small sample size
• Destructive
• Delayed results
‘PAT analytics’
• On-line/ in-line testing
– Starting materials
– Intermediates
– End products
• Sample size ~100 ++
• Non-destructive
• Immediate results
Enables dynamic feed-back and feed-forward controls15
Quality by DesignRegulatory guidance: ICH
Q8: Pharmaceutical development
– quality target product profile (QTPP)
– Design of experiments
– design space
– control strategy
16
9
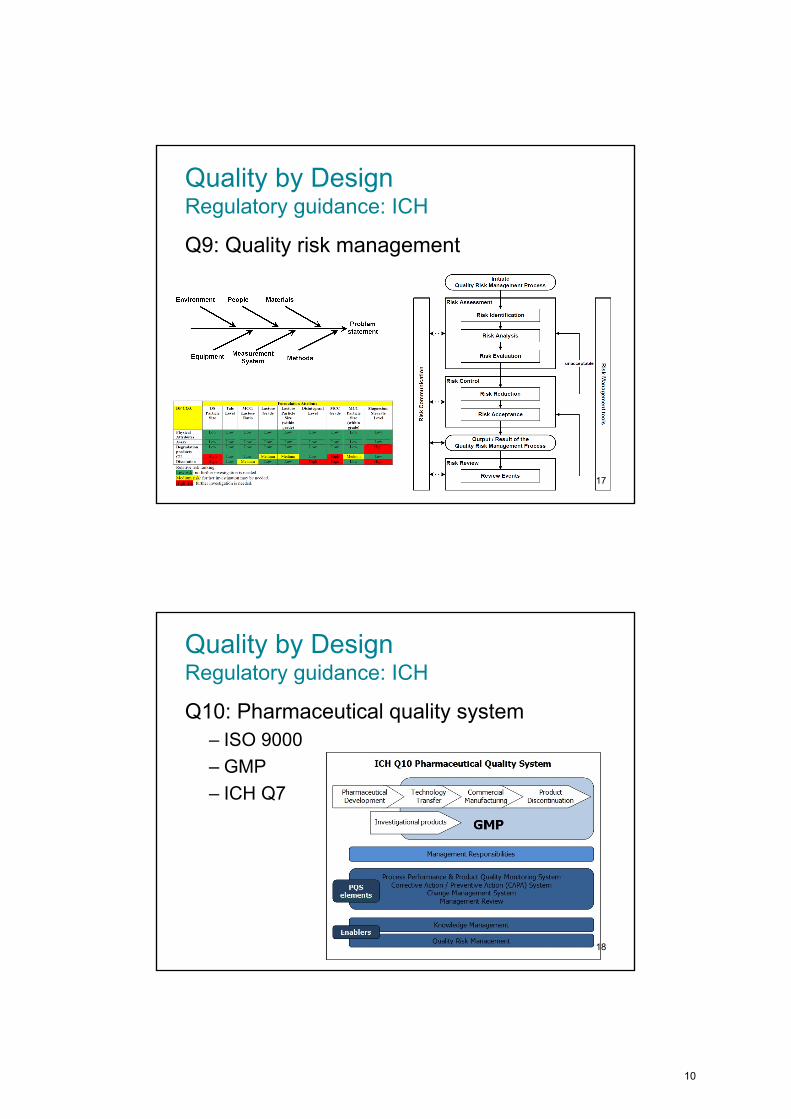
Quality by DesignRegulatory guidance: ICH
Q9: Quality risk management
17
Quality by DesignRegulatory guidance: ICH
Q10: Pharmaceutical quality system
– ISO 9000
– GMP
– ICH Q7
18
10

Quality by DesignRegulatory guidance: ICH
Q11: Dev. and manufacture of drug substances
– Chemical and biotechnological/ biological entities
– Q8, Q9 and Q10 applicable to drug substance
– Drug substance quality – link to drug product
– Starting material definition
19
Ph.Eur. 2.9.47Uniformity of dosage units using large sample sizes
• Details of the development and interpretation of themethod can be found in– Ø. Holte and M. Horvat 2011. Evaluation of uniformity of dosage
units using large sample sizes. Pharmeuropa 23(2): 286-293
– Ø. Holte and M. Horvat 2012. Uniformity of Dosage Units Using Large Sample Sizes. Pharm Sci Technol 36(10): 118-122
– Ø. Holte and M. Horvat 2014. Letter to the Editor: Statistical Properties of Large Sample Tests for Dose Content Uniformity. Therapeutic Innovation & Regulatory Science. DOI 10.1177/2168479014531761
20
11
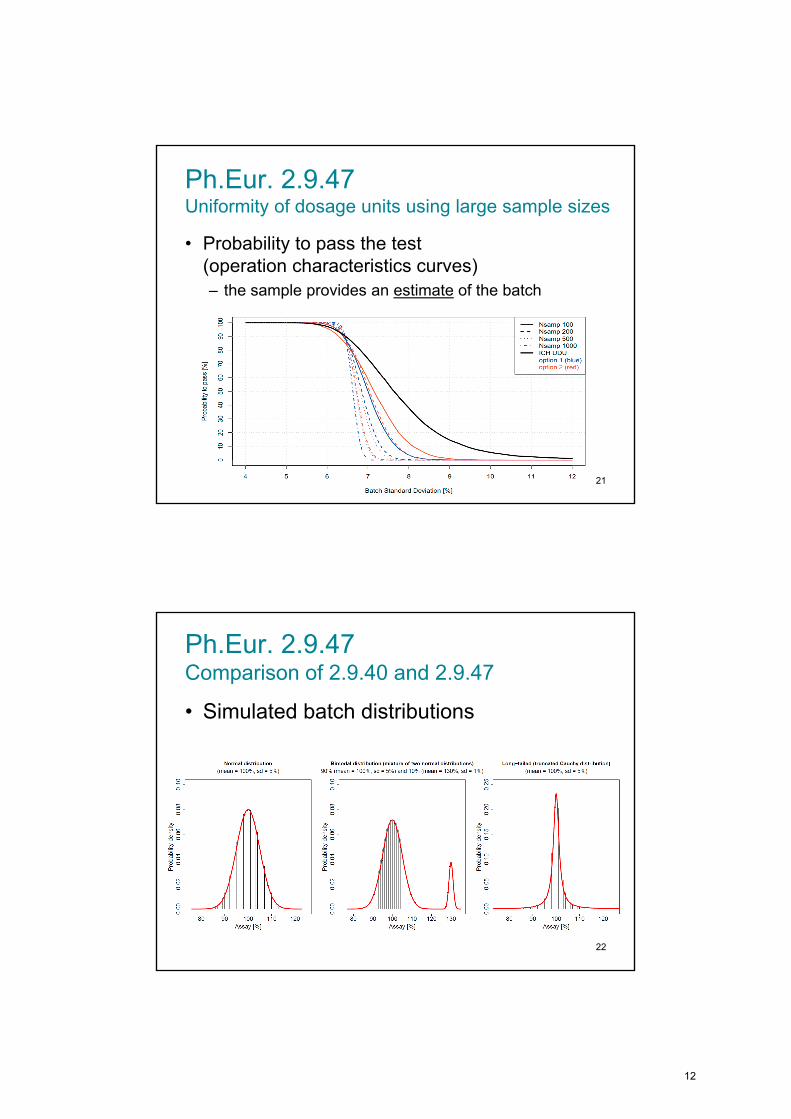
Ph.Eur. 2.9.47Uniformity of dosage units using large sample sizes
• Probability to pass the test (operation characteristics curves)
– the sample provides an estimate of the batch
21
Ph.Eur. 2.9.47 Comparison of 2.9.40 and 2.9.47
• Simulated batch distributions
22
12
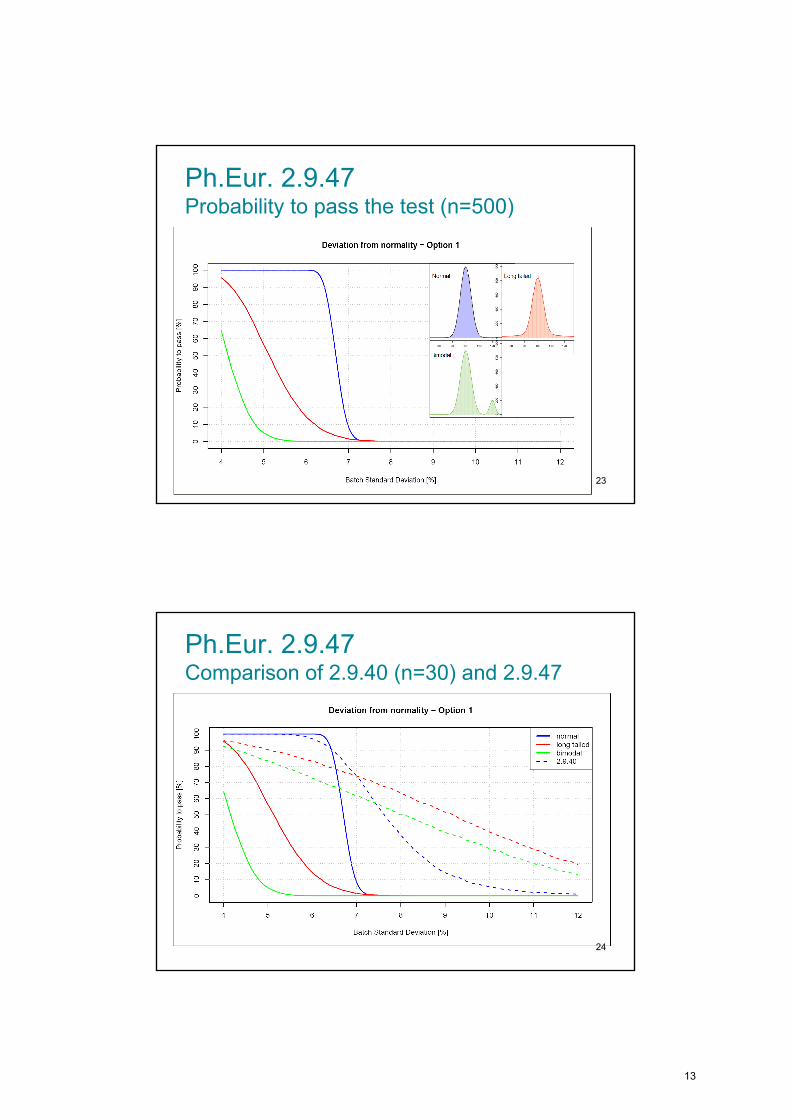
Ph.Eur. 2.9.47 Probability to pass the test (n=500)
23
Ph.Eur. 2.9.47 Comparison of 2.9.40 (n=30) and 2.9.47
24
13

Analytical QbD andPharmacopoeialMonographs– a vision
Dr. Oliver Grosche
Efpia TDOC Subteam on Analytical Design Space
Pittcon 1970 – first mentioning “HPLC”
1970’s:
• Particle size d= 10 µm
• pressure p<80 bar
1980’s:
• d= 5 µm
• p<250 bar
1990-Millenium:
• d= 3-4 µm
• p<400 bar
Now:
• d= <2 µm
• p<1000 bar
Modernization of analytical technology –Analytical Rocket Science versus HPLC
5 0 Y E A R S E D Q M W O R K S H O P 2
1964: Start ofELDO / ESRO
1970’s: ESA formed/ COS-B• measurement of gamma ray
emission of the universe
1980’s : Giotto Mission,
• spectral evidence of organic material on comet Halley
Millenium: ISO mission• most sensitive IR telescope at the time
Now: Rosetta Mission
• , e.g. TOF-MS for comet dust analysis
Majority of Ph. Eur.Monographs
14

3
No change – No risk?
Risks associated with or without analytical changes
no change change
Loss of data consistency
Marginal Innovation
Change to non-standard technology
Loss of long-term experience with method
Supply issues with “old” materials and parts
Outdated technology
Non- Compliance with newest regulations
Not adequate to reflect manufacturing process
changes
Change decisions must be made with respect to Patient safety Drug availability Time for
implementation Costs
Balanced Innovation
5 0 Y E A R S E D Q M W O R K S H O P
General chapter 1.1 – accelerating innovation?
Alternative methods. ◀The tests and assays described are the official methods upon which the standards of the Pharmacopoeia are based. With the agreement of the competent authority, alternative methods of analysis may be used for control purposes, provided that the methods used enable an unequivocal decision to be made as to whether compliance with the standards of the monographs would be achieved if the official methods were used. In the event of doubt or dispute, the methods of analysis of the Pharmacopoeia are alone authoritative.
5 0 Y E A R S E D Q M W O R K S H O P 4
How can this be demonstrated / ensured for an alternative analytical method?
Method performance standards as enabler
15
5
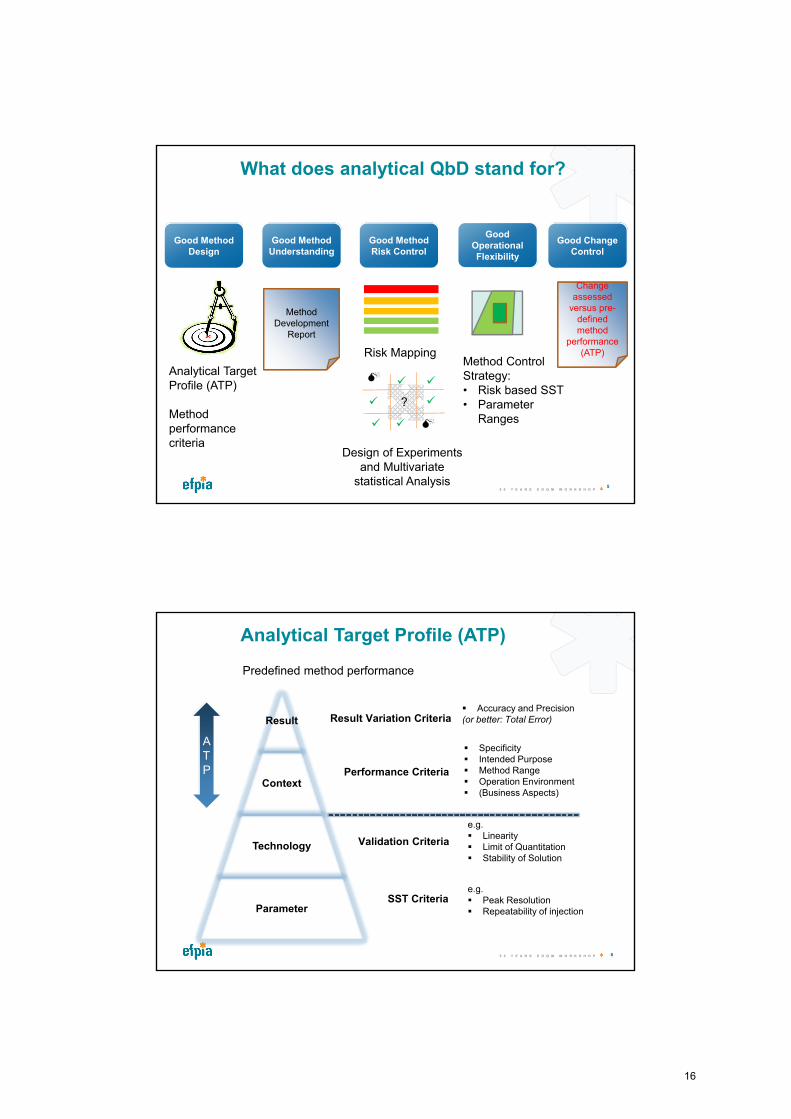
What does analytical QbD stand for?
Good Method Design
Good Method Understanding
Good Method Risk Control
Analytical Target Profile (ATP)
Method performance criteria
Risk Mapping
Design of Experiments and Multivariate
statistical Analysis
?
Method Control Strategy:• Risk based SST• Parameter
Ranges
Good Operational Flexibility
Good Change Control
Method Development
Report
ChChange assessed
versus pre-defined method
performance (ATP)
5 0 Y E A R S E D Q M W O R K S H O P
6
Analytical Target Profile (ATP)
Predefined method performance
Result Variation Criteria Accuracy and Precision(or better: Total Error)
Performance Criteria
Specificity Intended Purpose Method Range Operation Environment (Business Aspects)
Validation Criteria
e.g. Linearity Limit of Quantitation Stability of Solution
SST Criteriae.g. Peak Resolution Repeatability of injection
ATP
Result
Context
Technology
Parameter
5 0 Y E A R S E D Q M W O R K S H O P
16

ATP Example
5 0 Y E A R S E D Q M W O R K S H O P 7
Requirement Method performance criteriaAccuracy Impurity A: ≤ 80-120 % of true value
Impurity B: ≤ 75-125% of true value
Precision ofreportable result
Impurity A srel ≤ 10%
Impurity B srel ≤ 15%
Intended Purpose
Quantification of manufacturing process related impurity (not a degradation product)
Range Impurity A: at least 0.05%-0.6%
Impurity B: at least 0.05%-0.12%
Specificity No interference of the quantification of the specified impurities by
• other related substances C, D, and E
• the salt forming agent
with Impurity A,B or the API. Operating conditions and Environment
The analytical procedure must be applicable for use in a standard analytical QC laboratory environment of ADS Pharma for routine analysis. The test procedure must be stable for at least 24 h of consecutive analyses.
Advantages of the ATP concept for the Ph. Eur.
Facilitation of Assessment of Methods from multiple manufacturers
Enabling balance between cost/safety of public standards (costly high end technology standard techology)
Predefinition of Method Interchangeability with public standards (Pharmacopoeial Compliance)
Facilitation of change control for individual monographs
ATPs can be retrospectively defined for already existing monographs to anticipate future changes
May be used as trigger to evaluate monograph revisions
5 0 Y E A R S E D Q M W O R K S H O P 8
17
9
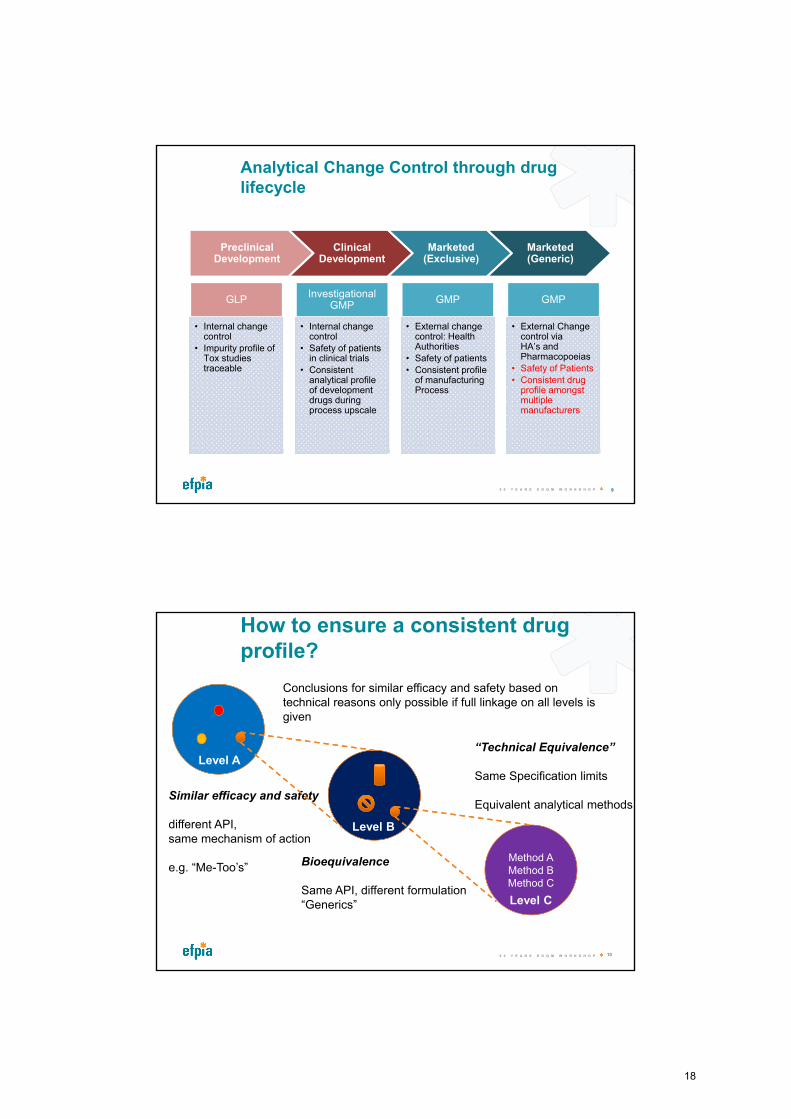
Analytical Change Control through drug lifecycle
Preclinical Development
Clinical Development
Marketed (Exclusive)
Marketed (Generic)
GLP
• Internal change control
• Impurity profile of Tox studies traceable
Investigational GMP
• Internal change control
• Safety of patients in clinical trials
• Consistent analytical profile of development drugs during process upscale
GMP
• External change control: Health Authorities
• Safety of patients
• Consistent profile of manufacturing Process
GMP
• External Change control via HA’s and Pharmacopoeias
• Safety of Patients
• Consistent drug profile amongst multiple manufacturers
5 0 Y E A R S E D Q M W O R K S H O P
How to ensure a consistent drug profile?
10
Method AMethod BMethod C
Similar efficacy and safety
different API, same mechanism of action
e.g. “Me-Too’s” Bioequivalence
Same API, different formulation“Generics”
“Technical Equivalence”
Same Specification limits
Equivalent analytical methods
Conclusions for similar efficacy and safety based on technical reasons only possible if full linkage on all levels is given
Level A
Level B
Level C
5 0 Y E A R S E D Q M W O R K S H O P
18
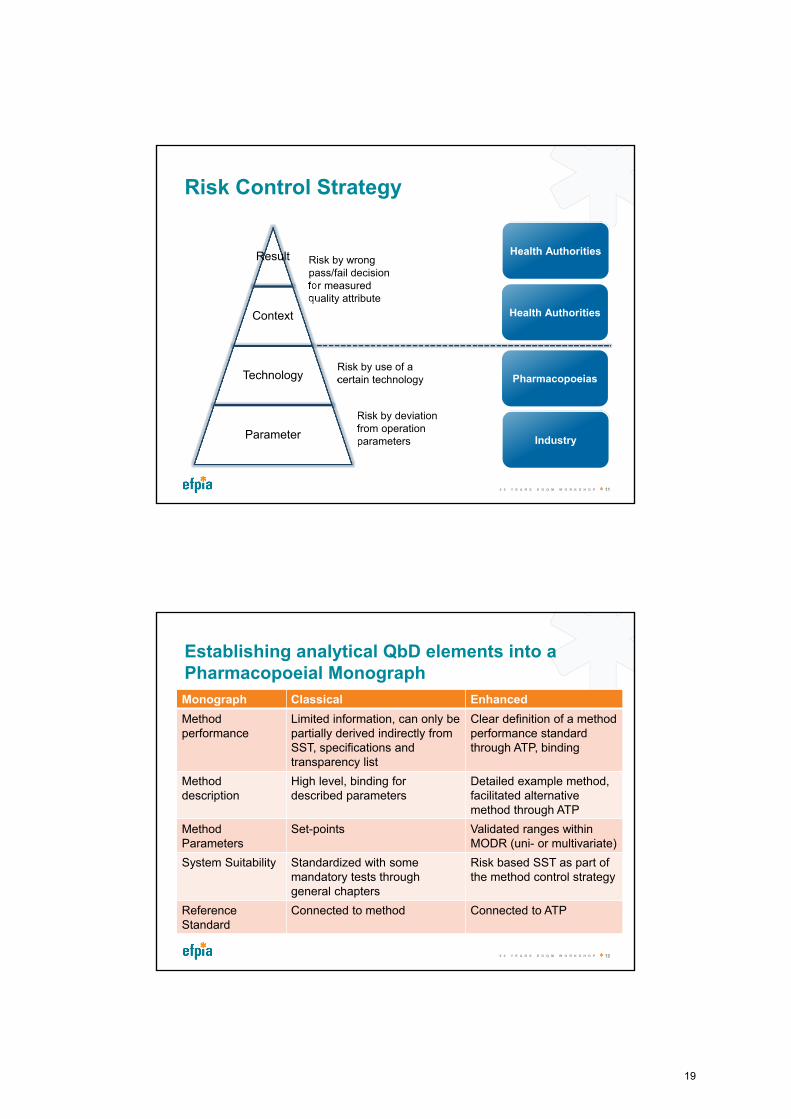
Risk Control Strategy
5 0 Y E A R S E D Q M W O R K S H O P 11
Risk by wrong pass/fail decision for measured quality attribute
Risk by deviation from operation parameters
Risk by use of a certain technology
Industry
Pharmacopoeias
Health Authorities
Health AuthoritiesResult
Context
Technology
Parameter
Establishing analytical QbD elements into a Pharmacopoeial Monograph
Monograph Classical Enhanced
Method performance
Limited information, can only be partially derived indirectly from SST, specifications and transparency list
Clear definition of a method performance standard through ATP, binding
Method description
High level, binding fordescribed parameters
Detailed example method, facilitated alternative method through ATP
MethodParameters
Set-points Validated ranges within MODR (uni- or multivariate)
System Suitability Standardized with some mandatory tests through general chapters
Risk based SST as part of the method control strategy
Reference Standard
Connected to method Connected to ATP
5 0 Y E A R S E D Q M W O R K S H O P 12
19

Conclusions
An analytical innovation lag of 10-30 years may exist for pharmacopoeial methods
Ph. Eur. general chapter 1.1 supportive for alternative innovative methods
No guidance is given in Phar. Eur. on how to demonstrate interchangeability
The ATP concept may help to balance establish method interchangeability
Enhanced Pharmacopoeial Monographs may help to adapt the analytical method standards to the operational environment of multiple manufacturers
5 0 Y E A R S E D Q M W O R K S H O P 13
Thank You
EDQM Efpia “Analytical Design Space” Subteam:Melissa Hanna Brown (Pfizer)Jörg Hoffmann (Merck KGaA)Joachim Ermer (Sanofi)Christof Finkler (Hoffmann-La Roche), Stefanie Katzenbach (AbbVie)Phil Nethercote (GSK)Andy Rignall (AstraZeneca)Thomas Uhlich (Bayer)Kieran McLaughlin (Merck)Thorsten Sokoliess (Boehringer Ingelheim)
5 0 Y E A R S E D Q M W O R K S H O P 14
20

Backup Slides
5 0 Y E A R S E D Q M W O R K S H O P 15
Failure Mode Effect Analysis of Methods
5 0 Y E A R S E D Q M W O R K S H O P 16
Overall risk
Frequency Impact Detectability0
10
0
10
0
10
Patient riskProcess risk
Occurrence per sample and/or test
Clarity/Detail of instructionIndication of system suitability
low
high high
highlow
lowhigh
low
Identification of Critical Parameters
FMEA methodology to help to define allowable variations in Standard Tests
Risk based SST criteria for Monographs
21

Multivariate Experimental Design
5 0 Y E A R S E D Q M W O R K S H O P 17
low set high
Gradient slope
Temperature
pH
Buffer conc.
Wavelength
Univariate 1 Univariate 2
Multivariate 1 Multivariate 2
Selection of a Method which runs robust in multiple companies and countries
Establishment of Parameters/Ranges that can be adjusted for specific methods to meet SST criteria
Risk based Impurity Profile / Selectivity
5 0 Y E A R S E D Q M W O R K S H O P 18
Component Definition RequirementSpecified components
Regularly monitored (and qualified) related substances (by- and degradation products), Specified process related impurities
Specificity for all individual specified components obvious/demonstrated.
Unspecified regularcomponents
Unspecified related substances (by- and degradation products) and unspecified process related impurities regularly present in some or all batches of current manufacturing route/process or known degradation pathway. Components used as critical attributes/indicators for process control.
Specificity for unspecified regularly controlled components obvious/demonstrated
Unspecified potential components
Related substances for which the test method has the potential to separate but did not occur in batches of current manufacturing route/process or known degradation pathway.
Demonstration of Specificity beneficial but not required if related substances did not occur
Clear categorization will support monograph specifications and transparency list
Support specificty requirements for ATP and future monograph changes
22

Description of Method Design Space
5 0 Y E A R S E D Q M W O R K S H O P 19
Increased clarity which method parameters may be varied to meet SST criteria
Moving from method set-points to method parameter ranges
Level Concept for Analytical Changes
5 0 Y E A R S E D Q M W O R K S H O P 20
RR
C
T
P
20
Within design space
Same Technology
Same performance
characteristics
Analytical method validation tests &
criteria
Method independent performance tests
and criteria
System Suitability test &
criteria
Level 0 Level 1 Level 2
Very similar or same method
Same Technology
Same performance
characteristics
Very similar or same method
Same Technology
Same performance
characteristics
Level of comparison
not possible
(Level 3: all others)
Change control and experiments are proportionate to the risk of change
Safe changes in-between techologies become manageble through ATP
Safe Monograph Modernizaton
ATP
23
21
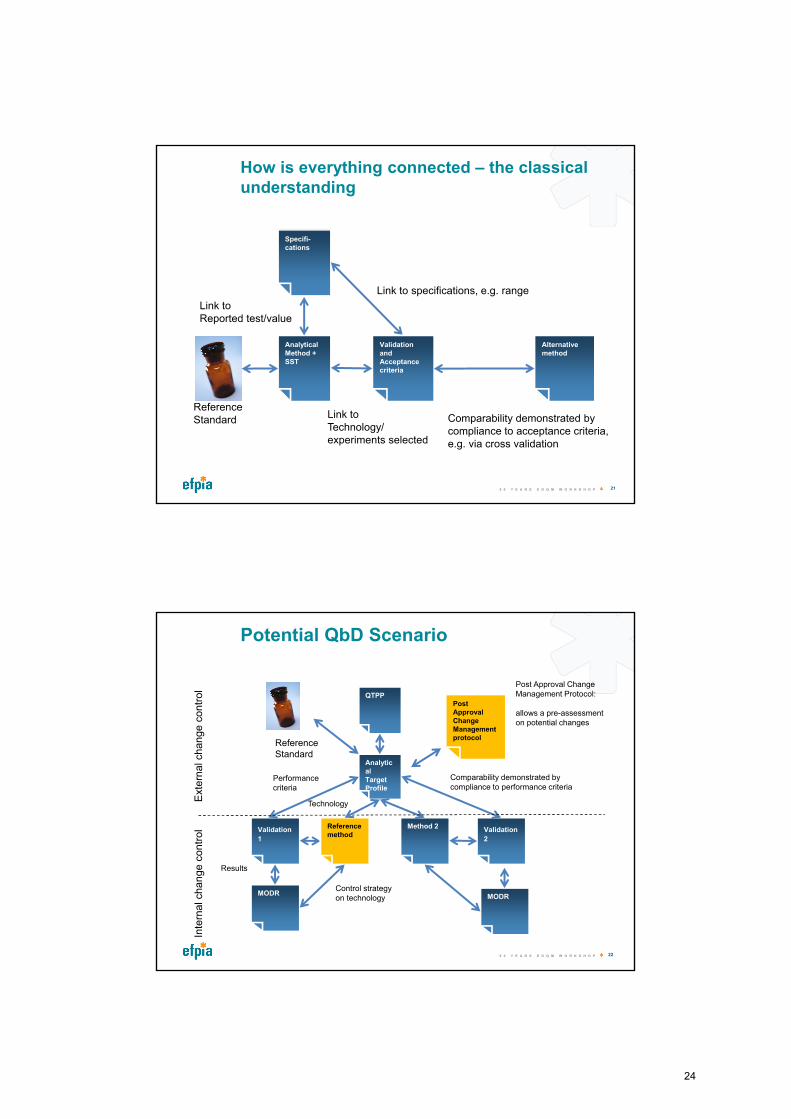
How is everything connected – the classical understanding
Specifi-cations
Link to specifications, e.g. range
Analytical Method + SST
Link to Reported test/value
Validation and Acceptance criteria
Link to Technology/experiments selected
Alternative method
Comparability demonstrated by compliance to acceptance criteria, e.g. via cross validation
Reference Standard
5 0 Y E A R S E D Q M W O R K S H O P
22
Potential QbD Scenario
Technology
QTPP
Analytical TargetProfile
Validation
1
Performancecriteria
Reference method
MODRMODR
Results
Control strategyon technology MODR
Method 2 Validation
2
Comparability demonstrated by compliance to performance criteria
Exte
rna
l ch
an
ge
co
ntr
ol
Inte
rna
l ch
an
ge
co
ntr
ol
Post Approval Change Management protocol
Post Approval Change Management Protocol:
allows a pre-assessment on potential changes
Reference Standard
5 0 Y E A R S E D Q M W O R K S H O P
24

EFPIA Brussels Office
Leopold Plaza BuildingRue du Trône 108B-1050 Brussels - Belgium Tel: +32 (0)2 626 25 55
www.efpia.eu
25

Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
Application of QbD to analytical methods - a regulatory perspective
Dr. Jobst Limberg- Bundesinstitut für Arzneimittel und
Medizinprodukte (BfArM)
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Views and opinions expressed in this presentation are those of the presenter
Disclaimer
2
26

Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Suitable for intended purpose
• Validation characteristics (ICH Q2) Specificity Accuracy (of the mean) Linearity & range Limit of Detection, Limit of Quantitation Precision & intermediate precision (ruggedness) Robustness
• Includes sample preparation, reference substances, etc…
Analytical validation - overview
3
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• No validation data required Pharmacopoeial test procedures
• Full validation necessary in the dossier for test procedures: used for batch release (end-product testing or real time release
testing) used for exploratory stability studies
• In case of variations When is a new validation necessary? What has to be done? What are the acceptance criteria?
Regulatory perspective on validation of analytical test procedures
4
27

Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Variation of a test procedure New methods should be equal or better concerning the validation
parameters
• Variation during ongoing stability studies equivalence of results in terms of accuracy of the mean and
intermediate precision
• Adding a second, alternatively used test procedure Different equipment at different production sites Real time release test procedure differs from reference test
procedure for batch release Both methods should be equal/equivalent/similar/not
significantly different
Comparison of two analytical test procedures intended for the same purpose / statements
5
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
Comparison of two analytical test procedures –four scenarios on a dartboard
6
similar resultssimilar precisionsame systematic error
similar resultsdifferent precisionsimilar systematic error
Different resultsdifferent precisionsystematic error – yes and no
Similar resultsSimilar precisionno systematic error
28
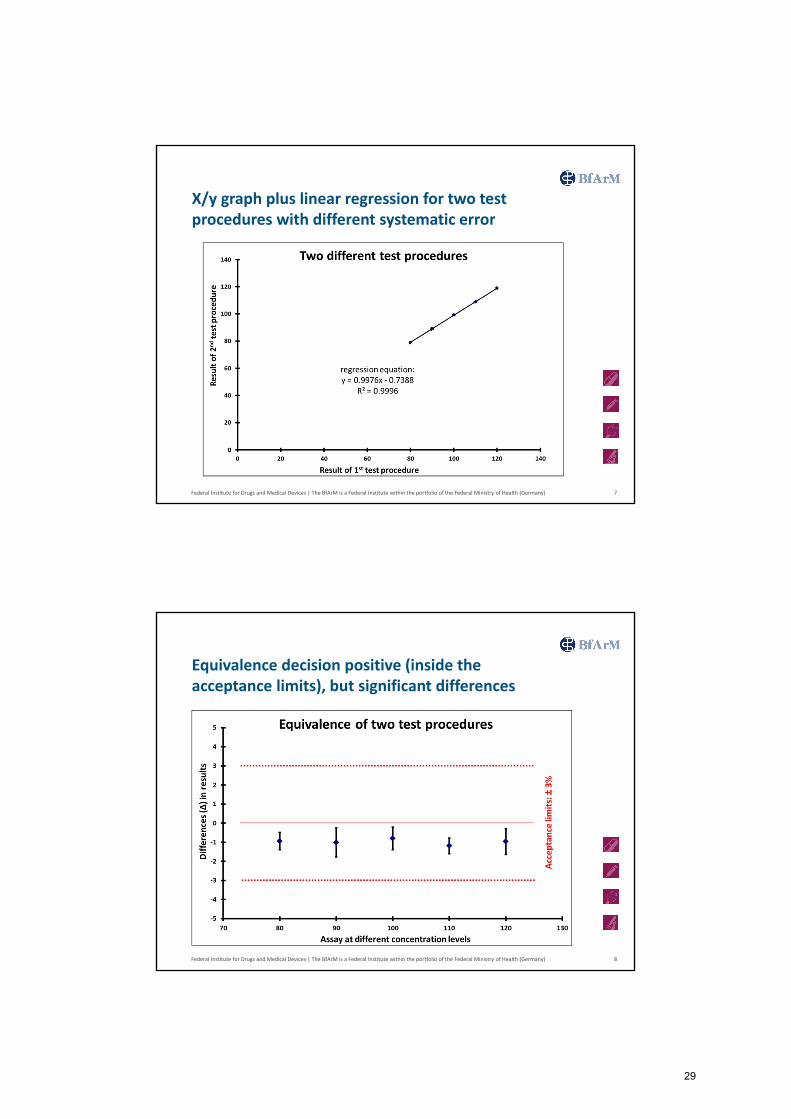
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
X/y graph plus linear regression for two test procedures with different systematic error
7
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
Equivalence decision positive (inside the acceptance limits), but significant differences
8
29

Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• The discriminatory power to distinguish between “good” and “bad” batches should be comparable. O.k. for regulators, best scenario
• Over-discriminatory power leads to rejection of good batches (type II error) Also o.k. with regulators, but may be costly for industry
• Under-discriminatory power leads to releasing of bad batches (type I error) Not o.k. for regulators
Comparison of analytical test procedures -parallel testing / regulatory view
9
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Systematic approach to method development
• Analytical target profile ≡ acceptance criteria of validation plan
• Method operable design region (MODR), i.e. limits for procedural parameters in which the test procedure is suitable for its intended use
EFPIA approach (white paper)
10
30

Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Analytical target profile should be restricted to test procedures based on the same physical methodology
• Method operable design region should be based on design of experiments
• Regulatory environment: Variation regulation has to be followed Post approval change management protocol allows up-front
information about potential future changes Assessment of analytical development and validation is part of
licensing and not of GMP inspections
Response to white paper by regulatory agencyEMA (joint QWP and BWP) (I)
11
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Approach restricted to specific quality attributes (?) Critical quality attributes Some test procedures are able to find the “unexpected”, which
may not be valid for another The discriminatory power to find new (unknown) impurities is not
predictable Knowledge gaps due to the complex nature of a test, e.g.
biological activity measurements In case of applying another physicochemical methodology, the
acceptance criteria for validation parameters may change
Response to white paper by regulatory agencyEMA (joint QWP and BWP) (II)
12
31

Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Comparability of results obtained by alternative tests
• Two alternative test procedures for batch release at one manufacturing sites will constitute GMP problems
• No testing into specification acceptable
• Change inside a stability study may be problematic due to its explorative nature
Response to white paper by regulatory agencyEMA (joint QWP and BWP) (III)
13
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Operable design space: Small ranges for analytical parameters are already acceptable
according to ICH Q2 – robustness Ranges for analytical parameters are already acceptable according
to the European Pharmacopoeia
• Acceptability is based on data of a single methodology, which has been evaluated
• How to transfer this flexibility of analytical parameters from HPLC to NIRS and vice versa
Response to white paper by regulatory agencyEMA (joint QWP and BWP) (IV)
14
32

Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
• Current guideline ICH Q2 allows small variability in analytical parameters – robustness
• Variation regulations allow flexibility for improvement
• Post approval change management protocol as a tool to implement planned changes in a shorter timeframe
• Once ICH Q2 will be under revision, some ideas may be incorporated
• Examples and requirements of ICH Q2 are focussed on chromatographic test procedures and should be extended to cover also spectroscopic in-line and on-line methodologies
Conclusions
15
Thank you for your attention!
Federal Institute for Drugs and Medical Devices | The BfArM is a Federal Institute within the portfolio of the Federal Ministry of Health (Germany)
33





























