Embed Size (px)
Citation preview

MOLECULAR AND CELLULAR BIOLOGY, Oct. 2010, p. 4767–4785 Vol. 30, No. 200270-7306/10/$12.00 doi:10.1128/MCB.01021-09Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Downregulation of Thrombomodulin, a Novel Target of Snail, InducesTumorigenesis through Epithelial-Mesenchymal Transition�†
Yuan-Chung Kao,1,2 Li-Wha Wu,3,4 Chung-Sheng Shi,4 Che-Hsien Chu,1 Chia-Wei Huang,1,2
Chou-Pin Kuo,1,4 Hamm-Ming Sheu,5 Guey-Yueh Shi,1,4* and Hua-Lin Wu1,4*Department of Biochemistry and Molecular Biology,1 Institute of Basic Medical Sciences,2 Institute of
Molecular Medicine,3 Cardiovascular Research Center,4 and Department of Dermatology,5
College of Medicine, National Cheng Kung University, 701 Tainan, Taiwan
Received 3 August 2009/Returned for modification 14 October 2009/Accepted 19 July 2010
The expression of thrombomodulin (TM), a calcium-dependent adhesion molecule, is frequently downregu-lated in various cancer types. However, the mechanism responsible for the low expression level of TM intumorigenesis is unknown. Here, an inverse expression of TM and Snail was detected in different cancer celllines. We further confirmed this inverse relation using the epithelial-mesenchymal transition cell model inHaCaT and A431 cells. We demonstrated that Snail suppressed TM expression by binding to E-box (CACCTG)in TM promoter. Moreover, TM knockdown by short hairpin RNA disrupted E-cadherin-mediated cell junc-tions and contributed to tumorigenesis. In the calcium switch assay, E-cadherin lost the ability to associatewith �-catenin and accumulated in cytoplasm in TM knockdown cells. Meanwhile, wound healing and invasiveassays showed that TM knockdown promoted cell motility. A subcutaneous injection of TM knockdowntransfectants into immunocompromised mice induced squamous cell carcinoma-like tumors. Besides, forcedexpression of murine TM in TM knockdown cells made the cells reassume epithelium-like morphology andincreased calcium-dependent association of E-cadherin and �-catenin. In conclusion, TM, a novel downstreamtarget of Snail in epithelial-mesenchymal transition, is required for maintaining epithelial morphology andfunctions as a tumor suppressor.
Thrombomodulin (TM), a type 1 transmembrane glycopro-tein, was first identified in endothelial cells and is well knownas an anticoagulant factor (12). TM consists of 557 amino acidresidues arranged in five distinct domains including an NH2-terminal lectin-like domain, a domain with six epidermalgrowth factor (EGF)-like structures that contain thrombinbinding sites, an O-glycosylation site-rich domain, a transmem-brane domain, and a cytoplasmic tail (43). Depletion of theTM gene leads to embryonic lethality due to an impairedcardiovascular system (18). TM was later found in humankeratinocytes and served as a differential biomarker for theclinical stages of skin cancers (36). Recent studies further re-vealed that TM has pleiotropic effects in both physiology andpathology via its different domains, including the calcium-de-pendent control of cell-cell adhesion by its lectin-like domain(20), angiogenic stimulation by its EGF domain (38), and anti-inflammatory effect by its lectin-like domain in sepsis via bind-ing to Lewis-Y, a tetrasaccharide expressed on the surface ofpathogens (39).
Mesenchymal-epithelial transition is characterized as a mor-phological change from fibroblast-like to epithelium-like cells,which is the reverse of epithelial-mesenchymal transition
(EMT). Transfection of human TM cDNA into A2058 mela-noma cells with fibroblast-like shape inhibited cell prolifera-tion in vitro and reduced xenograft tumor growth in immuno-compromised mice (20). We also found that A2058 cells stablyexpressing ectopic TM induced closely clustered colonies, rem-iniscent of mesenchymal-epithelial transition. The effect of TMin promoting epithelial morphogenesis is consistent with theclinical observations that reduced TM expression is associatedwith poor prognosis for patients with tumor metastases in lung(31), breast (24), and colorectal (16) cancer. These data sug-gest that TM may play a negative regulatory role in tumori-genesis by modulating the assembly of cell junctions. However,the exact mechanism underlying TM downregulation and thecorrelation between TM and E-cadherin involved in tumori-genesis have never been investigated.
E-cadherin is a major component of adherens junctions andmediates cell-cell adhesion in a calcium-dependent manner.Loss of E-cadherin expression was correlated with increasedinvasive potential of both carcinoma cell lines and tumor sam-ples (10). Reduced E-cadherin expression or altered subcellu-lar localization of E-cadherin protein has been reported in thecells undergoing EMT and different human cancers such asprimary tumors of esophagus, stomach (41), and pancreas (34).In contrast, E-cadherin overexpression increased cell-cell ad-hesion and suppressed gelatinase secretion and cell growth andthereby partially suppressed tumorigenesis in HaCa4 carci-noma cells (30). Moreover, E-cadherin expression in cells isrepressed by members of the Snail superfamily, includingSnail, Slug, and E12/47 (4). The suppression also causes epi-dermal cell lines, MCA3D and PDV cells, to assume a mes-enchymal phenotype and acquire tumorigenic properties (9).
* Corresponding author. Mailing address: Department of Biochem-istry and Molecular Biology, College of Medicine, National ChengKung University, No. 1, University Rd., 701 Tainan, Taiwan. Phone:886-6-2353535, ext. 5541. Fax: 886-6-3028037. E-mail for Hua-Lin Wu:[email protected]. E-mail for Guey-Yueh Shi: [email protected].
† Supplemental material for this article may be found at http://mcb.asm.org/.
� Published ahead of print on 16 August 2010.
4767
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.

Like E-cadherin, TM functioned as a calcium-dependent cell-to-cell adhesion molecule and its ectopic expression induced afibroblastic-to-epithelial morphological change in A2058 mel-anoma cells (20). Since both TM and E-cadherin mediated celladhesion and are expressed at low levels in metastatic tumors,downregulation of TM may also participate in tumorigenesisand Snail-mediated EMT.
EMT, which involves characteristic change in cellular mor-phology from an epithelial to a fibroblast-like phenotype, lossof cell-cell junctions, and increase in cell motility and cellproliferation, frequently takes place in embryonic development(42), cancer progression (22), and wound healing (1). Down-regulation of adhesion molecules is documented to induceEMT via either reducing E-cadherin expression or abolishingE-cadherin-mediated cell-cell contact. For instance, knock-down of claudin 7, the major component of intercellular tightjunctions, can directly lead to decreased E-cadherin expres-sion, cell morphology changes, and motility enhancement inesophageal squamous cell carcinoma (SCC) (26). In con-trast, knockdown of mucin (MUC-1), a human epithelialtumor antigen and tumor-associated glycoprotein, increasesE-cadherin/�-catenin complex formation and restores E-cadherin localization at the cell membrane in PANC1 pan-creatic adenocarcinoma cells (47). Together, these resultsindicate that a loss in the architecture of epithelial celljunctions might lead to the acquisition of mesenchymal cellbehavior.
Chemokine-mediated signaling pathways are involved in theprocess of EMT (14). Among many chemokines, transforminggrowth factor � (TGF-�) has dual functions in tumor growth,inhibiting tumor growth in the early stage but facilitating tumorinvasion and metastasis in the later stage of carcinogenesis(37). Normal murine mammary gland (NMuMG) and mousecortical tubular (MCT) epithelial cells are among a few pri-mary cultured cells and cell lines that are used as models toinvestigate the mechanism of TGF-� action in EMT. A mor-phological change of these cells induced by the treatment withTGF-�1 accompanies the loss of E-cadherin (8). Moreover,TGF-�1 together with EGF can synergistically induce EMT inHaCaT cells, spontaneously immortalized keratinocytes,through the activation of a mitogen-activated protein kinase-dependent signal transduction pathway (11). Furthermore,scatter factors, such as hepatocyte growth factor and EGF,modulate E-cadherin and induce EMT via different mecha-nisms. Hepatocyte growth factor disrupts cadherin-mediatedcell-cell adhesion between keratinocytes (45), whereas EGFmodulates the phosphorylation of the cadherin-catenin systemin the human esophageal cancer cell line TE-2R, which ex-presses E-cadherin and the EGF receptor (40).
Snail, a zinc finger transcriptional factor, functions as a reg-
ulator to suppress the expression of adhesion molecules and toassist the escape of tumor cells from cell death during EMT(44). Besides having a regulatory role in EMT, Snail also reg-ulates genes that are associated with EMT-independent bio-logical functions, including mesodermal determinants, cellmovement, and cell survival. Snail is frequently expressed inmany types of tumor cells in which E-cadherin expression isreduced. Moreover, this inverse relationship of Snail and E-cadherin was often observed in the invasive types of tumors(3). Later studies confirmed a suppressing effect of Snail ex-pression on E-cadherin promoter via a specific binding to thethree E-boxes (CACCTG) in the promoter region at �178 to�92 of the E-cadherin gene (2).
In this study, we investigated the relationship of TM andseveral EMT markers in various tumor cells and the in vitroEMT model induced by cotreatment of TGF-�1 and EGF. Adirect Snail binding to the proximal TM promoter was identi-fied by electrophoretic mobility shift assay (EMSA), chromatinimmunoprecipitation (ChIP), and promoter-driven luciferasereporter assay. The knockdown approach was used to examinethe role of TM in cell migration, invasion, and tumorigenesisand to confirm the inverse relationship of TM expression andSnail.
MATERIALS AND METHODS
Cell culture and treatment. The basal cell carcinoma (BCC) was kindly pro-vided by Hamm-Ming Sheu. TSGH8301 was purchased from Food IndustryResearch and Development Institute (Hsinchu, Taiwan). A2058, DU145, A431,HepG2, Huh7, HeLa, SiHa, Cx, and KB cells were purchased from the AmericanType Culture Collection. The culture conditions of HaCaT cells were the sameas previously described (5). HaCaT cells that overexpressed Snail were preparedby transfecting cells with pcDNA3-mm snail-HA, ectopic Snail with hemagglu-tinin (HA) (a kind gift of A. G. de Herreros), using Lipofectamine 2000 (In-vitrogen), and selecting them with 600 �g/ml G418. Silencing of TM gene ex-pression in HaCaT cells was accomplished using short hairpin RNA (shRNA)technology by the pSM2c vector system (Gendiscovery). Two shRNA sequencesspecific to TM (Table 1) were cloned into pSM2c vector. HaCaT cells weretransfected with these two constructs or with empty vector and selected with 1�g/ml puromycin. Cell morphology was observed with a phase-contrast micro-scope (Leica model DM). Images were obtained using a 40� objective (numer-ical aperture [NA], 1.4).
Constructs and transfections. Full-length cDNA of murine TM (mTM) wasconstructed in an adeno-associated virus (AAV) vector with green fluorescentprotein. This construct was transiently transfected into cells with an electropo-ration system as described by the manufacturer (Promega).
Western blotting. Cell extracts were subjected to SDS-PAGE, and proteinswere detected with antibodies that were specific to E-cadherin (Calbiochem),N-cadherin (Calbiochem), TM (D3; Santa Cruz), Snail (Abcam), or HA (F-7;Santa Cruz). The same membranes were also probed to detect �-tubulin(DM1A; Calbiochem) as loading controls. The images were captured with LAS-3000 (Fujifilm).
Immunofluorescent staining. Cells were fixed in 3.7% (vol/vol) formaldehydesolution, permeabilized with 0.1% (vol/vol) Triton X-100, blocked with 10% fetalbovine serum for 30 min at room temperature, and incubated with antibodiesspecific for TM (clone 1009; Dako), E-cadherin (Calbiochem), �-catenin (BD
TABLE 1. Short hairpin sequences specific to TM
Name Sequence (5�–3�)a
TM1.6 (specific target of sh-TM1) ......................................TGCTGTTGACAGTGAGCGACCAATTAGGGCCTAGCCTTAATAGTGAAGCCACAGATGTATTAAGGCTAGGCCCTAATTGGGTGCCTACTGCCTCGGA
TM1.9 (specific target of sh-TM2 and -3)..........................TGCTGTTGACAGTGAGCGACCCAATTAGGGCCTAGCCTTATAGTGAAGCCACAGATGTATAAGGCTAGGCCCTAATTGGGCTGCCTACTGCCTCGGA
a Boldface indicates the mir-30 loop construct sequence in the hairpin; underlining indicates the antisense/target sequence 19-mer; italics indicate the sense/targetsequence 19-mer.
4768 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.

Biosciences), Snail (Abcam), nonspecific mouse IgG (Rockland), or rabbit IgG(Rockland) for 2 h. This was followed by incubation of cells with secondaryantibody conjugated with Alexa Fluor 488 (Invitrogen) for 1 h or phalloidin (BDBiosciences) for 30 min. Images were obtained using either a fluorescence mi-croscope (DMIRE2; Leica) with a 40� objective (NA, 0.5) or a confocal micro-scope (SP2; Leica) with a 63� oil-immersion (NA, 1.4) objective.
EMSA. Forward and reverse oligonucleotides (synthesized by Life Technolo-gies, Invitrogen) were annealed and radiolabeled according to the manufactur-er’s instructions (Amersham Pharmacia Biotech). EMSAs were performed withnuclear extracts of TGF-�1- and EGF-cotreated HaCaT cells. The differentprobes used were (i) wild-type probe for TM and (ii) mutant probe for TM(Table 2). In competition assays, excess of the wild-type probe or mutant probe,either 20- or 50-fold molar, was added and the reaction mixture was incubated onice. DNA-protein complexes were separated on 6% nondenaturing polyacryl-amide gels in TBE (Tris-borate-EDTA) at room temperature. The gels weredried and visualized by autoradiography.
Reporter gene assay. TM promoter activity assays were carried out in Chinesehamster ovary (CHO) cells using 400 ng of human TM promoter (positionnumbers �1014/�745) cloned in the pcDNA3.1 basic vector (Promega). Cellswere cotransfected with pcDNA3-mm snail-HA and each of several different TMpromoter truncation-derived pGL3-based luciferase vectors. Ten nanograms ofpSG5-LacZ plasmid was the control for transfection efficiency. The expression offirefly and �-galactosidase was analyzed by a Dual-Light gene assay system(Promega) 48 h after transfection.
ChIP assay. ChIP assay was performed according to a published procedure(Millipore). The sequences for PCR primers are provided in Table 3. PCRconditions for TM were denaturation at 95°C for 30 s, annealing at 50°C for 30 s,and extension at 70°C for 30 s, for 35 cycles.
Wound healing assay and individual cell tracking. TM-expressing HaCaT cells(pSM2c and sh-TM1) and TM knockdown HaCaT cells (sh-TM2 and sh-TM3)were grown to full confluence and wounded with a p200 pipette tip. The imageswere recorded for 24 h by time-lapse microscopy (Leica DMIRE2) with a 5�objective (NA, 0.25). Percentage of wound closure was analyzed by Image-Jsoftware and calculated by the formula [(wound area at 0 h � wound area at24 h)/wound area at 0 h] � 100%. For tracking individual cells, the migrationpaths of 15 cells at the wound margin were recorded at 30-min intervals for 24 hby time-lapse microscopy and were analyzed by MetaMorph imaging software(Universal Imaging Corp.).
Invasion assay. In the invasion assay, a porous membrane was coated with 1mg/ml Matrigel (BD Biosciences) and 0.1% bovine serum albumin (BSA) inserum-free high-glucose Dulbecco modified Eagle medium (DMEM) at 4°Covernight. Cells were seeded in the upper compartment of the chamber contain-ing 2% serum; medium containing 10% serum was added to the lower chamberas chemoattractant. The chambers were incubated for 24 h at 37°C. Cells thathad migrated to the lower side were fixed and stained with Liu’s solution (HandSel Technologies, Inc.). Random fields of the migrated cells were counted usinga 20� (NA, 0.3) objective.
Coimmunoprecipitation assay. Cells were lysed with lysis buffer containing 50mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 mM CaCl2, 2% SDS, Triton X-100, andcomplete Mini protease inhibitor cocktail (Roche Applied Science). The cellextract lysates were clarified by centrifugation at 12,000 rpm for 10 min at 4°C,used immediately for immunoprecipitation, and precleaned with protein G-Sepharose 4 FastFlow beads (Amersham GE Healthcare Bio-Science) for 1 h,and the beads were removed by centrifugation at 12,000 rpm for 3 min. Pre-cleaned supernatants were incubated overnight either with pan-extracellularsignal-regulated kinase (pan-ERK) as a nonspecific mouse IgG control or withE-cadherin-specific antibody in the presence of the beads. Immunocomplexesbound to beads were pelleted by centrifugation at 1,600 rpm for 5 min, washedfive times with lysis buffer without protease inhibitors, eluted in SDS-PAGEloading buffer at 95°C for 10 min, and separated by SDS-PAGE.
Cell fractionation. Cells were scraped and homogenized in ice-cold homoge-nization buffer (20 mM Tris-HCl, pH 7.5, 5 mM EGTA, 2 mM EDTA, 10%glycerol, and protease inhibitors) using a sonicator (Misonix; S-3000). Cell lysateswere harvested by centrifugation at 4,000 rpm for 10 min. The cytoplasmicfraction was collected following the removal of membrane by centrifugation ofcell lysates at 13,000 rpm for 30 min. The membrane fractions were dissolved inhomogenization buffer with 1% NP-40. Membrane and cytoplasmic fractionswere analyzed by Western blotting.
Tumorigenesis of TM knockdown transfectants in NOD-SCID mice. pSM2c(vector control), sh-TM1, sh-TM2, and sh-TM3 cell suspensions (5 � 106/mouse)were subcutaneously injected into 8-week-old nonobese diabetic severe com-bined immunodeficiency (NOD-SCID) mice. Tumor growth was monitored ev-ery 2 days for 2 months. Tumors that grew to a volume over 500 mm3 and did notregress were considered to be established tumors. Tumors were not allowed togrow beyond 2,000 mm3 in accordance with Institutional Animal Care and UseCommittee regulations. Tumors were photographed and weighed after micewere sacrificed.
Immunohistochemistry staining. Paraffin-embedded blocks containing mousetumor tissue were fixed in 3.7% formalin and processed for hematoxylin-eosin(H&E) staining and immunohistochemical analysis. Tissue sections were washedin phosphate-buffered saline (PBS), and the primary antibodies bromodeoxyuri-dine (BrdU) (BD Biosciences) and loricrin (Covance) were applied to slides andincubated overnight. LSAB2 System horseradish peroxidase as secondary anti-body was applied to slides for 1 h. The immune complex was visualized afterincubation with �-amino-9-ethyl-carbazole (Dako) as a substrate.
Statistical analysis. All data are expressed as means � standard deviations(SD). Statistical significance of differences between any two groups was an-alyzed using an unpaired Student t test. Differences between more than twogroups were compared by one-way analysis of variance (ANOVA) followedby Bonferroni’s post hoc test, with a P value of 0.05 considered statisticallysignificant.
RESULTS
Inverse expression of TM and Snail. Whether TM down-regulation involves EMT-mediated tumorigenesis has neverbeen investigated. We first analyzed the expression levels ofEMT markers and TM in diverse epithelial cells and tumorcells. In 9/12 (75%) cells, Snail was highly expressed in BCC,TSGH8301, A2058, DU145, HepG2, Huh7, HeLa, SiHa, andKB cells that were accompanied by downregulation of bothE-cadherin and TM. In the same line of observation, N-cad-herin, a mesenchymal marker, was detected mostly in high-Snail-expressing cells, BCC, TSGH8301, A2058, HepG2,Huh7, and KB cells (50%). In contrast, low expression of Snailwas observed in HaCaT, A431, and Cx with high levels of�-catenin, E-cadherin, and TM (25%) (Fig. 1A). The 12 celllines were analyzed for their relative expression (fold) of�-catenin, E-cadherin, N-cadherin, TM, and Snail (Table 4).These results are consistent with the notion that adhesionmolecules are often downregulated in tumorigenesis (10, 26).Therefore, an inverse expression of TM and Snail suggests arole of TM in EMT.
Snail is a key transcription factor participating in EMT. It has
TABLE 3. ChIP assay primer sequences
Primer Direction Sequence (5�–3�)
1 Forward ACTCAGCGGGACGTTTGGReverse GGCCCAGTAGATCCAGGG
2 Forward GTGCAAGAAGCACCATCCTTReverse CCAGACCCCATCTCATCG
3 Forward GAGCTCTTGCAATCCAGGReverse CTGTAACAAGACGACTGT
TABLE 2. Probe sequences of TM in EMSA
Probe Sequence (5�–3�)a
Wild type......................S: 5�-GCAATTCACCTGCCACCGCCT-3�AS: 5�-AAGCGGTGGCAGGTGAATTGC-3�
Mutant..........................S: 5�-GCAATTAACCTACCACCGCCT-3�AS: 5�-AGGCGGTGGTAGGTTAATTGC-3�
a S, sense; AS, antisense.
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4769
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
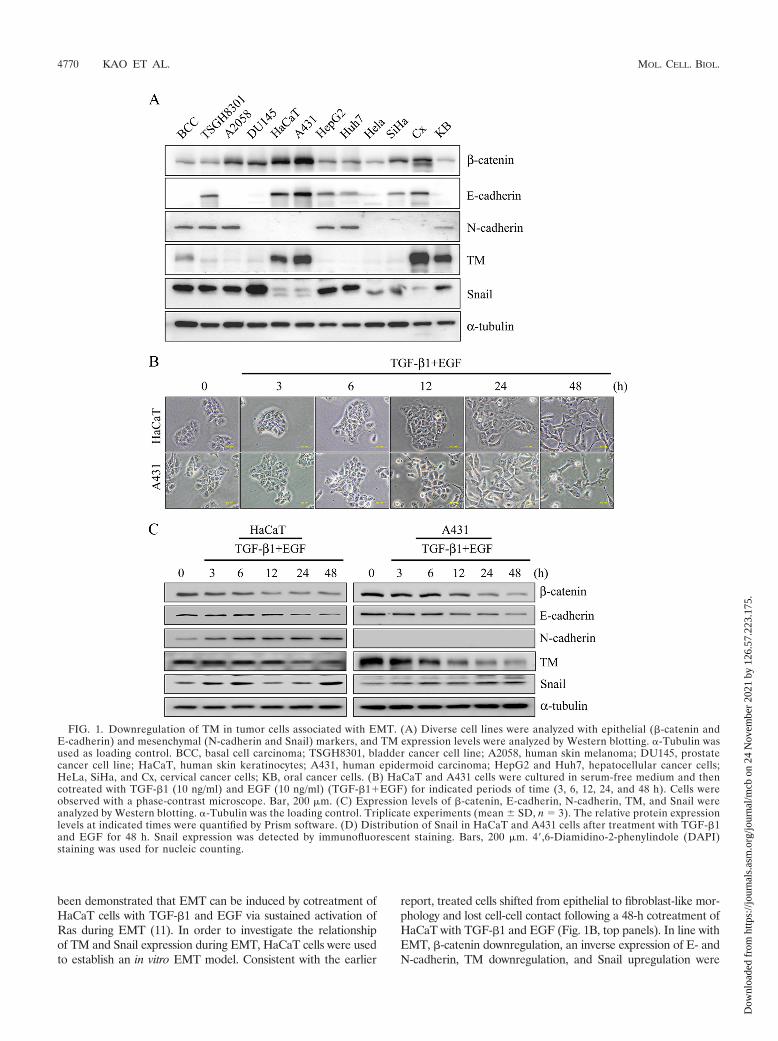
been demonstrated that EMT can be induced by cotreatment ofHaCaT cells with TGF-�1 and EGF via sustained activation ofRas during EMT (11). In order to investigate the relationshipof TM and Snail expression during EMT, HaCaT cells were usedto establish an in vitro EMT model. Consistent with the earlier
report, treated cells shifted from epithelial to fibroblast-like mor-phology and lost cell-cell contact following a 48-h cotreatment ofHaCaT with TGF-�1 and EGF (Fig. 1B, top panels). In line withEMT, �-catenin downregulation, an inverse expression of E- andN-cadherin, TM downregulation, and Snail upregulation were
FIG. 1. Downregulation of TM in tumor cells associated with EMT. (A) Diverse cell lines were analyzed with epithelial (�-catenin andE-cadherin) and mesenchymal (N-cadherin and Snail) markers, and TM expression levels were analyzed by Western blotting. �-Tubulin wasused as loading control. BCC, basal cell carcinoma; TSGH8301, bladder cancer cell line; A2058, human skin melanoma; DU145, prostatecancer cell line; HaCaT, human skin keratinocytes; A431, human epidermoid carcinoma; HepG2 and Huh7, hepatocellular cancer cells;HeLa, SiHa, and Cx, cervical cancer cells; KB, oral cancer cells. (B) HaCaT and A431 cells were cultured in serum-free medium and thencotreated with TGF-�1 (10 ng/ml) and EGF (10 ng/ml) (TGF-�1�EGF) for indicated periods of time (3, 6, 12, 24, and 48 h). Cells wereobserved with a phase-contrast microscope. Bar, 200 �m. (C) Expression levels of �-catenin, E-cadherin, N-cadherin, TM, and Snail wereanalyzed by Western blotting. �-Tubulin was the loading control. Triplicate experiments (mean � SD, n 3). The relative protein expressionlevels at indicated times were quantified by Prism software. (D) Distribution of Snail in HaCaT and A431 cells after treatment with TGF-�1and EGF for 48 h. Snail expression was detected by immunofluorescent staining. Bars, 200 �m. 4�,6-Diamidino-2-phenylindole (DAPI)staining was used for nucleic counting.
4770 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
observed in these cells by Western blotting (Fig. 1C, left panel).Immunofluorescent microscopy confirmed the nuclear expressionof Snail in the cotreated cells (Fig. 1D, left panels). Similar resultswere also observed in skin-derived A431 cells treated for 48 h withTGF-�1 and EGF (Fig. 1B, C, and D).
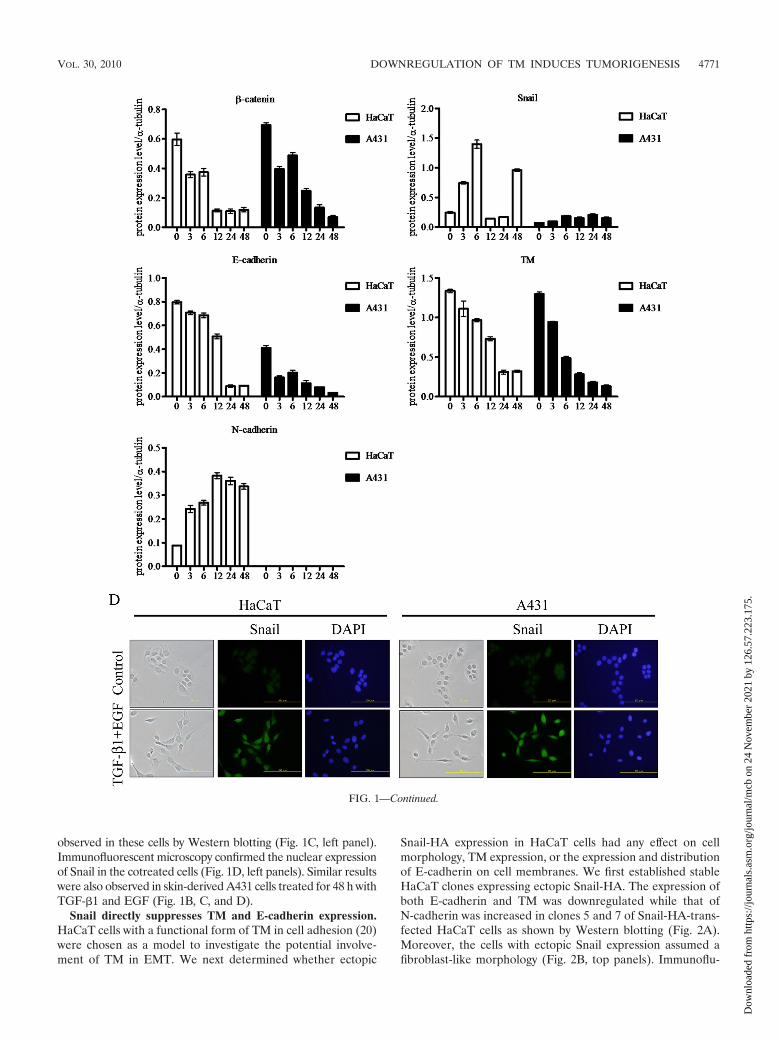
Snail directly suppresses TM and E-cadherin expression.HaCaT cells with a functional form of TM in cell adhesion (20)were chosen as a model to investigate the potential involve-ment of TM in EMT. We next determined whether ectopic
Snail-HA expression in HaCaT cells had any effect on cellmorphology, TM expression, or the expression and distributionof E-cadherin on cell membranes. We first established stableHaCaT clones expressing ectopic Snail-HA. The expression ofboth E-cadherin and TM was downregulated while that ofN-cadherin was increased in clones 5 and 7 of Snail-HA-trans-fected HaCaT cells as shown by Western blotting (Fig. 2A).Moreover, the cells with ectopic Snail expression assumed afibroblast-like morphology (Fig. 2B, top panels). Immunoflu-
FIG. 1—Continued.
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4771
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.

orescent staining confirmed the inverse expression of Snailwith TM or E-cadherin (Fig. 2B, panels a to i). �-Catenin is aninteracting protein of E-cadherin in the adherens junction andparticipates in maintaining epithelial cell-like morphology. Al-though there was no gross change in �-catenin expression, adiscontinuous rather than a continuous presence of �-cateninwas observed at the cell-cell boundary of Snail-HA-expressingcells (Fig. 2B, panels j to l). Together, these results indicatethat Snail may suppress TM expression in a fashion similar toits repressing effect on E-cadherin during EMT.
Binding of Snail to TM promoter suppresses TM expres-sion. Since TGF-�1 together with EGF induced an inverseexpression of TM and Snail during EMT of HaCaT and A431cells (Fig. 1B, C, and D), Snail, a transcription factor, maymodulate TM expression. By sequence alignment, a putativeSnail binding site (CACCTG) was identified between bp �828and �823 upstream from the transcription start site in thepromoter region of the TM gene (Fig. 3A). The sequence ofthe putative Snail binding site on the proximal TM promoterwas used to design wild-type and mutated probes for EMSA. Aprotein-DNA complex was observed in the reaction of thewild-type probe and nuclear extracts of TGF-�1- and EGF-cotreated HaCaT cells. Moreover, only excess cold wild-typeprobe, but not mutated probe, could effectively compete forthe protein binding to the radiolabeled wild-type probes, indi-cating a specific binding of the nuclear protein to the putativeSnail binding site (Fig. 3B). We next tested the exogenouseffect of Snail on the activity of TM promoter fragments usinga dual-luciferase reporter gene assay in CHO cells, which aremore susceptible than HaCaT cells to transient transfection.Consistent with the presence of a putative Snail binding site atbp �828 to �823, the promoter activity of TM (�1014) wassignificantly suppressed by cotransfected Snail-HA in CHOcells. When the TM promoter sequence was deleted to �745,Snail-HA showed significantly reduced ability to suppress thepromoter activity of the TM (�745) promoter construct (Fig.3C). Moreover, the in vivo direct binding of Snail-HA to theputative Snail binding site on the proximal TM promoter siteswas demonstrated by ChIP assay in ectopic Snail-expressingHaCaT cells (Fig. 3D, clone 7). Thus, the region between�1014 and �745 on the TM promoter harboring a Snail bind-
ing sequence is required for negative regulation of the TMpromoter by Snail.
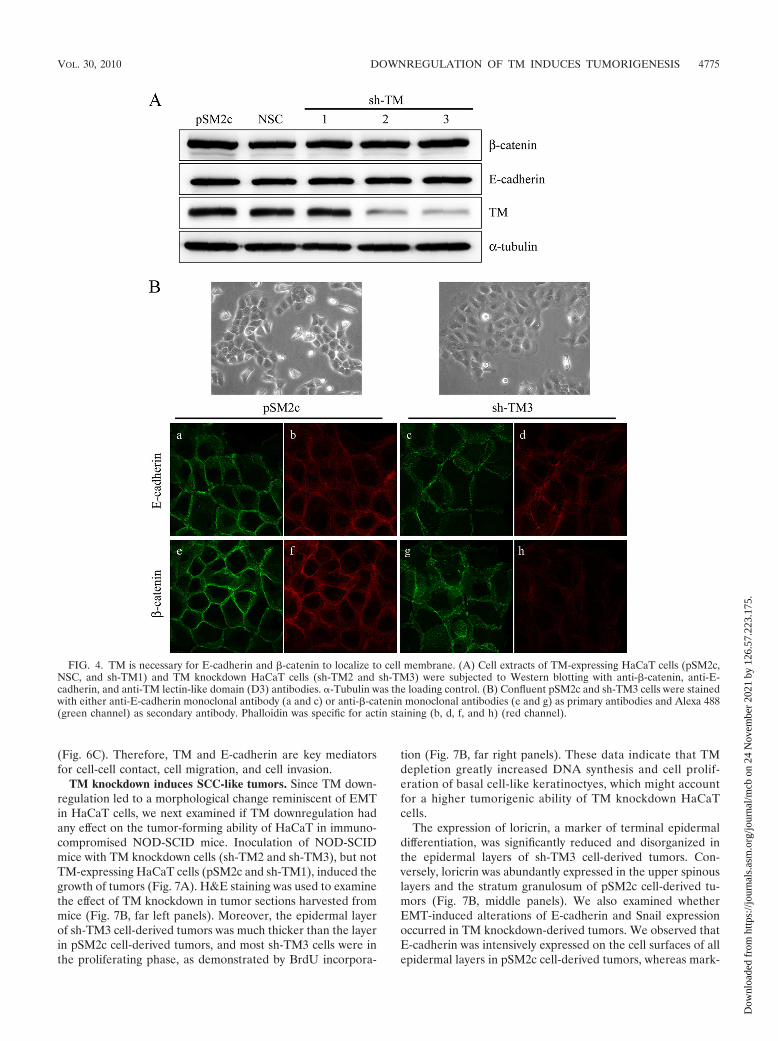
TM knockdown induces E-cadherin and �-catenin dissoci-ation from cell membrane. To investigate the role of TM in theassociation of E-cadherin and �-catenin in EMT, severalHaCaT cell lines were established, including pSM2c (vectorcontrol), NSC (nonsilencing control), sh-TM1 (TM nonknock-down transfectant), and sh-TM2 and sh-TM3 (TM knockdowntransfectants). Western blot analysis confirmed the decreasedTM expression in knockdown cells, and this knockdown had noeffect on the protein expression levels of �-catenin and E-cadherin (Fig. 4A). Interestingly, we observed that TM knock-down cells (sh-TM3) had flat cell morphology with overlappingcell-cell contact in comparison with the TM-expressing cells(pSM2c) (Fig. 4B). We also found that sh-TM3 cells hadhigher permeability than pSM2c cells, suggesting that TMknockdown induced loose cell-cell connections (see Fig. S1 inthe supplemental material). To evaluate whether the morpho-logical change induced by TM knockdown was involved inE-cadherin redistribution at cell contact, we cultured pSM2cand sh-TM3 cells to confluence and examined the subcellularlocalization of E-cadherin and �-catenin by immunofluores-cent staining. We found that both E-cadherin and �-catenincould regularly align at cell junctions in control pSM2c cells(Fig. 4B, panels a and e). In contrast, E-cadherin and �-cateninwere dislodged from cell-cell junctions in TM knockdown sh-TM3 cells (Fig. 4B, panels c and g). The F-actin aligned alongthe cell-cell junctions in the pSM2c cells (Fig. 4B, panels b andf) but disappeared in the sh-TM3 cells (Fig. 4B, panels d andh). Altogether, TM downregulation disrupted cell contactprobably by affecting the association of E-cadherin with �-cate-nin, indicating an essential role of TM in E-cadherin-mediatedcell-cell contact.
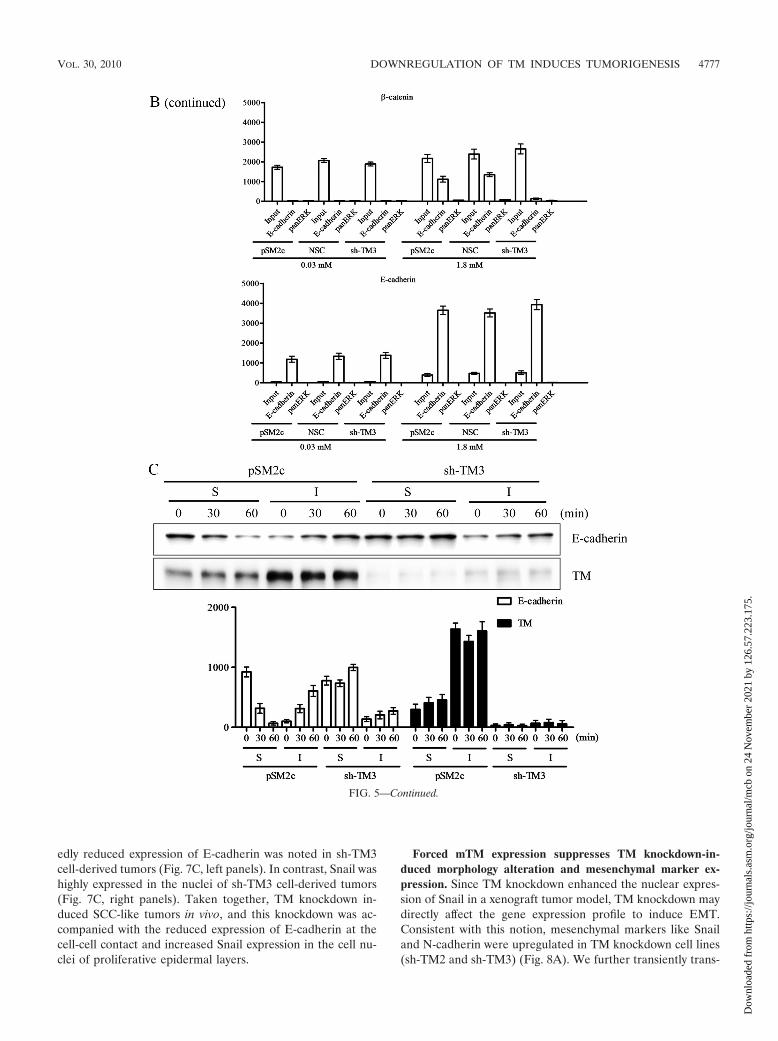
TM knockdown abolishes E-cadherin-mediated cell-cellcontact formation. Since TM knockdown had a negative effecton the integrity of cell-cell junctions, we next examined if TMknockdown also had an impact on cell junction rebuilding. Acalcium switch assay was used to test how TM regulated E-cadherin-mediated cell-cell contacts. TM-expressing HaCaTcells (pSM2c) and TM knockdown HaCaT cells (sh-TM3) werecultured in 0.03 mM calcium-containing medium for 24 h andthen in 1.8 mM calcium-containing medium for another 24 h.After calcium switch, control pSM2c cells reassumed epithelialmorphology. Conversely, sh-TM3 cells remained fibroblast-likeand the distribution of E-cadherin and �-catenin was diffusedin the cytoplasm even when they were exposed to 1.8 mMcalcium for 24 h (Fig. 5A). The coimmunoprecipitation assaydemonstrated that calcium switch reduced the association ofE-cadherin with �-catenin in sh-TM3 cells compared withpSM2c and NSC cells (Fig. 5B). Furthermore, we extractedcytoplasmic and membrane fractions to determine whetherTM is essential in E-cadherin dynamics. We found that E-cadherin in pSM2c cells, but not sh-TM3 cells, could translo-cate from cytoplasm to cell membrane after the calcium con-centration was switched from 0.03 mM to 1.8 mM (Fig. 5C).Based on these observations, we conclude that TM is requiredfor maintaining epithelial cell-cell adhesion by maintainingE-cadherin on cell membrane.
TM knockdown promotes cell motility. Since E-cadherin isimportant in contact inhibition, loss of E-cadherin leads to the
TABLE 4. Cell line phenotypes
Cell lineRelative expression (fold) ofa:
�-Catenin E-cadherin N-cadherin TM Snail
BCC 1.0 0 1.3 8.9 11.1TSGH8301 1.7 1.5 2.2 2.7 14.0A2058 3.6 0 1.6 1.5 9.7DU145 4.7 0 0 1.9 23.1HaCaT 9.0 2.2 0 35.3 1.3A431 15.4 3.4 0 49.9 1.0HepG2 3.3 1.7 3.1 3.1 21.8Huh7 3.1 1.0 2.8 1.0 15.5HeLa 1.3 0 0 1.4 2.8SiHa 3.6 0.6 0 2.0 4.7Cx 6.7 1.7 0 79.2 1.6KB 1.3 0 1.0 53.5 10.5
a The relative expression levels (fold) of �-catenin, E-cadherin, N-cadherin,TM, and Snail were normalized by �-tubulin.
4772 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
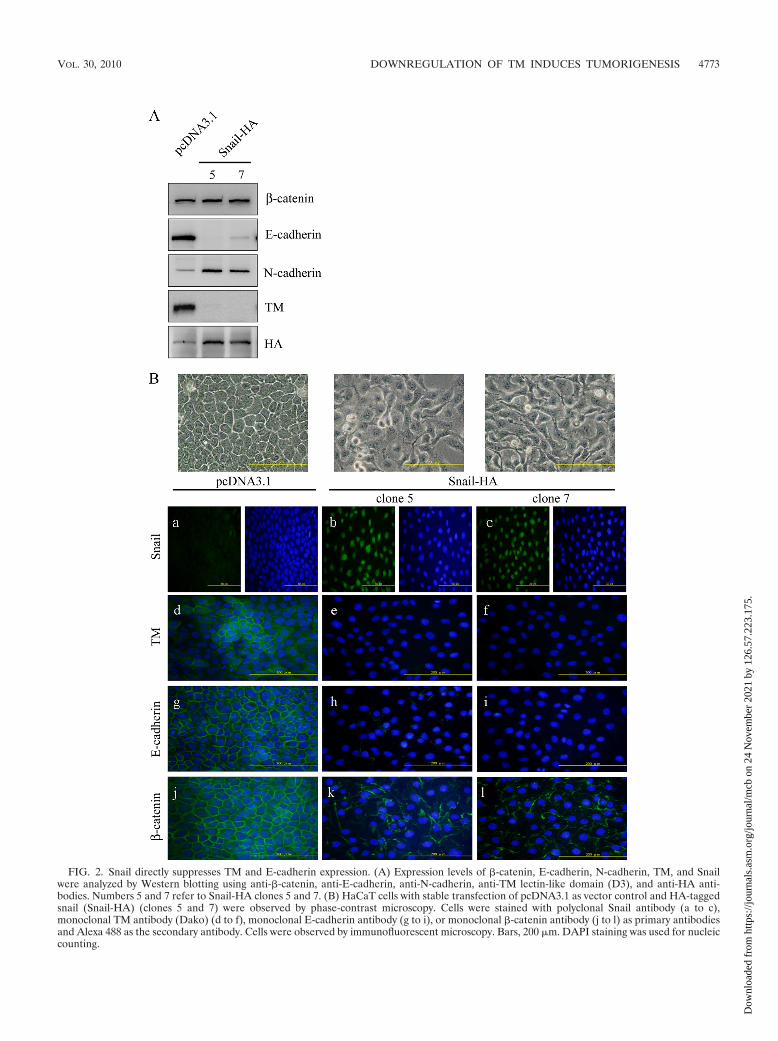
FIG. 2. Snail directly suppresses TM and E-cadherin expression. (A) Expression levels of �-catenin, E-cadherin, N-cadherin, TM, and Snailwere analyzed by Western blotting using anti-�-catenin, anti-E-cadherin, anti-N-cadherin, anti-TM lectin-like domain (D3), and anti-HA anti-bodies. Numbers 5 and 7 refer to Snail-HA clones 5 and 7. (B) HaCaT cells with stable transfection of pcDNA3.1 as vector control and HA-taggedsnail (Snail-HA) (clones 5 and 7) were observed by phase-contrast microscopy. Cells were stained with polyclonal Snail antibody (a to c),monoclonal TM antibody (Dako) (d to f), monoclonal E-cadherin antibody (g to i), or monoclonal �-catenin antibody (j to l) as primary antibodiesand Alexa 488 as the secondary antibody. Cells were observed by immunofluorescent microscopy. Bars, 200 �m. DAPI staining was used for nucleiccounting.
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4773
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
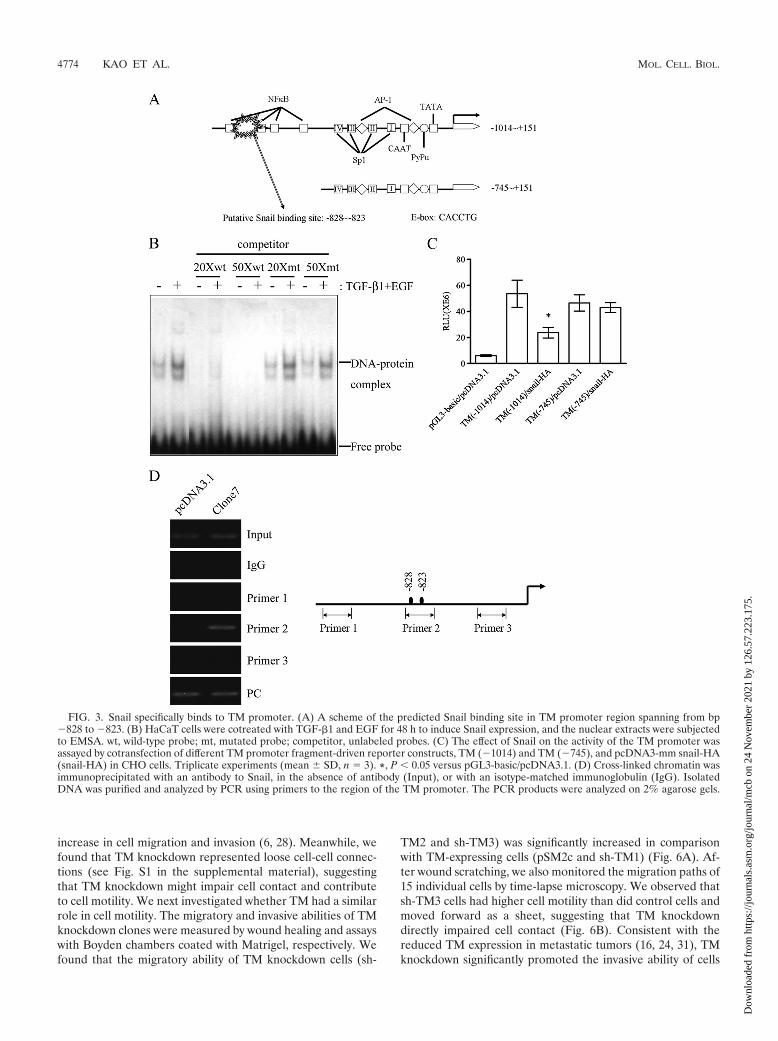
increase in cell migration and invasion (6, 28). Meanwhile, wefound that TM knockdown represented loose cell-cell connec-tions (see Fig. S1 in the supplemental material), suggestingthat TM knockdown might impair cell contact and contributeto cell motility. We next investigated whether TM had a similarrole in cell motility. The migratory and invasive abilities of TMknockdown clones were measured by wound healing and assayswith Boyden chambers coated with Matrigel, respectively. Wefound that the migratory ability of TM knockdown cells (sh-
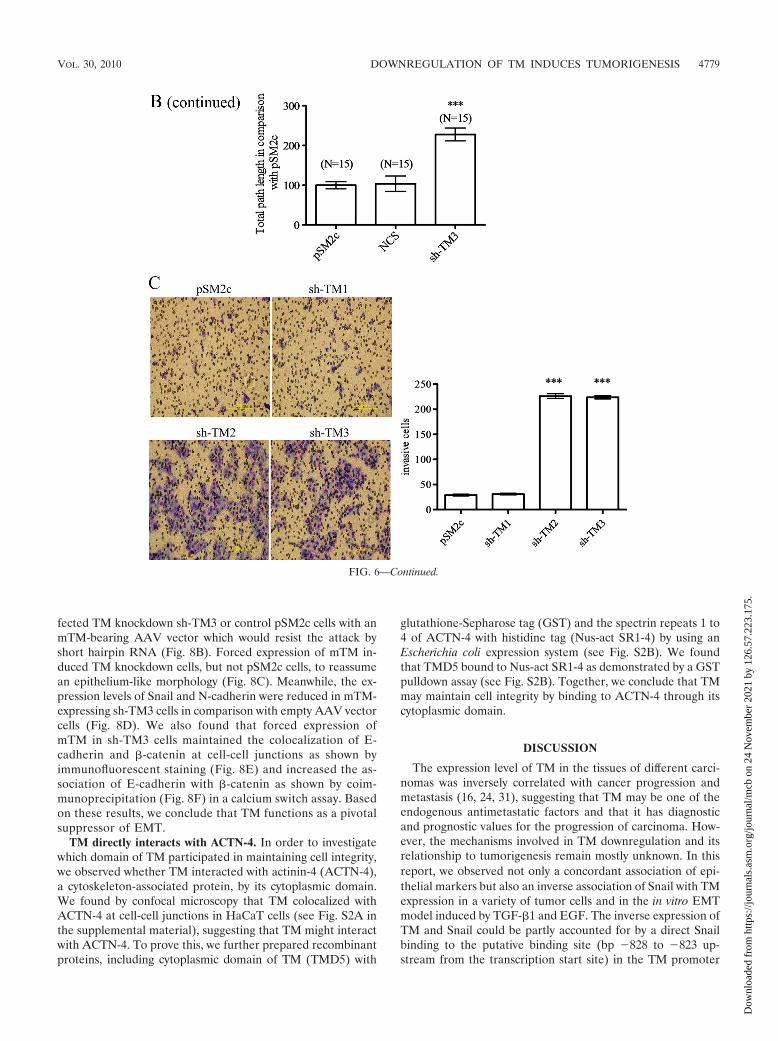
TM2 and sh-TM3) was significantly increased in comparisonwith TM-expressing cells (pSM2c and sh-TM1) (Fig. 6A). Af-ter wound scratching, we also monitored the migration paths of15 individual cells by time-lapse microscopy. We observed thatsh-TM3 cells had higher cell motility than did control cells andmoved forward as a sheet, suggesting that TM knockdowndirectly impaired cell contact (Fig. 6B). Consistent with thereduced TM expression in metastatic tumors (16, 24, 31), TMknockdown significantly promoted the invasive ability of cells
FIG. 3. Snail specifically binds to TM promoter. (A) A scheme of the predicted Snail binding site in TM promoter region spanning from bp�828 to �823. (B) HaCaT cells were cotreated with TGF-�1 and EGF for 48 h to induce Snail expression, and the nuclear extracts were subjectedto EMSA. wt, wild-type probe; mt, mutated probe; competitor, unlabeled probes. (C) The effect of Snail on the activity of the TM promoter wasassayed by cotransfection of different TM promoter fragment-driven reporter constructs, TM (�1014) and TM (�745), and pcDNA3-mm snail-HA(snail-HA) in CHO cells. Triplicate experiments (mean � SD, n 3). *, P 0.05 versus pGL3-basic/pcDNA3.1. (D) Cross-linked chromatin wasimmunoprecipitated with an antibody to Snail, in the absence of antibody (Input), or with an isotype-matched immunoglobulin (IgG). IsolatedDNA was purified and analyzed by PCR using primers to the region of the TM promoter. The PCR products were analyzed on 2% agarose gels.
4774 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
(Fig. 6C). Therefore, TM and E-cadherin are key mediatorsfor cell-cell contact, cell migration, and cell invasion.
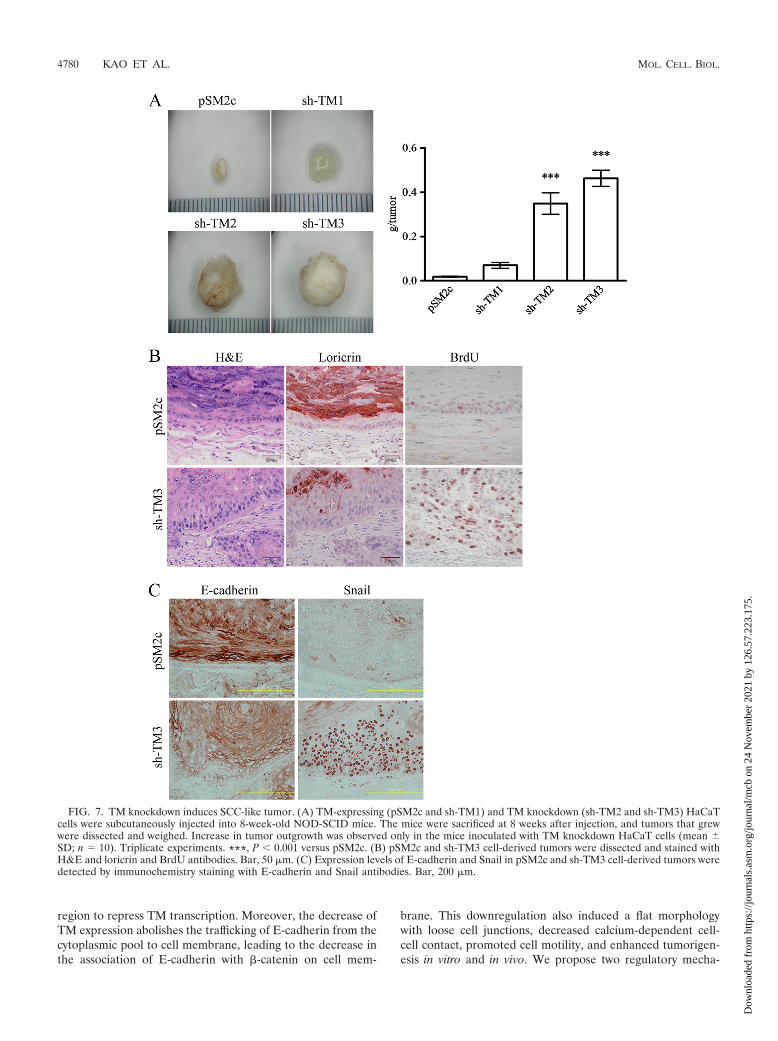
TM knockdown induces SCC-like tumors. Since TM down-regulation led to a morphological change reminiscent of EMTin HaCaT cells, we next examined if TM downregulation hadany effect on the tumor-forming ability of HaCaT in immuno-compromised NOD-SCID mice. Inoculation of NOD-SCIDmice with TM knockdown cells (sh-TM2 and sh-TM3), but notTM-expressing HaCaT cells (pSM2c and sh-TM1), induced thegrowth of tumors (Fig. 7A). H&E staining was used to examinethe effect of TM knockdown in tumor sections harvested frommice (Fig. 7B, far left panels). Moreover, the epidermal layerof sh-TM3 cell-derived tumors was much thicker than the layerin pSM2c cell-derived tumors, and most sh-TM3 cells were inthe proliferating phase, as demonstrated by BrdU incorpora-
tion (Fig. 7B, far right panels). These data indicate that TMdepletion greatly increased DNA synthesis and cell prolif-eration of basal cell-like keratinoctyes, which might accountfor a higher tumorigenic ability of TM knockdown HaCaTcells.
The expression of loricrin, a marker of terminal epidermaldifferentiation, was significantly reduced and disorganized inthe epidermal layers of sh-TM3 cell-derived tumors. Con-versely, loricrin was abundantly expressed in the upper spinouslayers and the stratum granulosum of pSM2c cell-derived tu-mors (Fig. 7B, middle panels). We also examined whetherEMT-induced alterations of E-cadherin and Snail expressionoccurred in TM knockdown-derived tumors. We observed thatE-cadherin was intensively expressed on the cell surfaces of allepidermal layers in pSM2c cell-derived tumors, whereas mark-
FIG. 4. TM is necessary for E-cadherin and �-catenin to localize to cell membrane. (A) Cell extracts of TM-expressing HaCaT cells (pSM2c,NSC, and sh-TM1) and TM knockdown HaCaT cells (sh-TM2 and sh-TM3) were subjected to Western blotting with anti-�-catenin, anti-E-cadherin, and anti-TM lectin-like domain (D3) antibodies. �-Tubulin was the loading control. (B) Confluent pSM2c and sh-TM3 cells were stainedwith either anti-E-cadherin monoclonal antibody (a and c) or anti-�-catenin monoclonal antibodies (e and g) as primary antibodies and Alexa 488(green channel) as secondary antibody. Phalloidin was specific for actin staining (b, d, f, and h) (red channel).
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4775
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
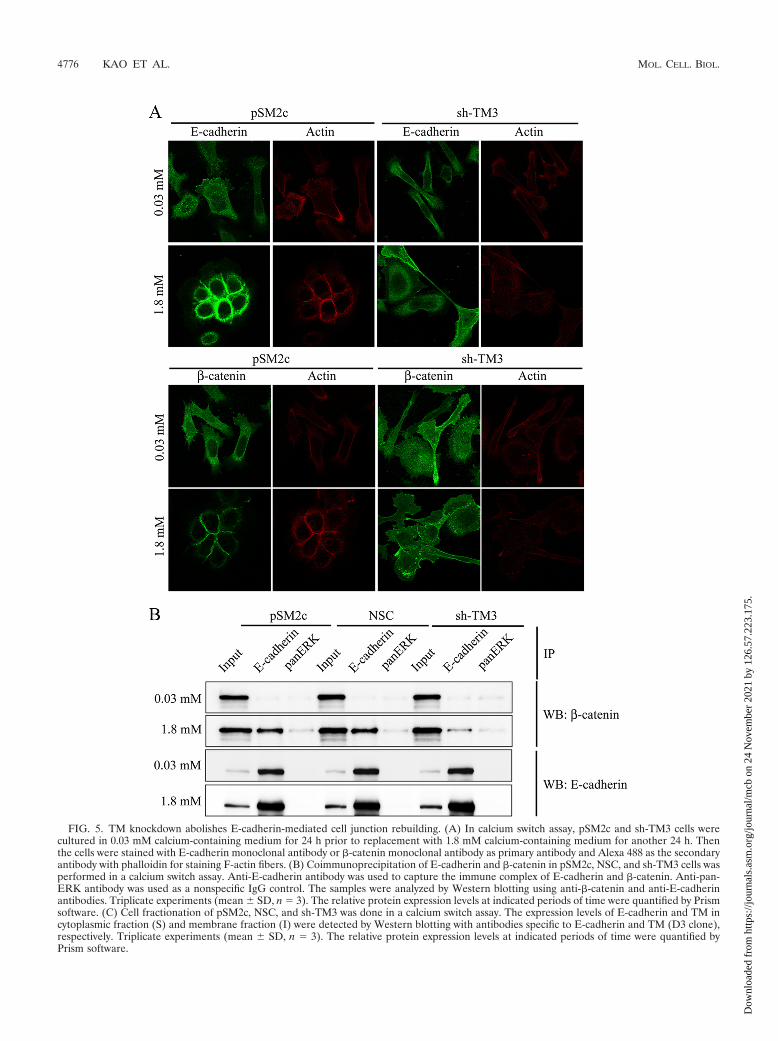
FIG. 5. TM knockdown abolishes E-cadherin-mediated cell junction rebuilding. (A) In calcium switch assay, pSM2c and sh-TM3 cells werecultured in 0.03 mM calcium-containing medium for 24 h prior to replacement with 1.8 mM calcium-containing medium for another 24 h. Thenthe cells were stained with E-cadherin monoclonal antibody or �-catenin monoclonal antibody as primary antibody and Alexa 488 as the secondaryantibody with phalloidin for staining F-actin fibers. (B) Coimmunoprecipitation of E-cadherin and �-catenin in pSM2c, NSC, and sh-TM3 cells wasperformed in a calcium switch assay. Anti-E-cadherin antibody was used to capture the immune complex of E-cadherin and �-catenin. Anti-pan-ERK antibody was used as a nonspecific IgG control. The samples were analyzed by Western blotting using anti-�-catenin and anti-E-cadherinantibodies. Triplicate experiments (mean � SD, n 3). The relative protein expression levels at indicated periods of time were quantified by Prismsoftware. (C) Cell fractionation of pSM2c, NSC, and sh-TM3 was done in a calcium switch assay. The expression levels of E-cadherin and TM incytoplasmic fraction (S) and membrane fraction (I) were detected by Western blotting with antibodies specific to E-cadherin and TM (D3 clone),respectively. Triplicate experiments (mean � SD, n 3). The relative protein expression levels at indicated periods of time were quantified byPrism software.
4776 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
edly reduced expression of E-cadherin was noted in sh-TM3cell-derived tumors (Fig. 7C, left panels). In contrast, Snail washighly expressed in the nuclei of sh-TM3 cell-derived tumors(Fig. 7C, right panels). Taken together, TM knockdown in-duced SCC-like tumors in vivo, and this knockdown was ac-companied with the reduced expression of E-cadherin at thecell-cell contact and increased Snail expression in the cell nu-clei of proliferative epidermal layers.
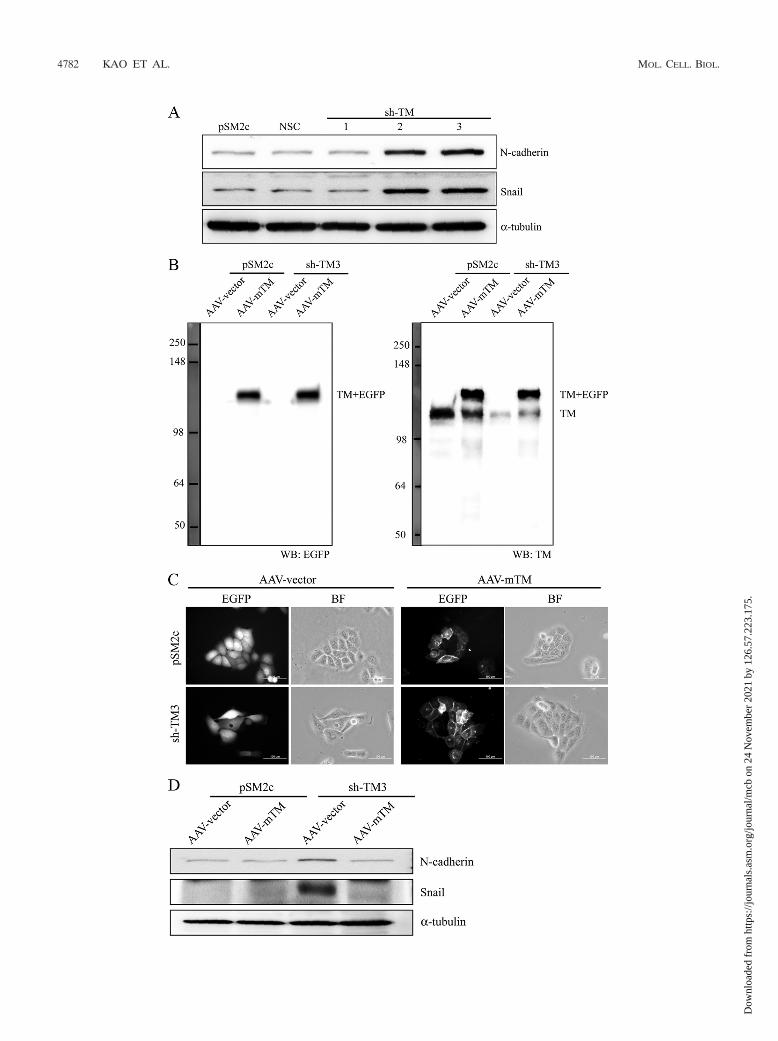
Forced mTM expression suppresses TM knockdown-in-duced morphology alteration and mesenchymal marker ex-pression. Since TM knockdown enhanced the nuclear expres-sion of Snail in a xenograft tumor model, TM knockdown maydirectly affect the gene expression profile to induce EMT.Consistent with this notion, mesenchymal markers like Snailand N-cadherin were upregulated in TM knockdown cell lines(sh-TM2 and sh-TM3) (Fig. 8A). We further transiently trans-
FIG. 5—Continued.
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4777
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
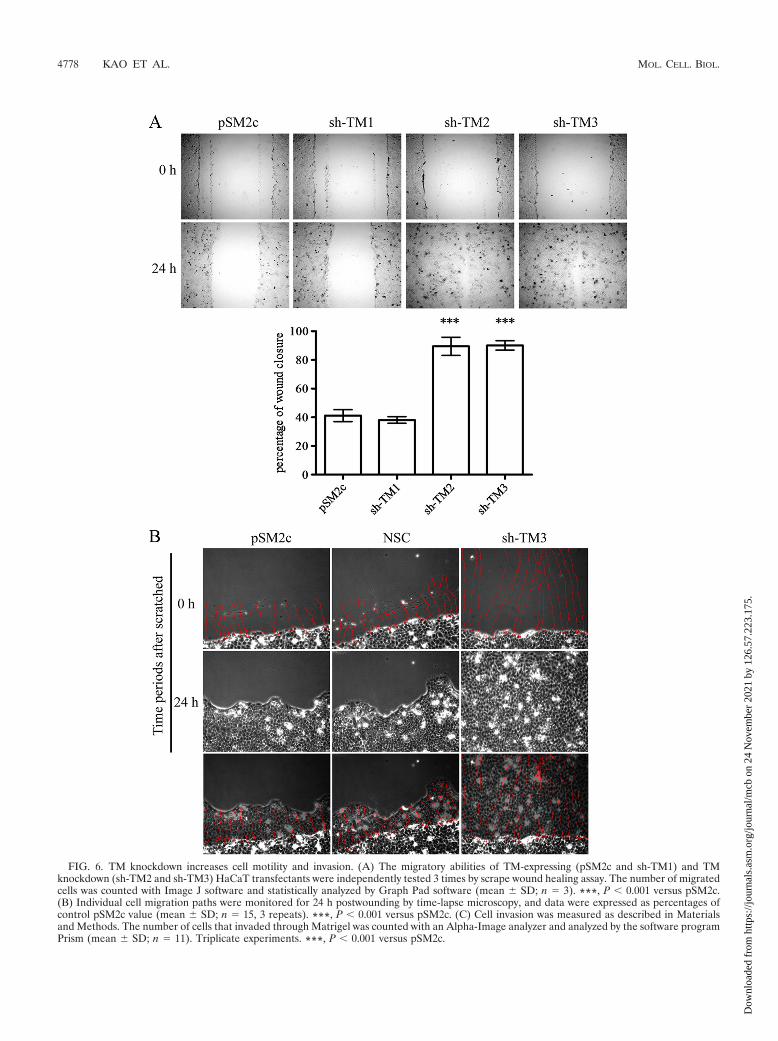
FIG. 6. TM knockdown increases cell motility and invasion. (A) The migratory abilities of TM-expressing (pSM2c and sh-TM1) and TMknockdown (sh-TM2 and sh-TM3) HaCaT transfectants were independently tested 3 times by scrape wound healing assay. The number of migratedcells was counted with Image J software and statistically analyzed by Graph Pad software (mean � SD; n 3). ***, P 0.001 versus pSM2c.(B) Individual cell migration paths were monitored for 24 h postwounding by time-lapse microscopy, and data were expressed as percentages ofcontrol pSM2c value (mean � SD; n 15, 3 repeats). ***, P 0.001 versus pSM2c. (C) Cell invasion was measured as described in Materialsand Methods. The number of cells that invaded through Matrigel was counted with an Alpha-Image analyzer and analyzed by the software programPrism (mean � SD; n 11). Triplicate experiments. ***, P 0.001 versus pSM2c.
4778 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
fected TM knockdown sh-TM3 or control pSM2c cells with anmTM-bearing AAV vector which would resist the attack byshort hairpin RNA (Fig. 8B). Forced expression of mTM in-duced TM knockdown cells, but not pSM2c cells, to reassumean epithelium-like morphology (Fig. 8C). Meanwhile, the ex-pression levels of Snail and N-cadherin were reduced in mTM-expressing sh-TM3 cells in comparison with empty AAV vectorcells (Fig. 8D). We also found that forced expression ofmTM in sh-TM3 cells maintained the colocalization of E-cadherin and �-catenin at cell-cell junctions as shown byimmunofluorescent staining (Fig. 8E) and increased the as-sociation of E-cadherin with �-catenin as shown by coim-munoprecipitation (Fig. 8F) in a calcium switch assay. Basedon these results, we conclude that TM functions as a pivotalsuppressor of EMT.
TM directly interacts with ACTN-4. In order to investigatewhich domain of TM participated in maintaining cell integrity,we observed whether TM interacted with actinin-4 (ACTN-4),a cytoskeleton-associated protein, by its cytoplasmic domain.We found by confocal microscopy that TM colocalized withACTN-4 at cell-cell junctions in HaCaT cells (see Fig. S2A inthe supplemental material), suggesting that TM might interactwith ACTN-4. To prove this, we further prepared recombinantproteins, including cytoplasmic domain of TM (TMD5) with
glutathione-Sepharose tag (GST) and the spectrin repeats 1 to4 of ACTN-4 with histidine tag (Nus-act SR1-4) by using anEscherichia coli expression system (see Fig. S2B). We foundthat TMD5 bound to Nus-act SR1-4 as demonstrated by a GSTpulldown assay (see Fig. S2B). Together, we conclude that TMmay maintain cell integrity by binding to ACTN-4 through itscytoplasmic domain.
DISCUSSION
The expression level of TM in the tissues of different carci-nomas was inversely correlated with cancer progression andmetastasis (16, 24, 31), suggesting that TM may be one of theendogenous antimetastatic factors and that it has diagnosticand prognostic values for the progression of carcinoma. How-ever, the mechanisms involved in TM downregulation and itsrelationship to tumorigenesis remain mostly unknown. In thisreport, we observed not only a concordant association of epi-thelial markers but also an inverse association of Snail with TMexpression in a variety of tumor cells and in the in vitro EMTmodel induced by TGF-�1 and EGF. The inverse expression ofTM and Snail could be partly accounted for by a direct Snailbinding to the putative binding site (bp �828 to �823 up-stream from the transcription start site) in the TM promoter
FIG. 6—Continued.
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4779
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
region to repress TM transcription. Moreover, the decrease ofTM expression abolishes the trafficking of E-cadherin from thecytoplasmic pool to cell membrane, leading to the decrease inthe association of E-cadherin with �-catenin on cell mem-
brane. This downregulation also induced a flat morphologywith loose cell junctions, decreased calcium-dependent cell-cell contact, promoted cell motility, and enhanced tumorigen-esis in vitro and in vivo. We propose two regulatory mecha-
FIG. 7. TM knockdown induces SCC-like tumor. (A) TM-expressing (pSM2c and sh-TM1) and TM knockdown (sh-TM2 and sh-TM3) HaCaTcells were subcutaneously injected into 8-week-old NOD-SCID mice. The mice were sacrificed at 8 weeks after injection, and tumors that grewwere dissected and weighed. Increase in tumor outgrowth was observed only in the mice inoculated with TM knockdown HaCaT cells (mean �SD; n 10). Triplicate experiments. ***, P 0.001 versus pSM2c. (B) pSM2c and sh-TM3 cell-derived tumors were dissected and stained withH&E and loricrin and BrdU antibodies. Bar, 50 �m. (C) Expression levels of E-cadherin and Snail in pSM2c and sh-TM3 cell-derived tumors weredetected by immunochemistry staining with E-cadherin and Snail antibodies. Bar, 200 �m.
4780 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.

nisms involved in the expression of E-cadherin, whichparticipate in Snail governing EMT (Fig. 9). First, Snail candirectly suppress E-cadherin expression at the transcriptionallevel. Second, Snail can also bind to the TM promoter todownregulate its expression. TM downregulation alters thebalance of E-cadherin dynamics and leads to the accumulationof E-cadherin in cytoplasmic pools. The impairment of cell-cellcontact and cytoskeletal integrity by the increase of Snail ex-pression is likely through downregulation of not only E-cad-herin but also TM. Altogether, TM functions as one key factorfor maintaining epithelial morphology.
A negative association of TM and Snail expression was firstdetected in immortalized HaCaT cells and a variety of carci-noma cell lines. We further confirmed the inverse relation ofTM and Snail during the EMT elicited by TGF-�1 and EGF incultured cells, TM knockdown sh-TM3 cells, and SCC-liketumors derived from sh-TM3 cells in the murine xenograftmodel. Although we report a novel pathway by which Snailrepresses TM expression via direct promoter binding duringEMT (Fig. 9), we also observed that ectopic TM expressionsuppressed Snail expression by transfection of shRNA-resis-tant mTM-EGFP (enhanced green fluorescent protein) fusion-expressing vector (Fig. 8D) and a cell type-specific induction(biphasic activation in HaCaT cells versus sustained activationin A431 cells) of Snail expression during EMT in cultured cells(Fig. 3C). To test whether forced expression of TM reversedthe effects elicited by TGF-�1 and EGF, mTM-EGFP-express-ing HaCaT cells were cotreated with TGF-�1 and EGF forEMT induction. We found that the cells with forced expressionof mTM still maintained epithelium-like morphology even un-der TGF-�1 and EGF stimulation for 48 h (see Fig. S3 in thesupplemental material). Meanwhile, we tested the expressionlevels of E-cadherin, TM, and Snail in these cells. A decreaseof endogenous TM and E-cadherin together with the increaseof Snail was observed. Taken together, mTM is able to main-tain epithelium-like morphology but fails to reverse the down-regulation of E-cadherin and upregulation of Snail elicited byTGF-�1 and EGF. Since mTM cDNA constructed in an AAVvector was driven by a cytomegalovirus (CMV) promoterwhich has no Snail binding site, TGF-�1- and EGF-inducedSnail could not suppress the expression of ectopic mTM.Therefore, ectopic mTM expression maintains epithelial cellmorphology of transfected cells even under TGF-�1 and EGFstimulation. This result is consistent with our previous reportthat TM functions as a cell-to-cell adhesion molecule even inthe absence of E-cadherin (20).
A tumor microenvironment rich in growth factors and cyto-kines may also play a role in the relation of TM and Snailexpression in vivo. More studies are needed to reveal the un-derlying mechanism responsible for the interrelationship ofTM and Snail, particularly how TM expression directly orindirectly suppresses Snail expression. TM promoter methyl-ation at CpG islands has been reported to be one mechanismresponsible for TM downregulation in melanoma cells (13).Although we cannot completely rule out this possibility, wewere able to demonstrate that TM expression could also benegatively regulated by Snail binding to its promoter region.Snail participates in modulating the expression of many genesassociated with tumor progression, including E-cadherin, ma-trix metalloproteinase 2, tissue inhibitor of metalloproteinase
1, tissue plasminogen activator, and ras homolog gene familymember A (25). We found that cotreatment of HaCaT andA431 cells with TGF-�1 and EGF induced Snail while reduc-ing the expression of E-cadherin and TM. A putative Snail-binding E-box (�828/�823) in the proximal TM promoterregion was confirmed by EMSA, promoter-driven luciferaseassay, and ChIP assay (Fig. 3B, C, and D). We demonstratedthat the E-box could be a functional element for Snail bindingto suppress TM expression. Along the same line, ectopic ex-pression of HA-tagged Snail induced a fibroblast-like pheno-type together with the reduction of TM and E-cadherin ex-pression in HaCaT cells (Fig. 2B). Therefore, we are the firstto demonstrate that Snail is a key suppressor of TM expressionduring EMT. Although there are other regulatory factors inthe Snail family, including Slug and the E12/47 transcriptionalfactors (33), more studies are needed to address the involve-ment of these factors in the modulation of TM expressionduring EMT. Together, our findings suggest that TM functionsas an “initiator” or a “stabilizer” in the formation of a stablecomplex, including E-cadherin and the associated proteins, atcell-cell junctions to maintain epithelial morphology.
Various adhesion molecules are involved in cell junctionformation with E-cadherin under different biological condi-tions. Loss of these molecules may lead to E-cadherin delocal-ization from the cell membrane rather than a decrease inexpression level of E-cadherin. For instance, Scribble is a con-served polarity protein required for neural differentiation andEMT. Disruption of E-cadherin-mediated cell adhesion byknockdown of Scribble increases cell migration and promotesa more fibroblast-like phenotype (35), which partially mimicsEMT in these cells. Similarly, AF6i3 is involved in signalingand organization of cell junctions during embryogenesis.Knockdown of AF6i3 leads to impaired E-cadherin-dependentcell adhesion due to the dissociation of E-cadherin and �-cate-nin (27). In line with our observations, we found that theprotein expression levels of typical adhesion molecules such asE-cadherin and �-catenin were not affected by knockdown ofTM expression (Fig. 4A). However, E-cadherin and �-cateninwere mostly in the cytoplasm rather than at the cell-cell bound-ary in TM knockdown HaCaT cells (Fig. 4B). Altogether, TMmay contribute to cell-cell adhesion by facilitating the localiza-tion of E-cadherin and �-catenin at the cell junctions.
Although initially identified in endothelial cells, TM was alsoexpressed in epithelial keratinocytes but limited to the supra-basal layer (36). The expression of TM was also increased inskin keratinocytes treated with high calcium (1 mM) but notwith reduced calcium (29). The expression of TM is thus pos-itively associated with keratinocyte differentiation. Consistentwith this notion, TM knockdown promoted the growth in miceof SCC-like tumors, where higher proliferative potential ac-counted for the larger tumors formed by these cells. Loricrin,a terminally differentiated marker, remained detectable al-though at a low level with uneven expression in the granularlayer around the nest of keratin pearls in the tumors of TMknockdown HaCaT cells (Fig. 7B, middle panels), indicatingthat this tumor was in a more poorly differentiated state be-cause of the knockdown. The reduced expression of E-cad-herin in the TM knockdown-derived tumors further supportedthe malignant potential of the tumors. The tumor-promotingeffect of TM knockdown observed in murine tumorigenesis was
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4781
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
4782 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.
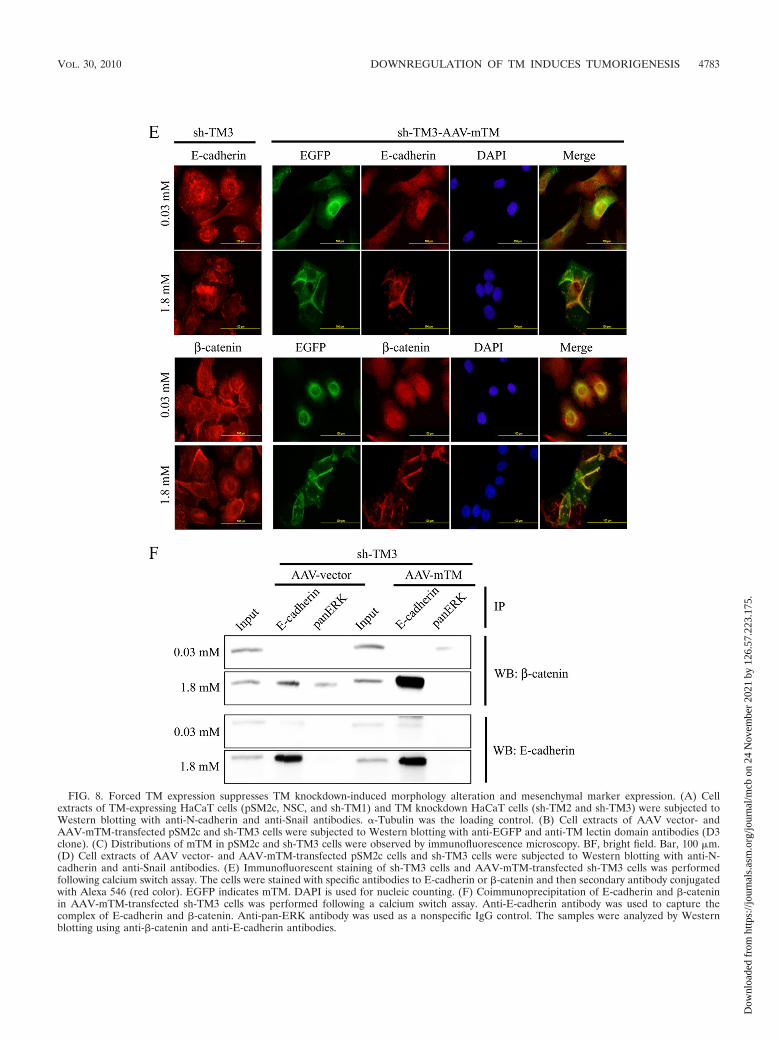
FIG. 8. Forced TM expression suppresses TM knockdown-induced morphology alteration and mesenchymal marker expression. (A) Cellextracts of TM-expressing HaCaT cells (pSM2c, NSC, and sh-TM1) and TM knockdown HaCaT cells (sh-TM2 and sh-TM3) were subjected toWestern blotting with anti-N-cadherin and anti-Snail antibodies. �-Tubulin was the loading control. (B) Cell extracts of AAV vector- andAAV-mTM-transfected pSM2c and sh-TM3 cells were subjected to Western blotting with anti-EGFP and anti-TM lectin domain antibodies (D3clone). (C) Distributions of mTM in pSM2c and sh-TM3 cells were observed by immunofluorescence microscopy. BF, bright field. Bar, 100 �m.(D) Cell extracts of AAV vector- and AAV-mTM-transfected pSM2c cells and sh-TM3 cells were subjected to Western blotting with anti-N-cadherin and anti-Snail antibodies. (E) Immunofluorescent staining of sh-TM3 cells and AAV-mTM-transfected sh-TM3 cells was performedfollowing calcium switch assay. The cells were stained with specific antibodies to E-cadherin or �-catenin and then secondary antibody conjugatedwith Alexa 546 (red color). EGFP indicates mTM. DAPI is used for nucleic counting. (F) Coimmunoprecipitation of E-cadherin and �-cateninin AAV-mTM-transfected sh-TM3 cells was performed following a calcium switch assay. Anti-E-cadherin antibody was used to capture thecomplex of E-cadherin and �-catenin. Anti-pan-ERK antibody was used as a nonspecific IgG control. The samples were analyzed by Westernblotting using anti-�-catenin and anti-E-cadherin antibodies.
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4783
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.

consistent with the poor prognosis of cancer patients withreduced TM expression (15, 21, 24, 31). Taken together, ourstudy further supports a tumor-suppressing role of TM.
There are two approaches commonly used to address poten-tial off-target effects caused by the use of shRNA. First, morethan one shRNA targeting different regions of the same targetgene could be used to examine if either one has the sameknockdown effect. Second, forced expression of a cDNA ofinterest without the shRNA targeting sites would prevent thecDNA from being attacked by the shRNA in the same cells. Inthis report, we overexpressed the mTM lack-of-recognitionsequence by TM1.9 (Table 1) in the cells expressing thisshRNA to examine any off-target effects. As predicted, only theexpression of endogenous TM rather than mTM fusion proteinwas decreased by TM1.9 (Fig. 8B, right panel), supporting aTM-specific shRNA used in this study. Moreover, ectopic ex-pression of mTM reversed TM knockdown-induced alterationof cell morphology (Fig. 8C) and maintained the colocalizationof E-cadherin and �-catenin at cell junctions in a calcium-dependent manner (Fig. 8E and F). Since we successfully re-versed the TM knockdown effects by introduction of mTMexpression, the observed phenotypic change induced by TMknockdown was unlikely due to off-target effects.
ACTN-4, a scaffold protein connecting the cytoskeleton tocell junctions, is a biomarker for cancer invasion and metasta-sis in several cancer types (7, 19, 23, 32, 46). During the processof tumor cell migration, the expression of ACTN-4 is increasedand mainly localized at the leading edge of migrating cells (19).Silencing the expression of ACTN-4 restores E-cadherin-me-diated cell-cell contact and reduces the cell motility of pancre-atic cancer cells (23). Knockdown of E-cadherin librates�-catenin from cell membrane, and the free �-catenin togetherwith ACTN-4 is recruited into cell protrusion in colorectalcancer cells (17). In our unpublished data, we found that over-
expression of TM in A2058 cells upregulated the expressionlevel of ACTN-4 and increased its association with TM asshown by using confocal microscopy and immunoprecipitation.Moreover, this association in HaCaT cells was highly concen-trated at cell-cell contact (see Fig. S2A in the supplementalmaterial). GST pulldown assay further confirmed that the cy-toplasmic tail of TM is required for its interaction withACTN-4 (see Fig. S2B and C). Altogether, TM holds E-cad-herin in check at the cell-cell junction and maintains epithelialmorphology. The loss of TM expression not only induced themesenchymal phenotype but also increased cell motility partlyvia the accumulation of E-cadherin in the cytoplasmic pool anddissociation of TM from ACTN-4, thereby increasing the poolof the free ACTN-4 and �-catenin to cell protrusions for cellmigration (Fig. 9).
In conclusion, the expression of TM and Snail in varioushuman tumor cells and during EMT is not only inversely cor-related but also interregulated. Snail is a novel transcriptionalrepressor of TM via a direct binding to the Snail-responsiveE-box in the TM proximal promoter. TM downregulation ei-ther by knockdown or by induction by EMT led to a dissocia-tion of E-cadherin with �-catenin at cell-cell junctions. There-fore, the expression level of TM may function as a gatekeeperto prevent epithelial cells from undergoing EMT and may beused as a prognostic or therapeutic target for cancer prognosisor treatment. Our studies provide another biological functionof TM besides its role in cell adhesion, angiogenesis, andanti-inflammation.
ACKNOWLEDGMENTS
This work was supported by National Science Council, ExecutiveYuan, Taipei, Taiwan, grants NSC 97-2752-B-006-003-PAE, NSC 97-2752-B-006-004-PAE, and NSC 97-2752-B-006-005-PAE.
REFERENCES
1. Barbour, W., S. Saika, T. Miyamoto, K. Ohkawa, H. Utsunomiya, and Y.Ohnishi. 2004. Expression patterns of beta1-related alpha integrin subunitsin murine lens during embryonic development and wound healing. Curr. EyeRes. 29:1–10.
2. Batlle, E., E. Sancho, C. Franci, D. Dominguez, M. Monfar, J. Baulida, andA. Garcia De Herreros. 2000. The transcription factor snail is a repressor ofE-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2:84–89.
3. Becker, K. F., E. Rosivatz, K. Blechschmidt, E. Kremmer, M. Sarbia, and H.Hofler. 2007. Analysis of the E-cadherin repressor Snail in primary humancancers. Cells Tissues Organs 185:204–212.
4. Bolos, V., H. Peinado, M. A. Perez-Moreno, M. F. Fraga, M. Esteller, and A.Cano. 2003. The transcription factor Slug represses E-cadherin expressionand induces epithelial to mesenchymal transitions: a comparison with Snailand E47 repressors. J. Cell Sci. 116:499–511.
5. Boukamp, P., R. T. Petrussevska, D. Breitkreutz, J. Hornung, A. Markham,and N. E. Fusenig. 1988. Normal keratinization in a spontaneously immor-talized aneuploid human keratinocyte cell line. J. Cell Biol. 106:761–771.
6. Bracke, M. E., H. Depypere, C. Labit, V. Van Marck, K. Vennekens, S. J.Vermeulen, I. Maelfait, J. Philippe, R. Serreyn, and M. M. Mareel. 1997.Functional downregulation of the E-cadherin/catenin complex leads to lossof contact inhibition of motility and of mitochondrial activity, but not ofgrowth in confluent epithelial cell cultures. Eur. J. Cell Biol. 74:342–349.
7. Broderick, M. J., and S. J. Winder. 2005. Spectrin, alpha-actinin, and dys-trophin. Adv. Protein Chem. 70:203–246.
8. Brown, K. A., M. E. Aakre, A. E. Gorska, J. O. Price, S. E. Eltom, J. A.Pietenpol, and H. L. Moses. 2004. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. BreastCancer Res. 6:R215–R231.
9. Cano, A., M. A. Perez-Moreno, I. Rodrigo, A. Locascio, M. J. Blanco, M. G.del Barrio, F. Portillo, and M. A. Nieto. 2000. The transcription factor snailcontrols epithelial-mesenchymal transitions by repressing E-cadherin expres-sion. Nat. Cell Biol. 2:76–83.
10. Conacci-Sorrell, M., J. Zhurinsky, and A. Ben-Ze’ev. 2002. The cadherin-catenin adhesion system in signaling and cancer. J. Clin. Invest. 109:987–991.
FIG. 9. Proposed role of TM in EMT process. Inducers (such asTGF-�1 and EGF) upregulate the expression level of Snail, whichleads to EMT directly or indirectly. Snail can either suppress expres-sion of E-cadherin at the transcriptional level (1, direct manner) orinhibit TM promoter activity to dissociate E-cadherin from �-catenin(2, indirect manner), which promotes cell motility. Finally, both directand indirect mechanisms result in EMT and tumorigenesis. Thedashed red line represents the mechanism of TM knockdown-inducedenhanced cell motility. The dashed black line shows an interrelation-ship between Snail and TM.
4784 KAO ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.

11. Davies, M., M. Robinson, E. Smith, S. Huntley, S. Prime, and I. Paterson.2005. Induction of an epithelial to mesenchymal transition in human immor-tal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad andAP-1 signalling pathways. J. Cell. Biochem. 95:918–931.
12. Esmon, C. T. 1987. The regulation of natural anticoagulant pathways. Sci-ence 235:1348–1352.
13. Furuta, J., A. Kaneda, Y. Umebayashi, F. Otsuka, T. Sugimura, and T.Ushijima. 2005. Silencing of the thrombomodulin gene in human malignantmelanoma. Melanoma Res. 15:15–20.
14. Grunert, S., M. Jechlinger, and H. Beug. 2003. Diverse cellular and molec-ular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev.Mol. Cell Biol. 4:657–665.
15. Hanly, A. M., A. Hayanga, D. C. Winter, and D. J. Bouchier-Hayes. 2005.Thrombomodulin: tumour biology and prognostic implications. Eur. J. Surg.Oncol. 31:217–220.
16. Hanly, A. M., M. Redmond, D. C. Winter, S. Brophy, J. M. Deasy, D. J.Bouchier-Hayes, and E. W. Kay. 2006. Thrombomodulin expression in colo-rectal carcinoma is protective and correlates with survival. Br. J. Cancer94:1320–1325.
17. Hayashida, Y., K. Honda, M. Idogawa, Y. Ino, M. Ono, A. Tsuchida, T. Aoki,S. Hirohashi, and T. Yamada. 2005. E-cadherin regulates the associationbetween beta-catenin and actinin-4. Cancer Res. 65:8836–8845.
18. Healy, A. M., H. B. Rayburn, R. D. Rosenberg, and H. Weiler. 1995. Absenceof the blood-clotting regulator thrombomodulin causes embryonic lethalityin mice before development of a functional cardiovascular system. Proc.Natl. Acad. Sci. U. S. A. 92:850–854.
19. Honda, K., T. Yamada, R. Endo, Y. Ino, M. Gotoh, H. Tsuda, Y. Yamada, H.Chiba, and S. Hirohashi. 1998. Actinin-4, a novel actin-bundling proteinassociated with cell motility and cancer invasion. J. Cell Biol. 140:1383–1393.
20. Huang, H. C., G. Y. Shi, S. J. Jiang, C. S. Shi, C. M. Wu, H. Y. Yang, andH. L. Wu. 2003. Thrombomodulin-mediated cell adhesion: involvement of itslectin-like domain. J. Biol. Chem. 278:46750–46759.
21. Iino, S., K. Abeyama, K. Kawahara, M. Yamakuchi, T. Hashiguchi, S. Mat-sukita, S. Yonezawa, S. Taniguchi, M. Nakata, S. Takao, T. Aikou, and I.Maruyama. 2004. The antimetastatic role of thrombomodulin expression inislet cell-derived tumors and its diagnostic value. Clin. Cancer Res. 10:6179–6188.
22. Kang, Y., and J. Massague. 2004. Epithelial-mesenchymal transitions: twistin development and metastasis. Cell 118:277–279.
23. Kikuchi, S., K. Honda, H. Tsuda, N. Hiraoka, I. Imoto, T. Kosuge, T. Umaki,K. Onozato, M. Shitashige, U. Yamaguchi, M. Ono, A. Tsuchida, T. Aoki,J. Inazawa, S. Hirohashi, and T. Yamada. 2008. Expression and gene am-plification of actinin-4 in invasive ductal carcinoma of the pancreas. Clin.Cancer Res. 14:5348–5356.
24. Kim, S. J., E. Shiba, H. Ishii, T. Inoue, T. Taguchi, Y. Tanji, Y. Kimoto, M.Izukura, and S. Takai. 1997. Thrombomodulin is a new biological andprognostic marker for breast cancer: an immunohistochemical study. Anti-cancer Res. 17:2319–2323.
25. Kuphal, S., H. G. Palm, I. Poser, and A. K. Bosserhoff. 2005. Snail-regulatedgenes in malignant melanoma. Melanoma Res. 15:305–313.
26. Lioni, M., P. Brafford, C. Andl, A. Rustgi, W. El-Deiry, M. Herlyn, and K. S.Smalley. 2007. Dysregulation of claudin-7 leads to loss of E-cadherin expres-sion and the increased invasion of esophageal squamous cell carcinoma cells.Am. J. Pathol. 170:709–721.
27. Lorger, M., and K. Moelling. 2006. Regulation of epithelial wound closureand intercellular adhesion by interaction of AF6 with actin cytoskeleton.J. Cell Sci. 119:3385–3398.
28. Mandal, M., J. N. Myers, S. M. Lippman, F. M. Johnson, M. D. Williams, S.Rayala, K. Ohshiro, D. I. Rosenthal, R. S. Weber, G. E. Gallick, and A. K.El-Naggar. 2008. Epithelial to mesenchymal transition in head and necksquamous carcinoma: association of Src activation with E-cadherin down-regulation, vimentin expression, and aggressive tumor features. Cancer 112:2088–2100.
29. Mizutani, H., T. Hayashi, N. Nouchi, S. Ohyanagi, K. Hashimoto, M.Shimizu, and K. Suzuki. 1994. Functional and immunoreactive thrombo-modulin expressed by keratinocytes. J. Invest. Dermatol. 103:825–828.
30. Navarro, P., M. Gomez, A. Pizarro, C. Gamallo, M. Quintanilla, and A.Cano. 1991. A role for the E-cadherin cell-cell adhesion molecule duringtumor progression of mouse epidermal carcinogenesis. J. Cell Biol. 115:517–533.
31. Ogawa, H., S. Yonezawa, I. Maruyama, Y. Matsushita, Y. Tezuka, H.Toyoyama, M. Yanagi, H. Matsumoto, H. Nishijima, T. Shimotakahara, T.Aikou, and E. Sato. 2000. Expression of thrombomodulin in squamous cellcarcinoma of the lung: its relationship to lymph node metastasis and prog-nosis of the patients. Cancer Lett. 149:95–103.
32. Otey, C. A., and O. Carpen. 2004. Alpha-actinin revisited: a fresh look at anold player. Cell Motil. Cytoskeleton 58:104–111.
33. Peinado, H., D. Olmeda, and A. Cano. 2007. Snail, Zeb and bHLH factors intumour progression: an alliance against the epithelial phenotype? Nat. Rev.Cancer 7:415–428.
34. Pignatelli, M., T. W. Ansari, P. Gunter, D. Liu, S. Hirano, M. Takeichi, G.Kloppel, and N. R. Lemoine. 1994. Loss of membranous E-cadherin expres-sion in pancreatic cancer: correlation with lymph node metastasis, highgrade, and advanced stage. J. Pathol. 174:243–248.
35. Qin, Y., C. Capaldo, B. M. Gumbiner, and I. G. Macara. 2005. The mam-malian Scribble polarity protein regulates epithelial cell adhesion and mi-gration through E-cadherin. J. Cell Biol. 171:1061–1071.
36. Raife, T. J., D. J. Lager, K. C. Madison, W. W. Piette, E. J. Howard, M. T.Sturm, Y. Chen, and S. R. Lentz. 1994. Thrombomodulin expression byhuman keratinocytes. Induction of cofactor activity during epidermal differ-entiation. J. Clin. Invest. 93:1846–1851.
37. Roberts, A. B., and L. M. Wakefield. 2003. The two faces of transforminggrowth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 100:8621–8623.
38. Shi, C. S., G. Y. Shi, Y. S. Chang, H. S. Han, C. H. Kuo, C. Liu, H. C. Huang,Y. J. Chang, P. S. Chen, and H. L. Wu. 2005. Evidence of human thrombo-modulin domain as a novel angiogenic factor. Circulation 111:1627–1636.
39. Shi, C. S., G. Y. Shi, S. M. Hsiao, Y. C. Kao, K. L. Kuo, C. Y. Ma, C. H. Kuo,B. I. Chang, C. F. Chang, C. H. Lin, C. H. Wong, and H. L. Wu. 2008.Lectin-like domain of thrombomodulin binds to its specific ligand Lewis Yantigen and neutralizes lipopolysaccharide-induced inflammatory response.Blood 112:3661–3670.
40. Shiozaki, H., T. Kadowaki, Y. Doki, M. Inoue, S. Tamura, H. Oka, T.Iwazawa, S. Matsui, K. Shimaya, M. Takeichi, et al. 1995. Effect of epider-mal growth factor on cadherin-mediated adhesion in a human oesophagealcancer cell line. Br. J. Cancer 71:250–258.
41. Shiozaki, H., H. Tahara, H. Oka, M. Miyata, K. Kobayashi, S. Tamura, K.Iihara, Y. Doki, S. Hirano, M. Takeichi, et al. 1991. Expression of immuno-reactive E-cadherin adhesion molecules in human cancers. Am. J. Pathol.139:17–23.
42. Shook, D., and R. Keller. 2003. Mechanisms, mechanics and function ofepithelial-mesenchymal transitions in early development. Mech. Dev. 120:1351–1383.
43. Suzuki, K., H. Kusumoto, Y. Deyashiki, J. Nishioka, I. Maruyama, M. Zushi,S. Kawahara, G. Honda, S. Yamamoto, and S. Horiguchi. 1987. Structureand expression of human thrombomodulin, a thrombin receptor on endo-thelium acting as a cofactor for protein C activation. EMBO J. 6:1891–1897.
44. Vega, S., A. V. Morales, O. H. Ocana, F. Valdes, I. Fabregat, and M. A. Nieto.2004. Snail blocks the cell cycle and confers resistance to cell death. GenesDev. 18:1131–1143.
45. Watabe, M., K. Matsumoto, T. Nakamura, and M. Takeichi. 1993. Effect ofhepatocyte growth factor on cadherin-mediated cell-cell adhesion. CellStruct. Funct. 18:117–124.
46. Yamagata, N., Y. Shyr, K. Yanagisawa, M. Edgerton, T. P. Dang, A. Gonza-lez, S. Nadaf, P. Larsen, J. R. Roberts, J. C. Nesbitt, R. Jensen, S. Levy, J. H.Moore, J. D. Minna, and D. P. Carbone. 2003. A training-testing approach tothe molecular classification of resected non-small cell lung cancer. Clin.Cancer Res. 9:4695–4704.
47. Yuan, Z., S. Wong, A. Borrelli, and M. A. Chung. 2007. Down-regulation ofMUC1 in cancer cells inhibits cell migration by promoting E-cadherin/cate-nin complex formation. Biochem. Biophys. Res. Commun. 362:740–746.
VOL. 30, 2010 DOWNREGULATION OF TM INDUCES TUMORIGENESIS 4785
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 24
Nov
embe
r 20
21 b
y 12
6.57
.223
.175
.





























