Embed Size (px)
Citation preview

DNA to Disease Examining phenotypic expression of DNA mutations
Jenny Kravitz (Methuen HS) and Kari Roberts (Tufts) Overview These lessons are designed to reinforce genetics concepts that define DNA structure and function the process of transcription and translation into a protein product and ultimately the effect on an individualrsquos observed phenotype Activities are designed in a case study format to provide an opportunity for students to apply fundamental genetics concepts (transcription translation gene expression mutations) so that they can better understand how mutations may or may not lead to phenotypic changes Students will be asked to serve as ldquodoctorsrdquo putting the previously learned genetics concepts into a medical context to increase student interest and relate material to personal experiences (newspaper articles family history newborn screening) Misconceptions
Human disease results directly from alterationsmutations in DNA (Students fail to recognize the intermediate steps involving RNA and protein expression)
All mutations are deleterious ldquobreakingrdquo the protein to completely prevent normal function (Students fail to recognize that affected proteins may have partial function or may be absent entirely)
Massachusetts Learning Standards Addressed Standard 3 Genes allow for the storage and transmission of genetic information They are a set of instructions encoded in the nucleotide sequence of each organism Genes code for the specific sequences of amino acids that comprise the proteins that are characteristic of that organism
Strand 32 Describe the basic process of DNA replication and how it relates to the transmission and conservation of the genetic code Explain the basic processes of transcription and translation and how they result in the expression of genes Distinguish among the end products of replication transcription and translation Strand 33 Explain how mutations in the DNA sequence of a gene may or may not result in phenotypic change in an organism Explain how mutations in gametes may result in phenotypic changes in offspring
Major Science Concepts Addressed
DNA structure and function RNA structure and function Protein structure and function Transcription Translation Mutations Phenotypes and genotypes Gene expression
- 1 -
Learning Objectives 1 Students will be able to explain the processes of transcription and translation 2 Students will be able to read and understand given data (graphs tables gels genetic
sequences) 3 Students will be able to interpret and apply data to explain how DNA mutations affect
each step in the process of gene expression 4 Students will be able to interpret and apply data to predict phenotype 5 Students will be able to communicate ideas effectively though visual aids and oral
presentation Materials
Patient analysis worksheet (for group work as teams of ldquodoctorsrdquo) Overhead teacher example worksheet (for modeling with Sickle Cell) Student example worksheet (for modeling with Sickle Cell) Patient summary sheet (description and data for each disease) Codon reference chart (ldquothe wheelrdquo) Rounds Analysis Worksheet (to use during presentations) Genetics formal assessment (given as pre- andor post-test) Posterboard (or large white sheets of paper) and markers
Duration Five class periods (50 minutes each) Prior Knowledge
DNA structure and function RNA structure and function Protein structure and function Transcription Translation Mutations Phenotypes and genotypes Gene expression Reading and understanding data (graphs tables DNA sequences gels)
Description Day One Modeling with Sickle Cell Anemia Learning Cycle Engage The instructor leads the class through an example using sickle cell anemia The instructor should put the Patient Analysis Worksheet up on the overhead (or using a projector) and give each student his or her own blank copy Students should also be given the Patient Summary Sheet for sickle cell anemia Through class discussion the instructor and the students should complete the patient analysis together The instructor should elicit answers from the students and the class should discuss the rationale behind each correct answer This discussion is likely to identify the specific misconceptions and points of confusion for each individual class which the
- 2 -
instructor should address as they come up Through this modeling students should understand how to complete the subsequent group activity and what is expected of them Day Two Analysis of Patient Learning Cycle Explore Students should be placed into groups of 4 Each group is a team of ldquodoctorsrdquo and they will be analyzing various medical case studies all of which are single gene disorders with variable penetrance Each group should be given one Patient Analysis Worksheet and one Patient Summary Sheet (various diseases) Each group of doctors should receive a different patient Using the sickle cell model as a guide teams should then analyze their own specific patient and complete the Patient Analysis Worksheet Day Three Preparation for Rounds Learning Cycle Explain Each team of doctors will be responsible for giving an oral presentation on their patient during ldquoroundsrdquo (next class) Requirements include a poster and an oral description of the genetic disease affecting the patient Two students in the group should be responsible for putting together the poster which should include a clear title the names of all doctors on the team a list of typical symptoms a flow chart describing the effects of the mutation (DNA rarr mRNA rarr protein rarr phenotype) and all applicable images associated with their disease (x-rays photographs gels etc) The other two students in the group will be responsible for orally describing the disease (patient presentation genetic cause applicable treatmenttherapy options) The two students should take turns Therefore during this planning day those students should split of the material to be presented and prepare notecards to use while presenting Thus half of the team is responsible for the visual component of the presentation and half of the team is responsible for the oral component of the presentation Day Four Rounds (Presentations) Learning Cycle Elaborate Each team should present their case to the all the other doctors (approx 5 mins) Before each presentation listening doctors should each be given a copy of the Rounds Analysis Worksheet While other teams are presenting all other doctors are responsible for paying close attention to the details of the disease being described At the conclusion of the presentation a short question and answer period (approx 2-3 min) should give all listening doctors a chance to probe for further information clarify information presented or simply provide feedback Listening doctors should also receive a short amount of time to complete and submit Rounds Analysis Worksheets (approx 2-3 min) Total time for each case is therefore approximately 10 minutes Day Five Quiz (formal summative assessment) Learning Cycle Evaluate The final component of the activity is a quiz (taken individually) which should directly target each of the learning objectives If desired by the instructor a version of this quiz can be given as a pre-assessment to determine both the studentsrsquo starting point (identify potential misconceptions) and the success of the activity in reinforcing the applicable genetics concepts
- 3 -
Assessment Formative The instructor should formatively assess students throughout the entire process in order to address areas of confusion and guide group progress The instructor will model disease analysis with the class by completing the disease worksheet interactively through class discussion The instructor should clarify as student confusion or misconceptions arise and the instructor should communicate expectations for subsequent group work The instructor should also formatively assess students during group work by circulating and interacting with each group as they analyze their patient or prepare for ldquoroundsrdquo Summative Instructor should summatively assess groups upon completion of ldquoroundsrdquo (oral presentation) Groups should turn in their patient analysis worksheet to be graded (see rubric A) and instructor should evaluate oral presentation (ldquoroundsrdquo) for groupindividual grade (see rubric B) Also students will have a formal summative assessment (quiz) on underlying concepts (learning objectives) for individual assessment (see Genetics formal assessment) Possible Extensions
Discuss examples of diseases in which proteins are over- or underexpressed (not just a ldquobrokenrdquo protein) examples of diseases which involve environmental factors and examples of disease that display less than 100 penetrance
Explore emerging genetic technologies which may be used to treat or correct the diseases studied in the cases
- 4 -
Implementation Report (2009) The ldquoDNA to Diseaserdquo lesson was implemented in two Honors freshman biology classes April 13-17 2009 In total there were 36 students 23 male and 13 female The classes had already formally learned all the applicable genetics concepts (see Prior Knowledge above) In addition they had just completed a protein gel electrophoresis lab in which they diagnosed patients as affected unaffected or carriers in respect to sickle cell anemia This provided an excellent segue into Day One of this lesson in which the classroom teacher modeled completion of the patient analysis worksheet using sickle cell anemia Class discussion during this modeling was extremely engaging for students who revealed their misconceptions as well as their personal interests in medical genetics At the conclusion of Day One the students had a good idea of what was expected of them during Day Two For Day Two the patient analysis students self-selected teams of doctors (4 students per group) and were assigned their cases No two teams of doctors received the same genetic disorder Using the sickle cell anemia sheet from Day One as a guide the students were able to successfully complete the patient analysis sheet Both the classroom teacher (Jenny Kravitz) and the higher education partner (Kari Roberts) circulated the room to work with students to clarify information answer questions and keep them on task In general the students appreciated analyzing ldquorealrdquo medical information (ie radiography films) even though the material was challenging at times At the conclusion of Day Two the majority of doctor teams had completed their patient analysis sheets On Day Three the student groups completed their patient analyses and prepared their presentations for rounds (Day Four) Students were given a large white sheet of paper and markers as well as a guide for setting up their poster (see Poster Format Guide) Again both the classroom teacher and the higher education partner circulated the room to assist and monitor students At the end of Day Three most of the doctor teams had completed their posters (a few groups took them home to complete) Day Four was when each class did rounds in which each team of doctors took approximately five minutes to give an overview of their patient (using their poster which was taped to the wall as a guide) The other students filled out the Rounds Analysis Worksheet during the presentation and used it as a guide to direct questioning after the formal presentation For most groups a significant question and answer period entirely student driven occurred after their 5-minute formal presentation Very little clarification was offered by the classroom teacher and the higher education partner and for the most part the students were able to field questions successfully The Rounds Analysis Worksheet appeared to drive students to participate in a thorough and interactive question and answer period which required more critical thinking for students to compare and contrast the various diseases and disorders On Day Five the students finished up rounds presentations that were overflow from Day Four After all presentations the students were given the post-test (Genetics Formal Assessment) Rounds posters were kept up on the walls of the classroom until the end of the school year which prompted discussions about genetic disorders in a variety of classes that did not participate in this lesson implementation further extending the impact
- 5 -
Analysis of pre- and post-test data The formal summative assessment tool was a 20 question (multiple choice) quiz This quiz was administered prior to the implementation comparison between the pre- and post-test scores are used to assess the strengths and weaknesses of our implementation Questions were drawn from both textbook and MCAS examinations Table 1 outlines the question source and the genetics concepts that are addressed
Table 1
Question Source Concepts 1 textbook 5 2 textbook 6 3 textbook 1 8 4 textbook 4 5 MCAS 5 6 textbook 2 7 MCAS 3 6 8 8 textbook 5 9 textbook 1 7 8
10 MCAS 3 4 8 11 MCAS 3 6 8 12 MCAS 1 6 7 13 textbook 1 2 14 textbook 1 2 15 textbook 1 16 textbook 6 17 MCAS 4 8 18 textbook 4 1 2 19 MCAS 1 20 textbook 5
Concept Definition (MA Learning Standard)
1 DNA structure and function (Mass 31 32) 2 RNA structure and function (Mass 32) 3 Protein structure and function (Mass 12 32) 4 Transcription (Mass 32) 5 Translation (Mass 32) 6 Mutations (Mass 33) 7 Phenotypes and genotypes (Mass 33) 8 Gene expression (Mass 32 33)
MCAS Questions (Table 2) Seven of the questions that we used were taken from the MCAS examination These questions tend to be integrative across concepts (average 229 concepts per questions) and therefore require the students to adopt a more critical thinking approach The range of percent correct answers for these questions was 25 - 92 Following implementation there were improvements for all questions however certain questions (10 and 12) were answered incorrectly by the majority of students
- 6 -
Table 2
Question
Pre Test Post Test
Improvement Correct Percent Correct Percent 5 27 075 31 089 015 7 19 053 22 063 016
10 9 025 17 049 049 11 13 036 25 071 049 12 13 036 15 043 016 17 17 047 19 054 013 19 33 092 35 1 008
Textbook Questions (Table 3) Thirteen of the assessment questions were drawn from textbook materials On average these questions focused on 154 concepts Following implementation student scores improved for all but three of the questions (6 8 and 15) Students had a particularly difficult time with questions 9 16 and 18 despite improvements on the post test these questions were answered correctly by less than 50 of students
Table 3
Question
Pre Test Post Test Improvement
Correct Percent Correct Percent 1 17 047 23 066 028 2 22 061 31 089 031 3 33 092 34 097 006 4 13 036 24 069 047 6 28 078 23 066 - 016 8 23 064 16 046 - 028 9 10 028 12 034 019
13 35 097 35 100 003 14 26 072 29 083 013 15 33 092 28 080 - 013 16 11 031 16 046 033 18 12 033 15 043 022 20 19 053 21 060 012
Genetics Concepts In order to determine if there were areas of knowledge that were underserved by our project we compared the average correct response rates with the questions clustered into eight main areas The Massachusetts state guidelines addressed by these questions are defined in Table 1 At the completion of this project students had a good working knowledge of the structure and function of DNA and RNA as well as an understanding of translation there were many concepts that they struggled with
- 7 -
- 8 -
Students baseline knowledge of Protein structure and function (Mass 12 and 32) was poor with only 41 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions with 61 of questions answered correctly Students baseline knowledge of Transcription (Mass 32) was poor with only 35 of the questions covering this material answered correctly at the Pre test The Post test demonstrated notable improvements for all of these questions however students clearly still struggled with the concept as only 54 of questions were answered correctly Students baseline knowledge of Mutations (Mass 33) was poor with only 43 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions however students clearly still struggled with the concept as only 62 of questions were answered correctly Students baseline knowledge of Phenotypes and genotypes (Mass 33) was poor with only 32 of the questions covering this material answered correctly at the Pre test The Post test did not demonstrate improvement for all of these questions as only 39 of questions were answered correctly Students baseline knowledge of Gene expression (Mass 32 and 33) was poor with only 47 of the questions covering this material answered correctly at the Pre test After implementation students were able to answer 61 of the questions accurately Summary Overall the implementation was quite successful Students enjoyed the lesson and showed a genuine interest in medical genetics Importantly test data showed improvement after completing the lesson suggesting that more traditional educational methods were enhanced by utilization of a case-study approach which may have more successfully addressed student misconceptions The assessments demonstrate that students still struggle to connect changes in DNA genotype with phenotype In retrospect some of the disease materials may have been too dense with medical terminology thus students were not able to focus on the genetics concepts Future revisions of the materials will contain embedded definitions of medical terminology In addition the workflow during classroom sessions felt somewhat rushed We feel that a one day extension of this curriculum will enable a more careful examination of the genetic concepts within the context of specific disease states We believe that this approach increased student interest in the topic and offered them an alternative method of learning With appropriate modifications in emphasis and support materials we expect that future implementations will demonstrate even greater improvements in student knowledge and understanding of the way in which alterations at the level of DNA result in human disease Both the classroom teacher and the higher education partner are committed to using this lesson next school year with incorporation of the aforementioned modifications
Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
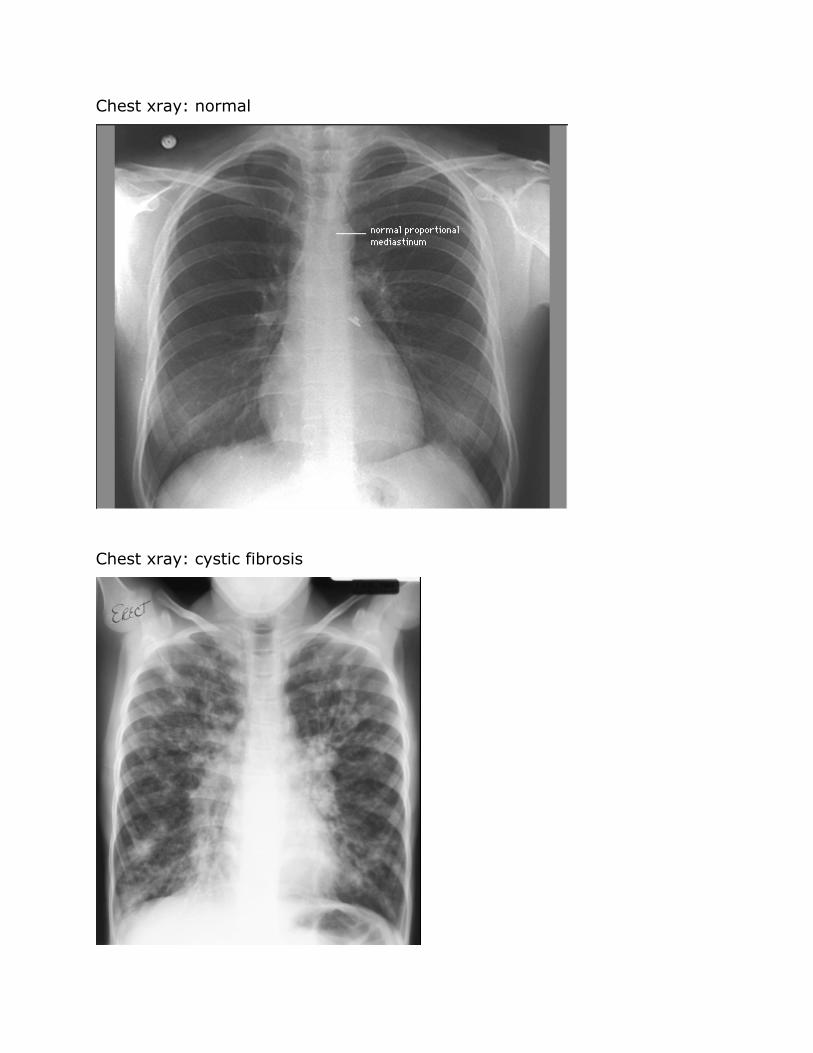
Chest xray normal
Chest xray cystic fibrosis
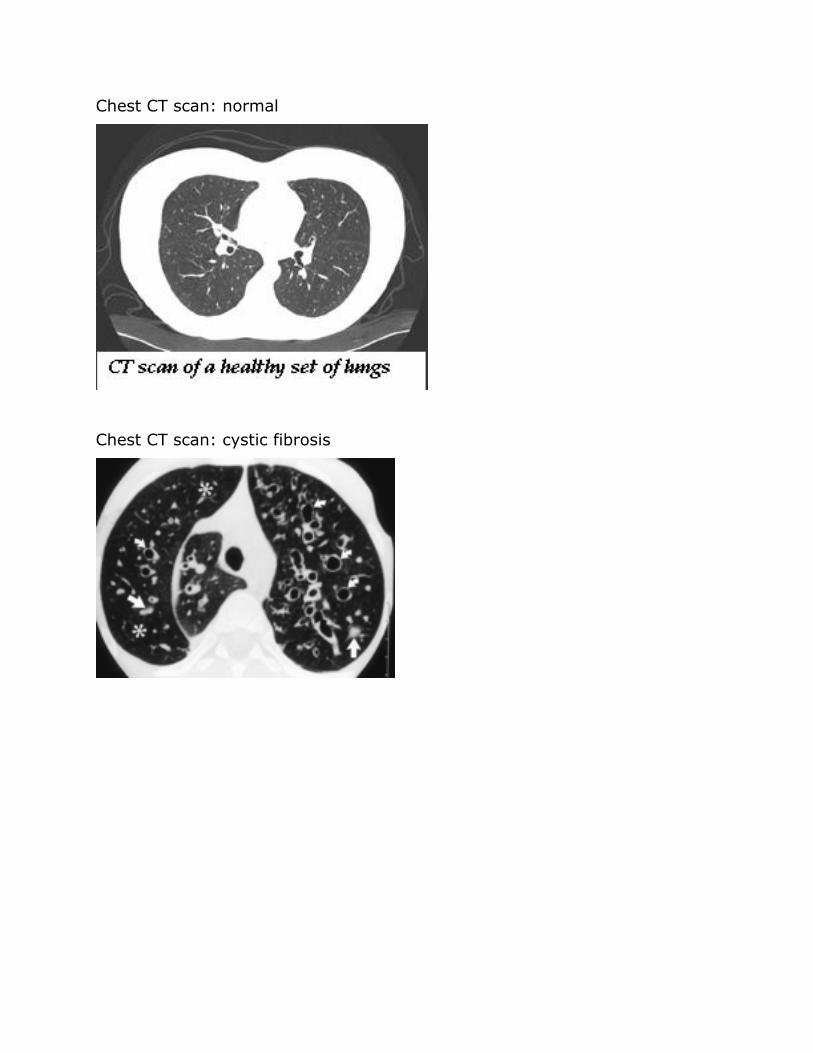
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
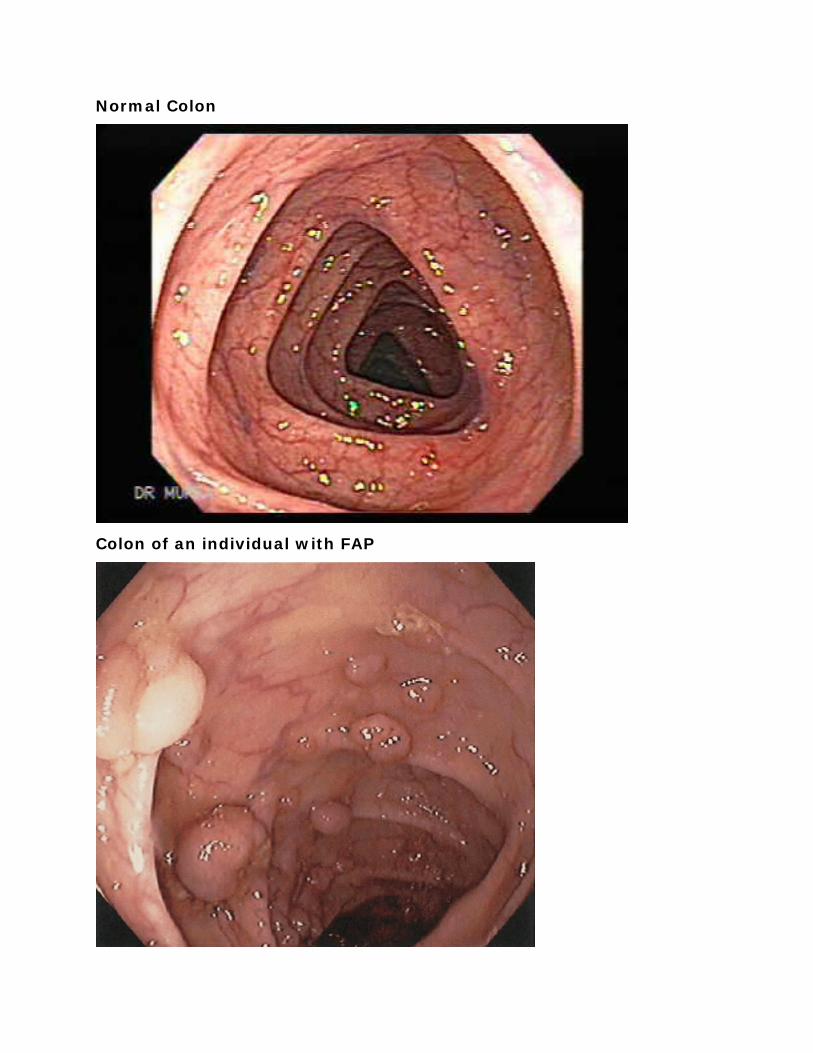
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
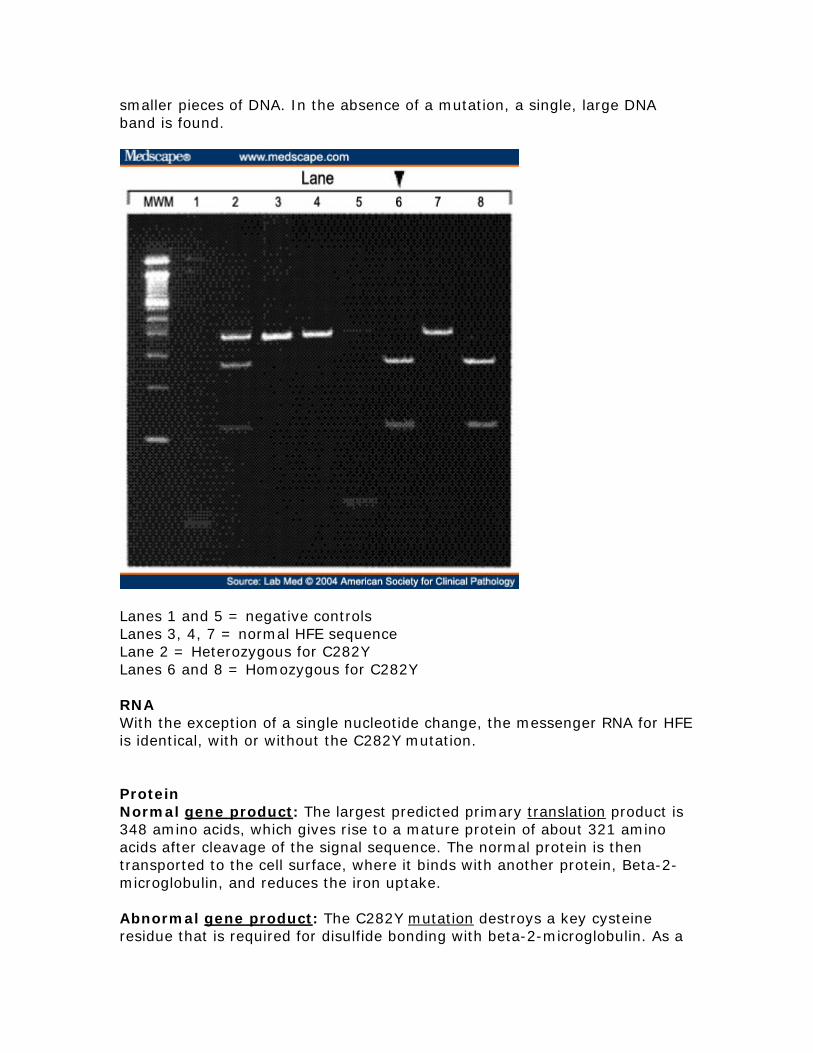
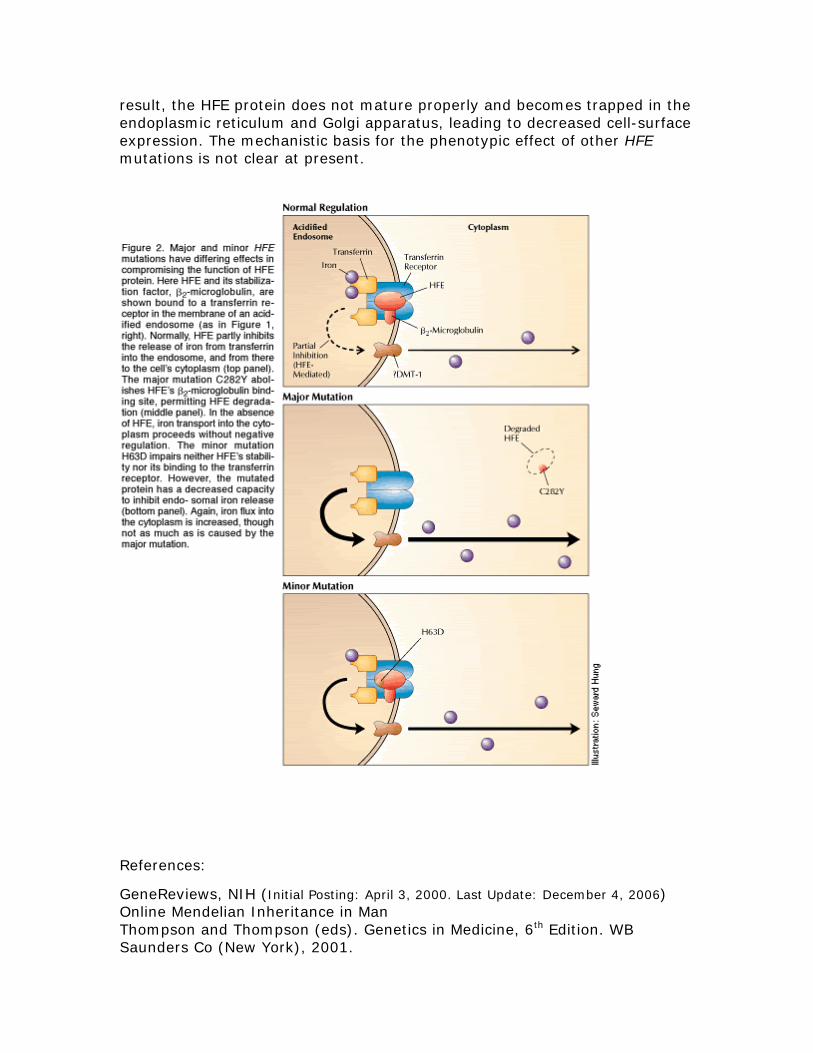
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
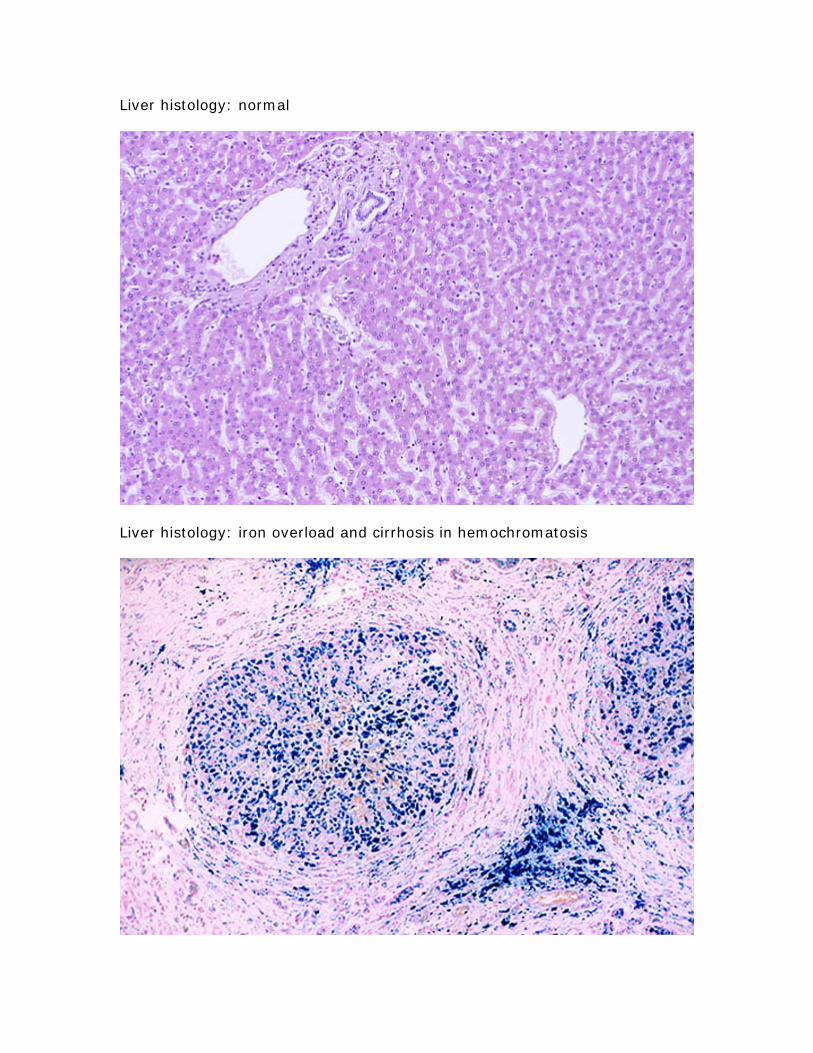
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Learning Objectives 1 Students will be able to explain the processes of transcription and translation 2 Students will be able to read and understand given data (graphs tables gels genetic
sequences) 3 Students will be able to interpret and apply data to explain how DNA mutations affect
each step in the process of gene expression 4 Students will be able to interpret and apply data to predict phenotype 5 Students will be able to communicate ideas effectively though visual aids and oral
presentation Materials
Patient analysis worksheet (for group work as teams of ldquodoctorsrdquo) Overhead teacher example worksheet (for modeling with Sickle Cell) Student example worksheet (for modeling with Sickle Cell) Patient summary sheet (description and data for each disease) Codon reference chart (ldquothe wheelrdquo) Rounds Analysis Worksheet (to use during presentations) Genetics formal assessment (given as pre- andor post-test) Posterboard (or large white sheets of paper) and markers
Duration Five class periods (50 minutes each) Prior Knowledge
DNA structure and function RNA structure and function Protein structure and function Transcription Translation Mutations Phenotypes and genotypes Gene expression Reading and understanding data (graphs tables DNA sequences gels)
Description Day One Modeling with Sickle Cell Anemia Learning Cycle Engage The instructor leads the class through an example using sickle cell anemia The instructor should put the Patient Analysis Worksheet up on the overhead (or using a projector) and give each student his or her own blank copy Students should also be given the Patient Summary Sheet for sickle cell anemia Through class discussion the instructor and the students should complete the patient analysis together The instructor should elicit answers from the students and the class should discuss the rationale behind each correct answer This discussion is likely to identify the specific misconceptions and points of confusion for each individual class which the
- 2 -
instructor should address as they come up Through this modeling students should understand how to complete the subsequent group activity and what is expected of them Day Two Analysis of Patient Learning Cycle Explore Students should be placed into groups of 4 Each group is a team of ldquodoctorsrdquo and they will be analyzing various medical case studies all of which are single gene disorders with variable penetrance Each group should be given one Patient Analysis Worksheet and one Patient Summary Sheet (various diseases) Each group of doctors should receive a different patient Using the sickle cell model as a guide teams should then analyze their own specific patient and complete the Patient Analysis Worksheet Day Three Preparation for Rounds Learning Cycle Explain Each team of doctors will be responsible for giving an oral presentation on their patient during ldquoroundsrdquo (next class) Requirements include a poster and an oral description of the genetic disease affecting the patient Two students in the group should be responsible for putting together the poster which should include a clear title the names of all doctors on the team a list of typical symptoms a flow chart describing the effects of the mutation (DNA rarr mRNA rarr protein rarr phenotype) and all applicable images associated with their disease (x-rays photographs gels etc) The other two students in the group will be responsible for orally describing the disease (patient presentation genetic cause applicable treatmenttherapy options) The two students should take turns Therefore during this planning day those students should split of the material to be presented and prepare notecards to use while presenting Thus half of the team is responsible for the visual component of the presentation and half of the team is responsible for the oral component of the presentation Day Four Rounds (Presentations) Learning Cycle Elaborate Each team should present their case to the all the other doctors (approx 5 mins) Before each presentation listening doctors should each be given a copy of the Rounds Analysis Worksheet While other teams are presenting all other doctors are responsible for paying close attention to the details of the disease being described At the conclusion of the presentation a short question and answer period (approx 2-3 min) should give all listening doctors a chance to probe for further information clarify information presented or simply provide feedback Listening doctors should also receive a short amount of time to complete and submit Rounds Analysis Worksheets (approx 2-3 min) Total time for each case is therefore approximately 10 minutes Day Five Quiz (formal summative assessment) Learning Cycle Evaluate The final component of the activity is a quiz (taken individually) which should directly target each of the learning objectives If desired by the instructor a version of this quiz can be given as a pre-assessment to determine both the studentsrsquo starting point (identify potential misconceptions) and the success of the activity in reinforcing the applicable genetics concepts
- 3 -
Assessment Formative The instructor should formatively assess students throughout the entire process in order to address areas of confusion and guide group progress The instructor will model disease analysis with the class by completing the disease worksheet interactively through class discussion The instructor should clarify as student confusion or misconceptions arise and the instructor should communicate expectations for subsequent group work The instructor should also formatively assess students during group work by circulating and interacting with each group as they analyze their patient or prepare for ldquoroundsrdquo Summative Instructor should summatively assess groups upon completion of ldquoroundsrdquo (oral presentation) Groups should turn in their patient analysis worksheet to be graded (see rubric A) and instructor should evaluate oral presentation (ldquoroundsrdquo) for groupindividual grade (see rubric B) Also students will have a formal summative assessment (quiz) on underlying concepts (learning objectives) for individual assessment (see Genetics formal assessment) Possible Extensions
Discuss examples of diseases in which proteins are over- or underexpressed (not just a ldquobrokenrdquo protein) examples of diseases which involve environmental factors and examples of disease that display less than 100 penetrance
Explore emerging genetic technologies which may be used to treat or correct the diseases studied in the cases
- 4 -
Implementation Report (2009) The ldquoDNA to Diseaserdquo lesson was implemented in two Honors freshman biology classes April 13-17 2009 In total there were 36 students 23 male and 13 female The classes had already formally learned all the applicable genetics concepts (see Prior Knowledge above) In addition they had just completed a protein gel electrophoresis lab in which they diagnosed patients as affected unaffected or carriers in respect to sickle cell anemia This provided an excellent segue into Day One of this lesson in which the classroom teacher modeled completion of the patient analysis worksheet using sickle cell anemia Class discussion during this modeling was extremely engaging for students who revealed their misconceptions as well as their personal interests in medical genetics At the conclusion of Day One the students had a good idea of what was expected of them during Day Two For Day Two the patient analysis students self-selected teams of doctors (4 students per group) and were assigned their cases No two teams of doctors received the same genetic disorder Using the sickle cell anemia sheet from Day One as a guide the students were able to successfully complete the patient analysis sheet Both the classroom teacher (Jenny Kravitz) and the higher education partner (Kari Roberts) circulated the room to work with students to clarify information answer questions and keep them on task In general the students appreciated analyzing ldquorealrdquo medical information (ie radiography films) even though the material was challenging at times At the conclusion of Day Two the majority of doctor teams had completed their patient analysis sheets On Day Three the student groups completed their patient analyses and prepared their presentations for rounds (Day Four) Students were given a large white sheet of paper and markers as well as a guide for setting up their poster (see Poster Format Guide) Again both the classroom teacher and the higher education partner circulated the room to assist and monitor students At the end of Day Three most of the doctor teams had completed their posters (a few groups took them home to complete) Day Four was when each class did rounds in which each team of doctors took approximately five minutes to give an overview of their patient (using their poster which was taped to the wall as a guide) The other students filled out the Rounds Analysis Worksheet during the presentation and used it as a guide to direct questioning after the formal presentation For most groups a significant question and answer period entirely student driven occurred after their 5-minute formal presentation Very little clarification was offered by the classroom teacher and the higher education partner and for the most part the students were able to field questions successfully The Rounds Analysis Worksheet appeared to drive students to participate in a thorough and interactive question and answer period which required more critical thinking for students to compare and contrast the various diseases and disorders On Day Five the students finished up rounds presentations that were overflow from Day Four After all presentations the students were given the post-test (Genetics Formal Assessment) Rounds posters were kept up on the walls of the classroom until the end of the school year which prompted discussions about genetic disorders in a variety of classes that did not participate in this lesson implementation further extending the impact
- 5 -
Analysis of pre- and post-test data The formal summative assessment tool was a 20 question (multiple choice) quiz This quiz was administered prior to the implementation comparison between the pre- and post-test scores are used to assess the strengths and weaknesses of our implementation Questions were drawn from both textbook and MCAS examinations Table 1 outlines the question source and the genetics concepts that are addressed
Table 1
Question Source Concepts 1 textbook 5 2 textbook 6 3 textbook 1 8 4 textbook 4 5 MCAS 5 6 textbook 2 7 MCAS 3 6 8 8 textbook 5 9 textbook 1 7 8
10 MCAS 3 4 8 11 MCAS 3 6 8 12 MCAS 1 6 7 13 textbook 1 2 14 textbook 1 2 15 textbook 1 16 textbook 6 17 MCAS 4 8 18 textbook 4 1 2 19 MCAS 1 20 textbook 5
Concept Definition (MA Learning Standard)
1 DNA structure and function (Mass 31 32) 2 RNA structure and function (Mass 32) 3 Protein structure and function (Mass 12 32) 4 Transcription (Mass 32) 5 Translation (Mass 32) 6 Mutations (Mass 33) 7 Phenotypes and genotypes (Mass 33) 8 Gene expression (Mass 32 33)
MCAS Questions (Table 2) Seven of the questions that we used were taken from the MCAS examination These questions tend to be integrative across concepts (average 229 concepts per questions) and therefore require the students to adopt a more critical thinking approach The range of percent correct answers for these questions was 25 - 92 Following implementation there were improvements for all questions however certain questions (10 and 12) were answered incorrectly by the majority of students
- 6 -
Table 2
Question
Pre Test Post Test
Improvement Correct Percent Correct Percent 5 27 075 31 089 015 7 19 053 22 063 016
10 9 025 17 049 049 11 13 036 25 071 049 12 13 036 15 043 016 17 17 047 19 054 013 19 33 092 35 1 008
Textbook Questions (Table 3) Thirteen of the assessment questions were drawn from textbook materials On average these questions focused on 154 concepts Following implementation student scores improved for all but three of the questions (6 8 and 15) Students had a particularly difficult time with questions 9 16 and 18 despite improvements on the post test these questions were answered correctly by less than 50 of students
Table 3
Question
Pre Test Post Test Improvement
Correct Percent Correct Percent 1 17 047 23 066 028 2 22 061 31 089 031 3 33 092 34 097 006 4 13 036 24 069 047 6 28 078 23 066 - 016 8 23 064 16 046 - 028 9 10 028 12 034 019
13 35 097 35 100 003 14 26 072 29 083 013 15 33 092 28 080 - 013 16 11 031 16 046 033 18 12 033 15 043 022 20 19 053 21 060 012
Genetics Concepts In order to determine if there were areas of knowledge that were underserved by our project we compared the average correct response rates with the questions clustered into eight main areas The Massachusetts state guidelines addressed by these questions are defined in Table 1 At the completion of this project students had a good working knowledge of the structure and function of DNA and RNA as well as an understanding of translation there were many concepts that they struggled with
- 7 -
- 8 -
Students baseline knowledge of Protein structure and function (Mass 12 and 32) was poor with only 41 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions with 61 of questions answered correctly Students baseline knowledge of Transcription (Mass 32) was poor with only 35 of the questions covering this material answered correctly at the Pre test The Post test demonstrated notable improvements for all of these questions however students clearly still struggled with the concept as only 54 of questions were answered correctly Students baseline knowledge of Mutations (Mass 33) was poor with only 43 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions however students clearly still struggled with the concept as only 62 of questions were answered correctly Students baseline knowledge of Phenotypes and genotypes (Mass 33) was poor with only 32 of the questions covering this material answered correctly at the Pre test The Post test did not demonstrate improvement for all of these questions as only 39 of questions were answered correctly Students baseline knowledge of Gene expression (Mass 32 and 33) was poor with only 47 of the questions covering this material answered correctly at the Pre test After implementation students were able to answer 61 of the questions accurately Summary Overall the implementation was quite successful Students enjoyed the lesson and showed a genuine interest in medical genetics Importantly test data showed improvement after completing the lesson suggesting that more traditional educational methods were enhanced by utilization of a case-study approach which may have more successfully addressed student misconceptions The assessments demonstrate that students still struggle to connect changes in DNA genotype with phenotype In retrospect some of the disease materials may have been too dense with medical terminology thus students were not able to focus on the genetics concepts Future revisions of the materials will contain embedded definitions of medical terminology In addition the workflow during classroom sessions felt somewhat rushed We feel that a one day extension of this curriculum will enable a more careful examination of the genetic concepts within the context of specific disease states We believe that this approach increased student interest in the topic and offered them an alternative method of learning With appropriate modifications in emphasis and support materials we expect that future implementations will demonstrate even greater improvements in student knowledge and understanding of the way in which alterations at the level of DNA result in human disease Both the classroom teacher and the higher education partner are committed to using this lesson next school year with incorporation of the aforementioned modifications
Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

instructor should address as they come up Through this modeling students should understand how to complete the subsequent group activity and what is expected of them Day Two Analysis of Patient Learning Cycle Explore Students should be placed into groups of 4 Each group is a team of ldquodoctorsrdquo and they will be analyzing various medical case studies all of which are single gene disorders with variable penetrance Each group should be given one Patient Analysis Worksheet and one Patient Summary Sheet (various diseases) Each group of doctors should receive a different patient Using the sickle cell model as a guide teams should then analyze their own specific patient and complete the Patient Analysis Worksheet Day Three Preparation for Rounds Learning Cycle Explain Each team of doctors will be responsible for giving an oral presentation on their patient during ldquoroundsrdquo (next class) Requirements include a poster and an oral description of the genetic disease affecting the patient Two students in the group should be responsible for putting together the poster which should include a clear title the names of all doctors on the team a list of typical symptoms a flow chart describing the effects of the mutation (DNA rarr mRNA rarr protein rarr phenotype) and all applicable images associated with their disease (x-rays photographs gels etc) The other two students in the group will be responsible for orally describing the disease (patient presentation genetic cause applicable treatmenttherapy options) The two students should take turns Therefore during this planning day those students should split of the material to be presented and prepare notecards to use while presenting Thus half of the team is responsible for the visual component of the presentation and half of the team is responsible for the oral component of the presentation Day Four Rounds (Presentations) Learning Cycle Elaborate Each team should present their case to the all the other doctors (approx 5 mins) Before each presentation listening doctors should each be given a copy of the Rounds Analysis Worksheet While other teams are presenting all other doctors are responsible for paying close attention to the details of the disease being described At the conclusion of the presentation a short question and answer period (approx 2-3 min) should give all listening doctors a chance to probe for further information clarify information presented or simply provide feedback Listening doctors should also receive a short amount of time to complete and submit Rounds Analysis Worksheets (approx 2-3 min) Total time for each case is therefore approximately 10 minutes Day Five Quiz (formal summative assessment) Learning Cycle Evaluate The final component of the activity is a quiz (taken individually) which should directly target each of the learning objectives If desired by the instructor a version of this quiz can be given as a pre-assessment to determine both the studentsrsquo starting point (identify potential misconceptions) and the success of the activity in reinforcing the applicable genetics concepts
- 3 -
Assessment Formative The instructor should formatively assess students throughout the entire process in order to address areas of confusion and guide group progress The instructor will model disease analysis with the class by completing the disease worksheet interactively through class discussion The instructor should clarify as student confusion or misconceptions arise and the instructor should communicate expectations for subsequent group work The instructor should also formatively assess students during group work by circulating and interacting with each group as they analyze their patient or prepare for ldquoroundsrdquo Summative Instructor should summatively assess groups upon completion of ldquoroundsrdquo (oral presentation) Groups should turn in their patient analysis worksheet to be graded (see rubric A) and instructor should evaluate oral presentation (ldquoroundsrdquo) for groupindividual grade (see rubric B) Also students will have a formal summative assessment (quiz) on underlying concepts (learning objectives) for individual assessment (see Genetics formal assessment) Possible Extensions
Discuss examples of diseases in which proteins are over- or underexpressed (not just a ldquobrokenrdquo protein) examples of diseases which involve environmental factors and examples of disease that display less than 100 penetrance
Explore emerging genetic technologies which may be used to treat or correct the diseases studied in the cases
- 4 -
Implementation Report (2009) The ldquoDNA to Diseaserdquo lesson was implemented in two Honors freshman biology classes April 13-17 2009 In total there were 36 students 23 male and 13 female The classes had already formally learned all the applicable genetics concepts (see Prior Knowledge above) In addition they had just completed a protein gel electrophoresis lab in which they diagnosed patients as affected unaffected or carriers in respect to sickle cell anemia This provided an excellent segue into Day One of this lesson in which the classroom teacher modeled completion of the patient analysis worksheet using sickle cell anemia Class discussion during this modeling was extremely engaging for students who revealed their misconceptions as well as their personal interests in medical genetics At the conclusion of Day One the students had a good idea of what was expected of them during Day Two For Day Two the patient analysis students self-selected teams of doctors (4 students per group) and were assigned their cases No two teams of doctors received the same genetic disorder Using the sickle cell anemia sheet from Day One as a guide the students were able to successfully complete the patient analysis sheet Both the classroom teacher (Jenny Kravitz) and the higher education partner (Kari Roberts) circulated the room to work with students to clarify information answer questions and keep them on task In general the students appreciated analyzing ldquorealrdquo medical information (ie radiography films) even though the material was challenging at times At the conclusion of Day Two the majority of doctor teams had completed their patient analysis sheets On Day Three the student groups completed their patient analyses and prepared their presentations for rounds (Day Four) Students were given a large white sheet of paper and markers as well as a guide for setting up their poster (see Poster Format Guide) Again both the classroom teacher and the higher education partner circulated the room to assist and monitor students At the end of Day Three most of the doctor teams had completed their posters (a few groups took them home to complete) Day Four was when each class did rounds in which each team of doctors took approximately five minutes to give an overview of their patient (using their poster which was taped to the wall as a guide) The other students filled out the Rounds Analysis Worksheet during the presentation and used it as a guide to direct questioning after the formal presentation For most groups a significant question and answer period entirely student driven occurred after their 5-minute formal presentation Very little clarification was offered by the classroom teacher and the higher education partner and for the most part the students were able to field questions successfully The Rounds Analysis Worksheet appeared to drive students to participate in a thorough and interactive question and answer period which required more critical thinking for students to compare and contrast the various diseases and disorders On Day Five the students finished up rounds presentations that were overflow from Day Four After all presentations the students were given the post-test (Genetics Formal Assessment) Rounds posters were kept up on the walls of the classroom until the end of the school year which prompted discussions about genetic disorders in a variety of classes that did not participate in this lesson implementation further extending the impact
- 5 -
Analysis of pre- and post-test data The formal summative assessment tool was a 20 question (multiple choice) quiz This quiz was administered prior to the implementation comparison between the pre- and post-test scores are used to assess the strengths and weaknesses of our implementation Questions were drawn from both textbook and MCAS examinations Table 1 outlines the question source and the genetics concepts that are addressed
Table 1
Question Source Concepts 1 textbook 5 2 textbook 6 3 textbook 1 8 4 textbook 4 5 MCAS 5 6 textbook 2 7 MCAS 3 6 8 8 textbook 5 9 textbook 1 7 8
10 MCAS 3 4 8 11 MCAS 3 6 8 12 MCAS 1 6 7 13 textbook 1 2 14 textbook 1 2 15 textbook 1 16 textbook 6 17 MCAS 4 8 18 textbook 4 1 2 19 MCAS 1 20 textbook 5
Concept Definition (MA Learning Standard)
1 DNA structure and function (Mass 31 32) 2 RNA structure and function (Mass 32) 3 Protein structure and function (Mass 12 32) 4 Transcription (Mass 32) 5 Translation (Mass 32) 6 Mutations (Mass 33) 7 Phenotypes and genotypes (Mass 33) 8 Gene expression (Mass 32 33)
MCAS Questions (Table 2) Seven of the questions that we used were taken from the MCAS examination These questions tend to be integrative across concepts (average 229 concepts per questions) and therefore require the students to adopt a more critical thinking approach The range of percent correct answers for these questions was 25 - 92 Following implementation there were improvements for all questions however certain questions (10 and 12) were answered incorrectly by the majority of students
- 6 -
Table 2
Question
Pre Test Post Test
Improvement Correct Percent Correct Percent 5 27 075 31 089 015 7 19 053 22 063 016
10 9 025 17 049 049 11 13 036 25 071 049 12 13 036 15 043 016 17 17 047 19 054 013 19 33 092 35 1 008
Textbook Questions (Table 3) Thirteen of the assessment questions were drawn from textbook materials On average these questions focused on 154 concepts Following implementation student scores improved for all but three of the questions (6 8 and 15) Students had a particularly difficult time with questions 9 16 and 18 despite improvements on the post test these questions were answered correctly by less than 50 of students
Table 3
Question
Pre Test Post Test Improvement
Correct Percent Correct Percent 1 17 047 23 066 028 2 22 061 31 089 031 3 33 092 34 097 006 4 13 036 24 069 047 6 28 078 23 066 - 016 8 23 064 16 046 - 028 9 10 028 12 034 019
13 35 097 35 100 003 14 26 072 29 083 013 15 33 092 28 080 - 013 16 11 031 16 046 033 18 12 033 15 043 022 20 19 053 21 060 012
Genetics Concepts In order to determine if there were areas of knowledge that were underserved by our project we compared the average correct response rates with the questions clustered into eight main areas The Massachusetts state guidelines addressed by these questions are defined in Table 1 At the completion of this project students had a good working knowledge of the structure and function of DNA and RNA as well as an understanding of translation there were many concepts that they struggled with
- 7 -
- 8 -
Students baseline knowledge of Protein structure and function (Mass 12 and 32) was poor with only 41 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions with 61 of questions answered correctly Students baseline knowledge of Transcription (Mass 32) was poor with only 35 of the questions covering this material answered correctly at the Pre test The Post test demonstrated notable improvements for all of these questions however students clearly still struggled with the concept as only 54 of questions were answered correctly Students baseline knowledge of Mutations (Mass 33) was poor with only 43 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions however students clearly still struggled with the concept as only 62 of questions were answered correctly Students baseline knowledge of Phenotypes and genotypes (Mass 33) was poor with only 32 of the questions covering this material answered correctly at the Pre test The Post test did not demonstrate improvement for all of these questions as only 39 of questions were answered correctly Students baseline knowledge of Gene expression (Mass 32 and 33) was poor with only 47 of the questions covering this material answered correctly at the Pre test After implementation students were able to answer 61 of the questions accurately Summary Overall the implementation was quite successful Students enjoyed the lesson and showed a genuine interest in medical genetics Importantly test data showed improvement after completing the lesson suggesting that more traditional educational methods were enhanced by utilization of a case-study approach which may have more successfully addressed student misconceptions The assessments demonstrate that students still struggle to connect changes in DNA genotype with phenotype In retrospect some of the disease materials may have been too dense with medical terminology thus students were not able to focus on the genetics concepts Future revisions of the materials will contain embedded definitions of medical terminology In addition the workflow during classroom sessions felt somewhat rushed We feel that a one day extension of this curriculum will enable a more careful examination of the genetic concepts within the context of specific disease states We believe that this approach increased student interest in the topic and offered them an alternative method of learning With appropriate modifications in emphasis and support materials we expect that future implementations will demonstrate even greater improvements in student knowledge and understanding of the way in which alterations at the level of DNA result in human disease Both the classroom teacher and the higher education partner are committed to using this lesson next school year with incorporation of the aforementioned modifications
Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Assessment Formative The instructor should formatively assess students throughout the entire process in order to address areas of confusion and guide group progress The instructor will model disease analysis with the class by completing the disease worksheet interactively through class discussion The instructor should clarify as student confusion or misconceptions arise and the instructor should communicate expectations for subsequent group work The instructor should also formatively assess students during group work by circulating and interacting with each group as they analyze their patient or prepare for ldquoroundsrdquo Summative Instructor should summatively assess groups upon completion of ldquoroundsrdquo (oral presentation) Groups should turn in their patient analysis worksheet to be graded (see rubric A) and instructor should evaluate oral presentation (ldquoroundsrdquo) for groupindividual grade (see rubric B) Also students will have a formal summative assessment (quiz) on underlying concepts (learning objectives) for individual assessment (see Genetics formal assessment) Possible Extensions
Discuss examples of diseases in which proteins are over- or underexpressed (not just a ldquobrokenrdquo protein) examples of diseases which involve environmental factors and examples of disease that display less than 100 penetrance
Explore emerging genetic technologies which may be used to treat or correct the diseases studied in the cases
- 4 -
Implementation Report (2009) The ldquoDNA to Diseaserdquo lesson was implemented in two Honors freshman biology classes April 13-17 2009 In total there were 36 students 23 male and 13 female The classes had already formally learned all the applicable genetics concepts (see Prior Knowledge above) In addition they had just completed a protein gel electrophoresis lab in which they diagnosed patients as affected unaffected or carriers in respect to sickle cell anemia This provided an excellent segue into Day One of this lesson in which the classroom teacher modeled completion of the patient analysis worksheet using sickle cell anemia Class discussion during this modeling was extremely engaging for students who revealed their misconceptions as well as their personal interests in medical genetics At the conclusion of Day One the students had a good idea of what was expected of them during Day Two For Day Two the patient analysis students self-selected teams of doctors (4 students per group) and were assigned their cases No two teams of doctors received the same genetic disorder Using the sickle cell anemia sheet from Day One as a guide the students were able to successfully complete the patient analysis sheet Both the classroom teacher (Jenny Kravitz) and the higher education partner (Kari Roberts) circulated the room to work with students to clarify information answer questions and keep them on task In general the students appreciated analyzing ldquorealrdquo medical information (ie radiography films) even though the material was challenging at times At the conclusion of Day Two the majority of doctor teams had completed their patient analysis sheets On Day Three the student groups completed their patient analyses and prepared their presentations for rounds (Day Four) Students were given a large white sheet of paper and markers as well as a guide for setting up their poster (see Poster Format Guide) Again both the classroom teacher and the higher education partner circulated the room to assist and monitor students At the end of Day Three most of the doctor teams had completed their posters (a few groups took them home to complete) Day Four was when each class did rounds in which each team of doctors took approximately five minutes to give an overview of their patient (using their poster which was taped to the wall as a guide) The other students filled out the Rounds Analysis Worksheet during the presentation and used it as a guide to direct questioning after the formal presentation For most groups a significant question and answer period entirely student driven occurred after their 5-minute formal presentation Very little clarification was offered by the classroom teacher and the higher education partner and for the most part the students were able to field questions successfully The Rounds Analysis Worksheet appeared to drive students to participate in a thorough and interactive question and answer period which required more critical thinking for students to compare and contrast the various diseases and disorders On Day Five the students finished up rounds presentations that were overflow from Day Four After all presentations the students were given the post-test (Genetics Formal Assessment) Rounds posters were kept up on the walls of the classroom until the end of the school year which prompted discussions about genetic disorders in a variety of classes that did not participate in this lesson implementation further extending the impact
- 5 -
Analysis of pre- and post-test data The formal summative assessment tool was a 20 question (multiple choice) quiz This quiz was administered prior to the implementation comparison between the pre- and post-test scores are used to assess the strengths and weaknesses of our implementation Questions were drawn from both textbook and MCAS examinations Table 1 outlines the question source and the genetics concepts that are addressed
Table 1
Question Source Concepts 1 textbook 5 2 textbook 6 3 textbook 1 8 4 textbook 4 5 MCAS 5 6 textbook 2 7 MCAS 3 6 8 8 textbook 5 9 textbook 1 7 8
10 MCAS 3 4 8 11 MCAS 3 6 8 12 MCAS 1 6 7 13 textbook 1 2 14 textbook 1 2 15 textbook 1 16 textbook 6 17 MCAS 4 8 18 textbook 4 1 2 19 MCAS 1 20 textbook 5
Concept Definition (MA Learning Standard)
1 DNA structure and function (Mass 31 32) 2 RNA structure and function (Mass 32) 3 Protein structure and function (Mass 12 32) 4 Transcription (Mass 32) 5 Translation (Mass 32) 6 Mutations (Mass 33) 7 Phenotypes and genotypes (Mass 33) 8 Gene expression (Mass 32 33)
MCAS Questions (Table 2) Seven of the questions that we used were taken from the MCAS examination These questions tend to be integrative across concepts (average 229 concepts per questions) and therefore require the students to adopt a more critical thinking approach The range of percent correct answers for these questions was 25 - 92 Following implementation there were improvements for all questions however certain questions (10 and 12) were answered incorrectly by the majority of students
- 6 -
Table 2
Question
Pre Test Post Test
Improvement Correct Percent Correct Percent 5 27 075 31 089 015 7 19 053 22 063 016
10 9 025 17 049 049 11 13 036 25 071 049 12 13 036 15 043 016 17 17 047 19 054 013 19 33 092 35 1 008
Textbook Questions (Table 3) Thirteen of the assessment questions were drawn from textbook materials On average these questions focused on 154 concepts Following implementation student scores improved for all but three of the questions (6 8 and 15) Students had a particularly difficult time with questions 9 16 and 18 despite improvements on the post test these questions were answered correctly by less than 50 of students
Table 3
Question
Pre Test Post Test Improvement
Correct Percent Correct Percent 1 17 047 23 066 028 2 22 061 31 089 031 3 33 092 34 097 006 4 13 036 24 069 047 6 28 078 23 066 - 016 8 23 064 16 046 - 028 9 10 028 12 034 019
13 35 097 35 100 003 14 26 072 29 083 013 15 33 092 28 080 - 013 16 11 031 16 046 033 18 12 033 15 043 022 20 19 053 21 060 012
Genetics Concepts In order to determine if there were areas of knowledge that were underserved by our project we compared the average correct response rates with the questions clustered into eight main areas The Massachusetts state guidelines addressed by these questions are defined in Table 1 At the completion of this project students had a good working knowledge of the structure and function of DNA and RNA as well as an understanding of translation there were many concepts that they struggled with
- 7 -
- 8 -
Students baseline knowledge of Protein structure and function (Mass 12 and 32) was poor with only 41 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions with 61 of questions answered correctly Students baseline knowledge of Transcription (Mass 32) was poor with only 35 of the questions covering this material answered correctly at the Pre test The Post test demonstrated notable improvements for all of these questions however students clearly still struggled with the concept as only 54 of questions were answered correctly Students baseline knowledge of Mutations (Mass 33) was poor with only 43 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions however students clearly still struggled with the concept as only 62 of questions were answered correctly Students baseline knowledge of Phenotypes and genotypes (Mass 33) was poor with only 32 of the questions covering this material answered correctly at the Pre test The Post test did not demonstrate improvement for all of these questions as only 39 of questions were answered correctly Students baseline knowledge of Gene expression (Mass 32 and 33) was poor with only 47 of the questions covering this material answered correctly at the Pre test After implementation students were able to answer 61 of the questions accurately Summary Overall the implementation was quite successful Students enjoyed the lesson and showed a genuine interest in medical genetics Importantly test data showed improvement after completing the lesson suggesting that more traditional educational methods were enhanced by utilization of a case-study approach which may have more successfully addressed student misconceptions The assessments demonstrate that students still struggle to connect changes in DNA genotype with phenotype In retrospect some of the disease materials may have been too dense with medical terminology thus students were not able to focus on the genetics concepts Future revisions of the materials will contain embedded definitions of medical terminology In addition the workflow during classroom sessions felt somewhat rushed We feel that a one day extension of this curriculum will enable a more careful examination of the genetic concepts within the context of specific disease states We believe that this approach increased student interest in the topic and offered them an alternative method of learning With appropriate modifications in emphasis and support materials we expect that future implementations will demonstrate even greater improvements in student knowledge and understanding of the way in which alterations at the level of DNA result in human disease Both the classroom teacher and the higher education partner are committed to using this lesson next school year with incorporation of the aforementioned modifications
Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Implementation Report (2009) The ldquoDNA to Diseaserdquo lesson was implemented in two Honors freshman biology classes April 13-17 2009 In total there were 36 students 23 male and 13 female The classes had already formally learned all the applicable genetics concepts (see Prior Knowledge above) In addition they had just completed a protein gel electrophoresis lab in which they diagnosed patients as affected unaffected or carriers in respect to sickle cell anemia This provided an excellent segue into Day One of this lesson in which the classroom teacher modeled completion of the patient analysis worksheet using sickle cell anemia Class discussion during this modeling was extremely engaging for students who revealed their misconceptions as well as their personal interests in medical genetics At the conclusion of Day One the students had a good idea of what was expected of them during Day Two For Day Two the patient analysis students self-selected teams of doctors (4 students per group) and were assigned their cases No two teams of doctors received the same genetic disorder Using the sickle cell anemia sheet from Day One as a guide the students were able to successfully complete the patient analysis sheet Both the classroom teacher (Jenny Kravitz) and the higher education partner (Kari Roberts) circulated the room to work with students to clarify information answer questions and keep them on task In general the students appreciated analyzing ldquorealrdquo medical information (ie radiography films) even though the material was challenging at times At the conclusion of Day Two the majority of doctor teams had completed their patient analysis sheets On Day Three the student groups completed their patient analyses and prepared their presentations for rounds (Day Four) Students were given a large white sheet of paper and markers as well as a guide for setting up their poster (see Poster Format Guide) Again both the classroom teacher and the higher education partner circulated the room to assist and monitor students At the end of Day Three most of the doctor teams had completed their posters (a few groups took them home to complete) Day Four was when each class did rounds in which each team of doctors took approximately five minutes to give an overview of their patient (using their poster which was taped to the wall as a guide) The other students filled out the Rounds Analysis Worksheet during the presentation and used it as a guide to direct questioning after the formal presentation For most groups a significant question and answer period entirely student driven occurred after their 5-minute formal presentation Very little clarification was offered by the classroom teacher and the higher education partner and for the most part the students were able to field questions successfully The Rounds Analysis Worksheet appeared to drive students to participate in a thorough and interactive question and answer period which required more critical thinking for students to compare and contrast the various diseases and disorders On Day Five the students finished up rounds presentations that were overflow from Day Four After all presentations the students were given the post-test (Genetics Formal Assessment) Rounds posters were kept up on the walls of the classroom until the end of the school year which prompted discussions about genetic disorders in a variety of classes that did not participate in this lesson implementation further extending the impact
- 5 -
Analysis of pre- and post-test data The formal summative assessment tool was a 20 question (multiple choice) quiz This quiz was administered prior to the implementation comparison between the pre- and post-test scores are used to assess the strengths and weaknesses of our implementation Questions were drawn from both textbook and MCAS examinations Table 1 outlines the question source and the genetics concepts that are addressed
Table 1
Question Source Concepts 1 textbook 5 2 textbook 6 3 textbook 1 8 4 textbook 4 5 MCAS 5 6 textbook 2 7 MCAS 3 6 8 8 textbook 5 9 textbook 1 7 8
10 MCAS 3 4 8 11 MCAS 3 6 8 12 MCAS 1 6 7 13 textbook 1 2 14 textbook 1 2 15 textbook 1 16 textbook 6 17 MCAS 4 8 18 textbook 4 1 2 19 MCAS 1 20 textbook 5
Concept Definition (MA Learning Standard)
1 DNA structure and function (Mass 31 32) 2 RNA structure and function (Mass 32) 3 Protein structure and function (Mass 12 32) 4 Transcription (Mass 32) 5 Translation (Mass 32) 6 Mutations (Mass 33) 7 Phenotypes and genotypes (Mass 33) 8 Gene expression (Mass 32 33)
MCAS Questions (Table 2) Seven of the questions that we used were taken from the MCAS examination These questions tend to be integrative across concepts (average 229 concepts per questions) and therefore require the students to adopt a more critical thinking approach The range of percent correct answers for these questions was 25 - 92 Following implementation there were improvements for all questions however certain questions (10 and 12) were answered incorrectly by the majority of students
- 6 -
Table 2
Question
Pre Test Post Test
Improvement Correct Percent Correct Percent 5 27 075 31 089 015 7 19 053 22 063 016
10 9 025 17 049 049 11 13 036 25 071 049 12 13 036 15 043 016 17 17 047 19 054 013 19 33 092 35 1 008
Textbook Questions (Table 3) Thirteen of the assessment questions were drawn from textbook materials On average these questions focused on 154 concepts Following implementation student scores improved for all but three of the questions (6 8 and 15) Students had a particularly difficult time with questions 9 16 and 18 despite improvements on the post test these questions were answered correctly by less than 50 of students
Table 3
Question
Pre Test Post Test Improvement
Correct Percent Correct Percent 1 17 047 23 066 028 2 22 061 31 089 031 3 33 092 34 097 006 4 13 036 24 069 047 6 28 078 23 066 - 016 8 23 064 16 046 - 028 9 10 028 12 034 019
13 35 097 35 100 003 14 26 072 29 083 013 15 33 092 28 080 - 013 16 11 031 16 046 033 18 12 033 15 043 022 20 19 053 21 060 012
Genetics Concepts In order to determine if there were areas of knowledge that were underserved by our project we compared the average correct response rates with the questions clustered into eight main areas The Massachusetts state guidelines addressed by these questions are defined in Table 1 At the completion of this project students had a good working knowledge of the structure and function of DNA and RNA as well as an understanding of translation there were many concepts that they struggled with
- 7 -
- 8 -
Students baseline knowledge of Protein structure and function (Mass 12 and 32) was poor with only 41 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions with 61 of questions answered correctly Students baseline knowledge of Transcription (Mass 32) was poor with only 35 of the questions covering this material answered correctly at the Pre test The Post test demonstrated notable improvements for all of these questions however students clearly still struggled with the concept as only 54 of questions were answered correctly Students baseline knowledge of Mutations (Mass 33) was poor with only 43 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions however students clearly still struggled with the concept as only 62 of questions were answered correctly Students baseline knowledge of Phenotypes and genotypes (Mass 33) was poor with only 32 of the questions covering this material answered correctly at the Pre test The Post test did not demonstrate improvement for all of these questions as only 39 of questions were answered correctly Students baseline knowledge of Gene expression (Mass 32 and 33) was poor with only 47 of the questions covering this material answered correctly at the Pre test After implementation students were able to answer 61 of the questions accurately Summary Overall the implementation was quite successful Students enjoyed the lesson and showed a genuine interest in medical genetics Importantly test data showed improvement after completing the lesson suggesting that more traditional educational methods were enhanced by utilization of a case-study approach which may have more successfully addressed student misconceptions The assessments demonstrate that students still struggle to connect changes in DNA genotype with phenotype In retrospect some of the disease materials may have been too dense with medical terminology thus students were not able to focus on the genetics concepts Future revisions of the materials will contain embedded definitions of medical terminology In addition the workflow during classroom sessions felt somewhat rushed We feel that a one day extension of this curriculum will enable a more careful examination of the genetic concepts within the context of specific disease states We believe that this approach increased student interest in the topic and offered them an alternative method of learning With appropriate modifications in emphasis and support materials we expect that future implementations will demonstrate even greater improvements in student knowledge and understanding of the way in which alterations at the level of DNA result in human disease Both the classroom teacher and the higher education partner are committed to using this lesson next school year with incorporation of the aforementioned modifications
Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
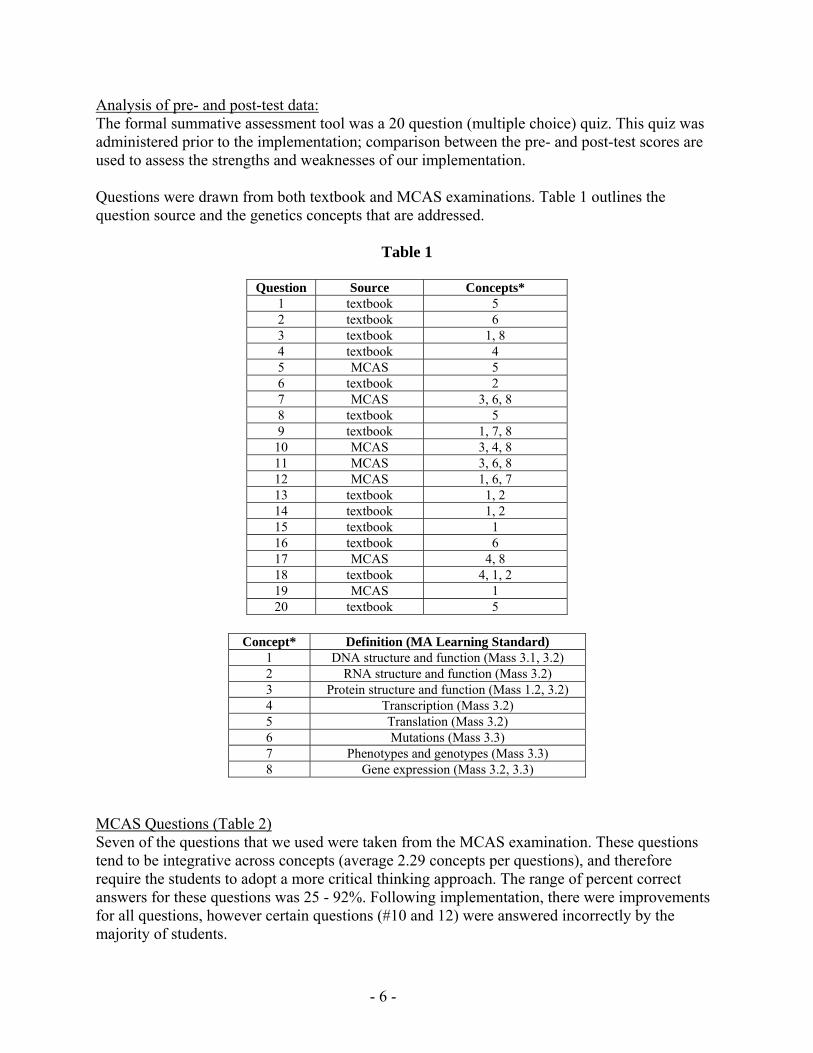
Analysis of pre- and post-test data The formal summative assessment tool was a 20 question (multiple choice) quiz This quiz was administered prior to the implementation comparison between the pre- and post-test scores are used to assess the strengths and weaknesses of our implementation Questions were drawn from both textbook and MCAS examinations Table 1 outlines the question source and the genetics concepts that are addressed
Table 1
Question Source Concepts 1 textbook 5 2 textbook 6 3 textbook 1 8 4 textbook 4 5 MCAS 5 6 textbook 2 7 MCAS 3 6 8 8 textbook 5 9 textbook 1 7 8
10 MCAS 3 4 8 11 MCAS 3 6 8 12 MCAS 1 6 7 13 textbook 1 2 14 textbook 1 2 15 textbook 1 16 textbook 6 17 MCAS 4 8 18 textbook 4 1 2 19 MCAS 1 20 textbook 5
Concept Definition (MA Learning Standard)
1 DNA structure and function (Mass 31 32) 2 RNA structure and function (Mass 32) 3 Protein structure and function (Mass 12 32) 4 Transcription (Mass 32) 5 Translation (Mass 32) 6 Mutations (Mass 33) 7 Phenotypes and genotypes (Mass 33) 8 Gene expression (Mass 32 33)
MCAS Questions (Table 2) Seven of the questions that we used were taken from the MCAS examination These questions tend to be integrative across concepts (average 229 concepts per questions) and therefore require the students to adopt a more critical thinking approach The range of percent correct answers for these questions was 25 - 92 Following implementation there were improvements for all questions however certain questions (10 and 12) were answered incorrectly by the majority of students
- 6 -
Table 2
Question
Pre Test Post Test
Improvement Correct Percent Correct Percent 5 27 075 31 089 015 7 19 053 22 063 016
10 9 025 17 049 049 11 13 036 25 071 049 12 13 036 15 043 016 17 17 047 19 054 013 19 33 092 35 1 008
Textbook Questions (Table 3) Thirteen of the assessment questions were drawn from textbook materials On average these questions focused on 154 concepts Following implementation student scores improved for all but three of the questions (6 8 and 15) Students had a particularly difficult time with questions 9 16 and 18 despite improvements on the post test these questions were answered correctly by less than 50 of students
Table 3
Question
Pre Test Post Test Improvement
Correct Percent Correct Percent 1 17 047 23 066 028 2 22 061 31 089 031 3 33 092 34 097 006 4 13 036 24 069 047 6 28 078 23 066 - 016 8 23 064 16 046 - 028 9 10 028 12 034 019
13 35 097 35 100 003 14 26 072 29 083 013 15 33 092 28 080 - 013 16 11 031 16 046 033 18 12 033 15 043 022 20 19 053 21 060 012
Genetics Concepts In order to determine if there were areas of knowledge that were underserved by our project we compared the average correct response rates with the questions clustered into eight main areas The Massachusetts state guidelines addressed by these questions are defined in Table 1 At the completion of this project students had a good working knowledge of the structure and function of DNA and RNA as well as an understanding of translation there were many concepts that they struggled with
- 7 -
- 8 -
Students baseline knowledge of Protein structure and function (Mass 12 and 32) was poor with only 41 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions with 61 of questions answered correctly Students baseline knowledge of Transcription (Mass 32) was poor with only 35 of the questions covering this material answered correctly at the Pre test The Post test demonstrated notable improvements for all of these questions however students clearly still struggled with the concept as only 54 of questions were answered correctly Students baseline knowledge of Mutations (Mass 33) was poor with only 43 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions however students clearly still struggled with the concept as only 62 of questions were answered correctly Students baseline knowledge of Phenotypes and genotypes (Mass 33) was poor with only 32 of the questions covering this material answered correctly at the Pre test The Post test did not demonstrate improvement for all of these questions as only 39 of questions were answered correctly Students baseline knowledge of Gene expression (Mass 32 and 33) was poor with only 47 of the questions covering this material answered correctly at the Pre test After implementation students were able to answer 61 of the questions accurately Summary Overall the implementation was quite successful Students enjoyed the lesson and showed a genuine interest in medical genetics Importantly test data showed improvement after completing the lesson suggesting that more traditional educational methods were enhanced by utilization of a case-study approach which may have more successfully addressed student misconceptions The assessments demonstrate that students still struggle to connect changes in DNA genotype with phenotype In retrospect some of the disease materials may have been too dense with medical terminology thus students were not able to focus on the genetics concepts Future revisions of the materials will contain embedded definitions of medical terminology In addition the workflow during classroom sessions felt somewhat rushed We feel that a one day extension of this curriculum will enable a more careful examination of the genetic concepts within the context of specific disease states We believe that this approach increased student interest in the topic and offered them an alternative method of learning With appropriate modifications in emphasis and support materials we expect that future implementations will demonstrate even greater improvements in student knowledge and understanding of the way in which alterations at the level of DNA result in human disease Both the classroom teacher and the higher education partner are committed to using this lesson next school year with incorporation of the aforementioned modifications
Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
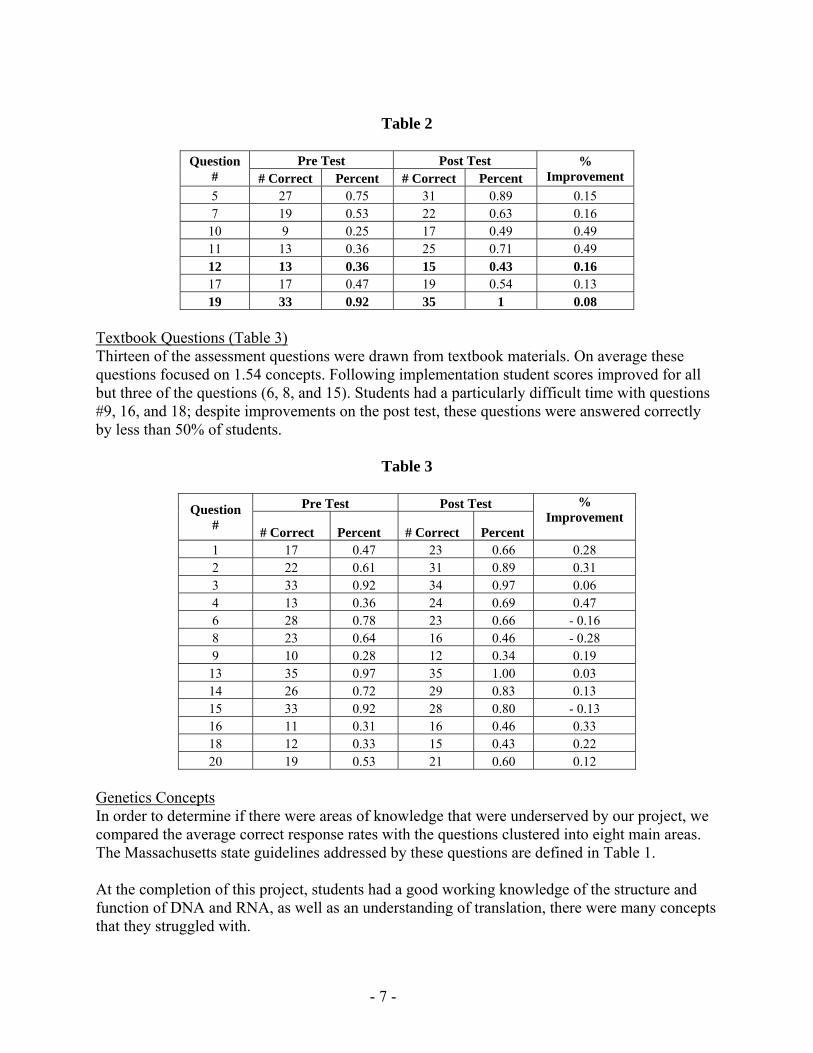
Table 2
Question
Pre Test Post Test
Improvement Correct Percent Correct Percent 5 27 075 31 089 015 7 19 053 22 063 016
10 9 025 17 049 049 11 13 036 25 071 049 12 13 036 15 043 016 17 17 047 19 054 013 19 33 092 35 1 008
Textbook Questions (Table 3) Thirteen of the assessment questions were drawn from textbook materials On average these questions focused on 154 concepts Following implementation student scores improved for all but three of the questions (6 8 and 15) Students had a particularly difficult time with questions 9 16 and 18 despite improvements on the post test these questions were answered correctly by less than 50 of students
Table 3
Question
Pre Test Post Test Improvement
Correct Percent Correct Percent 1 17 047 23 066 028 2 22 061 31 089 031 3 33 092 34 097 006 4 13 036 24 069 047 6 28 078 23 066 - 016 8 23 064 16 046 - 028 9 10 028 12 034 019
13 35 097 35 100 003 14 26 072 29 083 013 15 33 092 28 080 - 013 16 11 031 16 046 033 18 12 033 15 043 022 20 19 053 21 060 012
Genetics Concepts In order to determine if there were areas of knowledge that were underserved by our project we compared the average correct response rates with the questions clustered into eight main areas The Massachusetts state guidelines addressed by these questions are defined in Table 1 At the completion of this project students had a good working knowledge of the structure and function of DNA and RNA as well as an understanding of translation there were many concepts that they struggled with
- 7 -
- 8 -
Students baseline knowledge of Protein structure and function (Mass 12 and 32) was poor with only 41 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions with 61 of questions answered correctly Students baseline knowledge of Transcription (Mass 32) was poor with only 35 of the questions covering this material answered correctly at the Pre test The Post test demonstrated notable improvements for all of these questions however students clearly still struggled with the concept as only 54 of questions were answered correctly Students baseline knowledge of Mutations (Mass 33) was poor with only 43 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions however students clearly still struggled with the concept as only 62 of questions were answered correctly Students baseline knowledge of Phenotypes and genotypes (Mass 33) was poor with only 32 of the questions covering this material answered correctly at the Pre test The Post test did not demonstrate improvement for all of these questions as only 39 of questions were answered correctly Students baseline knowledge of Gene expression (Mass 32 and 33) was poor with only 47 of the questions covering this material answered correctly at the Pre test After implementation students were able to answer 61 of the questions accurately Summary Overall the implementation was quite successful Students enjoyed the lesson and showed a genuine interest in medical genetics Importantly test data showed improvement after completing the lesson suggesting that more traditional educational methods were enhanced by utilization of a case-study approach which may have more successfully addressed student misconceptions The assessments demonstrate that students still struggle to connect changes in DNA genotype with phenotype In retrospect some of the disease materials may have been too dense with medical terminology thus students were not able to focus on the genetics concepts Future revisions of the materials will contain embedded definitions of medical terminology In addition the workflow during classroom sessions felt somewhat rushed We feel that a one day extension of this curriculum will enable a more careful examination of the genetic concepts within the context of specific disease states We believe that this approach increased student interest in the topic and offered them an alternative method of learning With appropriate modifications in emphasis and support materials we expect that future implementations will demonstrate even greater improvements in student knowledge and understanding of the way in which alterations at the level of DNA result in human disease Both the classroom teacher and the higher education partner are committed to using this lesson next school year with incorporation of the aforementioned modifications
Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

- 8 -
Students baseline knowledge of Protein structure and function (Mass 12 and 32) was poor with only 41 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions with 61 of questions answered correctly Students baseline knowledge of Transcription (Mass 32) was poor with only 35 of the questions covering this material answered correctly at the Pre test The Post test demonstrated notable improvements for all of these questions however students clearly still struggled with the concept as only 54 of questions were answered correctly Students baseline knowledge of Mutations (Mass 33) was poor with only 43 of the questions covering this material answered correctly at the Pre test The Post test demonstrated improvements for all of these questions however students clearly still struggled with the concept as only 62 of questions were answered correctly Students baseline knowledge of Phenotypes and genotypes (Mass 33) was poor with only 32 of the questions covering this material answered correctly at the Pre test The Post test did not demonstrate improvement for all of these questions as only 39 of questions were answered correctly Students baseline knowledge of Gene expression (Mass 32 and 33) was poor with only 47 of the questions covering this material answered correctly at the Pre test After implementation students were able to answer 61 of the questions accurately Summary Overall the implementation was quite successful Students enjoyed the lesson and showed a genuine interest in medical genetics Importantly test data showed improvement after completing the lesson suggesting that more traditional educational methods were enhanced by utilization of a case-study approach which may have more successfully addressed student misconceptions The assessments demonstrate that students still struggle to connect changes in DNA genotype with phenotype In retrospect some of the disease materials may have been too dense with medical terminology thus students were not able to focus on the genetics concepts Future revisions of the materials will contain embedded definitions of medical terminology In addition the workflow during classroom sessions felt somewhat rushed We feel that a one day extension of this curriculum will enable a more careful examination of the genetic concepts within the context of specific disease states We believe that this approach increased student interest in the topic and offered them an alternative method of learning With appropriate modifications in emphasis and support materials we expect that future implementations will demonstrate even greater improvements in student knowledge and understanding of the way in which alterations at the level of DNA result in human disease Both the classroom teacher and the higher education partner are committed to using this lesson next school year with incorporation of the aforementioned modifications
Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Drs __________________________________________ Block _____ Date __________
DNA to Disease Patient Analysis Worksheet 1 What is the name of the disease that affects your patient 2 Examine the disease information sheet for your patient
A Who does this disease typically affect B What are the symptoms
C How common is it (prevalence)
D What gene is affected in your patient E What type of DNA mutation causes this disease F Can this disease be inherited If so how
3 How can this disorder be diagnosed 4 What treatmenttherapy options would be recommended for a patient with this disease
5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

5 Examine the DNA sequences for your patient and an unaffected individual A Write them here
Affected - Unaffected - B What differences do you see in the DNA nucleotides 6 Transcribe the DNA (patient and unaffected) into mRNA
A Write them here Affected ndash Unaffected - B What differences do you see in the mRNA nucleotides 7 Translate the mRNA into a polypeptide (Use your codon chart)
A Write them here Affected ndash Unaffected - B What differences do you see in the amino acids 8 How do all of these differences affect proteins in your patientrsquos body
Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Dr _____________________________________ Block_____ Date________________
DNA to Disease Rounds Analysis Worksheet Presenting Doctors ___________________________ ________________________ ___________________________ ________________________ Name of Disease _________________________________________________________ 1 What are the major symptoms of this disease (Give at least 3) 2 What is the name of the gene or protein that is involved in this disease 3 Describe the mutation that causes this disease A How does it affect the DNA B How does it affect the RNA C How does it affect the protein D How does this lead to the symptoms seen in the patient 4 Explain one way in which this diseasemutation is similar to your patientrsquos disease 5 Explain one way in which this diseasemutation is different from your patientrsquos disease How would you rate the rounds presentation given by these doctors (Choose one)
Excellent Very Good Good So-So Poor
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
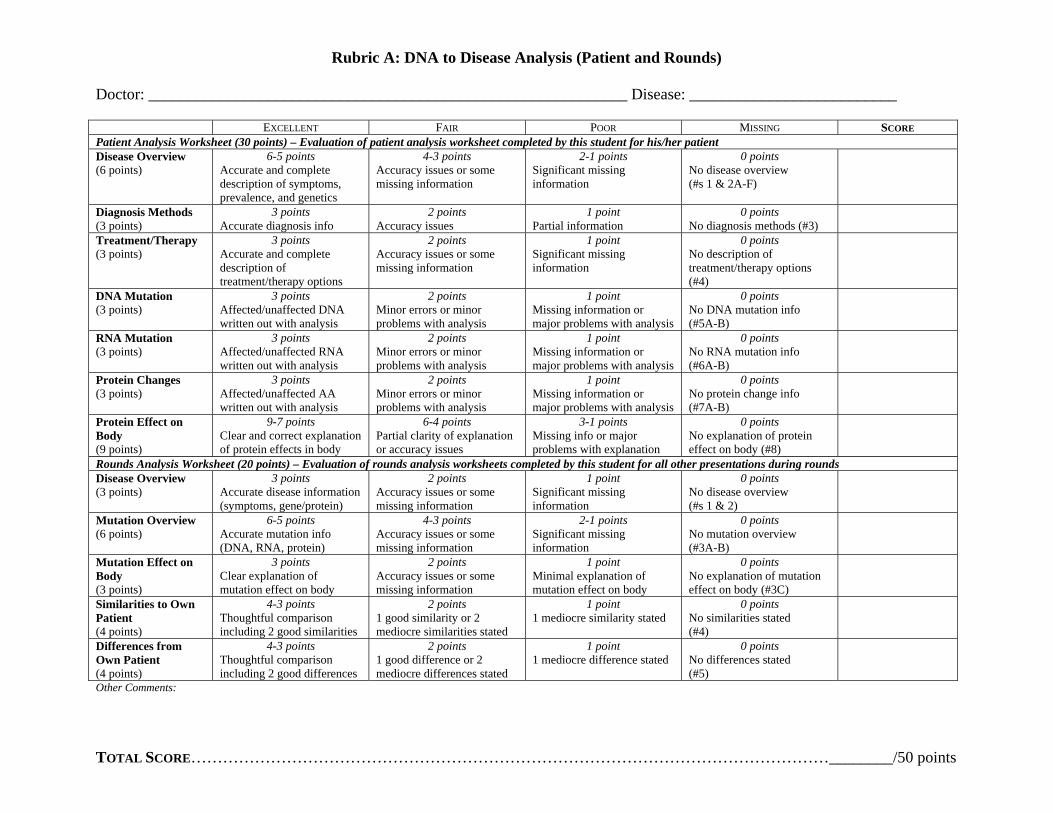
Rubric A DNA to Disease Analysis (Patient and Rounds)
Doctor ____________________________________________________________ Disease __________________________
E F P MXCELLENT AIR OOR ISSING SCORE Patient Analysis Worksheet (30 points) ndash Evaluation of patient analysis worksheet completed by this student for hisher patient Disease Overview (6 points)
6-5 points Accurate and complete description of symptoms prevalence and genetics
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No disease overview (s 1 amp 2A-F)
Diagnosis Methods (3 points)
3 points Accurate diagnosis info
2 points Accuracy issues
1 point Partial information
0 points No diagnosis methods (3)
TreatmentTherapy (3 points)
3 points Accurate and complete description of treatmenttherapy options
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No description of treatmenttherapy options (4)
DNA Mutation (3 points)
3 points Affectedunaffected DNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No DNA mutation info (5A-B)
RNA Mutation (3 points)
3 points Affectedunaffected RNA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No RNA mutation info (6A-B)
Protein Changes (3 points)
3 points Affectedunaffected AA written out with analysis
2 points Minor errors or minor problems with analysis
1 point Missing information or major problems with analysis
0 points No protein change info (7A-B)
Protein Effect on Body (9 points)
9-7 points Clear and correct explanation of protein effects in body
6-4 points Partial clarity of explanation or accuracy issues
3-1 points Missing info or major problems with explanation
0 points No explanation of protein effect on body (8)
Rounds Analysis Worksheet (20 points) ndash Evaluation of rounds analysis worksheets completed by this student for all other presentations during rounds Disease Overview (3 points)
3 points Accurate disease information (symptoms geneprotein)
2 points Accuracy issues or some missing information
1 point Significant missing information
0 points No disease overview (s 1 amp 2)
Mutation Overview (6 points)
6-5 points Accurate mutation info (DNA RNA protein)
4-3 points Accuracy issues or some missing information
2-1 points Significant missing information
0 points No mutation overview (3A-B)
Mutation Effect on Body (3 points)
3 points Clear explanation of mutation effect on body
2 points Accuracy issues or some missing information
1 point Minimal explanation of mutation effect on body
0 points No explanation of mutation effect on body (3C)
Similarities to Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good similarities
2 points 1 good similarity or 2 mediocre similarities stated
1 point 1 mediocre similarity stated
0 points No similarities stated (4)
Differences from Own Patient (4 points)
4-3 points Thoughtful comparison including 2 good differences
2 points 1 good difference or 2 mediocre differences stated
1 point 1 mediocre difference stated
0 points No differences stated (5)
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
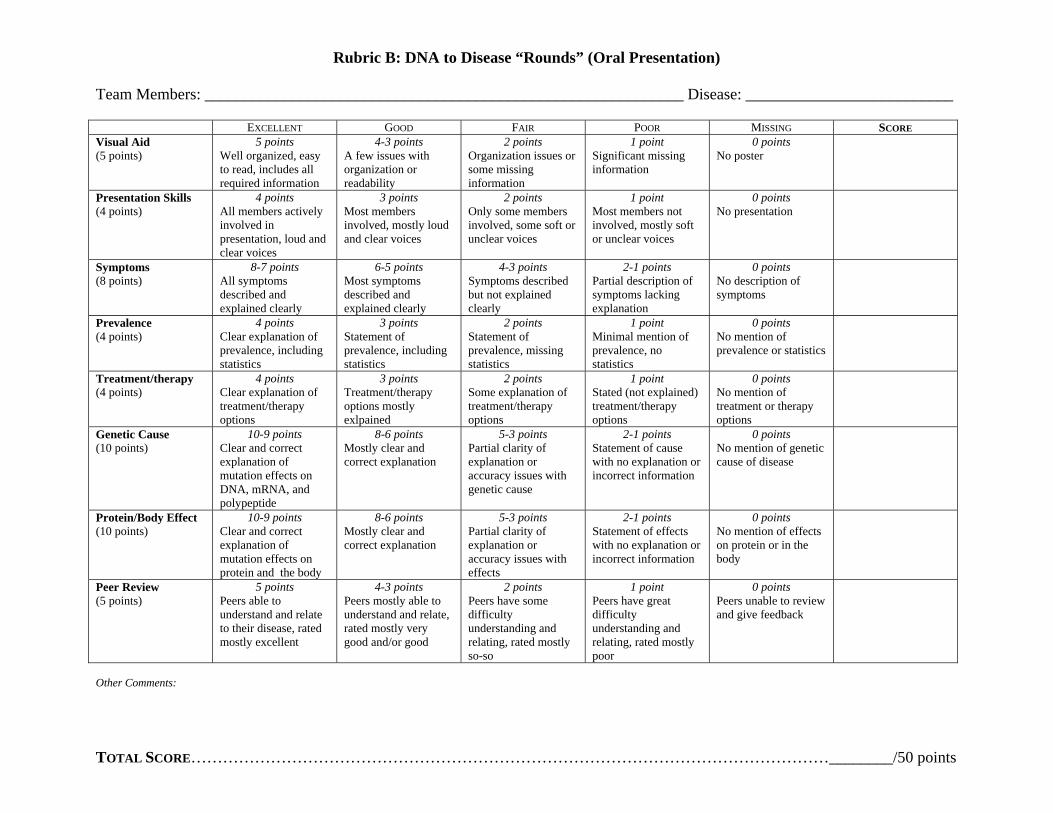
Rubric B DNA to Disease ldquoRoundsrdquo (Oral Presentation)
Team Members ____________________________________________________________ Disease __________________________
E G F P MXCELLENT OOD AIR OOR ISSING SCORE Visual Aid (5 points)
5 points Well organized easy to read includes all required information
4-3 points A few issues with organization or readability
2 points Organization issues or some missing information
1 point Significant missing information
0 points No poster
Presentation Skills (4 points)
4 points All members actively involved in presentation loud and clear voices
3 points Most members involved mostly loud and clear voices
2 points Only some members involved some soft or unclear voices
1 point Most members not involved mostly soft or unclear voices
0 points No presentation
Symptoms (8 points)
8-7 points All symptoms described and explained clearly
6-5 points Most symptoms described and explained clearly
4-3 points Symptoms described but not explained clearly
2-1 points Partial description of symptoms lacking explanation
0 points No description of symptoms
Prevalence (4 points)
4 points Clear explanation of prevalence including statistics
3 points Statement of prevalence including statistics
2 points Statement of prevalence missing statistics
1 point Minimal mention of prevalence no statistics
0 points No mention of prevalence or statistics
Treatmenttherapy (4 points)
4 points Clear explanation of treatmenttherapy options
3 points Treatmenttherapy options mostly exlpained
2 points Some explanation of treatmenttherapy options
1 point Stated (not explained) treatmenttherapy options
0 points No mention of treatment or therapy options
Genetic Cause (10 points)
10-9 points Clear and correct explanation of mutation effects on DNA mRNA and polypeptide
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with genetic cause
2-1 points Statement of cause with no explanation or incorrect information
0 points No mention of genetic cause of disease
ProteinBody Effect (10 points)
10-9 points Clear and correct explanation of mutation effects on protein and the body
8-6 points Mostly clear and correct explanation
5-3 points Partial clarity of explanation or accuracy issues with effects
2-1 points Statement of effects with no explanation or incorrect information
0 points No mention of effects on protein or in the body
Peer Review (5 points)
5 points Peers able to understand and relate to their disease rated mostly excellent
4-3 points Peers mostly able to understand and relate rated mostly very good andor good
2 points Peers have some difficulty understanding and relating rated mostly so-so
1 point Peers have great difficulty understanding and relating rated mostly poor
0 points Peers unable to review and give feedback
Other Comments
TOTAL SCOREhelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphelliphellip________50 points
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz
Multiple Choice Identify the letter of the choice that best completes the statement or answers the question
____ 1 What happens during the process of translation
a Messenger RNA is made from DNA b The cell uses information from messenger RNA to produce proteins c Transfer RNA is made from messenger RNA d Copies of DNA molecules are made
____ 2 A mutation that involves a single nucleotide is called a(an) a chromosomal mutation c point mutation b inversion d translocation
____ 3 Genes contain instructions for assembling a purines c proteins b nucleosomes d pyrimidines
____ 4 What is produced during transcription a RNA molecules c RNA polymerase b DNA molecules d proteins
____ 5 The diagram below represents part of a process that occurs in cells
Which process is represented a meiosis c replication b osmosis d translation
____ 6 Which type of RNA functions as a blueprint of the genetic code a rRNA c mRNA b tRNA d RNA polymerase
____ 7 In phenylketonuria (PKU) an enzyme that converts one amino acid into another does not work properly Which of the following is the most likely cause of this genetic condition a an error in the transcription of the gene for the enzyme b a mutation in the DNA sequence that codes for the enzyme c an excess of the amino acids necessary to produce the enzyme d a structural variation in the amino acid modified by the enzyme
Page 1
Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Name________________________________________ Block_____ Date____________________
____ 8 How many codons are needed to specify three amino acids a 3 c 9 b 6 d 12
____ 9 Which of the following statements is false a Some genes code for enzymes b The instructions for making some proteins are not specified by genes c An organismrsquos inherited traits depend on proteins d An organismrsquos genes determine its inherited traits
____ 10 Individuals with one form of lactose intolerance do not produce the enzyme lactase because the gene coding for the production of lactase is shut off in their cells This means that which of the following processes does not occur for the gene a hydrogenation c replication b mutation d transcription
____ 11 The diagram below shows the final steps of a biochemical pathway used by the bacterium Serratia marcescens to produce a red pigment molecule Letters X Y and Z represent intermediate molecules produced in the pathway Four enzymes are also involved in the pathway as shown
A mutant strain of S marcescens produces molecules X and Y but does not produce the red pigment molecule or molecule Z Based on this result it can be concluded that there must be a mutation in the gene coding for which enzyme a enzyme 1 c enzyme 3 b enzyme 2 d enzyme 4
____ 12 Which of the following best describes the result of a mutation in an organismrsquos DNA a The mutation may produce a zygote b The mutation may cause a phenotypic change c The mutation causes damage when it occurs d The mutation creates entirely new organisms
____ 13 Unlike DNA RNA contains a adenine c phosphate groups b uracil d thymine
____ 14 Which of the following is a nucleotide found in DNA a ribose + phosphate group + thymine b ribose + phosphate group + uracil c deoxyribose + phosphate group + uracil d deoxyribose + phosphate group + cytosine
____ 15 DNA is copied during a process called a replication c transcription b translation d transformation
____ 16 Which of the following is NEVER a frameshift mutation a substitution c deletion b insertion d point mutation
Page 2
Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Name________________________________________ Block_____ Date____________________
____ 17 The mold Aspergillus flavus grows on grain A flavus produces a toxin that binds to DNA in the bodies of animals that eat the grain The binding of the toxin to DNA blocks transcription so it directly interferes with the ability of an animal cell to do which of the following a transport glucose across the cell membrane into the cytoplasm b produce ATP using energy released from glucose and other nutrients c transfer proteins from the endoplasmic reticulum to Golgi complexes d send protein-building instructions from the nucleus to the cytoplasm and ribosomes
____ 18 During transcription an RNA molecule is formed a that is complementary to both strands of DNA b that is identical to part of a single strand of DNA c that is double-stranded d inside the nucleus
____ 19 Which of the following statements best describes a DNA molecule a It is a double helix c It is composed of amino acids b It contains the sugar ribose d It contains the nitrogenous base uracil
____ 20 During translation the type of amino acid that is added to the growing polypeptide depends on the a codon on the mRNA only b anticodon on the mRNA only c anticodon on the tRNA to which the amino acid is attached only d codon on the mRNA and the anticodon on the tRNA to which the amino acid is attached
Page 3
Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Name________________________________________ Block_____ Date____________________
DNA to Disease Quiz Answer Section
MULTIPLE CHOICE
1 ANS B
2 ANS C
3 ANS C
4 ANS A
5 ANS D 2008 Biology - High School Question 39 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
6 ANS C
7 ANS B 2007 Biology - High School Question 28 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
8 ANS A
9 ANS B
10 ANS D 2008 Biology - High School Question 11 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
11 ANS C 2008 Biology - High School Question 17 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
12 ANS B 2007 Biology - High School Question 3 Multiple-Choice Reporting Category Genetics Standard Genetics - B 33
13 ANS B
14 ANS D
15 ANS A
16 ANS A
17 ANS D 2007 Biology - High School Question 16 Multiple-Choice Reporting Category Genetics Standard Genetics - B 32
18 ANS D
19 ANS A 2008 Biology - High School Question 24 Multiple-Choice
Page 4
Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Name________________________________________ Block_____ Date____________________
Page 5
Reporting Category Genetics Standard Genetics - B 31
20 ANS D
Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Achondroplasia
Achondroplasia is a disorder of bone growth Although achondroplasia
literally means without cartilage formation the problem is not in forming
cartilage but in converting it to bone particularly in the long bones of the
arms and legs
All people with achondroplasia have short stature The average height of an
adult male with achondroplasia is 131 centimeters (4 feet 4 inches) and the
average height for adult females is 124 centimeters (4 feet 1 inch)
Characteristic features of achondroplasia include an average-size trunk
short arms and legs with particularly short upper arms and thighs limited
range of motion at the elbows and an enlarged head (macrocephaly) with a
prominent forehead Fingers are typically short and the ring finger and
middle finger may diverge giving the hand a three-pronged (trident)
appearance People with achondroplasia are generally of normal intelligence
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
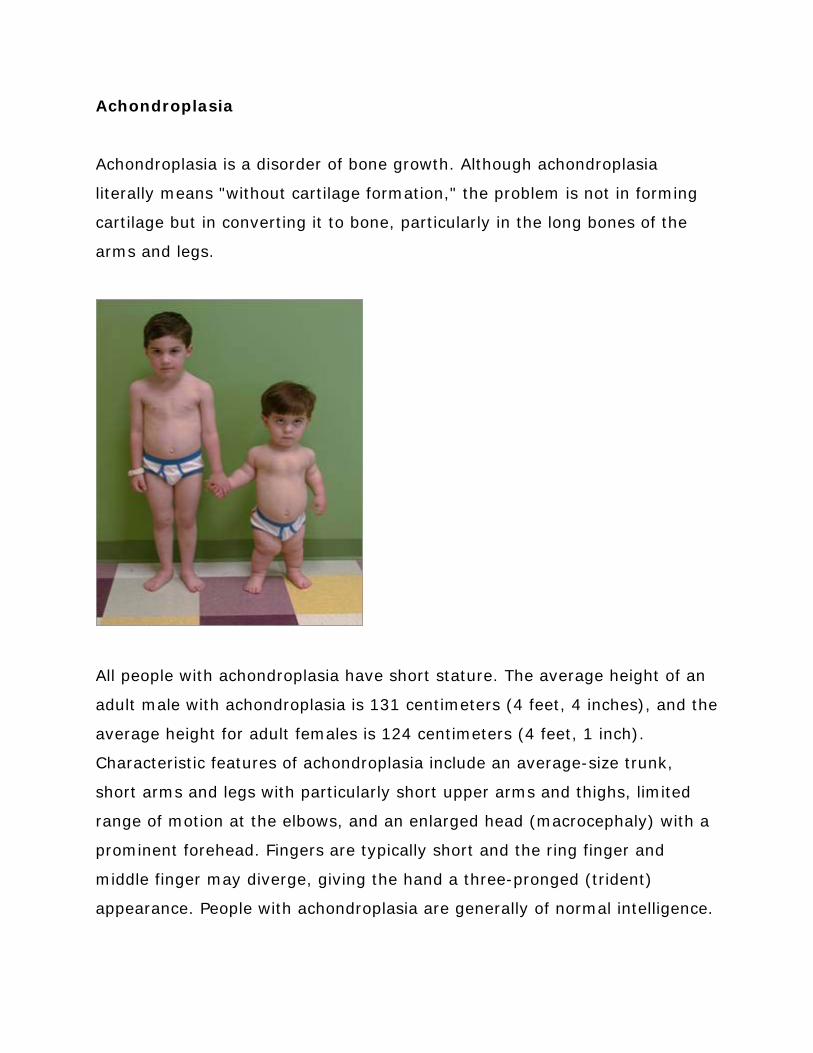
Health problems commonly associated with achondroplasia include episodes
in which breathing slows or stops for short periods (apnea) obesity and
recurrent ear infections In adulthood individuals with the condition usually
develop a pronounced and permanent sway of the lower back (lordosis) and
bowed legs Older individuals often have back pain which can cause
difficulty with walking Life span is usually normal although compression of
the spinal cord andor upper airway obstruction increases the risk of death in
infancy
Achondroplasia is the most common type of inherited disproportionate short
stature (short-limbed dwarfism) The condition occurs in 1 in 15000 to
40000 newborns
Diagnosistesting Achondroplasia can be diagnosed by characteristic
clinical and radiographic findings in most affected individuals In individuals
who may be too young to diagnose with certainty or in individuals with
atypical findings molecular genetic testing can be used to detect a mutation
in the FGFR3 gene
Molecular genetics
Mutations in the Fibroblast growth factor receptor 3 (FGFR3) gene cause
achondroplasia
More than 99 of individuals with achondroplasia have one of two mutations
in FGFR3 In about 98 of individuals the mutation is a G380R substitution
resulting from a G-to-A point mutation at nucleotide 1138 of the FGFR3
gene About 1 of individuals have a G-to-C point mutation at nucleotide
1138
The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

The penetrance of the gene is 100 meaning that all individuals who have
a single copy of the altered FGFR3 have achondroplasia
Normal DNA
CCG TAG GAG TCG ATG CCC CAC CCG AAG AAG GAC
Mutant DNA
CCG TAG GAG TCG ATG TCC CAC CCG AAG AAG GAC
FGFR3 gene product
The FGFR3 gene provides instructions for making a protein called fibroblast
growth factor receptor 3 This protein is part of a family of fibroblast growth
factor receptors that share similar structures and functions These proteins
play a role in several important cellular processes including regulation of cell
growth and division determination of cell type formation of blood vessels
wound healing and embryo development
The FGFR3 protein spans the cell membrane so that one end of the protein
remains inside the cell and the other end projects from the outer surface of
the cell This positioning of the protein allows it to interact with specific
growth factors outside the cell and to receive signals that control growth and
development When these growth factors attach to the FGFR3 protein the
protein triggers a cascade of chemical reactions inside the cell that instruct
the cell to undergo certain changes such as maturing to take on specialized
functions
The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

The FGFR3 gene provides instructions for making a protein that is involved
in the development and maintenance of bone and brain tissue This protein
limits the formation of bone from cartilage (a process called ossification)
particularly in the long bones
Normal gene product Proteins in the family of fibroblast growth factor
receptors (FGFRs) have a highly conserved structure The mature fibroblast
growth factor receptor 3 protein like all of the FGFRs is a membrane-
spanning tyrosine kinase receptor with an extracellular ligand-binding
domain consisting of three immunoglobulin subdomains a transmembrane
domain and a split intracellular tyrosine kinase domain
In people with achondroplasia the substitution of an arginine for a glycine
causes the protein to be overly active which interferes with skeletal
development and leads to the disturbances in bone growth seen with this
disorder
Abnormal gene product The G380R mutation has been shown to result in
constitutive activation of the FGF receptor Targeted disruption of the FGFR3
gene causes enhanced growth of long bones and vertebrae in mice
suggesting that fibroblast growth factor receptor 3 negatively regulates bone
growth Thus FGFR3 mutations in achondroplasia can be interpreted as
gain-of-function mutations that activate the fundamentally negative growth
control exerted by the FGFR3 pathway
Genetic counseling Achondroplasia is inherited in an autosomal dominant
manner which means one copy of the altered gene in each cell is sufficient to
cause the disorder About 80 percent of people with achondroplasia have
average-size parents these cases result from a new (de novo) mutation in
the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

the FGFR3 gene Such parents have a low risk of having another child with
achondroplasia
De novo gene mutations are more common when the father is over the age
of 35 (advanced paternal age) Studies have demonstrated that de novo
gene mutations causing achondroplasia are exclusively inherited from the
father These mutations appear to result from de novo events during
spermatogenesis in the unaffected father
In the remaining cases people with achondroplasia have inherited an altered
FGFR3 gene from one or two affected parents If an individual with
achondroplasia has a reproductive partner with normal stature there is a
50 risk in each pregnancy of having a child with achondroplasia When
both parents have achondroplasia the risk to their offspring of having
normal stature is 25 of having achondroplasia 50 and of having
homozygous achondroplasia (a lethal condition) 25 Individuals who
inherit two altered copies of this gene typically have very severe problems
with bone growth and are usually stillborn or die shortly after birth from
respiratory failure Prenatal molecular genetic testing is available
Management Recommendations for management of children with
achondroplasia include monitoring of height weight and head
circumference measures to avoid obesity MRI or CT for evaluation of signs
of spinal cord compression adenotonsillectomy continuous positive airway
pressure (CPAP) by nasal mask and tracheostomy to correct obstructive
sleep apnea surgery to correct spinal stenosis and educational support in
socialization and school adjustment
References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

References for mutation locations
Shiang et al Cell 1994 Bellus et al 1995 Rousseau et al 1996
Reference for normal gene product
Green et al 1996
Abnormal gene product
Colvin et al 1996 Deng et al Cell 1996
Inheritance from father
Wilkin et al 1998
Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

Hand xray Achondroplasia
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
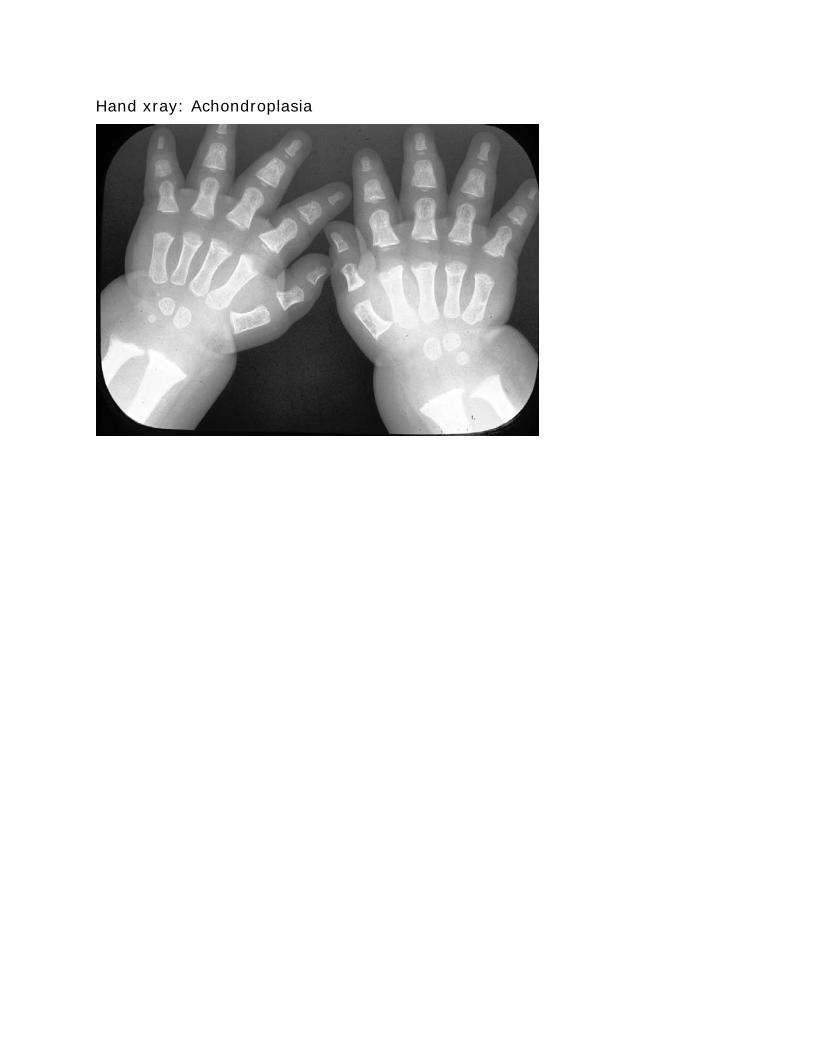
Hand xray normal
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
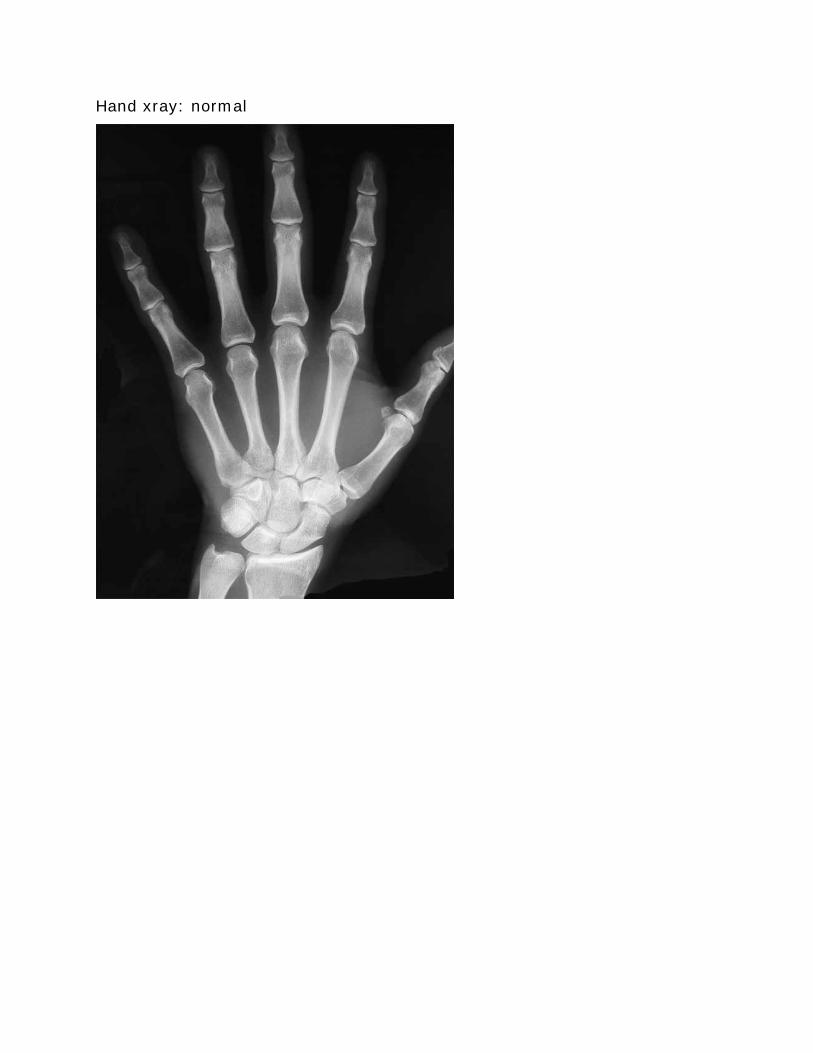
Cystic Fibrosis
ldquoSalty baby syndromerdquo
Cystic fibrosis (CF) affects epithelia of the respiratory tract exocrine
pancreas intestine male genital tract hepatobiliary system and exocrine
sweat glands resulting in complex multisystem disease The disorders most
common signs and symptoms include progressive damage to the respiratory
system and chronic digestive system problems
The epithelia or cells that line our internal organs normally produce mucus
Mucus is a slippery substance that lubricates and protects the linings of the
airways digestive system reproductive system and other organs and
tissues In people with cystic fibrosis the body produces mucus that is
abnormally thick and sticky As a result of this abnormal mucus affected
individuals have lower airway inflammation and chronic endobronchial
(airways of the lungs) infection Over time mucus buildup and infections
result in permanent lung damage including the formation of scar tissue
(fibrosis) and cysts in the lungs The result is to severe problems with
breathing and recurrent bacterial infections in the lungs These infections
cause chronic coughing wheezing and inflammation
Most people with cystic fibrosis also have digestive problems because thick
sticky mucus interferes with the function of the pancreas The pancreas is an
organ that produces insulin (a hormone that helps control blood sugar
levels) It also makes enzymes that help digest food In people with cystic
fibrosis mucus blocks the ducts of the pancreas preventing these enzymes
from reaching the intestines to aid digestion Problems with digestion can
lead to diarrhea malnutrition poor growth and weight loss Some babies
with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

with cystic fibrosis have meconium ileus a blockage of the intestine that
occurs shortly after birth
Cystic fibrosis used to be considered a fatal disease of childhood With
improved treatments and better ways to manage the disease many people
with cystic fibrosis now live well into adulthood Adults with cystic fibrosis
experience medical problems affecting the respiratory digestive and
reproductive systems For example most men with cystic fibrosis are unable
to father children (infertile) because the tubes that carry sperm (the vas
deferens) are blocked by mucus and do not develop properly This condition
is known as congenital bilateral absence of the vas deferens (CBAVD)
Infertility is also possible though less common in women with cystic
fibrosis
Today the overall median survival is 365 years (95 confidence intervals
337-400 years) Males with CF tend to survive longer than females
Prevalence
CF is the most common life-limiting autosomal recessive disorder in the
Caucasian population The disease incidence is 1 in 2500 to 3500 live births
among Caucasians Approximately 30000 affected persons live in the US
Cystic fibrosis is less common in other ethnic groups affecting about 1 in
17000 African Americans and 1 in 31000 Asian Americans
Molecular genetics
Mutations in the Cystic fibrosis transmembrane conductance regulator
(CFTR) gene cause cystic fibrosis CFTR gene is 230 kb long and contains 27
coding exons It produces a 65-kb mRNA product and encodes a 1480-
amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI

amino acid integral membrane protein that functions as a regulated chloride
channel in epithelial cells
The CFTR gene provides instructions for making a channel that transports
negatively charged chloride ions into and out of cells The flow of chloride
ions helps control the movement of water in tissues which is necessary for
the production of thin freely flowing mucus
Mutations in the CFTR gene disrupt the function of the chloride channels
preventing them from regulating the flow of chloride ions and water across
cell membranes As a result cells that line the passageways of the lungs
pancreas and other organs produce mucus that is unusually thick and
sticky This mucus clogs the airways and glands causing the characteristic
signs and symptoms of cystic fibrosis
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI
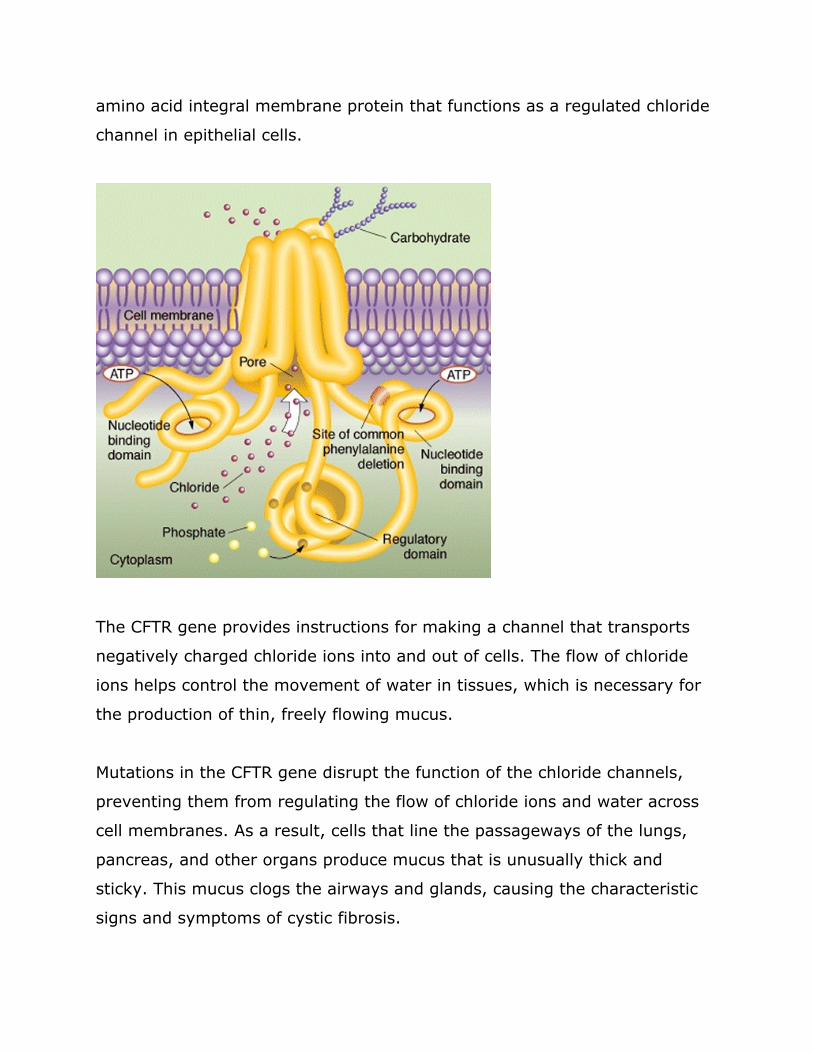
Pathologic allelic variants More than 1000 CFTR mutations are known to
be associated with CF almost all are point mutations or small (1-84 bp)
deletions The most common mutation is ΔF508 (Phe508del) or delta F508
which accounts for an estimated 30-80 (depending on the ethnic group)
of mutant alleles In the Caucasian population 1 in 22-28 people is
heterozygous for the ΔF508 allele
Normal DNA
TAG TAG AAA CCA CAA
Mutant DNA
TAG TAA CCA CCA
Delta F508 is a 3 basepair deletion in Exon 10 The result is a deletion of a
Phenylalanine residue from the CFTR protein The resultant segment of DNA
is smaller than the wildtype DNA and this difference can be detected by
electrophoresis The smaller DNA migrates faster through the agarose gel
This mutant protein which is only one amino acid short of the wild type
protein is retained in the endoplasmic reticulum and cannot reach the cell
surface where it normally resides
Cystic fibrosis (CF) Phenotypic features of CF include but are not limited
to the following
Respiratory Pulmonary disease remains the major cause of morbidity and
mortality in CF Affected individuals have lower airway inflammation and
chronic airway infection Failure of lung defenses leads to bacterial infection
of the lining of the airways Early manifestations are chronic cough
intermittent sputum production and exertional dyspnea As the lung disease
progresses as a result of chronic infection structural injury to the airways
occurs with resulting bronchiectasis End-stage lung disease is characterized
by extensive damage to the airways (cystsabscesses) and accompanying
scarring of lung adjacent to airways
Gastrointestinal 15-20 of newborns diagnosed with CF have a history
of meconium ileus at birth Individuals with CF also malabsorb fat in their
intestinal tracts leading to diarrhea poor growth and malnutrition and skin
rashes due to deficiencies of fat-soluble vitamins and zinc Acute or chronic
recurrent inflammation of the pancreas with pain fever nausea and
vomiting can be a presenting manifestation
Cystic fibrosis-related diabetes mellitus may present in adolescence It is
diagnosed in 7 of those age 11 to 17 years The prevalence increases in
adulthood
Liver scarring and failure can also be seen in CF Liver disease is second to
pulmonary disease as a cause of mortality in CF (17 of deaths)
Fertility More than 95 of males with CF are infertile as a result of
azoospermia caused by altered vas deferens which may be absent atrophic
or fibrotic Women with CF are fertile although a few females have
abnormal cervical mucus that may contribute to infertility
Diagnosistesting Most commonly the diagnosis of cystic fibrosis (CF) is
established in individuals with one or more characteristic phenotypic features
of CF plus evidence of an abnormality in cystic fibrosis transmembrane
conductance regulator (CFTR) function CFTR function can be assessed using
either a sweat chloride test or transepithelial nasal potential difference The
more common sweat chloride test measures the amount of chloride
produced by the skin in response to a chemical called pilocarpine In
individuals with CFTR mutations sweat chloride levels are abnormally high
(gt60 mEqL) because without a normally functioning CFTR they cannot
bring chloride into their cells
Genetic counseling CF is inherited in an autosomal recessive manner
Therefore siblings of an individual with cystic fibrosis have a 25 chance of
being affected a 50 chance of being asymptomatic carriers and a 25
chance of being unaffected and not carriers Molecular genetic testing for
disease-causing mutation(s) in the CFTR gene is used for carrier detection in
population screening programs Prenatal testing is available for pregnancies
at increased risk for CF if the disease-causing mutations in the family are
known
Parents of a proband with cystic fibrosis (CF)
bull The unaffected parents are obligate carriers (heterozygotes) and
have an alteration in one copy of the CFTR gene
o If the disease-causing CFTR alleles have been
identified in the proband it is most informative
to test parents by molecular genetic testing
bull Carriers are generally asymptomatic
bull On rare occasions a parent may be diagnosed as affected
subsequent to the diagnosis of the child
Newborn screening Newborn screening using immunoreactive trypsinogen
(IRT) assays performed on blood spots has been implemented in most of the
United States Trypsinogen is synthesized in the pancreas in CF its release
into the circulation appears to be enhanced by abnormal pancreatic duct
secretions Thus IRT levels are elevated in cystic fibrosis Newborns found
to have abnormal IRT results are evaluated through sweat testing andor
molecular genetic testing of CFTR
Treatment of Manifestations
Respiratory
bull Intervention to treat or prevent pulmonary complications may
include antibiotics bronchodilators anti-inflammatory agents
mucolytic agents and chest physiotherapy (postural drainage
with chest percussion)
bull Lung or heartlung transplantation is an option for selected
individuals with severe disease
bull Sinus symptoms may require antibiotics andor surgery to
correct
Gastrointestinal
bull Nutritional therapy may include special formulas for infants or
tuge feeding often via an indwelling gastric feeding catheter
bull Pancreatic insufficiency is treated with oral pancreatic enzyme
replacement with meals
Endocrine
bull Diabetes mellitus is treated with dietary restrictions andor
insulin
Physical activity exercise and conditioning A regular exercise
program is important to all individuals in maintaining overall health For
persons with CF regular exercise has the added benefits of maintaining
bone health and improving airway clearance Although exercise improves
clearance of airway secretions it should be used more as an adjunct rather
than as a replacement for the individuals prescribed airway clearance
regimen
References
CT Helbich Radiology 1999
Chest xray normal
Chest xray cystic fibrosis
Chest CT scan normal
Chest CT scan cystic fibrosis
Familial Adenomatous Polyposis (FAP)
Introduction
Familial adenomatous polyposis (FAP) is an inherited disorder characterized
by cancer of the large intestine (colon) and rectum Individuals with FAP
develop hundreds to thousands of precancerous colonic polyps beginning
on average at age 16 years (range 7-36 years) By age 35 years 95 of
individuals with FAP have polyps without colectomy (removal of the colon)
colon cancer is inevitable The average age of colon cancer diagnosis is 39
years Some people have a variant of the disorder called attenuated familial
adenomatous polyposis in which polyp growth is delayed The average age
of colorectal cancer onset for attenuated familial adenomatous polyposis is
55 years
In people with classic familial adenomatous polyposis the number of polyps
increases with age and hundreds to thousands of polyps can develop in the
colon Also of particular significance are noncancerous growths called
desmoid tumors These fibrous tumors usually occur in the tissue covering
the intestines and may be provoked by surgery to remove the colon
Desmoid tumors tend to recur after they are surgically removed In both
classic familial adenomatous polyposis and its attenuated variant benign
and malignant tumors are sometimes found in other places in the body
including the duodenum (a section of the small intestine) stomach bones
skin and other tissues People who have colon polyps as well as growths
outside the colon are sometimes described as having Gardner syndrome
A milder type of familial adenomatous polyposis called autosomal recessive
familial adenomatous polyposis has also been identified People with the
autosomal recessive type of this disorder have fewer polyps than those with
the classic type Fewer than 100 polyps typically develop rather than
hundreds or thousands The autosomal recessive type of this disorder is
caused by mutations in a different gene than the classic and attenuated
types of familial adenomatous polyposis
Prevalence
The reported incidence of familial adenomatous polyposis varies from 1 in
7000 to 1 in 22000 individuals FAP (and associated colon cancer
syndromes) historically accounted for about 05 of all colorectal cancers
this figure is declining as more at-risk family members undergo successful
treatment following early polyp detection and prophylactic colectomy
Diagnosistesting The diagnosis relies primarily on clinical findings
Familial adenomatous polyposis (FAP) is diagnosed clinically in an individual
with one of the following
bull One hundred or more colorectal adenomatous polyps
Note The diagnosis of FAP is generally considered in
individuals with polyposis occurring before age 40 years
bull Fewer than 100 adenomatous polyps and a relative with FAP
Note Individuals with 100 or more polyps occurring at
ldquoadvancedrdquo ages (35 to 40 years or older) may be found to
have attenuated FAP
bull A personal history of colorectal cancer before age 60 years and a
family history of multiple adenomatous polyps
Other features variably present in FAP
Adenomatous polyps of the small bowel are observed in 50-90 of
individuals with FAP The lifetime risk of small bowel malignancy is 4-12
the majority occurs in the duodenum
Extraintestinal manifestations
Osteomas are bony growths found most commonly on the skull and
mandible however they may occur in any bone of the body Osteomas do
not usually cause clinical problems and do not become malignant they may
appear in children prior to the development of colonic polyps
Dental abnormalities Unerupted teeth congenital absence of one or more
teeth and other dental abnormalities have been reported in approximately
17 of individuals with FAP compared to 1-2 of the general population
Benign skin lesions include epidermoid cysts and fibromas that may be
found on any part of the body including the face and are mainly of
cosmetic concern
Desmoid tumors develop in approximately 10 of children and adults with
FAP These poorly understood benign tumors are locally invasive but do not
metastasize Desmoid tumors form predominantly within the abdomen or in
the abdominal wall but may also occur extra-abdominally Desmoid tumors
may compress abdominal organs or complicate abdominal surgery About
5 of individuals with FAP experience morbidity andor mortality from
desmoid tumors
Cancer outside of the gastrointestinal tract Individuals with FAP have a
small but measurable increase in pancreatic liver thyroid and brain cancer
Molecular genetics
Mutations in the APC (adenomatous polyposis coli) gene cause both classic
and attenuated familial adenomatous polyposis APC is a tumor suppressor
gene that plays a role in cell signaling
Most cases of colon cancer are thought to develop slowly over a period of
several years usually arising within a benign polyp Every polyp has the
potential to develop into cancer therefore individuals with more polyps are
at a much higher risk for cancer Mutation in APC is thought to be an
important step in the initial formation of polyps Inactivation of the APC
gene located on chromosome 5 is thought to lead to increased cell
proliferation and contribute to the formation of colonic polyps Several
genetic alterations must occur during the conversion of normal colon cells
into cells capable of forming tumors In many cases mutation of the APC
gene is thought to be one of the first steps Although most people with
mutations in the APC gene will develop colorectal cancer the number of
polyps and the time frame in which they become malignant depend on the
location of the mutation in the gene
Normal allelic variants The gene is alternatively spliced in multiple coding
and noncoding regions the main transcript has 15 exons with 8532 base
pairs that code for 2843 amino acids and result in a 3118-kd protein Exon
15 is large and comprises over three-quarters of the coding region of the
gene
Pathologic allelic variants Over 826 different mutations have been found
in families with an APC-associated polyposis condition Mutations almost
always cause a premature truncation of the APC protein usually through
single amino-acid substitutions or frameshifts While mutations have been
found scattered throughout the gene they are predominantly located in the
5 end of the gene The most common APC mutation is a 5 basepair deletion
at codon 1309 as depicted below
Normal DNA
TTT CTT TTC TAA CCT TGA TC
Mutant DNA
TTT CTA ACC TTG ATC
Interestingly the location of APC mutation is linked to the average age of
symptom onset codon 1309 age 20 years between codon 168 and 1580
(excluding 1309) age 30 years and all others age 52 years
Normal gene product The APC protein product is a tumor suppressor The
presence of normal APC protein appears to maintain normal apoptosis (cell
death) and may also decrease cell proliferation probably through its
regulation of b-catenin
Abnormal gene product Disease-causing mutations in the APC gene most
often result in truncated protein products When the APC gene is mutated
and abnormal protein is present high levels of free cytosolic b-catenin
result Free b-catenin migrates to the nucleus binds to a transcription factor
Tcf-4 or Lef-1 (T cell factor-lymphoid enhancer factor) which leads to
increasing proliferation or decreasing apoptosis
Penetrance
In FAP the penetrance of colonic adenomatous polyposis and colon cancer is
virtually 100 in untreated individuals
Management
Surveillance
Recommended surveillance of individuals known to have FAP or an APC
disease-causing mutation and individuals at risk for FAP who have not
undergone molecular genetic testing or who are members of families in
which molecular genetic testing did not identify a disease-causing mutation
bull Sigmoidoscopy or colonoscopy every one to two years beginning
at age ten to 12 years
bull Colonoscopy once polyps are detected
bull Annual colonoscopy if colectomy is delayed more than a year
after polyps emerge In individuals age ten to 20 years in
whom adenomas are smaller than 60 mm and without villous
component delay in colectomy may be considered
bull Esophagogastroduodenoscopy (EGD) beginning by age 25 years
or prior to colectomy and repeated every one to three years
Treatment of manifestations Colectomy is advised when more than 20 or 30
adenomas or multiple adenomas with advanced histology have occurred
Endoscopic or surgical removal of duodenal adenomas is considered if polyps
exhibit villous change or severe dysplasia exceed one centimeter in
diameter or cause symptoms
Testing of relatives at risk Molecular genetic testing for early identification
of at-risk family members improves diagnostic certainty and reduces the
need for costly screening procedures in those at-risk family members who
have not inherited the disease-causing mutation
Clinically available molecular genetic testing of APC detects disease-causing
mutations in up to 90 of probands with typical FAP Molecular genetic
testing is most often used in the early diagnosis of at-risk family members
as well as in confirming the diagnosis of FAP or attenuated FAP in individuals
with equivocal findings (eg lt100 adenomatous polyps)
Pattern of InheritanceGenetic counseling
FAP is inherited in an autosomal dominant manner
Use of molecular genetic testing for early identification of at-risk family
members improves diagnostic certainty and reduces the need for costly
screening procedures in those at-risk family members who have not
inherited the disease-causing mutation Additionally individuals diagnosed
with APC-associated polyposis conditions as a result of having an affected
relative have a significantly greater life expectancy than those individuals
diagnosed on the basis of symptoms [Heiskanen et al 2000] As colon
screening for those at risk for classic FAP begins as early as age ten to12
years molecular genetic testing is generally offered to children at risk for
classic FAP by age ten years
Approximately 75-80 of individuals with FAP have an affected parent
The remaining 20-25 of individuals with an APC-associated polyposis
condition have the altered gene as the result of a de novo gene mutation
Offspring of an affected individual have a 50 risk of inheriting the altered
APC gene Prenatal testing and preimplantation genetic diagnosis are
possible if a disease-causing mutation is identified in an affected family
member
Normal Colon
Colon of an individual with FAP
Gross pathology specimen Colon from an individual with FAP
Hereditary hemochromatosis (HHC) ldquoBronze diabetesrdquo Introduction Hereditary hemochromatosis (HHC) is an inherited disorder that increases the amount of iron that the body absorbs from the gut Symptoms are caused by this excess iron being deposited in multiple organs of the body Most commonly excess iron in the liver causes cirrhosis which may develop into liver cancer Iron deposits in the pancreas can result in diabetes Similarly excess iron stores can cause cardiomyopathy (damage to the heart muscle) pigmentation of the skin and arthritis Without therapy males may develop symptoms between age 40 and 60 years and females after menopause
Iron is an essential nutrient found in many foods The greatest amount is found in red meat and iron-fortified breads and cereals In the body iron becomes part of hemoglobin a molecule in the blood that transports oxygen from the lungs to all body tissues Healthy people usually absorb about 10 percent of the iron contained in the food they eat which meets normal dietary requirements People with hemochromatosis absorb up to 30 percent of iron Over time they absorb and retain between five to 20 times more iron than the body needs Because the body has no natural way to rid itself of the excess iron it is stored in body tissues specifically the liver heart and pancreas HHC is caused by mutations in the HFE gene This gene is located on chromosome 6 and one mutation leads to the substitution of the 282nd amino acid Cysteine (C) becomes tyrosine (Y) therefore the mutation is called C282Y The switch of amino acids is thought to affect how the HFE protein interacts with the transferrin receptor (TFR1) which plays an important role in iron homeostasis A less common mutation H63D has also been identified in the HFE gene but will not be addressed here Prevalence
HHC is the most frequent autosomal recessive disorder among individuals of European descent The prevalence of individuals with two HFE mutant alleles (homozygote) is about 3-5 in 1000 or 1300 This means that 11 of Caucasians carry one mutant allele (heterozygote) HFE mutations are much less common in other ethnicracial groups Among African Americans the frequency of homozygotes for hemochromatosis is about 1 in 7000 with 23 of that population being heterozygotes Hispanics have homozygote and heterozygote frequencies of 0027 and 30 respectively Interestingly only a small proportion of people carrying HFE mutations suffer any symptoms This may be attributable to both environmental (diet and blood loss) and genetic factors Genetics HHC is inherited in an autosomal recessive manner Usually the genetic risk to the siblings of an affected individual (the proband) of having HHC would be 25 However the high carrier frequency for a mutant HFE allele in the general population of European origin (11 of the population or 19 persons) means that on occasion one parent has two abnormal HFE alleles usually in the absence of clinical findings In such instances the risk to each sibling of a proband of being homozygous for HFE is 50 Although prenatal testing would be technically feasible when both parents carry identified HFE mutations such requests would be highly unusual because HHC is an adult-onset treatable disease and the homozygous C282Y mutation has low clinical penetrance Diagnosis HHC should be suspected in any individual presenting with clinical signs of advanced iron overload including
bull Hepatomegaly (enlarged liver) bull Hepatic cirrhosis bull Liver cancer (hepatocellular carcinoma) bull Diabetes mellitus bull Heart failure bull Hypogonadism bull Arthritis bull Progressive increase in skin pigmentation (ldquobronzingrdquo)
The diagnosis of HHC in individuals with clinical symptoms consistent with HHC andor biochemical evidence of iron overload is typically based on the results of two blood screening tests
1 A transferrin-iron saturation greater than 45 2 Elevated serum ferritin concentration (gt 1000)
The confirmatory test is molecular genetic testing of the HFE gene
Liver biopsy is useful to confirm hepatic iron overload particularly in individuals with presumed hemochromatosis who lack the common HFE mutations associated with HHC but it is not diagnostic for HHC Magnetic resonance imaging (MRI) can estimate liver iron content by utilizing the paramagnetic properties of iron This method has been approved by the Food and Drug Association for the evaluation of individuals with iron overload Natural History Individuals with HFE-associated hereditary hemochromatosis (HHC) who express the phenotype clinically have inappropriately high intestinal absorption of iron from a normal diet resulting in excessive tissue storage of iron which may result in damage in a number of end-organs and potentially organ failure Although previous reports have suggested that males are ten times more likely than females to have symptoms of organ failure resulting from HHC recent studies show that among individuals with HHC women are half as likely as men to develop complications of end-stage organ failure Affected individuals may be identified because of signs and symptoms related to iron overload most frequently however they are identified before symptoms develop either through detection of abnormal iron-related studies or by evaluation as family members at risk for HHC Symptoms related to iron overload usually appear between age 40 and 60 years in males and after menopause in females Occasionally HHC manifests at an earlier age but hepatic fibrosis or cirrhosis is rare before age 40 years Often the first signs of clinically expressed HCC are an enlarged liver (hepatomegaly) arthritis involving the metacarpophalangeal joints (hand knuckles) a progressive increase in skin pigmentation resulting from deposits of melanin and iron diabetes mellitus resulting from pancreatic iron deposits and heart failure resulting from build up of iron in the heart muscle By the time cirrhosis or liver failure is recognized about 50 of individuals have diabetes mellitus and 15 have heart failure or irregular heart rhythms Hepatomegaly may or may not be present early in the disease however asymptomatic individuals can occasionally have hepatomegaly on physical examination With progression of the disease liver cirrhosis may develop and be complicated by cancer and end-stage liver disease Other common symptoms early in the disease are joint stiffness and pain Males may have impotence from pituitary dysfunction Abdominal pain weakness lethargy and weight loss are common but nonspecific findings When individuals with HHC are identified through iron studies or screening of at-risk family members most (75-90) do not have any symptoms Liver biopsy can be helpful to determine prognosis
bull Individuals diagnosed and treated prior to the
development of cirrhosis appear to have normal life expectancy
bull Those identified after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy
bull Individuals with cirrhosis who are treated have a better outcome than those who are not however treatment does not eliminate the 10-30 risk for liver cancer even years after successful iron depletion
Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign With iron depletion dysfunction of some affected organs (liver and heart) can improve however endocrine abnormalities and arthritis improve in only 20 of those treated Alcohol consumption causes worsening of symptoms in HHC In addition cirrhosis is much more common among C282Y homozygotes who consume excessive amounts of alcohol Death in clinically affected individuals with HHC is usually caused by liver failure liver cancer heart failure or an irregular heart rhythm However many individuals who are HFE mutant homozygotes (identified through screening) studies survive to old age Treatment of Manifestations Presence of characteristic clinical endpoints Treatment by phlebotomy is clearly indicated when clinical symptoms of hemochromatosis are present Therapeutic phlebotomy
bull The usual therapy is removal of the excess iron by weekly phlebotomy (ie removal of a unit of blood) until the serum ferritin concentration is 50 ngmL or lower Twice-weekly phlebotomy may be occasionally useful to accelerate iron depletion
bull Weekly phlebotomy is carried out until the hematocrit is 75 of the baseline hematocrit
bull Maintenance therapy is aimed at maintaining serum ferritin concentration below 50 ngmL and transferrin-iron saturation below 50 On average men require removal of twice as many units of blood as women Subsequent phlebotomies can be carried out to prevent reaccumulation of iron about every three to four months for men and once or twice a year for women
Treatment of iron overload
bull Periodic phlebotomy is a simple inexpensive safe and effective treatment Each unit of blood (400-500 mL) with a hematocrit of 40 contains about 160-200 mg of iron Each mL of packed red blood cells contains 1 mg of iron
bull Iron chelation (administration of the chemical that binds iron directly) is not recommended unless an individual has an elevated serum ferritin concentration and concomitant anemia that makes therapeutic phlebotomy impossible However this is uncommon in individuals with HHC
Liver transplantation Liver transplantation is the only treatment for end-stage liver disease from decompensated cirrhosis However the post-transplant survival among untreated individuals with HHC is poor Absence of characteristic clinical endpoints Current data suggest that even though serum ferritin concentration may rise over time development of end-organ damage from iron storage is rare Consequently there is no general agreement that phlebotomy treatment is indicated in the presence of biochemically defined abnormalities (ie elevated transferrin-iron saturation and elevated serum ferritin concentration) in the absence of characteristic clinical endpoints (ie diabetes mellitus cirrhosis and liver carcinoma) More long-term studies are required Molecular genetics Pathologic allelic variants At least 28 distinct HFE mutations have been reported most being missense or nonsense mutations One missense mutation accounts for the vast majority of disease-causing alleles in the population Cys282Tyr (C282Y nucleotide 845GgtA) This missense mutation removes a highly conserved cysteine residue that normally forms an intermolecular disulfide bond with beta-2-microglobulin and thereby prevents the protein from being expressed on the cell surface Nucleotide sequence Normal DNA TCT ATA TGC ACG GTC CAC CTC C282Y Mutant DNA TCT ATA TGC ATG GTC CAC CTC Detection of C282Y mutation Using a restriction enzyme digestion of the DNA sequence the presence of the C282Y mutation introduces a breakpoint in the DNA The result is two
smaller pieces of DNA In the absence of a mutation a single large DNA band is found
Lanes 1 and 5 = negative controls Lanes 3 4 7 = normal HFE sequence Lane 2 = Heterozygous for C282Y Lanes 6 and 8 = Homozygous for C282Y RNA With the exception of a single nucleotide change the messenger RNA for HFE is identical with or without the C282Y mutation Protein Normal gene product The largest predicted primary translation product is 348 amino acids which gives rise to a mature protein of about 321 amino acids after cleavage of the signal sequence The normal protein is then transported to the cell surface where it binds with another protein Beta-2-microglobulin and reduces the iron uptake Abnormal gene product The C282Y mutation destroys a key cysteine residue that is required for disulfide bonding with beta-2-microglobulin As a
result the HFE protein does not mature properly and becomes trapped in the endoplasmic reticulum and Golgi apparatus leading to decreased cell-surface expression The mechanistic basis for the phenotypic effect of other HFE mutations is not clear at present
References
GeneReviews NIH (Initial Posting April 3 2000 Last Update December 4 2006) Online Mendelian Inheritance in Man Thompson and Thompson (eds) Genetics in Medicine 6th Edition WB Saunders Co (New York) 2001
Skin image Lim R Kid Intl 2008
Liver histology normal
Liver histology iron overload and cirrhosis in hemochromatosis
Osteogenesis Imperfecta
ldquoBrittle bone syndromerdquo
Introduction
Osteogenesis imperfecta (OI) is a group of genetic disorders that mainly
affect the bones The term osteogenesis imperfecta means imperfect bone
formation People with this condition have bones that break easily often
from mild trauma or with no apparent cause Multiple fractures are common
and in severe cases can occur even before birth Milder cases may involve
only a few fractures over a persons lifetime
There are at least eight recognized forms of osteogenesis imperfecta
designated type I through type VIII The types can be distinguished by their
signs and symptoms although their characteristic features overlap Type I is
the mildest form of osteogenesis imperfecta and type II is the most severe
other types of this condition have signs and symptoms that fall somewhere
between these two extremes Increasingly genetic factors are used to define
the different forms of osteogenesis imperfecta
The milder forms of osteogenesis imperfecta including type I are
characterized by bone fractures during childhood and adolescence that often
result from minor trauma Fractures occur less frequently in adulthood
People with mild forms of the condition typically have a blue or grey tint to
the part of the eye that is usually white (the sclera) and may develop
hearing loss in adulthood Affected individuals are usually of normal or near
normal height
Other types of osteogenesis imperfecta are more severe characterized by
frequent bone fractures that may begin before birth and result from little or
no trauma Additional features of these conditions can include blue sclerae
short stature hearing loss respiratory problems and a disorder of tooth
development called dentinogenesis imperfecta The most severe forms of
osteogenesis imperfecta particularly type II can include an abnormally
small fragile rib cage and underdeveloped lungs Infants with these
abnormalities have life-threatening problems with breathing and often die
shortly after birth
Prevalence
Considering all types OI affects an estimated 6 to 7 per 100000 people
worldwide The two mildest forms OI type I and OI type IV account for
considerably more than half of all OI (prevalence of 4-5 per 100000) OI is
found in all racial and ethnic groups
Clinical Diagnosis
The clinical diagnosis of OI depends on the presence of a number of
features Features of OI
bull Fractures with minimal or no trauma in the absence of
other factors such as abuse or other known disorders of
bone
bull Short stature or stature shorter than predicted based on
stature of unaffected family members often with bone
deformity
bull Blue sclerae
bull Dentinogenesis imperfecta (DI) brown-grey
discoloration of the teeth
bull Progressive post-pubertal hearing loss
bull Additional clinical features including ligamentous laxity
and other signs of connective tissue abnormality
bull Family history of OI usually consistent with autosomal
dominant inheritance
Radiographic features of OI The radiographic features of OI change with
age Fractures of varying ages and stages of healing are seen often of the
long bones but may also include ribs and skull In addition Osteopenia
(thin bones) detected by dual energy X-ray absorptiometry (DEXA) or
regular xrays
Natural history
The severity of OI ranges from perinatal lethality to individuals with severe
skeletal deformities mobility impairments and very short stature to nearly
asymptomatic individuals with a mild predisposition to fractures normal
stature and normal life span Before the molecular basis of OI was
understood OI was classified into four types based on clinical presentation
radiographic features family history and natural history
OI type I OI type I is characterized by blue sclerae and normal stature A
small proportion of infants with OI type I have femoral bowing at birth The
first fractures may occur at birth or with diapering More often the first
fractures occur when the infant begins to walk and more importantly to fall
Fractures generally occur at a rate of a few to several per year and then
decrease in frequency after puberty Fracture frequency often increases
again late in adulthood especially after age 50 Affected individuals may
have anywhere from a few fractures to more than 100 but the fractures
usually heal normally with no resulting deformity
Most affected individuals have normal or near normal stature but may be
short compared to other members of their families
Joint hypermobility may contribute to morbidity
Progressive hearing loss occurs in about 50 of adults with OI type I
beginning as a conductive hearing loss but becoming a sensorineural hearing
loss in time
Other types of OI
OI type II Abnormalities characteristic of OI type II are evident at birth
Weight and length are small for gestational age The sclerae are dark blue
and connective tissue is extremely fragile The skull is large for the body size
and soft to palpation Extremities are short and bowed Hips are usually
flexed and abducted in a frog-leg position Although some fetuses with OI
type II die in utero or are spontaneously aborted more typically infants die
in the immediate perinatal period More than 60 of affected infants die on
the first day 80 die within the first week survival beyond one year is
exceedingly rare
OI type III The diagnosis of OI type III is readily apparent at birth
Fractures in the newborn period simply with handling of the infant are
common In some affected infants the number and severity of rib fractures
leads to death from pulmonary failure in the first few weeks of life Infants
who survive this period generally fare well although most do not walk
without assistance and usually use a wheel chair or other assistance for
mobility because of severe bone fragility and marked bone deformity
Affected individuals have as many as 200 fractures and progressive
deformity even in the absence of fracture Growth is extremely slow and
adults with OI type III are among the shortest individuals known with some
having adult stature of less than one meter (three feet) Usually sclerae are
blue in infancy but lighten with age Hearing loss generally begins in the
teenage years
OI type IV OI type IV is characterized by mild short stature adult-onset
hearing loss and normal-to-grey sclerae This is the most variable form of
OI ranging in severity from moderately severe to so mild that it may be
difficult to make the diagnosis Stature is variable and may vary markedly
within the family Sclerae are typically light blue or gray at birth but quickly
lighten to near normal Hearing loss occurs in some
Pregnancy Fertility is normal in OI For most women who have OI
pregnancy is uncomplicated The exception is women with OI type III who
may need to deliver by cesarian section due to a small pelvis
Diagnosistesting The clinical diagnosis of OI is based on family history
a history of fractures characteristic physical findings including blue sclerae
and radiographic findings Radiographic findings include fractures of varying
ages and stages of healing and osteopenia (thin bones) Analysis of type 1
collagen synthesized in vitro by culturing dermal fibroblasts obtained from a
small skin biopsy shows the structure and quantity of the collagen Such
biochemical testing detects abnormalities in 98 of individuals with OI type
II about 90 with OI type I about 84 with OI type IV and about 84
with OI type III
Molecular genetics
Genes COL1A1 and COL1A2 are the two genes associated with OI types I
II III and IV They encode the two chains pro αlpha1 and pro αlpha2
respectively of type I procollagen
Normal gene product and function
Collagens are a family of proteins that strengthen and support many tissues
in the body including cartilage bone tendon skin and the white part of the
eye (the sclera) Type 1 collagen is the most abundant form of collagen in
the human body Type 1 collagen creates layers of fibrils in the extracellular
matrix of bone giving bone its tensile strength and forming a template for
the deposition of inorganic minerals The type I procollagen molecule is
formed from of two pro alpha1 and one pro alpha2 collagen chains that
combine to form a helical structure The 21 ratio of pro alpha1 pro alpha2
is critical for normal assembly of collagen protein
Type 1 collagen owes its ability to form fibrils to its very specific amino acid
sequence Both the alpha1 chain encoded by COL1A1 and the alpha2 chain
encoded by COL1A2 are built from 338 uninterrupted repeats of the amino
acid sequence Glycine-X-Y where X is often proline and Y is often
hydroxyproline In order to form a Type 1 collagen protein two alpha1 and
one alpha2 chains associate assuming a triple helix formation (the fibril)
Presence of a glycine in every third position is critical for triple helix
formation since only glycine the smallest of the amino acids fits into the
confined space in the center of the triple helix
Individual collagen molecules are cross-linked to one another within these
fibrils The formation of cross-links results in very strong type I collagen
fibrils which are found in the spaces around cells The figure below
demonstrates the formation of Type 1 collagen translation of mRNA
assembly and processing of the triple helix crosslinking
Abnormal gene product
About 90 of individuals with OI types I II III and IV (but none with OI
types V VI and VII) have an identifiable mutation in either COL1A1 or
COL1A2 Mutations in the COL1A1 and COL1A2 genes are responsible for
more than 90 percent of all cases of osteogenesis imperfecta
Most of the mutations that cause osteogenesis imperfecta type I occur in the
COL1A1 gene The COL1A1 mutations that are responsible for osteogenesis
imperfecta type 1 reduce the production of pro-αlpha1 chains With fewer
pro-alpha1 chains available cells can make only half the normal amount of
type 1 collagen A shortage of this critical protein underlies the bone fragility
and other characteristic features of osteogenesis imperfecta type 1 These
genetic changes reduce the amount of type I collagen produced in the body
which causes bones to be brittle and to fracture easily
Sample mutation from patient with Type I OI
Normal DNA
CCA CTT GGG CCT GGG TGA CCG
Mutant DNA
CCA CTT GGG TCT GGG TCA CCG
Genetic counseling
Osteogenesis imperfecta types I-V are inherited in an autosomal dominant
manner For types I-IV the proportion of cases caused by a de novo
mutation in either COL1A1 or COL1A2 varies by the severity of disease
Approximately 60 of individuals with mild OI have de novo mutations
virtually 100 of individuals with lethal (type II) OI and a smaller proportion
of those with OI type II have a de novo mutation Each child of an individual
with a dominantly inherited form of OI has a 50 chance of inheriting the
mutation and of developing some manifestations of OI Prenatal testing in
at-risk pregnancies can be performed by analysis of collagen synthesized by
fetal cells obtained by chorionic villus sampling (CVS) at about 10-12 weeks
gestation if an abnormality of collagen has been identified in cultured cells
from the proband Prenatal testing in high-risk pregnancies can be
performed by molecular genetic testing of COL1A1 and COL1A2 if the
mutation has been identified in an affected relative Prenatal ultrasound
examination performed in a center with experience in diagnosing OI and
done at the appropriate gestational age can be valuable in the prenatal
diagnosis of the lethal form and most severe forms of OI prior to 20 weeks
gestation fetuses affected with milder forms may be detected later in
pregnancy when fractures or deformities occur
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and
maximize function minimize disability foster independence and maintain
overall health [Marini amp Gerber 1997] Ideally OI is managed by a
multidisciplinary team including specialists in medical therapy of OI
orthopedics and rehabilitation medicine Supportive therapy is individualized
depending upon the severity the degree of impairment and the age of the
patient Considerable support is generally required by medical personnel to
help parents feel comfortable caring for infants with OI type II
Normal lower extremity radiographs
Lower extremity radiographs from an infant with OI