Embed Size (px)
Citation preview

JOURNAL OF BACTERIOLOGY, Aug. 1991, p. 4717-4724 Vol. 173, No. 150021-9193/91/154717-08$02.00/0Copyright X 1991, American Society for Microbiology
Functional Analysis of the Pseudomonas putida Regulatory ProteinCatR: Transcriptional Studies and Determination of the CatR
DNA-Binding Site by Hydroxyl-Radical FootprintingRANDI KUBRICK ROTHMEL,t DEAN L. SHINABARGER, MATTHEW R. PARSEK, TERI L. ALDRICHJ
AND A. M. CHAKRABARTY*Department of Microbiology and Immunology, University of Illinois College of Medicine, Chicago, Illinois 60612
Received 25 February 1991/Accepted 22 May 1991
CatR, a LysR family protein, positively regulates the Pseudomonas putida catBC operon, which is requiredfor growth on benzoate as a sole carbon source. Transcriptional studies show that the catR and catBCpromoters are divergent and overlapping by 2 bp. A I8-galactosidase promoter probe vector was constructedto analyze expression from the catR and catBC promoters under induced and uninduced conditions. Aspredicted, the catBC promoter is expressed only under induced conditions, while the catR promoter isconstitutive. CatR has been shown to specifically bind the catRBC promoter region, and this property was usedto devise a purification protocol for CatR. Linear M13 DNA containing the catRBC control region wascovalently bound to cyanogen bromide-activated Sepharose in order to construct a DNA ainity column. Crudeextracts containing hyperproduced CatR protein were then incubated with the affinity resin under bindingconditions, and the CatR protein was eluted with 1 M NaCl. CatR was also purified by heparin-agarosechromatography. This highly purified protein was used for gel retardation and hydroxyl-radical footprintingstudies. From this analysis, it was shown that CatR binds upstream of the catBC promoter within thetranscribed region of catR.
Pseudomonads utilize many natural and man-made or-ganic compounds. In order to develop strains capable ofdissimilating recalcitrant compounds such as chlorinatedaromatics, it is important to understand how biodegradativepathways evolve in nature and how they are regulated.Pseudomonads provide a good model for studying howdegradative pathways evolve in order to expand the sub-strate range of a microorganism so that it may degrade morecomplex and toxic compounds. A simple model used for thisanalysis has been Pseudomonas putida, which is capable ofutilizing benzoate and can also degrade 3-chlorobenzoatewhen harboring plasmid pAC27 (10). While the structuralgenes encoding enzymes for the dissimilation of benzoateand 3-chlorobenzoate have been fairly well characterized (1,2, 13, 15, 43), the mechanism for regulation of these genes isonly now being investigated.The catBC operon encodes the genes for two enzymes:
cis,cis-muconate lactonizing enzyme I (EC 5.5.1.1) andmuconolactone isomerase (EC 5.3.3.4), respectively. Bothof these genes are required for the dissimilation of benzoate(28). The operon is coordinately regulated and requires theproduct of a regulatory gene for induction (42). As describedpreviously (32), the regulatory gene catR maps upstream ofthe catBC operon and is divergently transcribed from thecatBC operon (Fig. 1). The catR gene encodes a 32.2-kDapolypeptide that binds to the catBC promoter region in vitroin the presence or absence of the inducer cis,cis-muconate.The inducer, however, is required for in vivo transcriptionalactivation of the catBC operon. CatR was shown to be amember of a large class of procaryotic regulatory proteins,
* Corresponding author.t Present address: Envirogen, Inc., Princeton Research Center,
Lawrenceville, NJ 08648.t Present address: Department of Genetics, University of Wash-
ington, Seattle, WA 98195.
designated the LysR family, by homology studies (32). Thisfamily of regulatory proteins presumably acts by binding thepromoters of genes that they regulate to allow transcriptionin the presence of an inducer molecule. Many of theseproteins also regulate their own expression.
In this report, we describe further characterization of thecatR promoter region and the CatR protein. Reverse tran-scriptase mapping has localized the catR promoter to aregion that overlaps the catBC promoter by 2 bp. In addi-tion, the catR-catBC promoter region has been cloned into aP-galactosidase promoter probe vector for analysis of pro-moter activity. As expected, the catBC promoter promotesP-galactosidase activity only when cells have a functionalCatR protein and are grown in the presence of benzoate. Onthe other hand, expression from the catR promoter appearsto be constitutive and does not respond to the presence ofbenzoate in wild-type Pseudomonas putida.CatR has been purified by DNA affinity chromatography
or by heparin-agarose chromatography. Such purified prco-tein was shown to specifically bind a 200-bp AvaI fragmentcontaining the catRBC promoter region. Hydroxyl-radicdlfootprint analysis was also conducted. Results showed thatthe protein interacts with nucleotides at three closely linkedsites within a 25-bp region located upstream of the catBCpromoter and within the transcribed region of catR. Thebinding pattern was not significantly altered by the presenceof the inducer cis,cis-muconate. A model for catBC activa-tion and catR autoregulation is presented based on theidentification of the CatR binding site.
MATERIALS AND METHODS
Bacteria, plasmids, bacteriophage, and media. The Esche-richia coli strain used for general cloning procedures wasJM109 [A(pro-lac) recAl thi-J supE endA gyrA96 hsdRrelAJ(F' traD36 proAB lacIq lacZAM15)] (26). The promoter
4717
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
27
Janu
ary
2022
by
93.7
8.17
1.4.

4718 ROTHMEL ET AL.
a Benzoate
OH
Catechol
COCOOH
COOHcis.cis -muconate
,<c:OOH
Muconolactone
COOH>0
pEketoadipate enol-lactone
0C COOHCOOH
P-ketoadipate
Suocinate and acetyl CoA
CaMPyrocatechase I
CatoMuconatelactonizing enzyme I
'atcMuconolactoneisomerase
b136 bp
catR
(4 bp)CRT----CCTCC-GTR----GGRGG-4cat R
(114 bp)catBC
(10 bp) -GGR ----RTG
calDHydrolase I
FIG. 1. (a) Catechol biodegradative pathway. The enzymes and corresponding genes for catechol utilization are shown. Filled arrowsindicate the two enzymatic steps encoded by the catBC operon. (b) Structural organization of catR, catB, and catC. The gene order on thechromosome is catR-catB-catC, as indicated by the boxed genes. Arrows show the direction of transcription of catR and the catBC operon.The two translational start sites of catR and catBC are separated by 136 bp of intervening region containing the Shine-Dalgarno sequencesand promoters of these two genes.
probe constructs were initially transformed into the A(pro-lac) strain CSH50 (27) and then mated into P. putida strains.The P. putida strains used were PRS2000 (per-i 103) (42) andPRS3026 (per-1103 catR::TnS Kmr) (20). The plasmids andbacteriophage used are described in the text. Cultures weregrown either on LB or on basal salts medium (BSM) supple-mented with glucose and/or benzoate as described previ-ously (2). Antibiotics were added as required for selection:ampicillin (75 mg/liter), streptomycin (50 mg/liter), and ka-namycin (50 mg/liter) for E. coli and carbenicillin (1,000mg/liter), streptomycin (1,000 mg/liter), and kanamycin(1,000 mg/liter) for P. putida.
Nucleic acid preparations and DNA sequencing. PlasmidDNA was isolated from 40-ml cultures by alkaline lysis asdescribed by Maniatis et al. (24) for a large-scale preparationexcept that after isopropanol precipitation the DNA wasphenol-chloroform extracted twice, precipitated with etha-nol, and treated with RNase. Large-scale DNA isolation wasdone by CsCl centrifugation. Single-stranded DNA wasisolated as previously described (1). RNA was isolated bythe guanidinium isothiocyanate-hot phenol extractionmethod (3). Dideoxy-DNA sequencing was carried out underpreviously described conditions (3, 26). The Maxam-GilbertG+A reaction was carried out as described by Sambrook etal. (33).
Molecular cloning. Restriction enzymes were purchasedfrom Bethesda Research Laboratories, Gaithersburg, Md.,and used as instructed by the manufacturer. Vector DNAwas treated with calf intestinal alkaline phosphatase pur-chased from Boehringer Mannheim Biochemicals, Indianap-olis, Ind. Ligations using T4 DNA ligase (New England
BioLabs, Beverly, Mass.) were done at 15°C overnight.DNA was transformed into E. coli JM109 or CSH50 cells andscreened by miniprep analysis (17). Broad-host-range plas-mids were mated into P. putida strains, and exconjugantswere selected by antibiotic resistance as previously de-scribed (1, 2).Promoter probe construction. A f-galactosidase promoter
probe vector was constructed as follows. The ,B-galactosi-dase promoter probe vector pQF50 (21) was first cut withXbaI, and the ends were filled in. A second cut was madewith PstI, and the 3.2-kb fragment containing the P-galac-tosidase gene and the N-terminal region of the ,-lactamasegene was isolated. This fragment was ligated into the luxpromoter probe vector pUCD615 (31), which first had beencut with EcoRI blunt ended with T4 DNA polymerase, andcut at the PstI site. The resulting vector, pKRZ-1, is shownin Fig. 2. As indicated, the EcoRI site in the multiple cloningregion as well as the P-galactosidase gene are restored.
Reverse transcriptase mapping. RNA was isolated frombenzoate- or glucose-grown P. putida PRS2000 harboringthe pKT240 (4) clone pKR400 (32) or pTAAXH (3). pKR400has the catR promoter inserted upstream of the promoterlessaminoglycoside phosphotransferase gene, while pTAA&XHrepresents the catBC promoter cloned upstream of theaminoglycoside phosphotransferase gene. The RNA washybridized to a specific 5' 32P-end-labeled oligomer that wascomplementary to either the downstream region of the catRpromoter or the catBC promoter. Primer extension wasperformed as described previously (3). Dideoxy sequencingstandards were prepared by using the same labeled primer
Hcatc-
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
27
Janu
ary
2022
by
93.7
8.17
1.4.

FOOTPRINTING OF P. PUTIDA catR 4719
S X B Sm K Sc E H
Pv
Pv /
pKZK
Sc
FIG. 2. Promoter probe vector pKRZ-1. This vector was con-
structed by cloning the IacZ gene from PQF5O into the broad-host-
range vector pUCD615. Abbreviations: B, BamHI; E, EcoRI; H,
HindIII; K, KpnI; P, PstI; Pv, PvuII; S, SalI; Sc, SstI; Sm, SinaI;
X, XbaI.
and template DNA prepared from M13 clones containing the
catRBC promoter region in either orientation.
j3-Galactosidase activity. ~-Galactosidase activity was de-
termined as described by Miller (27). P. putida cells harbor-
ing the promoter probe constructs were grown at 30°C to
mid-log phase in 500 ml of BSM supplemented with either 20
mM glucose or glucose plus 5 mM benzoate. The cells were
harvested by centrifugation at 10,000 x g, sonicated in 100
mM phosphate buffer (pH 7.0) containing 5 mM ~-mercap-toethanol, and centrifuged at 10,000 x g for 15 min to pellet
the cells. The resulting supernatant was used as the source of
enzyme for the 3-galactosidase assays. Enzyme specific
activity was calculated by the method of Miller (27), and
protein was assayed by using the method of Bradford (6).
Protein purification. A DNA affinity column was prepared
as described by Wagner et al. (39), using CNBr-activated
Sepharose 4B (Sigma Chemical Co.). The affinity resin (1 g)
was washed three times with 50 ml of deionized water and
then three times with 200 mM morpholinepropanesulfonic
acid (MOPS), pH 6.0 (buffer A). Approximately 2 mg of
pTAAXH:18 DNA (3) containing the catRBC promoterregion was digested with EcoRI, extracted two times with
phenol-chloroform (1:1), and precipitated with ethanol. Thedried DNA pellet was dissolved in 500 uil of buffer A and
mixed with 10 ml of settled affinity resin overnight at 4°C in
a plastic tube with gentle stirring. The affinity resin was thencollected by centrifugation and washed three times with
buffer A. To block unreacted CNBr groups, the affinity resin
was incubated with 20 ml of 100 mM triethanolamine for 1 h
and then washed three times with 50 ml of buffer B (50 mMTris-HCl [pH 8.0], 100 mM NaCl, 5 mM ,B-mercaptoetha-nol).
Crude extracts containing overexpressed CatR proteinwere prepared by growing JM1O9/pKR119AHf (CatR expres-
sion vector; see Results) in LB containg 1 mM isopropyl-B-D-thiogalactopyranoside (IPTG) for 10 h. The cells were
harvested by centrifugation at 10,000 x g, washed once with0.9%o NaCl solution, and lysed by sonication. This crude
lysate was centrifuged at 20,000 x g for 20 min, and the
resulting supernatant was centrifuged at 100,000 x g for 60
min. The supernatant from this high-speed spin was utilized
for all CatR purification procedures.The resulting DNA affinity resin containing the catRBC
promoter region covalently linked to Sepharose 4B was thenmixed with 10 ml (200 mg) of crude extract containingoverexpressed CatR protein for 1 h at 30°C with gentleshaking. The slurry was then poured into a 10-by-100-mmcolumn and washed with 100 ml of buffer B. CatR proteinwas eluted from the column with buffer B supplemented with1 M NaCl, and fractions containing protein were analyzed bysodium dodecyl sulfate-polyacrylamide gel electrophoresis(SDS-PAGE) according to the method of Laemmli (22).
Heparin-agarose affinity chromatography was used as analternative to DNA affinity columns in order to partiallypurify large quantities of CatR. Approximately 300 mg ofcrude extract containing overexpressed CatR protein wasapplied to a 30-by-100-mm heparin-agarose column equili-brated with buffer containing 50mM Tris-HCl (pH 8.0) 5 mMdithiothreitol, 10% glycerol, and 10 mM MgCl2. After thecolumn was washed with 200 ml of this equilibration buffer,CatR was eluted with a 0 to 1 M NaCl gradient and thecollected fractions (4 ml) were assayed by the gel shift assayfor DNA binding and by SDS-PAGE for purity.
Radioactive labeling of DNA. A 200-bp AvaI fragmentcontaining the catRBC promoter was specifically labeled atone 5' end by the following method. The 200-bp fragmentwas first blunt-end ligated into the HincII site of M13 mpl8(45) to obtain two recombinant phages, RT4 and RT04,having the fragment in different orientations. A sequencing18-mer was end labeled as described by Sambrook et al. (33)with [32P]ATP (5,000 Ci/mmol), using polynucleotide kinasepurchased from United States Biochemical. Fifty nanogramsof end-labeled primer was annealed to either S jig of single-stranded DNA from RT4 or RT04 at 65°C with Sequenaseannealing buffer (40 mM Tris [pH 7.5], 10 mM MgCl2, 50 mMNaCl) in a total volume of 15 ,l. After slow cooling to roomtemperature, 1 ,ul of deoxynucleoside triphosphates (25 mMdGATC), 2 ,u of dithiothreitol (100 mM), and 1.5 U ofSequenase were added. The DNA was elongated for 5 to 10min at 37°C. The DNA was then extracted twice withphenol-chloroform and ethanol precipitated. The 200-bppromoter fragment was released by cutting with EcoRI. TheDNA was again extracted twice with phenol-chloroform andethanol precipitated. From 5 ,ug of template DNA, theapproximate yield of the 200-bp fragment is 0.14 jig. Becausethe DNA is labeled only at one end, it can be used directlyfor hydroxyl-radical footprinting Without being purified fromthe M13 DNA.DNA for gel retardation was labeled by a similar method
except that cold oligomer was used as a primer and[35S]dCTP(1,000 Ci/mmol) was included in the elongationreaction mix. The labeled fragment was released by diges-tion with both EcoRI and HindIll, and the fragment wasisolated by using GeneClean (purchased from Bio 101) froma 4% Nusieve agarose gel after staining with ethidiumbromide.
Gel retardation and hydroxyl-radical footprinting. Gel re-tardation was performed as described by Fried and Crothers(14). The assay conditions were modified as follows. A 30-,ureaction mix containing 3 RIl of lOx binding buffer (1% TritonX-100, 40% glycerol, 10 mM NaEDTA, 100 mM 2-mercap-toethanol, 100 mM Tris-HCl, pH 7.5), 5 ,u of poly(dI - dC) at1 mg/ml, 100 jig of bovine serum albumin, 50 ng of a labeledDNA fragment, and 0.001 to 10 jig of protein was incubatedat 30°C for 30 min. After incubation, the samples wereloaded onto a 5% polyacrylamide gel (50:1 acrylamide/N,N'-methylene bisacrylamide linkage). One lane was loaded witha loading buffer containing bromophenol blue. Electropho-resis was done at 120 V in Tris-borate-EDTA buffer until the
VOL. 173, 1991
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
27
Janu
ary
2022
by
93.7
8.17
1.4.

4720 ROTHMEL ET AL.
bromophenol blue had travelled three-fourths the length ofthe gel. The gel was then soaked in 10% glycerol, transferredonto Whatman paper, covered with plastic wrap, dried, andexposed to X-ray film.
Hydroxyl-radical footprinting was done as described byTullius et al. (37, 38). Approximately 10 ng of labeled DNAwas incubated with 1 to 5 p,g of partially purified CatRprotein in 170 ,ul of buffer containing 50 mM Tris-HCl (pH8.0), 50 mM KCl, 5 mM MgCl2, 1 mM dithiothreitol, 100 ,ugof bovine serum albumin per ml, and 0.5 ,ug of salmon spermDNA for 30 min at 30TC. Some reactions included cis,cis-muconate (a generous gift from Celgene Corp.) at a finalconcentration of 2 mM. The cutting reaction was initiated bythe addition of 10 ,u1 each of the following freshly madesolutions: 0.2 M Fe(NH4)2(SO4)2, 0.4 M EDTA, 0.6% H202,and 20 mM sodium ascorbate. After 2 min at room temper-ature, the reaction was quenched by the addition of 45 ,ul ofa solution containing 50 mM thiourea, 1.5 M sodium acetate(pH 6.0), and 2 mg of tRNA solution per ml. Proteins wereremoved from the DNA by phenol-chloroform extractionand ethanol precipitation. The final DNA pellet was dis-solved in 9 of a sequencing stop solution and boiled, anda 3-pI portion was loaded onto a 6% denatuting polyacryl-amide gel and run beside the Maxam-Gilbert G+A reactionconducted on the same labeled DNA fragment.
RESULTS
Analysis of the catR promoter. We have previously re-
ported (32) that the catBC operon and its regulatory genecatR are divergently transcribed. The catR promoter was
localized to a 385-bp fragment which also contains the catBCpromoter. The -35 region of the catBC promoter is sepa-rated by only 36 nucleotides from the catR translational start(32). In this study, the location of the catR promoter was
determined by reverse transcriptase mapping. Figure 3shows that the transcriptional start is at an A on the oppositestrand located 48 bp upstream of the transcriptional start forthe catBC operon. As indicated by Fig. 3, identification ofthe catR start site required overexposure of the X-ray film.Low transcript level due to autoregulation and probable highturnover rate made isolation of mRNA from catR difficult.Additional minor bands seen on the gel are probably decayproducts. The proximity of the two start sites indicates thatthe two promoters overlap each other. In fact, there is a
sequence upstream of the catR transcriptional start thatresembles an E. coli a70 promoter that overlaps the catBCpromoter by 2 bp within both the -10 and -35 regions. Thesequence of the catR promoter as indicated in Fig. 3 isCGCGTT at the -35 site and CTATAA at the -10 site. Thisregion containing the overlapping promoters of both catRand catBC has been designated catRBC.We have previously shown that the catBC promoter
functions only in cells grown in the presence of benzoatebecause of the production of the inducer molecule cis,cis-muconate (1, 32). These studies were performed by using thepromoterless aphC gene in plasmid pKT240 as a promoterprobe for determining streptotnycin resistance conferred tohost cells. To further investigate this induction phenomenon,a new promoter probe vector (pKRZ-1) containing a promot-erless ,B-galactosidase gene was constructed (Fig. 2). A485-bp EcoRI-HindIII fragment from the M13 vectorpTAAXH containing the overlapping catRBC promoters wasblunt ended into the SmaI site of pKRZ-1 to obtain plasmidpKRZ-BC. In this construct, the insert is oriented with thecatBC promoter 5' to the lacZ gene, thereby allowing the
a bGA TC I
..10.:
.i
.:,...
Vs
GA TC
cOatBC
+15'- AGATTCATTCGRTRTTGGACGGCTATCAGGGTCTCGCGCRRTCCTTGRACRIGCA -3
-35 -10
3'- TCTIRAGTRAGCTRTRACCTGCCGATRGTCCCAGAGCGCGTTAGGARCTTGTTCGT-5'+1 -10 -35
catR
FIG. 3. Reverse transcriptase mRNA mapping. RNA was iso-lated from P. putida PRS2000 grown with benzoate harboring thepKT240 construct pTAAXH (catBC operon) (a) or pKR400 (catRgene) (b). As indicated ih panel b (lane 1), the level of transcriptionfrom catR is very low. (c) Double-stranded DNA sequence of the55-bp catRBC promoter region showing the two transcriptional startsites (in outlined type) as well as the -10 and -35 promoter regions(underlined). The double-underlined bases represent promoter re-gions shared by catR and catBC. Note that the transcription ofcatBC is from left to right (top strand), while catR transcription isfrom right to left (bottom strand).
measurement of catBC promoter strength as a function of,3-galactosidase activity. Construct pKRZ-R represents thesame insert in the opposite orientation such that the catRpromoter controls transcription of the lacZ gene of pKRZ-1.These cohstructs were transformed into the E. coli lacmutanlt CSH50, and triparental matings were used to transferthe plasmids into P. putida as shown in Table 1.
P. putida strains harboring the promoter probe vectorpKRZ-1 and the two constructs pKRZ-BC and pKRZ-R
TABLE 1. Activation of the P. aeruginosa catBC promoterby benzoatea
1-GalactosidaseStrain Benzoate (5 mM) sp act (nmolmin1 mg of
protein-l)
PRS2000/pKRZ-BC - 15+ 235
PRS2000/pKRZ-R - 18+ 11
PRS3026/pKRZ-BC - 3+ 1
a Cells were grown for 12 h in BSM supplemented with either 20 mMglucose alone or glucose plus 5 mM benzoate. PRS2000 is wild-type P. putida;PRS3026 is the CatR- mutant derived from it.
J. BACTERIOL.
m .-m
100
*WI
n
No400
4ow
aM.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
27
Janu
ary
2022
by
93.7
8.17
1.4.
FOOTPRINTING OF P. PUTIDA catR 4721
were grown in BSM containing glucose or glucose andbenzoate as carbon sources. P-Galactosidase activity wasdetermined in the crude extracts of cells grown to mid-logphase as described in Materials and Methods. As indicatedin Table 1, there is a low level of catBC promoter activity inthe wild-type P. putida strain PRS2000 grown in the absenceof benzoate. However, the addition of 5 mM benzoate to thegrowth medium leads to a 16-fold activation of the catBCpromoter. There is very little catBC promoter activity in thecatR mutant PRS3026, confirming that CatR is required forcatBC activation by benzoate. The catR promoter was notactivated by benzoate and appears to be weakly constitutivein the wild-type background with a low level of expression(Fig. 3b), as might be expected of a regulatory gene.
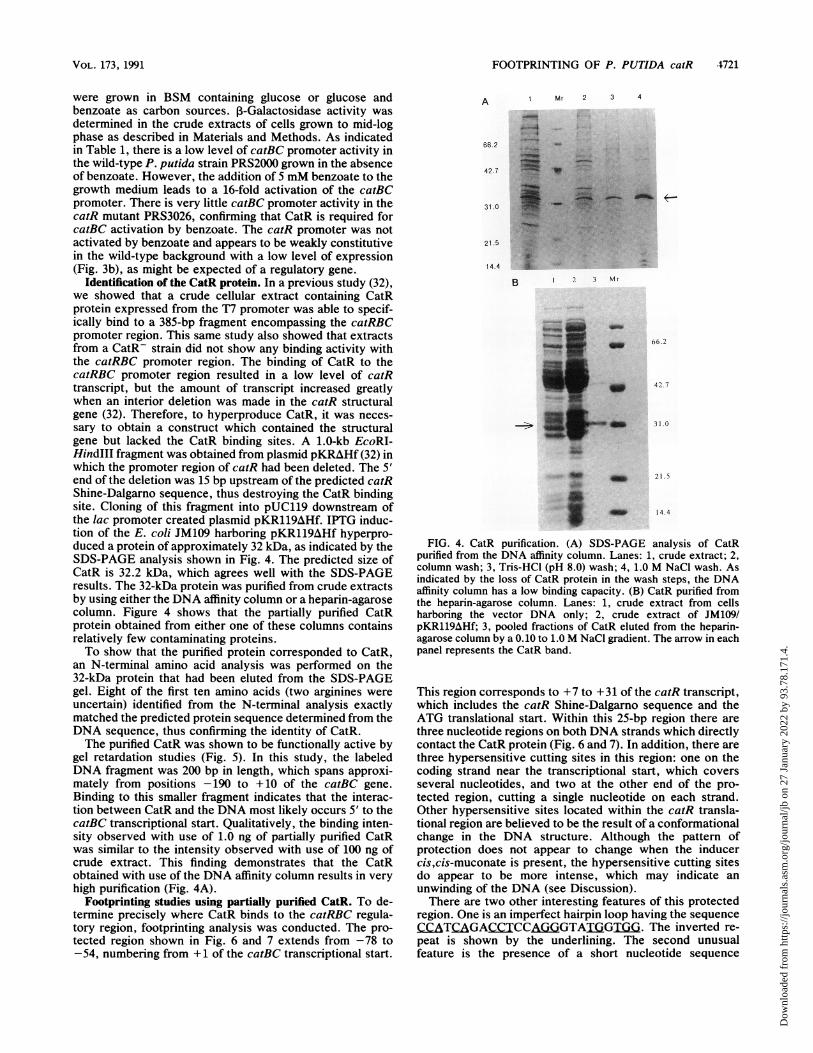
Identification of the CatR protein. In a previous study (32),we showed that a crude cellular extract containing CatRprotein expressed from the T7 promoter was able to specif-ically bind to a 385-bp fragment encompassing the catRBCpromoter region. This same study also showed that extractsfrom a CatR- strain did not show any binding activity withthe catRBC promoter region. The binding of CatR to thecatRBC promoter region resulted in a low level of catRtranscript, but the amount of transcript increased greatlywhen an interior deletion was made in the catR structuralgene (32). Therefore, to hyperproduce CatR, it was neces-sary to obtain a construct which contained the structuralgene but lacked the CatR binding sites. A 1.0-kb EcoRI-HindlIl fragment was obtained from plasmid pKRAHf (32) inwhich the promoter region of catR had been deleted. The 5'end of the deletion was 15 bp upstream of the predicted catRShine-Dalgarno sequence, thus destroying the CatR bindingsite. Cloning of this fragment into pUC119 downstream ofthe lac promoter created plasmid pKR119AHf. IPTG induc-tion of the E. coli JM109 harboring pKR119AHf hyperpro-duced a protein of approximately 32 kDa, as indicated by theSDS-PAGE analysis shown in Fig. 4. The predicted size ofCatR is 32.2 kDa, which agrees well with the SDS-PAGEresults. The 32-kDa protein was purified from crude extractsby using either the DNA affinity column or a heparin-agarosecolumn. Figure 4 shows that the partially purified CatRprotein obtained from either one of these columns containsrelatively few contaminating proteins.To show that the purified protein corresponded to CatR,
an N-terminal amino acid analysis was performed on the32-kDa protein that had been eluted from the SDS-PAGEgel. Eight of the first ten amino acids (two arginines wereuncertain) identified from the N-terminal analysis exactlymatched the predicted protein sequence determined from theDNA sequence, thus confirming the identity of CatR.The purified CatR was shown to be functionally active by
gel retardation studies (Fig. 5). In this study, the labeledDNA fragment was 200 bp in length, which spans approxi-mately from positions -190 to + 10 of the catBC gene.Binding to this smaller fragment indicates that the interac-tion between CatR and the DNA most likely occurs 5' to thecatBC transcriptional start. Qualitatively, the binding inten-sity observed with use of 1.0 ng of partially purified CatRwas similar to the intensity observed with use of 100 ng ofcrude extract. This finding demonstrates that the CatRobtained with use of the DNA affinity column results in veryhigh purification (Fig. 4A).
Footprinting studies using partially purified CatR. To de-termine precisely where CatR binds to the catRBC regula-tory region, footprinting analysis was conducted. The pro-tected region shown in Fig. 6 and 7 extends from -78 to-54, numbering from + 1 of the catBC transcriptional start.
A Mr 2 3 4
66.2
42.7
31.0
21.5
14.4
B I 3 Mr
66.2
42.7
31.0
= _ ~~~~~14.4
FIG. 4. CatR purification. (A) SDS-PAGE analysis of CatRpurified from the DNA affinity column. Lanes: 1, crude extract; 2,column wash; 3, Tris-HCI (pH 8.0) wash; 4, 1.0 M NaCl wash. Asindicated by the loss of CatR protein in the wash steps, the DNAaffinity column has a low binding capacity. (B) CatR purified fromthe heparin-agarose column. Lanes: 1, crude extract from cellsharboring the vector DNA only; 2, crude extract of JM109/pKR119AHf; 3, pooled fractions of CatR eluted from the heparin-agarose column by a 0.10 to 1.0 M NaCI gradient. The arrow in eachpanel represents the CatR band.
This region corresponds to +7 to +31 of the catR transcript,which includes the catR Shine-Dalgarno sequence and theATG translational start. Within this 25-bp region there arethree nucleotide regions on both DNA strands which directlycontact the CatR protein (Fig. 6 and 7). In addition, there arethree hypersensitive cutting sites in this region: one on thecoding strand near the transcriptional start, which coversseveral nucleotides, and two at the other end of the pro-tected region, cutting a single nucleotide on each strand.Other hypersensitive sites located within the catR transla-tional region are believed to be the result of a conformationalchange in the DNA structure. Although the pattern ofprotection does not appear to change when the inducercis,cis-muconate is present, the hypersensitive cutting sitesdo appear to be more intense, which may indicate anunwinding of the DNA (see Discussion).There are two other interesting features of this protected
region. One is an imperfect hairpin loop having the sequenceCCATCAGACCTCCAGGGTAIGT G. The inverted re-peat is shown by the underlining. The second unusualfeature is the presence of a short nucleotide sequence
VOL. 173, 1991
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
27
Janu
ary
2022
by
93.7
8.17
1.4.

4722 ROTHMEL ET AL.
co
1 2 3 4 5 6
FIG. 5. Gel retardation with crude and partially purified CatR.Crude CatR protein extract at 0.01, 0.1, and 1.0 ,ug (lanes 4 to 6,respectively) was incubated with a 200-bp 32P-labeled DNA frag-ment containing the catRBC promoter region. Partially purifiedCatR from either the DNA affinity column or the heparin-agarosecolumn (not shown) was used for gel retardation at protein concen-trations of 0.001, 0.01, and 0.1 ,ug (lanes 1 to 3, respectively). Thelast lane represents the mobility of the labeled DNA fragment in theabsence of protein. As indicated from the relative intensities of theretarded band, the CatR protein obtained from the DNA affinitycolumn was about 80% pure.
CTCCA read 5' to 3' and 3' to 5' bordered by the sequenceCAG at either end and separating the reverse repeatedsequence. Whether any of these sequences are significant isunknown. Site-directed mutational analysis of these se-quences would determine whether they are functionallysignificant.
DISCUSSION
The regulatory protein CatR, as proposed by Wu et al.(44), has been shown to be required for transcriptionalactivation of the catBC operon (1, 32). Results of experi-ments described in this report begin to address the questionof how CatR functions to regulate both its own expressionand the function of the catBC operon.Promoter probe analysis using a promoterless lacZ gene
supports previous results showing that CatR is required forcatBC transcription. Transcription from catR, however,appears to be constitutive in the wild-type P. putida strainPRS2000. From the magnitude of P-galactosidase specificactivities, it can be seen that the catBC promoter is strongwhen induced and that the catR promoter is relatively weak.A model of how CatR mechanistically regulates itself andhow it activates catBC can be inferred by looking at thecatRBC promoter structure as well as the footprinting resultsdiscussed below.We have identified the transcriptional start site of catR
and have predicted a likely promoter region that showsconsiderable homology to the r70-type promoter sequence.This predicted promoter overlaps the promoter for catBCtranscription. Like many other LysR proteins that regulatetheir own expression (9, 11, 23, 25, 30, 34-36, 41), CatRbinds to a regulatory region which includes its own pro-moter. It has been shown that binding leads to both autoreg-ulation and activation of divergent genes, in this case catBC,by stimulating RNA'polymerase binding downstream fromthe regulatory binding site (16). Advantages for gene regu-
ADA N G
A-A +
DN Q
B
A- A +.-
_2
.01'. . fdi
,;P::F:'ik. a_as-;w
i_
. m
FIG. 6. Hydroxyl-radical footprinting. A 32P-end-labeled 200-bpAvaI fragment containing the catRBC promoter region was used infootprinting analysis. CatR protein was added at a concentration of5 ,ug. There were three protected regions on both strands, as shownby the brackets. In addition, several nucleotides were hypersensi-tive to strand scission by the hydroxyl radical. There appears to belittle difference between reactions with the inducer cis,cis-muconate(+) or without inducer (-). The Maxam-Gilbert G+A reaction onthe same end-labeled fragments provided the size marker. Anexpanded view of the relevant part of the 200-bp region is shown inFig. 7.
lation using divergent promoters have been reviewed (5).Since the catR and catBC promoters overlap, it is likely thatthis is the mode of regulation which governs catechol utili-zation.The specific binding sequences of a number of LysR
proteins, including TrpI, NahR, NodD, AmpR, MetR, andIlvY, are known (7, 8, 18, 23, 30, 35, 40, 41, 46). Partiallypurified CatR binds to a 200-bp promoter/control region inthe presence or absence of inducer, as shown by gel retar-dation, as do many LysR regulatory proteins. Footprintanalysis has further defined this region to three protectedsites within a 25-bp region located near the transcriptionalstart of catR. The long stretch of DNA that appears to beprotected in the lower half of the gel in Fig. 6B is probably anexperimental artifact. CatR binding to this promoter regionresults in an extremely hypersensitive cutting site on thisstrand of DNA, as seen toward the top half of the gel. Theresult is that a large fraction of the labeled DNA is cut at thissite, which effectively reduces the concentration of labeledDNA available for hydroxyl-radical cleavage of shorter
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
27
Janu
ary
2022
by
93.7
8.17
1.4.

FOOTPRINTING OF P. PUTIDA catR 4723
'IooDoElUl 00 ON l Dlaooo l444'
RR6T66C6CR6CfC9 ARC97EEMST66S6GRGRTTCTTTCRCCCGTCGRC6Rg6tR# CCRCTCTCTMRGT
so catRcatR
transtinalstNwt
cat BC
TCUWMWY66RCGGCTRTCRG66TCTC_T6RRTCCTTG6RCAfCRRRGIUTTIIICCTGCCARTRGTCCCA6RGBWWIGGARCTT6TTCGTT
FIG. 7. Sequence of the catRBC promoter region. Sequence inoutlined lettering is the catR promoter and transcriptional start site.Underlined sequence shows the catBC promoter region and startsite. Boxed nucleotides indicate the bases that are protected byCatR. The arrows ( I, f ) show bases that are hypersensitive tocutting. There is a potential imperfect hairpin in this region, asindicated by the italicized nucleotides. In the protected area, there isa divergent repeated sequence CTCCA/ACCTC (U) separated bythe repeated sequence CAG (l).
lengths; thus, the band intensity in this region of the gel issignificantly reduced.Except for their general location near the transcriptional
start of the regulatory gene, the DNA-binding sites that havebeen identified so far for the LysR proteins show no appar-ent homology to each other. Some, such as the TrpI site,show the potential for forming a hairpin loop structure, whileother binding sites, such as the MetR and NahR sites, do notappear to form such structures. It is possible that the CatRDNA-binding site forms a hairpin loop structure. The lack ofany homology between LysR family binding sites is interest-ing when one considers that all of these proteins have verysimilar helix-turn-helix structures, located at the N-terminalregion, that is implicated in DNA binding (29). With suchhomology, it might be expected that there would be homol-ogy at the DNA-binding site. Although the primary nucleo-tide structures show no such homology, there may be a
secondary folding structure that is similar between the targetDNA sites.The mode of activation by LysR regulatory proteins is
believed to occur by a conformational change between theDNA-protein complex in the presence of an inducer mole-cule. This change thus primes the DNA for transcriptioninitiation (16, 41). Support for a conformational change inbinding due to an inducer was shown for TrpI (8). In theabsence of its inducer, indoleglycerol phosphate, TrpI bindsupstream of the trpBA operon overlapping its own promoterregion. The binding pattern alters in the presence of inducerso that the protected region extends downstream toward thetrpBA promoter region. Conformational changes in bindinghave also been indicated by an altered footprint pattern ofIlvY and its target DNA in the presence of inducer (40, 41).However, as with NahR and AmpR (23, 35), alteration inbinding patterns is not always indicated by gel retardation orfootprinting analysis. Results from footprinting with CatRdoes not indicate a substantial conformational change due tothe inducer cis,cis-muconate. The only apparent change isan enhanced cutting at either end of the protected region.This may indicate an unfolding of the DNA that would allowRNA polymerase to interact with the catBC promoter re-
gion. It is also possible that during our footprint studies wesaturated the DNA with CatR, thereby simulating the con-ditions that occur when cis,cis-muconate is present. Whenwe used lower concentrations of CatR (0.1 or 1.0 ,ug), wewere unable to observe protection of the DNA molecule.
Additional hypersensitive DNA cutting sites as seen in
Fig. 6A are likely to be the result of a DNA conformationalchange that occurs when CatR binds. These cut sites occurin the overlapping promoter region as well as the catBCtranscribed region. It is unclear why these sites occur onlyon one strand of the DNA, the coding strand for catBC. Thisunresolved problem is now being investigated. Other futureexperiments will investigate the in vitro interaction betweenP. putida RNA polymerase, CatR, and cis,cis-muconatewith this highly regulated promoter region. We hope thatthese results will help us to further understand the transcrip-tional activation process.The proposed conformational change in the DNA-protein
binding is thought to be mediated by the inducer interactingwith the regulatory protein. This interaction likely occurswithin either the C-terminal or central region of the regula-tory protein, as indicated by studies on several NodDregulatory proteins (19). Because this region shows signifi-cant divergence among LysR protein members, it is a likelycandidate for interaction with an individual inducer mole-cule. It is interesting that there are large regions of aminoacid similarities seen in the C-terminal region between CatRand ClcR, the regulatory protein needed for induction of thegenes required for 3-chlorocatechol degradation (12, 13).This is not surprising since the likely inducer for the clcABDoperon is 2-chloro-cis,cis-muconate, which structurally re-sembles cis,cis-muconate. We are currently devising exper-iments to look at inducer binding to the DNA regulatoryprotein complex for both CatR and ClcR as well as deter-mining whether there is any cross-talk between these tworegulatory proteins. Future experiments will be geared todetermine how much functional homology has been pre-served between CatR and ClcR, in hope of understandinghow these proteins have evolved to perform their particularrole in regulating catabolic genes. Understanding how regu-latory proteins have evolved to accommodate new inducerswill be invaluable for developing organisms that have ex-panded substrate (inducer) ranges for degrading more com-plex and toxic compounds.
ACKNOWLEDGMENTS
This investigation was supported by a National Science Founda-tion grant DMB-87 21743 and in part by Public Health Service grantES04050-06 from the National Institute of Environmental HealthSciences.We thank K.-L. Ngai for N-terminal amino acid sequencing.
REFERENCES1. Aldrich, T. L., and A. M. Chakrabarty. 1988. Transcriptional
regulation, nucleotide sequence, and localization of the pro-moter of the catBC operon in Pseudomonas putida. J. Bacteriol.170:1297-1304.
2. Aldrich, T. L., B. Frantz, J. F. Gill, J. J. Kilbane, and A. M.Chakrabarty. 1987. Cloning and complete nucleotide sequencedetermination of the catB gene encoding cis,cis-muconate lac-tonizing enzyme. Gene 52:185-195.
3. Aldrich, T. L., R. K. Rothmel, and A. M. Chakrabarty. 1989.Identification of nucleotides critical for activity of the Pseudo-monas putida catBC promoter. Mol. Gen. Genet. 218:266-271.
4. Bagdasarian, M. M., E. Amann, R. Lurz, B. Ruckert, and M.Bagdasarian. 1983. Activity of the hybrid trp-lac (tac) promoterof Escherichia coli in Pseudomonas putida. Construction ofbroad-host-range, controlled-expression vectors. Gene 26:273-282.
5. Beck, C. F., and R. A. J. Warren. 1988. Divergent promoters, acommon form of gene organization. Microbiol. Rev. 52:318-326.
6. Bradford, M. M. 1976. Rapid and sensitive method for quanti-fication of microgram quantities of protein utilizing the principle
VOL. 173, 1991
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
27
Janu
ary
2022
by
93.7
8.17
1.4.

4724 ROTHMEL ET AL.
of protein-dye binding. Anal. Biochem. 72:248-254.7. Cai, X.-Y., M. E. Maxon, B. Redfield, R. Glass, N. Brot, and H.
Weissbach. 1989. Methionine synthesis in Escherichia coli:effect of MetR protein on metE and metH expression. Proc.Natl. Acad. Sci. USA 86:4407-4411.
8. Chang, M., and I. P. Crawford. 1990. The roles of indoleglyc-erol phosphate and the TrpI protein in the expression of trpBAfrom Pseudomonas aeruginosa. Nucleic Acids Res. 18:979-988.
9. Chang, M., A. Hadero, and I. P. Crawford. 1989. Sequence ofthe Pseudomonas aeruginosa trpI activator gene and related-ness of trpl to other procaryotic regulatory genes. J. Bacteriol.171:172-183.
10. Chatterjee, D. K., and A. M. Chakrabarty. 1982. Geneticrearrangements in plasmids specifying total degradation ofchlorinated benzoic acids. Mol. Gen. Genet. 188:279-285.
11. Christman, M. F., G. Storz, and B. N. Ames. 1989. OxyR, apositive regulator of hydrogen peroxide-inducible genes inEscherichia coli and Salmonella typhimurium, is homologous toa family of bacterial regulatory proteins. Proc. Natl. Acad. Sci.USA 86:3483-3488.
12. Coco, W. M., R. K. Rothmel, and A. M. Chakrabarty. 1989.Program Abstr. Annu. Meet. Am. Soc. Microbiol., abstr. K-8.
13. Frantz, B., and A. M. Chakrabarty. 1987. Organization andnucleotide sequence determination of a gene cluster involved in3-chlorocatechol degradation. Proc. Natl. Acad. Sci. USA 84:4460-4464.
14. Fried, M., and D. M. Crothers. 1981. Equilibria and kinetics oflac repressor-operator interactions by polyacrylamide gel elec-trophoresis. Nucleic Acids Res. 9:6505-6525.
15. Ghosal, D., I.-S. You, D. K. Chatterjee, and A. M. Chakrabarty.1985. Microbial degradation of halogenated compounds. Sci-ence 228:135-142.
16. Henikoff, S., G. W. Haughn, J. M. Calvo, and J. C. Wallace.1988. A large family of bacterial activator proteins. Proc. Natl.Acad. Sci. USA 85:6602-6606.
17. Holmes, D. S., and M. Quigley. 1981. A rapid boiling method forpreparation of bacterial plasmids. Anal. Biochem. 114:193-197.
18. Hong, G. F., J. E. Burn, and A. W. Johnston. 1988. Evidencethat DNA involved in the expression of nodulation (nodD) genesin Rhizobium binds to the product of the regulatory gene nodD.Nucleic Acids Res. 15:9677-9690.
19. Horvath, B., C. W. B. Bachem, J. Schell, and A. Kondorosi.1987. Host-specific regulation of nodulation genes in Rhizobiumis mediated by a plant-signal, interacting with the nodD geneproduct. EMBO J. 6:841-848.
20. Houghton, J. E., E. J. Hughes, and L. N. Ornston. 1988.Program Abstr. Annu. Meet. Am. Soc. Microbiol., abstr. K-4.
21. Farinha, M. A., and A. M. Kropinski. 1990. Construction ofbroad-host-range plasmid vectors for easy visible selection andanalysis of promoters. J. Bacteriol. 172:3496-3499.
22. Laemmli, U. K. 1970. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature (London)227:680-685.
23. Linduist, S., F. Lindberg, and S. Normark. 1989. Binding of theCitrobacter freundii AmpR regulator to a single DNA siteprovides both autoregulation and activation of the inducibleampC P-lactamase gene. J. Bacteriol. 171:3746-3753.
24. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
25. Maxon, M. E., B. Redfield, X.-Y. Cai, R. Shoeman, K. Fujita,W. Fisher, G. Stauffer, H. Weissbach, and N. Brot. 1989.Regulation of methionine synthesis in Escherichia coli: effect ofthe MetR protein on the expression of the metE and metRgenes. Proc. Natl. Acad. Sci. USA 86:85-89.
26. Messing, J., R. Crea, and P. H. Seeburg. 1981. A system forshotgun DNA sequencing. Nucleic Acids Res. 9:309-321.
27. Miller, J. 1972. Experiments in molecular genetics. Cold SpringHarbor Laboratory, Cold Spring Harbor, N.Y.
28. Ornston, L. N., and D. Parke. 1976. Evolution of catabolicpathways. Biochem. Soc. Trans. 4:468-473.
29. Pabo, C. O., and R. J. Sauer. 1984. Protein DNA recognition.Annu. Rev. Biochem. 53:293-321.
30. Plaman, L. S., and G. V. Stauffer. 1987. Nucleotide sequence ofthe Salmonella typhimurium metR gene and the metR-metEcontrol region. J. Bacteriol. 169:3932-3937.
31. Rogowsky, P. M., T. J. Close, J. A. Chimera, J. J. Shaw, andC. I. Kado. 1987. Regulation of the vir genes of Agrobacteriumtumefaciens plasmid pTiC58. J. Bacteriol. 169:5101-5112.
32. Rothmel, R. K., T. L. Aldrich, J. E. Houghton, W. M. Coco,L. N. Ornston, and A. M. Chakrabarty. 1990. Nucleotide se-quencing and characterization of Pseudomonas putida catR: apositive regulator of the catBC operon is a member of the LysRfamily. J. Bacteriol. 172:922-931.
33. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
34. Schell, M. A., and E. F. Poser. 1989. Demonstration, character-ization, and mutational analysis of NahR protein binding to nahand sal promoters. J. Bacteriol. 171:837-846.
35. Schell, M. A., and M. Sukordhaman. 1989. Evidence that thetranscription activator encoded by the Pseudomonas putidanahR gene is evolutionarily related to the transcription activa-tors encoded by the Rhizobium nodD genes. J. Bacteriol.171:1952-1959.
36. Stragier, P., and J. C. Patte. 1983. Regulation of diaminopime-late decarboxylase synthesis in Escherichia coli. III. Nucleotidesequence and regulation of the lysR gene. J. Mol. Biol. 168:333-350.
37. Tullius, T. D., and B. A. Dombroski. 1986. Hydroxl-radical"footprinting": high-resolution information about DNA-proteincontacts and application to X repressor and Cro protein. Proc.Natl. Acad. Sci. USA 83:5469-5473.
38. Tullius, T. D., B. A. Dombroski, M. E. A. Churchill, and L.Kam. 1987. Hydroxyl radical footprinting: a high-resolutionmethod for mapping protein-DNA contacts. Methods Enzymol.155:537-558.
39. Wagner, A. F., R. L. Bugianesi, and T. Y. Shen. 1971. Prepara-tion of Sepharose-bound poly(rI:rC). Biochem. Biophys. Res.Commun. 45:184-189.
40. Wek, R. C., and G. W. Hatfield. 1986. Nucleotide sequence andin vivo expression of ilvY and ilvC genes in Escherichia coliK12. J. Biol. Chem. 261:2441-2450.
41. Wek, R. C., and G. W. Hatfield. 1988. Transcriptional activationat adjacent operators in the divergent-overlapping ilvY and ilvCpromoters of Escherichia coli. J. Mol. Biol. 203:643-663.
42. Wheelis, M. L., and L. N. Ornston. 1972. Genetic control ofenzyme induction in the 3-ketoadipate pathway of Pseudomo-nas putida: deletion mapping of cat mutations. J. Bacteriol.109:790-795.
43. Wheelis, M. L., and R. Y. Stanier. 1970. The genetic control ofdissimilatory pathways in Pseudomonas putida. Genetics 66:245-266.
44. Wu, C. H., M. K. Ornston, and L. N. Ornston. 1972. Geneticcontrol of enzyme induction in the beta-ketoadipate pathway ofPseudomonas putida: two-point crosses with a regulatory mu-tant strain. J. Bacteriol. 109:796-802.
45. Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. ImprovedM13 phage cloning vectors and host strains: nucleotide se-quences of M13mpl8 and pUC19 vectors. Gene 33:103-119.
46. You, I. S., D. Ghosal, and I. C. Gunsalus. 1988. Nucleotidesequence of plasmid NAH7 gene nahR and DNA binding of thenahR product. J. Bacteriol. 170:5409-5415.
J. BACTERIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
b on
27
Janu
ary
2022
by
93.7
8.17
1.4.





























