Embed Size (px)
Citation preview

Inhibition and Stimulation of Intestinal and Hepatic CYP3A Activity:Studies in Humanized CYP3A4 Transgenic Mice Using Triazolam□S
Robert A. B. van Waterschoot, Rogier W. Rooswinkel, Rolf W. Sparidans,Antonius E. van Herwaarden, Jos H. Beijnen, and Alfred H. Schinkel
Division of Molecular Biology, The Netherlands Cancer Institute, Amsterdam, The Netherlands (R.A.B.v.W., R.W.R., A.E.v.H.,A.H.S.); Department of Pharmaceutical Sciences, Utrecht University, Utrecht, The Netherlands (R.W.S., J.H.B.); and
Department of Pharmacy and Pharmacology, Slotervaart Hospital, Amsterdam, The Netherlands (J.H.B.)
Received July 13, 2009; accepted September 9, 2009
ABSTRACT:
CYP3A4 is an important determinant of drug-drug interactions. Inthis study, we evaluated whether cytochrome P450 3A knockoutmice [Cyp3a(�/�)] and CYP3A4 transgenic (CYP3A4-Tg) mice canbe used to study drug-drug interactions in the liver and intestine.Triazolam was used as a probe drug because it is a highly specificCYP3A substrate and not a P-glycoprotein substrate. Triazolammetabolism was profoundly reduced in Cyp3a(�/�) mice both invitro and in vivo. In vitro studies revealed clear species differencesin humans and mice, but triazolam metabolism in microsomesderived from CYP3A4-Tg “humanized” mice closely resembled thatin human microsomes. It is interesting to note that studies withtissue-specific CYP3A4-Tg mice revealed that intestinal CYP3A4has a major impact on oral triazolam exposure, whereas the effectof hepatic CYP3A4 was limited. To mimic a drug-drug interaction,
we coadministered triazolam with the prototypical CYP3A inhibitorketoconazole, which increased triazolam exposure in all theCYP3A-proficient mouse strains but not in Cyp3a(�/�) mice. Wefurther found that the anticancer drug gefitinib is a potent stimu-lator of 1�-OH triazolam formation in vitro. It is noteworthy that wecould also show in vivo stimulation of triazolam metabolism bygefitinib, resulting in a lower oral triazolam exposure. To ourknowledge, this is the first in vivo example of direct stimulation ofCYP3A4 activity after oral drug administration. Overall, this studyillustrates how Cyp3a(�/�) and CYP3A4-Tg mice can be usedto study drug-drug interactions. The data clarify that for drugsthat are not P-glycoprotein substrates, intestinal metabolismalso can be more important than hepatic metabolism after oraladministration.
CYP3A enzymes represent one of the most important drug-metab-olizing systems, affecting �50% of currently prescribed drugs(Guengerich, 1999). Because so many drugs are substrates and/orinhibitors for CYP3A, the enzyme is also an important determinant ofmany drug-drug interactions (Thummel and Wilkinson, 1998; Dresseret al., 2000). The most common type of CYP3A-mediated drug-druginteraction is that one drug inhibits CYP3A activity, which leads tohigher levels of other drugs metabolized by CYP3A, potentiallyleading to toxicity. For example, this was the case for the antihista-minic terfenadine and the antihypertensive mibefradil, which wereconsequently both withdrawn from the market. Although most drug-drug interactions are undesirable, in certain cases CYP3A can beinhibited on purpose to improve drug therapy. For example, the
CYP3A inhibitor ritonavir is given in combination with lopinavir,which improves the lopinavir oral bioavailability considerably (Ku-mar et al., 1999). Not only drugs but also food constituents can bepotent inhibitors of CYP3A. For example, components of grapefruitjuice potently inhibit CYP3A, and several clinically relevant drug-grapefruit juice interactions have been described previously (Dresserand Bailey, 2003; Paine and Oberlies, 2007).
Another possible mechanism of a drug-drug interaction is that onedrug directly increases the rate of CYP3A-mediated metabolism ofanother drug. This stimulation of the metabolism is also known asheterotropic positive cooperativity (Tang and Stearns, 2001; Hutzlerand Tracy, 2002; Houston and Galetin, 2005). Stimulation of metab-olism could be of clinical relevance as it could result in subtherapeuticdrug levels. A classic example of a drug that is known to stimulateseveral CYP3A-mediated reactions is 7,8-benzoflavone, which in-creases the metabolism of diazepam and aflatoxin B1 in vitro(Andersson et al., 1994; Ueng et al., 1997). It has to be noted thatalthough many in vitro examples of CYP3A stimulation have beenpublished, there is only very limited in vivo evidence for this partic-ular drug-drug interaction (Hutzler and Tracy, 2002; Wienkers andHeath, 2005).
This work was supported by the Technical Sciences Foundation of the Neth-erlands Organization for Scientific Research.
Article, publication date, and citation information can be found athttp://dmd.aspetjournals.org.
doi:10.1124/dmd.109.029397.□S The online version of this article (available at http://dmd.aspetjournals.org)
contains supplemental material.
ABBREVIATIONS: Cyp3a(�/�), cytochrome P450 3A knockout mice; Tg, transgenic; P-gp, P-glycoprotein; P450, cytochrome P450; Cyp3a(�/�)Tg-3A4Hep, Cyp3a knockout mice with liver-specific transgenic expression of human CYP3A4; Cyp3a(�/�)Tg-3A4Int, Cyp3a knockout mice withintestine-specific transgenic expression of human CYP3A4; HPLC, high-performance liquid chromatography; PEG, polyethylene glycol; LC/MS/MS, liquid chromatography/tandem mass spectrometry; AUC, area under the curve.
0090-9556/09/3712-2305–2313$20.00DRUG METABOLISM AND DISPOSITION Vol. 37, No. 12Copyright © 2009 by The American Society for Pharmacology and Experimental Therapeutics 29397/3533628DMD 37:2305–2313, 2009 Printed in U.S.A.
2305
http://dmd.aspetjournals.org/content/suppl/2009/09/14/dmd.109.029397.DC1Supplemental material to this article can be found at:
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from

Recently, we have generated Cyp3a knockout mice to study thisimportant drug-handling system in vivo (van Herwaarden et al.,2007). In addition, we created cytochrome P450 3A knockout[Cyp3a(�/�)] transgenic (Tg) mice with expression of humanCYP3A4 exclusively in the intestine or in the liver (van Herwaardenet al., 2007). We subsequently used these mice to determine therelative importance of intestinal versus hepatic CYP3A4 activity infirst-pass drug metabolism. These studies revealed that intestinalCYP3A4 alone was sufficient to virtually abrogate net docetaxel entryfrom the gut, whereas hepatic CYP3A4 was more important in sys-temic docetaxel clearance (van Herwaarden et al., 2007). This studyclarified the potential significance of intestinal metabolism, which hasbeen a matter of debate for decades (Lin et al., 1999; Doherty andCharman, 2002; Thummel, 2007).
Besides being a good CYP3A4 substrate, however, docetaxel isalso a very good substrate of the apical drug transporter P-glycopro-tein (P-gp) (Bardelmeijer et al., 2002). It has been hypothesized thatthe function of P-gp could prevent saturation of intestinal CYP3A andgive the enzyme repeated access to its substrates, resulting in highlyefficient intestinal metabolism (Benet and Cummins, 2001). We con-sidered that P-gp might have contributed greatly to the efficiency ofintestinal CYP3A4-mediated docetaxel metabolism. Hence, it is notclear whether intestinal CYP3A can also dominate the metabolism ofsubstrates that are not transported by P-gp, such as midazolam. It isunfortunate that useful analysis of midazolam pharmacokinetics in ourmouse models was precluded by the up-regulation of (midazolam-metabolizing) mouse CYP2C in the Cyp3a(�/�) mice (van Water-schoot et al., 2008).
In contrast to midazolam, the closely related drug triazolam is amore specific substrate for CYP3A compared with other mouse cy-tochrome P450 (P450) isoforms (Perloff et al., 2000). Therefore, itmight be a better probe drug to characterize our Cyp3a(�/�) andCYP3A4-Tg mouse lines. Similar to midazolam, triazolam is also nota P-gp substrate (von Moltke et al., 2004). Therefore, in this study weused triazolam to study the relative contribution of intestinal andhepatic CYP3A-mediated metabolism. Furthermore, we also assessedwhether our mouse models could be used to study in vivo drug-druginteractions such as inhibition and stimulation of CYP3A activity.
Materials and Methods
Materials. Triazolam and 1�-OH triazolam were obtained from Sigma-Aldrich (St. Louis, MO). D4-Triazolam and D4-1�-hydroxy-triazolam wereboth obtained as 100-�g/ml solutions in methanol from Cerilliant Corporation(Round Rock, TX). NADPH-generation system and pooled human liver andintestinal microsomes were obtained from BD Bioscience (Alphen aan denRijn, The Netherlands). Gefitinib, imatinib, erlotinib, dasatinib, sorafenib, andsunitinib were purchased from Sequoia Research Products Ltd. (Pangbourne,UK). Pentobarbital (Nembutal) was obtained from sanofi-aventis (Bridgewa-ter, NJ), and methoxyflurane (Metofane) was obtained from Medical Devel-opments Australia Pty. Ltd. (Springvale, VIC, Australia). All of the otherchemicals were of analytical grade and were obtained from commercialsources.
Animals. The mice used in this study were housed and handled accordingto institutional guidelines complying with Dutch legislation. The animals werekept in a temperature-controlled environment with a 12:12-h light/dark cycleand permitted ad libitum consumption of acidified water and a standard(AM-II) diet (Hope Farms, The Netherlands) unless indicated otherwise.Wild-type, Cyp3a knockout [Cyp3a(�/�)], or Cyp3a(�/�) mice with specificexpression of human CYP3A4 in either the liver [Cyp3a(�/�)Tg-3A4Hep,previously Cyp3a(�/�)A], intestine [Cyp3a(�/�)Tg-3A4Int, previouslyCyp3a(�/�)V], or both [Cyp3a(�/�)Tg-3A4Hep/Int, previously Cyp3a(�/�)AV] (van Herwaarden et al., 2007) were used. All the mouse strains wereof a homogeneous (�99%) FVB genetic background, and all the experimentswere done using male mice aged between 8 and 12 weeks old.
Microsomal Incubations. Mouse liver and intestinal microsomes wereprepared using the whole tissues as described previously (van Waterschoot etal., 2008). The incubations were carried out in a total volume of 200 �l,containing 100 mM potassium phosphate buffer, pH 7.4. Protein concentra-tions were 0.5 mg/ml for liver microsomes and 1 mg/ml for intestinal micro-somes unless indicated otherwise. Formation of triazolam metabolites waslinear with respect to incubation time and microsomal protein concentration.Control experiments without cofactor were performed to ascertain P450-dependent metabolism. Final concentration of methanol was 0.5% in all theincubations. After 5 min of preincubation at 37°C, the reactions were initiatedby addition of an NADPH-regenerating system. After 20 min, the reactionswere stopped by adding 100 �l of ice-cold acetonitrile and cooling on ice for5 min. After centrifugation (10 min at 6800g), 50 �l of the sample was injectedinto the high-performance liquid chromatography (HPLC) system.
The enzyme kinetics for both 1�-OH and 4-OH triazolam were investigatedusing GraphPad Software Inc. (San Diego, CA) Prism 4.0 nonlinear regressionanalysis. Most of the kinetic data could be fitted using a standard Michaelis-Menten equation (eq. 1):
V �Vmax � �s�
Km � �s�(1)
Where indicated, a Michaelis-Menten kinetics model with noncompetitivesubstrate inhibition (von Moltke et al., 1996) was used (eq. 2).
V �Vmax � �s�
Km � �s� � �1 ��s�
Ks� (2)
Stimulation of 1�-OH Triazolam Formation in Vitro. Microsomes werepreincubated for 4 min in the presence of gefitinib, imatinib, erlotinib, dasatinib,sorafenib, and sunitinib (12.5 �M final concentration) leading to 0.2% dimethylsulfoxide in the final incubation. Subsequently, triazolam was added and preincu-bated for 1 min after which the reaction was started by adding the NADPH-regenerating system. Other conditions were similar as described above.
Chemical and Immunoinhibition. For the inhibition experiments, twoantibodies targeting rat CYP2C11 were used. Polyclonal anti-CYP2C11 goatantibodies were obtained from Daiichi Pure Chemical Co. (Tokyo, Japan), andmonoclonal anti-CYP2C11 mouse antibodies were obtained from Invitrogen(Carlsbad, CA); they were referred to as anti-CYP2C-A and anti-CYP2C-B,respectively. After 10 min of preincubation with ketoconazole (2.5 �M finalconcentration) or one of two anti-CYP2C11 antibodies, the microsomal reac-tion mixture was incubated for 20 min. The final concentration of triazolam inthe incubations was 50 �M. All the other conditions were as described above.
HPLC. For determination of triazolam and its metabolites, the HPLCmobile phase consisted of 20% acetonitrile, 24% methanol, and 56% 5 mMphosphate buffer, pH 7.4 (0.15% triethylamine), with a flow rate of 0.4 ml/min.The analytical column was a reverse-phase C18 XBridge, 3.0 � 150 mm, 3.5�m (Waters, Milford, MA). Column effluent was monitored by UV absorptionat 230 nm.
Identity and quantity of 1�-OH and 4-OH triazolam metabolites wereverified using the retention time and standard curve of an authentic 1�-OHtriazolam standard. For determining triazolam in the presence of gefitinib, themobile phase consisted of two solutions. Solution A consisted of 20% aceto-nitrile, 30% methanol, and 50% phosphate buffer (5 mM), pH 7.4 (0.15%triethylamine). Solution B consisted of 60% acetonitrile and 40% H2O. From0 to 5 and from 8 to 20 min after injection, the mobile phase consisted of 100%A. Between 5 and 8 min after injection, the mobile phase consisted of a mixof A and B (10:90).
Triazolam Pharmacokinetics in Vivo. Thirty minutes before triazolamadministration, fasted mice (2 h) received orally either ketoconazole (35mg/kg) in polyethylene glycol (PEG) 400, gefitinib (25 mg/kg) in PEG 400, orvehicle (PEG 400). At t � 0, triazolam in ethanol and saline (1:99) wasadministered by oral gavage at 0.5 mg/kg. Ten, 20, 40, 80, 160, and 320 minafter triazolam administration, blood samples were collected by tail sampling.Blood was centrifuged for 6 min at 6800g, after which 25 �l of serum wascollected and stored at �20°C until further analysis.
Liquid Chromatography/Tandem Mass Spectrometry. Mouse plasmasamples were measured by liquid chromatography/tandem mass spectrometry
2306 VAN WATERSCHOOT ET AL.
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from
(LC/MS/MS). To a 20-�l plasma sample, 100 �l of 25% (v/v) methanol, 20 �lof 40-ng/ml D4-triazolam and 10-ng/ml D4-1�-hydroxy-triazolam in 25% (v/v)methanol, and 200 �l of 5 mM sodium hydroxide were added. The analyteswere extracted with 2 ml of diethyl ether, and the organic phase was evapo-rated under a stream of nitrogen at 30°C. The residue was reconstituted in 100�l of 50% (v/v) methanol before injection in the chromatographic system.
The LC/MS/MS equipment consisted of a DGU-14A degasser, a Sil-HTcautosampler, two LC10-ADvp-� pumps, and a CTO10-Avp column oven (allobtained from Shimadzu, Kyoto, Japan) and a Finnigan TSQ Quantum Dis-covery Max triple quadrupole mass spectrometer with electrospray ionization(Thermo Fisher Scientific, Waltham, MA). Data were processed using Finni-gan Xcalibur software (version 1.4; Thermo Fisher Scientific).
Twenty-microliter injections were made on a Polaris 3 C18-A column (50 �2 mm; dp, 3 �m; average pore diameter, 10 nm; Varian, Middelburg, TheNetherlands) with a Polaris 3 C18-A precolumn (10 � 2 mm; dp, 3 �m;Varian). The column temperature was maintained at 35°C, and the autosamplerwas maintained at 4°C. The flow rate was 0.3 ml/min, and the eluent wascomposed 35% (v/v) of 0.01% (v/v) formic acid in water and 65% (v/v) ofmethanol.
Mass transitions [collision energies (V)] were 3433308 (29), 239 (43), and315 (28) for triazolam; 3593331 (27), 239 (44), and 250 (38) for 1�-OHtriazolam; 3473312 (27) for D4-triazolam; and 3653337 (29) for D4-1�-OHtriazolam (transition of 37Cl-D4-1�-hydroxy-triazolam was used because ofisotopic interference, mainly by 37Cl2-1�-hydroxy-triazolam). The mass reso-lutions were set at 0.2 full at half height for the first quadrupole and at 0.7 fullat half height (unit resolution) for the third quadrupole for all the compounds.
Results
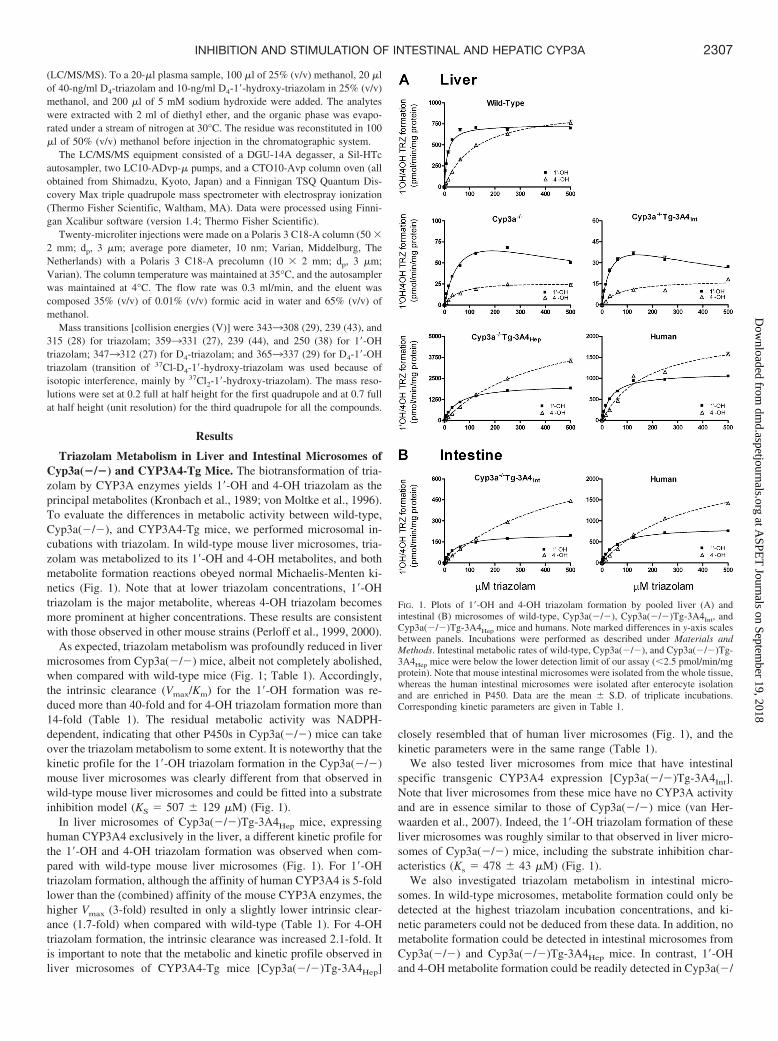
Triazolam Metabolism in Liver and Intestinal Microsomes ofCyp3a(�/�) and CYP3A4-Tg Mice. The biotransformation of tria-zolam by CYP3A enzymes yields 1�-OH and 4-OH triazolam as theprincipal metabolites (Kronbach et al., 1989; von Moltke et al., 1996).To evaluate the differences in metabolic activity between wild-type,Cyp3a(�/�), and CYP3A4-Tg mice, we performed microsomal in-cubations with triazolam. In wild-type mouse liver microsomes, tria-zolam was metabolized to its 1�-OH and 4-OH metabolites, and bothmetabolite formation reactions obeyed normal Michaelis-Menten ki-netics (Fig. 1). Note that at lower triazolam concentrations, 1�-OHtriazolam is the major metabolite, whereas 4-OH triazolam becomesmore prominent at higher concentrations. These results are consistentwith those observed in other mouse strains (Perloff et al., 1999, 2000).
As expected, triazolam metabolism was profoundly reduced in livermicrosomes from Cyp3a(�/�) mice, albeit not completely abolished,when compared with wild-type mice (Fig. 1; Table 1). Accordingly,the intrinsic clearance (Vmax/Km) for the 1�-OH formation was re-duced more than 40-fold and for 4-OH triazolam formation more than14-fold (Table 1). The residual metabolic activity was NADPH-dependent, indicating that other P450s in Cyp3a(�/�) mice can takeover the triazolam metabolism to some extent. It is noteworthy that thekinetic profile for the 1�-OH triazolam formation in the Cyp3a(�/�)mouse liver microsomes was clearly different from that observed inwild-type mouse liver microsomes and could be fitted into a substrateinhibition model (KS � 507 129 �M) (Fig. 1).
In liver microsomes of Cyp3a(�/�)Tg-3A4Hep mice, expressinghuman CYP3A4 exclusively in the liver, a different kinetic profile forthe 1�-OH and 4-OH triazolam formation was observed when com-pared with wild-type mouse liver microsomes (Fig. 1). For 1�-OHtriazolam formation, although the affinity of human CYP3A4 is 5-foldlower than the (combined) affinity of the mouse CYP3A enzymes, thehigher Vmax (3-fold) resulted in only a slightly lower intrinsic clear-ance (1.7-fold) when compared with wild-type (Table 1). For 4-OHtriazolam formation, the intrinsic clearance was increased 2.1-fold. Itis important to note that the metabolic and kinetic profile observed inliver microsomes of CYP3A4-Tg mice [Cyp3a(�/�)Tg-3A4Hep]
closely resembled that of human liver microsomes (Fig. 1), and thekinetic parameters were in the same range (Table 1).
We also tested liver microsomes from mice that have intestinalspecific transgenic CYP3A4 expression [Cyp3a(�/�)Tg-3A4Int].Note that liver microsomes from these mice have no CYP3A activityand are in essence similar to those of Cyp3a(�/�) mice (van Her-waarden et al., 2007). Indeed, the 1�-OH triazolam formation of theseliver microsomes was roughly similar to that observed in liver micro-somes of Cyp3a(�/�) mice, including the substrate inhibition char-acteristics (Ks � 478 43 �M) (Fig. 1).
We also investigated triazolam metabolism in intestinal micro-somes. In wild-type microsomes, metabolite formation could only bedetected at the highest triazolam incubation concentrations, and ki-netic parameters could not be deduced from these data. In addition, nometabolite formation could be detected in intestinal microsomes fromCyp3a(�/�) and Cyp3a(�/�)Tg-3A4Hep mice. In contrast, 1�-OHand 4-OH metabolite formation could be readily detected in Cyp3a(�/
FIG. 1. Plots of 1�-OH and 4-OH triazolam formation by pooled liver (A) andintestinal (B) microsomes of wild-type, Cyp3a(�/�), Cyp3a(�/�)Tg-3A4Int, andCyp3a(�/�)Tg-3A4Hep mice and humans. Note marked differences in y-axis scalesbetween panels. Incubations were performed as described under Materials andMethods. Intestinal metabolic rates of wild-type, Cyp3a(�/�), and Cyp3a(�/�)Tg-3A4Hep mice were below the lower detection limit of our assay (2.5 pmol/min/mgprotein). Note that mouse intestinal microsomes were isolated from the whole tissue,whereas the human intestinal microsomes were isolated after enterocyte isolationand are enriched in P450. Data are the mean S.D. of triplicate incubations.Corresponding kinetic parameters are given in Table 1.
2307INHIBITION AND STIMULATION OF INTESTINAL AND HEPATIC CYP3A
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from
�)Tg-3A4Int intestinal microsomes. Both 1�-OH triazolam and 4-OHtriazolam formation obeyed normal Michaelis-Menten kinetics andclosely resembled the kinetic profile of human intestinal microsomes(Fig. 1; Table 1). Furthermore, the metabolic kinetic profile inCyp3a(�/�)Tg-3A4Int intestinal microsomes was also very similar tothat observed in liver microsomes from Cyp3a(�/�)Tg-3A4Hep mice(Fig. 1). Note that we have used the whole small intestine for theisolation of mouse microsomes and that the inclusion of nonentero-cyte tissue will result in lower apparent Vmax values compared withliver. For the human intestinal microsomes, only mature enterocytes,which are enriched in P450s, were used, precluding a simple com-parison between the mouse and human intestinal Vmax values.
Collectively, these data indicate that there are clear species differ-ences between mice and humans in the in vitro metabolism of tria-zolam. Of importance, the data further show that mouse microsomeswith transgenic CYP3A4 expression [in a Cyp3a(�/�) background]closely resemble the triazolam metabolism in human hepatic andintestinal microsomes, both in terms of the metabolic kinetic profile(Fig. 1) and the intrinsic clearance (Table 1). Finally, they alsoconfirm the tissue-specific activity of CYP3A4 in the transgenicstrains.
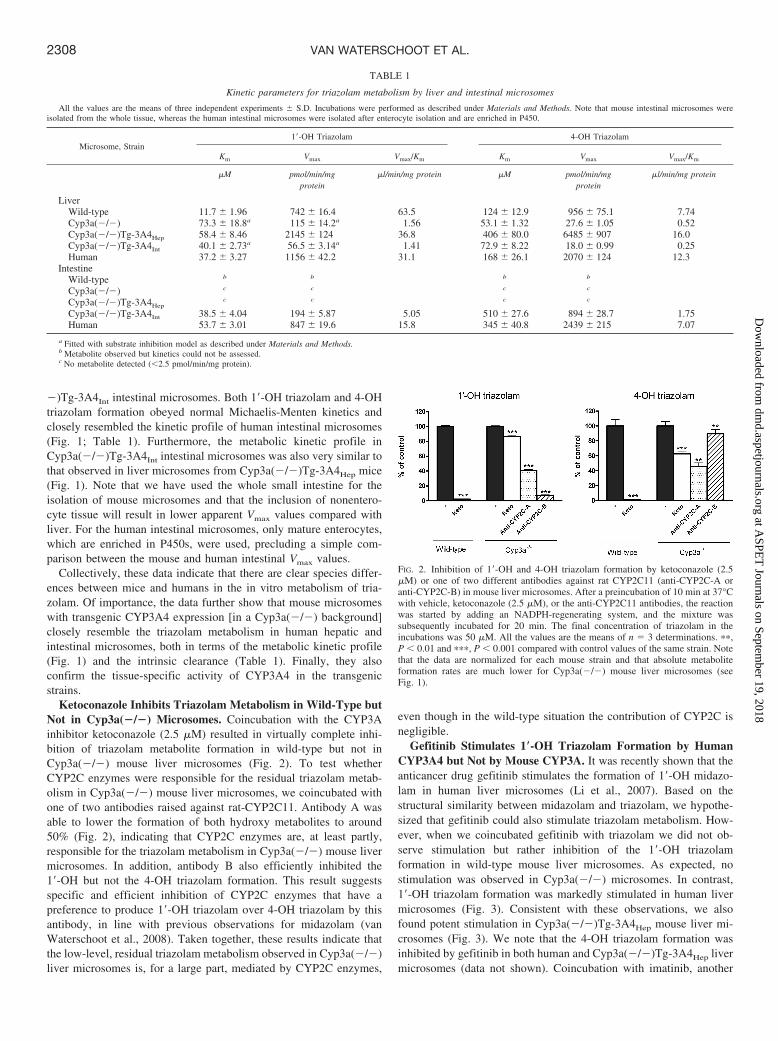
Ketoconazole Inhibits Triazolam Metabolism in Wild-Type butNot in Cyp3a(�/�) Microsomes. Coincubation with the CYP3Ainhibitor ketoconazole (2.5 �M) resulted in virtually complete inhi-bition of triazolam metabolite formation in wild-type but not inCyp3a(�/�) mouse liver microsomes (Fig. 2). To test whetherCYP2C enzymes were responsible for the residual triazolam metab-olism in Cyp3a(�/�) mouse liver microsomes, we coincubated withone of two antibodies raised against rat-CYP2C11. Antibody A wasable to lower the formation of both hydroxy metabolites to around50% (Fig. 2), indicating that CYP2C enzymes are, at least partly,responsible for the triazolam metabolism in Cyp3a(�/�) mouse livermicrosomes. In addition, antibody B also efficiently inhibited the1�-OH but not the 4-OH triazolam formation. This result suggestsspecific and efficient inhibition of CYP2C enzymes that have apreference to produce 1�-OH triazolam over 4-OH triazolam by thisantibody, in line with previous observations for midazolam (vanWaterschoot et al., 2008). Taken together, these results indicate thatthe low-level, residual triazolam metabolism observed in Cyp3a(�/�)liver microsomes is, for a large part, mediated by CYP2C enzymes,
even though in the wild-type situation the contribution of CYP2C isnegligible.
Gefitinib Stimulates 1�-OH Triazolam Formation by HumanCYP3A4 but Not by Mouse CYP3A. It was recently shown that theanticancer drug gefitinib stimulates the formation of 1�-OH midazo-lam in human liver microsomes (Li et al., 2007). Based on thestructural similarity between midazolam and triazolam, we hypothe-sized that gefitinib could also stimulate triazolam metabolism. How-ever, when we coincubated gefitinib with triazolam we did not ob-serve stimulation but rather inhibition of the 1�-OH triazolamformation in wild-type mouse liver microsomes. As expected, nostimulation was observed in Cyp3a(�/�) microsomes. In contrast,1�-OH triazolam formation was markedly stimulated in human livermicrosomes (Fig. 3). Consistent with these observations, we alsofound potent stimulation in Cyp3a(�/�)Tg-3A4Hep mouse liver mi-crosomes (Fig. 3). We note that the 4-OH triazolam formation wasinhibited by gefitinib in both human and Cyp3a(�/�)Tg-3A4Hep livermicrosomes (data not shown). Coincubation with imatinib, another
TABLE 1
Kinetic parameters for triazolam metabolism by liver and intestinal microsomes
All the values are the means of three independent experiments S.D. Incubations were performed as described under Materials and Methods. Note that mouse intestinal microsomes wereisolated from the whole tissue, whereas the human intestinal microsomes were isolated after enterocyte isolation and are enriched in P450.
Microsome, Strain1�-OH Triazolam 4-OH Triazolam
Km Vmax Vmax/Km Km Vmax Vmax/Km
�M pmol/min/mgprotein
�l/min/mg protein �M pmol/min/mgprotein
�l/min/mg protein
LiverWild-type 11.7 1.96 742 16.4 63.5 124 12.9 956 75.1 7.74Cyp3a(�/�) 73.3 18.8a 115 14.2a 1.56 53.1 1.32 27.6 1.05 0.52Cyp3a(�/�)Tg-3A4Hep 58.4 8.46 2145 124 36.8 406 80.0 6485 907 16.0Cyp3a(�/�)Tg-3A4Int 40.1 2.73a 56.5 3.14a 1.41 72.9 8.22 18.0 0.99 0.25Human 37.2 3.27 1156 42.2 31.1 168 26.1 2070 124 12.3
IntestineWild-type b b b b
Cyp3a(�/�) c c c c
Cyp3a(�/�)Tg-3A4Hepc c c c
Cyp3a(�/�)Tg-3A4Int 38.5 4.04 194 5.87 5.05 510 27.6 894 28.7 1.75Human 53.7 3.01 847 19.6 15.8 345 40.8 2439 215 7.07
a Fitted with substrate inhibition model as described under Materials and Methods.b Metabolite observed but kinetics could not be assessed.c No metabolite detected (2.5 pmol/min/mg protein).
FIG. 2. Inhibition of 1�-OH and 4-OH triazolam formation by ketoconazole (2.5�M) or one of two different antibodies against rat CYP2C11 (anti-CYP2C-A oranti-CYP2C-B) in mouse liver microsomes. After a preincubation of 10 min at 37°Cwith vehicle, ketoconazole (2.5 �M), or the anti-CYP2C11 antibodies, the reactionwas started by adding an NADPH-regenerating system, and the mixture wassubsequently incubated for 20 min. The final concentration of triazolam in theincubations was 50 �M. All the values are the means of n � 3 determinations. ��,P 0.01 and ���, P 0.001 compared with control values of the same strain. Notethat the data are normalized for each mouse strain and that absolute metaboliteformation rates are much lower for Cyp3a(�/�) mouse liver microsomes (seeFig. 1).
2308 VAN WATERSCHOOT ET AL.
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from
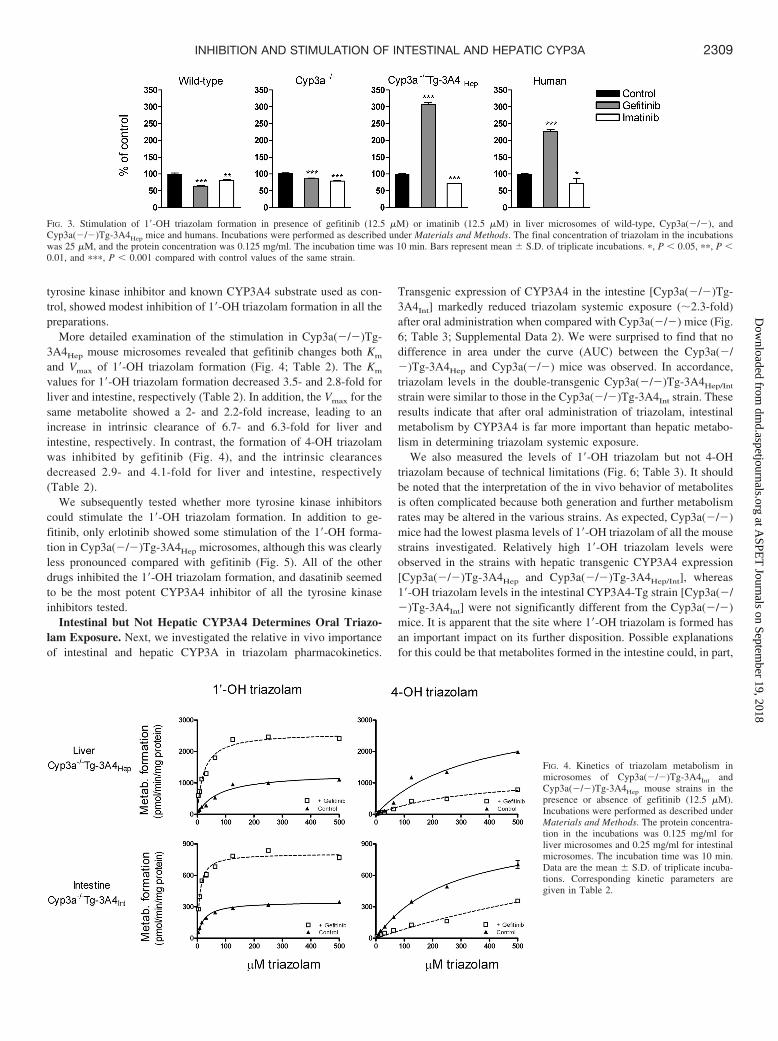
tyrosine kinase inhibitor and known CYP3A4 substrate used as con-trol, showed modest inhibition of 1�-OH triazolam formation in all thepreparations.
More detailed examination of the stimulation in Cyp3a(�/�)Tg-3A4Hep mouse microsomes revealed that gefitinib changes both Km
and Vmax of 1�-OH triazolam formation (Fig. 4; Table 2). The Km
values for 1�-OH triazolam formation decreased 3.5- and 2.8-fold forliver and intestine, respectively (Table 2). In addition, the Vmax for thesame metabolite showed a 2- and 2.2-fold increase, leading to anincrease in intrinsic clearance of 6.7- and 6.3-fold for liver andintestine, respectively. In contrast, the formation of 4-OH triazolamwas inhibited by gefitinib (Fig. 4), and the intrinsic clearancesdecreased 2.9- and 4.1-fold for liver and intestine, respectively(Table 2).
We subsequently tested whether more tyrosine kinase inhibitorscould stimulate the 1�-OH triazolam formation. In addition to ge-fitinib, only erlotinib showed some stimulation of the 1�-OH forma-tion in Cyp3a(�/�)Tg-3A4Hep microsomes, although this was clearlyless pronounced compared with gefitinib (Fig. 5). All of the otherdrugs inhibited the 1�-OH triazolam formation, and dasatinib seemedto be the most potent CYP3A4 inhibitor of all the tyrosine kinaseinhibitors tested.
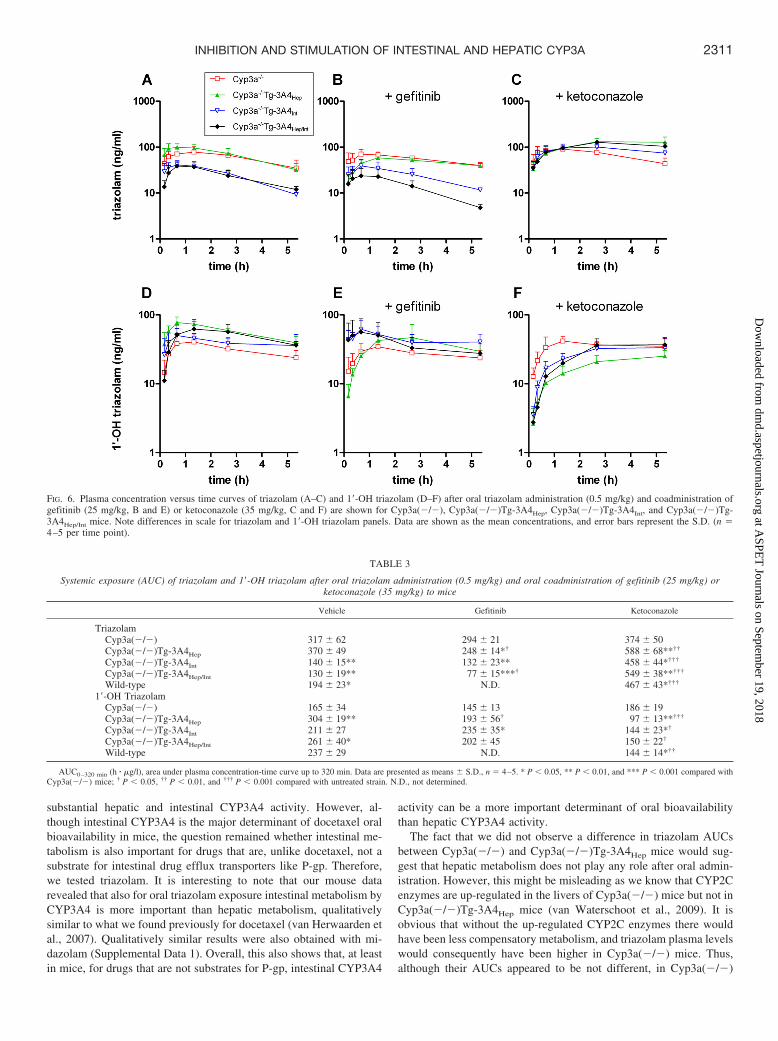
Intestinal but Not Hepatic CYP3A4 Determines Oral Triazo-lam Exposure. Next, we investigated the relative in vivo importanceof intestinal and hepatic CYP3A in triazolam pharmacokinetics.
Transgenic expression of CYP3A4 in the intestine [Cyp3a(�/�)Tg-3A4Int] markedly reduced triazolam systemic exposure (�2.3-fold)after oral administration when compared with Cyp3a(�/�) mice (Fig.6; Table 3; Supplemental Data 2). We were surprised to find that nodifference in area under the curve (AUC) between the Cyp3a(�/�)Tg-3A4Hep and Cyp3a(�/�) mice was observed. In accordance,triazolam levels in the double-transgenic Cyp3a(�/�)Tg-3A4Hep/Int
strain were similar to those in the Cyp3a(�/�)Tg-3A4Int strain. Theseresults indicate that after oral administration of triazolam, intestinalmetabolism by CYP3A4 is far more important than hepatic metabo-lism in determining triazolam systemic exposure.
We also measured the levels of 1�-OH triazolam but not 4-OHtriazolam because of technical limitations (Fig. 6; Table 3). It shouldbe noted that the interpretation of the in vivo behavior of metabolitesis often complicated because both generation and further metabolismrates may be altered in the various strains. As expected, Cyp3a(�/�)mice had the lowest plasma levels of 1�-OH triazolam of all the mousestrains investigated. Relatively high 1�-OH triazolam levels wereobserved in the strains with hepatic transgenic CYP3A4 expression[Cyp3a(�/�)Tg-3A4Hep and Cyp3a(�/�)Tg-3A4Hep/Int], whereas1�-OH triazolam levels in the intestinal CYP3A4-Tg strain [Cyp3a(�/�)Tg-3A4Int] were not significantly different from the Cyp3a(�/�)mice. It is apparent that the site where 1�-OH triazolam is formed hasan important impact on its further disposition. Possible explanationsfor this could be that metabolites formed in the intestine could, in part,
FIG. 3. Stimulation of 1�-OH triazolam formation in presence of gefitinib (12.5 �M) or imatinib (12.5 �M) in liver microsomes of wild-type, Cyp3a(�/�), andCyp3a(�/�)Tg-3A4Hep mice and humans. Incubations were performed as described under Materials and Methods. The final concentration of triazolam in the incubationswas 25 �M, and the protein concentration was 0.125 mg/ml. The incubation time was 10 min. Bars represent mean S.D. of triplicate incubations. �, P 0.05, ��, P 0.01, and ���, P 0.001 compared with control values of the same strain.
FIG. 4. Kinetics of triazolam metabolism inmicrosomes of Cyp3a(�/�)Tg-3A4Int andCyp3a(�/�)Tg-3A4Hep mouse strains in thepresence or absence of gefitinib (12.5 �M).Incubations were performed as described underMaterials and Methods. The protein concentra-tion in the incubations was 0.125 mg/ml forliver microsomes and 0.25 mg/ml for intestinalmicrosomes. The incubation time was 10 min.Data are the mean S.D. of triplicate incuba-tions. Corresponding kinetic parameters aregiven in Table 2.
2309INHIBITION AND STIMULATION OF INTESTINAL AND HEPATIC CYP3A
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from
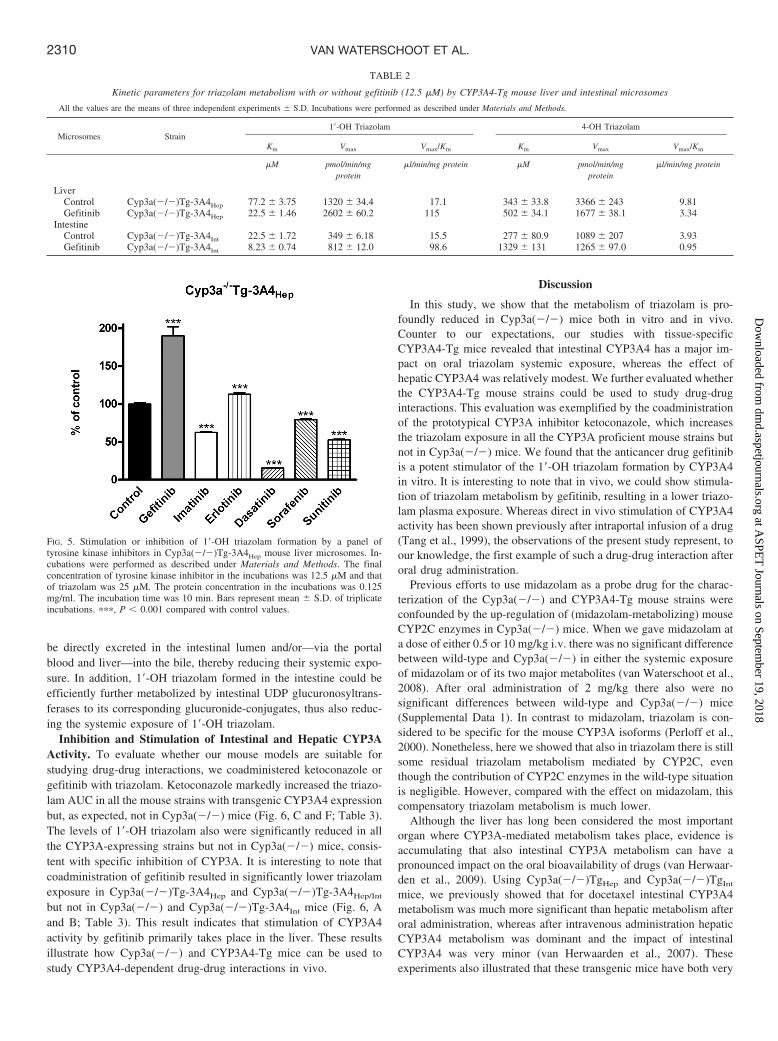
be directly excreted in the intestinal lumen and/or—via the portalblood and liver—into the bile, thereby reducing their systemic expo-sure. In addition, 1�-OH triazolam formed in the intestine could beefficiently further metabolized by intestinal UDP glucuronosyltrans-ferases to its corresponding glucuronide-conjugates, thus also reduc-ing the systemic exposure of 1�-OH triazolam.
Inhibition and Stimulation of Intestinal and Hepatic CYP3AActivity. To evaluate whether our mouse models are suitable forstudying drug-drug interactions, we coadministered ketoconazole orgefitinib with triazolam. Ketoconazole markedly increased the triazo-lam AUC in all the mouse strains with transgenic CYP3A4 expressionbut, as expected, not in Cyp3a(�/�) mice (Fig. 6, C and F; Table 3).The levels of 1�-OH triazolam also were significantly reduced in allthe CYP3A-expressing strains but not in Cyp3a(�/�) mice, consis-tent with specific inhibition of CYP3A. It is interesting to note thatcoadministration of gefitinib resulted in significantly lower triazolamexposure in Cyp3a(�/�)Tg-3A4Hep and Cyp3a(�/�)Tg-3A4Hep/Int
but not in Cyp3a(�/�) and Cyp3a(�/�)Tg-3A4Int mice (Fig. 6, Aand B; Table 3). This result indicates that stimulation of CYP3A4activity by gefitinib primarily takes place in the liver. These resultsillustrate how Cyp3a(�/�) and CYP3A4-Tg mice can be used tostudy CYP3A4-dependent drug-drug interactions in vivo.
Discussion
In this study, we show that the metabolism of triazolam is pro-foundly reduced in Cyp3a(�/�) mice both in vitro and in vivo.Counter to our expectations, our studies with tissue-specificCYP3A4-Tg mice revealed that intestinal CYP3A4 has a major im-pact on oral triazolam systemic exposure, whereas the effect ofhepatic CYP3A4 was relatively modest. We further evaluated whetherthe CYP3A4-Tg mouse strains could be used to study drug-druginteractions. This evaluation was exemplified by the coadministrationof the prototypical CYP3A inhibitor ketoconazole, which increasesthe triazolam exposure in all the CYP3A proficient mouse strains butnot in Cyp3a(�/�) mice. We found that the anticancer drug gefitinibis a potent stimulator of the 1�-OH triazolam formation by CYP3A4in vitro. It is interesting to note that in vivo, we could show stimula-tion of triazolam metabolism by gefitinib, resulting in a lower triazo-lam plasma exposure. Whereas direct in vivo stimulation of CYP3A4activity has been shown previously after intraportal infusion of a drug(Tang et al., 1999), the observations of the present study represent, toour knowledge, the first example of such a drug-drug interaction afteroral drug administration.
Previous efforts to use midazolam as a probe drug for the charac-terization of the Cyp3a(�/�) and CYP3A4-Tg mouse strains wereconfounded by the up-regulation of (midazolam-metabolizing) mouseCYP2C enzymes in Cyp3a(�/�) mice. When we gave midazolam ata dose of either 0.5 or 10 mg/kg i.v. there was no significant differencebetween wild-type and Cyp3a(�/�) in either the systemic exposureof midazolam or of its two major metabolites (van Waterschoot et al.,2008). After oral administration of 2 mg/kg there also were nosignificant differences between wild-type and Cyp3a(�/�) mice(Supplemental Data 1). In contrast to midazolam, triazolam is con-sidered to be specific for the mouse CYP3A isoforms (Perloff et al.,2000). Nonetheless, here we showed that also in triazolam there is stillsome residual triazolam metabolism mediated by CYP2C, eventhough the contribution of CYP2C enzymes in the wild-type situationis negligible. However, compared with the effect on midazolam, thiscompensatory triazolam metabolism is much lower.
Although the liver has long been considered the most importantorgan where CYP3A-mediated metabolism takes place, evidence isaccumulating that also intestinal CYP3A metabolism can have apronounced impact on the oral bioavailability of drugs (van Herwaar-den et al., 2009). Using Cyp3a(�/�)TgHep and Cyp3a(�/�)TgInt
mice, we previously showed that for docetaxel intestinal CYP3A4metabolism was much more significant than hepatic metabolism afteroral administration, whereas after intravenous administration hepaticCYP3A4 metabolism was dominant and the impact of intestinalCYP3A4 was very minor (van Herwaarden et al., 2007). Theseexperiments also illustrated that these transgenic mice have both very
FIG. 5. Stimulation or inhibition of 1�-OH triazolam formation by a panel oftyrosine kinase inhibitors in Cyp3a(�/�)Tg-3A4Hep mouse liver microsomes. In-cubations were performed as described under Materials and Methods. The finalconcentration of tyrosine kinase inhibitor in the incubations was 12.5 �M and thatof triazolam was 25 �M. The protein concentration in the incubations was 0.125mg/ml. The incubation time was 10 min. Bars represent mean S.D. of triplicateincubations. ���, P 0.001 compared with control values.
TABLE 2
Kinetic parameters for triazolam metabolism with or without gefitinib (12.5 �M) by CYP3A4-Tg mouse liver and intestinal microsomes
All the values are the means of three independent experiments S.D. Incubations were performed as described under Materials and Methods.
Microsomes Strain1�-OH Triazolam 4-OH Triazolam
Km Vmax Vmax/Km Km Vmax Vmax/Km
�M pmol/min/mgprotein
�l/min/mg protein �M pmol/min/mgprotein
�l/min/mg protein
LiverControl Cyp3a(�/�)Tg-3A4Hep 77.2 3.75 1320 34.4 17.1 343 33.8 3366 243 9.81Gefitinib Cyp3a(�/�)Tg-3A4Hep 22.5 1.46 2602 60.2 115 502 34.1 1677 38.1 3.34
IntestineControl Cyp3a(�/�)Tg-3A4Int 22.5 1.72 349 6.18 15.5 277 80.9 1089 207 3.93Gefitinib Cyp3a(�/�)Tg-3A4Int 8.23 0.74 812 12.0 98.6 1329 131 1265 97.0 0.95
2310 VAN WATERSCHOOT ET AL.
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from
substantial hepatic and intestinal CYP3A4 activity. However, al-though intestinal CYP3A4 is the major determinant of docetaxel oralbioavailability in mice, the question remained whether intestinal me-tabolism is also important for drugs that are, unlike docetaxel, not asubstrate for intestinal drug efflux transporters like P-gp. Therefore,we tested triazolam. It is interesting to note that our mouse datarevealed that also for oral triazolam exposure intestinal metabolism byCYP3A4 is more important than hepatic metabolism, qualitativelysimilar to what we found previously for docetaxel (van Herwaarden etal., 2007). Qualitatively similar results were also obtained with mi-dazolam (Supplemental Data 1). Overall, this also shows that, at leastin mice, for drugs that are not substrates for P-gp, intestinal CYP3A4
activity can be a more important determinant of oral bioavailabilitythan hepatic CYP3A4 activity.
The fact that we did not observe a difference in triazolam AUCsbetween Cyp3a(�/�) and Cyp3a(�/�)Tg-3A4Hep mice would sug-gest that hepatic metabolism does not play any role after oral admin-istration. However, this might be misleading as we know that CYP2Cenzymes are up-regulated in the livers of Cyp3a(�/�) mice but not inCyp3a(�/�)Tg-3A4Hep mice (van Waterschoot et al., 2009). It isobvious that without the up-regulated CYP2C enzymes there wouldhave been less compensatory metabolism, and triazolam plasma levelswould consequently have been higher in Cyp3a(�/�) mice. Thus,although their AUCs appeared to be not different, in Cyp3a(�/�)
FIG. 6. Plasma concentration versus time curves of triazolam (A–C) and 1�-OH triazolam (D–F) after oral triazolam administration (0.5 mg/kg) and coadministration ofgefitinib (25 mg/kg, B and E) or ketoconazole (35 mg/kg, C and F) are shown for Cyp3a(�/�), Cyp3a(�/�)Tg-3A4Hep, Cyp3a(�/�)Tg-3A4Int, and Cyp3a(�/�)Tg-3A4Hep/Int mice. Note differences in scale for triazolam and 1�-OH triazolam panels. Data are shown as the mean concentrations, and error bars represent the S.D. (n �4–5 per time point).
TABLE 3
Systemic exposure (AUC) of triazolam and 1�-OH triazolam after oral triazolam administration (0.5 mg/kg) and oral coadministration of gefitinib (25 mg/kg) orketoconazole (35 mg/kg) to mice
Vehicle Gefitinib Ketoconazole
TriazolamCyp3a(�/�) 317 62 294 21 374 50Cyp3a(�/�)Tg-3A4Hep 370 49 248 14*† 588 68**††
Cyp3a(�/�)Tg-3A4Int 140 15** 132 23** 458 44*†††
Cyp3a(�/�)Tg-3A4Hep/Int 130 19** 77 15***† 549 38**†††
Wild-type 194 23* N.D. 467 43*†††
1�-OH TriazolamCyp3a(�/�) 165 34 145 13 186 19Cyp3a(�/�)Tg-3A4Hep 304 19** 193 56† 97 13**†††
Cyp3a(�/�)Tg-3A4Int 211 27 235 35* 144 23*†
Cyp3a(�/�)Tg-3A4Hep/Int 261 40* 202 45 150 22†
Wild-type 237 29 N.D. 144 14*††
AUC0–320 min (h � �g/l), area under plasma concentration-time curve up to 320 min. Data are presented as means S.D., n � 4–5. * P 0.05, ** P 0.01, and *** P 0.001 compared withCyp3a(�/�) mice; † P 0.05, †† P 0.01, and ††† P 0.001 compared with untreated strain. N.D., not determined.
2311INHIBITION AND STIMULATION OF INTESTINAL AND HEPATIC CYP3A
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from

mice the triazolam metabolism could be mainly attributed to theup-regulated CYP2C enzymes, whereas in Cyp3a(�/�)Tg-3A4Hep
mice this can be primarily attributed to hepatic CYP3A4. This latterinterpretation is also supported by the fact that the CYP3A inhibitorketoconazole markedly increased the triazolam systemic exposure inCyp3a(�/�)Tg-3A4Hep mice but not in Cyp3a(�/�) mice (Table 3).
We found that the anticancer drug gefitinib is a potent stimulator ofthe 1�-OH triazolam formation but inhibits the 4-OH triazolam for-mation in vitro. Stimulation of CYP3A-mediated metabolism isthought to be attributable to presence of multiple binding sites withinthe enzyme’s active site (e.g., Tang and Stearns, 2001). Recent crystalstructures have verified the possible simultaneous binding of sub-strates to CYP3A4 and accompanying conformational changes (Ek-roos and Sjogren, 2006). Most likely, binding of gefitinib to the activesite of CYP3A4 results in a conformational change of the enzymesthat favors the 1�-OH but inhibits the 4-OH triazolam formation. Suchregioselectivity where one metabolic route is stimulated and the otheris inhibited has been observed previously. For example, testosteronecould stimulate the 4-OH midazolam formation by CYP3A, but itinhibited the 1�-OH metabolite formation (Wang et al., 2000). Incontrast, 7,8-benzoflavone stimulated 1�-OH but inhibited 4-OH mi-dazolam formation (Wang et al., 2000).
It is interesting to note that in vivo coadministration of gefitinibwith triazolam also resulted in significantly lower triazolam exposurein Cyp3a(�/�)Tg-3A4Hep and Cyp3a(�/�)Tg-3A4Hep/Int mice butnot in Cyp3a(�/�) and Cyp3a(�/�)Tg-3A4Int mice, indicating thatstimulation of CYP3A4 primarily takes place in the liver. Althoughcoadministration of gefitinib with triazolam resulted in lower triazo-lam plasma levels after oral administration, the effect was less pro-nounced than perhaps expected from the in vitro results. Clearly,because the in vivo situation is far more complex than an in vitrohomogenate, it is not surprising that there is not a simple one-to-onequantitative relationship between in vitro and in vivo quantitativeshifts. In the microsomal extract, the only route for removal oftriazolam is by metabolic conversion. In contrast, in vivo there aremany alternative routes for clearance of triazolam, including clearanceby transporters, diffusion, ultrafiltration, and so on. Thus, it is more orless expected that the in vivo impact of CYP3A4 stimulation will bemore limited than in vitro. The access of gefitinib to the metabolizingenzyme may be much more limited in vivo than in a simple in vitroextract because we are dealing with intact cells and organs. In addi-tion, as gefitinib itself is a substrate for CYP3A4 (McKillop et al.,2005), a substantial part of the gefitinib dose could already have beenmetabolized. Finally, administration of gefitinib, 30 min before tria-zolam administration, was used to obtain significant loading of thebody with gefitinib, but the concentration of gefitinib in intestinalepithelial cells may already have started to decrease after 30 min,resulting in levels too low to exert a marked stimulatory effect in theintestine.
Thus far, there are no recognized examples known of clinicallyrelevant drug-drug interactions that can be attributed to the directstimulation of CYP3A-mediated metabolism (as opposed to inductionof CYP3A expression, which is common). Clearly, because it typi-cally concerns only a specific combination of drugs, stimulation ofdrug metabolism by CYP3A4 is much more difficult to predict thaninhibition. We note that gefitinib does not only stimulate theCYP3A4-mediated metabolism of triazolam but also that of midazo-lam and the anticancer drug irinotecan in vitro (Fujita et al., 2005; Liet al., 2007). A recent clinical study also reported a drug-drug inter-action between gefitinib and sorafenib (Adjei et al., 2007). When bothdrugs were given simultaneously, the AUC of gefitinib was reduced(38%) compared with when the drug was given alone, suggesting that
sorafenib might stimulate gefitinib metabolism. In contrast, gefitinibhad no effect on sorafenib pharmacokinetics. It is clear that becauseanticancer drugs in general have narrow therapeutic windows and areoften combined, direct stimulation of CYP3A-mediated metabolismcould be of clinical relevance.
CYP3A4 is undoubtedly one of the most important players in manydrug-drug interactions. It is unfortunate that the prediction of in vivodrug-drug interactions based on in vitro data is not always straight-forward (Lin, 2000; Wienkers and Heath, 2005; Obach, 2009). Inaddition, as a result of species differences in CYP3A, animal studiesare not always representative of the human situation. The fact that forsome drugs the intestine and not the liver is the most important organfor CYP3A-mediated drug-drug interactions can further complicatepredictions. Therefore, humanized mouse models for CYP3A4, as wecharacterized and used here, could be of great value to better under-stand and predict drug-drug interactions, especially at a relativelyearly stage in drug development. Nonetheless, one should alwaysrealize that humanized mice are still not humans and that there willalways be limitations in using these models for quantitative predic-tions of drug exposure in humans.
Acknowledgments. We thank Dilek Iusuf, Jurjen Lagas, Evita vande Steeg, and Seng Chuan Tang for critical reading of the manuscript.
References
Adjei AA, Molina JR, Mandrekar SJ, Marks R, Reid JR, Croghan G, Hanson LJ, Jett JR, Xia C,Lathia C, and Simantov R (2007) Phase I trial of sorafenib in combination with gefitinib inpatients with refractory or recurrent non-small cell lung cancer. Clin Cancer Res 13:2684–2691.
Andersson T, Miners JO, Veronese ME, and Birkett DJ (1994) Diazepam metabolism by humanliver microsomes is mediated by both S-mephenytoin hydroxylase and CYP3A isoforms. Br JClin Pharmacol 38:131–137.
Bardelmeijer HA, Ouwehand M, Buckle T, Huisman MT, Schellens JH, Beijnen JH, and vanTellingen O (2002) Low systemic exposure of oral docetaxel in mice resulting from extensivefirst-pass metabolism is boosted by ritonavir. Cancer Res 62:6158–6164.
Benet LZ and Cummins CL (2001) The drug efflux-metabolism alliance: biochemical aspects.Adv Drug Deliv Rev 50 (Suppl 1):S3–S11.
Doherty MM and Charman WN (2002) The mucosa of the small intestine: how clinically relevantas an organ of drug metabolism? Clin Pharmacokinet 41:235–253.
Dresser GK and Bailey DG (2003) The effects of fruit juices on drug disposition: a new modelfor drug interactions. Eur J Clin Invest 33 (Suppl 2):10–16.
Dresser GK, Spence JD, and Bailey DG (2000) Pharmacokinetic-pharmacodynamic conse-quences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet38:41–57.
Ekroos M and Sjogren T (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4.Proc Natl Acad Sci U S A 103:13682–13687.
Fujita K, Ando Y, Narabayashi M, Miya T, Nagashima F, Yamamoto W, Kodama K, Araki K,Endo H, and Sasaki Y (2005) Gefitinib (Iressa) inhibits the CYP3A4-mediated formation of7-ethyl-10-(4-amino-1-piperidino)carbonyloxycamptothecin but activates that of 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino]carbonyloxycamptothecin from irinotecan. DrugMetab Dispos 33:1785–1790.
Guengerich FP (1999) Cytochrome P-450 3A4: regulation and role in drug metabolism. AnnuRev Pharmacol Toxicol 39:1–17.
Houston JB and Galetin A (2005) Modelling atypical CYP3A4 kinetics: principles and pragma-tism. Arch Biochem Biophys 433:351–360.
Hutzler JM and Tracy TS (2002) Atypical kinetic profiles in drug metabolism reactions. DrugMetab Dispos 30:355–362.
Kronbach T, Mathys D, Umeno M, Gonzalez FJ, and Meyer UA (1989) Oxidation of midazolamand triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol 36:89–96.
Kumar GN, Dykstra J, Roberts EM, Jayanti VK, Hickman D, Uchic J, Yao Y, Surber B, ThomasS, and Granneman GR (1999) Potent inhibition of the cytochrome P-450 3A-mediated humanliver microsomal metabolism of a novel HIV protease inhibitor by ritonavir: a positivedrug-drug interaction. Drug Metab Dispos 27:902–908.
Li J, Zhao M, He P, Hidalgo M, and Baker SD (2007) Differential metabolism of gefitinib anderlotinib by human cytochrome P450 enzymes. Clin Cancer Res 13:3731–3737.
Lin JH (2000) Sense and nonsense in the prediction of drug-drug interactions. Curr Drug Metab1:305–331.
Lin JH, Chiba M, and Baillie TA (1999) Is the role of the small intestine in first-pass metabolismoveremphasized? Pharmacol Rev 51:135–158.
McKillop D, McCormick AD, Millar A, Miles GS, Phillips PJ, and Hutchison M (2005)Cytochrome P450-dependent metabolism of gefitinib. Xenobiotica 35:39–50.
Obach RS (2009) Predicting drug-drug interactions from in vitro drug metabolism data: chal-lenges and recent advances. Curr Opin Drug Discov Devel 12:81–89.
Paine MF and Oberlies NH (2007) Clinical relevance of the small intestine as an organ of drugelimination: drug-fruit juice interactions. Expert Opin Drug Metab Toxicol 3:67–80.
Perloff MD, von Moltke LL, Cotreau MM, and Greenblatt DJ (1999) Unchanged cytochromeP450 3A (CYP3A) expression and metabolism of midazolam, triazolam, and dexamethasonein mdr(�/�) mouse liver microsomes. Biochem Pharmacol 57:1227–1232.
Perloff MD, von Moltke LL, Court MH, Kotegawa T, Shader RI, and Greenblatt DJ (2000)
2312 VAN WATERSCHOOT ET AL.
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from

Midazolam and triazolam biotransformation in mouse and human liver microsomes: relativecontribution of CYP3A and CYP2C isoforms. J Pharmacol Exp Ther 292:618–628.
Tang W and Stearns RA (2001) Heterotropic cooperativity of cytochrome P450 3A4 andpotential drug-drug interactions. Curr Drug Metab 2:185–198.
Tang W, Stearns RA, Kwei GY, Iliff SA, Miller RR, Egan MA, Yu NX, Dean DC, Kumar S,Shou M, et al. (1999) Interaction of diclofenac and quinidine in monkeys: stimulation ofdiclofenac metabolism. J Pharmacol Exp Ther 291:1068–1074.
Thummel KE (2007) Gut instincts: CYP3A4 and intestinal drug metabolism. J Clin Invest117:3173–3176.
Thummel KE and Wilkinson GR (1998) In vitro and in vivo drug interactions involving humanCYP3A. Annu Rev Pharmacol Toxicol 38:389–430.
Ueng YF, Kuwabara T, Chun YJ, and Guengerich FP (1997) Cooperativity in oxidationscatalyzed by cytochrome P450 3A4. Biochemistry 36:370–381.
van Herwaarden AE, van Waterschoot RA, and Schinkel AH (2009) How important is intestinalcytochrome P450 3A metabolism? Trends Pharmacol Sci 30:223–227.
van Herwaarden AE, Wagenaar E, van der Kruijssen CM, van Waterschoot RA, Smit JW, SongJY, van der Valk MA, van Tellingen O, van der Hoorn JW, Rosing H, et al. (2007) Knockoutof cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism.J Clin Invest 117:3583–3592.
van Waterschoot RA, Rooswinkel RW, Wagenaar E, van der Kruijssen CM, van HerwaardenAE, and Schinkel AH (2009) Intestinal cytochrome P450 3A plays an important role in theregulation of detoxifying systems in the liver. FASEB J 23:224–231.
van Waterschoot RA, van Herwaarden AE, Lagas JS, Sparidans RW, Wagenaar E, van derKruijssen CM, Goldstein JA, Zeldin DC, Beijnen JH, and Schinkel AH (2008) Midazolammetabolism in cytochrome P450 3A knockout mice can be attributed to up-regulated CYP2Cenzymes. Mol Pharmacol 73:1029–1036.
von Moltke LL, Granda BW, Grassi JM, Perloff MD, Vishnuvardhan D, and Greenblatt DJ(2004) Interaction of triazolam and ketoconazole in P-glycoprotein-deficient mice. DrugMetab Dispos 32:800–804.
von Moltke LL, Greenblatt DJ, Harmatz JS, Duan SX, Harrel LM, Cotreau-Bibbo MM, PritchardGA, Wright CE, and Shader RI (1996) Triazolam biotransformation by human liver micro-somes in vitro: effects of metabolic inhibitors and clinical confirmation of a predictedinteraction with ketoconazole. J Pharmacol Exp Ther 276:370–379.
Wang RW, Newton DJ, Liu N, Atkins WM, and Lu AY (2000) Human cytochrome P-450 3A4:in vitro drug-drug interaction patterns are substrate-dependent. Drug Metab Dispos 28:360–366.
Wienkers LC and Heath TG (2005) Predicting in vivo drug interactions from in vitro drugdiscovery data. Nat Rev Drug Discov 4:825–833.
Address correspondence to: Alfred H. Schinkel, Division of Molecular Biol-ogy, The Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam,The Netherlands. E-mail: [email protected]
2313INHIBITION AND STIMULATION OF INTESTINAL AND HEPATIC CYP3A
at ASPE
T Journals on Septem
ber 19, 2018dm
d.aspetjournals.orgD
ownloaded from





























