Embed Size (px)
Citation preview

MOLECULAR AND CELLULAR BIOLOGY, Apr. 2007, p. 3176–3186 Vol. 27, No. 80270-7306/07/$08.00�0 doi:10.1128/MCB.01652-06Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Disruption of the FEN-1/PCNA Interaction Results in DNAReplication Defects, Pulmonary Hypoplasia, Pancytopenia,
and Newborn Lethality in Mice�
Li Zheng,1 Huifang Dai,1 Junzhuan Qiu,1 Qin Huang,2 and Binghui Shen1*Departments of Radiation Biology1 and Pathology,2 City of Hope National Medical Center and
Beckman Research Institute, Duarte, California 91010
Received 4 September 2006/Returned for modification 3 October 2006/Accepted 22 January 2007
The interaction between flap endonuclease 1 (FEN-1) and proliferation cell nuclear antigen (PCNA) iscritical for faithful and efficient Okazaki fragment maturation. In a living cell, this interaction is probablyimportant for PCNA to load FEN-1 to the replication fork, to coordinate the sequential functions of FEN-1 andother enzymes, and to stimulate its enzyme activity. The FEN-1/PCNA interaction is mediated by the motif337QGRLDDFFK345 of FEN-1, such that an F343AF344A (FFAA) mutant cannot bind to PCNA but retains itsnuclease activities. To determine the physiological roles of the FEN-1/PCNA interaction in a mammaliansystem, we knocked the FFAA Fen1 mutation into the Fen1 gene locus of mice. FFAA/FFAA mouse embryofibroblasts underwent DNA replication and division at a slower pace, and FFAA/FFAA mutant embryosdisplayed significant defects in growth and development, particularly in the lung and blood systems. Allnewborn FFAA mutant pups died at birth, likely due to pulmonary hypoplasia and pancytopenia. Collectively,our data demonstrate the importance of the FEN-1/PCNA complex in DNA replication and in the embryonicdevelopment of mice.
Efficient and faithful Okazaki fragment maturation requireseffective recruitment of replication proteins involved in thisprocess and the coordination of enzyme-catalyzed reactions,such as DNA synthesis, RNA/DNA cleavage, and DNA liga-tion during nick translation of RNA primer processing. Ineukaryotic cells, this process requires the structure-specific flapendonuclease 1 (FEN-1). During replication of the lagging-strand DNA, the synthesis of an Okazaki fragment displacesthe RNA primer portion of the downstream Okazaki fragment.The resulting RNA primer flap structure is removed by FEN-1and other nucleases. Two models have been proposed to elu-cidate this process (2, 3, 37). In the first model, displacement ofthe Okazaki fragment generates a short flap structure of 1 to 10nucleotides (nt), which is recognized and efficiently cleaved byFEN-1 (2). In the second model, a long flap of approximately30 nt is generated during lagging-strand DNA synthesis. Thesingle-stranded DNA binding protein RPA binds to the longflap strand, preventing the cleavage of the RNA primer flap byFEN-1 (3). DNA2 nuclease can, instead, bind to RPA andremove the major portion of the long RNA primer flap, leavinga short flap of approximately 8 nt. This short flap is thencleaved by FEN-1 (3).
Assembly of DNA replication proteins at discrete replica-tion sites, called replication factories, has been postulated to becritical in DNA replication (18, 27, 28). The interaction ofFEN-1 and proliferation cell nuclear antigen (FEN-1/PCNA)enables FEN-1 to associate with the replication machinery forefficient RNA primer removal (27, 48). In agreement with this
suggestion, binding of PCNA significantly enhances FEN-1interaction with DNA flap substrates and strongly stimulatesthe FEN-1 cleavage activity of flap and nick substrates in vitro(44, 48). Biochemical characterization revealed that the337QGRLDDFFK345 motif in FEN-1 proteins from humansand other species is necessary for the high-affinity interactionwith PCNA (15, 47). Analysis using alanine scanning mutagen-esis further identified the fact that residues L340, D342, F343,and F344 are essential for the interaction in vitro (12). Re-placement of residues F343 and F344 with alanine residuescompletely eliminates the physical interaction in vitro (12, 15,16). Three-dimensional structure analysis of the FEN-1/PCNAcomplex revealed that other amino acid residues outside of the“QGRLDDFFK” motif also contribute to the protein-proteininteraction and activity stimulation (9, 38). This is in agree-ment with our previous study showing that deletion of the“LDDFF” motif from human FEN-1 abolishes the protein-protein interaction but does not affect the PCNA stimulationof FEN-1 nuclease activities (12).
Even though redundant nuclease activities are involved inthis process, a deficiency in the FEN-1/PCNA interactionchanges the dynamics of the FEN-1-mediated RNA primerremoval process. It affects the coordination of various reac-tions, leading to a delay in Okazaki fragment maturation, pro-gression of DNA replication, and cell proliferation. However,both Gary et al. and Jin et al. found that the disruption of theFEN-1/PCNA interaction had little effect on the growth ofSaccharomyces cerevisiae mutant cells (16, 20).
It is unclear whether a deficiency of the FEN-1/PCNA in-teraction will cause DNA replication defects in mammaliancells and subsequently lead to perturbations in the growth anddevelopment of mammals. The in vivo significance of FEN-1 inDNA replication in mammalian cells is different from that inyeast. A deletion of RAD27, the yeast FEN-1 homolog, does
* Corresponding author. Mailing address: City of Hope NationalMedical Center, Department of Molecular Biology, 1500 East DuarteRd., Duarte, CA 91010. Phone: (626) 301-8879. Fax: (626) 301-8280.E-mail: [email protected].
� Published ahead of print on 5 February 2007.
3176
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.

not result in complete lethality, whereas in mice, knockout ofFen1 causes cellular death and early embryonic lethality (22,23, 35, 41). Thus, the impact due to a disruption of the FEN-1/PCNA interaction on the growth and development of mam-mals is predicted to be more severe than that observed foryeast cells. To test this hypothesis, we used a gene targetingapproach to knock F343AF344A (FFAA) point mutations intothe Fen1 alleles of the mouse genome, which specifically elim-inates the PCNA binding activity of FEN-1. All newbornFFAA mutant pups died at birth, likely due to pulmonaryhypoplasia and pancytopenia. In this study, we outline themolecular events that explain how this point mutation causessuch a severe phenotype in mice.
MATERIALS AND METHODS
Generation of mice homozygous for the FFAA point mutation in Fen1 alleles.A DNA fragment encoding mouse FEN-1 was subcloned into the gene targetingvector PKO scrambler NTK (Invitrogen, Carlsbad, CA). The FFAA mutantmouse Fen1 gene in the PKO scrambler NTK vector was generated by site-directed mutagenesis (Stratagene, La Jolla, CA) using primers mFFAA-F andmFFAA-R (Table 1). The knock-in vector encoding the FFAA FEN-1 mutantwas electroporated into embryonic stem (ES) cells of the mouse 129S1 geneticbackground. FFAA ES cells were selected by neomycin marker and confirmed bySouthern blotting analysis using probes 1 and 2. DNA sequences of probes 1 and2 corresponded to the sequences from 19185 to 19673 and from 5155 to 5640,respectively, of bacterial artificial chromosome (BAC) RP22-325J22, chromo-some 19 of Mus musculus (129S1). Probes 1 and 2 were prepared by PCRamplification using primers described in Table 1. The neomycin selectionmarker, which is flanked with LoxP sequences, was removed by the transientexpression of Cre-recombinase in the mutant ES cells (13). Male chimeric micegenerated via FFAA ES cells (neo�) were crossed with female wild-type (wt)mice (129S1 genetic background) to transmit the mutation through the germ lineto produce heterozygous mice of 129S1 genetic background. By crossing theheterozygous mice, we obtained homozygous FFAA/FFAA mice, which alwaysdie at birth.
To confirm the correct genetic manipulation of the ES cells and mice, genomicDNA was extracted and purified from the ES cells or mouse tails and digestedwith HindIII or XhoI and resolved by agarose gel electrophoresis. The separatedDNA was then transferred to nitrocellulose membranes. Southern hybridizationwas performed with HindIII- and XhoI-digested genomic DNA, using probes 1and 2, respectively, to visualize exogenous DNA insertion and to estimate thesize of recombined DNA fragments. To genotype mice, Fen1 alleles were am-plified by PCR using genomic DNA isolated from mouse tails as the template
and primers mFFAA-1 and mFFAA-4 (Table 1). The PCR-amplified DNAfragment was then purified using a gel extraction DNA purification kit(QIAGEN, Valencia, CA) and subjected to sequencing to confirm the incorpo-ration of the mutation (City of Hope DNA sequencing facility, Duarte, CA).
FEN-1/PCNA binding assays. The interaction between recombinant FEN-1and PCNA was assayed following a published protocol (12). Briefly, His6-taggedFEN-1 or nontagged PCNA was expressed in Escherichia coli cells individually.The cell extract containing His6-tagged FEN-1 was mixed with the cell extract ofnontagged PCNA in a binding buffer containing 50 mM Tris-Cl (pH 7.5) and 150mM NaCl. The mixture was then incubated with Ni2� chelating agarose beads at4°C for 2 h. Agarose beads were then extensively washed with binding buffer andsuspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and boiled for 10 min. Proteins were resolved by 4 to 15%SDS-PAGE (Bio-Rad, Hercules, CA) and stained with Coomassie blue R250 forvisualization.
The interaction between FEN-1 and PCNA in mouse cellular extracts wasassayed by coimmunoprecipitation. Whole-cell extract was prepared by incuba-tion of cells in lysis buffer (20 mM HEPES [pH 7.5], 100 mM NaCl, 0.5 mMEDTA, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride [PMSF], and a pro-tease inhibitor cocktail [Roche, Indianapolis, IN]) on ice for 1 h. Whole-cellextracts were then incubated with monoclonal FEN-1 (mFEN-1) antibody boundto protein G agarose in binding buffer (20 mM HEPES [pH 7.5], 500 mM NaCl,0.5 mM EDTA, 0.5% NP-40, 1 mM PMSF, and a protease inhibitor cocktail) at4°C for 2 h. Agarose beads were then washed with binding buffer. Proteins boundto the agarose beads were eluted by boiling in SDS-PAGE loading buffer for 10min and resolved using 4 to 15% SDS-PAGE. PCNA was detected by Westernblotting using an antibody against PCNA.
Immunofluorescence analysis. The subnuclear localization sites of FEN-1,PCNA, and bromodeoxyuridine (BrdU) incorporation were determined by indi-rect immunofluorescence analysis as previously described (27). Cell cycles weresynchronized at the G1/S boundary by serum starvation for 48 h, followed bytreatment with 400 �M mimosine for 12 h (32). Cells were washed with phos-phate-buffered saline (PBS) and released into the S phase by incubation withfresh Dulbecco’s minimal essential medium (DMEM) containing 10% fetal bo-vine serum. After incubation for 4 h, typically more than 60% of cells were in Sphase. Flow cytometry was performed at 0, 2, 4, 6, and 8 h post-mimosinetreatment to monitor and confirm cell cycle progression. The cells at the G1/Sboundary or in S phase were fixed in methanol at �20°C for 30 min. To detectFEN-1 and PCNA, fixed cells were incubated with monoclonal anti-FEN-1 andrabbit polyclonal anti-PCNA antibodies (Santa Cruz Biotechnology, Inc., SantaCruz, CA). FEN-1 and PCNA were then detected with rhodamine-conjugateddonkey anti-mouse immunoglobulin G (IgG) and fluorescein isothiocyanate(FITC)-conjugated donkey anti-rabbit IgG (Invitrogen, Carlsbad, CA), respec-tively. The replication foci were detected using BrdU staining (26). Cells in theG1/S or S phase were incubated with DMEM containing 50 �M BrdU for 1 h.After FEN-1 or PCNA was stained, the slide was incubated with ice-cold meth-
TABLE 1. Oligonucleotide sequences and their applications in this study
Name Oligonucleotide sequenceb Application
mFFAA-F 5�-GGACGCCTCGATGATGCCGCCAAGGTGACAGGCTCAC-3� MutagenesismFFAA-R 5�-GTGAGCCTGTCACCTTGGCGGCATCATCGAGGCGTCC-3� MutagenesismFEN-86 5�-CAGACAATGTACCATCTTGTCACAGCTCTTACCCTTGG-3� Amplification for probe 1mFEN-106 5�-GTGCTGGGATTCACAGGAATATACAACTCCACCC-3� Amplification for probe 1mFEN-81 5�-CACAGGCCTACCACTTTCAGCTTAATCCTACGTTCCC-3� Amplification for probe 2mFEN-82 5�-GGGGTGACAAGGAGGCAGTCCTGTGGCTGG-3� Amplification for probe 2mFEN-125 5�-CCAGAACAGCTTCTGATTGTCAGGAG-3� PCR genotypingmFEN-166 5�-GGCTCCAGGAAGAGCTGCTGGGCTTCCTTGTGG-3� PCR genotypingmFFAA-1 5�-GGCTCCAGGAAGAGCTGCTGGGCTTCCTTGTGG-3� PCR amplificationmFFAA-4 5�-TGCTTTCTTCTTAGCAGGCCCCT-3� PCR amplificationmFFAA-3 5�-TTCCAGAGAACTGGCTCCAC-3� SequencingOL1 5�-GTTAAGATAGGTCTGCTTGGGATGTCAAGCAGTCCTAACTGGAAAT
CTAGCTCTGTGGAGTTGAGGCAGAGTCCTTAAGC-3�RNA/DNA substrates
OL2 5�-GCTTAAGGACTCTGCCTCAA-3� RNA/DNA substratesOL3 5�-AGTTAGGACTGCTTGACATCCCAAGCAGACCTATCTTAAC-3� RNA/DNA substratesOL4a 5�-aGTTAGGACTGCTTGACATCCCAAGCAGACCTATCTTAAC-3� RNA/DNA substratesOL5a 5�-agtTAGGACTGCTTGACATCCCAAGCAGACCTATCTTAAC-3� RNA/DNA substratesOL6 5�-CCGGTAGTTAGGACTGCTTGACATCCCAAGCAGACCTATCTTAAC-3� RNA/DNA substrates
a RNA-DNA hybrid oligonucleotides. The RNA portion is in lowercase, and the DNA portion is in uppercase.b Underlined bases are two codons in the Fen1 gene for replacement of phenylalanines with alanines.
VOL. 27, 2007 FEN-1/PCNA INTERACTION IN EMBRYONIC DEVELOPMENT 3177
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.

anol for 5 min. DNA was denatured by treatment with 2 M HCl for 1 h followedby neutralization with 0.1 M borate buffer (pH 8.5) for 30 min. Cells were thenincubated with FITC-conjugated anti-BrdU antibody (BD Biosciences, FranklinLakes, NJ). In all cases, nuclei were stained with 4�,6�-diamidino-2-phenylindoleas a control. All slides were analyzed using a Zeiss LSM510 confocal microscope(Carl Zeiss, Thornwood, NY). Images were processed using Adobe Photoshop7.0 (Adobe, San Jose, CA).
Nick translation assay. The reaction mixtures of gap filling, RNA/DNAprimer removal, and DNA ligation in Okazaki fragment maturation were assayedby nick translation reaction with four different gap substrates, following a mod-ified version of published protocols (33, 46). Briefly, the 3� end of a specificdownstream primer strand (OL3, OL4, OL5, or OL6; see Table 1) was labeledwith 32P. The labeled primers were annealed with oligonucleotides OL1 and OL2(Table 1) to make different substrates. Nuclear extracts from wt or FFAA/FFAAcells were prepared as previously described (49). To deplete FEN-1, wt nuclearextracts were incubated with monoclonal anti-FEN-1 antibody-conjugated pro-tein G-agarose (Santa Cruz Biotechnology Inc., CA) for 3 h. In control experi-ments, nuclear extracts from wt or FFAA/FFAA cells were incubated withnonspecific mouse IgG-conjugated protein G-agarose (Santa Cruz Biotechnol-ogy). Western blotting confirmed that FEN-1 was depleted by anti-FEN-1 aga-rose beads but not by nonspecific mouse IgG-agarose beads. After FEN-1 de-pletion, wt, FFAA, and FEN-1-depleted nuclear extracts were incubated withsubstrates in 20 mM HEPES (pH 7.5), 70 mM KCl, 5 mM MgCl2, 1 mMdithiothreitol, 1 mM ATP, and 200 �M of each deoxynucleotide triphosphate at37°C for 10, 20, and 40 min. Reactions were terminated by the addition of anequal volume of formamide loading buffer and resolved by denaturing PAGE(15%). The gel was visualized via autoradiography. To accurately quantify theamount of ligation products, bands of ligation products (80 nt) resolved indenaturing PAGE were cut and homogenized in PBS buffer, and the radioactivityof each band was determined using a liquid scintillation counter. A standardcurve (radioactivity versus concentration) was generated using the 32P-labeledDNA oligonucleotide of different concentrations resolved on the same denatur-ing polyacrylamide gel. The amount of ligation product was calculated based onthe radioactivity of the ligation product and the standard curve.
DNA replication assay. DNA replication efficiency in mouse embryo fibro-blasts (MEFs) was determined by monitoring the rate of thymidine incorpora-tion, as previously described (26). Briefly, MEFs were seeded onto a 6-cm dishin DMEM for 12 h. [3H]thymidine was added to a final concentration of1 �Ci/ml. Cells were incubated in [3H]thymidine containing DMEM for a specifictime period and washed with ice-cold PBS buffer. DNA was precipitated bytreating cells with 10% ice-cold trichloroacetic acid and 10 mM thymidine at 4°Cfor 15 min. After extensive washes with PBS buffer, DNA was solubilized in 0.5M NaOH. The amount of radioactivity in the sample was measured using a liquidscintillation counter.
Cell proliferation assay. To determine the cell proliferation rate, 2 � 105 cellsfrom wt, wt/FFAA, or FFAA/FFAA mice were seeded onto 6-cm dishes. Cellswere grown in DMEM at 37°C. Cell numbers were counted every day for 7 days.The cell proliferation rate is expressed as the increase in cell number in a giventime period.
Histopathology. Whole embryos and dissected tissues were fixed in 10% for-malin and stained with hematoxylin and eosin (H&E) stain. All examinationswere conducted in a double-blinded fashion.
RESULTS
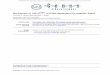
Generation of the FFAA FEN-1 mutation mouse model. Tostudy the in vivo requirement of the FEN-1/PCNA interaction, weused a gene targeting approach to replace a wt Fen1 allele with anallele carrying the F343AF344A (FFAA) Fen1 mutation (Fig.1A), which was previously shown to specifically abolish FEN-1binding to PCNA in vitro (15). ES cells carrying the FFAA mu-tant allele were selected for neomycin resistance, and the geno-types were confirmed by Southern blotting analysis (Fig. 1B). Theneomycin (neo) gene, which is flanked by Lox P sequences (Fig.1A), was then removed by transient expression of Cre-recombi-nase in the mutant ES cells (13). The remaining copy of the LoxP sequence was located in the intronic sequence 200 bp upstreamof the ATG start codon of the Fen1 open reading frame. Thec11orf10 gene (1810006K21Rik; RIKEN), which is located imme-
diately upstream of Fen1 in the reverse orientation (1), was notaffected by the insertion of the Lox P sequence 2.33 kb away fromthe c11orf10 gene. Male chimeric mice generated with the FFAAES cells (neo�) were crossed with wt female mice (129S1 geneticbackground) to transmit the mutation through the germ line andproduce mice heterozygous for the FFAA mutation (wt/FFAA)in a 129S1 genetic background. The wt/FFAA mice were indis-tinguishable from wt/wt mice by their phenotype. The genotypesof these mice were determined by PCR and direct DNA sequenc-ing analysis (Fig. 1C and D).
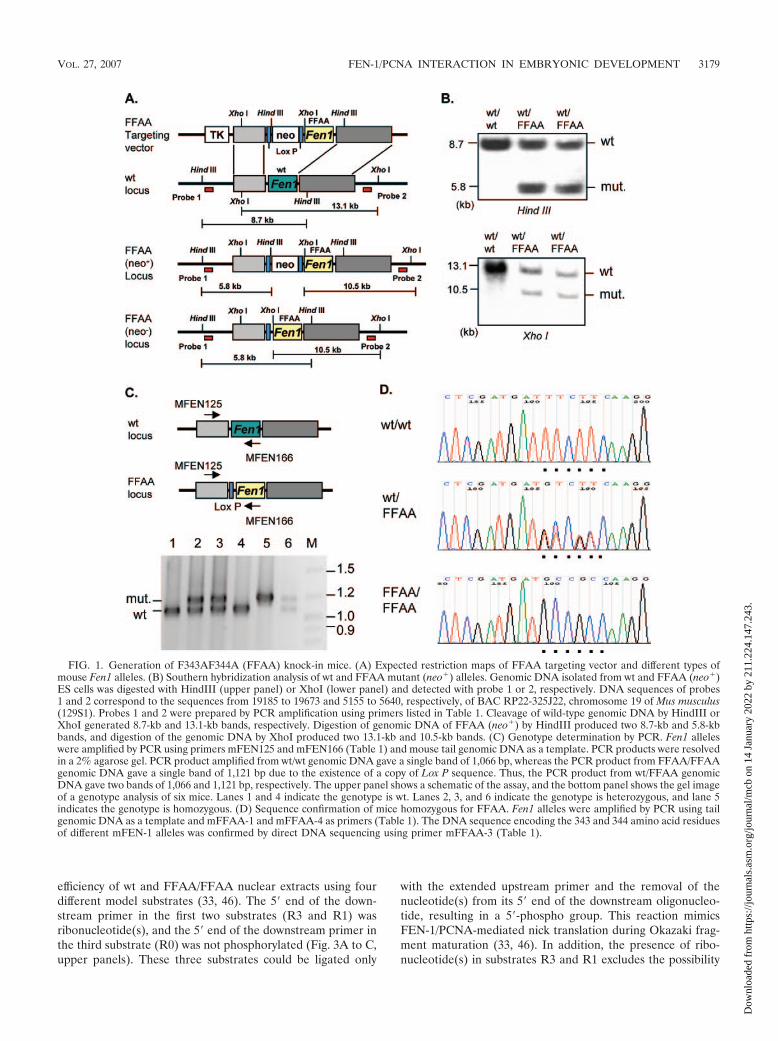
FFAA mutation specifically disrupts the FEN-1/PCNA phys-ical interaction and impairs the recruitment of FEN-1 to DNAreplication foci. The FFAA mutation was previously shown todisrupt human FEN-1/PCNA interaction in vitro (12, 15, 16).Likewise, the mouse FFAA FEN-1 mutation also eliminatedits PCNA binding activity in vitro (Fig. 2A). To further deter-mine whether the FFAA mutation abolished the FEN-1/PCNAinteraction, we crossbred wt/FFAA mice and establishedMEFs. The expression levels of FEN-1 and PCNA fromFFAA/FFAA MEFs were similar to those from wt cells (Fig.2B, lanes 1 and 2). The FEN-1/PCNA interaction was charac-terized by coimmunoprecipitation. We found that PCNA wascoprecipitated with wt FEN-1, whereas only trace amounts ofPCNA were pulled down with the FFAA FEN-1 mutant (Fig.2B, lanes 3 and 4), suggesting that the FFAA mutation impairsthe FEN-1/PCNA interaction.
Disruption of the FEN-1/PCNA interaction may impair therecruitment of FEN-1 to the DNA replication site. To test thishypothesis, we analyzed the subnuclear localization sites ofFEN-1, PCNA, and BrdU incorporation, which represent theDNA replication foci (28) in wt and FFAA MEFs. We firstexamined the localization of FEN-1 and PCNA in cells at theG1/S boundary and in cells in S phase. In wt and FFAA cells atthe G1/S boundary, no FEN-1 or PCNA foci were observedand no colocalization of FEN-1 with PCNA was found (Fig.2C). Interestingly, in both wt and FFAA cells at the G1/Sboundary, FEN-1 was typically enriched in two to three focalpoints (Fig. 2C), which were revealed to be nucleoli (L. Qianand B. Shen, unpublished data). In wt cells in S phase, FEN-1colocalized with PCNA foci (Fig. 2C). However, in FFAA cellsin S phase, most FFAA FEN-1 proteins did not colocalize withPCNA foci (Fig. 2C). Because PCNA foci are indicative ofDNA replication sites (5), our results suggested that FFAAFEN-1 mutant proteins failed to be recruited to DNA replica-tion foci. To validate this conclusion, we pulse-labeled wt andFFAA cells with BrdU and determined if FFAA FEN-1 mu-tant proteins were excluded from BrdU incorporation sites.We found that FEN-1 colocalized with BrdU incorporationsites in wt cells, whereas most of the mutant FEN-1 proteinsdid not colocalize with BrdU incorporation sites in FFAA cellsin S phase (Fig. 2D). Taken together, our data suggest thatthe localization of FEN-1 to replication foci is mediated by theinteraction between FEN-1 and PCNA. Disruption of theFEN-1/PCNA interaction impairs the recruitment of FEN-1 tothe DNA replication sites.
Disruption of the FEN-1/PCNA interaction affects the effi-ciency of DNA replication and cell proliferation. To determinewhether disruption of FEN-1/PCNA interaction caused anydefects in the function of FEN-1 in Okazaki fragment matu-ration in vitro, we assayed the Okazaki fragment maturation
3178 ZHENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.
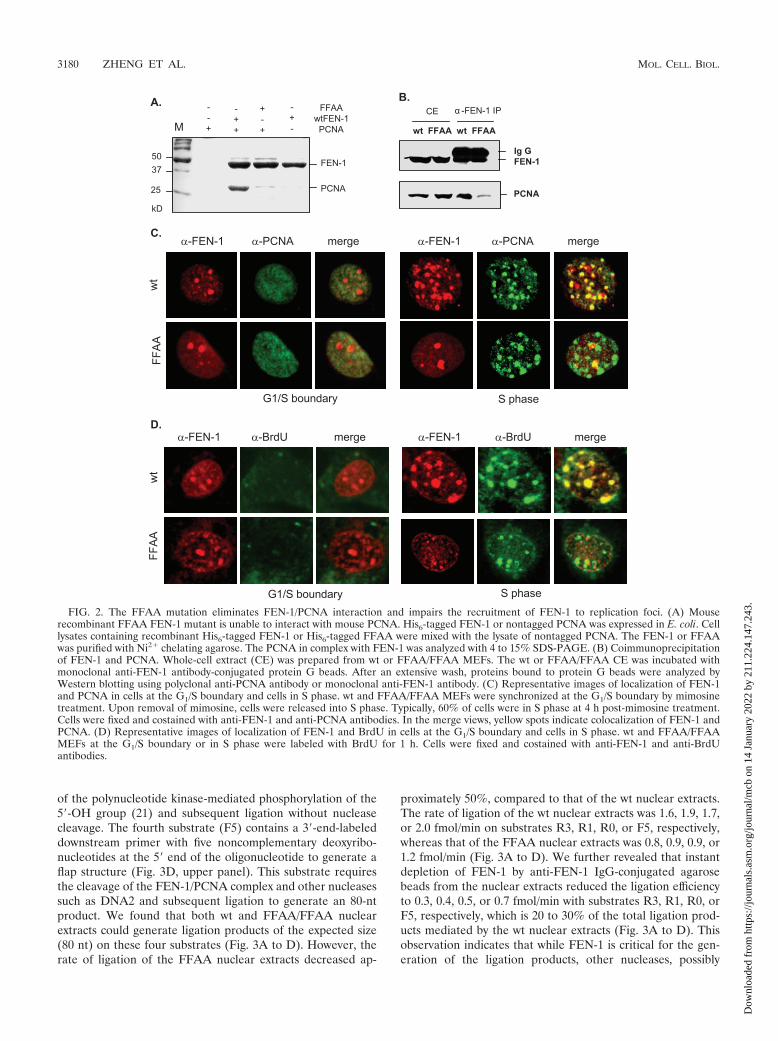
efficiency of wt and FFAA/FFAA nuclear extracts using fourdifferent model substrates (33, 46). The 5� end of the down-stream primer in the first two substrates (R3 and R1) wasribonucleotide(s), and the 5� end of the downstream primer inthe third substrate (R0) was not phosphorylated (Fig. 3A to C,upper panels). These three substrates could be ligated only
with the extended upstream primer and the removal of thenucleotide(s) from its 5� end of the downstream oligonucleo-tide, resulting in a 5�-phospho group. This reaction mimicsFEN-1/PCNA-mediated nick translation during Okazaki frag-ment maturation (33, 46). In addition, the presence of ribo-nucleotide(s) in substrates R3 and R1 excludes the possibility
FIG. 1. Generation of F343AF344A (FFAA) knock-in mice. (A) Expected restriction maps of FFAA targeting vector and different types ofmouse Fen1 alleles. (B) Southern hybridization analysis of wt and FFAA mutant (neo�) alleles. Genomic DNA isolated from wt and FFAA (neo�)ES cells was digested with HindIII (upper panel) or XhoI (lower panel) and detected with probe 1 or 2, respectively. DNA sequences of probes1 and 2 correspond to the sequences from 19185 to 19673 and 5155 to 5640, respectively, of BAC RP22-325J22, chromosome 19 of Mus musculus(129S1). Probes 1 and 2 were prepared by PCR amplification using primers listed in Table 1. Cleavage of wild-type genomic DNA by HindIII orXhoI generated 8.7-kb and 13.1-kb bands, respectively. Digestion of genomic DNA of FFAA (neo�) by HindIII produced two 8.7-kb and 5.8-kbbands, and digestion of the genomic DNA by XhoI produced two 13.1-kb and 10.5-kb bands. (C) Genotype determination by PCR. Fen1 alleleswere amplified by PCR using primers mFEN125 and mFEN166 (Table 1) and mouse tail genomic DNA as a template. PCR products were resolvedin a 2% agarose gel. PCR product amplified from wt/wt genomic DNA gave a single band of 1,066 bp, whereas the PCR product from FFAA/FFAAgenomic DNA gave a single band of 1,121 bp due to the existence of a copy of Lox P sequence. Thus, the PCR product from wt/FFAA genomicDNA gave two bands of 1,066 and 1,121 bp, respectively. The upper panel shows a schematic of the assay, and the bottom panel shows the gel imageof a genotype analysis of six mice. Lanes 1 and 4 indicate the genotype is wt. Lanes 2, 3, and 6 indicate the genotype is heterozygous, and lane 5indicates the genotype is homozygous. (D) Sequence confirmation of mice homozygous for FFAA. Fen1 alleles were amplified by PCR using tailgenomic DNA as a template and mFFAA-1 and mFFAA-4 as primers (Table 1). The DNA sequence encoding the 343 and 344 amino acid residuesof different mFEN-1 alleles was confirmed by direct DNA sequencing using primer mFFAA-3 (Table 1).
VOL. 27, 2007 FEN-1/PCNA INTERACTION IN EMBRYONIC DEVELOPMENT 3179
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.
of the polynucleotide kinase-mediated phosphorylation of the5�-OH group (21) and subsequent ligation without nucleasecleavage. The fourth substrate (F5) contains a 3�-end-labeleddownstream primer with five noncomplementary deoxyribo-nucleotides at the 5� end of the oligonucleotide to generate aflap structure (Fig. 3D, upper panel). This substrate requiresthe cleavage of the FEN-1/PCNA complex and other nucleasessuch as DNA2 and subsequent ligation to generate an 80-ntproduct. We found that both wt and FFAA/FFAA nuclearextracts could generate ligation products of the expected size(80 nt) on these four substrates (Fig. 3A to D). However, therate of ligation of the FFAA nuclear extracts decreased ap-
proximately 50%, compared to that of the wt nuclear extracts.The rate of ligation of the wt nuclear extracts was 1.6, 1.9, 1.7,or 2.0 fmol/min on substrates R3, R1, R0, or F5, respectively,whereas that of the FFAA nuclear extracts was 0.8, 0.9, 0.9, or1.2 fmol/min (Fig. 3A to D). We further revealed that instantdepletion of FEN-1 by anti-FEN-1 IgG-conjugated agarosebeads from the nuclear extracts reduced the ligation efficiencyto 0.3, 0.4, 0.5, or 0.7 fmol/min with substrates R3, R1, R0, orF5, respectively, which is 20 to 30% of the total ligation prod-ucts mediated by the wt nuclear extracts (Fig. 3A to D). Thisobservation indicates that while FEN-1 is critical for the gen-eration of the ligation products, other nucleases, possibly
FIG. 2. The FFAA mutation eliminates FEN-1/PCNA interaction and impairs the recruitment of FEN-1 to replication foci. (A) Mouserecombinant FFAA FEN-1 mutant is unable to interact with mouse PCNA. His6-tagged FEN-1 or nontagged PCNA was expressed in E. coli. Celllysates containing recombinant His6-tagged FEN-1 or His6-tagged FFAA were mixed with the lysate of nontagged PCNA. The FEN-1 or FFAAwas purified with Ni2� chelating agarose. The PCNA in complex with FEN-1 was analyzed with 4 to 15% SDS-PAGE. (B) Coimmunoprecipitationof FEN-1 and PCNA. Whole-cell extract (CE) was prepared from wt or FFAA/FFAA MEFs. The wt or FFAA/FFAA CE was incubated withmonoclonal anti-FEN-1 antibody-conjugated protein G beads. After an extensive wash, proteins bound to protein G beads were analyzed byWestern blotting using polyclonal anti-PCNA antibody or monoclonal anti-FEN-1 antibody. (C) Representative images of localization of FEN-1and PCNA in cells at the G1/S boundary and cells in S phase. wt and FFAA/FFAA MEFs were synchronized at the G1/S boundary by mimosinetreatment. Upon removal of mimosine, cells were released into S phase. Typically, 60% of cells were in S phase at 4 h post-mimosine treatment.Cells were fixed and costained with anti-FEN-1 and anti-PCNA antibodies. In the merge views, yellow spots indicate colocalization of FEN-1 andPCNA. (D) Representative images of localization of FEN-1 and BrdU in cells at the G1/S boundary and cells in S phase. wt and FFAA/FFAAMEFs at the G1/S boundary or in S phase were labeled with BrdU for 1 h. Cells were fixed and costained with anti-FEN-1 and anti-BrdUantibodies.
3180 ZHENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.
DNA2 nuclease and exonuclease 1, may also be involved in thisprocess.
Deficiency in recruitment of FEN-1 to DNA replication sites(Fig. 2C and D) and in Okazaki fragment maturation (Fig. 3)may affect DNA replication and cell proliferation of FFAAmutant cells. To determine if the FFAA mutation resulted in adeficiency in DNA replication in vivo, we evaluated the DNAreplication rate of wt/wt, wt/FFAA, and FFAA/FFAA MEFsby using a [3H]thymidine incorporation assay. The rate of[3H]thymidine incorporation in FFAA/FFAA cells was approx-imately 50% of that in wt/wt or wt/FFAA cells (Fig. 4A).Consistent with our biochemical data, the proliferation rate ofFFAA/FFAA MEFs was significantly reduced compared to
that of wt/wt or wt/FFAA MEFs (Fig. 4B). The cell doublingtime during log phase for wt/wt, wt/FFAA, or FFAA/FFAAMEFs was approximately 41, 44, and 60 h, respectively. Alltogether, these data suggest that disruption of the FEN-1/PCNA interaction reduces DNA replication efficiency.
Mice homozygous for the FFAA mutation displayed re-tarded embryonic development and died at birth. We nextinvestigated the consequences of the FFAA mutation in mousegrowth and development. Mice heterozygous for the FFAAmutation were fertile and developed similarly to wt mice. How-ever, mice homozygous for the FFAA mutation (FFAA/FFAA) always died at birth. These dead homozygous pupswere generally smaller than their wt and heterozygous litter-
FIG. 3. FFAA/FFAA nuclear extracts are deficient in Okazaki fragment maturation (OFM) in vitro. Four synthetic substrates that mimicvarious OFM intermediates were utilized in a nick translation assay to compare the OFM efficiency of nuclear extracts generated from wt withthose from homozygous FFAA mouse cells. As a control, wt nuclear extracts that were FEN-1 immunodepleted (�FEN-1) were assayed as well.Nick translation on gap substrates (A) R3, (B) R1, and (C) R0 having three, one, and zero 5� ribonucleotides, respectively. (D) Nick translationon a gap substrate with a 5-nt 5� flap (F5). In each panel, a schematic of the substrate used, a representative gel image from the experiment, anda bar graph depicting the quantitation of the product band are shown. Oligonucleotides used to make R3, R1, R0, and F5 are listed in Table 1.
VOL. 27, 2007 FEN-1/PCNA INTERACTION IN EMBRYONIC DEVELOPMENT 3181
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.
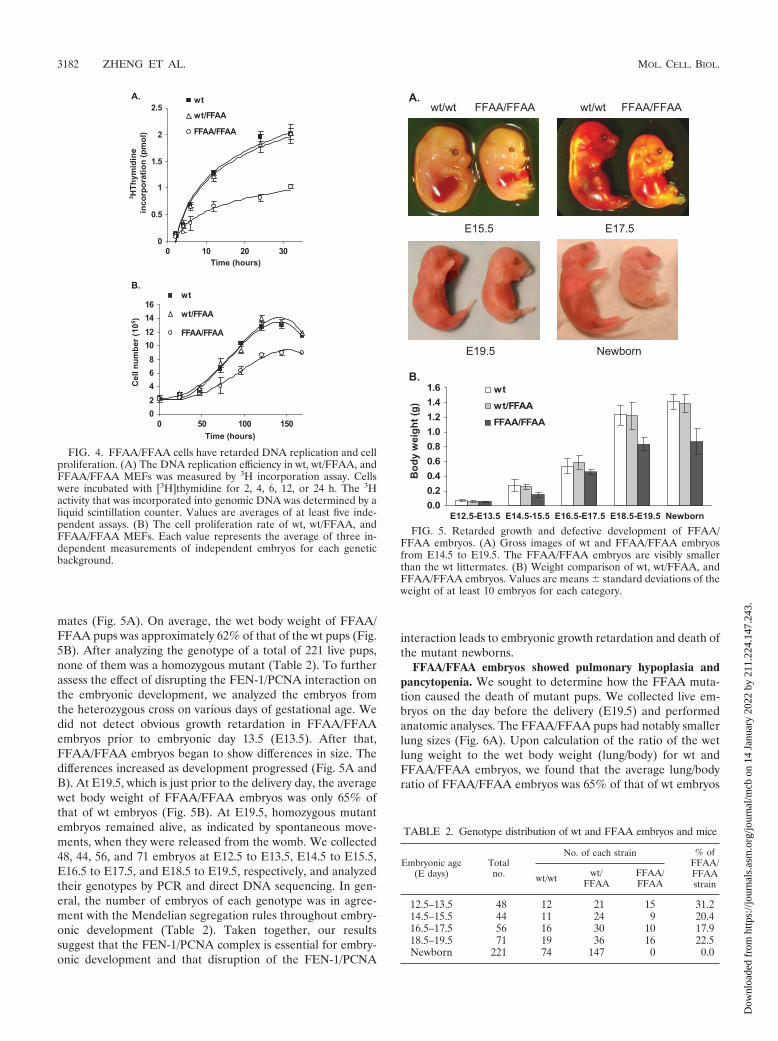
mates (Fig. 5A). On average, the wet body weight of FFAA/FFAA pups was approximately 62% of that of the wt pups (Fig.5B). After analyzing the genotype of a total of 221 live pups,none of them was a homozygous mutant (Table 2). To furtherassess the effect of disrupting the FEN-1/PCNA interaction onthe embryonic development, we analyzed the embryos fromthe heterozygous cross on various days of gestational age. Wedid not detect obvious growth retardation in FFAA/FFAAembryos prior to embryonic day 13.5 (E13.5). After that,FFAA/FFAA embryos began to show differences in size. Thedifferences increased as development progressed (Fig. 5A andB). At E19.5, which is just prior to the delivery day, the averagewet body weight of FFAA/FFAA embryos was only 65% ofthat of wt embryos (Fig. 5B). At E19.5, homozygous mutantembryos remained alive, as indicated by spontaneous move-ments, when they were released from the womb. We collected48, 44, 56, and 71 embryos at E12.5 to E13.5, E14.5 to E15.5,E16.5 to E17.5, and E18.5 to E19.5, respectively, and analyzedtheir genotypes by PCR and direct DNA sequencing. In gen-eral, the number of embryos of each genotype was in agree-ment with the Mendelian segregation rules throughout embry-onic development (Table 2). Taken together, our resultssuggest that the FEN-1/PCNA complex is essential for embry-onic development and that disruption of the FEN-1/PCNA
interaction leads to embryonic growth retardation and death ofthe mutant newborns.
FFAA/FFAA embryos showed pulmonary hypoplasia andpancytopenia. We sought to determine how the FFAA muta-tion caused the death of mutant pups. We collected live em-bryos on the day before the delivery (E19.5) and performedanatomic analyses. The FFAA/FFAA pups had notably smallerlung sizes (Fig. 6A). Upon calculation of the ratio of the wetlung weight to the wet body weight (lung/body) for wt andFFAA/FFAA embryos, we found that the average lung/bodyratio of FFAA/FFAA embryos was 65% of that of wt embryos
FIG. 4. FFAA/FFAA cells have retarded DNA replication and cellproliferation. (A) The DNA replication efficiency in wt, wt/FFAA, andFFAA/FFAA MEFs was measured by 3H incorporation assay. Cellswere incubated with [3H]thymidine for 2, 4, 6, 12, or 24 h. The 3Hactivity that was incorporated into genomic DNA was determined by aliquid scintillation counter. Values are averages of at least five inde-pendent assays. (B) The cell proliferation rate of wt, wt/FFAA, andFFAA/FFAA MEFs. Each value represents the average of three in-dependent measurements of independent embryos for each geneticbackground.
FIG. 5. Retarded growth and defective development of FFAA/FFAA embryos. (A) Gross images of wt and FFAA/FFAA embryosfrom E14.5 to E19.5. The FFAA/FFAA embryos are visibly smallerthan the wt littermates. (B) Weight comparison of wt, wt/FFAA, andFFAA/FFAA embryos. Values are means � standard deviations of theweight of at least 10 embryos for each category.
TABLE 2. Genotype distribution of wt and FFAA embryos and mice
Embryonic age(E days)
Totalno.
No. of each strain % ofFFAA/FFAAstrain
wt/wt wt/FFAA
FFAA/FFAA
12.5–13.5 48 12 21 15 31.214.5–15.5 44 11 24 9 20.416.5–17.5 56 16 30 10 17.918.5–19.5 71 19 36 16 22.5Newborn 221 74 147 0 0.0
3182 ZHENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.

(Fig. 6B), indicating that the mutant embryos developed pul-monary hypoplasia (39). In addition, the organs, particularlythe liver and the spleen, of FFAA/FFAA pups were pale,suggesting a defective blood system. Histological analysis wasperformed on all major organs. Wild-type and heterozygousembryos showed a normal progression of lung development,characterized by the formation of pre-alveoli and thinning ofmesenchyme (Fig. 6C), but the lungs of FFAA/FFAA embryoswere deficient in pre-alveoli and had thick mesenchyme (Fig.6D), clearly indicating a delay in lung development (45). Inaddition, FFAA/FFAA embryos displayed very poor embry-onic hematopoiesis compared to that of wt and heterozygousembryos. Extramedullary hematopoiesis in the liver of FFAA/FFAA embryos was largely impaired, with at least 50% fewerred blood precursor cells (Fig. 7A and B), which led to thedevelopment of marked peripheral pancytopenia, as mani-fested by a very low number of circulating red blood cells andthe virtual absence of white blood cells and platelets (Fig. 7Cand D). Failure of development of these two organ systemscould lead to impaired oxygen delivery and cause the death ofmutant newborns.
DISCUSSION
FEN-1 plays an essential role in Okazaki fragment matura-tion in mammalian cells. However, the mechanism by whichFEN-1 mediates RNA primer removal and coordinates withother enzymes in Okazaki fragment maturation remains un-clear. Previous biochemical studies have shown that FEN-1forms a complex with PCNA, suggesting that FEN-1 may beregulated by PCNA in Okazaki fragment maturation via three
possible mechanisms: (i) recruitment of FEN-1 to the site ofDNA replication (27); (ii) stimulation of the cleavage of RNAprimer flaps (44, 48); and (iii) coordination of highly orderedRNA primer processing and DNA ligation (9, 11, 36, 42, 43).
FIG. 6. FFAA/FFAA embryos develop pulmonary hypoplasia. Anatomical and histological analyses were performed on wt, wt/FFAA, andFFAA/FFAA embryos at E19.5. Because wt and wt/FFAA embryos exhibited identical phenotypes, the images of wt/FFAA embryos were notdisplayed. (A) Macrographic images of the lungs of a wt and an FFAA/FFAA embryo. (B) Lung/body ratios of the wild-type and FFAA mouseembryos. Values are means � standard deviations of measurements from at least 10 embryos on each genetic background. (C and D) Micrographicimages (�100) and higher-magnification views (insets, �400) of the lung of a wt (C) and an FFAA/FFAA (D) embryo (H&E stain).
FIG. 7. Impairment of embryonic hematopoiesis and peripheralpancytopenia. (A and B) Histological features of embryonic hemato-poiesis in the liver of a wt (A) and an FFAA/FFAA embryo (B) atE19.5 showing liver extramedullary hematopoiesis (H&E stain; �200).The FFAA/FFAA embryo displayed markedly impaired hematopoie-sis. (C and D) Blood smears of a wt (C) and an FFAA/FFAA embryo(D) at E19.5. Blood smears were prepared using blood sampling fromthe corresponding embryo hearts (Wright-Giemsa stain; �200).
VOL. 27, 2007 FEN-1/PCNA INTERACTION IN EMBRYONIC DEVELOPMENT 3183
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.

It has been demonstrated that two independent interactionsites exist in PCNA. The amino acids residues I126 and L128 inPCNA are essential for the physical interaction with FEN-1but not for the stimulation of FEN-1. Conversely, P252 andK253 of PCNA are required for the stimulation of FEN-1 butnot for the physical interaction with FEN-1 (17). Correspond-ingly, the stimulation site is also independent of the physicalinteraction site in FEN-1 (12). The physical FEN-1/PCNAinteraction is mediated by amino acids 337QGRLDDFFK345 ofFEN-1. Deletion or mutation of this motif eliminates the phys-ical interaction but does not affect the stimulation in vitro (12).Three-dimensional crystal structures of the human FEN-1/PCNA complex have revealed that besides the 337QGRLDDFFK345 sequence of FEN-1, PCNA also interacts with otherregions of FEN-1, including the core nuclease domain (9, 38),which may be responsible for the stimulation. The in vivosignificance of the physical interaction between FEN-1 andPCNA in mammalian DNA replication and cell proliferationwas previously unknown.
Here, we have generated a mutant mouse line harboring anF343AF344A (FFAA) double mutation in FEN-1, which spe-cifically eliminates the FEN-1/PCNA interaction. In FFAA/FFAA mutant MEFs, the FEN-1 mutant protein fails to belocalized to DNA replication sites, suggesting that the FEN-1/PCNA interaction is crucial for recruiting FEN-1 to the DNAreplication factory. This also supports the hypothesis thatPCNA binding is a common mechanism for recruiting PCNAinteraction proteins, including DNA ligase I and FEN-1, to thereplication site (27). It has been demonstrated that DNA rep-lication occurs at discrete sites of the nuclear matrix (18, 19,28). In order to execute their functions, DNA replication pro-teins must be assembled onto the replication foci (18, 19).Therefore, exclusion of FEN-1 from the replication foci shouldsignificantly affect the function of FEN-1 in DNA replication.Consistent with this view, we demonstrated that the FFAA/FFAA mutant MEFs have significantly reduced DNA replica-tion and cell proliferation rates. This, in turn, results in severedefects in embryonic development and causes the death ofmutant pups at birth.
In addition, disruption of the FEN-1/PCNA interaction af-fects the coordination of various reactions in Okazaki fragmentmaturation and further contributes to DNA replication defi-ciency. We have shown that compared to wt MEFs, the FFAAcells are significantly less efficient in generating ligation prod-ucts in a nick translation assay that mimics Okazaki fragmentmaturation. Because the FFAA mutation does not affect thenuclease activity of FEN-1 in the absence or presence ofPCNA (12), the deficiency in the generation of ligation prod-ucts is likely caused by the impaired coordination between theFEN-1-mediated RNA primer processing reaction and thesubsequent DNA ligase I-catalyzed DNA ligation reaction.PCNA has been implicated as a platform for sequential re-cruitment of FEN-1 and DNA ligase I to DNA replicationforks (9, 11, 42). FEN-1 and DNA ligase I share the samePCNA-binding motif, QXX(L/I)XXF(F/Y) (underlining indi-cates conserved amino acid residues) (12, 24). Both theQRSIESFFK motif in DNA ligase I and the QGRLDDFFKmotif in FEN-1 can bind to a subunit of the PCNA trimmer.This physical interaction is critical for the coordination ofFEN-1 and DNA ligase I during DNA replication (9, 11, 36, 42,
43). Two different models have been proposed to elucidatehow PCNA coordinates the actions of FEN-1 and DNA ligaseI (9, 11, 36, 43). In the first, a rotary handoff model (9, 11), bothFEN-1 and DNA ligase I recognize the PCNA-bound DNAsubstrate, which can rotate at the PCNA site. The rotationallows FEN-1 and DNA ligase I, which are bound to one ofthree binding sites on PCNA, to sequentially access interme-diate DNA substrates. By contrast, the second model proposesthat the PCNA binding by FEN-1 and that by DNA ligase I aremutually exclusive. The competition of DNA ligase I withFEN-1 for PCNA binding is crucial for the sequential loadingof FEN-1 and DNA ligase I onto the DNA replication fork(43). Supporting this model, the co-crystal of human DNAligase I-DNA complex indicates that DNA ligase I encirclesthe DNA substrate with a ring size and shape similar to thoseof the PCNA trimmer and PCNA binding of FEN-1, or DNAligase I mutually excludes the other to interact with PCNA(30). In either situation, the interaction of PCNA with FEN-1and DNA ligase I is critical for efficient transition from theFEN-1 cleavage reaction to the DNA ligase I-mediated DNAligation. However, the FFAA mutation in FEN-1 disrupts thephysical interaction with PCNA so that the association/disso-ciation of FEN-1 with/from the replication fork cannot bemediated by PCNA. The FFAA mutant may remain bound tothe replication fork after cleavage of the RNA primer flap andprevent the loading of DNA ligase I to the replication site. Thisin turn retards the ligation of DNA nicks and consequently theprogression of DNA replication and cell proliferation. In ad-dition, DNA nicks may potentially cause DNA double-strandbreaks, which activate checkpoint processes to arrest DNAreplication and cell division, further contributing to the retar-dation of cell proliferation. A lower cell proliferation ratesignificantly affects embryonic development; in particular, itcauses pulmonary hypoplasia and pancytopenia, which likelylead to the death of mutant newborns.
The drastic phenotype observed for FFAA/FFAA mutantmice contrasts to that found in the FFAA yeast mutant strainwith little phenotypical difference (16, 20). This observationmay reflect different in vivo requirements of FEN-1 in Okazakifragment maturation in yeast and in mammals. In yeast, unlikethat in mammals, FEN-1 is not essential in RNA primer re-moval. Deletion of FEN-1 results only in a slower-growth phe-notype, due to the existence of multiple backup pathways toremove RNA primers (35, 41). However, a knockout of theFen1 gene in mice suppresses cell proliferation and causesearly embryonic lethality, reflecting the essential role of Fen1in DNA replication and embryonic development in mammals(22, 23). In addition, the differences in Okazaki fragment mat-uration between yeast and mammals may contribute to theoutcome caused by FFAA mutation. Human cells contain 100-to 1,000-fold more Okazaki fragments than yeast (20), andtherefore, an efficient and precisely regulated system for Oka-zaki fragment maturation is likely more important in mamma-lian cells.
FEN-1 is a multifunctional nuclease (25, 40) that interactswith different proteins, including PCNA, RPA, WRN, poly-merase �, DNA2 nuclease, hnRNP A1, APE1, and Endo G (4,6–8, 10, 29, 31, 48). By interacting with various proteins, FEN-1executes its function in several DNA metabolic pathways (25,40). During lagging-strand DNA replication, FEN-1 interacts
3184 ZHENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.

with PCNA, RPA, and DNA2 nuclease. The concerted actionof these proteins efficiently removes the RNA primer (3). Inthe long patch base excision repair, FEN-1 interacts with poly-merase �, APE1, and PCNA (10, 14, 31, 34). These are co-stimulatory proteins functioning cohesively to efficiently andprecisely remove the modified base and generate a ligatableDNA nick. Recently, we proposed that in response to apop-totic stimuli, FEN-1 may interact with Endo G, which translo-cates from the mitochondria into the nucleus during apoptosisto cooperatively degrade DNA (29). Our data from this studyindicate that the FEN-1/PCNA complex is important in DNAreplication in mammalian cells. The FFAA FEN-1 mutationresults in inefficient Okazaki fragment maturation and retardsthe progression of cell proliferation. On the other hand, FFAAMEFs are only slightly more sensitive to methyl methane sul-fate than the wt cells (data not shown). This of course does notrule out the requirement of the FEN-1/PCNA complex inlong-patch base excision repair. We consider that the disrup-tion of the FEN-1/PCNA complex may also result in deficientbase excision repair, but this, by itself, would not lead directlyto the observed cell proliferation retardation and newbornlethality.
Our FFAA mutant mouse line provides an interesting modelthat may be useful to address the impact of Fen1 polymor-phisms on human diseases. We hypothesized that Fen1 poly-morphisms that severely affect its function in DNA replicationare rare but those that eliminate its function in DNA repair,apoptotic DNA degradation, and maintenance of stability ofdi- or tri-nucleotide repeat sequences or that have subtle de-fects in DNA replication can accumulate in a population andmay result in variations in the onset of diseases. Our currentstudy illustrates this point well.
ACKNOWLEDGMENTS
We acknowledge W. Tsark for technical assistance in the generationof transgenic mice. We thank J. Hurwitz for providing purified recom-binant RFC. We thank L. D. Finger and J. Stark for critical review ofthe manuscript and K. A. Justus for editorial assistance.
All protocols involved in animals were approved by the ResearchAnimal Care Committee of the City of Hope National Medical Centerand Beckman Research Institute in compliance with the Public HealthService Policy on Use of Laboratory Animals.
This work was supported by NIH grants R01 CA085344 andCA073764 to B.S.
REFERENCES
1. Adachi, N., Z. E. Karanjawala, Y. Matsuzaki, H. Koyama, and M. R. Lieber.2002. Two overlapping divergent transcription units in the human genome:the FEN1/C11orf10 locus. OMICS J. Integr. Biol. 6:273–279.
2. Ayyagari, R., X. V. Gomes, D. A. Gordenin, and P. M. Burgers. 2003.Okazaki fragment maturation in yeast. I. Distribution of functions betweenFEN1 and DNA2. J. Biol. Chem. 278:1618–1625.
3. Bae, S. H., K. H. Bae, J. A. Kim, and Y. S. Seo. 2001. RPA governs endo-nuclease switching during processing of Okazaki fragments in eukaryotes.Nature 412:456–461.
4. Biswas, E. E., F. X. Zhu, and S. B. Biswas. 1997. Stimulation of RTH1nuclease of the yeast Saccharomyces cerevisiae by replication protein A.Biochemistry 36:5955–5962.
5. Bravo, R., and H. Macdonald-Bravo. 1987. Existence of two populations ofcyclin/proliferating cell nuclear antigen during the cell cycle: association withDNA replication sites. J. Cell Biol. 105:1549–1554.
6. Brosh, R. M., Jr., C. von Kobbe, J. A. Sommers, P. Karmakar, P. L. Opresko,J. Piotrowski, I. Dianova, G. L. Dianov, and V. A. Bohr. 2001. Wernersyndrome protein interacts with human flap endonuclease 1 and stimulatesits cleavage activity. EMBO J. 20:5791–5801.
7. Budd, M. E., and J. L. Campbell. 1997. A yeast replicative helicase, Dna2helicase, interacts with yeast FEN-1 nuclease in carrying out its essentialfunction. Mol. Cell. Biol. 17:2136–2142.
8. Chai, Q., J. Qiu, B. R. Chapados, and B. Shen. 2001. Archaeoglobus fulgidusRNase HII in DNA replication: enzymological functions and activity regu-lation via metal cofactors. Biochem. Biophys. Res. Commun. 286:1073–1081.
9. Chapados, B. R., D. J. Hosfield, S. Han, J. Qiu, B. Yelent, B. Shen, and J. A.Tainer. 2004. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell 116:39–50.
10. Dianova, I. I., V. A. Bohr, and G. L. Dianov. 2001. Interaction of human APendonuclease 1 with flap endonuclease 1 and proliferating cell nuclear an-tigen involved in long-patch base excision repair. Biochemistry 40:12639–12644.
11. Dore, A. S., M. L. Kilkenny, S. A. Jones, A. W. Oliver, S. M. Roe, S. D. Bell,and L. H. Pearl. 2006. Structure of an archaeal PCNA1-PCNA2-FEN1complex: elucidating PCNA subunit and client enzyme specificity. NucleicAcids Res. 34:4515–4526.
12. Frank, G., J. Qiu, L. Zheng, and B. Shen. 2001. Stimulation of eukaryoticflap endonuclease-1 activities by proliferating cell nuclear antigen (PCNA) isindependent of its in vitro interaction via a consensus PCNA binding region.J. Biol. Chem. 276:36295–36302.
13. Fukushige, S., and B. Sauer. 1992. Genomic targeting with a positive-selec-tion lox integration vector allows highly reproducible gene expression inmammalian cells. Proc. Natl. Acad. Sci. USA 89:7905–7909.
14. Gary, R., K. Kim, H. L. Cornelius, M. S. Park, and Y. Matsumoto. 1999.Proliferating cell nuclear antigen facilitates excision in long-patch base ex-cision repair. J. Biol. Chem. 274:4354–4363.
15. Gary, R., D. L. Ludwig, H. L. Cornelius, M. A. MacInnes, and M. S. Park.1997. The DNA repair endonuclease XPG binds to proliferating cell nuclearantigen (PCNA) and shares sequence elements with the PCNA-bindingregions of FEN-1 and cyclin-dependent kinase inhibitor p21. J. Biol. Chem.272:24522–24529.
16. Gary, R., M. S. Park, J. P. Nolan, H. L. Cornelius, O. G. Kozyreva, H. T.Tran, K. S. Lobachev, M. A. Resnick, and D. A. Gordenin. 1999. A novel rolein DNA metabolism for the binding of Fen1/Rad27 to PCNA and implica-tions for genetic risk. Mol. Cell. Biol. 19:5373–5382.
17. Gomes, X. V., and P. M. Burgers. 2000. Two modes of FEN1 binding toPCNA regulated by DNA. EMBO J. 19:3811–3821.
18. Hozak, P., A. B. Hassan, D. A. Jackson, and P. R. Cook. 1993. Visualizationof replication factories attached to nucleoskeleton. Cell 73:361–373.
19. Hozak, P., D. A. Jackson, and P. R. Cook. 1994. Replication factories andnuclear bodies: the ultrastructural characterization of replication sites duringthe cell cycle. J. Cell Sci. 107:2191–2202.
20. Jin, Y. H., R. Ayyagari, M. A. Resnick, D. A. Gordenin, and P. M. Burgers.2003. Okazaki fragment maturation in yeast. II. Cooperation between thepolymerase and 3�–5�-exonuclease activities of Pol delta in the creation of aligatable nick. J. Biol. Chem. 278:1626–1633.
21. Karimi-Busheri, F., G. Daly, P. Robins, B. Canas, D. J. Pappin, J. Sgouros,G. G. Miller, H. Fakhrai, E. M. Davis, M. M. Le Beau, and M. Weinfeld.1999. Molecular characterization of a human DNA kinase. J. Biol. Chem.274:24187–24194.
22. Kucherlapati, M., K. Yang, M. Kuraguchi, J. Zhao, M. Lia, J. Heyer, M. F.Kane, K. Fan, R. Russell, A. M. Brown, B. Kneitz, W. Edelmann, R. D.Kolodner, M. Lipkin, and R. Kucherlapati. 2002. Haploinsufficiency of Flapendonuclease (Fen1) leads to rapid tumor progression. Proc. Natl. Acad. Sci.USA 99:9924–9929.
23. Larsen, E., C. Gran, B. E. Saether, E. Seeberg, and A. Klungland. 2003.Proliferation failure and gamma radiation sensitivity of Fen1 null mutantmice at the blastocyst stage. Mol. Cell. Biol. 23:5346–5353.
24. Levin, D. S., A. E. McKenna, T. A. Motycka, Y. Matsumoto, and A. E.Tomkinson. 2000. Interaction between PCNA and DNA ligase I is critical forjoining of Okazaki fragments and long-patch base-excision repair. Curr. Biol.10:919–922.
25. Liu, Y., H. I. Kao, and R. A. Bambara. 2004. Flap endonuclease 1: a centralcomponent of DNA metabolism. Annu. Rev. Biochem. 73:589–615.
26. Lu, R., and G. Serrero. 2000. Inhibition of PC cell-derived growth factor(PCDGF, epithelin/granulin precursor) expression by antisense PCDGFcDNA transfection inhibits tumorigenicity of the human breast carcinomacell line MDA-MB-468. Proc. Natl. Acad. Sci. USA 97:3993–3998.
27. Montecucco, A., R. Rossi, D. S. Levin, R. Gary, M. S. Park, T. A. Motycka,G. Ciarrocchi, A. Villa, G. Biamonti, and A. E. Tomkinson. 1998. DNA ligaseI is recruited to sites of DNA replication by an interaction with proliferatingcell nuclear antigen: identification of a common targeting mechanism for theassembly of replication factories. EMBO J. 17:3786–3795.
28. Nakayasu, H., and R. Berezney. 1989. Mapping replicational sites in theeukaryotic cell nucleus. J. Cell Biol. 108:1–11.
29. Parrish, J. Z., C. Yang, B. Shen, and D. Xue. 2003. CRN-1, a Caenorhabditiselegans FEN-1 homologue, cooperates with CPS-6/EndoG to promoteapoptotic DNA degradation. EMBO J. 22:3451–3460.
30. Pascal, J. M., P. J. O’Brien, A. E. Tomkinson, and T. Ellenberger. 2004.Human DNA ligase I completely encircles and partially unwinds nickedDNA. Nature 432:473–478.
31. Prasad, R., G. L. Dianov, V. A. Bohr, and S. H. Wilson. 2000. FEN1 stim-ulation of DNA polymerase beta mediates an excision step in mammalianlong patch base excision repair. J. Biol. Chem. 275:4460–4466.
VOL. 27, 2007 FEN-1/PCNA INTERACTION IN EMBRYONIC DEVELOPMENT 3185
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.

32. Qiu, J., X. Li, G. Frank, and B. Shen. 2001. Cell cycle-dependent and DNAdamage-inducible nuclear localization of FEN-1 nuclease is consistent withits dual functions in DNA replication and repair. J. Biol. Chem. 276:4901–4908.
33. Qiu, J., Y. Qian, P. Frank, U. Wintersberger, and B. Shen. 1999. Saccharo-myces cerevisiae RNase H(35) functions in RNA primer removal duringlagging-strand DNA synthesis, most efficiently in cooperation with Rad27nuclease. Mol. Cell. Biol. 19:8361–8371.
34. Ranalli, T. A., S. Tom, and R. A. Bambara. 2002. AP endonuclease 1 coor-dinates Flap endonuclease 1 and DNA ligase I activity in long patch baseexcision repair. J. Biol. Chem. 277:41715–41724.
35. Reagan, M. S., C. Pittenger, W. Siede, and E. C. Friedberg. 1995. Charac-terization of a mutant strain of Saccharomyces cerevisiae with a deletion ofthe RAD27 gene, a structural homolog of the RAD2 nucleotide excisionrepair gene. J. Bacteriol. 177:364–371.
36. Refsland, E. W., and D. M. Livingston. 2005. Interactions among DNA ligaseI, the flap endonuclease and proliferating cell nuclear antigen in the expan-sion and contraction of CAG repeat tracts in yeast. Genetics 171:923–934.
37. Rossi, M. L., V. Purohit, P. D. Brandt, and R. A. Bambara. 2006. Laggingstrand replication proteins in genome stability and DNA repair. Chem. Rev.106:453–473.
38. Sakurai, S., K. Kitano, H. Yamaguchi, K. Hamada, K. Okada, K. Fukuda,M. Uchida, E. Ohtsuka, H. Morioka, and T. Hakoshima. 2005. Structuralbasis for recruitment of human flap endonuclease 1 to PCNA. EMBO J.24:683–693.
39. Seegmiller, R. E., C. A. Cooper, M. J. Houghton, and J. C. Carey. 1986.Pulmonary hypoplasia in chondrodystrophic mice. Teratology 33:339–347.
40. Shen, B., P. Singh, R. Liu, J. Qiu, L. Zheng, L. D. Finger, and S. Alas. 2005.Multiple but dissectible functions of FEN-1 nucleases in nucleic acid pro-cessing, genome stability and diseases. Bioessays 27:717–729.
41. Sommers, C. H., E. J. Miller, B. Dujon, S. Prakash, and L. Prakash. 1995.
Conditional lethality of null mutations in RTH1 that encodes the yeastcounterpart of a mammalian 5�- to 3�-exonuclease required for laggingstrand DNA synthesis in reconstituted systems. J. Biol. Chem. 270:4193–4196.
42. Sporbert, A., P. Domaing, H. Leonhardt, and M. C. Cardoso. 2005. PCNAacts as a stationary loading platform for transiently interacting Okazakifragment maturation proteins. Nucleic Acids Res. 33:3521–3528.
43. Subramanian, J., S. Vijayakumar, A. E. Tomkinson, and N. Arnheim. 2005.Genetic instability induced by overexpression of DNA ligase I in buddingyeast. Genetics 171:427–441.
44. Tom, S., L. A. Henricksen, and R. A. Bambara. 2000. Mechanism wherebyproliferating cell nuclear antigen stimulates flap endonuclease 1. J. Biol.Chem. 275:10498–10505.
45. Tseng, B. S., S. T. Cavin, F. W. Booth, E. N. Olson, M. C. Marin, T. J.McDonnell, and I. J. Butler. 2000. Pulmonary hypoplasia in the myogeninnull mouse embryo. Am. J. Respir. Cell Mol. Biol. 22:304–315.
46. Turchi, J. J., and R. A. Bambara. 1993. Completion of mammalian laggingstrand DNA replication using purified proteins. J. Biol. Chem. 268:15136–15141.
47. Warbrick, E., D. P. Lane, D. M. Glover, and L. S. Cox. 1997. Homologousregions of Fen1 and p21Cip1 compete for binding to the same site on PCNA:a potential mechanism to co-ordinate DNA replication and repair. Onco-gene 14:2313–2321.
48. Wu, X., J. Li, X. Li, C. L. Hsieh, P. M. Burgers, and M. R. Lieber. 1996.Processing of branched DNA intermediates by a complex of human FEN-1and PCNA. Nucleic Acids Res. 24:2036–2043.
49. Zheng, L., M. Zhou, Q. Chai, J. Parrish, D. Xue, S. M. Patrick, J. J. Turchi,S. M. Yannone, D. Chen, and B. Shen. 2005. Novel function of the flapendonuclease 1 complex in processing stalled DNA replication forks. EMBORep. 6:83–89.
3186 ZHENG ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 14
Jan
uary
202
2 by
211
.224
.147
.243
.