Embed Size (px)
Citation preview

Difunctionalized N-Confused Porphyrins: Synthesis,Fluorescence, and Electrochemical Studies
Sudipta Das,A Naresh Balsukuri,A Praseetha E. Kesavan,A
and Iti GuptaA,B
AIndian Institute of Technology Gandhinagar, VGEC Campus,
Chandkheda, Ahmedabad-382424, India.BCorresponding author. Email: [email protected]
Seven di-substituted N-confused porphyrins (NCPs) 9–15 bearing two aryl functional groups (cis-A2B2 type) were
synthesized in 4–7% yields via [3þ1] approach. The corresponding five 5,10-diaryl-substituted symmetrical tripyrranes1–5were prepared and condensed with 2,4-bis(hydroxypentaflurophenyl)pyrrole 6. Two outer N-methyl type A2B2 NCPs14 and 15 were also prepared via a similar approach using a new key precursor 8. All the porphyrins 9–15 werecharacterized by high-resolutionmass spectrometry, NMR, infrared spectroscopy, UV–visible spectroscopy, fluorescence
spectroscopy, and cyclic voltammetry. Fluorescence studies of 9–15 showed blue-shifted emission maxima and lowerStokes shifts values when compared with N-confused tetraphenylporphyrin (NCTPP). Electrochemical studies indicatedeasier oxidation of N-methyl NCPs 14 and 15 when compared with remaining NCPs 9–13.
Manuscript received: 14 June 2014.Manuscript accepted: 28 August 2014.
Published online: 18 November 2014.
Introduction
N-Confused porphyrin (NCP)[1] is the isomer of normal porphyrin,whereby three pyrrole rings havea–a linkages andone pyrrole ringhas a a–b linkage within the porphyrin core.[2] NCPs are alsoknown as either carbaporphyrins or inverted porphyrins, andshow characteristic spectral[3] and metal coordination beha-
viours.[4] A typical A4-type NCP can exist in two different tauto-meric forms in solution;[3] they can be easily identified by theircolours in solution, brown in dichloromethane (DCM) and green inN,N-dimethylformamide (DMF). Due to the presence of the inner
CH moiety available for complexation with metal ions, NCPs canform organometallic complexes withmetals such as PdII, CdII, andZnII.[5] Furthermore, NCPs can stabilize metals in their unusual
oxidation states such asAgIII, CuIII, andRhIV.[6] NCPs have shownto be less aromatic and less symmetric[7] when compared withnormal porphyrins, thereby making them the chromophores
of choice for light-harvesting antenna systems. The syntheticstrategies used to prepare the various meso-substituted normalporphyrins such as cis-A2B2, trans-A2B2, A2BC, andABCD typesare well established.[8] On the other hand, little effort has been
made to prepare various types of meso-substituted N-confusedporphyrins. The A4-type NCPs can be easily synthesized by theone-pot condensation method by Lindsey et al.[9] The [2þ2]
approach has been used to make A4-type NCP, whereby thebiscarbinol derivative of N-confused dipyrrane is condensedwith normal dipyrrane under acidic conditions.[10] The partially
meso-free A2-type NCP and its AgIII complex were synthesizedby [3þ1] approach. The 2,4-bis(phenylhydroxymethyl)pyrrolewas condensed with 5,10-meso-free tripyrromethane in the pres-
ence of acid catalyst, followed by oxidation with chloranil.[11]
Modarelli and coworkers[12] have reported the synthesis ofA2B2-type NCPs by one-pot condensation method, whereby
3,5-di-tert-butylbenzaldehyde was reacted with para-substitutedaryl aldehyde and pyrrole. The authors proposed that the presenceof 3,5-di-tert-butylbenzaldehyde in the 5,20-meso positions of
NCP was crucial for their column chromatographic separationfrom other possible 10,15-difunctionalized isomeric NCPs. Thesame group also achieved the synthesis of A3B and AB2C-type
asymmetricNCPsbymixedcondensationofN-confuseddipyrraneand aryl aldehyde with functionalized dipyrrane.[13] A2B2-typemeso-aryl carbaporphyrins with appropriate functional groups canserve as potential building blocks for covalently linked symmet-
rical or asymmetrical porphyrin triads and other related systems.Herein, we report the synthesis, characterization, and emission andelectrochemical studiesof seven10,15-difunctionalizedA2B2-type
NCPs 9–15. These difunctionalized N-confused porphyrins 9–15contain pentafluorophenyl groups at the C5 and C20 positions, andthe substituents at the C10 and C15 positions are varied from
p-bromophenyl, p-nitrophenyl, p-iodophenyl, m-bromophenyl top-tolyl groups.
Results and Discussion
The seven A2B2-type difunctionalized N-confused porphyrins(9–15) were prepared using [3þ1] approach as shown in
Scheme 1. In order to prepare the aforementionedNCPs, we alsosynthesized symmetrical tripyrranes 1–5 shown in Chart 1containing p-bromophenyl, p-nitrophenyl, p-iodophenyl,
m-bromophenyl, and p-tolyl groups at their 5,10 positions byfollowing the literature reportedmethod.[14] The tripyrranes 1–3have been synthesized previously,[14] but 4 and 5 have been
prepared for the first time and characterized (Chart 1). TheTFA-catalyzed reaction of functional aryl aldehydes with pyr-role gave predominantly amixture of 5,10-diaryl tripyrranes and
CSIRO PUBLISHING
Aust. J. Chem.
http://dx.doi.org/10.1071/CH14383
Journal compilation � CSIRO 2014 www.publish.csiro.au/journals/ajc
Full Paper

5-aryl dipyrranes, and the mixture was separated by silica gelcolumn chromatography. The expected symmetrical tripyrranes
were collected in a 40–80% DCM/petroleum ether solventmixture. Evaporation of the solvent gave a dark brown oil thatturned into a flaky solid after vacuum drying, and the isolated
yields of tripyrranes 1–5were in the range of 4–15%. The otherstarting compound 2,4-bis(hydroxylpentaflurophenyl)pyrrole(6) was prepared in multi-gram quantities by literature reportedprocedure.[15] The 2,4-bisformyl-N-methylpyrrole (7) and its
corresponding diol 2,4-bis(hydroxylpentafluoro-phenyl)-N-methylpyrrole (8) were also prepared. The diols 6 and 8 wereused directly for the condensation reaction without any column
chromatographic purification. The NCPs 9–13 were then pre-
pared by reacting 1 equiv. of 2,4-bis (hydroxylpentafluorophenyl)pyrrole (7)with 1 equiv. of the appropriate 5,10-diaryl-substitutedsymmetrical tripyrranes 1–5 in the presence of catalytic amounts
of methanesulfonic acid (MSA) or trifluoroacetic acid (TFA) inDCM (Scheme 1). Oxidation with 3 equiv. of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) resulted in the formation of
NCPs, as judged by the greenish brown spot on the silica TLCplates. A quick silica gel column filtration was performed toremove excess DDQ from the crude reaction mixture; a smallamount of normal porphyrin (,1%) was collected in 100%
DCMand a small amount of impureNCPwas collected in 2–5%methanol/DCM mixture. The NCPs were then subjected toneutral alumina column for final purification. The sequential
column chromatographic purification (first with silica gel thenwith neutral alumina column) was necessary to get pure NCPs.The desired NCPs were observed as a brown band on the alu-
mina column and were collected in 10–15% DCM/petroleumether mixture; evaporation of the solvent afforded pure NCPs9–13 in 3–7% yields. The mixed condensation method to makeA2B2-type NCPs[12] has the advantage of ready to use aryl
aldehydes; however, its synthetic potential was not fullyexplored to prepare a variety of difunctionalized NCPs. Also, itgave an isomeric mixture of two NCPs: 3,5-di-tert-butyl-
benzaldehyde at 5,20-meso positions of NCP (4% isolatedyield) and 10,15-difunctionalized isomeric NCP (,1% yield).In contrast, the [3þ1] synthetic approach described here gives
only one isomer of difunctionalized NCP and the regular por-phyrin (,1% isolated yield). The difunctionalized NCPs 9–13contain pentafluorophenyl groups at their C5 and C20 positions
and the substituents at their C10 and C15 positions varied fromp-bromophenyl, p-nitrophenyl, p-iodophenyl, m-bromophenylto p-tolyl groups. Also, we have tested the [3þ1] condensationreaction of tripyrrane 1 (1mM) with pyrrolediol 6 in DCM in
the presence of three types of acids (Table 1). MSA gave lower
N
NH
N
HN
I
I
N
NH
N
HN
Br
BrN
NH
N
HN
NO2
N
NH
N
HN
Br
Br
N
NH
N
HN
Me
Me
NH
ANH
A
HNHN
OH
B
HO
BN
NH N
HN
A
A
B
B(i) TFA, DCM
(ii) DDQ�
B �A � Functional aryl group
C6F5
C6F5C6F5
C6F5
C6F5
C6F5
C6F5
C6F5
C6F5
O2N C6F5C6F5
910 11
12 13
1–5 69–13
Scheme 1. Synthesis of difunctionalized NCPs.
NH
Br Br
NH
Me Me
NH
O2N NO2
NH
I I
NH
HNNH
NH NHHN
NH HN
HN
NH HN
1
2 3
4 5
Br Br
Chart 1.
B S. Das et al.

yields of NCP 9 (entry 1) and no porphyrin was obtained whenBF3�Et2O was used as catalyst (entry 2). The isolated percentage
yield of NCP 9 in the presence of TFA gradually increased uponincreasing the equiv. amounts from 0.1 to 1.0 (entries 3–6).However, the yield of NCP 9 decreased drastically upon furtherincreasing the amount of TFA to 2 equiv. (entry 7). Thus, 1 equiv.
of TFA can be used as standard catalyst for [3þ1] condensationreaction to obtain NCP in moderate (6–7%) yields.
N-methyl NCPs 14 and 15were prepared in a similar fashion
by TFA-catalyzed condensation of 2,4-bis (hydroxylpentafluor-ophenyl)-N-methylpyrrole 8 with 1 equiv. of the symmetricaltripyrranes 4 and 5, respectively (Scheme 2). After oxidation
with DDQ, crude mixtures of NCPs 14 and 15 were filtered offby silica column in 5–7% methanol/DCM mixture and furtherpurified by neutral alumina column chromatography. Pure
NCPs 14 and 15 were collected as brown–green bands in25–30% DCM/petroleum ether mixture; evaporation of thesolvents produced violet–brown solids in 5–7% yields. Wehave also prepared the nickel complex of NCPs 9, 12, and 13
by refluxing nickel(II) acetate in ethanol/toluene with the corre-sponding porphyrin. Crude mixtures were purified by neutralalumina column chromatography in 70–80% DCM/petroleum
ether mixture. Removal of the solvents afforded 9-Ni, 12-Ni,and 13-Ni as green solids in 72–80% yields. All NCPs 9–15and their nickel complexes were identified using NMR, high-
resolution mass spectrometry (electrospray ionization quadru-pole time-of-flight; ESI-Q TOF), infrared (IR) spectroscopy,and UV–visible (UV–vis) spectroscopy. High-resolution massspectra were recorded on an ESI-Q-TOF mass spectrometer for
NCPs 9–15 in negative or positive ionmode. The [M�H]�1 was
the most abundant peak in the HRMS spectra of NCPs 9 and
11–13 (SupplementaryMaterial). HRMSanalyses of tripyrranes1–5 were conducted in positive ion mode (SupplementaryMaterial). Mass analysis of compound 8, NCP 10, 9-Ni,
12-Ni, and 13-Ni showed [MþH]þ peak in their HRMS spectra(Supplementary Material). Also, [MþH]þ or [M]þ were themost abundant peaks in the HRMS spectra of tripyrranes 1–5.The NMR data of the two new tripyrranes 4 and 5 were in
accordance with the previously reported tripyrrane.[14] 1H NMRspectra of NCPs 9–15 recorded in CDCl3 (see SupplementaryMaterial) reflected the aromatic nature of the macrocycles,
and the pattern of the proton signals matched the reportedN-confused tetraphenyl porphyrin.[3] The selected 1H NMRchemical shift values for the seven NCPs and their nickel
complexes are given in Table 2. The inner NH signal was shifteddownfield in NCP 10 (�1.74 ppm) and 12 (�1.90 ppm) contain-ing p-nitro and p-bromophenyl groups at their 10,15-meso-
positions, respectively, when compared with that of N-confusedtetraphenylporphyrin (NCTPP) (�2.41 ppm). The inner C21-Hwas observed at �4.89 and �5.02 ppm for NCP 10 and 12,respectively. For the rest of the NCPs (i.e. 9, 11, and 13), the
chemical shifts for both the inner NH and inner CH signals wereshifted upfield when compared with the NCTPP signals(Table 2). This may be attributed to the substituent effect of
the meso-aryl groups on the aromatic nature of the porphyrincore.[16] For all seven NCPs, the peripheral b-pyrrole protonsand meso-aryl protons appeared in the range of 6.14–8.98 ppm.
The representative NMR spectra of NCP 9 and its metalcomplex 9-Ni are shown in Fig. 1. In NCP 9, the inner two NHprotons and inner CH proton appeared at�2.76 and�5.48 ppm,
respectively. The NMR peaks for the b-H protons of normalpyrrole rings and meso-aryl protons were observed at ,7.66–8.88 ppm (Fig. 1a). The pyrrole a-H signal of the N-confusedpyrrole ring was observed at 8.97 ppm, which is very much in
accordance with the NCTPP NMR data.[12] Its metal complex9-Ni showed no signals corresponding to the inner NH and innerCH protons, reflecting the coordination of Ni metal in the
porphyrin core, and the outer NH and pyrrole a-H protonsshowed up at 10.11 and 8.45 ppm, respectively (Fig. 1b). Inthe 1H NMR spectra of N-methyl NCPs 14 and 15, Me-H
protons showed up as sharp singlets at 3.36 and 3.28 ppm. Thepyrrole inner C-H proton appeared as a sharp singlet at �5.38and �5.46 ppm for 14 and 15, respectively.
The UV–vis absorption spectra of all seven NCPs were
recorded in CHCl3 and showed the presence of inner-2H-type
N
OHHO
C6F5
C6F5
C6F5
C6F5
C6F5
C6F5
�
8 (i) TFA,DCM
(ii) DDQMe
NH
N
N
N
I
I
15
NH
N
N
N
Br
Br
14
Tripyrrane 1 or 5
Me Me
or
Scheme 2. Synthesis of N-methyl NCPs.
Table 2. Selected 1H NMR chemical shift (d) values for NCPs 9–15
and NCTPP recorded in CDCl3
Compound d Inner NH [ppm] d Inner CH [ppm] d Outer NH [ppm]
9 �2.76 �5.48 –
10 �1.74 �4.89 –
11 �2.49 �5.04 –
12 �1.90 �5.02 –
13 �2.78 �5.51 –
NCTPPA �2.41 �4.99 –
14 – �5.38 –
15 – �5.46 –
9-Ni – – 10.11
12-Ni – – 10.06
13-Ni – – 9.65
ADominant tautomer of NCTPP taken from ref. 3.
Table 1. Yields of NCP 9 in the presence of different acids. Reaction
was carried out at 1mM tripyrrane 1 in DCM
Entry Acid Equiv. Yield [%]
1 MSA 0.1 3.5
2 BF3�Et2O 0.1 Not detected
3 TFA 0.1 3
4 TFA 0.3 4
5 TFA 0.6 5
6 TFA 1.0 6
7 TFA 2.0 Trace
Difunctionalized N-Confused Porphyrins C

tautomer[12,17] in solution (the absorption data are summarized
in Table 3). With respect to NCTPP, a small hypsochromic shiftwas observed in the Soret bands of NCPs 9, 11, and 12, and therest of the Q bands were not altered much in their positions. The
absorption spectra for NCPs 14 and 15 showed slight batho-
chromic shifts (6–7 nm) in their Soret bands relative to that ofthe reported NCTPP data. In contrast, their first two Q bandsshowed blue shifts of,30 nmwith respect to NCTPP (Table 3).
�3 �4
1.34
0.84
0.82
0.69
0.82
2.21
4.33
3.67
1.98
1.00
0.87
0.86
1.69
1.70
1.65
1.76
1.73
2.00
�5 [ppm]
(a)
(b)
[ppm]10.0 9.5 9.0 8.5 8.0 7.5
[ppm]10.0 9.5 9.0 8.5 8.0 7.5
Fig. 1. 1H NMR spectra of (a) NCP 9 (inset shows up-field region) and (b) 9-Ni recorded in CDCl3.
Table 3. Summary of absorption data of NCPs 9–15 recorded in CHCl3
Compound Soret [nm] (e [� 105M�1 cm�1]) Q Bands [nm], (e [� 104M�1 cm�1])
9 436 (1.10) 534 (1.15), 573 (0.61), 656 (0.23), 722 (0.61)
10 439 (0.40) 528 (0.66), 574 (0.37), 657 (0.16), 720 (0.23)
11 435 (1.03) 534 (1.03), 576 (0.55), 661 (0.15), 724 (0.60)
12 436 (0.76) 534 (0.76), 575 (0.40), 657 (0.12), 722 (0.42)
13 438 (0.89) 533 (1.00), 575 (0.57), 656 (0.23), 723 (0.48)
NCTPPA 438 (1.59) 539 (0.78), 580 (1.08), 665 (0.27), 724 (1.04)
H2TPPB 419 (4.11) 515 (1.73), 550 (0.81), 590 (0.64), 645 (0.62)
14 443 (0.50) 535 (0.04), 575 (0.02), 633 (0.01), 694 (0.01)
15 444 (0.56) 535 (0.04), 576 (0.02), 633(0.01), 693 (0.01)
9-Ni 361 (0.49), 427 (0.88), 466 (0.38) 570 (0.13), 606 (0.13), 746 (0.06), 825 (0.06)
12-Ni 363 (0.42), 427 (0.79), 466 (0.32) 570 (0.11), 605 (0.11), 744 (0.05), 827 (0.05)
13-Ni 365 (0.57), 427 (1.09), 467 (0.43) 569 (0.12), 605 (0.12), 743 (0.04), 822 (0.03)
ADominant tautomer of N-confused tetraphenylporphyrin in DCM.BNormal tetraphenylporphyrin.
D S. Das et al.
These shifts in the UV–vis spectral peaks of 14 and 15 reflect
moderate aromaticity of the outer N-methyl derivatives whencompared with NCTPP.[18–20]
Steady-state fluorescence spectra were recorded and quan-
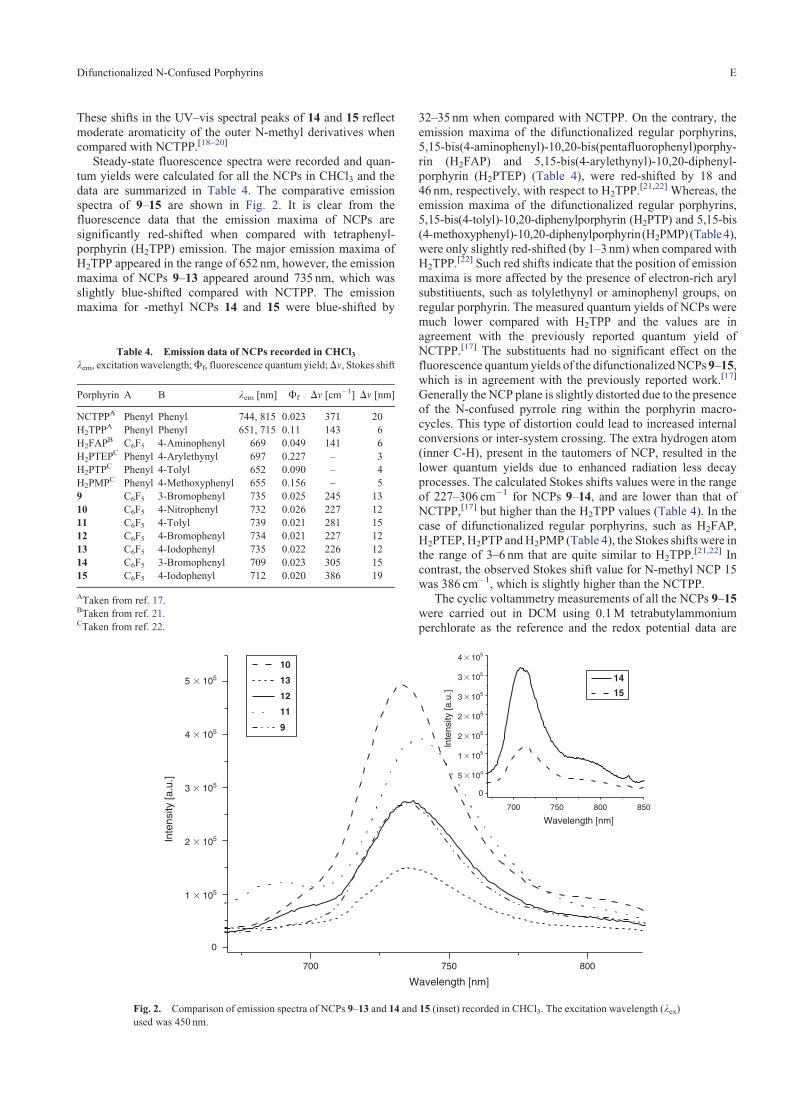
tum yields were calculated for all the NCPs in CHCl3 and thedata are summarized in Table 4. The comparative emissionspectra of 9–15 are shown in Fig. 2. It is clear from thefluorescence data that the emission maxima of NCPs are
significantly red-shifted when compared with tetraphenyl-porphyrin (H2TPP) emission. The major emission maxima ofH2TPP appeared in the range of 652 nm, however, the emission
maxima of NCPs 9–13 appeared around 735 nm, which wasslightly blue-shifted compared with NCTPP. The emissionmaxima for -methyl NCPs 14 and 15 were blue-shifted by
32–35 nm when compared with NCTPP. On the contrary, the
emission maxima of the difunctionalized regular porphyrins,5,15-bis(4-aminophenyl)-10,20-bis(pentafluorophenyl)porphy-rin (H2FAP) and 5,15-bis(4-arylethynyl)-10,20-diphenyl-
porphyrin (H2PTEP) (Table 4), were red-shifted by 18 and46 nm, respectively, with respect to H2TPP.
[21,22] Whereas, theemission maxima of the difunctionalized regular porphyrins,5,15-bis(4-tolyl)-10,20-diphenylporphyrin (H2PTP) and 5,15-bis
(4-methoxyphenyl)-10,20-diphenylporphyrin(H2PMP)(Table4),were only slightly red-shifted (by 1–3 nm) when compared withH2TPP.
[22] Such red shifts indicate that the position of emission
maxima is more affected by the presence of electron-rich arylsubstitiuents, such as tolylethynyl or aminophenyl groups, onregular porphyrin. The measured quantum yields of NCPs were
much lower compared with H2TPP and the values are inagreement with the previously reported quantum yield ofNCTPP.[17] The substituents had no significant effect on thefluorescence quantumyields of the difunctionalizedNCPs 9–15,
which is in agreement with the previously reported work.[17]
Generally the NCP plane is slightly distorted due to the presenceof the N-confused pyrrole ring within the porphyrin macro-
cycles. This type of distortion could lead to increased internalconversions or inter-system crossing. The extra hydrogen atom(inner C-H), present in the tautomers of NCP, resulted in the
lower quantum yields due to enhanced radiation less decayprocesses. The calculated Stokes shifts values were in the rangeof 227–306 cm�1 for NCPs 9–14, and are lower than that of
NCTPP,[17] but higher than the H2TPP values (Table 4). In thecase of difunctionalized regular porphyrins, such as H2FAP,H2PTEP, H2PTP andH2PMP (Table 4), the Stokes shifts were inthe range of 3–6 nm that are quite similar to H2TPP.
[21,22] In
contrast, the observed Stokes shift value for N-methyl NCP 15was 386 cm�1, which is slightly higher than the NCTPP.
The cyclic voltammetry measurements of all the NCPs 9–15
were carried out in DCM using 0.1M tetrabutylammoniumperchlorate as the reference and the redox potential data are
Table 4. Emission data of NCPs recorded in CHCl3lem, excitationwavelength;Ff, fluorescence quantumyield;Dn, Stokes shift
Porphyrin A B lem [nm] Ff Dn [cm�1] Dn [nm]
NCTPPA Phenyl Phenyl 744, 815 0.023 371 20
H2TPPA Phenyl Phenyl 651, 715 0.11 143 6
H2FAPB C6F5 4-Aminophenyl 669 0.049 141 6
H2PTEPC Phenyl 4-Arylethynyl 697 0.227 – 3
H2PTPC Phenyl 4-Tolyl 652 0.090 – 4
H2PMPC Phenyl 4-Methoxyphenyl 655 0.156 – 5
9 C6F5 3-Bromophenyl 735 0.025 245 13
10 C6F5 4-Nitrophenyl 732 0.026 227 12
11 C6F5 4-Tolyl 739 0.021 281 15
12 C6F5 4-Bromophenyl 734 0.021 227 12
13 C6F5 4-Iodophenyl 735 0.022 226 12
14 C6F5 3-Bromophenyl 709 0.023 305 15
15 C6F5 4-Iodophenyl 712 0.020 386 19
ATaken from ref. 17.BTaken from ref. 21.CTaken from ref. 22.
700 750 800
5 � 105
4 � 105
3 � 105
2 � 105
1 � 105
0
700 750 800 850
4 � 105
5 � 104
0
3 � 105
3 � 105
2 � 105
2 � 105
1 � 105
10
13
12
11
9
Inte
nsity
[a.u
.]
Inte
nsity
[a.u
.]
Wavelength [nm]
Wavelength [nm]
14
15
Fig. 2. Comparison of emission spectra of NCPs 9–13 and 14 and 15 (inset) recorded in CHCl3. The excitation wavelength (lex)used was 450 nm.
Difunctionalized N-Confused Porphyrins E

presented in Table 5. The representative cyclic voltammograms
(CV) of NCP 9 and 14 are shown in Fig. 3. CV plots for rest ofthe NCPs are given in the Supplementary Material. Typically,for NCPs 9–13, there is one irreversible reduction peak that
appeared in the range of �1.27 to �1.44V depending on thecompounds, and two reversible waves were observed onthe negative side (Table 5). In the case of NCP 10, two reversiblereduction waves were observed at �0.83 and �1.09V. The
NCPs 9, 11–13 showed one quasi-reversible anodic peak inthe range of 1.44–1.56V and two irreversible oxidation peaksaround 1.25 and 0.87V. The N-methyl NCP 14 showed one
quasi-reversible reduction peak at �1.48V and three irrevers-ible peaks in the range of�0.45 to�1.19V. The N-methyl NCP
15 showed four irreversible reduction peaks in the range of
�0.48 to �1.50V. Both NCPs 14 and 15 showed two irrevers-ible oxidation peaks in the range of 0.73–1.08V. Also, 14showed one reversible oxidation peak at 1.42V and 15 showed
one quasi-reversible oxidation peak. In general, all NCPs 9–15showed three oxidation peaks, whereas NCTPP showed fouroxidation peaks (Table 5). In contrast to NCTPP,[23] which onlyshowed one reduction wave at�0.87V, the NCPs 9–13 showed
three reduction peaks and NCPs 14 and 15 showed fourreduction peaks. Close inspection of Table 5 revealed that theN-methylated NCPs 14 and 15 have experienced anodic shifts
when compared with other free NCPs 9–13 and therefore theyare easy to oxidize.
Conclusion
A series of difunctionalized N-confused porphyrins (A2B2 type)have been synthesized by [3þ1] condensation reaction andnickel complexes of some of the A2B2-type porphyrins wereprepared. A new key precursor (N-methyl pyrrolediol 8) was
prepared and further utilized to prepare N-methyl A2B2-typeNCPs via a similar approach. Fluorescence studies showed blue-shifted emission maxima for reported NCPs when compared
with NCTPP, and quantum yields were in agreement with theNCTPP value. Electrochemical studies revealed easier oxida-tions of N-methyl NCPs 14 and 15 when compared with other
NCP analogues.
Experimental
General
Unless otherwise mentioned, all the reagents and solvents werepurchased from Aldrich, Across Organics, or Merck, and usedwithout further purification. Pyrrole was distilled under vacuum
before use. Silica gel (60–120 mesh size) and neutral alumina(Brockmann Grade I-II) that were used for column chroma-tography were procured from Merck. The tripyrranes 1–5 were
visualized upon exposure of the TLC plate to Br2 vapours.Solution NMR spectroscopy of compounds 9 and 10 was per-formed on a Varian 300MHz NMR instrument at SAIF, CentralDrug & Research Institute, Lucknow (India). Also, NMR
spectra of compounds 4–8 and 11–15were recorded on a BrukerAvance III 500MHz NMR spectrometer at IIT Gandhinagar.The HRMS data for all compounds 4, 5, and 8–15were obtained
(in negative or positive ionmode) on aWaters Synapt-G2S ESI-Q-TOF Mass instrument at IIT Gandhinagar. UV–Vis absorp-tion spectra were recorded on Shimadzu UV-1700 and IR
spectral data was obtained on Thermo Scientific Nicolet iS10 atIIT Gandhinagar. Cyclic voltammetry studies were carried outon an electrochemical system utilizing a three-electrode con-
figuration consisting of a glassy carbon (working electrode),platinum wire (auxiliary electrode), and saturated calomel(reference electrode; SCE) electrodes. The experiments weredone in dry dichloromethane using 0.1M tetrabutylammonium
perchlorate as supporting electrolyte. Half-wave potentials werecalculated manually by taking the average of the cathodic andanodic peak potentials. All potentials were calibrated versus
saturated calomel electrode by the addition of ferrocene as aninternal standard, taking E1/2 (Fc/Fc
þ)¼ 0.51V versus SCE.
General Procedure for Tripyrranes 1–5
The appropriate functional aryl aldehyde (1 equiv.) wasdissolved in pyrrole (5 equiv.) under inert atmosphere and stir-red for 5min at room temperature. Then, TFA (0.1 equiv.) was
�2.0 2.0
14
9
�1.5 1.5�1.0 1.0�0.5 0.50
Potential [V] versus SCE
Fig. 3. Cyclic voltammograms of NCPs 9 and 14 in DCM, containing
0.1M tetrabutylammoniumperchlorate (TBAP) as supporting electrolyte
recorded at a scan speed of 50mVs�1 (V versus SCE).
Table 5. Electrochemical redox data of NCPs 9–15 inDCMcontaining
0.1M TBAP as supporting electrolyte recorded at 50mV s�1
I–IV are first–fourth oxidation and reduction potentials respectively
Entry E1/2(oxidation) [V]
versus SCE
E1/2(reduction) [V]
versus SCE
I II III IV I II III IV
9 1.52 1.25 0.87 – �0.64 �1.24 �1.34 –
10 1.56 1.26 0.98 – �0.83 �1.09 �1.42 –
11 1.55 1.18 0.96 – �0.78 �1.28 �1.44 –
12 1.57 1.29 0.89 – �0.74 �1.16 �1.41 –
13 1.44 1.27 1.04 – �0.84 �1.10 �1.27 –
14 1.42 1.08 0.74 – �0.45 �0.93 �1.19 �1.48
15 1.32 1.04 0.73 – �0.48 �0.84 �1.16 �1.50
NCTPPA 1.85 1.47 1.25 0.87 – �0.87 – –
ATaken from ref. 21.
F S. Das et al.

added and stirred for 20min. Then, the reaction mixture was
diluted with DCM (50mL) followed by addition of 0.1Naqueous NaOH solution. The mixture was extracted with DCMand washed with brine, and the organic layer was dried over
Na2SO4. The solvent was evaporated using a rotary evaporatorand excess pyrrole was removed under vacuum. The mixturethat predominantly contained dipyrrane and tripyrrane wassubjected to silica gel column chromatography and the desired
tripyrranes 1–5 were collected in 40–80% DCM/petroleumether solvent mixture. Evaporation of the solvent resulted in adark brown oil that turned into a brown flaky solid of tripyrranes
(4–15% yield) upon vacuum drying.
5,10-Bis(m-bromophenyl)tripyrromethane (1)
m-Bromobenzaldehyde (2.0 g, 10.809mmol), pyrrole(11.24mL, 54.048mmol), and TFA (241 mL, 1.081mmol) werereacted as per the aforementioned procedure to obtain the title
compound (900.0mg, 15%). nmax (neat)/cm�1 3425 (NH),2360, 1694, 1589, 1566, 1471, 1423, 1297, 1264, 1165, 1090,997, 883, 717. dH (CDCl3, 500MHz) 7.93 (2H, br s, pyrrole
N-H), 7.75 (1H, br s, pyrrole N-H), 7.39–7.11 (8H, m, Ar-H),6.72 (2H, s, pyrrole b-H), 6.15 (2H, q, J 3, pyrrole b-H), 5.90(2H, d, pyrroleb-H), 5.75 (2H, s, pyrrole a-H), 5.38–5.30 (2H, s,C-H). dC (CDCl3, 125MHz) 151.74, 144.32, 144.30, 131.90,
131.51, 131.37, 130.17, 127.02, 122.71, 117.61, 108.56, 107.83,107.40. m/z 536.1661. HRMS (ESI-Q TOF) Anal. Calc. forC26H22Br2N3
þ [MþH]þ 536.2810.
5,10-Bis(p-nitrophenyl)tripyrromethane (2)
p-Nitrobenzaldehyde (3.0 g, 19.852mmol), pyrrole (6.9mL,99.259mmol), and TFA (148mL, 1.985mmol) were reacted by
following the general procedure to afford the title compound(1.1 g, 12%). nmax (neat)/cm
�1 3406 (NH), 1594, 1514, 1111,861, 773, 728. dH (CDCl3, 500MHz) 8.13 (4H, d, J 8.5, Ar-H),
8.03 (2H, br s, pyrrole N-H), 7.94 (1H, br s, pyrrole N-H), 7.34(4H, d, J 8.5, Ar-H), 6.74 (2H, s, pyrrole b-H), 6.16 (2H, m,pyrrole b-H), 5.85 (2H, s, pyrrole b-H), 5.71 (2H, t, J 2.5,pyrrole a-H), 5.49 (2H, s, C-H). dC (CDCl3, 125MHz) 149.91,
149.62, 146.83, 131.60, 130.93, 130.75, 129.79, 129.25, 126.89,123.77, 118.61, 117.74, 111.80, 108.73, 108.15. m/z 467.1566.HRMS (ESI-Q TOF) Anal. Calc. for C26H21N5O4
þ [M]þ
467.1594.
5,10-Bis(p-tolyl)tripyrromethane (3)
p-Tolualdehyde (6.8mL, 57.654mmol), pyrrole (20.0mL,
288.269mmol), and TFA (420 mL, 5.765mmol) were mixed asper the general procedure to afford the title compound (1.0 g,4%. nmax (neat)/cm�1 3425 (NH), 2360, 1694, 1589, 1566,
1471, 1423, 1297, 1264, 1165, 1090, 997, 883, 717. dH (CDCl3,500MHz) 7.93 (2H, br s, pyrrole N-H), 7.75 (1H, br s, pyrroleN-H), 7.39–7.11 (8H, m, Ar-H), 6.72 (2H, s, pyrrole b-H), 6.15(2H, q, J 3, pyrrole b-H), 5.90 (2H, d, pyrrole b-H), 5.75 (2H, s,pyrrole a-H), 5.38–5.30 (2H, s, C-H). dC (CDCl3, 125MHz)139.18, 136.47, 132.83, 132.48, 132.37, 129.26, 129.17, 129.05,128.95, 128.27, 117.04, 108.30, 107.23, 107.13, 106.98, 21.06.
m/z 405.2982. HRMS (ESI-Q TOF) Anal. Calc. for C28H28N3þ
[M]þ 405.2205.
5,10-Bis(p-bromophenyl)tripyrromethane (4)
p-Bromobenzaldehyde (4.0 g, 21.745mmol), pyrrole (7.5mL,108.725mmol), and TFA (161 mL, 2.162mmol) were reactedby following the general procedure to afford the title compound
(1.1 g, 10%). nmax (neat)/cm�1 3427 (NH), 1581, 1485, 1402,
1264, 1160, 1114, 1071, 1010, 958, 884, 847, 761. dH (CDCl3,500MHz) 7.91 (2H, br s, pyrrole N-H), 7.75 (1H, br s, pyrroleN-H), 7.42 (4H, d, J 8.5, Ar-H), 7.05 (4H, m, Ar-H), 6.69 (2H,
s, pyrrole b-H), 6.13 (2H, t, J 3, pyrrole b-H), 5.83 (2H, s,pyrrole b-H), 5.72 (2H, s, pyrrole a-H), 5.31 (2H, d, C-H). dC(CDCl3, 125MHz) 151.75, 141.2, 132.99, 132.73, 130.65,121.29, 121.08, 118.47, 117.58, 111.41, 108.53, 107.67, 107.41,
107.04. m/z 536.2322. HRMS (ESI-Q TOF) Anal. Calc. forC26H22Br2N3
þ [MþH]þ 536.2810.
5,10-Bis(p-iodophenyl)tripyrromethane (5)
p-Iodobenzaldehyde (2.0 g, 8.623mmol), pyrrole (3.0mL,43.115mmol), and TFA (64mL, 0.862mmol) were mixedtogether as per the general procedure to afford the title
compound (433.0mg, 8%). nmax (neat)/cm�1 3366 (NH), 2954,
2923, 2852, 1706, 1560, 1581, 1482, 1399, 1060, 1006, 964,846, 776, 760, 722. dH (CDCl3, 500MHz) 7.90 (3H, br s, pyrrole
N-H), 7.62 (4H, d, J 8,Ar-H), 6.92 (4H, d, J 8.5,Ar-H), 6.69 (2H,s, pyrrole b-H), 6.13 (2H, s, pyrrole b-H), 5.83 (2H, s, pyrroleb-H), 5.72 (2H, s, pyrrole a-H), 5.31 (2H, s, C-H). dC (CDCl3,125MHz) 141.08, 132.00, 131.88, 131.67, 131.34, 130.51,
130.08, 129.60, 129.21, 120.86, 120.18, 117.62, 108.52, 108.29,107.65. m/z 630.1163. HRMS (ESI-Q TOF) Anal. Calc. forC26H22I2N3
þ [MþH]þ 629.9903.
N-Methylpyrrole-2,4-pentafluorophenylbiscarbinol (8)
Mg turnings (875.093mg, 36.485mmol) were added to THF(50mL) in a three-neck flask under nitrogen atmosphere.Bromopentafluorobenzene (4.55mL, 36.485mmol), dissolvedin THF (40mL), was added dropwise to the reaction mixture at
08C. The reaction mixture was stirred for 6 h under nitrogenatmosphere at room temperature. The reaction flask wasprotected from light. 2,4-Bisformyl-N-methylpyrrole (1 g,
7.297mmol), dissolved in THF (50mL), was added dropwise tothe reaction mixture at 08C under protective atmosphere. Thereaction mixture was stirred at 08C for 12 h under nitrogen
atmosphere away from light. After that, aqueous NH4Cl wasadded at 08C and stirred for 30min. The entire reaction mixturewas filtered through celite and the filtrate was extracted with
diethyl ether. After drying over anhydrous Na2SO4, the reactionmixture was dried in a rotary evaporator to afford diol 8 (1.59 g,46%). dH (CDCl3, 500MHz) 6.57 (2H, d, J 10, pyrrole a-H),6.08 (1H, s, pyrrole b-H), 6.00 (1H, s, C-H), 5.86 (1H, s, C-H),
3.73 (3H, s, pyrrole Me-H). dC (CDCl3, 125MHz) 151.75,138.72, 136.69, 131.26, 122.96, 122.26, 106.95, 34.31. m/z474.2077. HRMS (ESI-Q TOF) Anal. Calc. for C19H10F10NO2
[MþH]þ 474.0552.
5,20-Bis(pentafluorophenyl)-10,15-bis(m-bromophenyl)N-Confused Porphyrin (9)
In a 1000mL two-neck flask, 5,10-bis(m-bromophenyl)tripyrromethane (400.0mg, 0.747mmol) and pyrrole-2,4-
pentafluorophenylbiscarbinol (0.747mmol, 340.6mg) weredissolved in DCM (748mL). Methanesulfonic acid (17 mL) wasadded in the reaction flask and the mixture was stirred under
nitrogen for 1.5 h in the dark. After 1.5 h, DDQ (2.242mmol,508.9mg) was added and stirred for 15min under aerobic con-ditions. Then, the reaction mixture was passed through a small
silica gel column to remove excess DDQ using 2–5%methanol/DCM as the solvent mixture. The desired NCP was furtherpurified by neutral alumina column and collected with 20–30%
Difunctionalized N-Confused Porphyrins G

DCM/petroleum ether. Evaporation of the solvent using a rotary
evaporator afforded the title NCP (36.0mg, 5%). nmax (neat)/cm�1 3357 (NH), 3059, 2927, 1651, 1589, 1559, 1518, 1496,1149, 1026, 988, 893, 920, 798. dH (CDCl3, 300MHz) 8.97 (1H,
s, pyrrole a-H), 8.88 (1H, d, J 4.8, pyrrole b-H), 8.81 (1H, d, J 6,pyrrole b-H), 8.72 (2H, t, J 5.1, Ar-H), 8.60 (2H, q, J 4.8, Ar-H),8.31 (2H, d, J 4.2, Ar-H), 8.08 (2H, dd, J 7.2, 6.3, Ar-H), 7.95 (2H, d, J 7.8, Ar-H), 7.66 (2H, m, Ar-H), �2.76 (2H, br s,
pyrrole inner N-H), �5.48 (1H, s, pyrrole inner C-H). dC(CDCl3, 125MHz) 147.05, 145.04, 142.82, 138.75, 137.08,137.04, 136.99, 136.65, 135.89, 135.26, 133.05, 133.00, 132.94,
131.60, 131.54, 128.26, 128.13, 127.23, 121.55, 120.08, 118.33,109.17. m/z 950.9655. HRMS (ESI-Q TOF) Anal. Calc. forC44H17Br2F10N4
� [M�H]� 951.4246.
5,20-Bis(pentafluorophenyl)-10,15-bis(p-nitrophenyl)N-Confused Porphyrin (10)
5,10-Bis(p-nitrophenyl)tripyrromethane (200.0mg, 0.428mmol)
and pyrrole-2,4-pentafluorophenylbiscarbinol (196.5mg,0.428mmol) were dissolved in DCM (430mL) in a 500mLflask. Methanesulfonic acid (8 mL) was added to the flask and
the mixture was stirred for 1.5 h in the dark. DDQ(291.3mg, 1.283mmol) was added and stirred for 20min underatmospheric conditions. A quick silica gel column chromatog-raphy was performed to filter off excess DDQ. The desired NCP
was obtained following further purification by alumina columnchromatography and elution with 20–30% DCM/petroleumether (12.0mg, 3%). nmax (neat)/cm
�1 3385 (NH), 2927, 2854,
1702, 1651, 1594, 1583, 1519, 1497, 1345, 1108, 987, 847, 797,775. dH (CDCl3, 500MHz) 8.65 (6H, m, pyrrole-H), 8.45 (3H,m, pyrrole b-H), 8.34 (8H, m, Ar-H), 6.95 (1H, s, pyrrole b-H),�1.74 (2H, br s, pyrrole inner N-H),�4.89 (1H, s, pyrrole innerC-H). dC (CDCl3, 125MHz) 150.79, 148.13, 148.08, 147.59,147.51, 146.98, 145.04, 139.61, 138.83, 137.3, 136.79, 135.63,
135.16, 135.03, 127.85, 127.60, 127.25, 126.74, 122.37, 122.33,119.11, 117.27, 110.04, 105.68, 100.69. m/z 883.1116. HRMS(ESI-Q TOF) Anal. Calc. for C44H17F10N6O4
� [M�H]�
883.6276.
5,20-Bis(pentafluorophenyl)-10,15-bis(p-tolyl)N-Confused Porphyrin (11)
In a 1000mL two-neck flask, to a mixture of 5,10-bis(p-tolyl)tripyrromethane (220.0mg, 0.543mmol) and pyrrole-2,4-
pentafluorophenylbiscarbinol (249.2mg, 0.543mmol), DCM(540mL) was added. Methanesulfonic acid (12mL) was addedto the reaction flask and the mixture was stirred under nitrogen
for 1.5 h in the dark. Then, DDQ (369.7mg, 1.629mmol) wasadded and stirred for 15min under aerobic conditions. Then, thereaction mixture was passed through a short pad of silica gel to
filter off excess DDQ using 2–5% methanol/DCM solventmixture. The solvent was evaporated using a rotary evaporator.Subsequent alumina column chromatography of the reactionmixture in 30%DCM/petroleum ether solvent mixture afforded
the desired NCP (19.0mg, 4%). nmax (neat)/cm�1 3414 (NH),
2923, 2854, 1605, 1574, 1519, 1497, 1248, 1174, 1036, 987,799. dH (CDCl3, 500MHz) 8.99 (1H, s, pyrrole a-H), 8.85 (1H,s, pyrrole b-H), 8.75 (3H, s, pyrrole b-H), 8.62 (2H, s, pyrroleb-H), 8.02 (4H, s, Ar-H), 7.56 (4H, s, Ar-H), 2.69 (6H, s,Me-H),�2.48 (2H, br s, pyrrole inner N-H),�5.49 (1H, s, pyrrole inner
C-H). dC (CDCl3, 125MHz) 149.21, 147.68, 147.09, 145.47,145.09, 138.95, 138.72, 138.54, 137.47, 135.92, 135.31, 134.49,130.92, 128.85, 127.86, 124.48, 123.99, 122.27, 120.54, 119.11,
119.02, 118.88, 99.26, 21.51. m/z 821.1779. HRMS (ESI-Q
TOF) Anal. Calc. for C46H23F10N4� [M�H]� 821.6857.
5,20-Bis(pentafluorophenyl)-10,15-bis(p-bromophenyl)N-Confused Porphyrin (12)
In a 500mL two-neck flask, to a mixture of 5,10-bis(p-bromo-phenyl)tripyrromethane (200.0mg, 0.374mmol) and pyrrole-
2,4-pentafluorophenylbiscarbinol (171.6mg, 0.374mmol) DCM(374mL) was added. Methanesulfonic acid (8 mL) was added tothe reaction flask and the reaction mixture was stirred under
nitrogen for 1.5 h at room temperature. After this, DDQ(264.5mg, 1.121mmol) was added and stirred for 15min underatmospheric conditions. A quick silica filtration column chro-
matography was performed to filter off excess DDQ and thedesired porphyrin was eluted with 5% methanol/DCM solventmixture. Subsequent alumina column chromatography wasperformed to purify the compound and the desired NCP was
collected in 20–30%DCM/petroleum ether (26.0mg, 7%). nmax
(neat)/cm�1 3416 (NH), 2923, 2850, 2360, 1577, 1519, 1127,987, 930, 909, 798. dH (CDCl3, 300MHz) 8.64 (2H, m, pyrrole-
H), 8.50 (2H, m, pyrrole N-H), 8.42 (1H, d, J 4.5, pyrrole a-H),8.00 (4H, d, J 8.1, Ar-H), 7.89 (4H, d, J 8.1, Ar-H), 6.14 (2H, m,pyrrole b-H),�1.90 (2H, br s, pyrrole inner N-H),�5.02 (1H, s,
pyrrole inner C-H). dC (CDCl3, 125MHz) 157.74, 156.50,147.09, 145.03, 139.85, 138.71, 137.55, 136.63, 135.78, 135.13,132.48, 130.89, 130.41, 128.82, 128.07, 127.29, 126.77, 124.48,
123.99, 123.07, 120.52, 119.11, 118.71, 109.05, 99.66. m/z950.9665. HRMS (ESI-Q TOF) anal. Calc. for C44H17Br2F10N4
�
[M–H]� 951.4246.
5,20-Bis(pentafluorophenyl)-10,15-bis(p-iodophenyl)N-Confused Porphyrin (13)
In a 1000mL two-neck flask, 5,10-bis(p-iodophenyl)tripyrro-methane (350.0mg, 0.556mmol) and pyrrole-2,4-pentafluoro-phenylbiscarbinol (255.4mg, 0.556mmol) were dissolved in
DCM (556mL). Methanesulfonic acid (13mL) was added to thereaction flask and the reaction mixture was stirred under nitrogenfor 1.5 h at room temperature. After this, DDQ (378.8mg,1.669mmol) was added and stirred for 15min under atmospheric
conditions. Then, the reactionmixturewas passed through a smallsilica gel column to filter off excess DDQ with 4% methanol/DCM solvent mixture. The compound was further purified by
alumina column chromatography and the desired NCP waseluted with 20–30% DCM/petroleum ether. Evaporation of thesolvent afforded pure NCP (29.0mg, 5%). nmax (neat)/cm�1
3414 (NH), 3046, 2955, 2922, 2852, 1702, 1651, 1518, 1472,1387, 1361, 1345, 1206, 1081, 987, 960, 798, 743. dH (CDCl3,500MHz) 8.96 (1H, s, pyrrole a-H), 8.88 (1H, d, J 4.5, pyrrole
b-H), 8.80 (1H, d, J 5.0, pyrrole b-H), 8.72 (2H, dd, J 5.0, 5.0,pyrrole b-H), 8.59 (2H, q, J 4.5, 4.5, pyrrole b-H), 8.11 (4H, m,Ar-H), 7.87 (4H, m, Ar-H),�2.78 (2H, br s, pyrrole inner N-H),�5.51 (1H, s, pyrrole inner C-H). dC (CDCl3, 125MHz) 147.67,
147.62, 147.09, 146.29, 138.53, 138.46, 137.27, 136.34, 136.13,136.03, 135.98, 132.31, 130.91, 129.70, 128.84, 128.07, 124.47,123.98, 119.10, 86.04. m/z 1044.9266. HRMS (ESI-Q TOF)
Anal. Calc. for C44H17F10N6O4� [M�H]� 1045.4256.
5,20-Bis(pentafluorophenyl)-10,15-bis(m-bromophenyl)N-Methyl N-Confused Porphyrin (14)
5,10-Bis(m-bromophenyl)tripyrromethane (100mg, 0.188mmol)was dissolved in DCM (188mL), followed by the addition ofN-methylpyrrole-2,4-pentafluorophenylbiscarbinol (86.298mg,
H S. Das et al.

0.188mmol).After 5min, trifluoroacetic acid (13mL, 0.188mmol,
dissolved in DCM) was added and the reaction was allowed tostir at room temperature for 1.5 h under nitrogen and away fromlight. Then, DDQ (128.034mg, 0.564mmol) was added and the
reaction mixture was stirred for 15min under atmosphericconditions. The entire reactionmixture was then filtered througha small silica gel column to filter off excess DDQ. The desiredcompound was eluted with 30% DCM/petroleum ether using
neutral alumina column chromatography. Removal of thesolvents gave the desired NCP (9.6mg, 5.3%). dH (CDCl3,500MHz) 8.66 (2H, br s, pyrrole b-H), 8.52 (3H, d, J 14.0,
Ar-H), 8.40 (1H, s, pyrroleb-H), 8.30 (2H, d, J 16.5, Ar-H), 8.07(2H, t, J 6, J 6.5, pyrrole b-H), 7.93 (2H, s, pyrrole b-H), 7.63(3H, s, Ar-H), 3.36 (3H, s, pyrrole Me-H),�5.38 (1H, s, pyrrole
inner C-H). dC (CDCl3, 125MHz) 154.27, 143.00, 142.88,139.16, 138.31, 137.56, 137.11, 136.86, 136.16, 134.75, 133.81,131.57, 131.45, 130.54, 128.57, 128.24, 127.72, 126.94, 125.20,123.33, 122.03, 119.12, 101.32, 29.71. m/z 966.9523. HRMS
(ESI-Q TOF) Anal. Calc. for C45H21Br2F10N4 [Mþ2]þ
966.9953.
5,20-Bis(pentafluorophenyl)-10,15-bis(p-iodophenyl)N-methyl N-Confused Porphyrin (15)
5,10-Bis(p-iodophenyl)tripyrromethane (200mg, 0.318mmol)and N-methylpyrrole-2,4-pentafluorophenylbiscarbinol (150.429mg, 0.318mmol) was dissolved in DCM (320mL). TFA
(14 mL, 0.191mmol) was added to the reaction mixture andstirred for 2 h under nitrogen and away from light. Then, DDQ(216.568mg, 0.954mmol) was added to the reaction flask and
stirred for 20min under atmospheric conditions. The reactionmixture was filtered through a small silica gel column to filteroff excess DDQ using 2–5% methanol/DCM mixture. Thecrude compound was subjected to column chromatography
using neutral alumina for purification. The desired compoundwas eluted with 25–30%DCM/petroleum ether. Evaporation ofthe solvent mixture afforded the desired NCP (14.2mg, 4.2%).
dH (CDCl3, 500MHz) 8.57 (2H, br s, pyrrole b-H), 8.44 (3H, m,pyrrole b-H), 8.02 (4H, t, J 6, J 5.5, Ar-H), 7.79 (4H, dd, J 7, J 8,Ar-H), 7.18 (2H, s, pyrrole b-H), 3.28 (3H, s, pyrrole Me-H),
�5.46 (1H, s, pyrrole inner C-H). dC (CDCl3, 125MHz) 148.13,148.08, 147.58, 147.50, 147.08, 145.01, 139.62, 138.84, 137.63,136.79, 135.64, 135.14, 135.03, 135.01, 127.85, 127.60, 127.26,
126.74, 124.47, 123.99, 122.37, 119.10, 117.27, 110.04, 105.68,100.68, 29.70.m/z 1060.9418. HRMS (ESI-Q TOF) Anal. Calc.for C45H21F10I2N4 [M]þ 1060.9696.
Nickel(II) Complex of N-Confused Porphyrin (9-Ni)
NCP 9 (80mg, 0.085mmol) was dissolved in toluene (16mL)and ethanol (5mL) and stirred for 5min. Nickel(II) acetate
(212.357mg, 0.85mmol) was added to the reaction mixture thatwas refluxed for 4 h. Completion of the metallation reaction wasconfirmed by TLC, which showed a new spot corresponding tothe nickel complex and traces of free NCP. The reactionmixture
was dried on a rotary evaporator and the crude compoundwas loaded on neutral alumina column. The desired nickelcompound was eluted with 80% DCM/petroleum ether mixture
and solvent removal afforded 9-Ni (70.0mg, 83%). dH (CDCl3,500MHz) 10.11 (1H, br s, pyrrole N-H), 8.45 (1H, s, pyrrolea-H), 7.97 (2H, d, J 13.5, Ar-H), 7.85 (6H, m, Ar-H and pyrrole
b-H), 7.75 (4H, m, Ar-H), 7.47 (2H, d, J 6.5, pyrrole b-H).dC (CDCl3, 125MHz) 154.60, 152.58, 150.76, 149.16, 145.94,144.64, 143.39, 142.68, 135.79, 135.76, 134.57, 133.05, 132.57,
131.90, 131.01, 130.93, 130.57, 128.74, 128.64, 126.56, 121.55,
121.46, 118.13, 116.67, 115.53. m/z 1008.9069. HRMS (ESI-QTOF) Anal. Calc. for C44H19Br2F10N4Ni [Mþ4]þ 1008.8857.
Nickel(II) Complex of N-Confused Porphyrin (12-Ni)
NCP 12 (50mg, 0.053mmol) was dissolved in toluene (10mL)and ethanol (4mL) and stirred for 5min. Nickel(II) acetate(131.89mg, 0.53mmol) was added to the reaction mixture thatwas refluxed for 4 h. Then, the crude reaction mixture was
subjected to neutral alumina column chromatography and thedesired compound was eluted with 80% DCM/petroleum ethermixture. Removal of the solvents afforded 12-Ni (37.0mg,
70%). dH (CDCl3, 500MHz) 10.06 (1H, s, pyrrole N-H), 8.47(1H, s, pyrrole a-H), 7.87 (4H, m, pyrrole b-H), 7.84 (1H, d,J 5.0, pyrroleb-H), 7.79 (1H, d, J 5.0, pyrroleb-H), 7.73 (4H,m,
Ar-H), 7.69–7.64 (4H, dd, J 8.5, 8.0, Ar-H). dC (CDCl3,125MHz) 154.77, 152.75, 151.76, 150.82, 149.25, 146.07,144.71, 143.00, 142.98, 139.54, 134.58, 134.46, 133.06, 132.37,
130.50, 126.55, 124.10, 123.53, 122.25, 118.41, 116.96, 115.93,114.08, 113.38, 106.30. m/z 1008.0396. HRMS (ESI-Q TOF)Anal. Calc. for C44H19Br2F10N4Ni [Mþ4]þ 1008.8857.
Nickel(II) Complex of N-Confused Porphyrin (13-Ni)
NCP 13 (36mg, 0.034mmol) was dissolved in toluene (9mL)and ethanol (4mL) and stirred for 5min. Nickel(II) acetate(84.606mg, 0.34mmol) was added to the reaction mixture that
was refluxed for 4 h. After completion of the reaction, the sol-vent was removed with a rotary evaporator and neutral aluminacolumn chromatography (70% DCM/petroleum ether) was
performed to isolate the desired compound (27.0mg, 72%). dH(CDCl3, 500MHz) 9.65 (1H, br s, pyrrole N-H), 8.56 (1H, s,pyrrole a-H), 7.93 (4H, m, Ar-H), 7.88 (2H, d, J 5.0, pyrroleb-H), 7.85 (2H, s, pyrrole b-H), 7.76 (2H, s, pyrrole b-H), 7.54(4H, dd, J 8.0, 8.0, Ar-H). dC (CDCl3, 125MHz) 140.46, 139.30,137.61, 136.43, 136.34, 134.79, 130.42, 129.74, 128.81, 126.33,126.07, 125.13, 125.01, 124.32, 123.98, 122.56, 119.33, 118.97,
117.63, 114.08, 111.31, 92.99. m/z 1101.8424. HRMS (ESI-QTOF) Anal. Calc. for C44H16F10I2N4Ni [MþH]þ 1101.858.
Supplementary Material
Characterization data including ESI-MS, 1H-NMR spectra, and
cyclic voltammograms of selected compounds are available assupporting information on the Journal’s website.
Acknowledgements
IG gratefully acknowledges DST (New Delhi), CSIR, (New Delhi), and IIT
Gandhinagar for financial support. IG thanks IIT Bombay, Chemistry
Department for the cyclic voltammetry studies. PEK thanksCSIR for project
fellowship. SD andNB are thankful to IITGandhinagar for their fellowships.
References
[1] H. Furuta, T. Asano, T. Ogawa, J. Am. Chem. Soc. 1994, 116, 767.doi:10.1021/JA00081A047
[2] P. J. Chmielewski, L. Latos-Grazynski, K. Rachlewicz, T. Głowiak,
Angew. Chem., Int. Ed. Engl. 1994, 33, 779. doi:10.1002/ANIE.199407791
[3] H. Furuta, T. Ishizuka, A. Osuka, H. Dejima, H. Nakagawa,
Y. Ishikawa, J. Am. Chem. Soc. 2001, 123, 6207. doi:10.1021/JA010237A
[4] J. D. Harvey, C. J. Ziegler, Coord. Chem. Rev. 2003, 247, 1.doi:10.1016/J.CCT.2003.07.001
[5] M. J. Chmielewski, M. Pawlicki, N. Sprutta, L. Szterenberg, L. L.
Grazynski, Inorg. Chem. 2006, 45, 8664. doi:10.1021/IC061091P
Difunctionalized N-Confused Porphyrins I

[6] M. Toganoh, H. Furuta, Chem. Commun. 2012, 48, 937. doi:10.1039/C1CC14633E
[7] J. P. Belair, C. J. Ziegler, C. S. Rajesh, D. A.Modarelli, J. Phys. Chem.
A 2002, 106, 6445. doi:10.1021/JP025569W[8] J. S. Lindsey,Acc. Chem. Res. 2010, 43, 300. doi:10.1021/AR900212T[9] G. R. Geier III, M. H. Denise, J. S. Lindsey, Org. Lett. 1999, 1, 1455.
doi:10.1021/OL9910114[10] H. Maeda, A. Osuka, Y. Ishikawa, I. Aritome, Y. Hisaeda, H. Furuta,
Org. Lett. 2003, 5, 1293. doi:10.1021/OL034227L[11] H. Furuta, T. Morimoto, A. Osuka, Org. Lett. 2003, 5, 1427.
doi:10.1021/OL034172N[12] S. A. Wolff, E. A. Alema’n, D. Banerjee, P. L. Rinaldi,
D. A. Modarelli, J. Org. Chem. 2004, 69, 4571. doi:10.1021/JO049621R
[13] R. Acharya, L. Paudel, J. Joseph, C. E. McCarthy, V. R. Dudipala,
J. M. Modarelli, D. A. Modarelli, J. Org. Chem. 2012, 77, 6043.doi:10.1021/JO300810N
[14] J.-W. Ka, C.-H. Lee, Tetrahedron Lett. 2000, 41, 4609. doi:10.1016/S0040-4039(00)00672-9
[15] S. Cadamuro, I. Degani, S. Dughera, R. Fochi, A. Gatti, L. Piscopo,
J. Chem. Soc., Perkin Trans. 1 1993, 273. doi:10.1039/P19930000273[16] C. J. Medforth, in The Porphyrin Handbook (Eds K. M. Kadish,
K. M. Smith, R. Guilard) 2000, Vol. 5, Ch. 35, pp. 1–80. (Academic
Press: San Diego)
[17] J. L. Shaw, S. A. Garrison, E. A. Alema’n, C. J. Ziegler, D. A.
Modarelli, J. Org. Chem. 2004, 69, 7423. doi:10.1021/JO049199E[18] P. J. Chmielewski, L. L. Grazynski, J. Chem. Soc., Perkin Trans. 2
1995, 503. doi:10.1039/P29950000503[19] W. Qu, T. Ding, A. Cetin, J. D. Harvey, M. J. Taschner, C. J. Ziegler,
J. Org. Chem. 2006, 71, 811. doi:10.1021/JO052188G[20] W.-P. Chang, W.-C. Lin, J.-H. Chen, S.-S. Wang, J.-Y. Tung, Dalton
Trans. 2012, 41, 13454. doi:10.1039/C2DT31426F[21] S. Cho, Y. Lee, H. S. Han, H. K. Lee, S. Jeon, J. Phys. Chem. A 2014,
118, 4995. doi:10.1021/JP505072X[22] P. K. Goldberg, T. J. Pundsack, K. E. Splan, J. Phys. Chem. A 2011,
115, 10452. doi:10.1021/JP205309F[23] H. Furuta, T. Ogawa, Y. Uwatoko, K. Araki, Inorg. Chem. 1999, 38,
2676. doi:10.1021/IC9901416
J S. Das et al.





























