Embed Size (px)
Citation preview

DIAGNOSTICO Y MANEJO DE LOS ERRORES
INNATOS DEL METABOLISMO
Dr. Manuel Saborío Rocafort
SERVICIO DE GENÉTICA MÉDICA Y METABOLISMO
PROGRAMA NACIONAL DE TAMIZAJE
HOSPITAL NACIONAL DE NIÑOS
“Dr. Carlos Sáenz Herrera”
0
5000
10000
15000
20000
25000N
úm
ero
de
en
tra
da
s e
n M
IM
Año
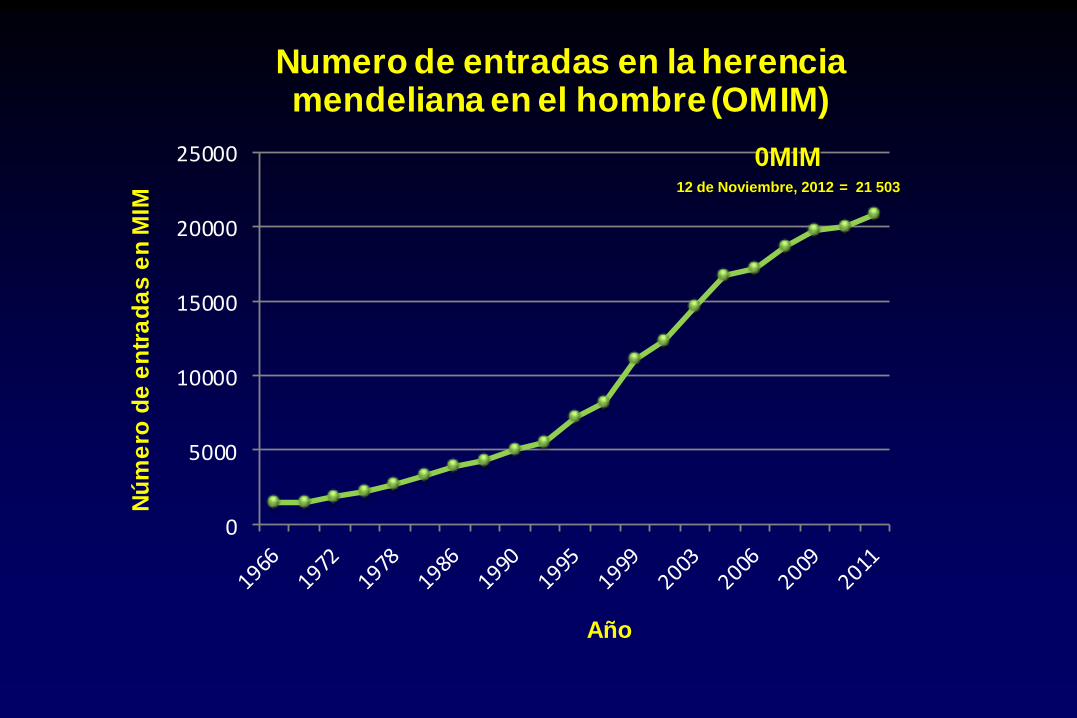
Numero de entradas en la herencia mendeliana en el hombre (OMIM)
0MIM 12 de Noviembre, 2012 = 21 503


• Metabolismo es la suma total de todas las reacciones químicas que
ocurren como parte del proceso continuo de renovación y
eliminación en el cuerpo:
• Conversión Degradación
• Producción Transporte
• Más de 600 errores congénitos del metabolismo se conocen hoy en
día, cada uno poco frecuente pero que colectivamente tienen una
frecuencia estimada de 1:650 y tienen un gran impacto en la
morbilidad y mortalidad del ser humano
• Nuestro conocimiento de los genes involucrados no es completo, a
la fecha el análisis molecular no puede sustituir al estudio
bioquímico en el diagnóstico de estas enfermedades
A Fuera A Dentro
1
B
Apoenzima
+
cofactor
Holoenzima
4
3
D
E F
6
2
5
-
C
Membrana
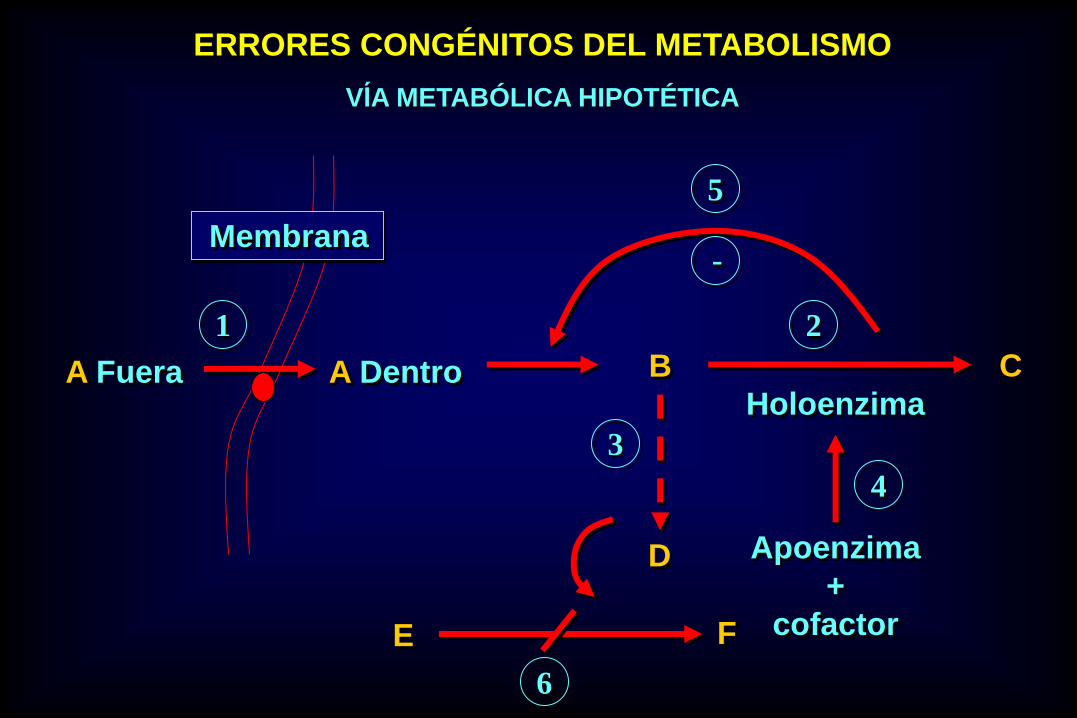
ERRORES CONGÉNITOS DEL METABOLISMO
VĺA METABÓLICA HIPOTÉTICA
1900 1910 1920 1930 1940 1950 1960 1970 1980 1990 2000 2010
Concepto de error innato
Era del Analito
Era Enzimática
Era de las Alteraciones Enzimáticas
Era de los Anticuerpos
Era del ADN
Era Genómica
Ratones Knockout
Era Proteomica
Era de los Microensayos
Futuro
?
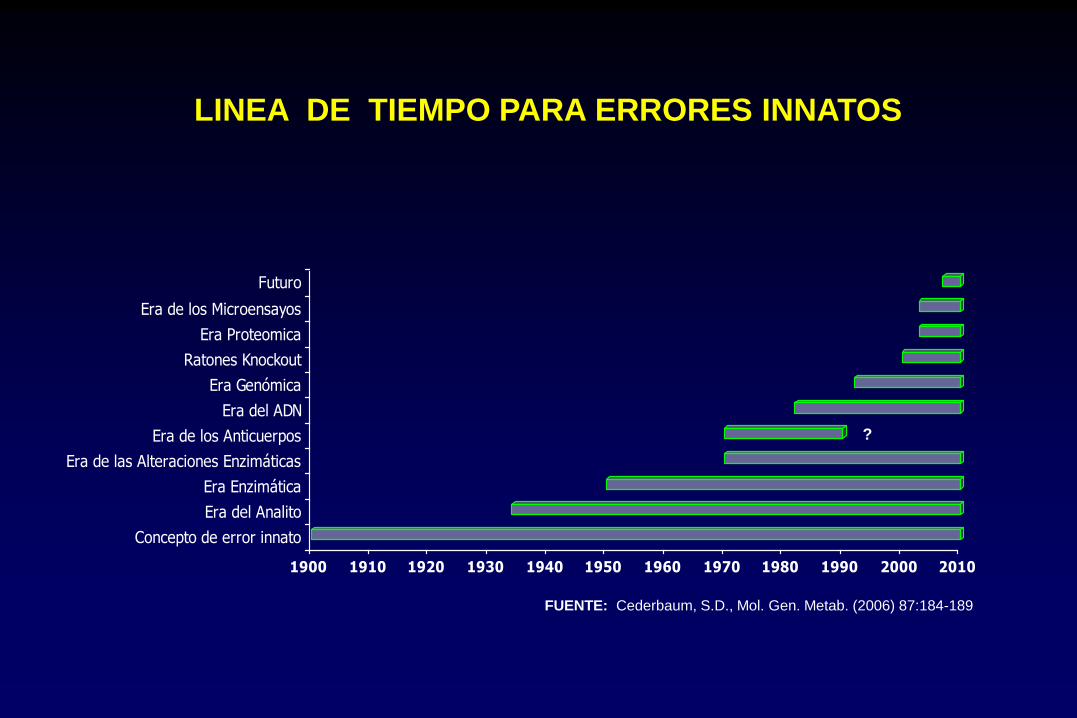
LINEA DE TIEMPO PARA ERRORES INNATOS
FUENTE: Cederbaum, S.D., Mol. Gen. Metab. (2006) 87:184-189
250
170
137
9580
67
20 15 109,46 9,62 7,86
0
50
100
150
200
250
1920 1930 1940 1950 1960 1970 1980 1990 2000 2010 2011 2012
Individuos
Años
Mortalidad Infantil
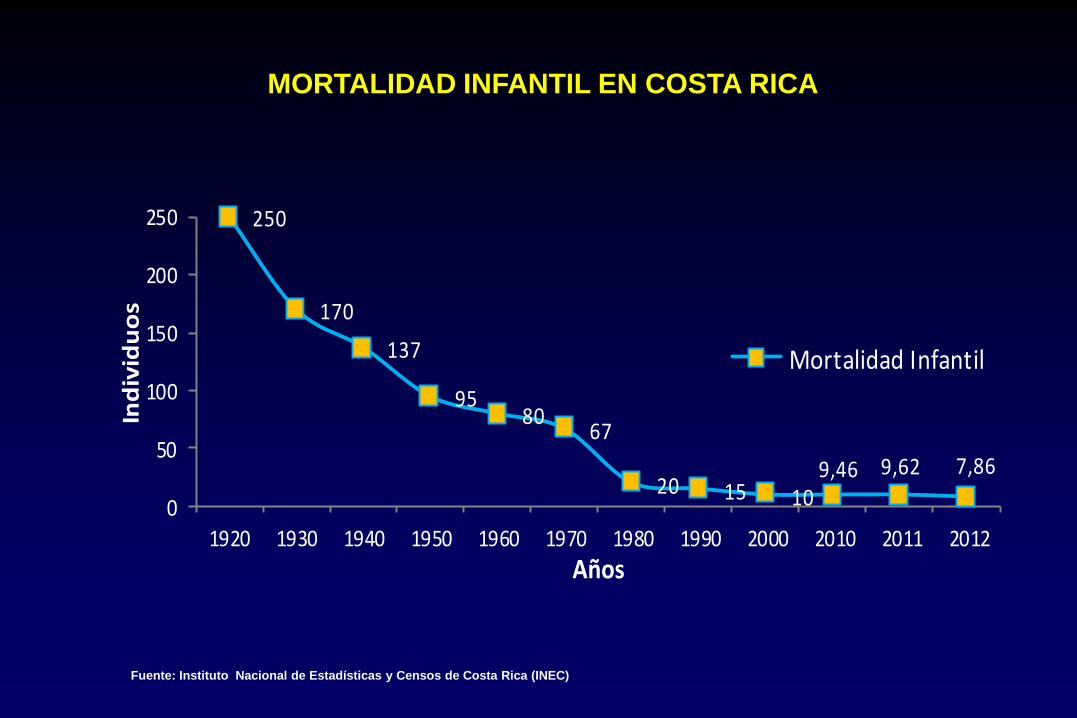
MORTALIDAD INFANTIL EN COSTA RICA
Fuente: Instituto Nacional de Estadísticas y Censos de Costa Rica (INEC)
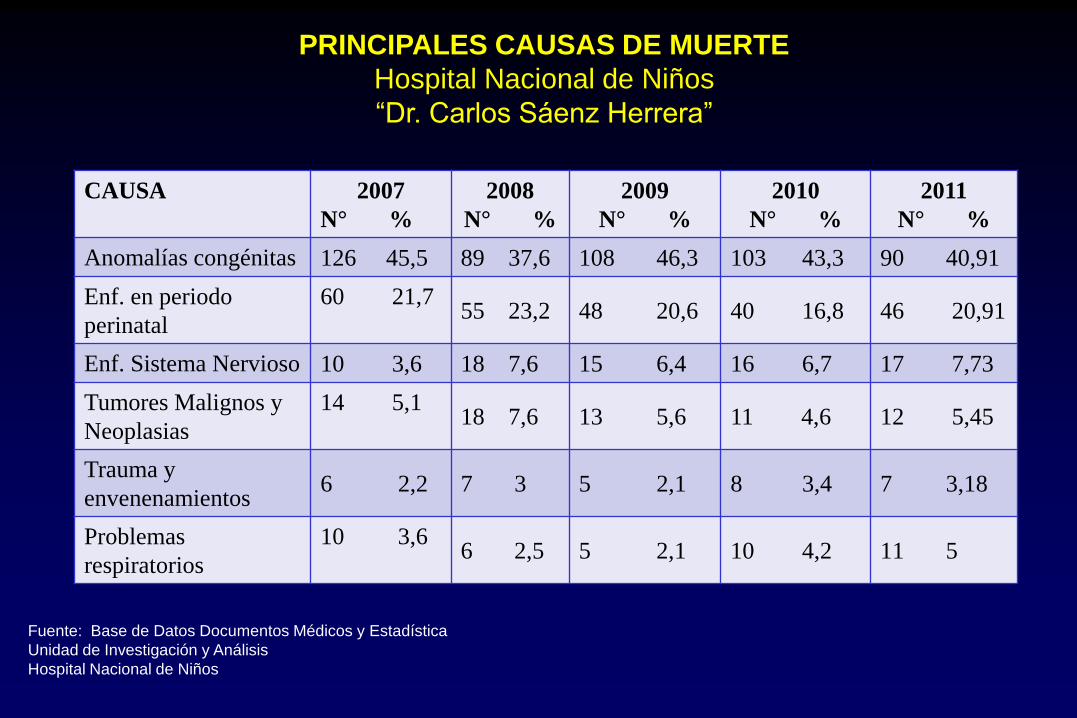
CAUSA 2007
N° %
2008
N° %
2009
N° %
2010
N° %
2011
N° %
Anomalías congénitas 126 45,5 89 37,6 108 46,3 103 43,3 90 40,91
Enf. en periodo
perinatal
60 21,7
55 23,2 48 20,6 40 16,8 46 20,91
Enf. Sistema Nervioso 10 3,6 18 7,6 15 6,4 16 6,7 17 7,73
Tumores Malignos y
Neoplasias
14 5,1
18 7,6 13 5,6 11 4,6 12 5,45
Trauma y
envenenamientos 6 2,2 7 3 5 2,1 8 3,4 7 3,18
Problemas
respiratorios
10 3,6
6 2,5 5 2,1 10 4,2 11 5
PRINCIPALES CAUSAS DE MUERTE
Hospital Nacional de Niños
“Dr. Carlos Sáenz Herrera”
Fuente: Base de Datos Documentos Médicos y Estadística
Unidad de Investigación y Análisis
Hospital Nacional de Niños

SISTEMA DE SALUD NACIONAL
Hospitales de Referencia Nacional 7
Hospitales 28
Clínicas Multifuncionales 14
EBAIS 915
* Inversión Nacional en Salud 9.3% PIB
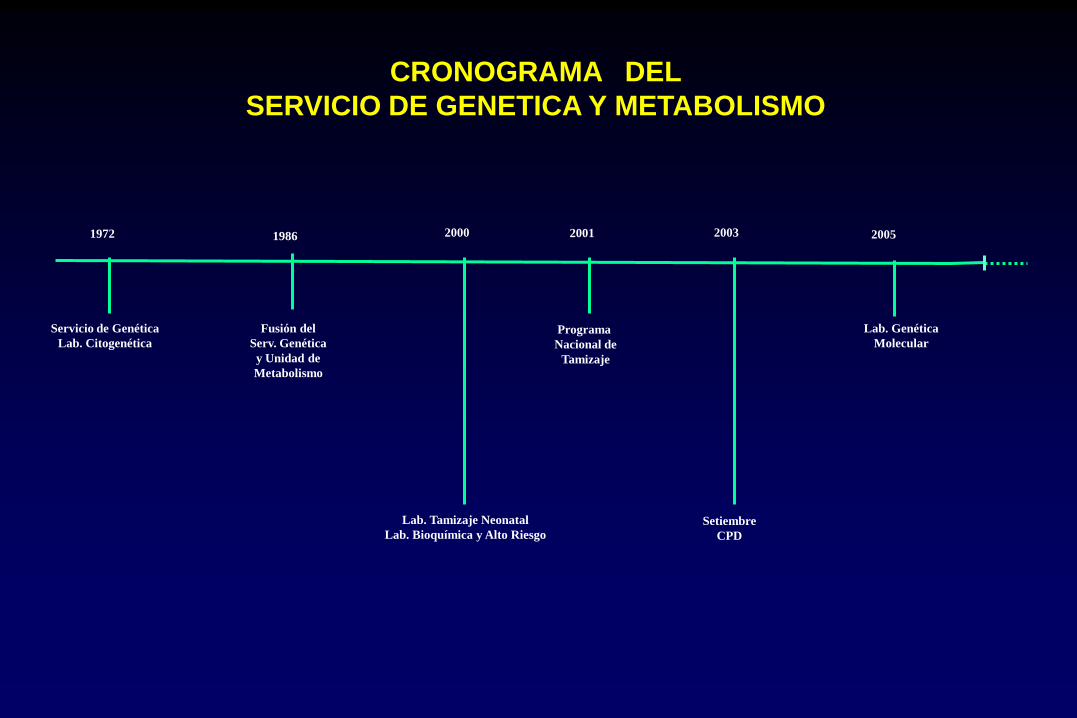
CRONOGRAMA DEL
SERVICIO DE GENETICA Y METABOLISMO
Servicio de Genética
Lab. Citogenética
Lab. Tamizaje Neonatal
Lab. Bioquímica y Alto Riesgo
Programa
Nacional de
Tamizaje
Setiembre
CPD
1972 2003 2000 2001
Lab. Genética
Molecular
2005 1986
Fusión del
Serv. Genética
y Unidad de
Metabolismo
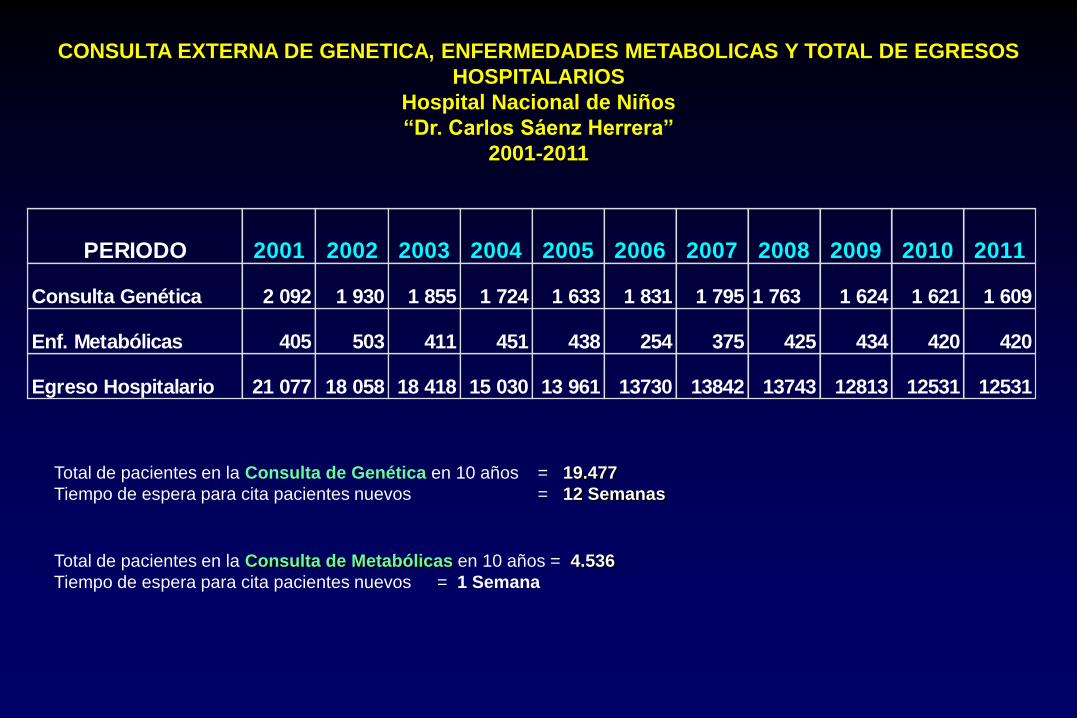
CONSULTA EXTERNA DE GENETICA, ENFERMEDADES METABOLICAS Y TOTAL DE EGRESOS
HOSPITALARIOS
Hospital Nacional de Niños
“Dr. Carlos Sáenz Herrera”
2001-2011
PERIODO 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011
Consulta Genética 2 092 1 930 1 855 1 724 1 633 1 831 1 795 1 763 1 624 1 621 1 609
Enf. Metabólicas 405 503 411 451 438 254 375 425 434 420 420
Egreso Hospitalario 21 077 18 058 18 418 15 030 13 961 13730 13842 13743 12813 12531 12531
Total de pacientes en la Consulta de Genética en 10 años = 19.477
Tiempo de espera para cita pacientes nuevos = 12 Semanas
Total de pacientes en la Consulta de Metabólicas en 10 años = 4.536
Tiempo de espera para cita pacientes nuevos = 1 Semana
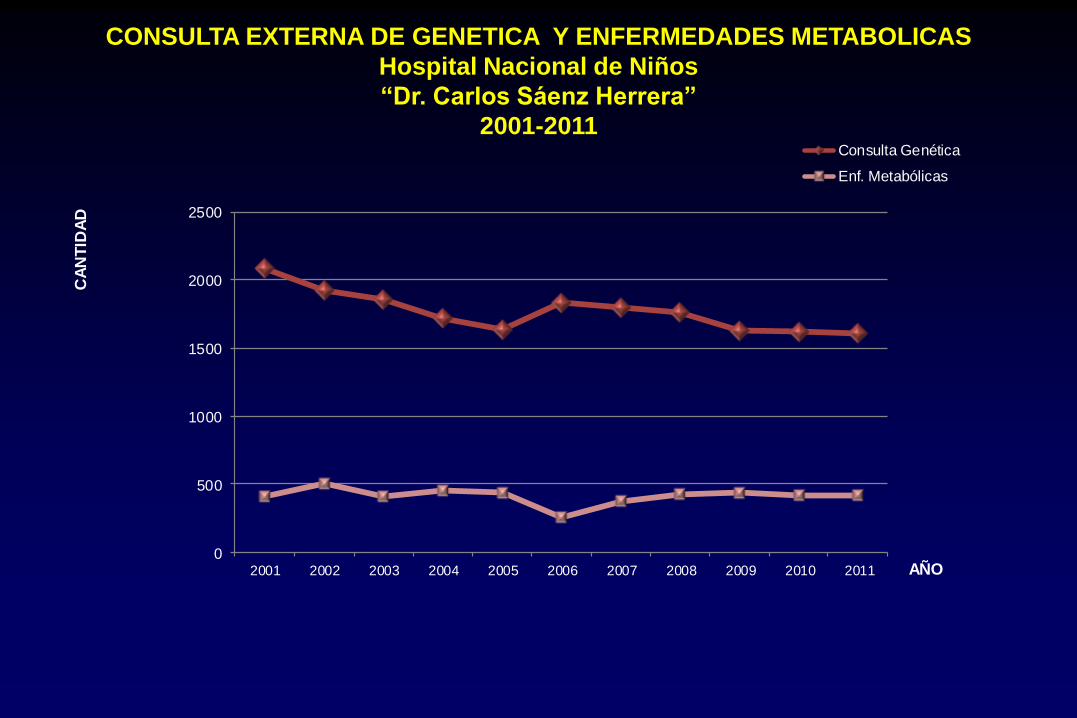
CONSULTA EXTERNA DE GENETICA Y ENFERMEDADES METABOLICAS
Hospital Nacional de Niños
“Dr. Carlos Sáenz Herrera”
2001-2011
0
500
1000
1500
2000
2500
2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011
CA
NT
IDA
D
AÑO
Consulta Genética
Enf. Metabólicas
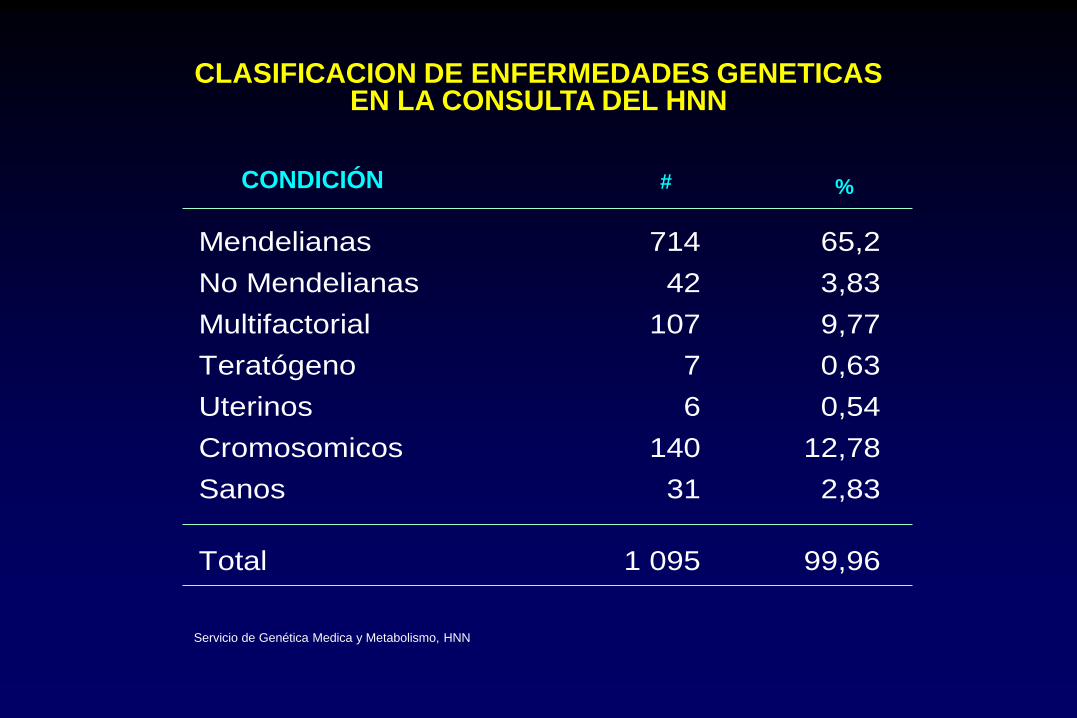
CLASIFICACION DE ENFERMEDADES GENETICAS EN LA CONSULTA DEL HNN
CONDICIÓN # %
Mendelianas 714 65,2
No Mendelianas 42 3,83
Multifactorial 107 9,77
Teratógeno 7 0,63
Uterinos 6 0,54
Cromosomicos 140 12,78
Sanos 31 2,83
Total 1 095 99,96
Servicio de Genética Medica y Metabolismo, HNN

ORGANIZACIÓN DE LA CONSULTA GENETICA
• SINDROMES
• TRASTORNOS CROMOSOMICOS
• DISPLASIAS ESQUELETICAS
• TRASTORNOS EN TEJIDO CONECTIVO
• ENFERMEDADES METABOLICAS
•Defecto del metabolismo de CHO
•Defecto del metabolismo de lipidos
•Defecto del metabolismo de AA y AO
•Defecto del metabolismo de organelas
• ASESORIA GENETICA
• CONSULTA NUEVOS
•PRECONSULTAS
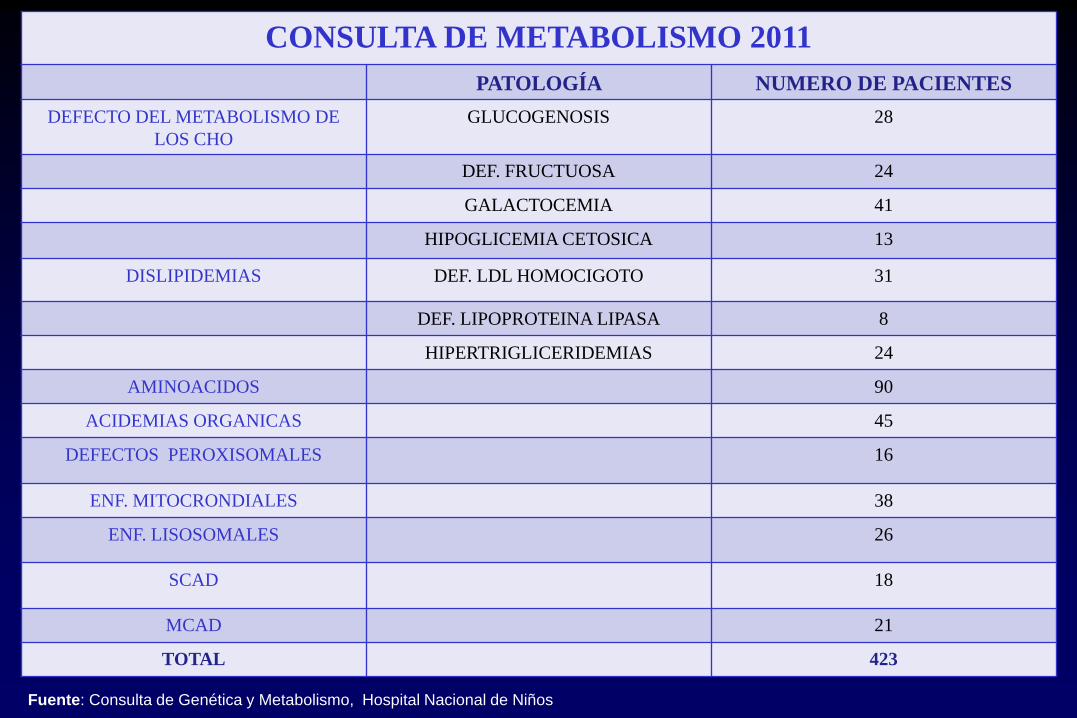
CONSULTA DE METABOLISMO 2011
PATOLOGÍA NUMERO DE PACIENTES
DEFECTO DEL METABOLISMO DE
LOS CHO
GLUCOGENOSIS 28
DEF. FRUCTUOSA 24
GALACTOCEMIA 41
HIPOGLICEMIA CETOSICA 13
DISLIPIDEMIAS DEF. LDL HOMOCIGOTO 31
DEF. LIPOPROTEINA LIPASA 8
HIPERTRIGLICERIDEMIAS 24
AMINOACIDOS 90
ACIDEMIAS ORGANICAS 45
DEFECTOS PEROXISOMALES 16
ENF. MITOCRONDIALES 38
ENF. LISOSOMALES 26
SCAD 18
MCAD 21
TOTAL 423
Fuente: Consulta de Genética y Metabolismo, Hospital Nacional de Niños
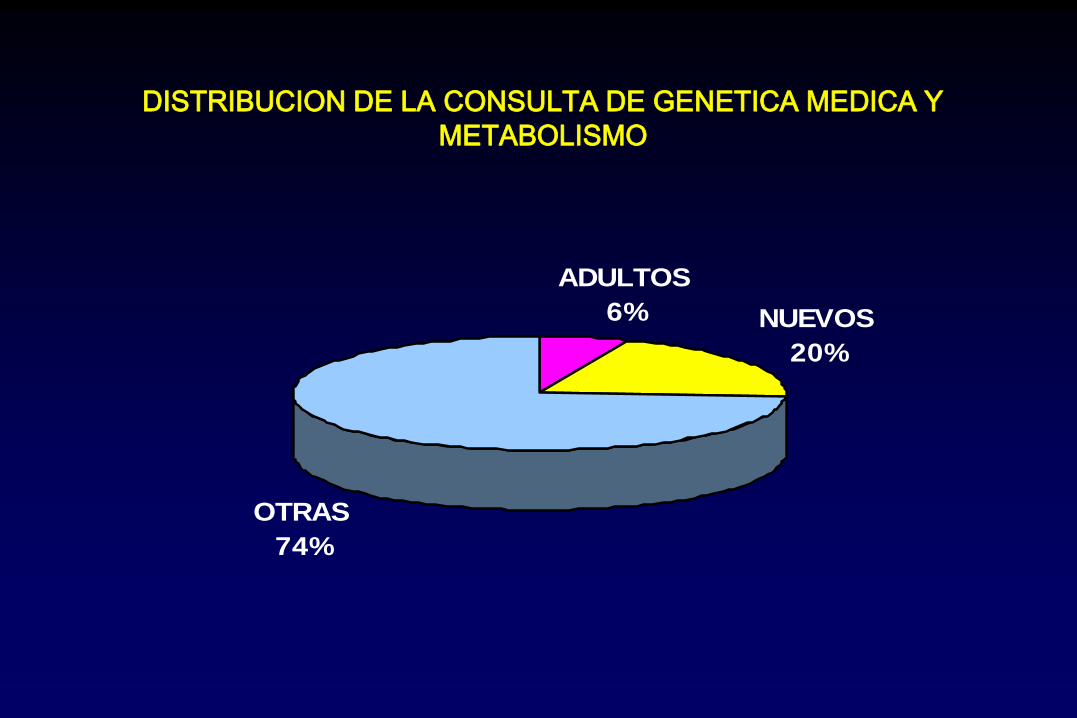
ADULTOS
6% NUEVOS
20%
OTRAS
74%
DISTRIBUCION DE LA CONSULTA DE GENETICA MEDICA Y
METABOLISMO

Incluye EIM del metabolismo intermedio que produce intoxicaciones agudas o progresivas
Metabolismo de aminoácidos
La mayoría de Ac. Orgánicos
T. del ciclo de la Urea
Intolerancia a azúcares
Intoxicación metálica
Porfirias
Características: A.- No interfieren con el desarrollo embrionario-fetal
B.- Todos tienen períodos libres de síntomas
C.- Presentación puede ser temprana y tardía
D.- Factor desintoxicante (fiebre, enf. Ingesta de comidas)
Todos ellos requieren de remoción del tóxico por:
- Dieta especial
- Procedimiento extracorpóreo (diálisis)
- Medicamentos de limpieza (Benzoato, sodio, carnitina, penicilina, vitaminas)
ENFERMEDADES QUE PRODUCEN INTOXICACION
Saudubray, J.M., et al. J Inherit Metab Dis (2006) 29:261-274

Los síntomas son consecuencia de una producción o utilización deficiente de la
energía por parte del hígado, miocardio, músculo, cerebro y otros
A.- Defectos Mitocondriales
Son los más severos
Lactacidemias congénitas
Defectos de cadena respiratoria: - def. CoQ10
- P.C.
Oxidación de Ácidos Grasos y cuerpos cetónicos
B.- Defectos Citoplasmáticos
Def. de Glucogenolisis
Def. de Gluconeogénesis
Def. Metabolismo del Glucógeno
Def. Metabolismo de creatina
Def. del Metabolismo de pentosa fosfato
Algunos del los metabolitos intermedios pueden afectar el desarrollo embrio-fetal
ENFERMEDADES QUE INVOLUCRAN
EL METABOLISMO DE ENERGIA
Saudubray, J.M., et al. J Inherit Metab Dis (2006) 29:261-274

Este grupo incluye defectos de organelas celulares y trastornos en la síntesis o
catabolismo de moléculas complejas
Los síntomas son permanentes, progresivos, constantes y no relacionados con
ingestas
Enf. Lisosomales
Enf. Peroxisomales
Enf. del transporte y procesamiento intracelular
Ej. Def. 1 - antitripsina, EIM del colesterol y glicoproteína deficiente de
carbohidratos
ENFERMEDADES QUE INVOLUCRAN MOLECULAS COMPLEJAS
Saudubray, J.M., et al. J Inherit Metab Dis (2006) 29:261-274

DIFERENCIACION CLINICA ENTRE ENFERMEDAD DE
ORGANELAS Y DE MOLECULAS PEQUEÑAS
CARACTERISTICA
Presentación
Evolución
Hallazgos físicos
Histopatología
Respuesta a tx
ENF. ORGANELA
Gradual
Progresiva
Características
Cambios esp.
Pobre
ENF. MOL PEQ.
Súbito
Intermitente
Inespecífico
Inespecífico
Súbito

• Hipotiroidismo Congénito (HC)
• Fenilcetonuria (PKU)
• Jarabe de Arce (MSUD)
• Hiperplasia suprarrenal congénita (HSC)
- Deficiencia de 21 – Hydroxilasa perdedora de sal
- Deficiencia de 21 – Hydroxilasa virilizante simple
• Galactosemia (GAO)
- Deficiencia de Galactoquinasa
- Deficiencia de Galactosa-1 Fosfato Uridiltransferasa
- Deficiencia de Galactosa-4-Epimerasa
Defectos de -Oxidación
• Deficiencia de la Acil CoA deshidrogenasa de cadena mediana (MCAD)
• Deficiencia de la Acil CoA deshidrogenasa de cadena
muy larga VLCAD)
• Deficiencia de la Acil CoA deshidrogenasa de cadena
larga (LCAD)
• Deficiencia de la Acil CoA deshidrogenasa de cadena
corta (SCAD)
Acidemias Orgánicas
• Deficiencia de la palmitoil transferasa transportadora (CPT II)
• Acidemia Glutárica II (GA II)
• Acideia Propiónica (PA)
• Acidemia Glutárica tipo I (GA I)
• Acidemia Metil Malónica (MMA)
• Acidemia Isovalérica (IVA)
• Deficiencia múltiple de carboxilasas (3 MMC)
• Deficiencia de la ketotiosase (b-KT)
• Acidemia Hidroxinetil glutarica (HMGCoA Lyase
deficiency)
Hemoglobinopatias
• variantes de cadenas
- Talasemias
• variantes de cadenas
- Hb C, Hb S, Hb E, Hb D, Talasemias
Plan Piloto
- Fibrosis Quística del Páncreas
- Def. de Biotinidasa

SÍNTOMAS INESPECÍFICOS DE UN EIM
Lactante que súbitamente se enferma sin causa aparente
Convulsiones - Hipotonía sin causa aparente
Olor y color peculiar de la orina
SÍNTOMAS REALMENTE SUGESTIVOS DE UN EIM
Vómitos recurrentes o persistentes
Falla para progresar de causa no determinada
Apneas o dificultad respiratoria
Organomegalia
Letargo y coma
Alteraciones en tono (hipotonia/hipertono)
Movimientos anormales, mioclonias
Neutropenia,Trombocitopenia
Cetosis
Consanguinidad, Antecedente familiar
Infección E. Coli
Síndrome de muerte súbita
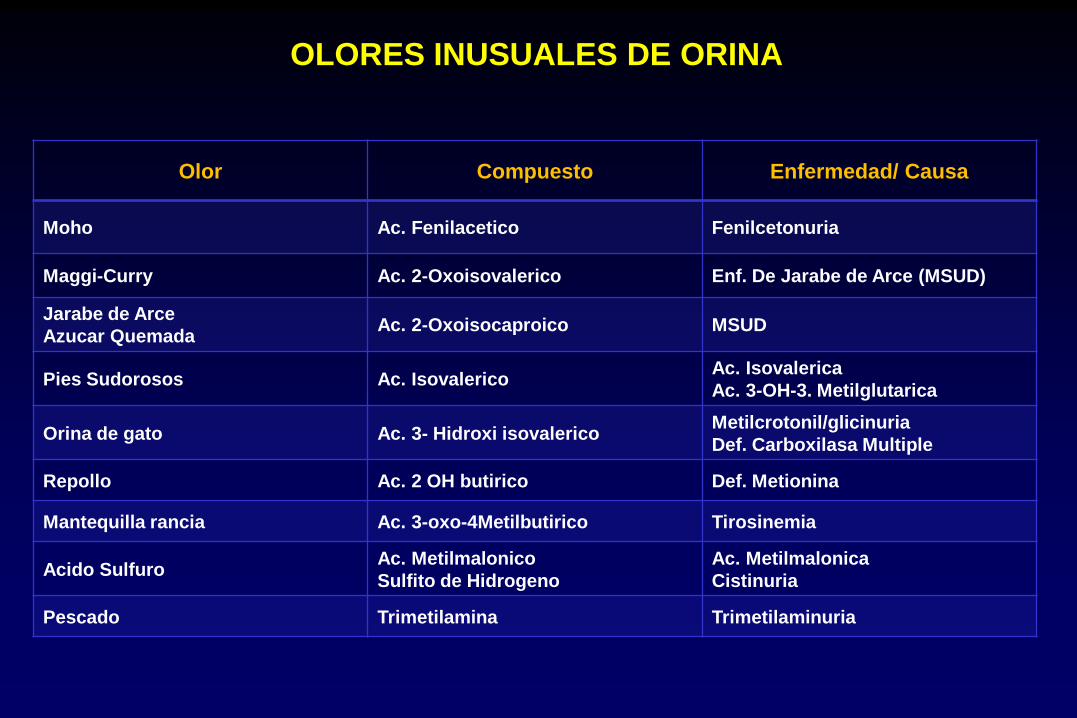
Olor Compuesto Enfermedad/ Causa
Moho Ac. Fenilacetico Fenilcetonuria
Maggi-Curry Ac. 2-Oxoisovalerico Enf. De Jarabe de Arce (MSUD)
Jarabe de Arce
Azucar Quemada Ac. 2-Oxoisocaproico MSUD
Pies Sudorosos Ac. Isovalerico Ac. Isovalerica
Ac. 3-OH-3. Metilglutarica
Orina de gato Ac. 3- Hidroxi isovalerico Metilcrotonil/glicinuria
Def. Carboxilasa Multiple
Repollo Ac. 2 OH butirico Def. Metionina
Mantequilla rancia Ac. 3-oxo-4Metilbutirico Tirosinemia
Acido Sulfuro Ac. Metilmalonico
Sulfito de Hidrogeno
Ac. Metilmalonica
Cistinuria
Pescado Trimetilamina Trimetilaminuria
OLORES INUSUALES DE ORINA
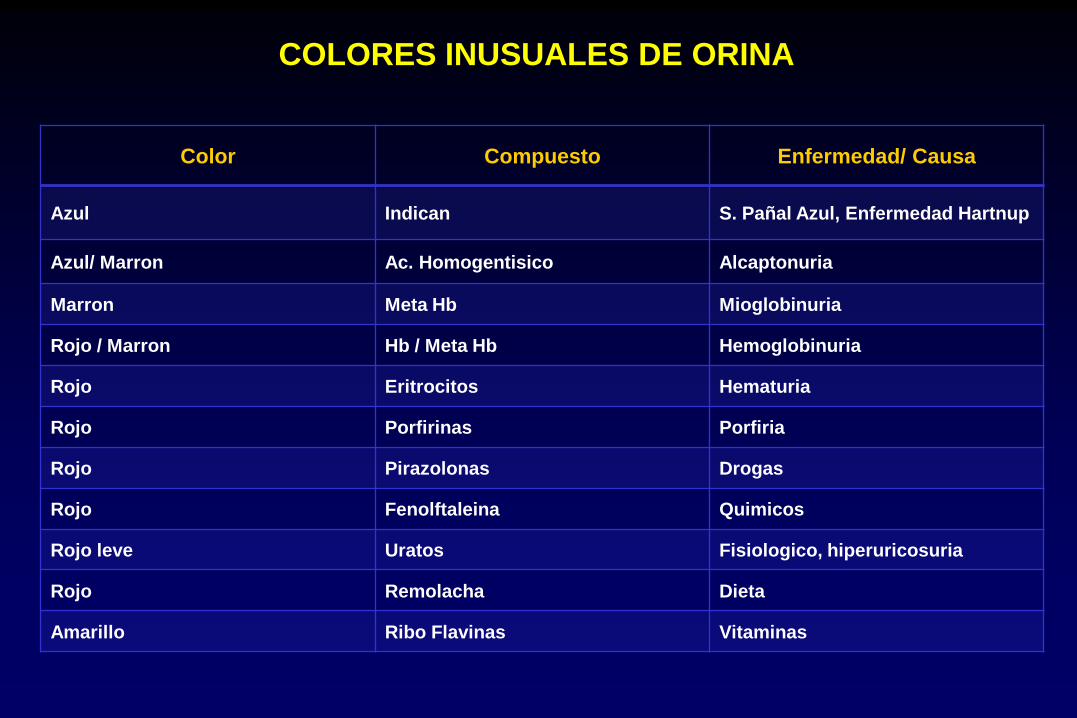
COLORES INUSUALES DE ORINA
Color Compuesto Enfermedad/ Causa
Azul Indican S. Pañal Azul, Enfermedad Hartnup
Azul/ Marron Ac. Homogentisico Alcaptonuria
Marron Meta Hb Mioglobinuria
Rojo / Marron Hb / Meta Hb Hemoglobinuria
Rojo Eritrocitos Hematuria
Rojo Porfirinas Porfiria
Rojo Pirazolonas Drogas
Rojo Fenolftaleina Quimicos
Rojo leve Uratos Fisiologico, hiperuricosuria
Rojo Remolacha Dieta
Amarillo Ribo Flavinas Vitaminas

MANIFESTACIONES CLÍNICAS EN NEONATOS Y < DE 3 MESES
Clínica Sx
Neurológica Enf. Metabólica (coma, mov. Anormales)
Convulsiones
Microcefalia
Hepática Insuf. Hepática
Ictericia, colestasis
Hepato-esplenomegalia
Cardiaca Insuf. Cardiaca
Cardiomiopatía
Trast. del ritmo cardiaco
Hipoglicemia Hepatomegalia
Infusiones con altas concentraciones de glucosa
Compromiso hepático/cardiaco

MANIFESTACIONES CLINICAS TARDIAS Y DEL ADULTO
Clínica Sx
Coma metabólicos sin focalización Acidosis
Ataxias agudas con letargo Hiperamonemia
Hipoglicemia
Ac. Láctico
Coma metabólico con focalización Edema cerebral
Sx extrapiramidales
Sincope
Accidente Tromboembólicos
Coma hepático Hiperamonemia
Ac. Láctico
Ictericia hemolítica
Enteropatía
Hipoglicemia
Síntomas Psiquiátricos Hiperamonemia
Cetoacidosis
Hiperhomocisteinemia
Orina en Vino Tinto

HALLAZGOS CLINICOS ASOCIADOS A ECM
Hepatomegalia GSD, def. fructosa 1-6 difosfatasa
Hepatoesplenomegalia Gaucher, N. Pick, Sialidosis
Macrocefalia Canavan
Microcefalia T. de electrones, Leigh
Cara grotesca I Cell, Sialidosis. MPS
Macroglosia Pompe, GM1
Distonía o signos extrapiramidales Gaucher, Krabbe, PKU
Manchas rojo cereza N. Pick, Tay-Sachs
Retinitis pigmentaria Enf. mitocondrial, Zellweger
Atrofia ótica Zellweger, Piruvato Deshidrogenasa
Opacidad corneal I cell, Def. Esteroide Sulfatasa
HALLAZGO ENFERMEDAD
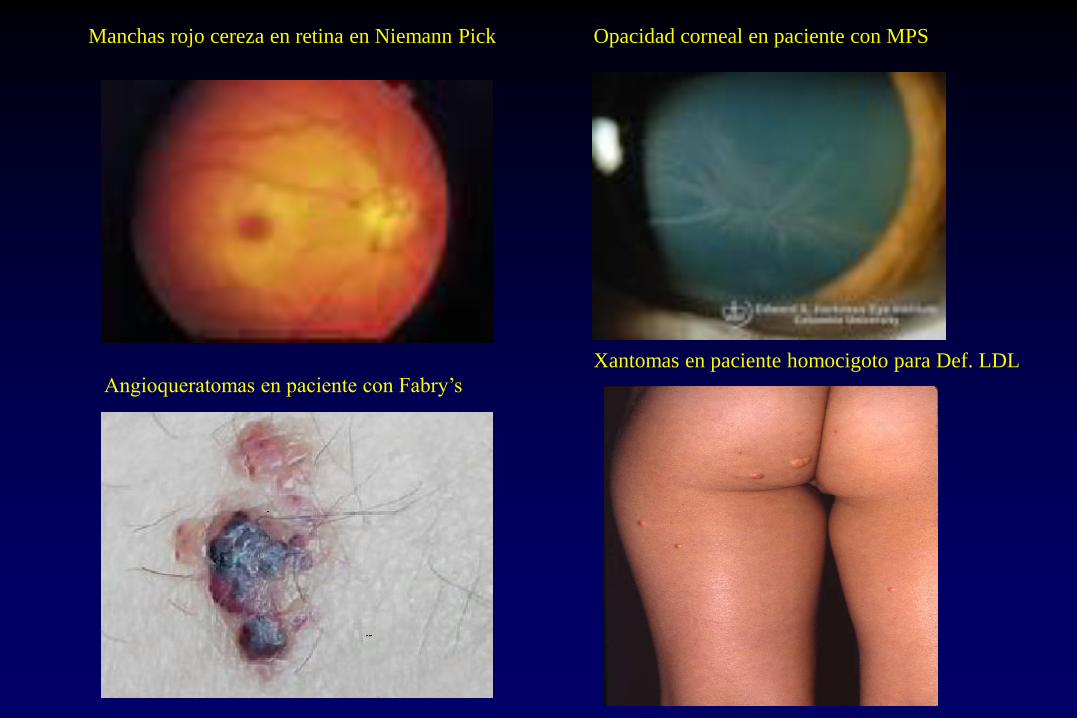
Manchas rojo cereza en retina en Niemann Pick Opacidad corneal en paciente con MPS
Angioqueratomas en paciente con Fabry’s
Xantomas en paciente homocigoto para Def. LDL

ESTUDIOS INICIALES EN UN ECM
SANGRE ORINA OTROS
-Hemograma completo -Pruebas metabólicas en orina -Rx de tórax
-Gases venosos -Cetosis -Líquido céfalo raquídeo
-Electrolitos sanguíneos -Electrolitos -Microbiología
-Glicemia, calcio -Ac. Úrico -ECO, EKG
-Amonio -US de cerebro y abdomen, EGG
-Acido úrico -Tóxicos de sangre
-UN -Análisis por malformaciones
-Pruebas función hepática congénitas
-Acido láctico y ácido pirúvico
en plasma

PRUEBAS METABOLICAS EN ORINA
Cloruro Ferrico
Color Compuesto Enf./ Causa
Azul-Verde Ac. Fenilpiruvico Fenilcetonuria
Catecolaminas
Ac. Imidazolpirunico
Sustancias Reductoras
Color Compuesto Enf./Causa
Verde- Amarentado Galactosa Galactosemia, Fanconi-Bikel
Enf. de Wilson
Fructosa Intolerancia
Fructosa
Glucosa Diabetes/Fanconi
Salicilatos Drogas
Ac. Úrico Hiperuricosuria

Dinitrofenilhidracina (DNPM)
Color Compuesto Enf./ Causa
Azul-Verde Ac. Fenilpiruvico Fenilcetonuria
2-oxoisovalerico MSDU
2- Metilaceto Acetato Ac. Propionica
Ac. p- Hidroxifenilpiruvico Hiperuricosuria
Nitroprusiato
Color Compuesto Enf./Causa
Rosado-Morado Cistina Cistinuria
S. de Fanconi
Homocistina Homocistinuria
Def. B-12
Glutation Glutationuria
PRUEBAS METABOLICAS EN ORINA

PRUEBAS METABOLICAS EN ORINA

ENFOQUE DIAGNOSTICO EN CASO DE “INTOXICACION METABOLICA”
Condición Acido-Base
Acido-Base normal
Acidosis
Renal
Ac. Metabólica
Brecha aniónica ↑
ESTUDIOS BASICOS EN SANGRE DE SINTOMAS POR INTOXICACIÓN
- Gases Venosos, Brecha Aniónica
- Glicemia, Cetonas, Lactato, Amonio

ENFOQUE DIAGNOSTICO EN CASO DE
“INTOXICACION METABOLICA”
Ac. Metabólica
Brecha Aniónica
Glicemia N
Amonio
Ac láctico N
Cetosis
Ac. Orgánicos
Glicemia
Amonio N
Ac láctico N
Cetosis N
Def.
oxidación
Glicemia N
Amonio N
Ac láctico
Cetosis N
Def. cadena
respiratoria

ENFOQUE DIAGNOSTICO EN CASO DE
“INTOXICACION METABOLICA”
Condición Acido-Base
normal
Glicemia N
Amonio N
Ac láctico N
Glicemia N
Amonio ↑↑↑
Ac láctico N
Cetosis N
T. ciclo de la
urea
Cetosis
MSUD
Cetosis N
Sulfitest +
NKH
Def. SO

ENFOQUE DIAGNOSTICO EN CASO DE
“INTOXICACION METABOLICA”
Condición Acido-Base
Acidosis renal
Brecha aniónica N o
Glicemia
Amonio N
Ac láctico N
Cetosis N ↑
Def. met. carbohidratos
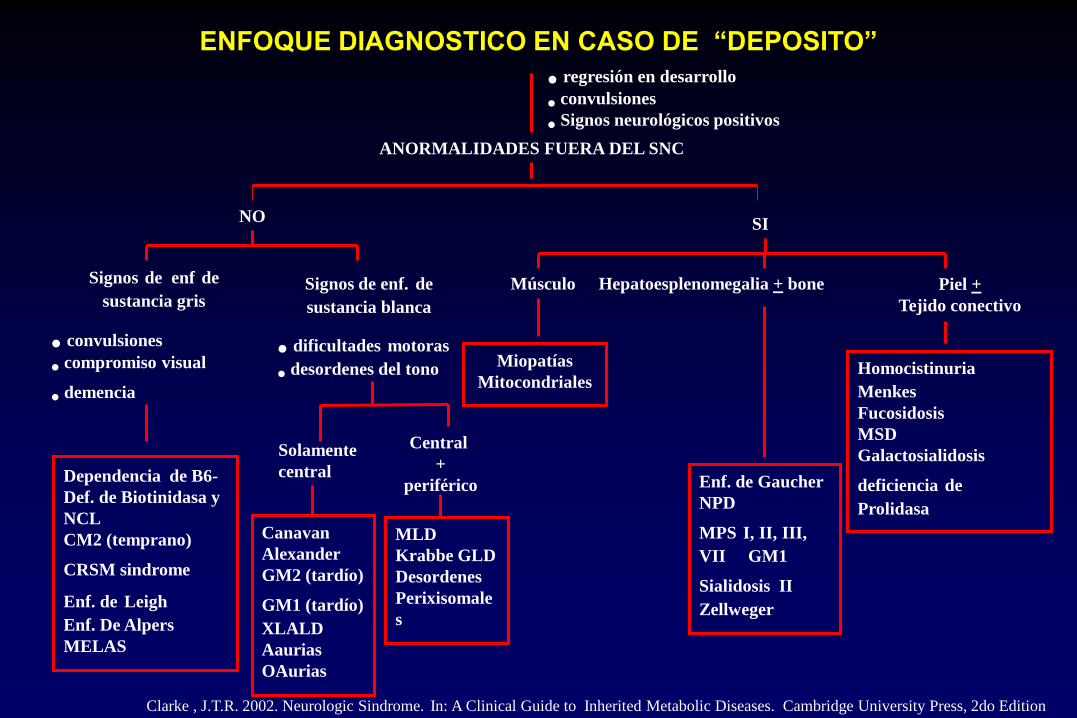
ENFOQUE DIAGNOSTICO EN CASO DE “DEPOSITO”
• regresión en desarrollo
• convulsiones
• Signos neurológicos positivos
ANORMALIDADES FUERA DEL SNC
NO SI
Signos de enf de
sustancia gris
• convulsiones
• compromiso visual
• demencia
Dependencia de B6-
Def. de Biotinidasa y
NCL
CM2 (temprano)
CRSM sindrome Enf. de Leigh
Enf. De Alpers
MELAS
Signos de enf. de
sustancia blanca
• dificultades motoras
• desordenes del tono
Solamente
central
Canavan
Alexander
GM2 (tardío)
GM1 (tardío) XLALD
Aaurias
OAurias
Central
+
periférico
MLD
Krabbe GLD
Desordenes
Perixisomale
s
Músculo
Miopatías
Mitocondriales
Hepatoesplenomegalia + bone
Enf. de Gaucher
NPD
MPS I, II, III,
VII GM1
Sialidosis II Zellweger
Piel +
Tejido conectivo
Homocistinuria Menkes
Fucosidosis
MSD
Galactosialidosis
deficiencia de
Prolidasa
Clarke , J.T.R. 2002. Neurologic Sindrome. In: A Clinical Guide to Inherited Metabolic Diseases. Cambridge University Press, 2do Edition

ESTUDIOS ESPECIALIZADOS EN DX ECM
- Aminoácidos en plasma y LCR
– Aminoácidos en orina.
– Acidos orgánicos urinarios GS - MS
– Acylcarmitina plasmática
– Acylglicinas urinarias
– Purinas plasmáticas, pirimidinas urinarias
– Galactosa (Beutler)
ESTUDIOS ALTAMENTE ESPECIALIZADOS EN EL DX ECM
– Determinación de actividad enzimática según tejido en que se estudia
– Análisis de mutaciones
– Ensayo de metabolitos específicos (VLCFA)
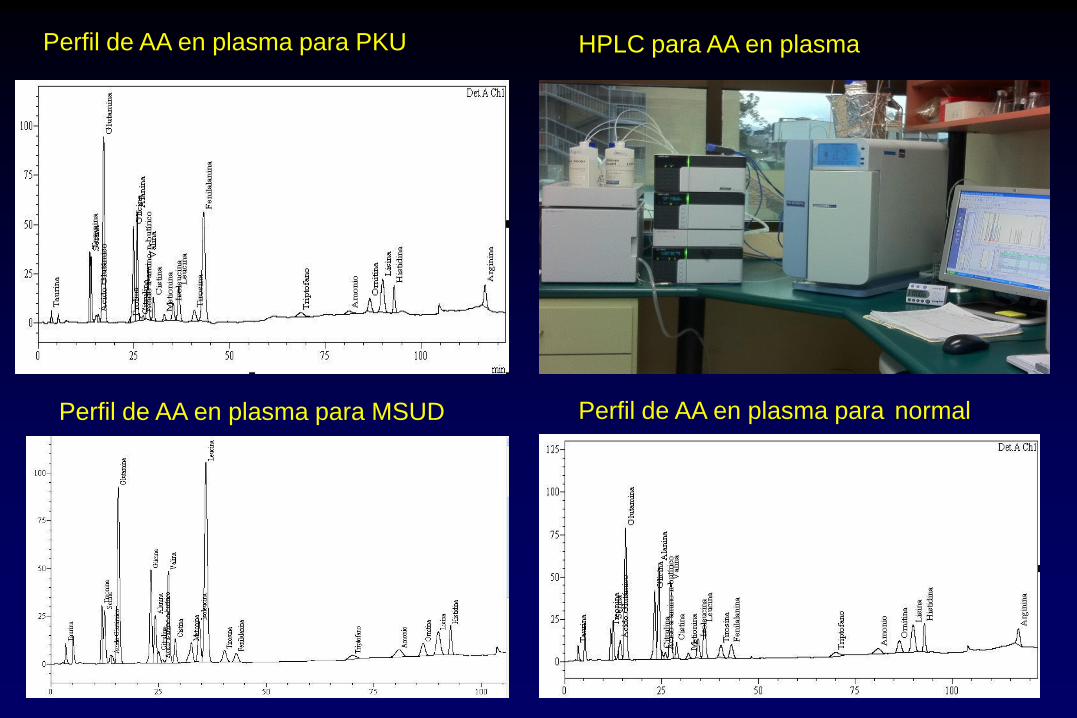
HPLC para AA en plasma Perfil de AA en plasma para PKU
Perfil de AA en plasma para MSUD Perfil de AA en plasma para normal
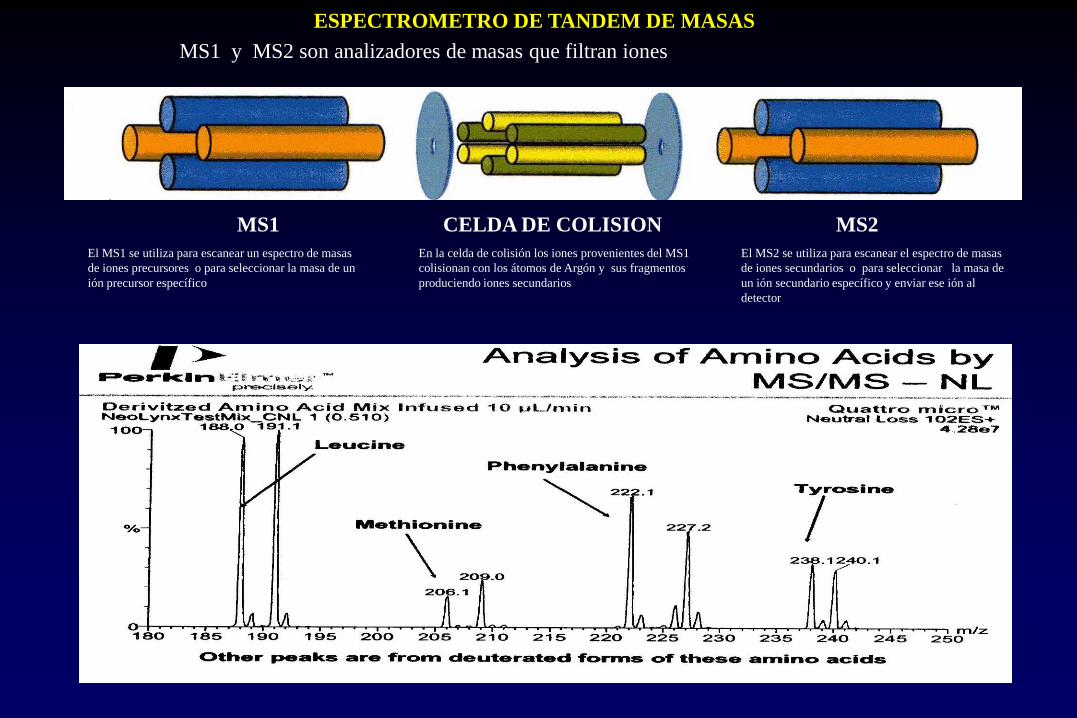
ESPECTROMETRO DE TANDEM DE MASAS
MS1 y MS2 son analizadores de masas que filtran iones
MS1 CELDA DE COLISION MS2
El MS1 se utiliza para escanear un espectro de masas
de iones precursores o para seleccionar la masa de un
ión precursor específico
En la celda de colisión los iones provenientes del MS1
colisionan con los átomos de Argón y sus fragmentos
produciendo iones secundarios
El MS2 se utiliza para escanear el espectro de masas
de iones secundarios o para seleccionar la masa de
un ión secundario específico y enviar ese ión al
detector

LABORATORIO DE MS/MS EN TANDEM
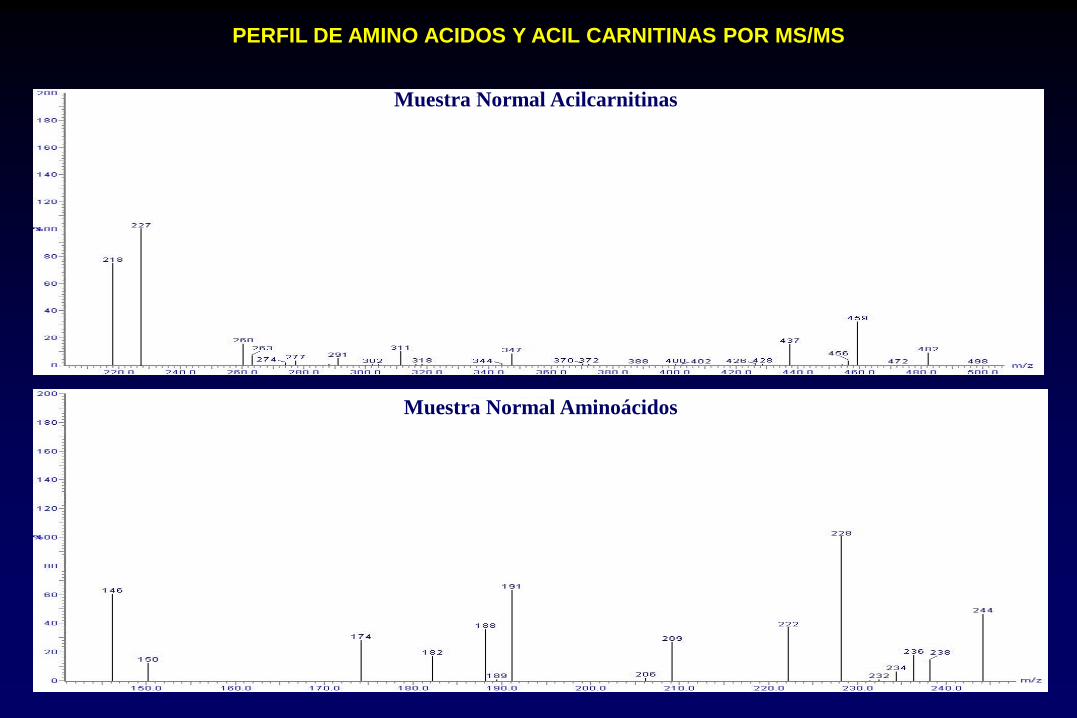
Muestra Normal Acilcarnitinas
Muestra Normal Aminoácidos
PERFIL DE AMINO ACIDOS Y ACIL CARNITINAS POR MS/MS
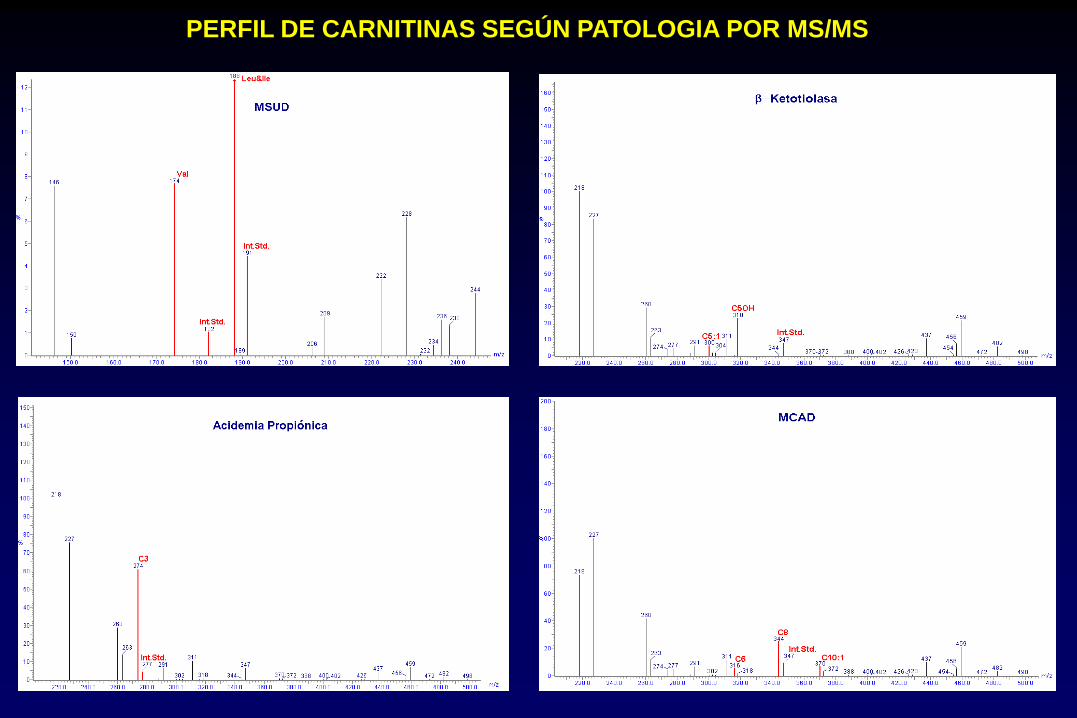
PERFIL DE CARNITINAS SEGÚN PATOLOGIA POR MS/MS
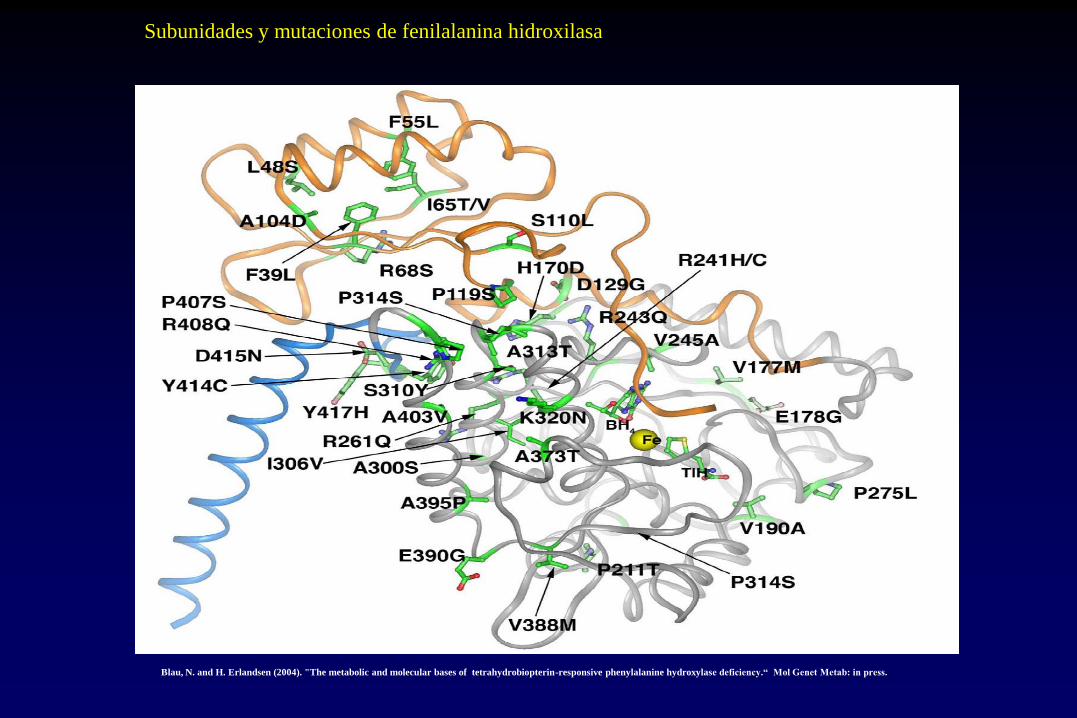
Blau, N. and H. Erlandsen (2004). "The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency.“ Mol Genet Metab: in press.
Subunidades y mutaciones de fenilalanina hidroxilasa
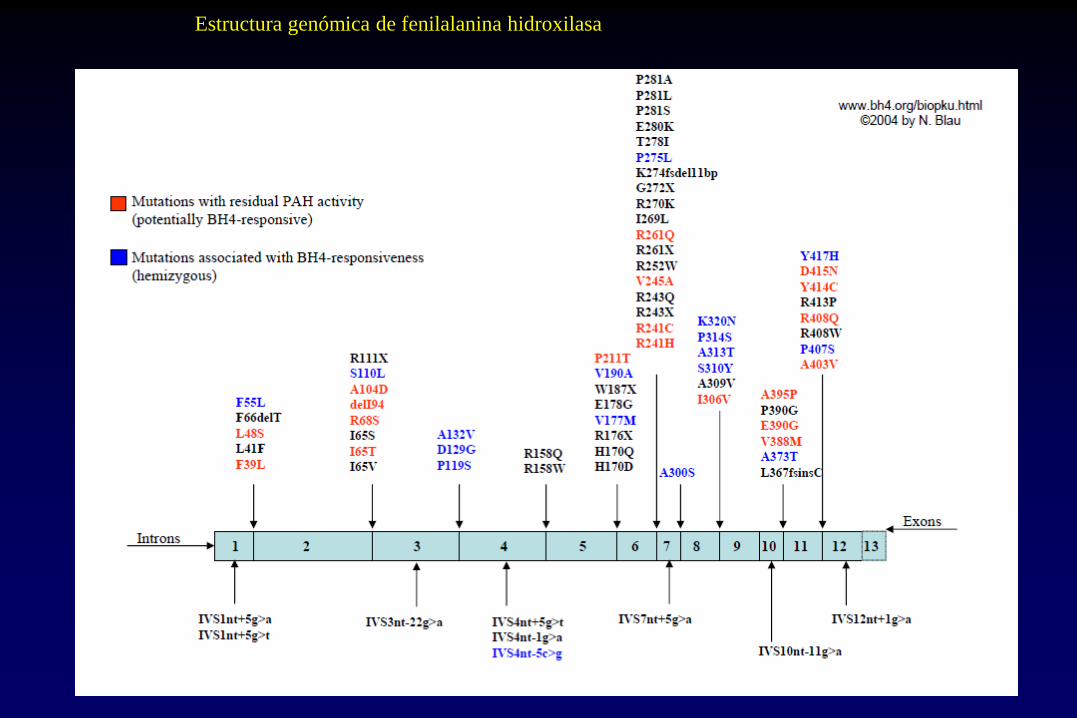
Estructura genómica de fenilalanina hidroxilasa
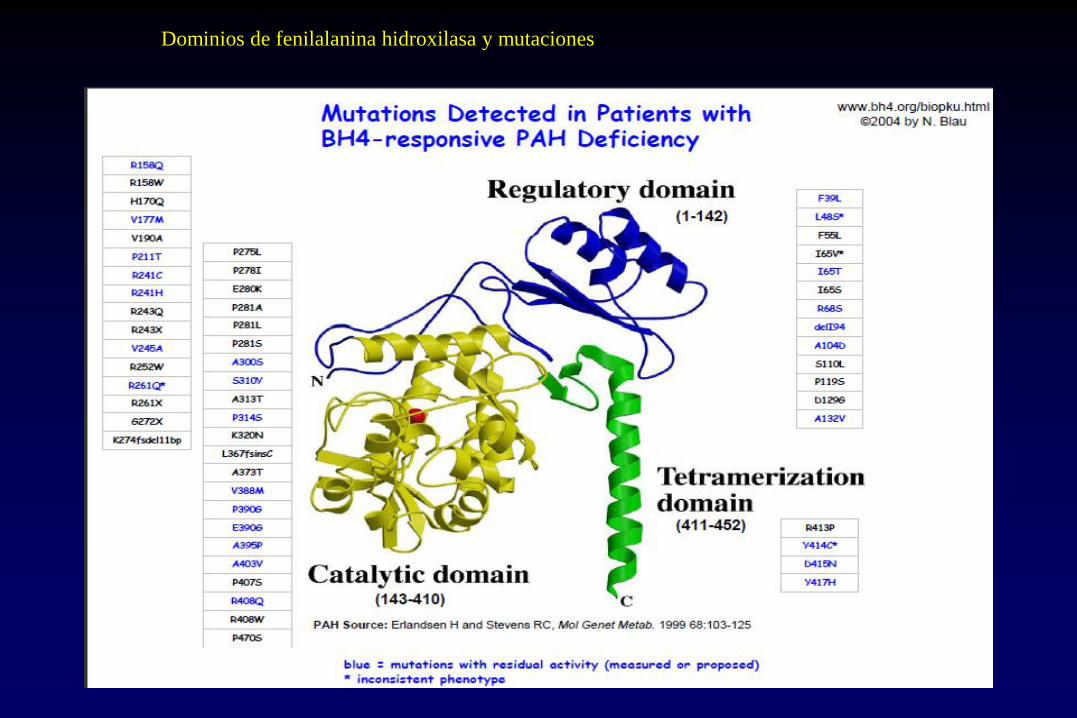
Dominios de fenilalanina hidroxilasa y mutaciones

– Plasma 3- 5 ml y plasma congelado
– Sangre total para DNA 3 - 5 ml EDTA
– Sangre en papel de filtro 3 - 5 gotas para estudio de Acilcarnitinas.
– Orina - congelada
– Biopsia de piel para cultivo de fibroblastos.
– Biopsia de músculo 1 cm3 congelado en hielo seco y por microscopio
electrónico.
– Biopsia de hígado 1 cm3 congelado en hielo seco
PROTOCOLO DE ESTUDIO EN CASO DE FALLECER

PROTOCOLO GENERAL DE TRATAMIENTO INICIAL DE UN ECM
SINTOMATICO
1. Eliminar la ingesta de compuesto agresor y sus precursores.
2. Instituir medidas de eliminación del tóxico (líquidos, electrolitos, hemodiálisis,
diálisis peritoneal, etc.)
3. Proveer un aporte calórico mínimo de 75 a 100 Kca/Kg/ día por vía I.V. Y oral.
4. Iniciar manejo farmacológico y vitamínico específico.
5. Ajustar los requerimientos mínimos de agentes tóxicos por medio de
monitorización constante.
6. Ajustar los líquidos, calorías y requerimientos mínimos en forma individual
usando como parámetros el crecimiento del paciente y los niveles plasmáticos
del agente agresor.
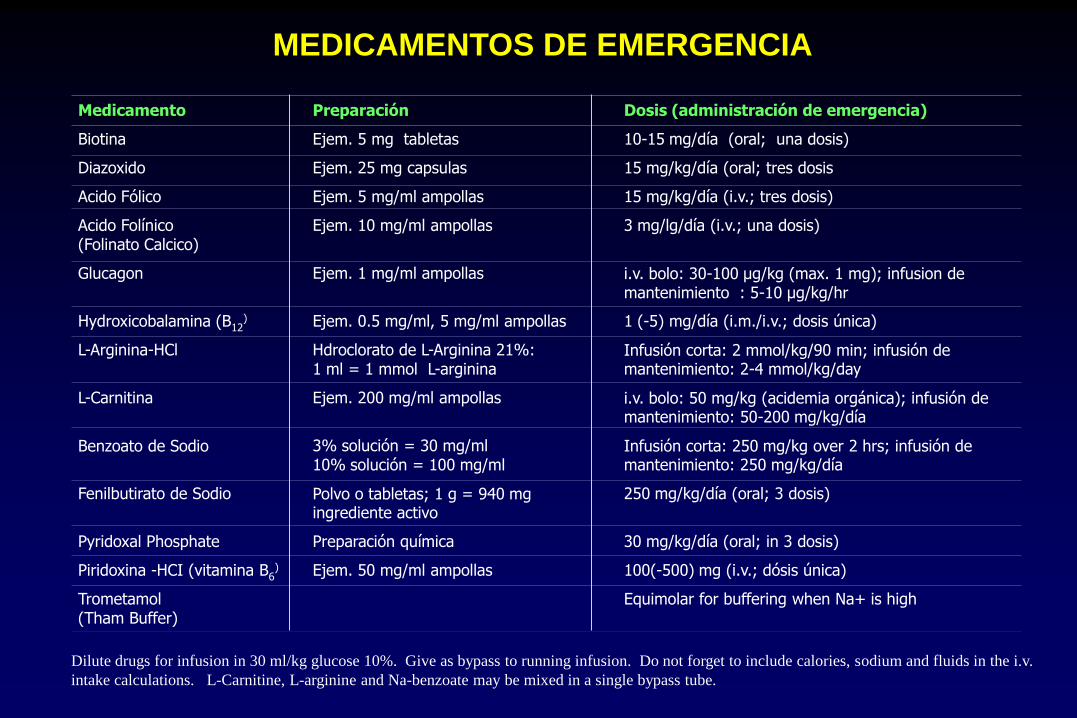
Medicamento Preparación Dosis (administración de emergencia)
Biotina Ejem. 5 mg tabletas 10-15 mg/día (oral; una dosis)
Diazoxido Ejem. 25 mg capsulas 15 mg/kg/día (oral; tres dosis
Acido Fólico Ejem. 5 mg/ml ampollas 15 mg/kg/día (i.v.; tres dosis)
Acido Folínico
(Folinato Calcico) Ejem. 10 mg/ml ampollas 3 mg/lg/día (i.v.; una dosis)
Glucagon Ejem. 1 mg/ml ampollas i.v. bolo: 30-100 µg/kg (max. 1 mg); infusion de mantenimiento : 5-10 µg/kg/hr
Hydroxicobalamina (B12) Ejem. 0.5 mg/ml, 5 mg/ml ampollas 1 (-5) mg/día (i.m./i.v.; dosis única)
L-Arginina-HCl Hdroclorato de L-Arginina 21%: 1 ml = 1 mmol L-arginina
Infusión corta: 2 mmol/kg/90 min; infusión de mantenimiento: 2-4 mmol/kg/day
L-Carnitina Ejem. 200 mg/ml ampollas i.v. bolo: 50 mg/kg (acidemia orgánica); infusión de mantenimiento: 50-200 mg/kg/día
Benzoato de Sodio 3% solución = 30 mg/ml 10% solución = 100 mg/ml
Infusión corta: 250 mg/kg over 2 hrs; infusión de mantenimiento: 250 mg/kg/día
Fenilbutirato de Sodio Polvo o tabletas; 1 g = 940 mg ingrediente activo
250 mg/kg/día (oral; 3 dosis)
Pyridoxal Phosphate Preparación química 30 mg/kg/día (oral; in 3 dosis)
Piridoxina -HCI (vitamina B6) Ejem. 50 mg/ml ampollas 100(-500) mg (i.v.; dósis única)
Trometamol (Tham Buffer)
Equimolar for buffering when Na+ is high
Dilute drugs for infusion in 30 ml/kg glucose 10%. Give as bypass to running infusion. Do not forget to include calories, sodium and fluids in the i.v.
intake calculations. L-Carnitine, L-arginine and Na-benzoate may be mixed in a single bypass tube.
MEDICAMENTOS DE EMERGENCIA
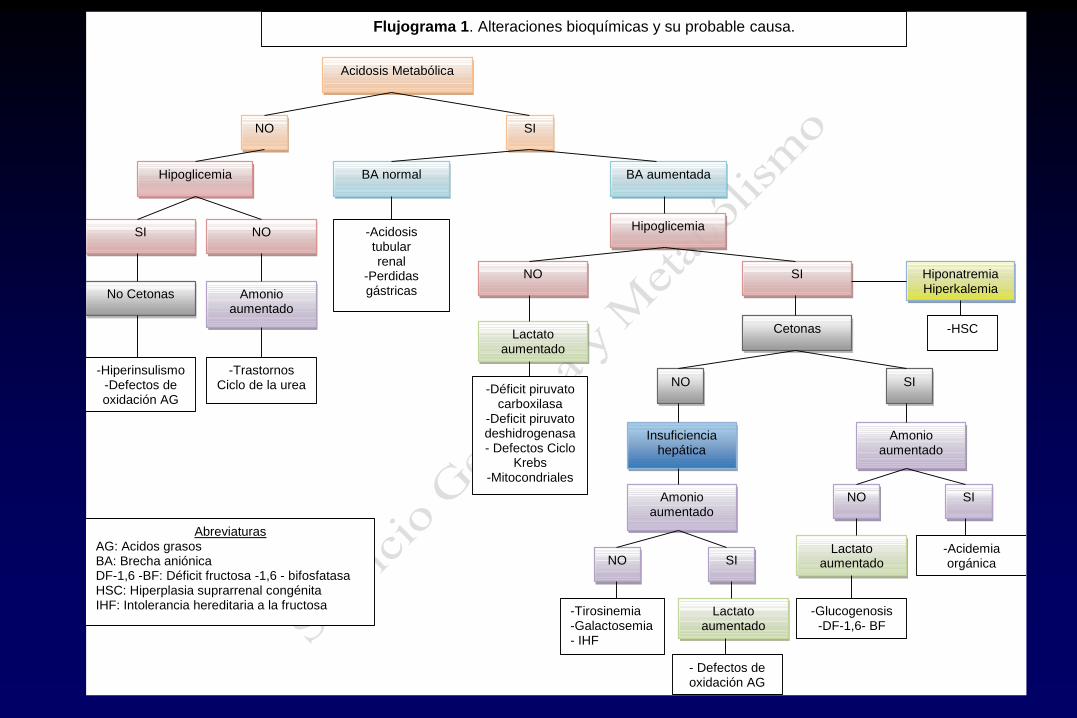
Acidosis Metabólica
NO SI
NO
SI NO
Hipoglicemia
Cetonas
BA aumentada BA normal
-Acidemia orgánica
Hiponatremia Hiperkalemia
Lactato aumentado
SI
-Glucogenosis -DF-1,6- BF
-Acidosis tubular renal
-Perdidas gástricas
-Déficit piruvato carboxilasa
-Deficit piruvato deshidrogenasa - Defectos Ciclo
Krebs -Mitocondriales
Insuficiencia hepática
-Hiperinsulismo -Defectos de oxidación AG
-Tirosinemia -Galactosemia - IHF
Abreviaturas AG: Acidos grasos BA: Brecha aniónica DF-1,6 -BF: Déficit fructosa -1,6 - bifosfatasa HSC: Hiperplasia suprarrenal congénita IHF: Intolerancia hereditaria a la fructosa
NO SI
Amonio aumentado
-Trastornos Ciclo de la urea
Flujograma 1. Alteraciones bioquímicas y su probable causa.
SI NO
-HSC
Lactato aumentado
Amonio aumentado
No Cetonas
Hipoglicemia
Amonio aumentado
NO SI
- Defectos de oxidación AG
Lactato aumentado
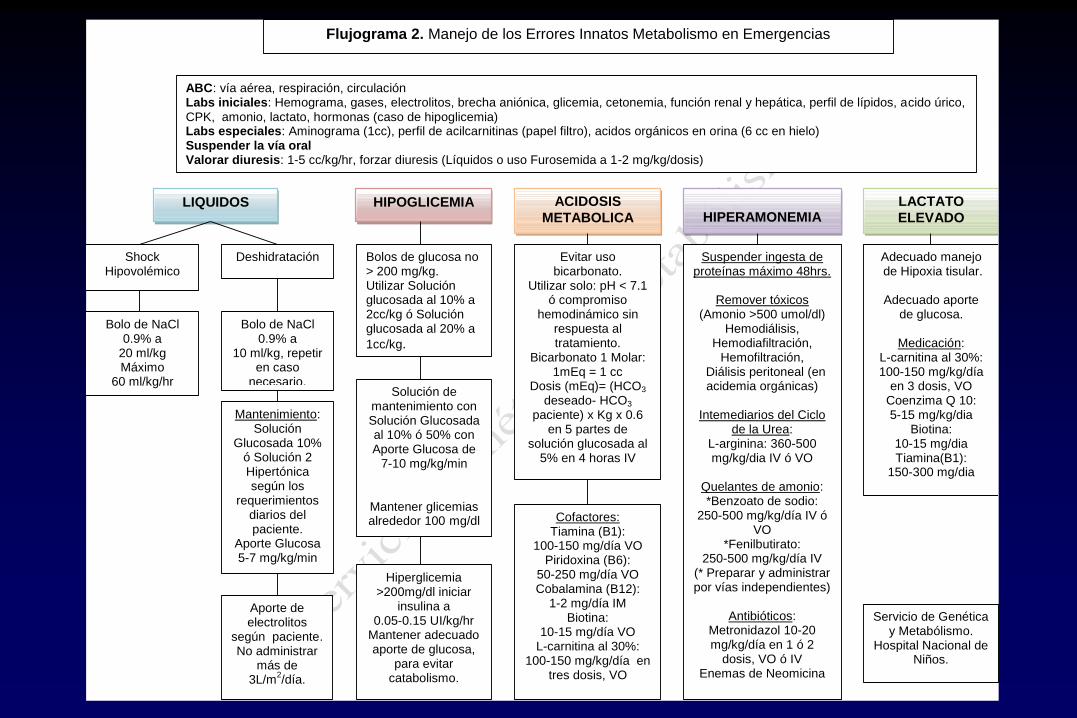
ABC: vía aérea, respiración, circulación Labs iniciales: Hemograma, gases, electrolitos, brecha aniónica, glicemia, cetonemia, función renal y hepática, perfil de lípidos, acido úrico, CPK, amonio, lactato, hormonas (caso de hipoglicemia) Labs especiales: Aminograma (1cc), perfil de acilcarnitinas (papel filtro), acidos orgánicos en orina (6 cc en hielo) Suspender la vía oral Valorar diuresis: 1-5 cc/kg/hr, forzar diuresis (Líquidos o uso Furosemida a 1-2 mg/kg/dosis)
Solución de mantenimiento con Solución Glucosada al 10% ó 50% con Aporte Glucosa de
7-10 mg/kg/min
Mantener glicemias alrededor 100 mg/dl
Evitar uso bicarbonato.
Utilizar solo: pH < 7.1 ó compromiso
hemodinámico sin respuesta al tratamiento.
Bicarbonato 1 Molar: 1mEq = 1 cc
Dosis (mEq)= (HCO3 deseado- HCO3
paciente) x Kg x 0.6 en 5 partes de
solución glucosada al 5% en 4 horas IV
Suspender ingesta de proteínas máximo 48hrs.
Remover tóxicos
(Amonio >500 umol/dl) Hemodiálisis,
Hemodiafiltración, Hemofiltración,
Diálisis peritoneal (en acidemia orgánicas)
Intemediarios del Ciclo
de la Urea: L-arginina: 360-500 mg/kg/dia IV ó VO
Quelantes de amonio: *Benzoato de sodio:
250-500 mg/kg/día IV ó VO
*Fenilbutirato: 250-500 mg/kg/día IV
(* Preparar y administrar por vías independientes)
Antibióticos:
Metronidazol 10-20 mg/kg/día en 1 ó 2
dosis, VO ó IV Enemas de Neomicina
Flujograma 2. Manejo de los Errores Innatos Metabolismo en Emergencias
LIQUIDOS
Shock Hipovolémico
Deshidratación
Bolo de NaCl 0.9% a
20 ml/kg Máximo
60 ml/kg/hr
Mantenimiento: Solución
Glucosada 10% ó Solución 2 Hipertónica según los
requerimientos diarios del paciente.
Aporte Glucosa 5-7 mg/kg/min
Bolo de NaCl 0.9% a
10 ml/kg, repetir en caso
necesario.
Aporte de electrolitos
según paciente. No administrar
más de 3L/m
2/día.
HIPERAMONEMIA
ACIDOSIS
METABOLICA
Cofactores: Tiamina (B1):
100-150 mg/día VO Piridoxina (B6):
50-250 mg/día VO Cobalamina (B12):
1-2 mg/día IM Biotina:
10-15 mg/día VO L-carnitina al 30%:
100-150 mg/kg/día en tres dosis, VO
HIPOGLICEMIA LACTATO
ELEVADO
Bolos de glucosa no > 200 mg/kg. Utilizar Solución glucosada al 10% a 2cc/kg ó Solución glucosada al 20% a
1cc/kg.
Hiperglicemia >200mg/dl iniciar
insulina a 0.05-0.15 UI/kg/hr
Mantener adecuado aporte de glucosa,
para evitar catabolismo.
Adecuado manejo de Hipoxia tisular.
Adecuado aporte
de glucosa.
Medicación: L-carnitina al 30%: 100-150 mg/kg/día
en 3 dosis, VO Coenzima Q 10: 5-15 mg/kg/dia
Biotina: 10-15 mg/dia Tiamina(B1):
150-300 mg/dia
Servicio de Genética y Metabólismo.
Hospital Nacional de Niños.





























