Embed Size (px)
Citation preview

Determination of the Barrier Height for Acetyl Radical Dissociation from Acetyl ChloridePhotodissociation at 235 nm Using Velocity Map Imaging†
Xiaonan Tang, Britni J. Ratliff, Benjamin L. FitzPatrick, and Laurie J. Butler*The James Franck Institute and Department of Chemistry, UniVersity of Chicago, Chicago, Illinois 60637
ReceiVed: June 30, 2008; ReVised Manuscript ReceiVed: August 15, 2008
This work uses velocity map imaging to determine the barrier height for acetyl radical, CH3CO, dissociationto CH3 + CO. Photodissociation of acetyl chloride at 235 nm generates acetyl radicals with an internal energydistribution spanning this barrier. We determine the velocity and internal energy distribution of all nascentacetyl radicals, stable and unstable, by measuring the velocities of the Cl(2P3/2) and Cl(2P1/2) cofragments.These Cl cofragments are detected with 2 + 1 resonance-enhanced multiphoton ionization (REMPI) in aspin-orbit branching ratio Cl(2P3/2):Cl(2P1/2) of 3.3 ( 0.2. Using 157 nm photoionization, we then detect therecoil velocities of the energetically stable acetyl radicals. The radicals and momentum matched Cl atomsevidence parallel angular distributions. Comparison of the total recoil translational energy distribution P(ET)for all radicals to that obtained from the detection of stable radicals yields an onset for dissociation at atranslational energy of 25.0 ( 0.4 kcal/mol. From this onset we can calculate the barrier height for CH3COf CH3 + CO, but this relies on prior determinations of the C-Cl bond energy of acetyl chloride. Using anexperimental bond dissociation energy of 83.4 ( 0.2 kcal/mol yields a dissociation barrier of 14.2 ( 0.5kcal/mol. Our data evidence that a portion of the acetyl radicals formed with total internal energy above thebarrier are stable due to the partitioning of energy into rotation during the C-Cl bond fission of the precursor.Thus, the internal energy onset for dissociation is not as sharp as was assumed in prior determinations of thebarrier. The experimentally determined onset is compared with that predicted from electronic structurecalculations at the G3//B3LYP and CCSD(T) levels of theory.
I. IntroductionNumerous groups have studied the photodissociation dynam-
ics of acetyl chloride1-6 and the unimolecular decompositionreaction dynamics of the acetyl radical, CH3CO.5-11 Thephotodissociation of acetyl chloride at 248 nm occurs via a 1[nO,π*CdO] excitation and results in the breaking of the C-Cl bondrather than the C-C bond.2,6 The angular distribution of the Cland CH3CO photofragments is anisotropic with � valuesreported between 0.9-1.0.1,6,7,10 This fast R-bond cleavageindicates that the dissociation occurs adiabatically on a singletA′′ electronic state rather than undergoing intersystem crossingto the triplet state.1,8
Numerous experimental and computed barrier heights5-7,9,10,12-14
for this dissociation channel have been published with valuesranging widely from 11.1 to 21.1 kcal/mol. A summary of selectrecent values are listed in Tables 1 and 2. Some of theseexperiments measured a kinetic energy threshold for CH3COradicals produced photolytically from acetyl chloride, so theirbarrier height determinations rely on an assumed value for theC-Cl bond dissociation energy. For comparison with the presentwork, Table 1 recalculates the reported barriers in prior workusing an updated experimental dissociation energy of 83.4 (0.2 kcal/mol for the C-Cl bond energy calculated from the heatsof formation15-17 at 298 K and the appropriate enthalpyincrements needed to adjust these values to 0 K.16,18 We alsouse a different assumption regarding the internal energy of thephotolytic precursor.
Using photofragment translational spectroscopy, North et al.5,6
report two measurements of the acetyl dissociation barrier
height, one using acetyl chloride and the other using acetoneas the photolytic precursors. Their data show that the acetylradicals produced in the lower recoil kinetic energy photolysisevents are lost due to their high internal energy and resultingsecondary dissociation to CH3 + CO. Thus they derive a barrierfor the secondary dissociation process from the experimentallydetermined kinetic energy threshold for loss of acetyl radicals.In their analysis, however, they assume that the recoil kineticenergy distribution, P(ET), used to fit the stable acetyl productis sharply truncated at the observed threshold. Though this givesa good fit to their data, their analysis assumes that all radicalswith internal energy above the threshold dissociate, not ac-counting for the fact that some of the available energy may bepartitioned into rotational energy of the radical or into spin-orbitexcitation of the Cl cofragment. Radicals formed in conjunctionwith spin-orbit excited Cl(2P1/2) have 2.52 kcal/mol less internalenergy at a given recoil kinetic energy, thereby causing themto have a higher ET threshold than radicals formed in conjunctionwith Cl(2P3/2). They also assume the internal energy of the acetylradical precursor is substantially cooled in the supersonicexpansion. We analyze these effects further in this paper.
Subsequent work by Suzuki and co-workers10 sought to timeresolve the unimolecular dissociation of acetyl radicals producedfrom the photolysis of acetyl chloride. Interestingly, when theybinned the acetyl radicals by recoil velocity, and thus crudelyby internal energy, they found the measured dissociation rateswere markedly slower than those predicted by statistical theories.Again, they did not account for the possibility that significantrotational energy is imparted to the acetyl radical duringphotolysis. However, their experiments do offer a firm upperbound on the CH3 + CO dissociation barrier. About a quarter
† Part of the “Karl Freed Festschrift”.* Corresponding author. E-mail: [email protected].
J. Phys. Chem. B 2008, 112, 16050–1605816050
10.1021/jp8057417 CCC: $40.75 2008 American Chemical SocietyPublished on Web 10/31/2008

of the CH3CO acetyl radicals formed in the lowest internalenergy range (this is reported as 16-18 kcal/mol for acetylradicals formed in conjunction with Cl(2P3/2), assumingD0(C-Cl) ) 83 kcal/mol and the internal energy of thephotolytic precursor is zero) dissociate after 600 fs. Thus, theyreport an upper limit of 17 ( 1 kcal/mol for the CH3CO fCH3 + CO barrier. Finally Watkins and Word9 performed akinetics experiment on the addition of methyl radicals to carbonmonoxide which included an elementary step involving thedecomposition of acetyl radicals. Their analysis derived anactivation energy of 17.2 kcal/mol ( 0.5 kcal/mol at 298 K.
In this study we obtain a more accurate measurement of thebarrier height for acetyl radical dissociation to CH3+ CO andresolve the distribution of acetyl radicals that are stable despitehaving a total internal energy above the barrier. The method issimilar to that of our earlier work on determining the barrierfor acrolyl radical decomposition.19 The photodissociation ofacetyl chloride at 235 nm produces acetyl radicals spanning therange of theoretical and experimental barriers for the dissociationof CH3CO to CH3 + CO. Using [2 + 1] REMPI, the Cl atomsin each spin-orbit state, Cl(2P3/2) and Cl(2P1/2), are selectivelyionized and detected. Using momentum conservation, the totalrecoil translational energy distribution from photodissociationis obtained from the detection of the Cl photofragments. Oneexpects the dissociation lifetime of acetyl radicals formed withvibrational energy even 0.1 kcal/mol above the dissociationbarrier to be short; so only acetyl radicals with vibrational energybelow the barrier are detected. Since the P(ET) obtained fromthe nascent Cl products is the recoil kinetic energy distributionof Cl recoiling from both stable and unstable acetyl radicals,comparison with the energy distribution obtained from the
detection of stable CH3CO radicals reveals the recoil kineticenergy threshold for producing unstable radicals. In this waywe directly measure the barrier height for CH3CO dissociationto CH3 + CO.
II. Experimental Methods
A. Velocity Map Imaging. This experimental setup has beenpreviously described in detail,20-24 so only a brief outline willbe provided here. A 10% molecular beam was created bybubbling helium gas through a cooled liquid sample of acetylchloride (Aldrich, 98%) at -20 °C. At a total stagnation pressureof 550 Torr, the molecular beam was supersonically expandedthrough a room temperature pulsed valve with an orificediameter of 0.8 mm into the photodissociation region via askimmer. The parent molecules in the long (∼300 µs) stableregion of the expansion were photodissociated with a focused235 nm beam generated by tripling the output of a Lamda PhysikFL 3002 dye laser pumped by the second harmonic of a Nd:YAG laser operating at 21 Hz. Using 2 + 1 REMPI, theCl(2P3/2) or Cl(2P1/2) fragments were selectively ionized byscanning the Doppler profiles centered at 235.33 nm(4p 2D3/2r3p 2P3/2) or 235.20 nm (4p 2P1/2r3p 2P1/2), respec-tively.24 The laser power was reduced to render Coulombrepulsion between the ions insignificant. The acetyl radicals wereionized with a 157 nm beam generated from a GAM (EX10F/300) F2 laser which was fired 40 ns after the photolysis laser.RRKM calculations indicate that CH3CO radicals with energy0.1 kcal/mol above the barrier have a lifetime of ≈5 × 10-10
s.8 As predicted by the short lifetime of the radical, themagnitude of this delay was found to have negligible effect onthe radical signal when varied between 10 and 400 ns. The pathof the 157 nm beam was evacuated to minimize the absorptionby O2 and N2 en route to the high vacuum chamber. Theelectrostatic lens optics with a repeller voltage of 2000 V andextractor voltage of 1424 V accelerated the spherically expand-ing ions down a ∼577 mm grounded time-of-flight tube towardthe detector. The detector consisted of a position sensitiveChevron microchannel plate assembly (MCP) coupled to a P20phosphor screen. The voltage of the front plate of the MCPwas pulsed to -750 V for 80 ns to mass selectively detect ionsbased on their arrival time. The phosphor screen was maintainedat 3.3 kV above the potential of the rear MCP plate. A cooledcharge-coupled device (CCD) camera with a standard 35 mmlens was used to grab images of the ions and store them in the
TABLE 1: Select Experimental Measurements of the Barrier Height for Acetyl Radical Dissociation to CH3 + CO
experimental methodλ (nm)
threshold ET
(kcal/mol)reported barrier
(kcal/mol)recalculated
barriera
photofragmenttranslational spectroscopyb
248 15.0 17 ( 1 18.2 ( 1c
photofragmenttranslational spectroscopyd
248 13.5 17.8 ( 3
kinetic measuremente Ea ) 17.2 ( 0.5subpicosecond time
clocked photofragment imagingf255 e17 ( 1 e17.7 ( 1g
velocity mapimaging (this work)
235 25.0 ( 0.4 14.2 ( 0.5 14.2 ( 0.5
a Values recalculated by accounting for the thermal vibrational energy in the precursor and using the updated16b experimental C-Cl bondenergy of 83.41 kcal/mol. The updated experimental bond energy is 0.19 kcal/mol lower16b than the best prior value of 83.6 kcal/mol. The priorvalue was calculated using the ∆Hf
°(CH3COCl) at 298 K of -58.03 ( 0.2 kcal/mol,15 an ATcT value of -2.48 ( 0.21 kcal/mol at 298 K forCH3CO,15b,16b and ∆Hf
°(Cl) at 298 K of 28.992 kcal/mol.16a,17 The enthalpies of formation at 298 K were then corrected to 0 K values usingenthalpy increments for CH3CO of 2.984 kcal/mol,15b,16b Cl of 1.499 kcal/mol,16a,b and gaseous CH3COCl of 3.529 kcal/mol.18 b From the workof North et al.5 c The nozzle temperature was 353 K giving ⟨Evib(CH3COCl)⟩ ) 1.6 kcal/mol. d From the work of North et al.6 This work usedacetone as the photolytic precursor and Do(C-Cl) ) 83.7 kcal/mol. e From the work of Watkins and Word.9 f From the work of Shibata et al.10
g The reported work did not give the nozzle temperature, so we assume it to be 298 K giving ⟨Evib(CH3COCl)⟩ ) 1.1 kcal/mol.
TABLE 2: Select Theoretical Predictions of the BarrierHeight for Acetyl Radical Dissociation to CH3 + CO
theoretical method barriera (kcal/ mol)
MP2/cc-pVTZb 18.7G2//G2Qc 17.3MP2/6-31 g(d)d 18.5G3//B3LYP/6-31 g(d)e 15.2G3//B3LYP/6-311 g(d,p)f 15.6G3//B3LYP/6-311++g(d,p)f 15.73CCSD(T)f 16.77
a Including zero-point corrections. b From the work of Deshmukhet al.7 c From the work of Osborn et al.12 d From the work of Liu etal.13 e From the work of Miller et al.14 f This work.
Barrier Height for Acetyl Radical Dissociation J. Phys. Chem. B, Vol. 112, No. 50, 2008 16051

computer using the ion-counting method.25 The resolution ofthe image is ∆V/V ) 0.048 at 1153 m/s and 0.036 at 1450 m/swhen calibrated with O2. The raw images were symmetrizedabout the vertical and horizontal axes in the data analysis.
B. Theoretical Methods. Calculations of the C-Cl bondenergy for acetyl chloride and the transition state for acetylradicals dissociation were performed at both G3//B3LYP/6-311++G(d,p) and UCCSD(T)/CBS//UCCSD(T)/aug-cc-pVTZlevels of theory, where CBS stands for complete basis set. Forthe G3//B3LYP results, we used the Gaussian 03 program,revision E.01.26 Optimized molecular geometries and vibrationalfrequencies were found using the B3LYP density functional withthe 6-311++G(d,p) basis set. The geometries were convergedto a root-mean-square (rms) force below 1 × 10-6 and an rmsdisplacement below 4 × 10-6, where both are in atomic units.The G3//B3LYP calculations27 were performed on B3LYP/6-311++G(d,p) optimized geometries. To compute the zero-pointvibrational energies, the B3LYP vibrational frequencies werescaled by 0.96 as recommended by Scott and Radom28 andrequired by the G3//B3LYP method. Wave functions for doubletspecies were spin-unrestricted, and wave functions for singletspecies were spin-restricted. The transition-state structure wasconfirmed by having only one imaginary frequency with motionalong the reaction coordinate. An intrinsic reaction coordinate(IRC) calculation was performed as an additional check. Allenergies are presented as zero-point corrected enthalpies offormation at zero Kelvin.
The coupled cluster geometry optimizations and single-pointcalculations were performed with MOLPRO29 using a UCCS-D(T)30/aug-cc-pVTZ31 with an ROHF reference. On advice,32
we chose unrestricted open-shell coupled cluster instead ofrestricted open-shell and checked that the spin contaminationwas minimal. No symmetry was assumed and multiple optimi-zations were carried out (e.g., CH3CO(Cl) was optimized havingeither O or Cl staggered with respect to the hydrogen atoms onthe methyl group) in order to find the structures correspondingto the global minima. Again, the geometries converged to a root-mean-square (rms) force below 1 × 10-6 and an rms displace-ment below 4 × 10-6, where both are in atomic units. Intrinsicreaction coordinate calculations were performed at the UCCSD(T)/aug-cc-pVDZ level of theory for the transition state to makesure it corresponds to the correct reactant and product wells.Single-point frozen-core UCCSD(T)30 energies were calculatedusing the augmented correlation-consistent basis sets (fragmentscontaining chlorine atoms utilized the updated aug-cc-pV(N+d)zbasis sets33 containing an additional tight d function), and ROHFand correlation energies were converged to less than onemicrohartree. A two-point CBS extrapolation devised by Hel-gaker34 was applied independently to the singles and doublescorrelation energy (CCSD - ROHF) and to the triples energyusing quadruple- and 5-� basis set results. The reference energiesutilized the 6-� basis set and were not extrapolated. Weaccounted for core-valence correlation by extrapolating thedifferences between two all-electron calculations (using the aug-cc-pwCVTZ and aug-cc-pwCVQZ basis sets,35 with the 1sorbital on chlorine frozen) and the triple- and quadruple-� froze-core calculations. Additionally, we included relativistic effectsusing sixth-order Douglas-Kroll-Hess (DKH6) calculations withthe aug-cc-pV5Z_DK basis sets (chlorine was again augmentedwith tight d functions),36 and the correction was taken asthe difference between the DKH6 UCCSD(T) energy and theanalogous calculation used in the extrapolations. Both of theabove basis sets were taken from the EMSL basis setexchange.37,38 The single-point energies include a zero-point
energy (ZPE) correction based on unscaled UCCSD(T)/aug-cc-pVTZ frequencies with anharmonic corrections from MP2/aug-cc-pVTZ for the closed-shell photolytic precursor andUMP2/aug-cc-pVTZ for the open-shell species using the Gauss-ian 0326 program. It should be noted that the method forgenerating the anharmonic corrections is very crude, but itshould yield qualitatively accurate corrections at the very least.The spin-orbit energy correction for chlorine was taken fromMoore’s tables24 and applied when calculating the bond dis-sociation energy of the precursor. All of the relevant structures,energies, unscaled vibrational frequencies, anharmonic correc-tions, and rotational constants of the precursor, CH3CO and Clradicals, and transition state leading to CH3 + CO calculatedat the G3//B3LYP/6-311++G(d,p) and UCCSD(T)/CBS//UCCSD(T)/aug-cc-pVTZ levels of theory are given in theSupporting Information.
III. Results
A. Recoil Translational Energy Distribution from AcetylChloride Photodissociation. The total recoil translationalenergy distribution for C-Cl bond fission from the 235 nmphotodissociation of acetyl chloride, generating both energeti-cally stable and unstable radicals, is determined by detectingthe Cl cofragments. Ion images of the 35Cl(2P1/2) and 35Cl-(2P3/2) photofragments are shown in Figure 1 with the laserpolarization along the vertical axis. Each image is used toreconstruct a three-dimensional scattering distribution using an
Figure 1. Raw images of (a) Cl(2P3/2) obtained with dissociation andprobe at a vacuum wavelength of 235.33 nm via the 4p 2D3/2 r 3p2P3/2 transition; (b) Cl(2P1/2) obtained with dissociation and probe at avacuum wavelength of 235.20 nm via the 4p 2P1/2r 3p 2P1/2 transition.The laser polarization is along the vertical direction in the plane of theimages as shown with the arrows. The image depicted is 901 × 901pixels with a linear intensity scale shown in the top right corner ofeach image. The images appear oval in shape due to the parallel angulardistribution and speed dependence (see text) of the anisotropy.
16052 J. Phys. Chem. B, Vol. 112, No. 50, 2008 Tang et al.
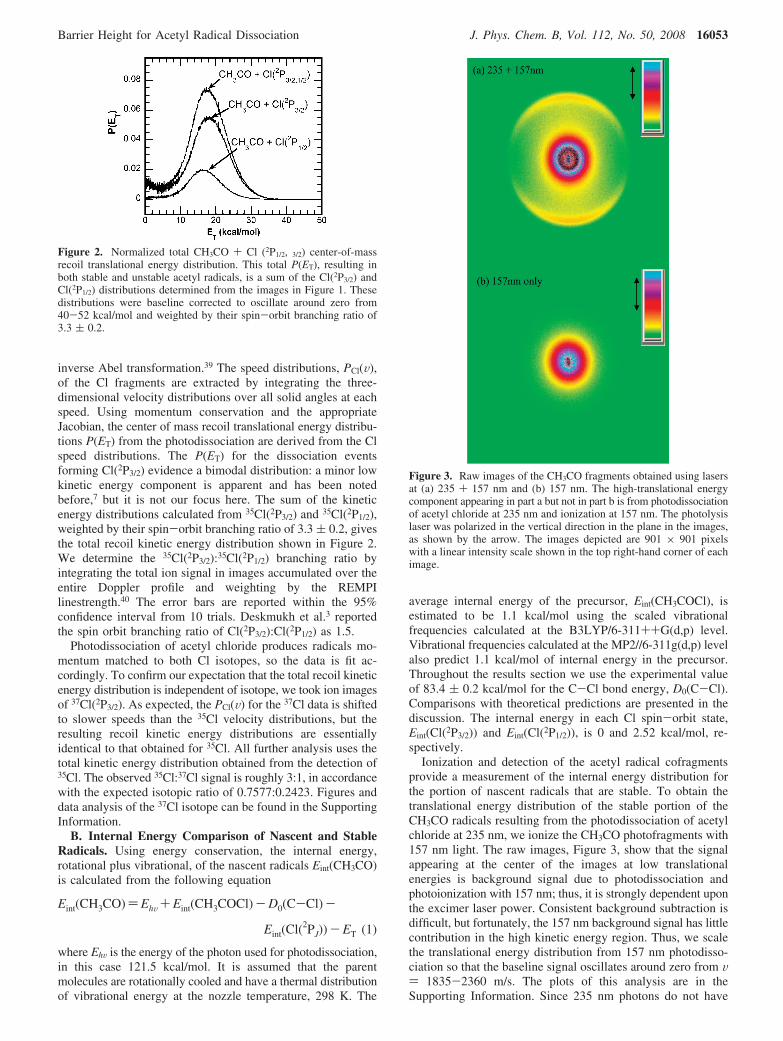
inverse Abel transformation.39 The speed distributions, PCl(V),of the Cl fragments are extracted by integrating the three-dimensional velocity distributions over all solid angles at eachspeed. Using momentum conservation and the appropriateJacobian, the center of mass recoil translational energy distribu-tions P(ET) from the photodissociation are derived from the Clspeed distributions. The P(ET) for the dissociation eventsforming Cl(2P3/2) evidence a bimodal distribution: a minor lowkinetic energy component is apparent and has been notedbefore,7 but it is not our focus here. The sum of the kineticenergy distributions calculated from 35Cl(2P3/2) and 35Cl(2P1/2),weighted by their spin-orbit branching ratio of 3.3 ( 0.2, givesthe total recoil kinetic energy distribution shown in Figure 2.We determine the 35Cl(2P3/2):35Cl(2P1/2) branching ratio byintegrating the total ion signal in images accumulated over theentire Doppler profile and weighting by the REMPIlinestrength.40 The error bars are reported within the 95%confidence interval from 10 trials. Deskmukh et al.3 reportedthe spin orbit branching ratio of Cl(2P3/2):Cl(2P1/2) as 1.5.
Photodissociation of acetyl chloride produces radicals mo-mentum matched to both Cl isotopes, so the data is fit ac-cordingly. To confirm our expectation that the total recoil kineticenergy distribution is independent of isotope, we took ion imagesof 37Cl(2P3/2). As expected, the PCl(V) for the 37Cl data is shiftedto slower speeds than the 35Cl velocity distributions, but theresulting recoil kinetic energy distributions are essentiallyidentical to that obtained for 35Cl. All further analysis uses thetotal kinetic energy distribution obtained from the detection of35Cl. The observed 35Cl:37Cl signal is roughly 3:1, in accordancewith the expected isotopic ratio of 0.7577:0.2423. Figures anddata analysis of the 37Cl isotope can be found in the SupportingInformation.
B. Internal Energy Comparison of Nascent and StableRadicals. Using energy conservation, the internal energy,rotational plus vibrational, of the nascent radicals Eint(CH3CO)is calculated from the following equation
Eint(CH3CO))EhV +Eint(CH3COCl)-D0(C-Cl)-
Eint(Cl(2PJ))-ET (1)
where EhV is the energy of the photon used for photodissociation,in this case 121.5 kcal/mol. It is assumed that the parentmolecules are rotationally cooled and have a thermal distributionof vibrational energy at the nozzle temperature, 298 K. The
average internal energy of the precursor, Eint(CH3COCl), isestimated to be 1.1 kcal/mol using the scaled vibrationalfrequencies calculated at the B3LYP/6-311++G(d,p) level.Vibrational frequencies calculated at the MP2//6-311g(d,p) levelalso predict 1.1 kcal/mol of internal energy in the precursor.Throughout the results section we use the experimental valueof 83.4 ( 0.2 kcal/mol for the C-Cl bond energy, D0(C-Cl).Comparisons with theoretical predictions are presented in thediscussion. The internal energy in each Cl spin-orbit state,Eint(Cl(2P3/2)) and Eint(Cl(2P1/2)), is 0 and 2.52 kcal/mol, re-spectively.
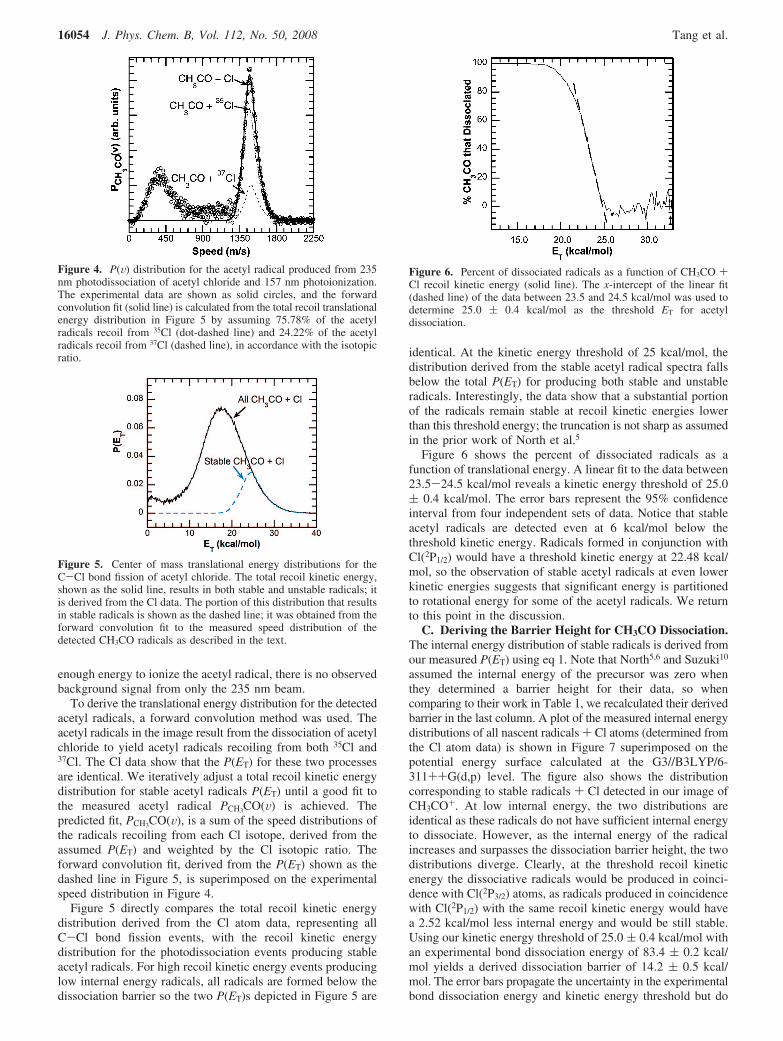
Ionization and detection of the acetyl radical cofragmentsprovide a measurement of the internal energy distribution forthe portion of nascent radicals that are stable. To obtain thetranslational energy distribution of the stable portion of theCH3CO radicals resulting from the photodissociation of acetylchloride at 235 nm, we ionize the CH3CO photofragments with157 nm light. The raw images, Figure 3, show that the signalappearing at the center of the images at low translationalenergies is background signal due to photodissociation andphotoionization with 157 nm; thus, it is strongly dependent uponthe excimer laser power. Consistent background subtraction isdifficult, but fortunately, the 157 nm background signal has littlecontribution in the high kinetic energy region. Thus, we scalethe translational energy distribution from 157 nm photodisso-ciation so that the baseline signal oscillates around zero from V) 1835-2360 m/s. The plots of this analysis are in theSupporting Information. Since 235 nm photons do not have
Figure 2. Normalized total CH3CO + Cl (2P1/2, 3/2) center-of-massrecoil translational energy distribution. This total P(ET), resulting inboth stable and unstable acetyl radicals, is a sum of the Cl(2P3/2) andCl(2P1/2) distributions determined from the images in Figure 1. Thesedistributions were baseline corrected to oscillate around zero from40-52 kcal/mol and weighted by their spin-orbit branching ratio of3.3 ( 0.2.
Figure 3. Raw images of the CH3CO fragments obtained using lasersat (a) 235 + 157 nm and (b) 157 nm. The high-translational energycomponent appearing in part a but not in part b is from photodissociationof acetyl chloride at 235 nm and ionization at 157 nm. The photolysislaser was polarized in the vertical direction in the plane in the images,as shown by the arrow. The images depicted are 901 × 901 pixelswith a linear intensity scale shown in the top right-hand corner of eachimage.
Barrier Height for Acetyl Radical Dissociation J. Phys. Chem. B, Vol. 112, No. 50, 2008 16053
enough energy to ionize the acetyl radical, there is no observedbackground signal from only the 235 nm beam.
To derive the translational energy distribution for the detectedacetyl radicals, a forward convolution method was used. Theacetyl radicals in the image result from the dissociation of acetylchloride to yield acetyl radicals recoiling from both 35Cl and37Cl. The Cl data show that the P(ET) for these two processesare identical. We iteratively adjust a total recoil kinetic energydistribution for stable acetyl radicals P(ET) until a good fit tothe measured acetyl radical PCH3CO(V) is achieved. Thepredicted fit, PCH3CO(V), is a sum of the speed distributions ofthe radicals recoiling from each Cl isotope, derived from theassumed P(ET) and weighted by the Cl isotopic ratio. Theforward convolution fit, derived from the P(ET) shown as thedashed line in Figure 5, is superimposed on the experimentalspeed distribution in Figure 4.
Figure 5 directly compares the total recoil kinetic energydistribution derived from the Cl atom data, representing allC-Cl bond fission events, with the recoil kinetic energydistribution for the photodissociation events producing stableacetyl radicals. For high recoil kinetic energy events producinglow internal energy radicals, all radicals are formed below thedissociation barrier so the two P(ET)s depicted in Figure 5 are
identical. At the kinetic energy threshold of 25 kcal/mol, thedistribution derived from the stable acetyl radical spectra fallsbelow the total P(ET) for producing both stable and unstableradicals. Interestingly, the data show that a substantial portionof the radicals remain stable at recoil kinetic energies lowerthan this threshold energy; the truncation is not sharp as assumedin the prior work of North et al.5
Figure 6 shows the percent of dissociated radicals as afunction of translational energy. A linear fit to the data between23.5-24.5 kcal/mol reveals a kinetic energy threshold of 25.0( 0.4 kcal/mol. The error bars represent the 95% confidenceinterval from four independent sets of data. Notice that stableacetyl radicals are detected even at 6 kcal/mol below thethreshold kinetic energy. Radicals formed in conjunction withCl(2P1/2) would have a threshold kinetic energy at 22.48 kcal/mol, so the observation of stable acetyl radicals at even lowerkinetic energies suggests that significant energy is partitionedto rotational energy for some of the acetyl radicals. We returnto this point in the discussion.
C. Deriving the Barrier Height for CH3CO Dissociation.The internal energy distribution of stable radicals is derived fromour measured P(ET) using eq 1. Note that North5,6 and Suzuki10
assumed the internal energy of the precursor was zero whenthey determined a barrier height for their data, so whencomparing to their work in Table 1, we recalculated their derivedbarrier in the last column. A plot of the measured internal energydistributions of all nascent radicals + Cl atoms (determined fromthe Cl atom data) is shown in Figure 7 superimposed on thepotential energy surface calculated at the G3//B3LYP/6-311++G(d,p) level. The figure also shows the distributioncorresponding to stable radicals + Cl detected in our image ofCH3CO+. At low internal energy, the two distributions areidentical as these radicals do not have sufficient internal energyto dissociate. However, as the internal energy of the radicalincreases and surpasses the dissociation barrier height, the twodistributions diverge. Clearly, at the threshold recoil kineticenergy the dissociative radicals would be produced in coinci-dence with Cl(2P3/2) atoms, as radicals produced in coincidencewith Cl(2P1/2) with the same recoil kinetic energy would havea 2.52 kcal/mol less internal energy and would be still stable.Using our kinetic energy threshold of 25.0 ( 0.4 kcal/mol withan experimental bond dissociation energy of 83.4 ( 0.2 kcal/mol yields a derived dissociation barrier of 14.2 ( 0.5 kcal/mol. The error bars propagate the uncertainty in the experimentalbond dissociation energy and kinetic energy threshold but do
Figure 4. P(V) distribution for the acetyl radical produced from 235nm photodissociation of acetyl chloride and 157 nm photoionization.The experimental data are shown as solid circles, and the forwardconvolution fit (solid line) is calculated from the total recoil translationalenergy distribution in Figure 5 by assuming 75.78% of the acetylradicals recoil from 35Cl (dot-dashed line) and 24.22% of the acetylradicals recoil from 37Cl (dashed line), in accordance with the isotopicratio.
Figure 5. Center of mass translational energy distributions for theC-Cl bond fission of acetyl chloride. The total recoil kinetic energy,shown as the solid line, results in both stable and unstable radicals; itis derived from the Cl data. The portion of this distribution that resultsin stable radicals is shown as the dashed line; it was obtained from theforward convolution fit to the measured speed distribution of thedetected CH3CO radicals as described in the text.
Figure 6. Percent of dissociated radicals as a function of CH3CO +Cl recoil kinetic energy (solid line). The x-intercept of the linear fit(dashed line) of the data between 23.5 and 24.5 kcal/mol was used todetermine 25.0 ( 0.4 kcal/mol as the threshold ET for acetyldissociation.
16054 J. Phys. Chem. B, Vol. 112, No. 50, 2008 Tang et al.

not account for the uncertainty in the internal energy of thephotolytic precursor. We compare these results to two theoreticalpredictions in the discussion.
D. Dynamics of Acetyl Chloride Photodissociation. Thelinear polarization of the photolysis laser allows for the studyof the angular distribution41-44 of acetyl chloride photodisso-ciation at 235 nm. The recoil angular distributions are shownin Figure 8. The Cl(2P3/2) angular distribution is fit using
I(θ) ∝ 1+ �2P2(cos θ)+ �4P4(cos θ) (2)
as the signal depends both on the anisotropy of the photodis-sociation process and on the REMPI detection of the J ) 3/2
atoms. The Cl(2P1/2) and CH3CO angular distributions are fitwith
I(θ) ∝ 14π
[1+ �P2(cos θ)] (3)
where θ is the angle between the recoiling fragment’s velocityvector and the electric vector of the photolysis laser. � is theanisotropy parameter, P2 is the second-order Legendre polyno-mial, and P4 is the fourth-order Legendre polynomial.
We examine the anisotropy of two kinetic energy rangeswhich correspond roughly to the formation of unstable versusstable radicals. The signal appearing in the Cl images in therange of 200-275 pixels from the center of the imagecorresponds to a recoil kinetic energy range (for verticallyrecoiling fragments) of 10.3-19.5 kcal/mol. This is the rangein which momentum matched radicals are unstable. In this range,the �2 ) 1.00 ( 0.07 and �4 ) -0.04 ( 0.01 for Cl(2P3/2)while � ) 1.01 ( 0.01 for Cl(2P1/2). As the recoil kinetic energyincreases, the radicals are formed with decreasing internalenergy. Thus, the signal appearing in the Cl images in the rangeof 275-350 pixels from the center of the image, whichcorresponds to a vertical recoil kinetic energy range of19.5-31.6 kcal/mol, is due primarily to stable radicals. In thishigher kinetic energy range, Cl(2P3/2) atoms recoil with anisot-ropy �2 ) 1.32 ( 0.11 and �4 ) 0.10 ( 0.02 while Cl(2P1/2)atoms have � of 1.34 ( 0.03. The anisotropy of the Cl atomsincreases gradually as a function of recoil kinetic energy. Theseanisotropy parameters confirm previous results indicating thatthe dissociation of acetyl chloride is prompt relative to molecularrotation and that the transition is predominantly parallel.
On the basis of momentum conservation, the angular distribu-tion of the CH3CO radicals should be an average of theanisotropies from the two spin-orbit Cl cofragments in the high-velocity portion of the distribution, weighted by the experi-
mentally determined spin-orbit branching ratio. Since �4 isessentially 0, we calculate the expected anisotropy of the acetylradical to be 1.33 ( 0.03. Integrating the CH3CO signal overthe pixel range of 250-315, which corresponds to a maximumvertical recoiling energy of 19.8-31.4 kcal/mol, we find theCH3CO anisotropy parameter � of 1.35 ( 0.03. The polarizationof the 157 nm probe light in the plane perpendicular to the 235nm beam and the molecular beam does not seem to significantlyaffect the anisotropy measurement. Plots of the angular distribu-tion for the higher kinetic energy range are shown in Figure 8.All error bars are reported within the 95% confidence intervalusing results from four trials. These experimental results arewithinreasonableagreementwithpreviouslypublishedresults.1,7,8,10
IV. Discussion
To compare the best prior measurement, that by North, etal.,5 of the CH3CO f CH3 + CO barrier to the presentdetermination, we first note that North’s analysis assumed theinternal energy of the acetyl chloride precursor was effectivelycooled in the supersonic expansion. However, North’s data wastaken with a continuous beam of acetyl chloride seeded inhelium at a very low stagnation pressure, P0, of 50 Torr and
Figure 7. Measured internal energy distribution (solid line) of all thenascent radicals + Cl atoms (determined from the Cl atom data) issuperimposed on the potential energy surface calculated at the G3//B3LYP/6-311++G(d,p) level. The distribution corresponding to stableradicals + Cl is also shown (dashed line).
Figure 8. Angular distributions for Cl(2P3/2), Cl(2P1/2), and CH3CO.Raw data is shown as open circles, and the fit is shown as a solid line.
Barrier Height for Acetyl Radical Dissociation J. Phys. Chem. B, Vol. 112, No. 50, 2008 16055

small nozzle diameter d (P0d ) 18.5 Torr-mm). For such amodest expansion in helium one can expect substantial coolingin the rotational degrees of freedom but not in the vibrationaldegrees of freedom. In our analysis, we assume the followingcompromise: the rotational degrees of freedom are completelycooled and the vibrational degrees of freedom are not cooled.Because the barrier is determined from a threshold measurement,if a portion of the precursors are vibrationally hot the thresholdwould reflect the radicals formed from the hotter precursors.Thus, correcting North’s barrier determination to account forthe 1.6 kcal/mol of vibrational energy in their uncooledprecursors (at a nozzle temperature of 80 °C) and the 0.41 kcal/mol update to the bond energy, the barrier determined fromtheir data would have been reported as 18.2 kcal/mol as opposedto 17 kcal/mol. Note this barrier determination is substantiallylarger than ours.
The present data allows us to determine a more accuratebarrier height for the dissociation of the acetyl radical to CH3
+ CO because we were able to resolve two other features ofthe data. Previous experiments by North and co-workers5 wereunable to measure the Cl atom spin-orbit distributions andassumed the energy imparted into rotation of the radical wasnegligible and therefore fit their data with a sharply truncatedP(ET). While North’s5 primary data is roughly consistent withours, we are able to resolve the production of Cl(2P3/2) vs Cl(2P1/2) and we have the resolution needed to detect thedistribution of radicals below the kinetic energy threshold whichare stable due to their rotational energy. As expected, thisresolution shifts our threshold kinetic energy higher and thusdecreases our derived barrier height. The radicals detected atkinetic energies below our threshold kinetic energy result onlyin a small part because some radicals are produced in conjunc-tion with excited-state Cl. It is clear that there is rotationalexcitation in the acetyl radical because this region is much widerthan 2.52 kcal/mol. While the impact parameter for thedissociation of acetyl chloride is small (≈0.1 Å) if one assumesit ensues from the ground-state geometry, it is well-known thatnO, π* excitation promotes the molecule to an excited state thatis pyramidal about the carbonyl carbon. Indeed, crude excited-state calculations2 suggest that the TS on the excited state ispyramidal, resulting in a larger impact parameter which wouldimpart significant rotational energy45 to the acetyl radicals. Ourdata evidence the production of some stable acetyl radicals atrecoil kinetic energies extending 6 kcal/mol below the kineticenergy threshold. Thus North’s barrier of 17 kcal/mol (correctedto 18.19 kcal/mol as described herein) must be an overestimateas it used a sharply truncated kinetic energy threshold.
The energy imparted to rotation in the photodissociation ofacetyl chloride also affects the analysis of Suzuki and co-workers.10 Suzuki’s RRKM rates assumed all internal energyis partitioned to vibrational degrees of freedom of the radical.They noted that the calculated rates are much faster than thoseexperimentally observed. One possible reason for the discrep-ancy between the measured unimolecular dissociation rate ofSuzuki and their RRKM estimates may be that they neglectedto account for some of the internal energy of the acetyl radicalbeing partitioned into rotational energy. This would, of course,result in their predicted RRKM rates being too fast.
Our experimental barrier height is directly dependent on theC-Cl bond dissociation energy of the precursor. The experi-mentally determined ET cutoff was 25.0 ( 0.4 kcal/mol and,using the best experimental bond energy, we obtained adissociation barrier height of 14.2 ( 0.5 kcal/mol. Because allof the most accurate experimental determinations of the barrier,
including ours, rely on an assumed C-Cl bond energy, weshould compare the experimental results with different levelsof electronic structure theory by summing the theoretical barrierenergy for decomposition, Ebarrier(CH3CO), and the theoreticallypredicted C-Cl bond energy of the precursor.
Ebarrier(CH3CO)+D0(C-Cl))EhV +Eint(CH3COCl)-
Eint(Cl(2P3/2))-ET,threshold (4)
On the right side of eq 4, the experimental kinetic energythreshold, ET,threshold, and the other experimental parameters havebeen described in the results and analysis sections. Our dataand analysis thus determine that the sum of the precursor C-Clbond energy and the acetyl decomposition barrier is 97.6 kcal/mol. Comparing this to an electronic structure prediction at theG3//B3LYP/6-311++G(d,p) level of theory, we sum thetheoretically predicted Ebarrier(CH3CO) ) 15.73 kcal/mol andD0(C-Cl) ) 83.01 kcal/mol and obtain 98.74 kcal/mol. As theestimated uncertainty in each is ∼1 kcal/mol, the prediction isin agreement with the experimental results. In contrast, a veryhigh level ab initio prediction at the CCSD(T) level of theorydoes not agree as well with the experimental result. The ab initioCCSD(T) prediction for the C-Cl bond energy described atlength in the theoretical methods subsection is 83.42 kcal/molwhile the acetyl dissociation barrier is predicted to be 16.77kcal/mol. The sum 100.19 kcal/mol is somewhat larger thanthe experimental sum of 97.6 kcal/mol. This discrepancy doesnot appear to be caused by the bond dissociation energy of 83.42kcal/mol, which is in fortuitously excellent agreement with theexperimental value, but it may be in part due to the ad hocnature of the anharmonic correction that is quite large for thebarrier height estimate. The difference may also reflect thedifficulty of calculating an accurate CH3CO f CH3 + COtransition-state energy. The possible multireference nature atthe transition state is yet to be investigated. We hope that thesenew experimental results will stimulate further considerationof these comparisons.
The CCSD(T) calculations presented here include three cor-rections which are worth analyzing further: an anharmoniccorrection to the zero-point energy, a core-valence correlationcorrection, and a relativistic correction. The contribution of eachimprovement is listed in Table 3. An initial assessment showsthat most of the corrections decrease the bond dissociationenergy/barrier height, except for the complete basis set extrapo-lation and the core-valence correlation corrections. This signdifference sets up a delicate interplay between the variouscorrections. While the magnitude of some entries, such as thelarge ZPE term, is unsurprising, others are not intuitivelyobvious when comparing the relative contribution to the bonddissociation energy versus the barrier height. Namely, theanharmonic contribution to the ZPE is miniscule for the bonddissociation energy, but it is sizable for the barrier height, at0.15 kcal/mol. A second example arises from the core-valencecorrelation and relativistic corrections. They are relatively smallfor the bond dissociation energy and almost cancel out oneanother, but in the case of the barrier height the core-valencecorrelation correction is four times larger than the relativisticcorrection. Hence, the net result from the core-valence cor-relation and relativistic corrections is approximately 0.33 kcal/mol, constituting a sizable adjustment. In contrast, the completebasis set extrapolation makes a sizable correction of almost 0.6kcal/mol to the bond dissociation energy, but the correspondingcorrection for the barrier height is much less. It should be notedthat an alternate extrapolation scheme, where the entirety ofthe correlation was extrapolated, was also investigated, and it
16056 J. Phys. Chem. B, Vol. 112, No. 50, 2008 Tang et al.

gave identical results for ∆E(CBS). Table 3 also contains thenet result of the higher-level corrections (HLCs) used in theG3 calculations for deriving the bond dissociation energy. Asis apparent, the HLC is approximately three times larger thanany of the corrections, excluding ZPE and Eso because theseare explicitly included in G3 as well, used in the coupled-clustercalculations. In summary, accurate bond dissociation energiesare affected more by the complete basis set extrapolation thanthe other, less often used, corrections. The barrier height studiedin this system showed less dependence on the complete basisset extrapolation, while anharmonic ZPE corrections andcore-valence correlation corrections could play a large role inbarrier height accuracy.
Acknowledgment. This work was funded by NSF renewalgrant #CHE-0746050 (Butler). B.R. acknowledges the supportof an NSF Graduate Research Fellowship. We thank BrankoRuscic for analyzing the primary experimental data on C-Clbond energies and incorporating that analysis in the ActiveThermochemical Tables (ATcT) to obtain a more accurateexperimental value. We also thank Kirk Peterson for hisilluminating and useful discussions regarding many aspects ofthe coupled-cluster calculations. Warmest wishes to our wonder-ful teacher and colleague, Karl Freed.
Supporting Information Available: We provide in theSupporting Information the G3//B3LYP and CCSD(T) resultsused to calculate the C-Cl bond energy and the CH3CO fCH3 + CO transition state. These include the structures, energies(with and without a harmonic zero-point correction), vibrationalfrequencies, and rotational constants. We also provide figuresof the PCl(V) and P(ET) obtained from data on the mass 35 and37 Cl(2P3/2) isotopes and also of the 157 nm light backgroundsubtraction used when analyzing the CH3CO data. This materialis available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) Shibata, T.; Suzuki, T. Chem. Phys. Lett. 1996, 262, 115.(2) Sumathi, R.; Chandra, A. K. J. Chem. Phys. 1993, 99, 6531.
(3) Deshmukh, S.; Hess, W. P. J. Chem. Phys. 1994, 100, 6429.(4) Martin, X.; Moreno, M.; Lluch, J. M. J. Phys. Chem. 1993, 97,
12186.(5) North, S.; Blank, D. A.; Lee, Y. T. Chem. Phys. Lett. 1994, 224,
38.(6) North, S. W.; Blank, D. A.; Gezelter, J. D.; Longfellow, C. A.;
Lee, Y. T. J. Chem. Phys. 1995, 102, 4447.(7) Deshmukh, S.; Myers, J. D.; Xantheas, S. S.; Hess, W. P. J. Phys.
Chem. 1994, 98, 12535.(8) Person, M. D.; Kash, P. W.; Butler, L. J. J. Phys. Chem. 1992, 96,
2021.(9) Watkins, K. W.; Word, W. W. Int. J. Chem. Kinet. 1974, 6, 855.
(10) Shibata, T.; Li, H.; Katayanagi, H.; Suzuki, T. J. Phys. Chem. A1998, 102, 3643.
(11) Somnitz, H.; Fida, M.; Ufer, T.; Zellner, R. Phys. Chem. Chem.Phys. 2005, 7, 3342.
(12) Osborn, D. L.; Choi, H.; Mordaunt, D. H.; Bise, R. T.; Neumark,D. M.; Rohlfing, C. M. J. Chem. Phys. 1997, 106, 3049.
(13) Liu, D.; Fang, W-H.; Fu, X-Y. Chem. Phys. Lett. 2000, 325, 86.(14) Miller, J. L.; McCunn, L. R.; Krisch, M. J.; Butler, L. J. J. Chem.
Phys. 2004, 121, 1830.(15) (a) Pedley, J. B.; Naylor, R. D.; Kirby, S. P. Thermochemical Data
of Organic Compounds, 2nd ed.; Capman and Hall: London, 1986. (b)Ruscic, B.; Boggs, J. E.; Burcat, A.; Csaszar, A. G.; Demaison, J.; Janoschek,R.; Martin, J. M. L.; Morton, M. L.; Rossi, M. J.; Stanton, J. F.; Szalay,P. G.; Westmoreland, P. R.; Zabel, F.; Berces, T. J. Phys. Chem. Ref. Data2005, 34, 573–656.
(16) (a) Ruscic, B.; Pinzon, R. E.; Morton, M. L.; von Laszewski, G.;Bittner, S.; Nijsure, S. G.; Amin, K. A.; Minkoff, M.; Wagner, A. F. J.Phys. Chem. A 2004, 108, 9979–9997. (b) Ruscic, B. Private communicationof unpublished interim results from Active Thermochemical Tables (ATcT)ver. 1.37 and the Core (Argonne) Thermochemical Network ver. 1.072 (July2008). The analysis in ver. 1.37 corrected a potential weak link in thebackground data supporting the conversion of the experimentally determinedenthalpy of hydrolysis of liquid acetyl chloride to the enthalpy of formationof gaseous acetyl chloride. This corrects the prior best value for D0(CH3C(O)-Cl) of 83.6 kcal/mol, calculated as described in the footnotes to Table 1, tothe current value of 83.41.
(17) Chase, M. W., Jr. NIST-JANAF Thermochemical Tables, 4thedition. J. Phys. Chem. Ref. Data, Monograph 9, 1-1951, 1998.
(18) Overend, J.; Nyquist, R. A.; Evans, J. C.; Potts, J. Spectrochem.Acta 1961, 17, 1205.
(19) Lau, K.-C.; Liu, Y.; Butler, L. J. J. Chem. Phys. 2005, 123, 054322.(20) Heck, A. J. R.; Chandler, D. W. Annu. ReV. Phys. Chem. 1995,
46, 335.(21) Eppink, A. T. J. B.; Parker, D. H. ReV. Sci. Instrum. 1997, 68,
3477.(22) Sato, Y.; Matsumi, Y.; Kawasaki, M.; Tsukiyama, K.; Bersohn, R.
J. Phys. Chem. 1995, 99, 16307.(23) Liu, Y.; Butler, L. J. J. Chem. Phys. 2004, 121, 11016.(24) Note that most authors label the upper state for detection of
Cl(2P1/2) as the 4p 2P1/2 state from C. E. Moore, “Atomic Energy Levels”NSRDS-NBS 35, 1, 1971. However, the NIST Atomic Spectra Databasehas adopted the reassignment of this upper state at 85917.937 cm-1 toa 2S state, as recommended in the comprehensive re-analysis of thechlorine spectrum by Radziemski, L. J.; Kaufman, V. J. Opt. Soc. Am.1969, 59, 424. To avoid confusion, we give the original C. E. Mooreassignment here. For the energy of the transition, we use the NIST valueof 85917.937-882.3515 cm-1.
(25) Chang, B.; Hoetzlein, R. C.; Mueller, J. A.; Geiser, J. D.; Houston,P. L. ReV. Sci. Instrum. 1998, 69, 1665.
(26) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb,M. A.; Cheeseman, J. R.; Montgomery, J. J. A.; Vreven, T.; Kudin, K. N.;Burant, J. C.; Milliam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.;Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.;Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.;Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li,X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.;Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.;Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.;Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich,S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.;Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.;Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz,P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.;Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson,B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03,ReVision E.01; Gaussian, Inc.: Wallingford, CT, 2004.
(27) Baboul, A. G.; Curtiss, L. A.; Redfern, P. C.; Raghavachari, K.J. Chem. Phys. 1999, 110, 7650.
(28) Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502.(29) Werner, H.-J.; Knowles, P. J.; Lindh, R.; Manby, F. R.; Schutz,
M.; Celani, P.; Korona, T.; Rauhut, G.; Amos, R. D.; Bernhardsson, A.;
TABLE 3: Summary of Corrections Made to CCSD(T)Calculations (in kcal/mol)
energy or correction Dea TSe
a
E(initial)b 86.27 19.36∆E(CBS)c 0.58 0.15∆E(ZPE)d -2.61 -3.21∆E(anharmonic)e 0.007 -0.148∆E(spin-orbit)f -0.84 -∆E(core-valence)g 0.21 0.44∆E(relativistic)h -0.20 -0.11∆E(sum)i -2.85 -2.58E(corrected)j 83.42 16.77∆E(HLC)k 1.37 -
a ZPE has intentionally been left out. b Reference energy usingaug-cc-pV6Z basis and correlation energy using aug-cc-pV5Z.c Energy difference between the above scheme and one where theCCSD and (T) energies are extrapolated separately from calculationsusing the aug-cc-pVQZ and aug-cc-pV5Z bases. d Zero-point energycorrection using harmonic frequencies from CCSD(T)/aug-cc-pVTZcalculations. e Anharmonic correction to the zero-point energydifference using MP2/aug-cc-pVTZ. f Spin-orbit correction for Cl asnoted in the computational section. g Core-valence correlationcorrection using energy differences extrapolated from aug-cc-pVTZ andaug-cc-pVQZ calculations. h Relativistic correction using sixth-orderDKH calculations with an aug-cc-pV5Z_DK basis. i Sum of the abovecorrections. j Sum of the above value and the value in the first row,E(initial). k Using parameter values found in the paper by Baboul, etal.46
Barrier Height for Acetyl Radical Dissociation J. Phys. Chem. B, Vol. 112, No. 50, 2008 16057

Berning, A.; Cooper, D. L.; Deegan, M. J. O.; Dobbyn, A. J.; Eckert, F.;Hampel, C.; Hetzer, G.; Lloyd, A. W.; McNicholas, S. J.; Meyer, W.; Mura,M. E.; Nicklass, A.; Palmieri, P.; Pitzer, R.; Schumann, U.; Stoll, H.; Stone,A. J.; Tarroni, R.; Thorsteinsson, T. MOLPRO, version 2006.1, a packageof ab initio programs; see http://www.molpro.net.
(30) Knowles, P. J.; Hampel, C.; Werner, H.-J. J. Chem. Phys. 1993,99, 5219. (a) Erratum: J. Chem. Phys. 2000, 112, 3106.
(31) Kendall, R. A., Jr.; Harrison, R. J. J. Chem. Phys. 1992, 96, 6796.(32) Peterson, K. A. Private communication (July 10, 2008).(33) Dunning, T. H., Jr.; Peterson, K. A.; Wilson, A. K. J. Chem. Phys.
2001, 114, 9244.(34) Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. J. Chem. Phys. 1997,
106, 9639.(35) Peterson, K. A., Jr J. Chem. Phys. 2002, 117, 10548.(36) de Jong, W. A.; Harrison, R. J.; Dixon, D. A. J. Chem. Phys. 2001,
114, 48.(37) Feller, D. J. Comput. Chem. 1996, 17, 1571.(38) Schuchardt, K. L.; Didier, B. T.; Elsethagen, T.; Sun, L.; Guru-
moorthi, V.; Chase, J.; Li, J.; Windus, T. L. J. Chem. Inf. Model 2007, 47,1045.
(39) Dribinski, V.; Ossadtchi, A.; Mandelshtam, V. A.; Reisler, H. ReV.Sci. Instrum. 2002, 73, 2634.
(40) Liyanage, R.; Yang, Y. A.; Hashimoto, S.; Gordon, R. J.; Field,R. W. J. Chem. Phys. 1995, 103, 6811.
(41) Rakitzis, T. P.; Zare, R. N. J. Chem. Phys. 1999, 110, 3341.(42) Rakitzis, T. P.; Kandel, S. A.; Alexander, A. J.; Kim, Z. H.; Zare,
R. N. J. Chem. Phys. 1999, 110, 3351.(43) Samartzis, P. C.; Bakker, B. L. G.; Rakitzis, P.; Parker, D. H.;
Kitsopoulos, T. N. J. Chem. Phys. 1999, 110, 5201.(44) Bass, M. J.; Brouard, M.; Clark, A. P.; Vallance, C.; Martinez,
Haya. Phys. Chem. Chem. Phys. 2003, 5, 856.(45) From the excited-state geometry reported by Martin et al.,4 we
calculate an impact parameter of 0.43 Å. Using an impulsive model, radicalsformed with ET ) 25 kcal/mol have 13 kcal/mol imparted to rotationalenergy.
(46) Baboul, A. G.; Curtiss, L. A.; Redfern, P. C.; Raghavachari, K.J. Chem. Phys. 1999, 110, 7650.
JP8057417
16058 J. Phys. Chem. B, Vol. 112, No. 50, 2008 Tang et al.





























