Embed Size (px)
Citation preview

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Sept. 1993, p. 2765-27700099-2240/93/092765-06$02.00/0Copyright X 1993, American Society for Microbiology
Vol. 59, No. 9
Detection of Hepatitis A Virus in Mercenaria mercenaria byCoupled Reverse Transcription and Polymerase
Chain ReactionBISWENDU B. GOSWAMI,* WALTER H. KOCH, AND THOMAS A. CEBULA
Division ofMolecular Biological Research and Evaluation, Food and Drug Administration,200 C Street, SW, Washington, D.C. 20204
Received 21 December 1992/Accepted 9 June 1993
Hepatitis A virus (HAV) is a major cause of infectious hepatitis in humans. In this respect, bivalve molluskspose a major health concern because they are filter feeders and can concentrate the virus up to 900-fold fromcontaminated water. Detection ofHAV has been hampered because wild-type HAV grows poorly if at all in cellculture. Here we describe a technique for the detection of HAV in shellfish based on reverse transcriptioncoupled with the polymerase chain reaction. RNA is isolated from hard-shell clam tissue and reversetranscribed with avian myeloblastosis virus reverse transcriptase. A portion of the cDNA pool is then amplifiedwith primers specific for HAV. In experiments with an in vitro-synthesized HAV transcript, we were able todetect HAV sequence in the presence ofa 200-million-fold excess of shellfish RNA. When intact virus was addedto shellfish tissue before the isolation of RNA, the method was capable of detecting 10 viral RNA molecules ina reaction mixture.
Hepatitis A virus (HAV) is a positive-strand RNA virusbelonging to the hepatovirus group of the picornavirusfamily. Worldwide, HAV causes a high percentage of theinfectious hepatitis cases reported each year (6, 11). A smallbut significant portion (approximately 7% by various esti-mates) of the reported cases of infectious hepatitis areattributed to the consumption of raw or inadequately cookedshellfish (1, 5). Detection of HAV in clinical samples orenvironmental samples such as shellfish is not routinelypossible because wild-type HAV grows very poorly in cellcultures (20). Except for virus preparations that have beenadapted for rapid growth in cell culture, HAV does notproduce a detectable cytopathic effect in infected cells (16).Even with cell culture-adapted virus, detection of viralantigens by techniques such as immunofluorescence or ra-dioimmunofocus assays requires 4 to 6 weeks of incubationof infected cell cultures (9, 12). Because of cytotoxicity, onlylimited quantities of virus-containing extracts from shellfishcan be tested in cell cultures. Hence, there is a need todevelop a more direct, reliable, and sensitive method for thedetection of HAV in environmental samples.
Recently, other investigators have developed methodsthat utilize radiolabeled DNA or RNA probes to detect HAVin environmental samples, which (unlike clinical samples, inwhich the viral load may be quite high) contain limitedquantities of virus (9, 10, 21). By using RNA probes in a dotblot assay, it was possible to detect 107 physical particles ofHAV, which were used to seed 20 g of oyster or clam tissue.However, in parallel experiments, the detection limit forHAV RNA isolated from purified virus was 100 ng or 4 x 109physical particles. The reason for a two-order-of-magnitudedecrease in sensitivity in detecting purified viral RNA is notclear. The procedure for the treatment of tissue samplesbefore RNA extraction was labor-intensive and time-con-suming (21).A simple technique based on the capture of virus particles
* Corresponding author.
from clinical material by antibody followed by reversetranscription (RT) and subsequent polymerase chain reac-tion (PCR) amplification has recently been described (8).During a recent multistate outbreak of hepatitis A, thismethod was used to detect HAV RNA in oyster samplesimplicated in the outbreak (4).
In this article, we describe an RT-PCR-based method forthe detection of HAV in environmental samples; the methodstarts with direct isolation of RNA from shellfish tissue. Themethod can detect 400 copies of an in vitro-synthesizedHAV transcript in the presence of a 200-million-fold excessof shellfish RNA when random hexamers are used to primecDNA synthesis. By use of oligo(dT)16 in the RT step, thesensitivity can be further augmented to detect 10 copies ofthe viral genomic RNA.
MATERIALS AND METHODS
Avian myeloblastosis virus reverse transcriptase (20 to 25U/,l) from several suppliers (Promega, Madison, Wis.;Boehringer Mannheim Biochemicals, Indianapolis, Ind.; andAmersham Corporation, Arlington Heights, Ill.) was diluted10-fold in dilution buffer (10 mM potassium phosphate buffer[pH 7.5], 2 mM dithiothreitol, 0.2% Triton X-100, 10%glycerol) and incubated on ice for 30 min before being addedto the RT reaction mixture. Taq polymerase was obtainedfrom Promega. 3'-oligonucleotide labeling and detection kitswere obtained from Amersham. Random hexamers, oli-go(dT)16, competent cells, restriction enzymes, and T4 DNAligase were from Bethesda Research Laboratories (Gaithers-burg, Md.). SP6 RNA polymerase was obtained from NewEngland Biolabs (Beverly, Mass.). RNase-free DNase wasfrom U.S. Biochemicals (Cleveland, Ohio).
Oligonucleotides for PCR. Two primer pairs were used inthis study. The sequences of all four primers were based onthe sequence of wild-type HAV (strain HM-175) (2). Primerpair 1 amplified a 276-bp region (nucleotides 6850 to 7125).The sequence of the forward primer was 5'CCTCTGGGTCTCCTTGTACAGC3'. The sequence of the reverse primer
2765
on January 25, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from

2766 GOSWAMI ET AL.
was 5'CCGAAACTGGTTlCAGCTGAGG3'. Primer pair 2amplified a 489-bp region (nucleotides 6256 to 6744). Thesequence of the forward primer was 5'ATGCTATCAACATGGATTCATCTCCTGG3'. The sequence of the re-verse primer was 5'CACTCATGATTCTACCTGCTTCTCTAATC3'. A fifth oligonucleotide, with the sequence5'TGTCAGGCTITGAAGATTCTCTG3' (nucleotides 6951to 6974), was used as an internal probe in hybridizationexperiments when primer pair 1 was used for amplification.All oligonucleotides were synthesized by M. Trucksess atthe Food and Drug Administration.
Synthesis of in vitro HAV transcript. A full-length cDNAclone of HAV in pBR322 was kindly provided by S. Emer-son (2). A BgllI fragment (nucleotides 6214 to 7143) wasisolated and cloned into BamHI-digested pGem4 (Promega).Crude plasmid was isolated from 100-ml cultures by alkalinelysis and further purified by polyethylene glycol precipita-tion (7). The plasmid was linearized with HindIII; RNA wassynthesized from S ,ug of linearized plasmid with 50 U of SP6polymerase in a total volume of 100 ,ul as previously de-scribed (13). The DNA template was removed by digestionwith RNase-free DNase. The sample was adjusted to 0.7 Mammonium acetate, and the RNA was recovered by precip-itation with 2.5 volumes of ethanol. The ethanol precipita-tion step was performed a total of three times to removecontaminating nucleoside triphosphates.
Isolation of tissue RNA. Hard-shell clams, identified asMercenana mercenaria by the Northeast Seafood Labora-tory, Davisville, R.I., were obtained from a local seafoodmarket. Fresh or frozen clam tissue was directly homoge-nized in a mixture of 10 volumes of 50 mM sodium acetatebuffer (pH 5.5)-2 mM EDTA-1% sodium dodecyl sulfate(SDS) and 10 volumes of water-saturated phenol in a Waringblender for 1 min at full speed. The mixture was shakenvigorously at room temperature for 10 to 15 min and centri-fuged to separate the phases. The cloudy upper aqueousphase was reextracted with an equal volume of water-saturated phenol as described above. The still-cloudy aque-ous phase was adjusted to 0.2 M sodium acetate, and totalnucleic acids were precipitated with 2.5 volumes of ethanol.The bulky precipitate was collected by centrifugation. Con-taminating DNA, tRNA, and polysaccharides were removedby three successive washes with ice-cold 3 M sodiumacetate, pH 5. These washes were carried out by thoroughlyresuspending the pellet with a glass rod in 3 M sodiumacetate, incubating the suspension on ice for 10 to 30 min,and centrifuging it at 10,000 x g for 15 to 20 min. The finalpellet was completely dissolved in water, and high-molecu-lar-weight RNA was precipitated from a mixture of 0.2 Msodium acetate and 2.5 volumes of ethanol. Any remainingDNA was digested by dissolving the RNA in 10mM Tris (pH7.5)-10 mM magnesium chloride-100 mM sodium chlorideand adding vanadyl ribonucleoside complex (to 5 mM) and50 U of RNase-free DNase per ml. After 30 min of incuba-tion at 37°C, the material was extracted twice with phenol-chloroform (1:1), and RNA was precipitated as describedabove. The final RNA pellet was dissolved in a suitablevolume of water and quantitated byA260. The yield of RNAwas approximately 1 mg/g of tissue with anA26/A280 ratio ofat least 1.8. When virus was added to shellfish tissue beforeRNA isolation, virus particles (kindly provided by B. H.Robertson, Centers for Disease Control and Prevention,Atlanta, Ga.) were added to 0.5 ml of a 10% homogenate ofclam tissue in extraction buffer only. RNA was then isolatedas described above by phenol extraction, except that allextractions were done in Eppendorf tubes. After addition of
phenol, the tubes were vortexed for 1 min and centrifuged toseparate the phases. The 3 M sodium acetate extraction wascarried out with 0.5 ml of solution each time, and the DNasedigestion was omitted. These modifications greatly reducedthe time needed to prepare the samples for RT-PCR, typi-cally requiring only 6 to 8 h from tissue homogenization tofinal RNA preparations.
Quantitation of virus. Quantitation of virus to be used forthe seeding experiments was carried out by quantitativecompetitive PCR, by using an in vitro-synthesized mutantRNA as a competitor (7a). The mutant competitor had a63-bp deletion in the region amplified by primer pair 2. Inorder to eliminate possible losses during isolation of RNAfrom virus, viral genomic RNA was directly quantitated bydiluting the virus preparation in water and heating it at 95°Cfor 5 min to dissociate RNA-protein complexes. A fixedamount of heat-dissociated virus was then reverse tran-scribed in the presence of several different concentrations ofthe mutant competitor and subsequently amplified withprimer pair 2. The amplification was carried out in thepresence of [ca-32P]dCTP. Radioactivity incorporated intoboth wild-type and mutant products was quantitated byscintillation counting, and the values for the mutant productwere corrected for the loss of G+C residues due to thedeletion. The ratio of incorporation into the two differentproducts was then plotted against the known concentrationof mutant competitor RNA added to obtain a regressioncurve. The concentration of viral genomic RNA moleculeswas the same as the number of mutant competitor moleculesadded when the ratio of incorporation into the two PCRproducts was 1.RT. RT of RNA was carried out in 25-,u reaction mixtures
which contained 20 fg to 1 pg of the in vitro HAV transcriptand 4 ,ug of shellfish RNA or various amounts of total RNA(approximately 50 ,ug of RNA from 0.5 ml of a 10% tissuehomogenate) isolated from clam tissue and which wereseeded with wild-type HAV, 50 mM Tris HCl (pH 8.3), 75mM KCl, 10 mM MgCl2, 1 mM concentrations of each of thefour deoxyribonucleoside triphosphates (dNTPs), 10 mMdithiothreitol, 1 U of RNase inhibitor per ,ul, either 50 to 200ng of random hexamers per ,ug of RNA or 0.5 ,ug ofoligo(dT)16, and 15 U of avian myeloblastosis virus reversetranscriptase. The reaction mixture was incubated for 10 minat 22°C and then for 50 min at 42°C. The reaction wasterminated by heating the mixture at 990C for 5 min, and themixture was chilled on ice. After centrifugation for 5 min, 5to 10 ,ul of the supernatant was used for PCR amplification ina total volume of 50 to 100 RI.PCR. PCR amplification was carried out in a 50- to 100-,u
volume which contained 5 to 10 RI of the cDNA pool fromRT reactions, 3 mM magnesium chloride, 200 ,uM eachdNTP, 0.5 p.M each amplification primer, and 1.5 U of Taqpolymerase. When RT reaction mixtures were diluted toobtain a specific concentration ofcDNA template in the PCRmixture, 5 to 10 p.l of a RT reaction mix without the RNAtemplate was added to the PCR mixture to achieve therequired magnesium chloride and dNTP concentrations. Themixture was denatured initially for 3 min at 95°C and thensubjected to 40 PCR cycles, each consisting of 90 s at 94°C,90 s at 630C, and 120 s at 720C. A final extension was carriedout at 720C for 10 min. Twenty microliters of each samplewas analyzed by agarose gel electrophoresis (1.6% agarose)in the presence of 1 ,ug of ethidium bromide per ml.
Southern blot hybridization. DNA from gels was trans-ferred to Zetaprobe membranes (Bio-Rad) with 0.2 M so-dium hydroxide as the transfer solution, as described in the
APPL. ENVIRON. MICROBIOL.
on January 25, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from

DETECTION OF HAV IN SHELLFISH BY PCR 2767
FIG. 1. Effect of tissue RNA isolated from M. mercenaria on
RT-PCR detection of in vitro-synthesized HAV transcript. The invitro transcript (20 fg to 1 pg) was reverse transcribed in thepresence of 4 ,ug of tissue RNA with random hexamers as describedin Materials and Methods. Portions of the cDNA pool representing4 fg (lane 3), 20 fg (lane 4), or 50 fg (lane 5) of the original RNA were
amplified with primer pair 2 in a total volume of 50 Lane 1 doesnot contain any template, and lane 2 contains 4 ,ug of tissue RNAonly. Lanes 6 to 8, cDNA pool representing 20 fg of original RNAamplified for 30, 35, and 40 cycles, respectively; lanes 9 to 11, cDNApool representing 4 fg of original RNA amplified for 30, 35, and 40cycles, respectively; lane M, 1-kb DNA marker. Sizes are indicatedon the left in base pairs.
manufacturer's protocol. The membranes were prehybrid-ized for 4 to 6 h and then hybridized overnight in a solutioncontaining 5x SSPE (lx SSPE is 0.18 M sodium chloride,0.01 M sodium phosphate buffer [pH 7.4], and 1 mM EDTA),7% SDS, 100 Vg of denatured and sonicated salmon spermDNA per ml, and 0.5% BLOTTO (nonfat dry milk). Afterprehybridization, the oligonucleotide probe was added to theprehybridization solution at a final concentration of 5 ng/ml.The probe was labeled with fluorescein-dUTP by usingterminal transferase according to the protocol of the manu-
facturer (ECL 3' Oligolabelling System; Amersham). Hy-bridization was carried out at 56°C, which is 10°C below themelting temperature (Tm) of this probe. After hybridization,the blots were washed twice for 30 min each time in 3 xSSPE-5% SDS-0.5% BLOTTO and twice for 30 min eachtime in lx SSPE-1% SDS. All washes were carried out at56°C with constant shaking. The hybridized probe was
detected by using an ECL detection kit (Amersham); 5%BLOTTO was used for blocking the membrane before theaddition of antibody, which was diluted in buffer containing1% BLOTTO. After the addition of enzyme substrate, theblots were exposed to Kodak X-Omat film for 30 s to 60 min.
RESULTS
The effect of addition of shellfish RNA during RT on
detection of the in vitro-synthesized HAV transcript byRT-PCR was investigated. Amounts of in vitro RNA rangingfrom 20 fg to 1 pg were reverse transcribed in the presence of4 ,ug of shellfish RNA with random hexamers as primers. AcDNA pool representing 4, 20, or 50 fg of initial RNA inputin the RT reaction mixture was then amplified in a totalvolume of 50 p.1. The addition of a 200-million-fold excess ofclam tissue RNA (20 fg in 4 p,g) during RT did not compro-mise the ability of this method to detect the 489-bp HAV-specific product (Fig. 1). Since 4 fg of the in vitro transcript(the input for each PCR) represents 8 x 103 molecules of a
0.9-kb transcript, 8 x 103 molecules of the HAV transcriptscould be detected in the presence of 1.9 x 1012 unrelated
FIG. 2. Southern hybridization of cDNA with an internal oligo-nucleotide probe. In lanes 1 to 5, a cDNA pool representing 2 pg,200 fg, 20 fg, 2 fg, and 0.2 fg, respectively, of the original transcriptwas amplified for 40 cycles with primer pair 1 and separated in anagarose gel. The DNA was transferred to a Zetaprobe membraneand hybridized to an internal probe as described in Materials andMethods. The position of the 276-bp amplified product is indicatedby the arrowhead. Lane 6 is a longer exposure of lane 5.
RNA molecules, assuming an average size of 4 kb. In orderto investigate the lower limit of detection of HAV sequencesby this method, cDNA pools corresponding to inputamounts ranging from 200 to 0.2 fg were amplified in 50-,ulreaction mixtures. In this experiment, primer pair 1, which isspecific for a 276-bp region of the wild-type HAV genome,was used. Portions (20 p,l) of each reaction mixture wereanalyzed by agarose gel electrophoresis and Southern hy-bridization with an internal oligonucleotide probe (Fig. 2).As little as 0.2 fg of input RNA, representing 400 molecules,could be detected after a 60-min exposure. Subsequently, wewere able to routinely detect 400 molecules of in vitro RNAin ethidium bromide-stained gels without the Southern hy-bridization step (data not shown). Previous reports haveindicated that coupled RT-PCR detection techniques arecapable of detecting 104 copies of an in vitro transcript bystaining in an agarose gel (15, 19). The method we havedescribed is at least as sensitive as those described inprevious reports, even in the presence of an overwhelmingamount of tissue RNA. With 2P-labeled primers, as few as50 to 60 copies of a specific transcript have reportedly beendetected (14). We should note that when we used oligo(dT)16rather than random hexamers in the RT reaction, as few as10 copies of purified viral genomic RNA could be detectedvisually in agarose gels (see below). We are currentlyexploring whether the sensitivity of the detection of the invitro transcript can be augmented to this level by synthesiz-ing a poly(A)-containing transcript and reverse transcribingit with oligo(dT)16.The effect of the number of PCR cycles on detection of the
HAV transcript in the presence of clam tissue RNA was alsoinvestigated (Fig. 1, lanes 6 to 11). Although a 489-bpproduct could be detected after 30 cycles of PCR, at least 35cycles were necessary to obtain a clear signal when westarted with cDNA from either 4 or 20 fg of the HAVtranscript.
In order to investigate whether the RNA extractionmethod would be able to effectively isolate viral RNA fromsmall amounts of intact virus present in contaminated tissue,a virus preparation was used to seed tissue extract before theisolation of RNA. RNA was isolated from 0.5 ml of a 10%extract of clam tissue in RNA isolation buffer after theaddition of 800 or 8,000 viral jparticles. These quantitiesrepresent 1.6 x 104 and 1.6 x 10 virus particles per gram ofclam tissue, respectively. As a control, RNA was alsoisolated from clam tissue that had not been seeded withHAV. Portions of the RNA samples representing 50, 250,500, and 2,500 particles of the original virus used for seedingwere then reverse transcribed in 25-p,l reaction volumes with
VOL. 59, 1993
on January 25, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from
2768 GOSWAMI ET AL.
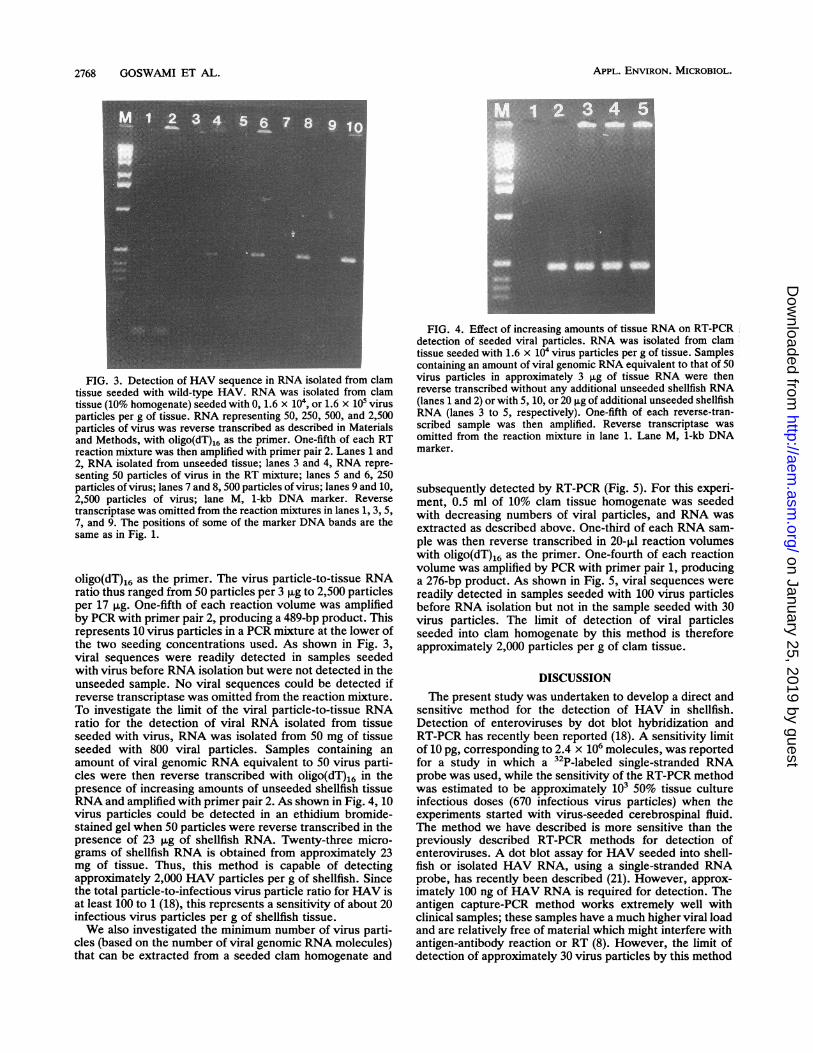
FIG. 3. Detection of HAV sequence in RNA isolated from clamtissue seeded with wild-type HAV. RNA was isolated from clamtissue (10% homogenate) seeded with 0, 1.6 x 104, or 1.6 x 105 virusparticles per g of tissue. RNA representing 50, 250, 500, and 2,500particles of virus was reverse transcribed as described in Materialsand Methods, with oligo(dT)16 as the primer. One-fifth of each RTreaction mixture was then amplified with primer pair 2. Lanes 1 and2, RNA isolated from unseeded tissue; lanes 3 and 4, RNA repre-senting 50 particles of virus in the RT mixture; lanes 5 and 6, 250particles of virus; lanes 7 and 8, 500 particles of virus; lanes 9 and 10,2,500 particles of virus; lane M, 1-kb DNA marker. Reversetranscriptase was omitted from the reaction mixtures in lanes 1, 3, 5,7, and 9. The positions of some of the marker DNA bands are thesame as in Fig. 1.
oligo(dT)16 as the primer. The virus particle-to-tissue RNAratio thus ranged from 50 particles per 3 jig to 2,500 particlesper 17 ,ug. One-fifth of each reaction volume was amplifiedby PCR with primer pair 2, producing a 489-bp product. Thisrepresents 10 virus particles in a PCR mixture at the lower ofthe two seeding concentrations used. As shown in Fig. 3,viral sequences were readily detected in samples seededwith virus before RNA isolation but were not detected in theunseeded sample. No viral sequences could be detected ifreverse transcriptase was omitted from the reaction mixture.To investigate the limit of the viral particle-to-tissue RNAratio for the detection of viral RNA isolated from tissueseeded with virus, RNA was isolated from 50 mg of tissueseeded with 800 viral particles. Samples containing anamount of viral genomic RNA equivalent to 50 virus parti-cles were then reverse transcribed with oligo(dT)16 in thepresence of increasing amounts of unseeded shellfish tissueRNA and amplified with primer pair 2. As shown in Fig. 4, 10virus particles could be detected in an ethidium bromide-stained gel when 50 particles were reverse transcribed in thepresence of 23 ,ug of shellfish RNA. Twenty-three micro-grams of shellfish RNA is obtained from approximately 23mg of tissue. Thus, this method is capable of detectingapproximately 2,000 HAV particles per g of shellfish. Sincethe total particle-to-infectious virus particle ratio for HAV isat least 100 to 1 (18), this represents a sensitivity of about 20infectious virus particles per g of shellfish tissue.We also investigated the minimum number of virus parti-
cles (based on the number of viral genomic RNA molecules)that can be extracted from a seeded clam homogenate and
FIG. 4. Effect of increasing amounts of tissue RNA on RT-PCRdetection of seeded viral particles. RNA was isolated from clamtissue seeded with 1.6 x 104 virus particles per g of tissue. Samplescontaining an amount of viral genomic RNA equivalent to that of 50virus particles in approximately 3 pLg of tissue RNA were thenreverse transcribed without any additional unseeded shellfish RNA(lanes 1 and 2) or with 5, 10, or 20 ,ug of additional unseeded shellfishRNA (lanes 3 to 5, respectively). One-fifth of each reverse-tran-scribed sample was then amplified. Reverse transcriptase wasomitted from the reaction mixture in lane 1. Lane M, 1-kb DNAmarker.
subsequently detected by RT-PCR (Fig. 5). For this experi-ment, 0.5 ml of 10% clam tissue homogenate was seededwith decreasing numbers of viral particles, and RNA wasextracted as described above. One-third of each RNA sam-ple was then reverse transcribed in 20-,ul reaction volumeswith oligo(dT)16 as the primer. One-fourth of each reactionvolume was amplified by PCR with primer pair 1, producinga 276-bp product. As shown in Fig. 5, viral sequences werereadily detected in samples seeded with 100 virus particlesbefore RNA isolation but not in the sample seeded with 30virus particles. The limit of detection of viral particlesseeded into clam homogenate by this method is thereforeapproximately 2,000 particles per g of clam tissue.
DISCUSSIONThe present study was undertaken to develop a direct and
sensitive method for the detection of HAV in shellfish.Detection of enteroviruses by dot blot hybridization andRT-PCR has recently been reported (18). A sensitivity limitof 10 pg, corresponding to 2.4 x 106 molecules, was reportedfor a study in which a 32P-labeled single-stranded RNAprobe was used, while the sensitivity of the RT-PCR methodwas estimated to be approximately 103 50% tissue cultureinfectious doses (670 infectious virus particles) when theexperiments started with virus-seeded cerebrospinal fluid.The method we have described is more sensitive than thepreviously described RT-PCR methods for detection ofenteroviruses. A dot blot assay for HAV seeded into shell-fish or isolated HAV RNA, using a single-stranded RNAprobe, has recently been described (21). However, approx-imately 100 ng of HAV RNA is required for detection. Theantigen capture-PCR method works extremely well withclinical samples; these samples have a much higher viral loadand are relatively free of material which might interfere withantigen-antibody reaction or RT (8). However, the limit ofdetection of approximately 30 virus particles by this method
APPL. ENvIRON. MICROBIOL.
on January 25, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from

DETECTION OF HAV IN SHELLFISH BY PCR 2769
FIG. 5. Limit of detection of HAV seeded into tissue homoge-nate. Portions (0.5 ml) of a 10% clam tissue homogenate in RNAextraction buffer were seeded with the genomic RNA equivalent of1,000, 300, 100, or 30 viral particles (lanes B to E, respectively).RNA was isolated as described in Materials and Methods. One-thirdof each RNA sample was reverse transcribed in a 20-,ul reactionvolume, and one-fourth of each reaction volume was amplified withprimer pair 1 to produce a 276-bp product. Lane A contains RNAisolated from an unseeded tissue homogenate that was similarlyreverse transcribed and amplified. Lane M is a 123-bp DNA ladderfrom Bethesda Research Laboratories. The position of the 276-bpband is indicated by the arrowhead.
was achieved only by using Southern hybridization with a
32P-labeled probe. Moreover, the inclusion of the antigencapture step did not enhance the sensitivity of this methodover the conventional RT-PCR method, which uses RNAisolated from fecal samples by conventional methods. Theantigen capture-PCR method was used to detect HAV inoysters during a multistate outbreak of hepatitis, althoughquantitative data are not available (4). The RT-PCR methodwe have described appears to be at least as sensitive, sinceit is able to detect as few as 10 copies of HAV RNA in thepresence of a large excess of unrelated RNA. The methoddescribed here uses RNA isolated from whole tissue. Ittherefore avoids the time-consuming procedures for theconcentration of virus from shellfish and minimizes potentialloss of virus (3).
In presenting our data and making extrapolations aboutthe sensitivity of our method of detection of HAV inshellfish, we have assumed that reverse transcribing 50particles of the virus and amplifying a portion of the cDNApool representing 10 particles of the original virus areequivalent to reverse transcribing 10 particles of the virusand amplifying all the cDNA derived therefrom. This work-ing assumption has been used by several laboratories in theirstudies to quantitate specific mRNA molecules in a popula-tion of total RNA (14, 19). We point this out since theefficiency of RT is never 100% and since the presence of alarge number of unrelated transcripts can presumably affectthe RT of a particular transcript. In the experiments de-scribed here, the efficiency of RT of viral RNA did not seemto be adversely affected by the presence of an overwhelmingnumber of shellfish RNA molecules. One apparent reasonfor this is that since more than 95% of the RNA is rRNA, the
vast majority of the RNA is not primed by oligo(dT) duringthe RT.A second assumption we have made is that virus added to
tissue homogenate is a reasonable approximation of shellfishcontaminated with virus. We have no direct evidence tosupport this assumption. Several clam samples obtainedfrom an outdoor market were negative for HAV by our test.In the absence of shellfish samples that are known to becontaminated by HAV, we are unable to prove this assump-tion. However, since our method for detection of HAV doesnot depend on isolation or concentration of virus particlesbefore the isolation of RNA and thus minimizes potentialloss of HAV, this assumption seems reasonable.A third assumption we have made pertains to the sequence
of the primers. While it is impossible to predict the nucle-otide sequence divergence between different HAV isolatesfrom the environment, studies done thus far have demon-strated that human HAV strains show remarkable sequencehomology, typically exceeding 92% (17). The primers wehave selected are both from the putative RNA replicaseregion of the HAV genome, which is highly conservedamong different strains (2). In addition, primer pair 2, whichwas used in most of the experiments, was designed to havea high Tm, 80°C. Since the primer-annealing temperature inour PCR protocol is 63°C, which is 17°C below the Tm, aconsiderable amount of mismatch (up to three mismatchedbase pairs in a total of 28 nucleotides) will be tolerated,unless a mutation occurs at a base complementary to the 3'end of the primer. It should be pointed out that at thisannealing temperature, we have found no evidence of falsepriming with shellfish RNA (Fig. 3 and 4) or other unrelatedRNAs (data not shown).
In conclusion, the lack of requirement for any specialreagents such as specific antibodies should make this methodgenerally applicable for the detection of HAV in shellfish.
REFERENCES1. Cliver, D. O., R. D. Ellender, and M. D. Sobsey. 1983. Methods
to detect viruses in foods: testing and interpretation of results. J.Food Prot. 46:345-357.
2. Cohen, J. I., J. R. Ticehurst, R. H. Purcell, A. Buckler-White,and B. M. Baroudy. 1987. Complete nucleotide sequence ofwild-type hepatitis A virus: comparison with different strains ofhepatitis A virus and other picornaviruses. J. Virol. 61:50-59.
3. Cole, M. T., M. B. Kilgen, and C. R. Hackney. 1986. Evaluationof methods for extraction of enteric virus from Louisianaoysters. J. Food Prot. 49:592-595.
4. Desenclos, J.-C. A., K. C. Klontz, M. H. Wilder, 0. V. Nainan,H. S. Margolis, and R. A. Gunn. 1991. A multistate outbreak ofhepatitis A caused by the consumption of raw oysters. Am. J.Public Health 81:1268-1272.
5. Gerba, C. P., and S. M. Goyal. 1978. Detection and occurrenceof enteric viruses in shellfish: a review. J. Food Prot. 41:743-754.
6. Gerety, R. J. 1984. Hepatitis A, p. 1-8. In R. J. Gerety (ed.),Hepatitis A. Academic Press, San Diego, Calif.
7. Goswami, B. B., and R. I. Glazer. 1991. A simplified method forthe production of recombinant baculovirus. BioTechniques 10:626-630.
7a.Goswami, B. B., W. H. Koch, and T. A. Cebula. Competitortemplate RNA for detection and quantitation of hepatitis A virusby polymerase chain reaction. BioTechniques, in press.
8. Jansen, R. W., G. Siegl, and S. M. Lemon. 1990. Molecularepidemiology of human hepatitis A virus defined by an antigen-capture polymerase chain reaction method. Proc. Natl. Acad.Sci. USA 87:2867-2871.
9. Jiang, X., M. K. Estes, and T. G. Metcalf. 1987. Detection ofhepatitis A virus by hybridization with single-stranded RNAprobes. Appl. Environ. Microbiol. 53:2487-2495.
VOL. 59, 1993
on January 25, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from

APPL. ENVIRON. MICROBIOL.
10. Jiang, X., M. K. Estes, T. G. Metcalf, and J. L. Melnick. 1986.Detection of hepatitis A virus in seeded estuarine samples byhybridization with cDNA probes. Appl. Environ. Microbiol.52:711-717.
11. Lemon, S. M. 1985. Type A viral hepatitis: new developments inan old disease. N. Engl. J. Med. 313:1059-1067.
12. Lemon, S. M., L. N. Binn, and R. H. Marchwicki. 1983.Radioimmunofocus assay for quantitation of hepatitis A virus incell cultures. J. Clin. Microbiol. 17:834-839.
13. Melton, D. A. 1987. Translation of messenger RNA in injectedfrog oocytes. Methods Enzymol. 152:288-296.
14. Murphy, L. D., C. E. Herzog, J. B. Rudick, A. T. Fojo, and S. E.Bates. 1990. Use of the polymerase chain reaction in thequantitation of mdr-1 gene expression. Biochemistry 29:10351-10356.
15. Myers, T. W., and D. H. Gelfand. 1991. Reverse transcriptionand DNA amplification by a 77ternus thermophilus DNA poly-merase. Biochemistry 30:7661-7666.
16. Nasser, A. M., and T. G. Metcalf. 1987. Production of cytopa-thology in FRhK-4 cells by BS-C-1-passaged hepatitis A virus.
Appl. Environ. Microbiol. 53:2967-2971.17. Robertson, B. H., R. W. Jansen, B. Khanna, A. Totsuka, 0. V.
Nainan, G. Siegl, A. Widell, H. S. Margolis, S. Isomura, K. Ito,T. Ishizu, Y. Moritsugu, and S. M. Lemon. 1992. Geneticrelatedness of hepatitis A virus strains recovered from differentgeographical regions. J. Gen. Virol. 73:1365-1377.
18. Rotbart, H. A. 1990. Enzymatic RNA amplification of theenteroviruses. J. Clin. Microbiol. 28:438-442.
19. Wang, A. M., M. V. Doyle, and D. F. Mark. 1989. Quantitationof mRNA by the polymerase chain reaction. Proc. Natl. Acad.Sci. USA 86:9717-9721.
20. Wheeler, C. M., H. A. Fields, C. A. Schable, W. J. Meinke, andJ. E. Maynard. 1986. Adsorption, purification, and growthcharacteristics of hepatitis A virus strain HAS-15 propagated infetal rhesus monkey kidney cells. J. Clin. Microbiol. 23:434-440.
21. Zhou, Y.-J., M. K. Estes, X. Jiang, and T. G. Metcalf. 1991.Concentration and detection of hepatitis A virus and rotavirusfrom shellfish by hybridization tests. Appl. Environ. Microbiol.57:2963-2968.
2770 GOSWAMI ET AL.
on January 25, 2019 by guesthttp://aem
.asm.org/
Dow
nloaded from



![[PPT]Clam Dissection · Web viewDissection of the Clam Venus mercenaria * copyright cmassengale * * * * * * copyright cmassengale Resource * * * * * * * * * * * * * * * * * * * * *](https://img.pdfslide.us/doc/110x75/5aa6232f7f8b9a7c1a8e5555/pptclam-dissection-viewdissection-of-the-clam-venus-mercenaria-copyright-cmassengale.jpg)

























