Embed Size (px)
Citation preview

Designing 3D COFs with Enhanced HydrogenStorage CapacityEmmanouel Klontzas,† Emmanuel Tylianakis,‡ and George E. Froudakis†,*†Department of Chemistry, University of Crete, P.O. Box 2208, 71003 Heraklion, Crete, Greece, and ‡MaterialsScience and Technology Department, University of Crete, P.O. Box 2208, 71409 Heraklion, Crete, Greece
ABSTRACT Hydrogen storage properties have been studied on newly designed three-dimensional covalent-organic framework (3D-COF). The design of these materials was based on the ctn network of the ultralow density COF-102. The structures were optimizedby multiscale techniques and the optimized structures were checked for their storage capacities by grand canonical Monte Carlosimulations. Our simulations demonstrate that the gravimetric uptake of one of these new COFs can overpass the value of 25 wt %in 77 K and reach the Department of Energy’s target of 6 wt % in room temperature, classifying them between the top hydrogenstorage materials.
KEYWORDS COF, hydrogen storage, multiscale, ab initio, GCMC
The world’s increasing demands for energy and theneed for the reduction of the air pollution have led tothe quest of new alternative energy sources that
would also be environmental friendly. Hydrogen has beenrecognized as an ideal energy carrier especially for automo-tive applications, but it has not been available for extensivecommercial use yet. One of the major reasons for this hasbeen the difficulty to create efficient storage materials tosatisfy the established gravimetric and volumetric storagetargets. Nowadays there is an intensive research in thisscientific field and has been proposed that the solution ofthis problem will become from the design, synthesis of newtargeted materials, or the development of already existingmaterials with specific properties.
Following both our previous experience and the litera-ture,1-6 some general guidelines can be proposed for newpotential materials for hydrogen storage. The main factorsthat enhance the ability of storing hydrogen in the case ofphysisorption are surface area, pore volume, and enthalpyof adsorption. These factors can be enhanced by extendedaromaticity, unsaturated metal sites, and point charges inthe framework of the proposed materials. The design ofnovel materials for hydrogen storage following the above-mentioned rules has been a major task nowadays.
A new family of materials that fulfill many of the previousdemands was recently synthesized by Yaghi and co-workers,named as three-dimensional covalent-organic frameworks(3D-COFs).7 These three-dimensional periodic covalent struc-tures were synthesized by targeting two nets (ctn and bor)based on the selection of appropriate triangular and tetra-hedral nodes. From the first look, COFs present all the
advantages of metal-organic frameworks (MOFs) consider-ing hydrogen storage (surface area, pore volume, rigidity ofthe structure) while their molecular framework is composedof light elements that lower the relative weight of thestructure compared to the MOFs. The main advantage of 3D-COF with respect to other light porous organic materials isthe crystalline framework, which reflects high surface area.Our previous theoretical investigations of these materials8
have shown exceptional storage properties where similarresults were reported from other theoretical9-11 and experi-mental studies.12
Taking into account our previous experience in MOFs andthe strategies to enhance hydrogen uptake in these materialslisted earlier, we have designed four novel 3D-COF in sucha way that their storage properties would be superior to theparent framework. Particularly, we substitute the phenylenemoieties of COF-102 with other extended aromatic moietieswithout changing the net topology. We used diphenyl,triphenyl, napthalene, and pyrene molecules (Figure 1) asthe new aromatic moieties and produced the so-called COF-102-2, COF-102-3, COF-102-4, and COF-102-5, respectively.
The construction of these new structures was based onthe carbon nitride (C3N4) net topology. This network is themost symmetric for the combination of tetrahedral andtriangular building blocks to form a 3D structure, which wasproven also experimentally by the formation of three struc-tures based on ctn network against 1 structure that wasbased on bor network. This was done in two steps. First wesubstituted every nitrogen atom with a B3O3 ring andafterward we replaced every C-N bond of original net witha new organic moiety. For every new structure, we read-justed the lattice parameters of the C3N4 net to take a morereliable initial structure, and we checked for the right con-nectivity for the bonds between the boron and tetrahedralcarbon atoms with the corresponding carbon atoms of thenew organic moieties.
* To whom correspondence should be addressed. Tel: +302810545055. Fax:+302810545001. E-mail: [email protected] for review: 09/16/2009Published on Web: 00/00/0000
pubs.acs.org/NanoLett
© XXXX American Chemical Society A DOI: 10.1021/nl903068a | Nano Lett. XXXX, xxx, 000-000
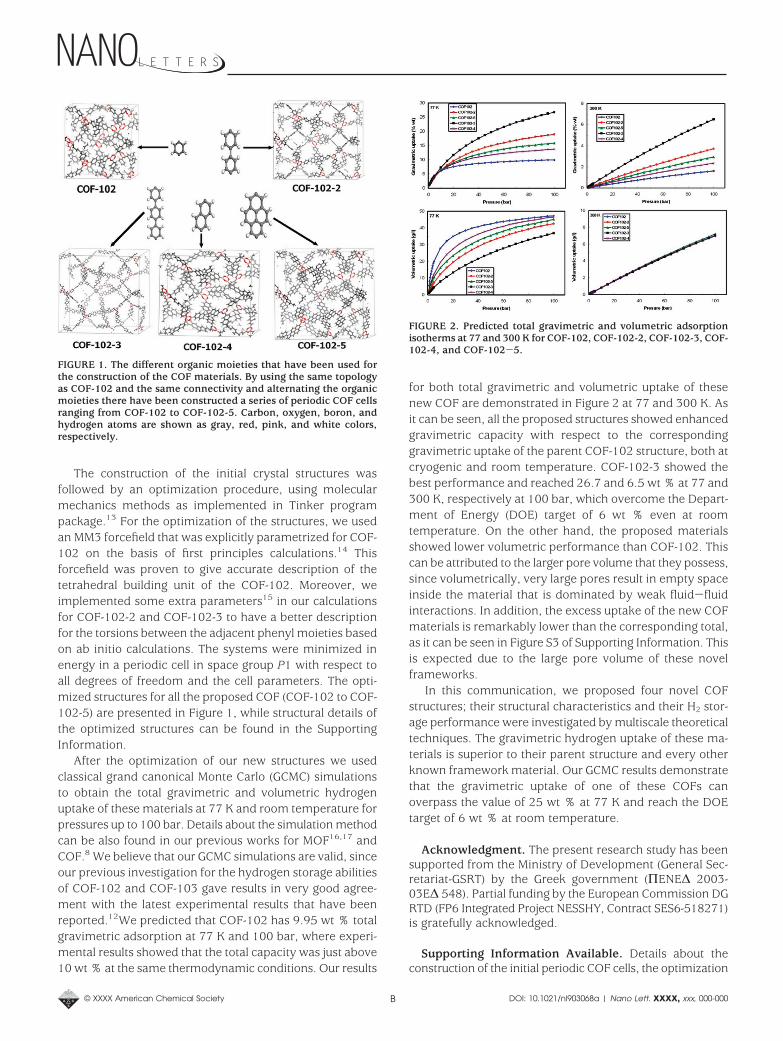
The construction of the initial crystal structures wasfollowed by an optimization procedure, using molecularmechanics methods as implemented in Tinker programpackage.13 For the optimization of the structures, we usedan MM3 forcefield that was explicitly parametrized for COF-102 on the basis of first principles calculations.14 Thisforcefield was proven to give accurate description of thetetrahedral building unit of the COF-102. Moreover, weimplemented some extra parameters15 in our calculationsfor COF-102-2 and COF-102-3 to have a better descriptionfor the torsions between the adjacent phenyl moieties basedon ab initio calculations. The systems were minimized inenergy in a periodic cell in space group P1 with respect toall degrees of freedom and the cell parameters. The opti-mized structures for all the proposed COF (COF-102 to COF-102-5) are presented in Figure 1, while structural details ofthe optimized structures can be found in the SupportingInformation.
After the optimization of our new structures we usedclassical grand canonical Monte Carlo (GCMC) simulationsto obtain the total gravimetric and volumetric hydrogenuptake of these materials at 77 K and room temperature forpressures up to 100 bar. Details about the simulation methodcan be also found in our previous works for MOF16,17 andCOF.8 We believe that our GCMC simulations are valid, sinceour previous investigation for the hydrogen storage abilitiesof COF-102 and COF-103 gave results in very good agree-ment with the latest experimental results that have beenreported.12We predicted that COF-102 has 9.95 wt % totalgravimetric adsorption at 77 K and 100 bar, where experi-mental results showed that the total capacity was just above10 wt % at the same thermodynamic conditions. Our results
for both total gravimetric and volumetric uptake of thesenew COF are demonstrated in Figure 2 at 77 and 300 K. Asit can be seen, all the proposed structures showed enhancedgravimetric capacity with respect to the correspondinggravimetric uptake of the parent COF-102 structure, both atcryogenic and room temperature. COF-102-3 showed thebest performance and reached 26.7 and 6.5 wt % at 77 and300 K, respectively at 100 bar, which overcome the Depart-ment of Energy (DOE) target of 6 wt % even at roomtemperature. On the other hand, the proposed materialsshowed lower volumetric performance than COF-102. Thiscan be attributed to the larger pore volume that they possess,since volumetrically, very large pores result in empty spaceinside the material that is dominated by weak fluid-fluidinteractions. In addition, the excess uptake of the new COFmaterials is remarkably lower than the corresponding total,as it can be seen in Figure S3 of Supporting Information. Thisis expected due to the large pore volume of these novelframeworks.
In this communication, we proposed four novel COFstructures; their structural characteristics and their H2 stor-age performance were investigated by multiscale theoreticaltechniques. The gravimetric hydrogen uptake of these ma-terials is superior to their parent structure and every otherknown framework material. Our GCMC results demonstratethat the gravimetric uptake of one of these COFs canoverpass the value of 25 wt % at 77 K and reach the DOEtarget of 6 wt % at room temperature.
Acknowledgment. The present research study has beensupported from the Ministry of Development (General Sec-retariat-GSRT) by the Greek government (ΠENE∆ 2003-03E∆ 548). Partial funding by the European Commission DGRTD (FP6 Integrated Project NESSHY, Contract SES6-518271)is gratefully acknowledged.
Supporting Information Available. Details about theconstruction of the initial periodic COF cells, the optimization
FIGURE 1. The different organic moieties that have been used forthe construction of the COF materials. By using the same topologyas COF-102 and the same connectivity and alternating the organicmoieties there have been constructed a series of periodic COF cellsranging from COF-102 to COF-102-5. Carbon, oxygen, boron, andhydrogen atoms are shown as gray, red, pink, and white colors,respectively.
FIGURE 2. Predicted total gravimetric and volumetric adsorptionisotherms at 77 and 300 K for COF-102, COF-102-2, COF-102-3, COF-102-4, and COF-102-5.
© XXXX American Chemical Society B DOI: 10.1021/nl903068a | Nano Lett. XXXX, xxx, 000-000

procedure of the initial cells, and the lattice parameters ofthe optimized cells are presented. This material is availablefree of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES(1) Collins, D. J.; Zhou, H.-C. J. Mater. Chem. 2007, 17, 3154.(2) Bhatia, S. K.; Myers, A. L. Langmuir 2006, 22, 1688.(3) Rowsell, J. L. C.; Yaghi, O. M. J. Am. Chem. Soc. 2006, 128, 1304.(4) Froudakis, G. E. Nano Lett. 2001, 1, 531.(5) Mpourmpakis, G.; Froudakis, G. E.; Lithoxoos, G. C.; Samios, J.
Nano Lett. 2006, 6, 1581.(6) Klontzas, E.; Mavrandonakis, A.; Froudakis, G. E.; Carissan, Y.;
Klopper, W. J. Phys. Chem. C 2007, 111, 13635.(7) El-Kaderi, H. M.; Hunt, J. R.; Mendoza-Cortes, J. L.; Cote, A. P.;
Taylor, R. E.; O’Keeffe, M.; Yaghi, O. M. Science 2007, 316, 268.
(8) Klontzas, E.; Tylianakis, E.; Froudakis, G. E. J. Phys. Chem. C 2008,112, 9095.
(9) Garberoglio, G. Langmuir 2007, 23, 12154.(10) Han, S. S.; Furukawa, H.; Yaghi, O. M.; Goddard, W. A., III. J. Am.
Chem. Soc. 2008, 130, 11580.(11) Cao, D.; Lan, J.; Wang, W.; Smit, B. Angew. Chem., Int. Ed. 2009,
48, 1.(12) Furukawa, H.; Yaghi, O. M. J. Am. Chem. Soc. 2009, 131 (25),
8875–8883.(13) Ponder, J. W.; Richards, F. M. J. Comput. Chem. 1987, 8, 1016;
Tinker version 4.2, June 2004; http://dasher.wustl.edu/tinker/.(14) Schmid, R.; Tafipolsky, M. J. Am. Chem. Soc. 2008, 130, 12600.(15) Amirjalayer, S.; Schmid, R. J. Phys. Chem. C 2008, 112, 14980.(16) Klontzas, E.; Mavrandonakis, A.; Tylianakis, E.; Froudakis, G. E.
Nano Lett. 2008, 8, 1572.(17) Mavrandonakis, A.; Tylianakis, E.; Stubos, A. K.; Froudakis, G. E.
J. Phys. Chem. C 2008, 112, 7290.
© XXXX American Chemical Society C DOI: 10.1021/nl903068a | Nano Lett. XXXX, xxx, 000-000





























