Embed Size (px)
Citation preview

of September 26, 2018.This information is current as
HLA Transgene MiceSpecific T Cells in−of Optimal NY-ESO-1
Definition of Key Variables for the Induction
Sanjiv A. Luther and Immanuel F. LuescherAlexandre Johannsen, Raphaël Genolet, Daniel F. Legler,
http://www.jimmunol.org/content/185/6/3445doi: 10.4049/jimmunol.1001397August 2010;
2010; 185:3445-3455; Prepublished online 23J Immunol
MaterialSupplementary
7.DC1http://www.jimmunol.org/content/suppl/2010/08/23/jimmunol.100139
Referenceshttp://www.jimmunol.org/content/185/6/3445.full#ref-list-1
, 23 of which you can access for free at: cites 50 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2010 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Definition of Key Variables for the Induction of OptimalNY-ESO-1–Specific T Cells in HLA Transgene Mice
Alexandre Johannsen,* Raphael Genolet,* Daniel F. Legler,† Sanjiv A. Luther,‡ and
Immanuel F. Luescher*
An attractive treatment of cancer consists in inducing tumor-eradicating CD8+ CTL specific for tumor-associated Ags, such as NY-
ESO-1 (ESO), a strongly immunogenic cancer germ line gene-encoded tumor-associated Ag, widely expressed on diverse tumors.
To establish optimal priming of ESO-specific CTL and to define critical vaccine variables and mechanisms, we used HLA-A2/
DR1 H-22/2 transgenic mice and sequential immunization with immunodominant DR1- and A2-restricted ESO peptides. Immu-
nization of mice first with the DR1-restricted ESO123–137 peptide and subsequently with mature dendritic cells (DCs) presenting
this and the A2-restriced ESO157–165 epitope generated abundant, circulating, high-avidity primary and memory CD8+ T cells that
efficiently killed A2/ESO157–165+ tumor cells. This prime boost regimen was superior to other vaccine regimes and required strong
Th1 cell responses, copresentation of MHC class I and MHC class II peptides by the same DC, and resulted in upregulation of
sphingosine 1-phosphate receptor 1, and thus egress of freshly primed CD8+ T cells from the draining lymph nodes into circu-
lation. This well-defined system allowed detailed mechanistic analysis, which revealed that 1) the Th1 cytokines IFN-g and
IL-2 played key roles in CTL priming, namely by upregulating on naive CD8+ T cells the chemokine receptor CCR5; 2) the
inflammatory chemokines CCL4 (MIP-1b) and CCL3 (MIP-1a) chemoattracted primed CD4+ T cells to mature DCs and
activated, naive CD8+ T cells to DC–CD4 conjugates, respectively; and 3) blockade of these chemokines or their common
receptor CCR5 ablated priming of CD8+ T cells and upregulation of sphingosine 1-phosphate receptor 1. These findings provide
new opportunities for improving T cell cancer vaccines. The Journal of Immunology, 2010, 185: 3445–3455.
From previous cancer vaccine trials, it has emerged that inorder for tumor-associated Ag (TAA)-specific CD8+ T cellresponses to be tumoricidal, they need to be persistent,
have high titers and high avidity, and they must infiltrate tumors(1–5). Although, in some cases, tumor regression was achieved,correlation between objective tumor control and vaccine-inducedT cell responses remained modest, stressing the need to vigorouslyinvestigate key variables that determine the efficacy of T cell-based cancer vaccines.An efficient cancer vaccine must induce maturation of dendritic
cells (DCs). Mature DCs highly express MHC class I (MHC I) andMHC class II (MHC II) molecules, CD40, and ligands for cos-timulatory molecules, such as CD80 (B7.1), CD86 (B7.2), andCD70 (1, 6). While triggering of CD28 on naive T cells by CD80and CD86 promotes T cell differentiation and IL-2 production,triggering of CD27 by CD70 drives the generation and maintenance
of CD8 memory (1, 6, 7). DCs naturally mature upon infectionswith bacteria or viruses in which inflammatory cytokines (e.g.,IFN type I, TNF-a, IL-6, and IL-1b) act on DCs, in addition totriggering various receptors. In cancer vaccines, various adjuvantshave been tested to promote adequate DC maturation. Good re-sults have been obtained with the TLR9 agonists CpG combinedwith emulsion-forming agents such as IFA or Montanide (1–3, 8).DCs play diverse and preeminent roles in tumor immunology, andsome tumors escape T cell immunity by inhibiting DCmaturation,thus favoring the induction of regulatory T cell and T cell toler-ance rather than of adaptive T cell immunity (1, 9). Moreover,there exist different types of DCs, and the CD8+ T cell responsesthey induce can be strikingly different (10).Induction of effective and persistent anti-TAA CD8+ T cell
responses typically requires adequate help from Th1 cells (2, 11–15). In mice, it has been shown that in the absence of Th1 cells (e.g.,in MHC II or CD4 knockout mice), formation of protective andpersistent CD8+ T cell responses is seriously compromised (13,14). Upon interaction with mature, cognate DCs, Th1 cells secretesubstantial amounts of Th1 cytokines (e.g., IFN-g, TNF-a, IL-2)and stimulate DC cytokine production (1, 6, 16). WhereasIL-2 promotes the proliferation and differentiation of Ag-selectedcytotoxic T cells and CD8 memory (17, 18), IFN-g upregulates theexpression of IL-12, adhesion molecules, and MHC I and MHC IImolecules and inhibits Th2 cell activities (6, 16).There are different views and models on the mechanism by
which Th1 cells assist priming of naive CD8+ T cells. According toone concept, Th1 cells help CD8+ T cell priming by licensing DCsfor efficient priming of naive CD8+ T cells (6, 15). This reliesprimarily on DC activation by CD40 triggering upon engagementwith CD40L (CD154) expressed on activated CD4+ T cells, and itresults in IL-12 production, a cytokine that supports the generationof CTL (1, 6, 16). In support of this model, it has been shown that
*Ludwig Institute for Cancer Research, Lausanne Branch and ‡Department of Bio-chemistry, University of Lausanne, Epalinges; and †Biotechnology Institute Thurgau,University of Konstanz, Kreuzlingen, Switzerland
Received for publication April 28, 2010. Accepted for publication July 13, 2010.
This work was supported by Grants 310030-125330 (to I.F.L.) and 31003A-127474 /1(to D.F.L.) from the Swiss National Foundation and Grant OCS 01421-08-2003 fromthe Swiss Cancer League.
Address correspondence and reprint requests to Dr. Immanuel F. Luescher, LudwigInstitute for Cancer Research, Chemin des Boveresses 155, 1066 Epalinges, Switzer-land. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this paper: DC, dendritic cell; ESO, NY-ESO-1; LN, lymphnode; MFI, mean fluorescence intensity; MHC I, MHC class I; MHC II, MHC classII; 87p, NY-ESO-187–101; 123p, NY-ESO-1123–137; 157p, NY-ESO-1157–165 withmutation C165V; + pep, stimulated; 2 pep, not stimulated; S1P1, sphingosine 1-phosphate receptor 1; Sp, splenocyte/spleen; TAA, tumor-associated Ag.
Copyright� 2010 by The American Association of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1001397
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

CD40 agonists strongly boost CD8+ T cell priming and can sub-stitute for T cell help at least in some systems (9, 14, 15). Anothermodel on Th1 cell help stipulates that primed CD4+ and naiveCD8+ T cells interact simultaneously with mature DCs presentingMHC I and MHC II peptides (19, 20). Against this model it hasbeen argued that the probability of the formation of three-cellclusters is scant due to the low frequency of Ag-specific naiveCD8+ T cells, estimated to be ∼1 out of 106 (21). However, an in-travital two-photon imaging study has shown that the inflammatorychemokines CCL3 (MIP-1a) and CCL4 (MIP-1b) chemoattractactivated naive CD8+ T cells to DC–CD4+ T cell conjugates,thereby favoring three-cell cluster formation (19). Such clusters havealso been observed and enumerated in another intravital imagingstudy (20). Interestingly, another study demonstrated that freshlyprimed CD8+ T cells, as CD4+ Th1 cells, can attract naive CD8+
T cells in a CCR5-dependent manner and promote their priming(22).Although these studies are compelling, the use of adoptive transfer
of T cells from TCR transgenic mice is artificial and leaves opencritical issues, such as: 1) What is the mechanism by which activatedCD4+ T cells are guided to mature cognate DCs and naive CD8+
T cells to DC–Th1 cell conjugates; 2) What are the factors andmechanisms that induce naive CD8+ T cells to surface expressCCR5; 3) What precise roles do Th1 cytokines play in CD8+ T cellpriming; and 4) It has been shown that primed CD8+ T cells exitlymph nodes upon expression of sphingosine 1-phosphate receptor 1(S1P1) (23, 24), but it is not known what factors govern S1P1 ex-pression during CD8+ T cell priming.To conclusively investigate these matters, we used as a pre-
clinical mouse model HLA-DRB1*0101, HLA-A*0201 (with a3from Db), H-22/2 transgenic mice (A2/DR1) (25). As TAA weused the cancer germ line Ag NY-ESO-1 (ESO), which has beenwidely used in cancer vaccine trials, is broadly expressed on di-verse tumor cells, and is highly immunogenic (3, 26–28). It is acytosolic protein of 180 aa, and MHC I and MHC II epitopeshave been extensively mapped (http://www.cancerimmunity.org/peptidedatabase/Tcellepitopes.htm). For A2 the immunodomi-nant epitope is 157–165 (157p), which is expressed by mosttumor cells (2, 3, 26), and for DR1 various epitopes have beenreported, for example, 123–137 (123p), 87–101 (87p), and 87–111 (27, 28).Our investigations in this highly defined system showed that 1)
efficient priming of CD8+ T cells in draining lymph nodes requiresavailable Th1 cells and copresentation of strongly immunogenicMHC I and MHC II epitopes by the same mature DC; 2) the Th1cytokines IL-2 and IFN-g, namely in combination, induce CCR5surface expression on naive CD8+ T cells; 3) the chemokinesCCL4 and CCL3 play differential roles in guiding Th1 cells toDCs and activated naive CD8+ T cells to DC–CD4 T cellconjugates, respectively; and 4) these chemoattractions and thestrength of Th1 cell help determine the upregulation of S1P1 onfreshly primed CD8+ T cells and hence their exit from the draininglymph nodes into circulation.
Materials and MethodsMice and cells
A2/DR1 mice have been described previously (19). AJ-H1 fibrosarcomacells were obtained from mice 6 mo previously injected s.c with 3-methylcholanthrene (100 mg/mouse). Single-cell suspensions were derivedfrom solid tumors and cultured in RPMI 1640 supplemented with 10%FCS and antibiotics. A 123p-specific Th1 CD4+ T cell line was derivedfrom lymph node CD4+ T cells from mice immunized with 123p bystimulation with 123p (1 mM) at day 0 and rIL-2 each 2 d (10 IU/ml;PeproTech, London, U.K.) and tetramer-guided sorting after 1 mo.
Immunizations
Groups of mice (n = 5) were immunized by s.c. injections of 50 mlemulsion containing the indicated peptides (50 mg peptide emulsified in 50ml PBS and 50 ml CFA [Difco Laboratories, Detroit, MI] or IFA and CpGat the base of the tail). DCs were obtained by culturing bone marrow cellsfor 8–10 d with GM-CSF (20 ng/ml; PeproTech) and activated with LPS(0.5 mg/ml; Sigma-Adrich, St. Louis, MO) overnight. After pulsing with 1mM indicated peptides at 37˚C for 1 h, DCs were washed, resuspended,and injected s.c. in the hind foot pads (0.5 3 106 cells/mouse); in someexperiments blocking anti-CCR5, CCL3, and CCL4 Abs (50 mg/mouse)were coinjected.
Peptides and tetramer
Peptides included 123p (LKEFTVSGNILTIRL), 87p (LLEFYLAMPFATP-ME), 157p with mutation C165V (SLLMWITQV), collagen type II261–273(AGFKGEQGPKGEP), and CMV pp65495–503 (NLVPMVATV). The fluo-rescent peptides Alexa 555–ESO157–165 (C[Alexa 555]LLMWITQV) andESO123–137 (C[Alexa 488]-GG-LKEFTVSGNILTIRL]) were prepared byreacting the thiol peptides with the corresponding Alexa maleimides as rec-ommended by the supplier (Molecular Probes/Invitrogen, Paisley, U.K.).HLA-A2a1,a2/Kb a3 tetramers (PE labeled) were prepared by refoldingusing human b2-microglobulin and 157p.
Flow cytometry and Abs
Popliteal and inguinal lymph nodes and spleens were homogenized, cellswere washed, and for flow cytometric detection of IL-2– or IFN-g–producing cells stimulated with the indicated peptides (1 mM) for 4 h inthe presence of brefeldin A (10 mg/ml; BD Biosciences, San Jose, CA),cells were fixed (5% paraformaldehyde), permeabilized with 0.5% saponin(Sigma-Aldrich), and stained with cytokine Abs (BD Biosciences). Fortetramer staining, CD8+ T cells (1 3 106) were incubated in 50 ml FACSbuffer with 10 nM A2/157p tetramer for 30 min at room temperaturefollowed by 30 min with FITC anti-mouse CD8 (clone 5H10; BioLegend,San Diego, CA) and Cy5-PE–labeled anti-CD4 (GK1.5) Abs. Anti-mouseS1P1 Ab (clone 11424) was from Abcam (Cambridge, U.K.), PE-conjugated anti-mouse CCL3 (clone 39624) was from R&D Systems(Oxon, U.K.), and PE anti-mouse CCR5 Ab (clone HM-CCR5) was fromBioLegend. All samples were analyzed on a FACSCalibur (BD Bioscien-ces) and data were analyzed with the FlowJo 8.7.1 software (Tree Star,Ashland, OR).
ELISPOT and multicytokine detection assay
ELISPOT was performed as described previously (29). All Abs were pur-chased from Mabtech (Nacka, Sweden). The plates were analyzed byZellNet Consulting (Fort Lee, NJ). For the detection of multicytokines,cytometric bead array kits were used on supernatants, obtained as describedfor ELISPOT, following the manufacturer’s instructions (BD Biosciences).
Cytotoxic assays
For in vitro cytotoxic assays, 51Cr-labeled AJ-H1 cells (5 3 103 cells/well)were incubated with CD8+ splenic T cells from immunized mice (E:T ratioof 1:10 or as indicated) for 4 h at 37˚C with the indicated concentrations of157p. Specific lysis was calculated from released 51Cr as: 100 3 [(exper-imental 2 spontaneous release)/(total 2 spontaneous release)]. In vivo cy-totoxic assay was performed as described previously (30). In brief, AJ-H1 cells or syngeneic splenocytes were labeled with 0.1 mM CFSE andpulsed with 157p (1 mM) or with 1 mM CFSE, mixed 1/1, and transferredi.v. in immune or control mice (1 3 107 cells). The specific lysis (%) wascalculated as: (1 2 ratio immunized/ratio nonimmunized) 3 100, wherebythe ratio indicates the percentage CFSEhigh/percentage CFSElow cells.
In vitro priming of CD8+ T cells and ELISA
Mature DCs (2 3 104) were incubated in 24-well plates with the differentpeptides (1 mM) at 37˚C for 1 h and washed, resulting in the removal ofmost nonadhered cells. CD4+ T cells (2.5 3 105), obtained by negativeselection (.90%) using MACS and Ab-coated magnetic beads (StemCellTechnologies, Grenoble, France) from lymph node cells from 123p-immunized mice and suspended in Iscove’s IMDM medium (Life Tech-nologies, Paisley, U.K.), supplemented with 10% FCS, b-mercapthanol,and antibiotics, were added, and after 12 h, conjugates were enumerated.Alternatively, naive CD8+ T cells (53 105), obtained by negative selectionfrom splenocytes, were added and after 6 d of incubation, the percentage ofA2/ESO157–165 tetramer+ CD8+ T cells was assessed by flow cytometry.For blocking or neutralization experiments, Abs (10 mg/ml) specific formouse IL-2 (clone JES6-5H4), IFN-g (H22), TNF-a (TN3-19.12), CD40
3446 OPTIMAL NY-ESO-1–SPECIFIC CD8+ T CELL VACCINE
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
(clone HM40), IL-12 (C17.8), CCR5 (HM-CCR5), anti-CD40L (cloneMR1) were from BioLegend, CCL3 (ab10382) and CCL4 (ab10386) werefrom Abcam, and CD70 (clone 118510) and CCL3 (ab-450-NA) werefrom R&D Systems. Isotype-matched irrelevant Abs, as recommendedby the suppliers, were used as negative controls. Recombinant IL-2, IFN-g,and IL-12 were from PeproTech. For ELISA, supernatants from cultureswere assayed using ELISArray kits as recommended by the supplier(SABiosciences, Frederick, MD).
Assessment of CCL3 and CCL4 messages by quantitative PCR
Experiments were performed as described previously (31). CD4+ T cells (13 106) from a 123p-specific line and mature 123p-pulsed DC (1 3 106)were coincubated and after 6 h, FACS sorted after pipetting in cold PBSand EDTA (5 mM). Total RNAwas extracted from CD4+ T cells (5 3 105
cells) and DCs (5 3 105 cells) using TRIzol (0.5 mM; Invitrogen). Thevalues of CCL3 and CCL4 were normalized to the expression of TATAbinding protein.
Fluorescence microscopy
Mature DCs were labeled with Cy5 succinimidyl ester (GE Healthcare,Otelfingen Switzerland) (5 mM, 10 min), pulsed or not with peptides (1 h at37˚C), and plated at the center of glass-bottom dishes (MatTek, Ashland,MA) (5–10 3 103 cells in 50 ml). After 3 h of incubation at 37˚C, the bulkof nonadherent DCs was washed off. Purified 123p-primed CD4+ T cells (53 105) were labeled with CFSE (Invitrogen) (5 mM, 8 min) and added inthe center of the dish. After 16 h of incubation in the absence or presenceof Abs, the bulk of nonadherent cells was washed off. Purified naive CD8+
T cells (5 3 105), preincubated for 6 h with IL-2 and IFN-g (10 IU/ml and100 ng/ml, respectively), were labeled with CellTracker Orange CMTMR(Invitrogen) (2 mM, 15 min) and added to the dishes. Images were acquiredon a Zeiss Axioplan microscope equipped with an AxioCam MRm cameraand analyzed on AxioVision LE 4.5 software. DCs and conjugates were
quantified using images (320) from the four quadrants near the center.Analyses and imaging of draining lymph nodes were performed as described(32).
Chemotaxis assay
Migration of lymphocytes was performed as previously described (33).Briefly, supernatants were obtained from mature DCs cultured alone ortogether with CD4+ T cells from 123p-immunized mice in the absence orpresence of 123p. After 6 h, supernatants were placed on the lower cham-ber of Transwell plates (96 wells, 5-mm pores) (ChemoTx; NeuroProbe,Gaithersburg, MD). Cells (2.5 3 105) with or without Abs specific forCCL3, CCL4, and CCR5 were added on the upper side of the membraneand incubated for 4 h at 37˚C. The numbers of migrated cells in the lowerwells were analyzed by flow cytometry.
Statistical analyses
An unpaired Student t test was used to determine the statistical significanceof the data. The level of significance was set at p values of ,0.05.
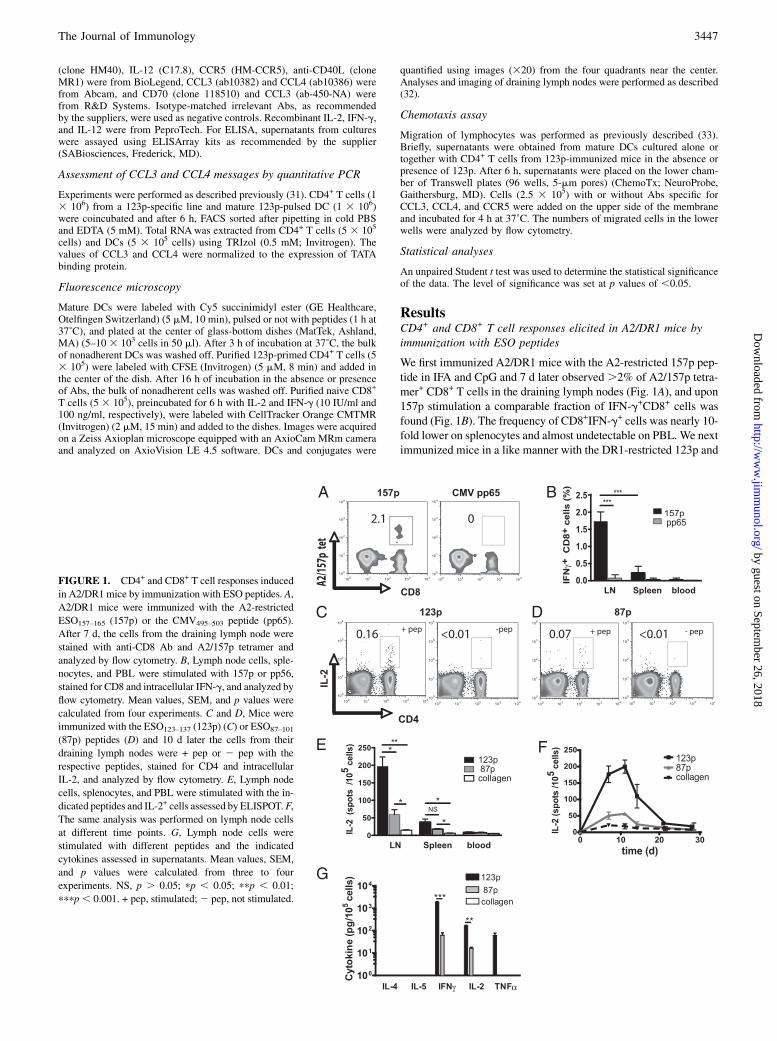
ResultsCD4+ and CD8+ T cell responses elicited in A2/DR1 mice byimmunization with ESO peptides
We first immunized A2/DR1 mice with the A2-restricted 157p pep-tide in IFA and CpG and 7 d later observed.2% of A2/157p tetra-mer+ CD8+ T cells in the draining lymph nodes (Fig. 1A), and upon157p stimulation a comparable fraction of IFN-g+CD8+ cells wasfound (Fig. 1B). The frequency of CD8+IFN-g+ cells was nearly 10-fold lower on splenocytes and almost undetectable on PBL. We nextimmunized mice in a like manner with the DR1-restricted 123p and
FIGURE 1. CD4+ and CD8+ T cell responses induced
in A2/DR1mice by immunization with ESO peptides. A,
A2/DR1 mice were immunized with the A2-restricted
ESO157–165 (157p) or the CMV495–503 peptide (pp65).
After 7 d, the cells from the draining lymph node were
stained with anti-CD8 Ab and A2/157p tetramer and
analyzed by flow cytometry. B, Lymph node cells, sple-
nocytes, and PBL were stimulated with 157p or pp56,
stained for CD8 and intracellular IFN-g, and analyzed by
flow cytometry. Mean values, SEM, and p values were
calculated from four experiments. C and D, Mice were
immunized with the ESO123–137 (123p) (C) or ESO87–101
(87p) peptides (D) and 10 d later the cells from their
draining lymph nodes were + pep or 2 pep with the
respective peptides, stained for CD4 and intracellular
IL-2, and analyzed by flow cytometry. E, Lymph node
cells, splenocytes, and PBL were stimulated with the in-
dicated peptides and IL-2+ cells assessed by ELISPOT.F,
The same analysis was performed on lymph node cells
at different time points. G, Lymph node cells were
stimulated with different peptides and the indicated
cytokines assessed in supernatants. Mean values, SEM,
and p values were calculated from three to four
experiments. NS, p . 0.05; pp , 0.05; ppp , 0.01;
pppp, 0.001. + pep, stimulated;2 pep, not stimulated.
The Journal of Immunology 3447
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
10 d later found 0.16% IL-2+CD4+ cells in the draining lymph nodes(Fig. 1C). When 87p was used, only 0.07% IL-2+CD4+ T cells wereobserved, indicating that this peptide is less immunogenic (Fig. 1D).When using the more sensitive ELISPOT assay an ∼4-fold differ-ence in IL-2 response was observed (Fig. 1E). Most IL-2+CD4+
T cells were in the draining lymph nodes. The IL-2 responses weremaximal 7–11 d after immunization and contracted thereafter (Fig.1F). Lymph node cells 10 d after 123p immunization produced upon123p stimulation high amounts of IFN-g, IL-2, and TNF-a, but nodetectable IL-4 and IL-5 (i.e., they were typical Th1 cells) (Fig. 1G).Conversely, 87p elicited ∼10-fold lower IFN-g and IL-2 responsesand no detectable TNF-a.
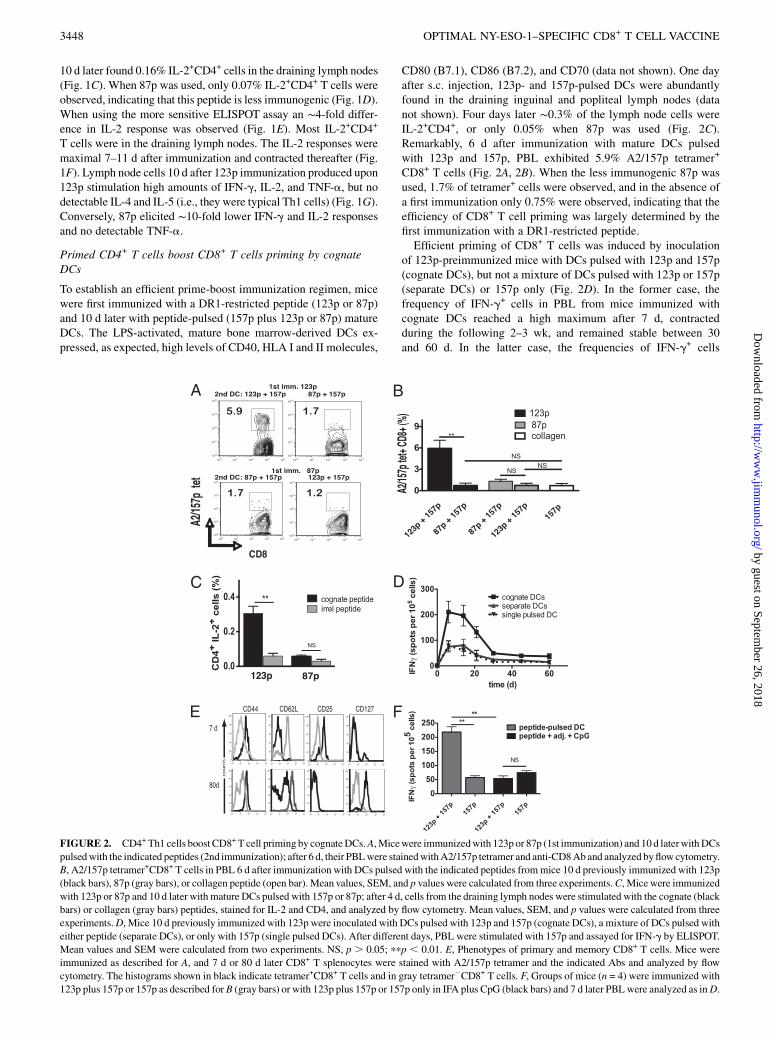
Primed CD4+ T cells boost CD8+ T cells priming by cognateDCs
To establish an efficient prime-boost immunization regimen, micewere first immunized with a DR1-restricted peptide (123p or 87p)and 10 d later with peptide-pulsed (157p plus 123p or 87p) matureDCs. The LPS-activated, mature bone marrow-derived DCs ex-pressed, as expected, high levels of CD40, HLA I and II molecules,
CD80 (B7.1), CD86 (B7.2), and CD70 (data not shown). One dayafter s.c. injection, 123p- and 157p-pulsed DCs were abundantlyfound in the draining inguinal and popliteal lymph nodes (datanot shown). Four days later ∼0.3% of the lymph node cells wereIL-2+CD4+, or only 0.05% when 87p was used (Fig. 2C).Remarkably, 6 d after immunization with mature DCs pulsedwith 123p and 157p, PBL exhibited 5.9% A2/157p tetramer+
CD8+ T cells (Fig. 2A, 2B). When the less immunogenic 87p wasused, 1.7% of tetramer+ cells were observed, and in the absence ofa first immunization only 0.75% were observed, indicating that theefficiency of CD8+ T cell priming was largely determined by thefirst immunization with a DR1-restricted peptide.Efficient priming of CD8+ T cells was induced by inoculation
of 123p-preimmunized mice with DCs pulsed with 123p and 157p(cognate DCs), but not a mixture of DCs pulsed with 123p or 157p(separate DCs) or 157p only (Fig. 2D). In the former case, thefrequency of IFN-g+ cells in PBL from mice immunized withcognate DCs reached a high maximum after 7 d, contractedduring the following 2–3 wk, and remained stable between 30and 60 d. In the latter case, the frequencies of IFN-g+ cells
FIGURE 2. CD4+ Th1 cells boostCD8+T cell priming by cognateDCs.A,Micewere immunizedwith 123p or 87p (1st immunization) and 10 d laterwithDCs
pulsedwith the indicated peptides (2nd immunization); after 6 d, their PBLwere stainedwithA2/157p tetramer and anti-CD8Aband analyzed byflowcytometry.
B, A2/157p tetramer+CD8+ T cells in PBL 6 d after immunization with DCs pulsed with the indicated peptides frommice 10 d previously immunized with 123p
(black bars), 87p (gray bars), or collagen peptide (open bar). Mean values, SEM, and p values were calculated from three experiments.C, Micewere immunized
with 123p or 87p and 10 d later with mature DCs pulsed with 157p or 87p; after 4 d, cells from the draining lymph nodes were stimulated with the cognate (black
bars) or collagen (gray bars) peptides, stained for IL-2 and CD4, and analyzed by flow cytometry. Mean values, SEM, and p values were calculated from three
experiments.D, Mice 10 d previously immunized with 123p were inoculated with DCs pulsed with 123p and 157p (cognate DCs), a mixture of DCs pulsed with
either peptide (separate DCs), or only with 157p (single pulsed DCs). After different days, PBL were stimulated with 157p and assayed for IFN-g by ELISPOT.
Mean values and SEM were calculated from two experiments. NS, p . 0.05; ppp , 0.01. E, Phenotypes of primary and memory CD8+ T cells. Mice were
immunized as described for A, and 7 d or 80 d later CD8+ T splenocytes were stained with A2/157p tetramer and the indicated Abs and analyzed by flow
cytometry. The histograms shown in black indicate tetramer+CD8+ T cells and in gray tetramer2CD8+ T cells. F, Groups of mice (n = 4) were immunized with
123p plus 157p or 157p as described for B (gray bars) or with 123p plus 157p or 157p only in IFA plus CpG (black bars) and 7 d later PBLwere analyzed as inD.
3448 OPTIMAL NY-ESO-1–SPECIFIC CD8+ T CELL VACCINE
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
were ∼4-fold lower, similar to 157p only-pulsed DCs. A2/157ptetramer+CD8+ T cells 7 d after immunization exhibited a typicaleffector T cell phenotype, characterized by increased expressionof CD44 and CD25, and decreased expression of CD62L (Fig.2E). After 80 d, the tetramer+ cells showed a typical memoryphenotype, characterized by biphasic expression of CD62L corre-sponding to central and effector memory subsets, increased CD44and CD127 expression, and CD25 background levels (Fig. 2E).For comparison we immunized mice with 123p plus 157p or only157p in IFA plus CpG. Upon challenge with 157p the frequency ofIFN-g+ cells was .4-fold lower in PBL from mice immunizedwith 123p plus 157p in IFA plus CpG than in PBL from miceimmunized using the prime-boost regimen (Fig. 2F). Note, how-ever, that while peptide-pulsed mature DCs induced powerfulCD8+ T cell responses in mice with preexisting Th1 cellsresponses, they were clearly less efficient at inducing these ascompared with immunization with peptide in adjuvant.
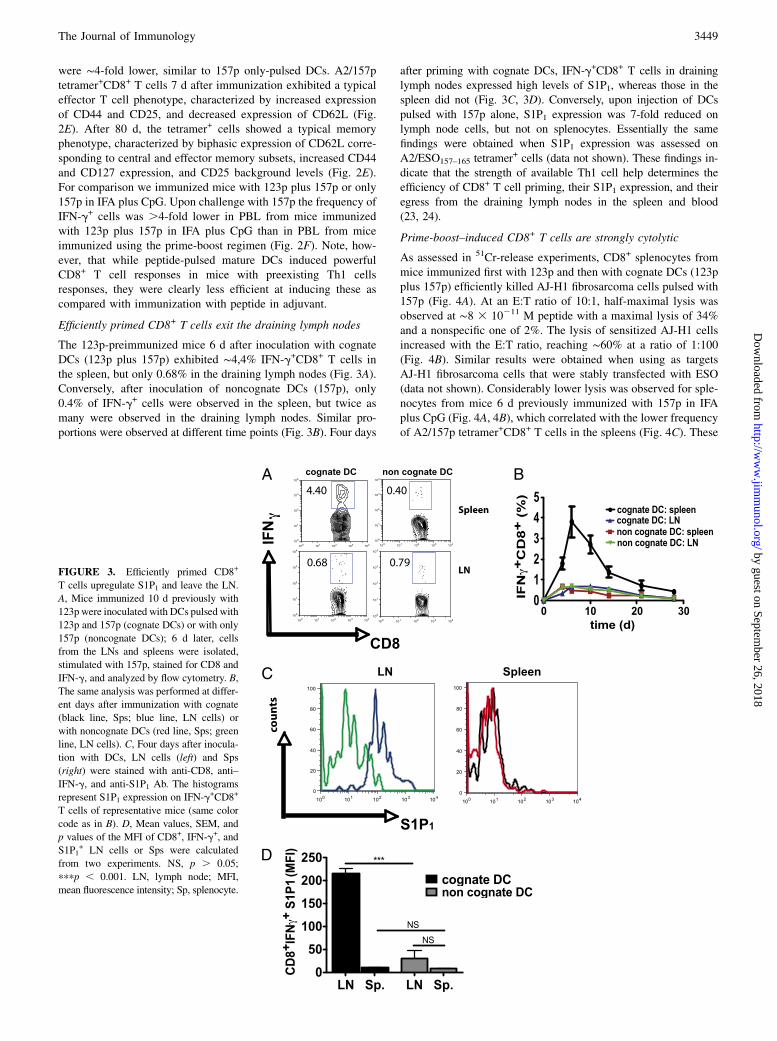
Efficiently primed CD8+ T cells exit the draining lymph nodes
The 123p-preimmunized mice 6 d after inoculation with cognateDCs (123p plus 157p) exhibited ∼4,4% IFN-g+CD8+ T cells inthe spleen, but only 0.68% in the draining lymph nodes (Fig. 3A).Conversely, after inoculation of noncognate DCs (157p), only0.4% of IFN-g+ cells were observed in the spleen, but twice asmany were observed in the draining lymph nodes. Similar pro-portions were observed at different time points (Fig. 3B). Four days
after priming with cognate DCs, IFN-g+CD8+ T cells in draininglymph nodes expressed high levels of S1P1, whereas those in thespleen did not (Fig. 3C, 3D). Conversely, upon injection of DCspulsed with 157p alone, S1P1 expression was 7-fold reduced onlymph node cells, but not on splenocytes. Essentially the samefindings were obtained when S1P1 expression was assessed onA2/ESO157–165 tetramer+ cells (data not shown). These findings in-dicate that the strength of available Th1 cell help determines theefficiency of CD8+ T cell priming, their S1P1 expression, and theiregress from the draining lymph nodes in the spleen and blood(23, 24).
Prime-boost–induced CD8+ T cells are strongly cytolytic
As assessed in 51Cr-release experiments, CD8+ splenocytes frommice immunized first with 123p and then with cognate DCs (123pplus 157p) efficiently killed AJ-H1 fibrosarcoma cells pulsed with157p (Fig. 4A). At an E:T ratio of 10:1, half-maximal lysis wasobserved at ∼8 3 10211 M peptide with a maximal lysis of 34%and a nonspecific one of 2%. The lysis of sensitized AJ-H1 cellsincreased with the E:T ratio, reaching ∼60% at a ratio of 1:100(Fig. 4B). Similar results were obtained when using as targetsAJ-H1 fibrosarcoma cells that were stably transfected with ESO(data not shown). Considerably lower lysis was observed for sple-nocytes from mice 6 d previously immunized with 157p in IFAplus CpG (Fig. 4A, 4B), which correlated with the lower frequencyof A2/157p tetramer+CD8+ T cells in the spleens (Fig. 4C). These
FIGURE 3. Efficiently primed CD8+
T cells upregulate S1P1 and leave the LN.
A, Mice immunized 10 d previously with
123p were inoculated with DCs pulsed with
123p and 157p (cognate DCs) or with only
157p (noncognate DCs); 6 d later, cells
from the LNs and spleens were isolated,
stimulated with 157p, stained for CD8 and
IFN-g, and analyzed by flow cytometry. B,
The same analysis was performed at differ-
ent days after immunization with cognate
(black line, Sps; blue line, LN cells) or
with noncognate DCs (red line, Sps; green
line, LN cells). C, Four days after inocula-
tion with DCs, LN cells (left) and Sps
(right) were stained with anti-CD8, anti–
IFN-g, and anti-S1P1 Ab. The histograms
represent S1P1 expression on IFN-g+CD8+
T cells of representative mice (same color
code as in B). D, Mean values, SEM, and
p values of the MFI of CD8+, IFN-g+, and
S1P1+ LN cells or Sps were calculated
from two experiments. NS, p . 0.05;
pppp , 0.001. LN, lymph node; MFI,
mean fluorescence intensity; Sp, splenocyte.
The Journal of Immunology 3449
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
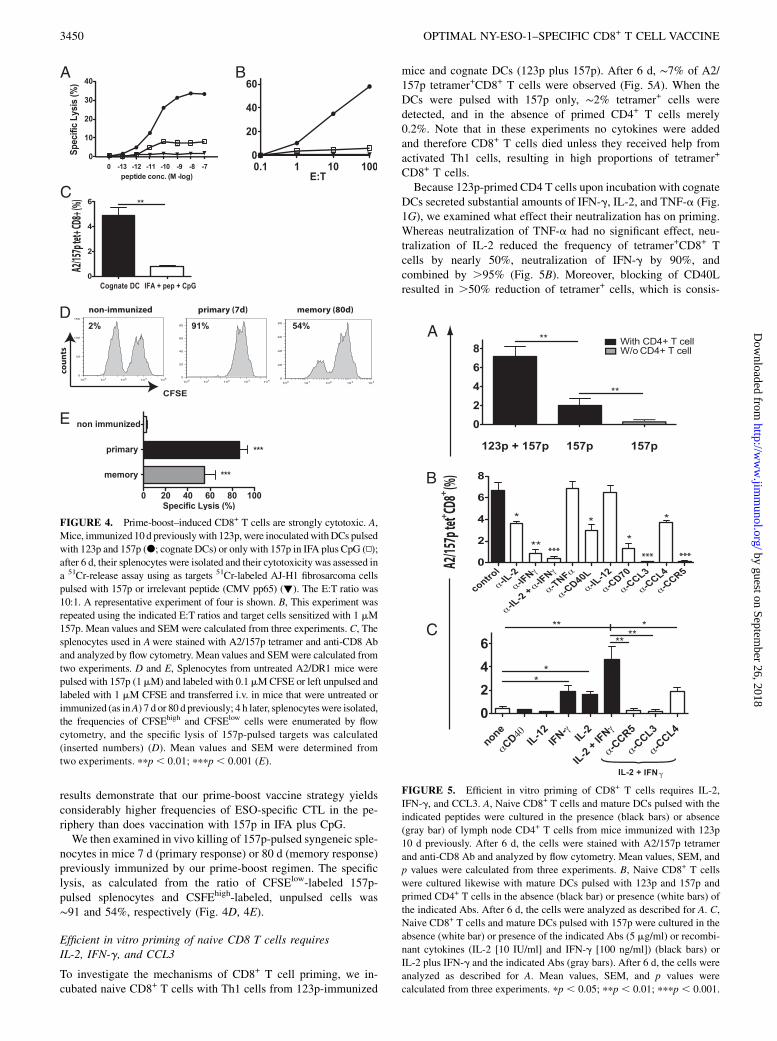
results demonstrate that our prime-boost vaccine strategy yieldsconsiderably higher frequencies of ESO-specific CTL in the pe-riphery than does vaccination with 157p in IFA plus CpG.We then examined in vivo killing of 157p-pulsed syngeneic sple-
nocytes in mice 7 d (primary response) or 80 d (memory response)previously immunized by our prime-boost regimen. The specificlysis, as calculated from the ratio of CFSElow-labeled 157p-pulsed splenocytes and CSFEhigh-labeled, unpulsed cells was∼91 and 54%, respectively (Fig. 4D, 4E).
Efficient in vitro priming of naive CD8 T cells requiresIL-2, IFN-g, and CCL3
To investigate the mechanisms of CD8+ T cell priming, we in-cubated naive CD8+ T cells with Th1 cells from 123p-immunized
mice and cognate DCs (123p plus 157p). After 6 d, ∼7% of A2/157p tetramer+CD8+ T cells were observed (Fig. 5A). When theDCs were pulsed with 157p only, ∼2% tetramer+ cells weredetected, and in the absence of primed CD4+ T cells merely0.2%. Note that in these experiments no cytokines were addedand therefore CD8+ T cells died unless they received help fromactivated Th1 cells, resulting in high proportions of tetramer+
CD8+ T cells.Because 123p-primed CD4 T cells upon incubation with cognate
DCs secreted substantial amounts of IFN-g, IL-2, and TNF-a (Fig.1G), we examined what effect their neutralization has on priming.Whereas neutralization of TNF-a had no significant effect, neu-tralization of IL-2 reduced the frequency of tetramer+CD8+ Tcells by nearly 50%, neutralization of IFN-g by 90%, andcombined by .95% (Fig. 5B). Moreover, blocking of CD40Lresulted in .50% reduction of tetramer+ cells, which is consis-
FIGURE 4. Prime-boost–induced CD8+ T cells are strongly cytotoxic. A,
Mice, immunized 10 d previouslywith 123p, were inoculatedwithDCs pulsed
with 123p and 157p (d; cognate DCs) or only with 157p in IFA plus CpG (N);
after 6 d, their splenocytes were isolated and their cytotoxicity was assessed in
a 51Cr-release assay using as targets 51Cr-labeled AJ-H1 fibrosarcoma cells
pulsed with 157p or irrelevant peptide (CMV pp65) (▼). The E:T ratio was
10:1. A representative experiment of four is shown. B, This experiment was
repeated using the indicated E:T ratios and target cells sensitized with 1 mM
157p. Mean values and SEM were calculated from three experiments. C, The
splenocytes used in Awere stained with A2/157p tetramer and anti-CD8 Ab
and analyzed by flow cytometry. Mean values and SEM were calculated from
two experiments. D and E, Splenocytes from untreated A2/DR1 mice were
pulsed with 157p (1mM) and labeled with 0.1mMCFSE or left unpulsed and
labeled with 1 mM CFSE and transferred i.v. in mice that were untreated or
immunized (as inA) 7 d or 80 d previously; 4 h later, splenocyteswere isolated,
the frequencies of CFSEhigh and CFSElow cells were enumerated by flow
cytometry, and the specific lysis of 157p-pulsed targets was calculated
(inserted numbers) (D). Mean values and SEM were determined from
two experiments. ppp , 0.01; pppp , 0.001 (E).
FIGURE 5. Efficient in vitro priming of CD8+ T cells requires IL-2,
IFN-g, and CCL3. A, Naive CD8+ T cells and mature DCs pulsed with the
indicated peptides were cultured in the presence (black bars) or absence
(gray bar) of lymph node CD4+ T cells from mice immunized with 123p
10 d previously. After 6 d, the cells were stained with A2/157p tetramer
and anti-CD8 Ab and analyzed by flow cytometry. Mean values, SEM, and
p values were calculated from three experiments. B, Naive CD8+ T cells
were cultured likewise with mature DCs pulsed with 123p and 157p and
primed CD4+ T cells in the absence (black bar) or presence (white bars) of
the indicated Abs. After 6 d, the cells were analyzed as described for A. C,
Naive CD8+ T cells and mature DCs pulsed with 157p were cultured in the
absence (white bar) or presence of the indicated Abs (5 mg/ml) or recombi-
nant cytokines (IL-2 [10 IU/ml] and IFN-g [100 ng/ml]) (black bars) or
IL-2 plus IFN-g and the indicated Abs (gray bars). After 6 d, the cells were
analyzed as described for A. Mean values, SEM, and p values were
calculated from three experiments. pp , 0.05; ppp , 0.01; pppp , 0.001.
3450 OPTIMAL NY-ESO-1–SPECIFIC CD8+ T CELL VACCINE
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
tent with reports showing that CD40 triggering promotes CD8+
T cell priming (1, 6, 15). In contrast, blocking of IL-12 showed nosignificant inhibition (Fig. 5B). Blocking of CD70 caused a 6- to 7-fold reduction of tetramer+ cells, which is in agreement with stud-ies showing that CD27 provides crucial costimulation for CD8+
T cell differentiation and memory formation (7, 34, 35). Remark-ably, CD8 T cell priming was abolished by blockade of CCL3 orits receptor CCR5, whereas neutralization of CCL4 inhibited prim-ing by only ∼40%. As negative controls, isotype-matched irrele-vant Abs were used, which in all cases exhibited no effect. In thecase of CCL3 blockade, the same results were obtained whenusing two different Abs (data not shown). Furthermore, the block-ing Abs specific for CCL3, CCL4, CCR5, and IFN-g did not affectthe proliferation of CD8+ T cell proliferation driven by anti-CD3and anti-CD28 Abs in IL-2 containing medium, ruling out non-specific effects.After 6 d of incubation of naive CD8+ T cells with 157p-pulsed
DCs, only 0.1% A2/157p tetramer+CD8+ T cells were observed(Fig. 5C). Although addition of agonist anti-CD40 Ab, rIL-12, orrTNF-a had no significant effects, in the presence of rIL-2 orrIFN-g, ∼1.8 and 2%, respectively, tetramer+ cells were observed,and in the presence of IL-2 and IFN-g combined, 4.7% tetramer+
cells were observed (Fig. 5C and data not shown). The IL-2 plusIFN-g–promoted CD8+ T cell priming was ablated by Abs specificfor CCR5 and CCL3, but it was only partially inhibited by anti-CCL4 Ab.
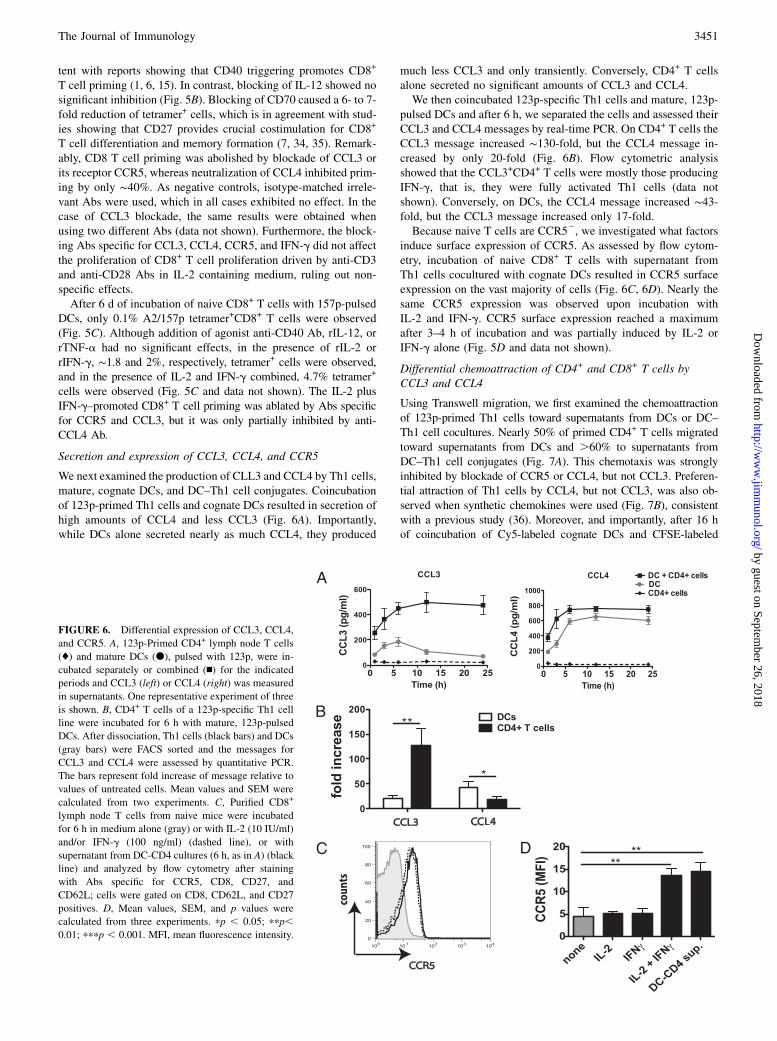
Secretion and expression of CCL3, CCL4, and CCR5
We next examined the production of CLL3 and CCL4 by Th1 cells,mature, cognate DCs, and DC–Th1 cell conjugates. Coincubationof 123p-primed Th1 cells and cognate DCs resulted in secretion ofhigh amounts of CCL4 and less CCL3 (Fig. 6A). Importantly,while DCs alone secreted nearly as much CCL4, they produced
much less CCL3 and only transiently. Conversely, CD4+ T cellsalone secreted no significant amounts of CCL3 and CCL4.We then coincubated 123p-specific Th1 cells and mature, 123p-
pulsed DCs and after 6 h, we separated the cells and assessed theirCCL3 and CCL4 messages by real-time PCR. On CD4+ T cells theCCL3 message increased ∼130-fold, but the CCL4 message in-creased by only 20-fold (Fig. 6B). Flow cytometric analysisshowed that the CCL3+CD4+ T cells were mostly those producingIFN-g, that is, they were fully activated Th1 cells (data notshown). Conversely, on DCs, the CCL4 message increased ∼43-fold, but the CCL3 message increased only 17-fold.Because naive T cells are CCR52, we investigated what factors
induce surface expression of CCR5. As assessed by flow cytom-etry, incubation of naive CD8+ T cells with supernatant fromTh1 cells cocultured with cognate DCs resulted in CCR5 surfaceexpression on the vast majority of cells (Fig. 6C, 6D). Nearly thesame CCR5 expression was observed upon incubation withIL-2 and IFN-g. CCR5 surface expression reached a maximumafter 3–4 h of incubation and was partially induced by IL-2 orIFN-g alone (Fig. 5D and data not shown).
Differential chemoattraction of CD4+ and CD8+ T cells byCCL3 and CCL4
Using Transwell migration, we first examined the chemoattractionof 123p-primed Th1 cells toward supernatants from DCs or DC–Th1 cell cocultures. Nearly 50% of primed CD4+ T cells migratedtoward supernatants from DCs and .60% to supernatants fromDC–Th1 cell conjugates (Fig. 7A). This chemotaxis was stronglyinhibited by blockade of CCR5 or CCL4, but not CCL3. Preferen-tial attraction of Th1 cells by CCL4, but not CCL3, was also ob-served when synthetic chemokines were used (Fig. 7B), consistentwith a previous study (36). Moreover, and importantly, after 16 hof coincubation of Cy5-labeled cognate DCs and CFSE-labeled
FIGURE 6. Differential expression of CCL3, CCL4,
and CCR5. A, 123p-Primed CD4+ lymph node T cells
(♦) and mature DCs (d), pulsed with 123p, were in-
cubated separately or combined (n) for the indicated
periods and CCL3 (left) or CCL4 (right) was measured
in supernatants. One representative experiment of three
is shown. B, CD4+ T cells of a 123p-specific Th1 cell
line were incubated for 6 h with mature, 123p-pulsed
DCs. After dissociation, Th1 cells (black bars) and DCs
(gray bars) were FACS sorted and the messages for
CCL3 and CCL4 were assessed by quantitative PCR.
The bars represent fold increase of message relative to
values of untreated cells. Mean values and SEM were
calculated from two experiments. C, Purified CD8+
lymph node T cells from naive mice were incubated
for 6 h in medium alone (gray) or with IL-2 (10 IU/ml)
and/or IFN-g (100 ng/ml) (dashed line), or with
supernatant from DC-CD4 cultures (6 h, as in A) (black
line) and analyzed by flow cytometry after staining
with Abs specific for CCR5, CD8, CD27, and
CD62L; cells were gated on CD8, CD62L, and CD27
positives. D, Mean values, SEM, and p values were
calculated from three experiments. pp , 0.05; ppp,0.01; pppp , 0.001. MFI, mean fluorescence intensity.
The Journal of Immunology 3451
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
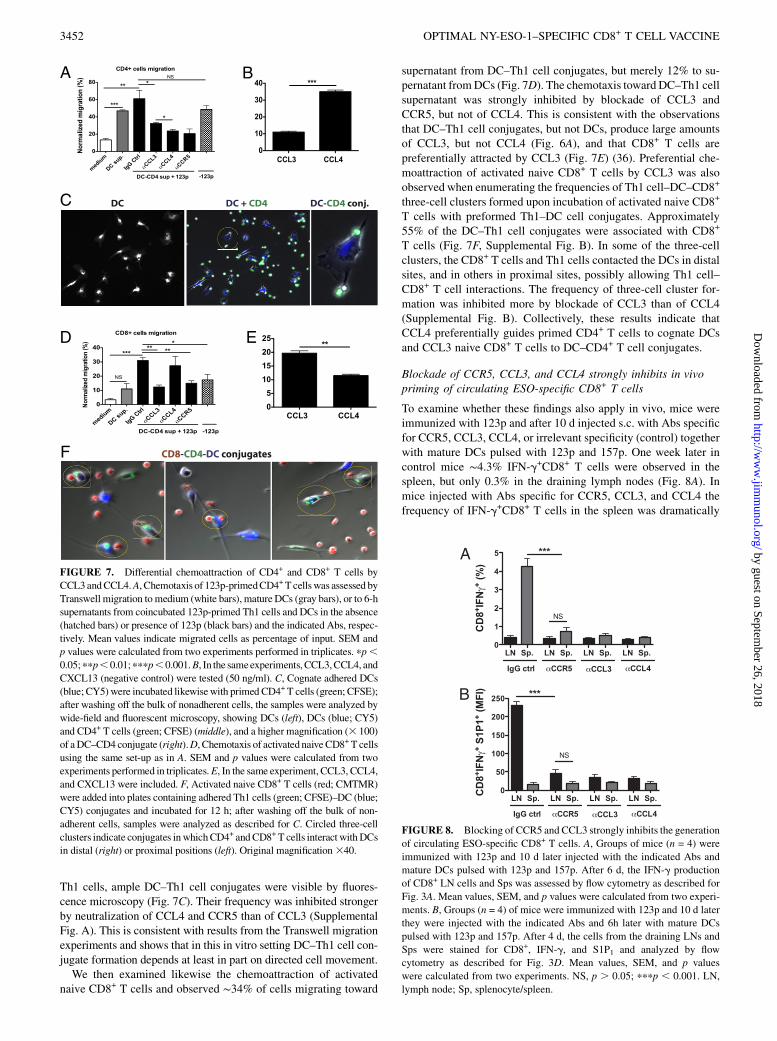
Th1 cells, ample DC–Th1 cell conjugates were visible by fluores-cence microscopy (Fig. 7C). Their frequency was inhibited strongerby neutralization of CCL4 and CCR5 than of CCL3 (SupplementalFig. A). This is consistent with results from the Transwell migrationexperiments and shows that in this in vitro setting DC–Th1 cell con-jugate formation depends at least in part on directed cell movement.We then examined likewise the chemoattraction of activated
naive CD8+ T cells and observed ∼34% of cells migrating toward
supernatant from DC–Th1 cell conjugates, but merely 12% to su-pernatant fromDCs (Fig. 7D). The chemotaxis toward DC–Th1 cellsupernatant was strongly inhibited by blockade of CCL3 andCCR5, but not of CCL4. This is consistent with the observationsthat DC–Th1 cell conjugates, but not DCs, produce large amountsof CCL3, but not CCL4 (Fig. 6A), and that CD8+ T cells arepreferentially attracted by CCL3 (Fig. 7E) (36). Preferential che-moattraction of activated naive CD8+ T cells by CCL3 was alsoobserved when enumerating the frequencies of Th1 cell–DC–CD8+
three-cell clusters formed upon incubation of activated naive CD8+
T cells with preformed Th1–DC cell conjugates. Approximately55% of the DC–Th1 cell conjugates were associated with CD8+
T cells (Fig. 7F, Supplemental Fig. B). In some of the three-cellclusters, the CD8+ T cells and Th1 cells contacted the DCs in distalsites, and in others in proximal sites, possibly allowing Th1 cell–CD8+ T cell interactions. The frequency of three-cell cluster for-mation was inhibited more by blockade of CCL3 than of CCL4(Supplemental Fig. B). Collectively, these results indicate thatCCL4 preferentially guides primed CD4+ T cells to cognate DCsand CCL3 naive CD8+ T cells to DC–CD4+ T cell conjugates.
Blockade of CCR5, CCL3, and CCL4 strongly inhibits in vivopriming of circulating ESO-specific CD8+ T cells
To examine whether these findings also apply in vivo, mice wereimmunized with 123p and after 10 d injected s.c. with Abs specificfor CCR5, CCL3, CCL4, or irrelevant specificity (control) togetherwith mature DCs pulsed with 123p and 157p. One week later incontrol mice ∼4.3% IFN-g+CD8+ T cells were observed in thespleen, but only 0.3% in the draining lymph nodes (Fig. 8A). Inmice injected with Abs specific for CCR5, CCL3, and CCL4 thefrequency of IFN-g+CD8+ T cells in the spleen was dramatically
FIGURE 7. Differential chemoattraction of CD4+ and CD8+ T cells by
CCL3andCCL4.A, Chemotaxis of 123p-primedCD4+Tcellswas assessed by
Transwell migration to medium (white bars), matureDCs (gray bars), or to 6-h
supernatants from coincubated 123p-primed Th1 cells and DCs in the absence
(hatched bars) or presence of 123p (black bars) and the indicated Abs, respec-
tively. Mean values indicate migrated cells as percentage of input. SEM and
p values were calculated from two experiments performed in triplicates. pp,0.05;ppp,0.01;pppp,0.001.B, In the sameexperiments,CCL3,CCL4, and
CXCL13 (negative control) were tested (50 ng/ml). C, Cognate adhered DCs
(blue; CY5)were incubated likewisewith primed CD4+ T cells (green; CFSE);
after washing off the bulk of nonadherent cells, the samples were analyzed by
wide-field and fluorescent microscopy, showing DCs (left), DCs (blue; CY5)
and CD4+ T cells (green; CFSE) (middle), and a higher magnification (3 100)
of aDC–CD4 conjugate (right).D, Chemotaxis of activatednaiveCD8+T cells
using the same set-up as in A. SEM and p values were calculated from two
experiments performed in triplicates.E, In the same experiment, CCL3, CCL4,
and CXCL13 were included. F, Activated naive CD8+ T cells (red; CMTMR)
were added into plates containing adhered Th1 cells (green; CFSE)–DC (blue;
CY5) conjugates and incubated for 12 h; after washing off the bulk of non-
adherent cells, samples were analyzed as described for C. Circled three-cell
clusters indicate conjugates inwhichCD4+ andCD8+ T cells interact withDCs
in distal (right) or proximal positions (left). Original magnification340.
FIGURE 8. Blocking of CCR5 and CCL3 strongly inhibits the generation
of circulating ESO-specific CD8+ T cells. A, Groups of mice (n = 4) were
immunized with 123p and 10 d later injected with the indicated Abs and
mature DCs pulsed with 123p and 157p. After 6 d, the IFN-g production
of CD8+ LN cells and Sps was assessed by flow cytometry as described for
Fig. 3A. Mean values, SEM, and p values were calculated from two experi-
ments. B, Groups (n = 4) of mice were immunized with 123p and 10 d later
they were injected with the indicated Abs and 6h later with mature DCs
pulsed with 123p and 157p. After 4 d, the cells from the draining LNs and
Sps were stained for CD8+, IFN-g, and S1P1 and analyzed by flow
cytometry as described for Fig. 3D. Mean values, SEM, and p values
were calculated from two experiments. NS, p . 0.05; pppp , 0.001. LN,
lymph node; Sp, splenocyte/spleen.
3452 OPTIMAL NY-ESO-1–SPECIFIC CD8+ T CELL VACCINE
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

decreased 0.3–0.75%) and in the draining lymph nodes the fre-quency of primed cells was low in all groups (∼0.3%). Blockadeof CCL4 exhibited markedly stronger inhibition in vivo thanin vitro (Figs. 5B, 8A). The S1P1 expression on IFN-g+CD8+ Tlymph node cells from mice injected with control Ab was ∼10-fold higher than on splenocytes (Fig. 8B). Blockade of CCR5,CCL3, and CCL4 resulted in a 7- to 8-fold lower S1P1 expressionon freshly primed lymph node cells (Fig. 8B). The S1P1 expres-sion on splenocytes was similarly low in all cases, which isexplained, at least in part, by internalization of S1P1 on cells uponexposure to the high S1P concentrations in blood and spleen (23,24). Taken collectively, these results indicate that the CCR5–CCL3/CCL4 axes are crucial for in vivo priming of ESO-specificCD8+ T cells and S1P1 upregulation and thus exit from the draininglymph nodes into circulation.
DiscussionA major finding of the present study is that the magnitude, quality,and localization of the prime-boost vaccine-induced ESO-specificCD8+ T cell response in A2/DR1 transgenic mice was largely de-termined by available Th1 cell help (Fig. 2). A2/DR1 mice immu-nized with 123p exhibited much higher frequencies of ESO-specificCD4+ T cells than did animals immunized with the ESO peptides87–111, 84–101, 89–98, or 87–101 (Figs. 1, 2 and unpublisheddata). This is at variance with studies showing that in patients withESO+ malignancies the frequencies of CD4+ T cells specific forthese epitopes are similar (27, 28, 37). This might be explained bythe fact that the DCs that prime CD4+ T cells generate and presentbiased repertoires of ESO peptides, thus obscuring the actual im-munogenicity of the peptides. Because this is ruled out in oursystem, immunization with synthetic peptides allows conclusiveidentification of their immunogenicities, which is crucial knowl-edge for the design of efficient vaccines.Our results indicate that efficient priming requires that DCs
copresent the MHC I- and MHC II-restricted peptides (Fig. 2). Thisis consistent with a three-cell cluster model for CD8+ T cell prim-ing, in which activated CD4+ T cells and naive CD8+ T cellsinteract simultaneously with cognate DCs (Figs. 2, 3A, 4B, 5A,5B). Such three-cell clusters were observed in vitro and in vivo,and their formation relies on chemoattraction of activated naiveCD8+ T cells by CCL3 produced by Th1 cells upon conjugationwith cognate DCs (Figs. 7, 8, Supplemental Fig.) (19, 20). Three-cell clusters also provide integral stimulation and costimulationfor naive CD8+ T cells by DCs and CD4+ T cells. The 6-foldreduction of ESO-specific CD8+ T cells observed upon blockingof CD70 demonstrates that in our system triggering of CD27(on naive CD8+ T cells) by CD70 (on mature DCs) is important,which is in accordance with previous in vivo studies demonstrat-ing that the CD27–CD70 axis is crucial for the generation andmaintenance of CD8+ T cell responses (Fig. 5) (34, 35). Interest-ingly, primed CD8+ T cells, as Th1 cells, can attract naive CD8+
T cells in a CCR5-dependent manner and promote their primingif the DCs copresent the respective peptides (22). Because primedCD8+ T cells upon interaction with cognate DCs produce CCL3,as Th1 cells, an analogous mechanism is suggested (22).Although in our in vitro CD8+ T cell-priming experiments neu-
tralization of CCL4 was less inhibitory than was blocking ofCCL3, in the in vivo experiments neutralization of CCL4 andCCL3 exhibited similar effects (Fig. 8A) (19). We argue that thisdivergence is explained by the fact that CCL3 and CCL4 playdifferent roles in CD8+ T cell priming, which is not discerniblein in vivo experiments. The observations that mature DCs secretedhigh amounts of CCL4, but not CCL3, and that primed CD4+
T cells were strongly chemoattracted by CCL4, but not CCL3,
convincingly argues that CCL4 preferentially attracted Th1 cellsto mature DCs (Figs. 6A, 7A, 7B, Supplemental Fig. A) (36).Because the density of purified, primed, CD4+ T cells and cognatemature DCs in our in vitro experiments was high (Fig. 1C), theymay find cognate DCs by random cell movements with significantprobability. Conversely, in lymph nodes the probability of acti-vated CD4+ T cells to find rare cognate mature DCs by scanning incomplex three-dimensional structures is expected to be more de-pendent on CCL4-mediated chemoattraction, hence the strongerinhibition observed upon CCL4 blockade (Fig. 8A). In a secondstage, primed CD4+ T cells upon conjugation with cognate DCssecrete high amounts of CCL3, but not CCL4, which in turnchemoattracts activated naive CD8+ T cells to DC–CD4 conju-gates (Figs. 6A, 6B, 7D–F, Supplemental Fig. B) (36); becauseof the very low frequency of naive Ag-specific CD8+ T cells (es-timated at 1 out of 1026; see Ref. 21), this is crucial, as seen by thedeleterious effect that CCL3 neutralization had on CD8+ T cellpriming in vitro and in vivo (Figs. 5B, 5C, 8A).It is noteworthy that CCL3 can promote CD8+ T cell priming
also in other ways than chemoattraction. For example, it canpromote Th1 cell formation and IFN-g production in an IL-12–independent manner, which is consistent with the observation thatin vitro CD8+ T cell priming required IFN-g but not IL-12 (Fig.5B, 5C) (38–40). Moreover, CCL3 has been shown to have adju-vant effects on CD8+ T cell priming, to increase CD8+ T celleffector functions, and to be involved (together with other chemo-kines) in recruiting CD8+ T cells to inflamed sites (39, 41–43).Although CCL3 and CCL4 both bind to CCR5, the former, but notthe latter, also binds to CCR1, which may account for their di-verging biological effects (36, 39).Considering that CCR5 expression of CD8+ T cells plays key
roles in their priming (Figs. 5, 8) (19, 22), but also in directingprimed cells to tumors (44) or to sites of inflammation (45), sur-prisingly little is known about the factors and mechanisms thatgovern CCR5 expression. Our results demonstrate for the firsttime that the Th1 cytokines IFN-g and IL-2, namely when com-bined, induce within hours CCR5 surface expression on naiveCD8+ T cells (Fig. 6C, 6D). Importantly, this occurs in the absenceof Ag or TCR triggering as has been described previously (19, 22,46), and hence is a plausible mechanism by which naive CD8+
T cells become CCR5+ on entering sites where Th1 cytokines areproduced, becoming thus susceptible to chemotaxis (Fig. 5B, 5C).The importance of this is demonstrated by the dramatic inhibitionof CD8+ T cell priming by neutralization of IFN-g plus IL-2 or ofCCL3 or CCR5 and their ability to partially substitute primedCD4+ T cells in in vitro priming of CD8+ T cells, which againrelies on CCR5 expression (Fig. 5C).The strength of available Th1 cell help also determined the S1P1
expression on primed CD8+ T cells and thus their egress from thedraining lymph nodes in the circulation (Fig. 3) (23, 24, 47). Thisis of importance, because an efficient cancer vaccine must induceCD8+ effector T cells that leave the sites of priming and can ac-cess tumors in the periphery (44). Our observation that in miceimmunized with 157p in adjuvants, primed CD8+ T cells remainedmostly in the draining lymph nodes, cautions that such peptidevaccines may fall short in inducing circulating CTL. Interestingly,even when strong Th1 cell help was available, blockade of CCR5-CCL3/4 severely inhibited upregulation of S1P1 (Fig. 8B). Be-cause this blockade equally strongly inhibited the priming ofESO-specific CD8+ T cells in lymph nodes and in vitro, it appearsthat CCR5-CCL3/4 orchestrate interactions between Th1 cells,DCs, and naive activated CD8+ T cells and that this is essentialfor efficient CD8+ T cell priming, S1P1 expression, and CD8+
T cell memory formation (Figs. 5B, 8A) (19, 47, 48). S1P1 is
The Journal of Immunology 3453
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

downregulated upon initial T cell activation, but it is stronglyexpressed after extensive T cell differentiation and proliferation,and subsequent S1P1 expression seems to be programmed duringthe initial priming events (47). Although this needs to be investi-gated in further detail, it is interesting that CCR5 can be recruitedin the immunological synapse and contribute to the activation ofdistinct T cell functions (49).In conclusion, the present study describes a versatile prime-boost
vaccine strategy that allows the generation of powerful, high-avidity TAA-specific CD8+ CTL responses. By first inducingTh1 cells by immunization with a MHC II-restricted peptideand then priming CD8+ T cells with peptide-pulsed mature DCs,this allowed us to define key variables for efficient cancer vac-cines, such as: 1) the strength of available Th1 cell help deter-mines the frequency of primed CD8+ T cells and, via their S1P1expression, their egress from the lymph nodes in the circulation,which is required for their efficient interaction with tumors; and 2)efficient priming of CD8+ CTL requires coordinated interactionsof activated naive CD8+ T cells, cognate mature DCs, and primedTh1 cells, which are orchestrated by CCL4 and CCL3, guidingTh1 cells to mature DCs and activated naive CD8+ T cells toDC–Th1 cell conjugates, respectively. Our study advocates theuse of defined, strongly immunogenic MHC II- and MHC I-restricted peptides for cancer vaccines, because this circumventsthe caveat that TAA epitopes cross-presented by DCs may not bethe most immunogenic ones and/or may not be presented bytumor cells. Note, however, that ESO is not expressed in mice,but it is expressed in humans on cancer cells, but also on testis andat low levels in a limited number of somatic tissues (50); there-fore, conclusions obtained in mouse models should be temperedwith regard to applicability in human vaccination of cancerpatients, due to potential tolerance in humans. Advances in TAAdiscovery and peptide predictions make available an ever-growingrange of TAA epitopes restricted by diverse HLA molecules,which can be combined to broaden the range of T cell recognition.
AcknowledgmentsWe thank Drs. Yu-Chun Lone (INSERM U542, Hopital Paul Brousse,
Villejuif, France) for providing the A2/DR1 mice and Philippe Guillaume
(Ludwig Institute for Cancer Research facility, Lausannne, Switzerland)
for A2/157p tetramers.
DisclosuresThe authors have no financial conflicts of interest.
References1. Melief, C. J. 2008. Cancer immunotherapy by dendritic cells. Immunity 29: 372–
383.2. Pilla, L., L. Rivoltini, R. Patuzzo, A. Marrari, R. Valdagni, and G. Parmiani. 2009.
Multipeptide vaccination in cancer patients. Expert Opin. Biol. Ther. 9: 1043–1055.3. Karbach, J., S. Gnjatic, A. Bender, A. Neumann, E. Weidmann, J. Yuan,
C. A. Ferrara, E. Hoffmann, L. J. Old, N. K. Altorki, and E. Jager. 2010. Tumor-reactive CD8+ T-cell responses after vaccination with NY-ESO-1 peptide, CpG7909 and Montanide ISA-51: association with survival. Int. J. Cancer 126: 909–918.
4. Pittet, M. J., V. Rubio-Godoy, G. Bioley, P. Guillaume, P. Batard, D. Speiser,I. Luescher, J. C. Cerottini, P. Romero, and A. Zippelius. 2003. a3 Domainmutants of peptide/MHC class I multimers allow the selective isolation ofhigh avidity tumor-reactive CD8 T cells. J. Immunol. 171: 1844–1849.
5. Boissonnas, A., A. Scholer-Dahire, L. Fetler, and S. Amigorena. 2009.Multiphoton imaging of cytotoxic T lymphocyte-mediated antitumor immuneresponses. Curr. Top. Microbiol. Immunol. 334: 265–287.
6. Fujii, S., K. Liu, C. Smith, A. J. Bonito, and R. M. Steinman. 2004. The linkageof innate to adaptive immunity via maturing dendritic cells in vivo requiresCD40 ligation in addition to antigen presentation and CD80/86 costimulation. J.Exp. Med. 199: 1607–1618.
7. Hendriks, J., L. A. Gravestein, K. Tesselaar, R. A. van Lier, T. N. Schumacher,and J. Borst. 2000. CD27 is required for generation and long-term maintenanceof T cell immunity. Nat. Immunol. 1: 433–440.
8. Krieg, A. M. 2007. Development of TLR9 agonists for cancer therapy. J. Clin.Invest. 117: 1184–1194.
9. Wurzenberger, C., V. H. Koelzer, S. Schreiber, D. Anz, A. M. Vollmar,M. Schnurr, S. Endres, and C. Bourquin. 2009. Short-term activation inducesmultifunctional dendritic cells that generate potent antitumor T-cell responsesin vivo. Cancer Immunol. Immunother. 58: 901–913.
10. Watchmaker, P. B., E. Berk, R. Muthuswamy, R. B. Mailliard, J. A. Urban,J. M. Kirkwood, and P. Kalinski. 2010. Independent regulation of chemokineresponsiveness and cytolytic function versus CD8+ T cell expansion bydendritic cells. J. Immunol. 184: 591–597.
11. BenMohamed, L., R. Krishnan, J. Longmate, C. Auge, L. Low, J. Primus, andD. J. Diamond. 2000. Induction of CTL response by a minimal epitope vaccinein HLA A*0201/DR1 transgenic mice: dependence on HLA class II restricted TH
response. Hum. Immunol. 61: 764–779.12. Nakanishi, Y., B. Lu, C. Gerard, and A. Iwasaki. 2009. CD8+ T lymphocyte mo-
bilization to virus-infected tissue requires CD4+ T-cell help. Nature 462: 510–513.13. Gerner, M. Y., K. A. Casey, and M. F. Mescher. 2008. Defective MHC class II
presentation by dendritic cells limits CD4 T cell help for antitumor CD8 T cellresponses. J. Immunol. 181: 155–164.
14. Shedlock, D. J., and H. Shen. 2003. Requirement for CD4 T cell help in gen-erating functional CD8 T cell memory. Science 300: 337–339.
15. Schoenberger, S. P., R. E. Toes, E. I. van der Voort, R. Offringa, and C. J. Melief.1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40Linteractions. Nature 393: 480–483.
16. Mescher, M. F., J. M. Curtsinger, P. Agarwal, K. A. Casey, M. Gerner,C. D. Hammerbeck, F. Popescu, and Z. Xiao. 2006. Signals required for pro-gramming effector and memory development by CD8+ T cells. Immunol. Rev.211: 81–92.
17. Wilson, E. B., and A. M. Livingstone. 2008. Cutting edge: CD4+ T cell-derivedIL-2 is essential for help-dependent primary CD8+ T cell responses. J. Immunol.181: 7445–7448.
18. Williams, M. A., A. J. Tyznik, and M. J. Bevan. 2006. Interleukin-2 signalsduring priming are required for secondary expansion of CD8+ memoryT cells. Nature 441: 890–893.
19. Castellino, F., A. Y. Huang, G. Altan-Bonnet, S. Stoll, C. Scheinecker, andR. N. Germain. 2006. Chemokines enhance immunity by guiding naive CD8+
T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 440: 890–895.20. Beuneu, H., Z. Garcia, and P. Bousso. 2006. Cutting edge: cognate CD4 help
promotes recruitment of antigen-specific CD8 T cells around dendritic cells. J.Immunol. 177: 1406–1410.
21. Rizzuto, G. A., T. Merghoub, D. Hirschhorn-Cymerman, C. Liu, A. M. Lesokhin,D. Sahawneh, H. Zhong, K. S. Panageas, M. A. Perales, G. Altan-Bonnet, et al.2009. Self-antigen-specific CD8+ T cell precursor frequency determines the qualityof the antitumor immune response. J. Exp. Med. 206: 849–866.
22. Hugues, S., A. Scholer, A. Boissonnas, A. Nussbaum, C. Combadiere,S. Amigorena, and L. Fetler. 2007. Dynamic imaging of chemokine-dependentCD8+ T cell help for CD8+ T cell responses. Nat. Immunol. 8: 921–930.
23. Grigorova, I. L., S. R. Schwab, T. G. Phan, T. H. Pham, T. Okada, andJ. G. Cyster. 2009. Cortical sinus probing, S1P1-dependent entry and flow-basedcapture of egressing T cells. Nat. Immunol. 10: 58–65.
24. Pham, T. H., T. Okada, M. Matloubian, C. G. Lo, and J. G. Cyster. 2008. S1P1receptor signaling overrides retention mediated by Gai-coupled receptors topromote T cell egress. Immunity 28: 122–133.
25. Pajot, A., M. L. Michel, N. Fazilleau, V. Pancre, C. Auriault, D. M. Ojcius,F. A. Lemonnier, and Y. C. Lone. 2004. A mouse model of human adaptiveimmune functions: HLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur. J. Immunol. 34: 3060–3069.
26. Chen, J. L., P. R. Dunbar, U. Gileadi, E. Jager, S. Gnjatic, Y. Nagata, E. Stockert,D. L. Panicali, Y. T. Chen, A. Knuth, et al. 2000. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactiveCTL. J. Immunol. 165: 948–955.
27. Mandic, M., F. Castelli, B. Janjic, C. Almunia, P. Andrade, D. Gillet, V. Brusic,J. M. Kirkwood, B. Maillere, and H. M. Zarour. 2005. One NY-ESO-1-derivedepitope that promiscuously binds to multiple HLA-DR and HLA-DP4 moleculesand stimulates autologous CD4+ T cells from patients with NY-ESO-1-expressing melanoma. J. Immunol. 174: 1751–1759.
28. Gnjatic, S., D. Atanackovic, E. Jager, M. Matsuo, A. Selvakumar, N. K. Altorki,R. G. Maki, B. Dupont, G. Ritter, Y. T. Chen, et al. 2003. Survey of naturallyoccurring CD4+ T cell responses against NY-ESO-1 in cancer patients: correla-tion with antibody responses. Proc. Natl. Acad. Sci. USA 100: 8862–8867.
29. Lalvani, A., R. Brookes, S. Hambleton, W. J. Britton, A. V. Hill, andA. J. McMichael. 1997. Rapid effector function in CD8+ memory T cells. J. Exp.Med. 186: 859–865.
30. Barber, D. L., E. J. Wherry, and R. Ahmed. 2003. Cutting edge: rapid in vivokilling by memory CD8 T cells. J. Immunol. 171: 27–31.
31. Jing, H., J. H. Yen, and D. Ganea. 2004. A novel signaling pathway mediates theinhibition of CCL3/4 expression by prostaglandin E2. J. Biol. Chem. 279:55176–55186.
32. Link, A., T. K. Vogt, S. Favre, M. R. Britschgi, H. Acha-Orbea, B. Hinz,J. G. Cyster, and S. A. Luther. 2007. Fibroblastic reticular cells in lymphnodes regulate the homeostasis of naive T cells. Nat. Immunol. 8: 1255–1265.
33. Britschgi, M. R., A. Link, T. K. Lissandrin, and S. A. Luther. 2008. Dynamicmodulation of CCR7 expression and function on naive T lymphocytes in vivo. J.Immunol. 181: 7681–7688.
34. Keller, A. M., A. Schildknecht, Y. Xiao, M. van den Broek, and J. Borst. 2008.Expression of costimulatory ligand CD70 on steady-state dendritic cells breaksCD8+ T cell tolerance and permits effective immunity. Immunity 29: 934–946.
3454 OPTIMAL NY-ESO-1–SPECIFIC CD8+ T CELL VACCINE
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

35. Keller, A. M., Y. Xiao, V. Peperzak, S. H. Naik, and J. Borst. 2009. Cos-timulatory ligand CD70 allows induction of CD8+ T-cell immunity by immaturedendritic cells in a vaccination setting. Blood 113: 5167–5175.
36. Taub, D. D., K. Conlon, A. R. Lloyd, J. J. Oppenheim, and D. J. Kelvin. 1993.Preferential migration of activated CD4+ and CD8+ T cells in response toMIP-1a and MIP-1b. Science 260: 355–358.
37. Chen, Q., H. Jackson, P. Parente, T. Luke, M. Rizkalla, T. Y. Tai, H. C. Zhu,N. A. Mifsud, N. Dimopoulos, K. A. Masterman, et al. 2004. ImmunodominantCD4+ responses identified in a patient vaccinated with full-length NY-ESO-1 formulated with ISCOMATRIX adjuvant. Proc. Natl. Acad. Sci. USA101: 9363–9368.
38. Zou, W., J. Borvak, F. Marches, S. Wei, P. Galanaud, D. Emilie, and T. J. Curiel.2000. Macrophage-derived dendritic cells have strong Th1-polarizing potentialmediated by b-chemokines rather than IL-12. J. Immunol. 165: 4388–4396.
39. Luther, S. A., and J. G. Cyster. 2001. Chemokines as regulators of T cell dif-ferentiation. Nat. Immunol. 2: 102–107.
40. Karpus, W. J., N. W. Lukacs, K. J. Kennedy, W. S. Smith, S. D. Hurst, andT. A. Barrett. 1997. Differential CC chemokine-induced enhancement ofT helper cell cytokine production. J. Immunol. 158: 4129–4136.
41. Jones, E., D. A. Price, M. Dahm-Vicker, V. Cerundolo, P. Klenerman, andA. Gallimore. 2003. The influence of macrophage inflammatory protein-1a onprotective immunity mediated by antiviral cytotoxic T cells. Immunology 109:68–75.
42. Trifilo, M. J., and T. E. Lane. 2004. The CC chemokine ligand 3 regulatesCD11c+CD11b+CD8a2 dendritic cell maturation and activation following viral
infection of the central nervous system: implications for a role in T cell acti-vation. Virology 327: 8–15.
43. Song, R., S. Liu, and K. W. Leong. 2007. Effects of MIP-1a, MIP-3a, andMIP-3b on the induction of HIV Gag-specific immune response with DNAvaccines. Mol. Ther. 15: 1007–1015.
44. Harlin, H., Y. Meng, A. C. Peterson, Y. Zha, M. Tretiakova, C. Slingluff,M. McKee, and T. F. Gajewski. 2009. Chemokine expression in melanoma me-tastases associated with CD8+ T-cell recruitment. Cancer Res. 69: 3077–3085.
45. Karpus, W. J., and K. J. Kennedy. 1997. MIP-1a and MCP-1 differentiallyregulate acute and relapsing autoimmune encephalomyelitis as well as Th1/Th2 lymphocyte differentiation. J. Leukoc. Biol. 62: 681–687.
46. Iwasaki, M., T. Mukai, P. Gao, W. R. Park, C. Nakajima, M. Tomura,H. Fujiwara, and T. Hamaoka. 2001. A critical role for IL-12 in CCR5 inductionon T cell receptor-triggered mouse CD4+ and CD8+ T cells. Eur. J. Immunol. 31:2411–2420.
47. Cyster, J. G. 2005. Chemokines, sphingosine-1-phosphate, and cell migration insecondary lymphoid organs. Annu. Rev. Immunol. 23: 127–159.
48. Castellino, F., and R. N. Germain. 2007. Chemokine-guided CD4+ T cell helpenhances generation of IL-6RahighIL-7Rahigh prememory CD8+ T cells. J. Immunol.178: 778–787.
49. Contento, R. L., B. Molon, C. Boularan, T. Pozzan, S. Manes, S. Marullo, andA. Viola. 2008. CXCR4-CCR5: a couple modulating T cell functions. Proc. Natl.Acad. Sci. USA 105: 10101–10106.
50. Caballero, O. L., and Y. T. Chen. 2009. Cancer/testis (CT) antigens: potentialtargets for immunotherapy. Cancer Sci. 100: 2014–2021.
The Journal of Immunology 3455
by guest on September 26, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from





























