Embed Size (px)
Citation preview

�
RESEARCH ARTICLE
Deletions in 14q24.1q24.3 are AssociatedWith Congenital Heart Defects, Brachydactyly,and Mild Intellectual Disability
Barbara Oehl-Jaschkowitz,1 Olivier M. Vanakker,2 Anne De Paepe,2 Bjorn Menten,2 Thomas Martin,1Georg Weber,1 Alexander Christmann,1 Romain Krier,3 Simone Scheid,3 Susan E. McNerlan,4
Shane McKee,4 and Andreas Tzschach5*1Practice of Human Genetics, Homburg (Saar), Germany2Center for Medical Genetics, Ghent University Hospital, Ghent, Belgium3Practice of Pediatrics, Wittlich, Germany4Northern Ireland Regional Genetics Service, Belfast City Hospital, Belfast, UK5Institute of Medical Genetics and Applied Genomics, University of Tuebingen, Tuebingen, Germany
Manuscript Received: 29 April 2013; Manuscript Accepted: 14 September 20
13How to Cite this Article:Oehl-Jaschkowitz B, Vanakker OM, De
Paepe A, Menten B, Martin T, Weber G,
Christmann A, Krier R, Scheid S, McNerlan
SE, McKee S, Tzschach A. 2014. Deletions
in 14q24.1q24.3 are associated with
congenital heart defects, brachydactyly, and
mild intellectual disability.
Am J Med Genet Part A 164A:620–626.
Conflict of interest: none.
Grant sponsor: Wellcome Trust.
Interstitial deletions of chromosome band 14q24.1q24.3 are
apparently very rare. We report on three unrelated patients
with overlapping de novo deletions of sizes 5.4, 2.8, and
2.3Mb in this region. While some clinical problems such as
intestinal malrotation, cryptorchidism, and ectopic kidney were
only observed in single patients, all three patients had mild
intellectual disability, congenital heart defects (truncus arterio-
sus, pulmonary atresia, atrial septal defect, and/or ventricular
septal defect), brachydactyly, hypertelorism, broad nasal bridge,
and thin upper lips. Likely haploinsufficiency of one or several
of the 19 genes in the common deleted interval (ACTN1,
DCAF5, EXD2, GALNTL1, ERH, SLC39A9, PLEKHD1,
CCDC177, KIAA0247, LOC100289511, SRSF5, SLC10A1,
SMOC1, SLC8A3, ADAM21P1, COX16, SYNJ2BP, SYNJ2BP-
COX16, ADAM21) was responsible for these manifestations,
but apart from SMOC1, mutations in which cause autosomal
recessive Waardenburg anophthalmia syndrome, and ACTN1,
mutations in which are associated with congenital macrothrom-
bocytopenia, no disease associations have so far been reported
for the other genes. Functional studies and a systematic search
for mutations or chromosome aberrations in this region will
elucidate the role of individual genes in the clinical manifesta-
tions and will provide insight into the underlying biological
mechanisms. � 2013 Wiley Periodicals, Inc.
Key words: del14q24.1q24.3; array CGH; intellectual disability;
congenital heart disease; brachydactyly; SMOC1; ACTN1;
DCAF5
�Correspondence to:Andreas Tzschach, M.D., Institute of Medical Genetics and Applied
Genomics, University of Tuebingen, Calwerstr. 7, 72076 Tuebingen,
Germany. E-mail: [email protected]
Article first published online in Wiley Online Library
(wileyonlinelibrary.com): 19 December 2013
DOI 10.1002/ajmg.a.36321
INTRODUCTION
The widespread application of array technologies in routine cyto-
genetic diagnostics in recent years has provided the basis for an ever
more detailed correlation of small chromosomal deletions or
2013 Wiley Periodicals, Inc.
duplications with the accompanying clinical consequences. There
are, however, still “white spots” on this map, that is, chromosome
regions for which no well-characterized aberrations have
been described. One of those regions is chromosome band
14q24.1q24.3.
Here,we report on three unrelatedpatientswithmild intellectual
disability, congenital heart defects, brachydactyly and additional
abnormalities in whom overlapping interstitial de novo deletions
were detected in 14q24.1q24.3.
620

OEHL-JASCHKOWITZ ET AL. 621
CLINICAL REPORTS
Patient 1This boy was born after an uneventful pregnancy at term to healthy
andnonconsanguineousGermanparents.Atbirth, small hands and
a small atrial septal defect (which did not require surgery) were
noted, but he had no other malformations (birth measurements:
length 48 cm (10–25th centile), weight 2,560 g (<3rd centile), OFC
34 cm (10–25th centile). Motor development was normal (he
started walking at age 10 months), but language acquisition was
delayed (first words at age 24 months). On examination at age
10 years, height (138 cm; 25th centile) and OFC (52 cm; 25th
centile) were normal. He had mild intellectual disability and
attended a school for children with learning problems. Facial
dysmorphic features included high nasal bridge, apparent hyper-
telorism, long and flat philtrum and thin upper lip vermilion
(Fig. 1a). He had bilateral hypoplastic thumbs, short and tapering
fingers and cutaneous syndactyly of the fingers (Fig. 2). Extension
and supinationof the elbowswere limited.Hehadahoarse anddeep
voice and suffered from asthma. Neurological, ophthalmic and
ultrasound examinations of the heart and abdominal organs failed
to reveal any additional abnormalities. Screening for metabolic
disorders, conventional chromosome analysis and electrocardiog-
raphy detected no pathologic results. The patient had a healthy
older sister and a healthy younger brother.
Patient 2This boy (DECIPHER 251926) is the third of four children to
healthy non-consanguineous Northern Irish parents. A paternal
aunt had died as a neonate with congenital cardiac anomalies. He
a b
c
FIG. 1. Facial features of the three patients. a: Patient 1 at age 10 year
thin upper lip. b: Patient 2 at age 22 months and (c) at age 4 years. No
lip and downslanting palpebral fissures. d: Patient 3 at age 2 years and
downslanting palpebral fissures, and thin lips.
was born at 38 weeks gestation, weighing 3,100 g (25th centile). His
mother felt less fetal movement than in her other pregnancies, but
antenatal scanswere reportedlynormal.Hebecamecyanosed at one
hour of age and echocardiography revealed pulmonary atresia with
a ventricular septal defect, anteriorly-set aorta and severe stenosis of
the pulmonary arterial confluence. This was treated with a Blalock-
Taussig shunt procedure, and subsequently by a Rastelli-Senning
procedure. He was also found to have malrotation of the small
bowel and undescended testes. Echocardiography on the father was
normal. Dysmorphic features included hypertelorism with down-
slanting palpebral fissures, a broad nasal bridge and short nose,
narrow eyebrows, full plethoric cheeks, low-set abnormal ears and a
thin upper lip vermilion with down-turned corners (Fig. 1b,c). He
had a short neck with mild neck webbing, narrow shoulders, short
arms with very small hands that were narrow across the metacar-
pophalangeal joints and proximally-set thumbs. The nipples were
wide-set and the scrotum mildly hypoplastic. The feet were small
with minimal 2–3 toe syndactyly and an over-curved 4th toenail
bilaterally. There was limitation of elbow extension with bilateral
dislocation of the radial heads, but other joints were significantly
hypermobile.
Height and weight progressed along the 25th centile and head
circumference on the 2nd centile. Development was slower than his
siblings, with significant speech delay, but he remains in main-
stream education with some help at the age of 11 years.
Patient 3This girl (DECIPHER250043 and 253983)was born at termafter an
uneventful pregnancy with normal birth measurements to healthy
e
d
s. Note hypertelorism, broad nasal bridge, long and flat philtrum, and
te apparent hypertelorism, small nose, broad nasal bridge, thin upper
(e) at age 5 years. Note hypertelorism, broad nasal bridge,

FIG. 2. Photos and X-ray images of the hands and feet of Patient 1 at the age of 10 years [(a) left and (b) right hand, (c) left and (d) right
foot]. Note short fingers and toes.
622 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
and non-consanguineous parents from Belgium. In the neonatal
period, truncus arteriosus was diagnosed and surgically corrected.
Facial dysmorphisms included hypertelorism, mild synophrys,
epicanthic folds and downslanting palpebral fissures (Fig. 1d–e).
Midface hypoplasia became a more prominent feature over the
years. Joint hyperlaxity, primarily of the fingers and elbows, was
initially noted but improved when she got older. Additional
examinations revealed the presence of an ectopic kidney with
dilated ureter. Early psychomotor development was normal with
sitting at age 9months and walking at age 17months. The first sign
of developmental delay was a delayed speech development; also
grossmotor developmentwas delayed. At the age of 6 years, she had
been diagnosed with a borderline IQ of 81 (verbal IQ of 78,
performance IQ of 91). The patient has a healthy brother and a
healthy sister.
MATERIALS AND METHODS
DNA samples of the patients and their parents were isolated from
peripheral blood leukocytes using routine procedures. For Patient 1
andhis parents, whole-genome array comparative genomic hybrid-
ization (array CGH) analysis was performed using a 180K oligo-
nucleotide array (CytoChip Oligo 4X180K array, Bluegnome,
Cambridge, UK) according to protocols provided by the manufac-
turer. Laser scanningwasperformedby anAgilentDNAMicroarray
Scanner, and the BlueFuse Multi v2.6 software was employed for
data analysis. Patient 2 and his parents had been analysed using an
Agilent 44K array CGH chip, and for patient 3 and her parents, an
Agilent 60k array CGH chip was employed.
RESULTS
In all three patients, interstitial de novo deletions of the long
arm of chromosome 14 (14q24.1q24.3) were detected. The
genomic positions are given according to GRCh37/hg19. In
Patient 1, the deletion had a size of approximately 5.4Mb, and
its genomic position was chr14:69,379,727–74,826,674. The
deletion of Patient 2 (DECIPHER 251926) had a size of 2.3Mb
(genomic position chr14:68,609,319–70,919,016), and the deletion
of Patient 3 (DECIPHER 250043 and 253983) was 2.8Mb in size
and had the genomic coordinates chr14:68,816,535–71,659,567
(Fig. 3). Minimum deletion sizes were given for all three
patients.
The complete region covered by these deletions harbors more
than 50 RefSeq genes (Fig. 3 and Table I).The 1.5Mb interval that is
deleted in all three patients includes 19 genes: ACTN1, DCAF5,
EXD2, GALNTL1, ERH, SLC39A9, PLEKHD1, CCDC177,
KIAA0247, LOC100289511, SRSF5, SLC10A1, SMOC1, SLC8A3,
ADAM21P1, COX16, SYNJ2BP, SYNJ2BP-COX16, and ADAM21
(these genes are marked with a in Table I).
DISCUSSION
This is the first report of interstitial 14q24.1q24.3 deletions charac-
terized by high-resolution array CGH. A large 14q23.3q31.1 dele-
tion of an estimated size between 9.6 and 13.7Mb had previously
beendescribed inamalepatientwith clinical featuresofHolt–Oram
syndrome (which is clinically characterized by radial defects and
congenital heart defects and associated with mutations in TBX5

FIG. 3. Overview of the region 14q24.1q24.3 and its gene content according to the UCSC Genome Browser (GRCh37/hg19 assembly). The
deletions of Patients 1–3 are indicated by red bars.
OEHL-JASCHKOWITZ ET AL. 623
in 12q24.21), cleft palate and facial dysmorphisms [Le Meur
et al., 2005]. Comparison of this boy to the patients reported
here is, however, restricted by the fact that his deletion had only
been characterized by fluorescence in situ hybridization (FISH)
analysis, and neither additional submicroscopic aberrations nor
molecular defects had been excluded.
The clinical features shared by all three patients reported here are
congenital heart defects (truncus arteriosus, pulmonary atresia,
atrial septal defect, and/or ventricular septal defect), brachydactyly,
mild intellectual disability, and facial dysmorphic signs (hyper-
telorism, broad nasal bridge, and thin upper lip vermilion)
(Table II). It appears likely that these findings were caused by
haploinsufficiency of one or several of the 19 genes in the common
deleted interval.
The only genes in this interval with known disease associations
are SMOC1 and ACTN1. Mutations in SMOC1 cause autosomal
recessive Waardenburg anophthalmia syndrome (also known as
microphthalmia with limb anomalies or ophthalmo-acromelic
syndrome, OMIM 206920) that is characterized by severe
eye malformations and syndactyly, poly- or oligodactyly and
synostosis of hands and feet [Abouzeid et al., 2011; Okada
et al., 2011; Raingern et al., 2011]. Although no abnormalities of
the fingers have been reported in heterozygous carriers of SMOC1
mutations so far, for want of a better candidate gene in this
interval one might speculate on a role of this gene in the formation
of brachydactyly. However, this assumption needs to be corrobo-
rated by similar observations in other patients with SMOC1
haploinsufficiency.
Dominant negativemissensemutations inACTN1were recently
reported in patients with congenital macrothrombocytopenia
(OMIM 615193) [Kunishima et al., 2013]. The three patients
reported here had neither bleeding disorders nor abnormal plate-
lets, which supports the assumption that haploinsufficiency of this
gene does not cause platelet disorders. ACTN1 encodes for alpha-
actinin-1 and is also expressed in cardiac muscle and in the brain,
which may indicate roles in normal heart structure and cognitive
function [Kremerskothen et al., 2002].
The product of the brain-expressed DCAF5 (WDR22) gene is
DDB1-and-CUL4 associated factor 5, which forms a component of
the ubiquitin ligase complex DDB1-CUL4 [Angers et al., 2006; Lee
andZhou, 2007].Amajor part of this complex isCUL4B,mutations
in which cause X-linked intellectual disability [Tarpey et al., 2007].
Haploinsufficiency of its binding partner DCAF5 might have
similar consequences on cognitive function.
In conclusion, the patients reported here provide detailed infor-
mation on the clinical consequences of deletions in chromosome
bands 14q24.1q24.3. Functional studies of the deleted genes and
reports of pointmutationswhich are likely to be identified bywhole
exome studies in the near future will elucidate the role of individual
genes for the clinical manifestations and the underlying biological
mechanisms.
ACKNOWLEDGMENTS
We thank the parents of the patients for their support. This study
made use of data generated by the DECIPHER Consortium. A full

TABLE I. RefSeq Genes in the del14q24.1q24.3 Region Covered by the Three Deletions
Gene Description/functionRAD51B RAD51 homolog B (S. cerevisiae) (OMIM 602948)ZFP36L1 ZFP36 ring finger protein-like 1 (OMIM 601064)ACTN1a actinin, alpha 1 (OMIM 102575)DCAF5a DDB1-CUL4 associated factor 5 (OMIM 603812)EXD2a Exonuclease 30–50 domain containing 2GALNTL1a UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase-like 1ERHa Enhancer of rudimentary homolog (Drosophila) (OMIM 601191)SLC39A9a Solute carrier family 39 (zinc transporter), member 9; ZIP9PLEKHDa Pleckstrin homology domain containing, family D (with coiled-coil domains) member 1CCDC177a Coiled-coil domain containing 177 (C14orf162)KIAA0247a KIAA0247LOC100289511a Uncharacterized LOC100289511 (LOC100289511), non-coding RNASRSF5a Serine/arginine-rich splicing factor 5 (OMIM 600914)SLC10A1a Solute carrier family 10 (sodium/bile acid cotransporter family) (OMIM 182396)SMOC1a SPARC related modular calcium binding 1 (OMIM 608488), homozygous mutations cause autosomal
recessive microphthalmia with limb anomalies syndrome (OMIM 206920)SLC8A3a Solute carrier family 8 (sodium/calcium exchanger), member 3 (NCX3) (OMIM 607991)ADAM21P1a ADAM metallopeptidase domain 21 pseudogene 1COX16a COX16 cytochrome c oxidase assembly homolog (S. cerevisiae)SYNJ2BPa Synaptojanin 2 binding protein (OMIM 609411)SYNJ2BP-COX16a SYNJ2BP-COX16 readthroughADAM21a ADAM metallopeptidase domain 21 (OMIM 603713)ADAM20 ADAM metallopeptidase domain 20 (OMIM 603712)ADAM20P1 ADAM metallopeptidase domain 20 pseudogene 1MED6 Mediator complex subunit 6 (OMIM 602984)TTC9 Tetratricopeptide repeat domain 9 (OMIM 610488)MAP3K9 Mitogen-activated protein kinase kinase kinase 9 (OMIM 600136)PCNX Pecanex homolog (Drosophila)SNORD56B Small nucleolar RNA, C/D box 56BLOC145474 Uncharacterized LOC145474 (LOC145474), non-coding RNASIPA1L1 Signal-induced proliferation-associated 1 like 1RGS6 Regulator of G-protein signaling 6 (OMIM 603894)DPF3 D4, zinc and double PHD fingers, family 3 (OMIM 601672)DCAF4 DDB1 and CUL4 associated factor 4ZFYVE1 Zinc finger, FYVE domain containing 1 (OMIM 605471)RBM25 RNA binding motif protein 25 (OMIM 612427)PSEN1 Presenilin 1 (OMIM 104311). Missense mutations are associated with early onset familial Alzheimer disease
type 3 (OMIM 607822), and possibly dilated cardiomyopathy (OMIM 613694). A frameshift mutation wasreported in a family with autosomal dominant familial acne inversa-3 (ACNINV3, OMIM 613737).
PAPLN Papilin, proteoglycan-like sulfated glycoproteinNUMB Numb homolog (Drosophila) (OMIM 603728)HEATR4 HEAT repeat containing 4C14orf169 Chromosome 14 open reading frame 169 (OMIM 611919)ACOT1 Acyl-CoA thioesterase 1 (OMIM 614313)ACOT2 Acyl-CoA thioesterase 2 (OMIM 609972)ACOT4 Acyl-CoA thioesterase 4 (OMIM 614314)ACOT6 Acyl-CoA thioesterase 6 (OMIM 614267)DNAL1 Dynein, axonemal, light chain 1 (OMIM 610062). A homozygous mutation has been reported in a family with
autosomal recessive primary ciliary dyskinesia-16 (CILD16, OMIM 614017).PNMA1 Paraneoplastic Ma antigen 1 (OMIM 604010)ELMSAN1 ELM2 and Myb/SANT-like domain containing 1PTGR2 Prostaglandin reductase 2 (PTGR2) (OMIM 608642)ZNF410 Zinc finger protein 410FAM161B Family with sequence similarity 161, member BCOQ6 Coenzyme Q6 homolog, monooxygenase (S. cerevisiae) (OMIM 614647). Mutations in this gene are associated
with autosomal recessive coenzyme Q10 deficiency-6 (COQ10D6, OMIM 614650).ENTPD5 Ectonucleoside triphosphate diphosphohydrolase 5 (OMIM 603162)CCDC176 Coiled-coil domain containing 176
624 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
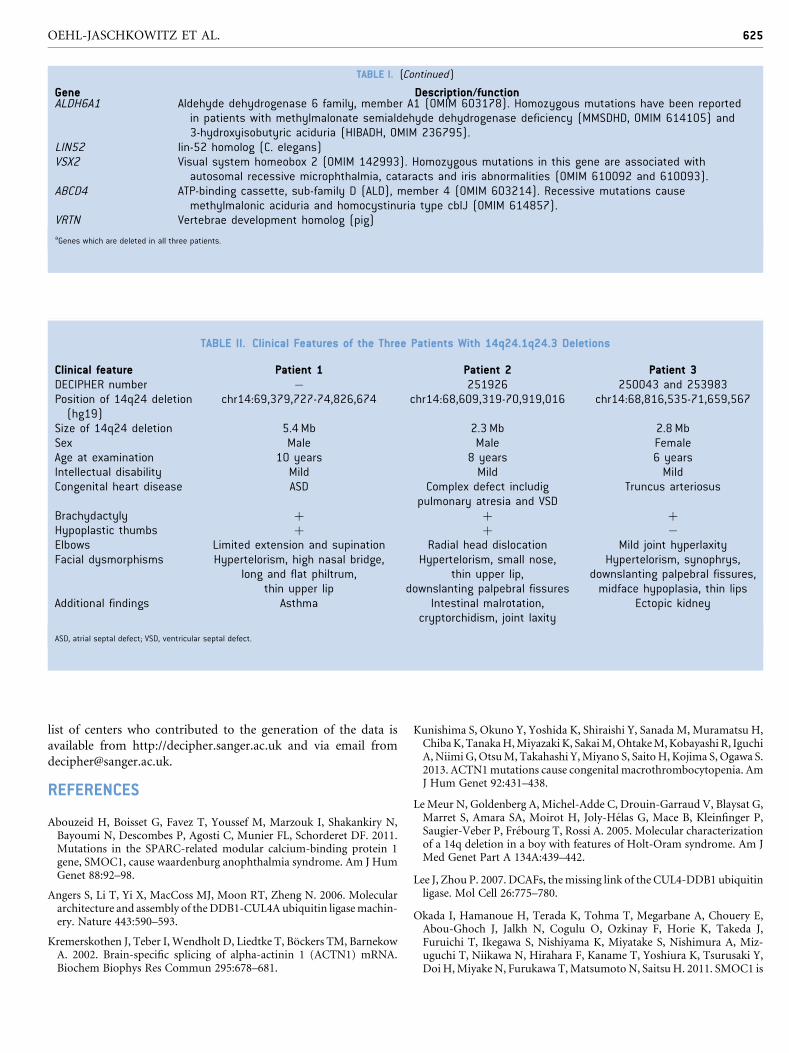
TABLE II. Clinical Features of the Three Patients With 14q24.1q24.3 Deletions
Clinical feature Patient 1 Patient 2 Patient 3DECIPHER number � 251926 250043 and 253983Position of 14q24 deletion(hg19)
chr14:69,379,727-74,826,674 chr14:68,609,319-70,919,016 chr14:68,816,535-71,659,567
Size of 14q24 deletion 5.4 Mb 2.3 Mb 2.8 MbSex Male Male FemaleAge at examination 10 years 8 years 6 yearsIntellectual disability Mild Mild MildCongenital heart disease ASD Complex defect includig
pulmonary atresia and VSDTruncus arteriosus
Brachydactyly þ þ þHypoplastic thumbs þ þ �Elbows Limited extension and supination Radial head dislocation Mild joint hyperlaxityFacial dysmorphisms Hypertelorism, high nasal bridge,
long and flat philtrum,thin upper lip
Hypertelorism, small nose,thin upper lip,
downslanting palpebral fissures
Hypertelorism, synophrys,downslanting palpebral fissures,midface hypoplasia, thin lips
Additional findings Asthma Intestinal malrotation,cryptorchidism, joint laxity
Ectopic kidney
ASD, atrial septal defect; VSD, ventricular septal defect.
TABLE I. (Continued)
Gene Description/functionALDH6A1 Aldehyde dehydrogenase 6 family, member A1 (OMIM 603178). Homozygous mutations have been reported
in patients with methylmalonate semialdehyde dehydrogenase deficiency (MMSDHD, OMIM 614105) and3-hydroxyisobutyric aciduria (HIBADH, OMIM 236795).
LIN52 lin-52 homolog (C. elegans)VSX2 Visual system homeobox 2 (OMIM 142993). Homozygous mutations in this gene are associated with
autosomal recessive microphthalmia, cataracts and iris abnormalities (OMIM 610092 and 610093).ABCD4 ATP-binding cassette, sub-family D (ALD), member 4 (OMIM 603214). Recessive mutations cause
methylmalonic aciduria and homocystinuria type cblJ (OMIM 614857).VRTN Vertebrae development homolog (pig)aGenes which are deleted in all three patients.
OEHL-JASCHKOWITZ ET AL. 625
list of centers who contributed to the generation of the data is
available from http://decipher.sanger.ac.uk and via email from
REFERENCES
Abouzeid H, Boisset G, Favez T, Youssef M, Marzouk I, Shakankiry N,Bayoumi N, Descombes P, Agosti C, Munier FL, Schorderet DF. 2011.Mutations in the SPARC-related modular calcium-binding protein 1gene, SMOC1, cause waardenburg anophthalmia syndrome. Am J HumGenet 88:92–98.
Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. 2006. Moleculararchitecture and assembly of theDDB1-CUL4A ubiquitin ligasemachin-ery. Nature 443:590–593.
Kremerskothen J, Teber I, Wendholt D, Liedtke T, Bockers TM, BarnekowA. 2002. Brain-specific splicing of alpha-actinin 1 (ACTN1) mRNA.Biochem Biophys Res Commun 295:678–681.
Kunishima S, Okuno Y, Yoshida K, Shiraishi Y, Sanada M, Muramatsu H,ChibaK,TanakaH,Miyazaki K, SakaiM,OhtakeM,Kobayashi R, IguchiA,Niimi G,OtsuM, Takahashi Y,Miyano S, SaitoH, Kojima S,Ogawa S.2013. ACTN1mutations cause congenital macrothrombocytopenia. AmJ Hum Genet 92:431–438.
Le Meur N, Goldenberg A, Michel-Adde C, Drouin-Garraud V, Blaysat G,Marret S, Amara SA, Moirot H, Joly-Helas G, Mace B, Kleinfinger P,Saugier-Veber P, Frebourg T, Rossi A. 2005. Molecular characterizationof a 14q deletion in a boy with features of Holt-Oram syndrome. Am JMed Genet Part A 134A:439–442.
Lee J, Zhou P. 2007. DCAFs, themissing link of the CUL4-DDB1 ubiquitinligase. Mol Cell 26:775–780.
Okada I, Hamanoue H, Terada K, Tohma T, Megarbane A, Chouery E,Abou-Ghoch J, Jalkh N, Cogulu O, Ozkinay F, Horie K, Takeda J,Furuichi T, Ikegawa S, Nishiyama K, Miyatake S, Nishimura A, Miz-uguchi T, Niikawa N, Hirahara F, Kaname T, Yoshiura K, Tsurusaki Y,DoiH,Miyake N, Furukawa T,MatsumotoN, SaitsuH. 2011. SMOC1 is

626 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
essential for ocular and limb development in humans and mice. Am JHum Genet 88:30–41.
Raingern J, van Beusekom E, Ramsay JK, McKie L, Al-Gazali L, PallottaR, Saponari A, Branney P, Fisher M, Morrison H, Bicknell L,Gautier P, Perry P, Sokhi K, Sexton D, Bardakjian TM, Schneider AS,Elcioglu N, Ozkinay F, Koenig R, Megarbane A, Semerci CN, Khan A,Zafar S, Hennekam R, Sousa SB, Ramos L, Garavelli L, Furga AS,Wischmeijer A, Jackson IJ, Gillessen-Kaesbach G, Brunner HG, Wiec-zorek D, van Bokhoven H, Fitzpatrick DR. 2011. Loss of the BMPantagonist, SMOC-1, causes Ophthalmo-acromelic (WaardenburgAnophthalmia) syndrome in humans andmice. PLoS Genet 7:e1002114.
Tarpey PS, Raymond FL, O’Meara S, Edkins S, Teague J, Butler A, Dicks E,StevensC,ToftC,AvisT, Barthorpe S, BuckG,Cole J,GrayK,HallidayK,Harrison R, Hills K, Jenkinson A, Jones D, Menzies A, Mironenko T,Perry J, Raine K, Richardson D, Shepherd R, Small A, Varian J, West S,Widaa S, Mallya U, Moon J, Luo Y, Holder S, Smithson SF, Hurst JA,Clayton-Smith J, Kerr B, Boyle J, Shaw M, Vandeleur L, Rodriguez J,Slaugh R, Easton DF, Wooster R, Bobrow M, Srivastava AK, StevensonRE, Schwartz CE, Turner G, Gecz J, Futreal PA, StrattonMR, PartingtonM. 2007. Mutations in CUL4B, which encodes a ubiquitin E3 ligasesubunit, cause an X-linkedmental retardation syndrome associated withaggressive outbursts, seizures, relative macrocephaly, central obesity,hypogonadism, pes cavus, and tremor. Am J Hum Genet 80:345–352.





























