Embed Size (px)
Citation preview

A Defect in Sodium-dependent Amino Acid Uptakein Diabetic Rabbit Peripheral NerveCorrection by an Aldose Reductase Inhibitor or myo-Inositol Administration
Douglas A. Greene, Sarah A. Lattimer, Patricia B. Carroll, John D. Fernstrom, and David N. FinegoldDiabetes Research Laboratories of the Department of Medicine, and the Western Psychiatric Institute and Clinic, School of Medicine,University of Pittsburgh, Pittsburgh, Pennsylvania 15261; and the Diabetes Research and Training Center and theDepartment of Internal Medicine of the University of Michigan, Ann Arbor, Michigan 48109
Abstract
A myo-inositol-related defect in nerve sodium-potassium ATP-ase activity in experimental diabetes has been suggested as apossible pathogenetic factor in diabetic neuropathy. Becausethe sodium-potassium ATPase is essential for other sodium-cotransport systems, and because myo-inositol-derived phos-phoinositide metabolites regulate multiple membrane trans-port processes, sodium gradient-dependent amino acid uptakewas examined in vitro in endoneurial preparations derived fromnondiabetic and 14-d alloxan diabetic rabbits. Untreated al-loxan diabetes reduced endoneurial sodium-gradient dependentuptake of the nonmetabolized amino acid 2-aminoisobutyricacid by > 50%. Administration of an aldose reductase inhibitorprevented reductions in both nerve myo-inositol content andendoneurial sodium-dependent 2-aminoisobutyric acid uptake.Myo-inositol supplementation that produced a transient phar-macological elevation in plasma myo-inositol concentration,but did not raise nerve myo-inositol content, reproduced theeffect of the aldose reductase inhibitor on endoneurial sodium-dependent 2-aminoisobutyric acid uptake. Phorbol myristateacetate, which acutely normalizes sodium-potassium ATPaseactivity in diabetic nerve, did not acutely correct 2-aminoiso-butyric uptake when added in vitro. These data suggest thatdepletion of a small myo-inositol pool may be implicated in thepathogenesis of defects in amino acid uptake in diabetic nerveand that rapid correction of sodium-potassium ATPase activitywith protein kinase Cagonists in vitro does not acutely normal-ize sodium-dependent 2-aminoisobutyric acid uptake. (J. Clin.Invest. 1990. 85:1657-1665.) amino acid transport * aminoacid metabolism * diabetic neuropathy * polyol pathway . sor-bitol pathway
Introduction
Derangements in peripheral nerve metabolism resulting frominsulin deficiency and/or hyperglycemia are associated withrapidly reversible abnormalities in nerve function in diabeticanimals, and are thought to contribute to progressively less
Address reprint requests to Dr. Greene, Room 5570, MSRB-2, Box0678, 1150 West Medical Center Drive, University of Michigan, AnnArbor, MI 48109.
Receivedfor publication 21 April 1989 and in revisedform 5 Sep-tember 1989.
reversible structurally based functional defects in animal andpossibly human diabetic neuropathy (1-3). A reduction inneural free myo-inositol (MI)' content that accompanies bothacute spontaneous and experimentally induced diabetes in ro-dents appears to underlie a cascade of metabolic and func-tional defects in nerve, including reduced sodium-potassiumATPase activity and impulse conduction velocity (2, 3). In theacutely diabetic rat, these abnormalities are completely pre-vented or reversed by MI supplementation or aldose reductaseinhibitors, both of which correct the fall in nerve MI level.Thus, increased polyol (sorbitol) pathway activity appears tocontribute to MI depletion and its consequences in diabeticnerve (2). The MI-related decrease in sodium-potassium ATP-ase activity has been proposed as the basis for the early andreversible slowing of nerve conduction velocity and paranodalaxonal swelling that are also reversed by aldose reductase in-hibitors or MI supplementation in spontaneously diabetic BBrats (3). In vitro exposure to protein kinase C activating phor-bol esters or functionally related phosphoinositide-deriveddiacylglycerols acutely normalizes the MI-related defect inouabain-sensitive ATPase activity in plasma membranes fromdiabetic rat sciatic nerve (4), as well as the diminished oua-bain-sensitive respiration (5) and ATPase activity (6) in nervepreparations from alloxan diabetic rabbits, thereby suggestingthat protein kinase C is a possible link between MI depletionand its associated sodium-potassium ATPase defect (2).
The relevance of these metabolic defects to the functionaland structural abnormalities underlying human diabetic pe-ripheral neuropathy (and perhaps other chronic complicationsof diabetes [7, 81) remains controversial. Aldose reductase in-hibitors improve nerve conduction in nonneuropathic diabeticpatients (9) and ameliorate the sorbitol accumulation (10, I 1)and structural fiber abnormalities (10) in the peripheral nervesof neuropathic diabetic subjects. MI administration also ap-pears to marginally improve nerve conduction in highly se-lected diabetic neuropathic patients (12-15), but not in moreheterogeneous populations of diabetic subjects (16, 17). (Theclinical efficacy of aldose reductase inhibitors or MI supple-mentation await large scale, long-term, randomized, con-trolled clinical trials with these drugs [10, 16, 17]). The con-flicting and fragmentary reports of measurements of wholenerve MI content in diabetic humans (11, 18-21) and thereported lack of an effect thereon of aldose reductase inhibitortherapy (1 1, 21) has recently been used to argue that altered
1. Abbreviations used in this paper: AIB, 2-aminoisobutyric acid;BCAA, branched-chain amino acid; KRB, Krebs Ringer bicarbonatebuffer; Kt, transport constant; MeAIB, 2-methyl-aminoisobutyric acid;MI, myo-inositol; PMA, phorbol myristate acetate; s, substrate con-centration; v, uptake velocity; Vm,,, maximum transport velocity.
Amino Acid Uptake in Diabetic Nerve 1657
J. Clin. Invest.© The American Society for Clinical Investigation, Inc.0021-9738/90/05/1657/09 $2.00Volume 85, May 1990, 1657-1665

MI metabolism is irrelevant to the pathogenesis of diabeticneuropathy (1 1, 22, 23). This view has been disputed (24, 25)partly on the basis of the fact that MI metabolism is nowthought to be highly compartmentalized, and that depletion ofa putative small metabolically labile MI pool by glucose duringin vitro incubation of aortic intima-media preparations im-pairs tissue function (including sodium-potassium ATPase ac-tivity) in the absence of detectable changes in tissue MI con-tent (26). One may therefore speculate that pathogeneticallysignificant depletion or repletion in nerve MI could occur indiabetes without detectable alterations in tissue MI content.This communication reports that MI treatment corrects a sor-bitol-related defect in amino acid transport in peripheral nervewithout correcting the diminished nerve MI content, suggest-ing that nerve MI content may be an insensitive indicator ofpolyol pathway-induced defects in MI metabolism.
Methods
Experimental animal model. Male, white NewZealand rabbits weigh-ing 1.5-2.0 kg were fed Wayne Rabbit Ration (Continental Grain Co.,Chicago, IL) and fasted overnight before the study or the induction ofalloxan diabetes as described previously (27). Alloxan monohydrate,90 mg/kg i.v. (Sigma Chemical Co, St. Louis, MO) in 0.73% sodiumchloride solution was administered by rapid injection to induce experi-mental diabetes. Nonfasting plasma glucose concentrations were mea-sured 48 h later, and rabbits with values < 300 mg/dl were excluded. 2wk thereafter, surviving animals were fasted overnight, and only rab-bits with plasma glucose concentrations 2 300 mg/dl were studied.Nondiabetic saline-injected control animals were maintained for 14 dunder similar environmental conditions (27). Sorbinil (Pfizer CentralResearch, Groton, CT) was suspended in water and administered bygavage daily at a dose of 15 mg/kg beginning 48 h after alloxan admin-istration. MI was similarly administered daily by gavage at a dose of100 mg/kg.
In vitro peripheral nerve preparations. Endoneurial preparationswere derived from the tibial division of the sciatic nerve as previouslydescribed in detail (28). After an overnight fast, diabetic and nondia-betic rabbits were sedated with diazepam, 2 mg/kg i.m. After inductionof anesthesia (sodium pentabarbital 24-36 mg/kg i.v.), a defined seg-ment of each sciatic nerve was surgically exposed, ligated, removedwith preservation of hemostasis, and transferred to a 125-ml erlen-meyer flask containing 70 mgof collagenase (Type I; Sigma ChemicalCo.) dissolved in 10 ml of 4.5% dialyzed defatted bovine serum albu-min (BSA) (Sigma fraction V powder) and 20 mMglucose in Krebs-Ringer bicarbonate buffer medium (KRB), pH 7.4 equilibrated with5%C02/95% 02 at 37°C. A separate portion of sciatic nerve proximalto the resected segment was removed and rapidly frozen in liquidnitrogen partially evacuated to its freezing point for analysis of MIcontent. After controlled digestion for 12-14 min, the ligated nervesegments were rinsed in 30 ml of similar medium containing aprotinin,200 kallikrein inactivation U/ml (Trasylol; FBA Pharmaceuticals,New York). A branch-free length of the major fascicle of the tibialdivision of each ligated sciatic nerve segment was further ligated andmicrodissected to produce the endoneurial preparation (28).
Standard incubation medium for the endoneurial preparation was4.5% dialyzed defatted BSAand 5 mMglucose in KRB, pH 7.4, equili-brated with 5%C02/95% 02 as previously described (28). The endo-neurial preparation was routinely equilibrated for 10 min in 3 ml ofstandard medium in a Dubnoff metabolic shaker set at 88 cycles/minand 37°C. The equilibrated endoneurial preparation was transferred toa new flask containing standard medium for incubation studies lastingas long as 2 h. Phorbol myristate acetate (PMA) was prepared indimethylsulfoxide (DMSO) (300 ug/ml) and 10 ul added to the incu-bation flasks for a final PMAconcentration of 2 nmol/ml for 1 h
(controls received 10 ul of DMSOalone). Other modifications in theincubation conditions for specific studies are described in the relevantportions of the text and figures.
2-Aminoisobutyric acid (AIB) uptake measurements. The preincu-bated endoneurial preparation was transferred to a new flask contain-ing standard medium with 0.5 ACi/ml of 1-["'Clmannitol 45 mCi/mmol, and 1 MCi/ml [3H]AIB 20 Ci/mmol (New England Nuclear,Boston, MA) to which unlabeled AIB or MeAIB was added to producethe specified concentrations. To assess the effects of sodium ion in theextracellular fluid on AIB uptake, sodium was replaced by equimolarconcentrations of choline during the last 15 min of preincubation andduring the exposure to isotopically labeled compounds. Sodium-de-pendent AIB uptake was defined as the difference in the intracellularuptake in the presence or absence of sodium under each experimentalcondition.
After brief, timed exposure to isotopically labeled medium theendoneurial preparation was rapidly rinsed in AIB-free standard me-dium, and frozen in liquid nitrogen. The frozen endoneurial prepara-tion was rapidly weighed on a microbalance, powdered in liquid nitro-gen, and homogenized in 6%perchloric acid. Incubation medium wasquantitatively transferred to a volumetric flask and diluted to a 5-mlvolume with water. Aliquots of diluted medium were deproteinizedwith equal volumes of 12% perchloric acid. After centrifugation at2,500 g for 15 min at 40C, the supernatant of the tissue or mediumperchloric acid extract was neutralized with KOHand recentrifuged. A0.4-ml aliquot of the resulting supernatant was added to 15 ml ofAquasol II (New England Nuclear) and counted for 3H and 14C in aliquid scintillation spectrometer (Packard Tri-Carb; Packard Instru-ment Co., Downers Grove, IL) with automatic external standard ratiosto correct for quenching. Cross-over of 14C and 3H radioactivity wasdetermined at varying automatic external standard ratios in vials con-taining varying amounts of acetone in water as a quenching agent.Recovery of [3H]AIB added to endoneurial homogenates was > 95%on a volume basis when compared with recovery after neutralizationand centrifugation of [3H]AIB added to 6%perchloric acid. The extra-cellular tissue space in milliliter per gram wet weight was calculated as:(dpm '4C/g wet weight of tissue) +. (dpm '4C/ml of incubation me-dium). Tissue and medium 3H was partially characterized by Iyophili-zation and thin-layer chromatography. Recovery of 3H from tissue ormedium filtrates postincubation was > 95% after lyophilization com-pared to fresh [3HJAIB suspended in 6% perchloric acid and neutral-ized with KOH, suggesting that 3H from AIB was not volatilized duringtissue incubation. Tritiated AIB-containing incubation medium ex-posed to endoneurial preparations for 2 h also had no detectable 3H20by microdiffusion compared to a medium blank incubated withouttissue. Water soluble 3H from AIB-exposed endoneurial filtrates co-migrated with [3H]AIB when chromatographed on cellulose thin-layerplates developed in butanol/acetic acid/water (25:4:10). Endoneurialouabain-sensitive uptake was essentially irreversible: there was no sig-nificant difference in the recovery of 3H effluxed over 10 min intoAIB-free medium from rinsed paired endoneurial preparations prela-beled for 10 min in 2.5 mM[3H]AIB in the presence or absence ofsodium for endoneurial preparations derived from either nondiabeticor untreated diabetic rabbits.
Analytical methods. Plasma glucose was determined in a BeckmanGlucose Analyzer II (Beckman Instruments, Fullerton, CA). Plasmaand sciatic nerve MI was determined by gas-liquid chromatography inprotein-free Somogyi filtrates of venous blood plasma or sciatic nervehomogenates and expressed per milliliter of plasma or per gram wetweight of whole nerve (29). The determinations of MI were performedon trimethylsilyl derivatives of Iyophilized filtrates containing alpha-o-methyl mannopyranoside as an internal standard in a Varian 3700gas-liquid chromatograph (Varian Associates, Inc., Palo Alto, CA) on a6 ft X 4 mm3% SE-30 Gaschrom Q glass column at 185°C with anitrogen carrier-gas flow rate of 40 ml/min using a flame ionizationdetector maintained at 250°C. Standard curves were determined daily,and the recovery of added MI to nerve or plasma samples consistentlyexceeded 95%.
1658 D. A. Greene, S. A. Lattimer, P. B. Carroll, J. D. Fernstrom, and D. N. Finegold

Plasma and sciatic nerve amino acids were quantitated using aBeckman Amino Acid Analyzer (model 6300; Beckman Instruments,Palo Alto, CA) equipped with a fluorometric detector. Plasma sampleswere precipitated using sulfosalicylic acid (50 mg/ml plasma) and cen-trifuged. 500 pl of each resulting supernatant were added to 1.0 ml of0.1 % trifluoroacetic acid containing 30% methanol and filteredthrough a Waters Sep-Pak (Waters Division, Millipore Corp., Milford,MA) as previously described (30), and 50 gl of the filtrate injecteddirectly onto the amino acid analyzer. Nerve samples were homoge-nized in 10 vol of 6%trichloracetic acid, centrifuged, and the superna-tants extracted three times with equal volumes of diethylether. Thefinal aqueous phases were lyophilized, reconstituted in 1 ml of 0.1%trifluoroacetic acid/methanol (80:20 vol/vol), filtered through WatersSep-Paks (30), lyophilized, reconstituted in 125 ,ul of starting buffer forthe amino acid analyzer, and small aliquots or dilutions injected.Computations were performed using the external standard method.
Statistical methods. Data were calculated as mean±SEM. Signifi-cance of difference between groups was analyzed by Student's two-tailed t test. In instances where comparisons were made among multi-ple groups, a more conservative test for significance would be to com-pare each Pvalue with 0.050 divided by the number of groups, which isthe Bonferroni adjustment to the P value. The differences betweenpaired samples were analyzed by the t test for paired comparisons.Kinetic analysis of AIB uptake was performed by the Eadie-Hofsteeand Lineweaver-Burk transformations of the Michaelis-Menten equa-tion. Linear regression analysis was performed by the method of leastsquares.
Results
AIB uptake in endoneurial preparations fromnondiabetic rabbitsLinearity with respect to time (Fig. 1). The intracellular uptakeof 2.5 mMAIB was linear with time for 15 min after preincu-bation in AIB-free medium for 1 h (Fig. 1) or 2 h (data notshown). When medium sodium was replaced with equimolarconcentrations of choline, AIB uptake remained linear withtime for 15 min, but uptake was reduced by about half. Paired10-min incubations in medium containing sodium or cholinewere routinely employed to estimate the initial rate of compos-ite (sodium-dependent + sodium-independent) and sodium-dependent AIB uptake.
Effect of medium AIB concentration (Fig. 2). Compositeintracellular AIB uptake was assessed during 10-min incuba-tions in the presence of sodium ion at AIB concentrationsbetween 0.5 and 12.5 mM. Composite AIB uptake increasedrapidly with AIB concentration below 2.5 mM, after whichAIB uptake continued to increase but at a slower rate (Fig. 2,solid line). This pattern was consistent with the presence of twotransport components, a higher affinity one that became fullysaturated and a lower affinity one, which demonstrated little orno saturation over the AIB concentration range tested.
Effect of sodium gradient (Fig. 2 and 3, Table I). To deter-mine which component constituted sodium-dependent co-transport, uptake of 0.5-12.5 mMAIB was studied in theabsence of extracellular sodium. The initial rate of AIB uptakeincreased linearly with concentration (r = 0.994, y-intercept= 33 nmol/kg per min, slope = 253 nmol/kg per min per mM;Fig. 2, dotted line), suggesting that sodium-independent up-take did not saturate within the concentration range tested.Sodium-dependent uptake velocity (composite uptake - so-dium-independent uptake) increased as a function of AIBconcentration, but at a progressively decreasing rate, suggest-ing a saturable uptake process (Fig. 2, dashed line). Therefore,
2000
EC/Lw 1500
F-D 1000co
I 500
00 5 10 15
TIME (minutes)
Figure 1. Endoneurial intracellular AIB uptake as a function of time.Paired endoneurial preparations were derived by microdissection andlimited collagenase digestion from rabbit sciatic nerve as described inMethods, and incubated for various time periods with 2.5 mM[3H]-AIB and tracer concentrations of I-['4C]mannitol (as a marker forextracellular space) in KRBpH 7.4 containing 4.5% dialyzed BSAand 5 mMglucose equilibrated with 5%C02/95% 02 at 370C in thepresence (solid line, filled circles) or absence (dotted line, triangles) ofsodium. Aliquots of neutralized perchloric acid filtrates of incubationmedium and endoneurial homogenates were counted for 3H and 14Cby liquid scintillation spectrometry, and endoneurial 3H accumula-tion, corrected for AIB specific activity in the incubation mediumand for extracellular contamination, was expressed per unit wetweight of endoneurial tissue.
AIB uptake consisted of two transport components, one so-dium dependent and saturable, and the other sodium indepen-dent and unsaturable below 12.5 mM.To confirm dependencyof AIB uptake on the transmembrane sodium gradient, so-
- Total AIB Uptake...... Na+-independent AIB Uptake
4000 - o--o Na+-dependent AIB Uptake
.k 3000 .*
-C
0~~~~~~~~~~~~~~~~~~Efi2000_ ..
> _ / .... ~~0 -----
1000 7 /.--e__-_o__
0 2000 4000 6000 8000 10,000 12,000S (PM)
Figure 2. Initial rate of intracellular AIB uptake by endoneurial prep-arations derived from nondiabetic animals as a function of mediumAIB concentration. Intracellular AIB uptake was measured at vary-ing medium AIB concentrations after 10-min incubations as de-scribed in Methods in the presence (solid line, filled circles) or ab-sence (dotted line, triangles) of sodium. Sodium-dependent uptake(dashed lines, open circles) was computed by subtracting the uptakein sodium-free medium from the composite uptake measured in thepresence of sodium. v, rate of intracellular AIB uptake. s, mediumAIB concentration. Each data point represents the mean of at leastthree determinations.
Amino Acid Uptake in Diabetic Nerve 1659
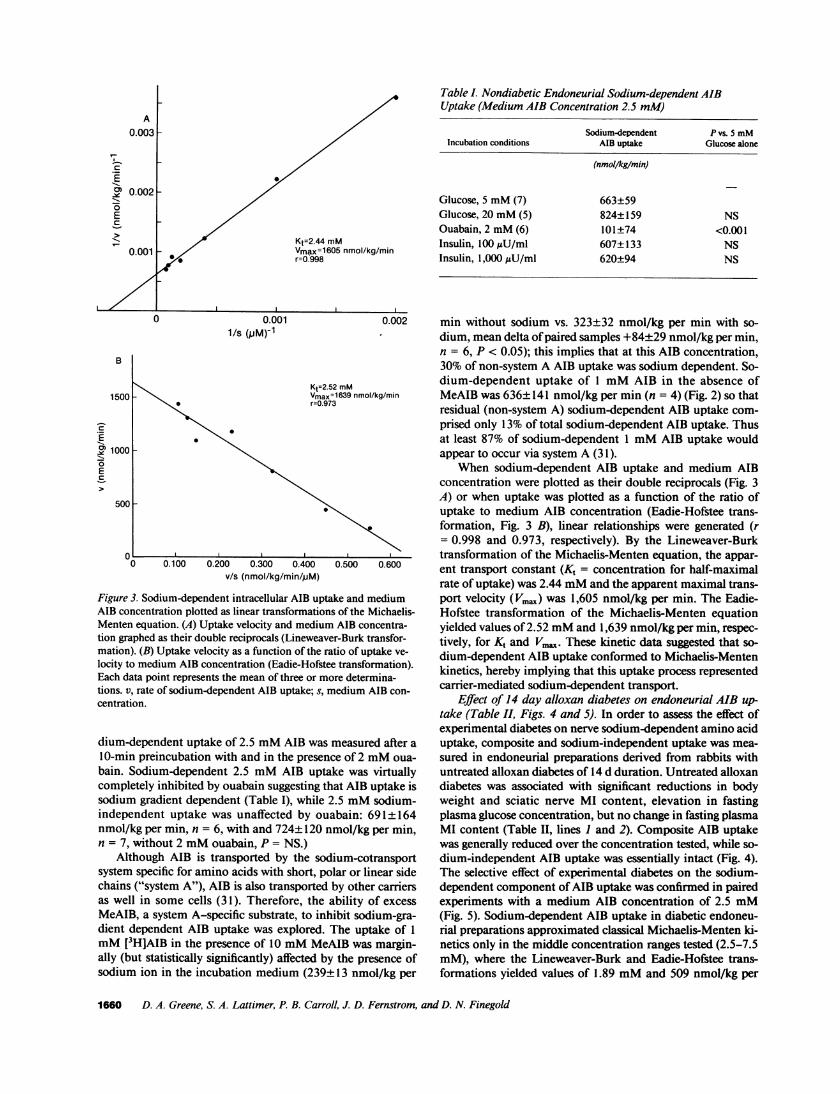
Kt=2.44 mMVmax=16O5 nmol/kg/minr=O.998
Table I. Nondiabetic Endoneurial Sodium-dependent AIBUptake (Medium AIB Concentration 2.5 mM)
Sodium-dependent P vs. 5 mMIncubation conditions AIB uptake Glucose alone
(nmol/kg/min)
Glucose, 5 mM(7) 663±59Glucose, 20 mM(5) 824±159 NSOuabain, 2 mM(6) 101±74 <0.001Insulin, 100 AU/ml 607±133 NSInsulin, 1,000 AU/ml 620±94 NS
B
0.0011/s (PM)-1
Kt=2.52 mMVmax=1639 nmol/kg/minr=0.973
1000 _
0
E
500
0 0.100 0.200 0.300 0.400 0.500 0.600v/s (nmol/kg/min/pM)
Figure 3. Sodium-dependent intracellular AIB uptake and mediumAIB concentration plotted as linear transformations of the Michaelis-Menten equation. (A) Uptake velocity and medium AIB concentra-tion graphed as their double reciprocals (Lineweaver-Burk transfor-mation). (B) Uptake velocity as a function of the ratio of uptake ve-
locity to medium AIB concentration (Eadie-Hofstee transformation).Each data point represents the mean of three or more determina-tions. v, rate of sodium-dependent AIB uptake; s, medium AIB con-centration.
dium-dependent uptake of 2.5 mMAIB was measured after a
10-min preincubation with and in the presence of 2 mMoua-
bain. Sodium-dependent 2.5 mMAIB uptake was virtuallycompletely inhibited by ouabain suggesting that AIB uptake issodium gradient dependent (Table I), while 2.5 mMsodium-independent uptake was unaffected by ouabain: 691±164nmol/kg per min, n = 6, with and 724±120 nmol/kg per min,n = 7, without 2 mMouabain, P = NS.)
Although AIB is transported by the sodium-cotransportsystem specific for amino acids with short, polar or linear sidechains ("system A"), AIB is also transported by other carriersas well in some cells (31). Therefore, the ability of excessMeAIB, a system A-specific substrate, to inhibit sodium-gra-dient dependent AIB uptake was explored. The uptake of 1
mM(3H]AIB in the presence of 10 mMMeAIB was margin-
ally (but statistically significantly) affected by the presence ofsodium ion in the incubation medium (239±13 nmol/kg per
min without sodium vs. 323±32 nmol/kg per min with so-
dium, mean delta of paired samples +84±29 nmol/kg per min,n = 6, P < 0.05); this implies that at this AIB concentration,30% of non-system A AIB uptake was sodium dependent. So-dium-dependent uptake of 1 mMAIB in the absence ofMeAIB was 636±141 nmol/kg per min (n = 4) (Fig. 2) so thatresidual (non-system A) sodium-dependent AIB uptake com-prised only 13% of total sodium-dependent AIB uptake. Thusat least 87% of sodium-dependent 1 mMAIB uptake wouldappear to occur via system A (31).
When sodium-dependent AIB uptake and medium AIBconcentration were plotted as their double reciprocals (Fig. 3A) or when uptake was plotted as a function of the ratio ofuptake to medium AIB concentration (Eadie-Hofstee trans-formation, Fig. 3 B), linear relationships were generated (r= 0.998 and 0.973, respectively). By the Lineweaver-Burktransformation of the Michaelis-Menten equation, the appar-ent transport constant (K, = concentration for half-maximalrate of uptake) was 2.44 mMand the apparent maximal trans-port velocity (Vma.) was 1,605 nmol/kg per min. The Eadie-Hofstee transformation of the Michaelis-Menten equationyielded values of 2.52 mMand 1,639 nmol/kg per min, respec-tively, for Kt and Vm.x. These kinetic data suggested that so-dium-dependent AIB uptake conformed to Michaelis-Mentenkinetics, hereby implying that this uptake process representedcarrier-mediated sodium-dependent transport.
Effect of 14 day alloxan diabetes on endoneurial AIB up-take (Table II, Figs. 4 and 5). In order to assess the effect ofexperimental diabetes on nerve sodium-dependent amino aciduptake, composite and sodium-independent uptake was mea-sured in endoneurial preparations derived from rabbits withuntreated alloxan diabetes of 14 d duration. Untreated alloxandiabetes was associated with significant reductions in bodyweight and sciatic nerve MI content, elevation in fastingplasma glucose concentration, but no change in fasting plasmaMI content (Table II, lines 1 and 2). Composite AIB uptakewas generally reduced over the concentration tested, while so-dium-independent AIB uptake was essentially intact (Fig. 4).The selective effect of experimental diabetes on the sodium-dependent component of AIB uptake was confirmed in pairedexperiments with a medium AIB concentration of 2.5 mM(Fig. 5). Sodium-dependent AIB uptake in diabetic endoneu-rial preparations approximated classical Michaelis-Menten ki-netics only in the middle concentration ranges tested (2.5-7.5mM), where the Lineweaver-Burk and Eadie-Hofstee trans-formations yielded values of 1.89 mMand 509 nmol/kg per
1660 D. A. Greene, S. A. Lattimer, P. B. Carroll, J. D. Fernstrom, and D. N. Finegold
A
ECD
-I0EC-
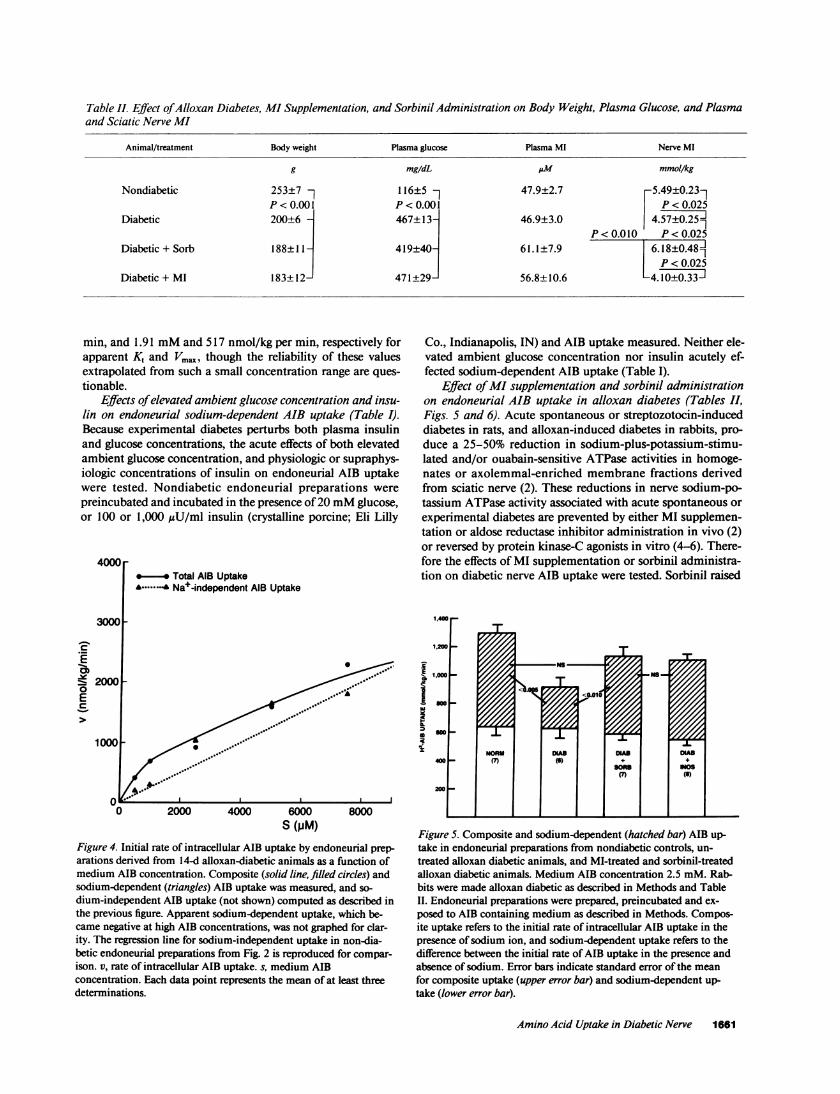
Table II. Effect of Alloxan Diabetes, MI Supplementation, and Sorbinil Administration on Body Weight, Plasma Glucose, and Plasmaand Sciatic Nerve MI
Animal/treatment Body weight Plasma glucose Plasma MI Nerve MI
g mg/dL AM mmol/kg
Nondiabetic 253±7 116±5 - 47.9±2.7 5.49±0.23-P<0.001 P<0.001 P<0.025
Diabetic 200±6 A 467±13 46.9±3.0 4.57±0.25fIIP<0.010 P<0.025
Diabetic + Sorb 188±11 - 419±40j 61.1±7.9 6.18±0.481P < 0.025
Diabetic + MI 183+ 12- 471 +29- 56.8+±10.6 -4.10±0.33J
min, and 1.91 mMand 517 nmol/kg per min, respectively forapparent K, and Vm,,a, though the reliability of these valuesextrapolated from such a small concentration range are ques-tionable.
Effects of elevated ambient glucose concentration and insu-lin on endoneurial sodium-dependent AIB uptake (Table I).Because experimental diabetes perturbs both plasma insulinand glucose concentrations, the acute effects of both elevatedambient glucose concentration, and physiologic or supraphys-iologic concentrations of insulin on endoneurial AIB uptakewere tested. Nondiabetic endoneurial preparations werepreincubated and incubated in the presence of 20 mMglucose,or 100 or 1,000 uU/ml insulin (crystalline porcine; Eli Lilly
4000*- *Total AIB Uptake
........ Nae-independent AIB Uptake
3000
._
Z__2000EC
0 2000 4000 6000 8000S (PM)
Figure 4. Initial rate of intracellular AIB uptake by endoneurial prep-arations derived from 14-d alloxan-diabetic animals as a function ofmedium AIB concentration. Composite (solid line, filled circles) andsodium-dependent (triangles) AIB uptake was measured, and so-dium-independent AIB uptake (not shown) computed as described inthe previous figure. Apparent sodium-dependent uptake, which be-came negative at high AIB concentrations, was not graphed for clar-ity. The regression line for sodium-independent uptake in non-dia-betic endoneurial preparations from Fig. 2 is reproduced for compar-ison. v, rate of intracellular AIB uptake. s, medium AIBconcentration. Each data point represents the mean of at least threedeterminations.
Co., Indianapolis, IN) and AIB uptake measured. Neither ele-vated ambient glucose concentration nor insulin acutely ef-fected sodium-dependent AIB uptake (Table I).
Effect of MI supplementation and sorbinil administrationon endoneurial AIB uptake in alloxan diabetes (Tables II,Figs. S and 6). Acute spontaneous or streptozotocin-induceddiabetes in rats, and alloxan-induced diabetes in rabbits, pro-duce a 25-50% reduction in sodium-plus-potassium-stimu-lated and/or ouabain-sensitive ATPase activities in homoge-nates or axolemmal-enriched membrane fractions derivedfrom sciatic nerve (2). These reductions in nerve sodium-po-tassium ATPase activity associated with acute spontaneous orexperimental diabetes are prevented by either MI supplemen-tation or aldose reductase inhibitor administration in vivo (2)or reversed by protein kinase-C agonists in vitro (4-6). There-fore the effects of MI supplementation or sorbinil administra-tion on diabetic nerve AIB uptake were tested. Sorbinil raised
1,400
1,00 _P"Om
NS1,000 N
NORM Mas DiA" DAls400 (7 (6) + +
sons NMo(7)()
200
FigureS5. Composite and sodium-dependent (hatched bar) AIB up-take in endoneurial preparations from nondiabetic controls, un-treated alloxan diabetic animals, and MI-treated and sorbinil-treatedalloxan diabetic animals. Medium AIB concentration 2.5 mM. Rab-bits were made alloxan diabetic as described in Methods and TableII. Endoneurial preparations were prepared, preincubated and ex-posed to AIB containing medium as described in Methods. Compos-ite uptake refers to the initial rate of intracellular AIB uptake in thepresence of sodium ion, and sodium-dependent uptake refers to thedifference between the initial rate of AIB uptake in the presence andabsence of sodium. Error bars indicate standard error of the meanfor composite uptake (upper error bar) and sodium-dependent up-take (lower error bar).
Amino Acid Uptake in Diabetic Nerve 1661
300r
*- 200E
coEcn'o 100a-
I-
0O 1gavage100 mg/kg Ml
2 3 4 5Hours After Gavage
6 24
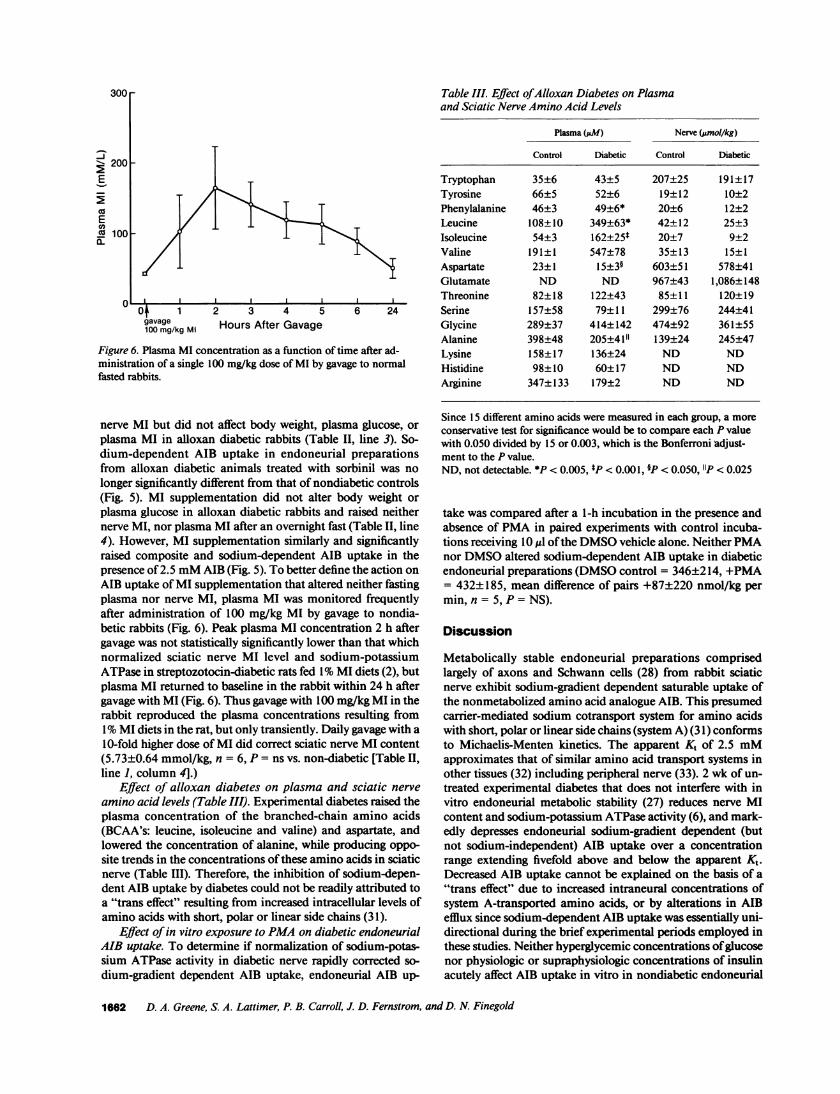
Figure 6. Plasma MI concentration as a function of time after ad-ministration of a single 100 mg/kg dose of MI by gavage to normalfasted rabbits.
nerve MI but did not affect body weight, plasma glucose, orplasma MI in alloxan diabetic rabbits (Table II, line 3). So-dium-dependent AIB uptake in endoneurial preparationsfrom alloxan diabetic animals treated with sorbinil was nolonger significantly different from that of nondiabetic controls(Fig. 5). MI supplementation did not alter body weight orplasma glucose in alloxan diabetic rabbits and raised neithernerve MI, nor plasma MI after an overnight fast (Table II, line4). However, MI supplementation similarly and significantlyraised composite and sodium-dependent AIB uptake in thepresence of 2.5 mMAIB (Fig. 5). To better define the action onAIB uptake of MI supplementation that altered neither fastingplasma nor nerve MI, plasma MI was monitored frequentlyafter administration of 100 mg/kg MI by gavage to nondia-betic rabbits (Fig. 6). Peak plasma MI concentration 2 h aftergavage was not statistically significantly lower than that whichnormalized sciatic nerve MI level and sodium-potassiumATPase in streptozotocin-diabetic rats fed 1%MI diets (2), butplasma MI returned to baseline in the rabbit within 24 h aftergavage with MI (Fig. 6). Thus gavage with 100 mg/kg MI in therabbit reproduced the plasma concentrations resulting from1%MI diets in the rat, but only transiently. Daily gavage with a10-fold higher dose of MI did correct sciatic nerve MI content(5.73±0.64 mmol/kg, n = 6, P = ns vs. non-diabetic [Table II,line 1, column 4].)
Effect of alloxan diabetes on plasma and sciatic nerveamino acid levels (Table III). Experimental diabetes raised theplasma concentration of the branched-chain amino acids(BCAA's: leucine, isoleucine and valine) and aspartate, andlowered the concentration of alanine, while producing oppo-site trends in the concentrations of these amino acids in sciaticnerve (Table III). Therefore, the inhibition of sodium-depen-dent AIB uptake by diabetes could not be readily attributed toa "trans effect" resulting from increased intracellular levels ofamino acids with short, polar or linear side chains (31).
Effect of in vitro exposure to PMAon diabetic endoneurialAIB uptake. To determine if normalization of sodium-potas-sium ATPase activity in diabetic nerve rapidly corrected so-dium-gradient dependent AIB uptake, endoneurial AIB up-
Table III. Effect of Alloxan Diabetes on Plasmaand Sciatic Nerve Amino Acid Levels
Plasma (jW) Nerve (jumol/kg)
Control Diabetic Control Diabetic
Tryptophan 35±6 43±5 207±25 191±17Tyrosine 66±5 52±6 19±12 10±2Phenylalanine 46±3 49±6* 20±6 12±2Leucine 108±10 349±63* 42±12 25±3Isoleucine 54±3 162±25* 20±7 9±2Valine 191±1 547±78 35±13 15±1Aspartate 23±1 15±3§ 603±51 578±41Glutamate ND ND 967±43 1,086± 148Threonine 82±18 122±43 85±11 120±19Serine 157±58 79±11 299±76 244±41Glycine 289±37 414±142 474±92 361±55Alanine 398±48 205±4 1" 139±24 245±47Lysine 158±17 136±24 ND NDHistidine 98±10 60±17 ND NDArginine 347± 133 179±2 ND ND
Since 15 different amino acids were measured in each group, a moreconservative test for significance would be to compare each P valuewith 0.050 divided by 15 or 0.003, which is the Bonferroni adjust-ment to the P value.ND, not detectable. *P < 0.005, *P < 0.00 1, §P < 0.050, '1P < 0.025
take was compared after a 1-h incubation in the presence andabsence of PMAin paired experiments with control incuba-tions receiving 10 jd of the DMSOvehicle alone. Neither PMAnor DMSOaltered sodium-dependent AIB uptake in diabeticendoneurial preparations (DMSOcontrol = 346±214, +PMA= 432±185, mean difference of pairs +87±220 nmol/kg permin, n = 5, P = NS).
Discussion
Metabolically stable endoneurial preparations comprisedlargely of axons and Schwann cells (28) from rabbit sciaticnerve exhibit sodium-gradient dependent saturable uptake ofthe nonmetabolized amino acid analogue AIB. This presumedcarrier-mediated sodium cotransport system for amino acidswith short, polar or linear side chains (system A) (31) conformsto Michaelis-Menten kinetics. The apparent Kt of 2.5 mMapproximates that of similar amino acid transport systems inother tissues (32) including peripheral nerve (33). 2 wk of un-treated experimental diabetes that does not interfere with invitro endoneurial metabolic stability (27) reduces nerve MIcontent and sodium-potassium ATPase activity (6), and mark-edly depresses endoneurial sodium-gradient dependent (butnot sodium-independent) AIB uptake over a concentrationrange extending fivefold above and below the apparent K,.Decreased AIB uptake cannot be explained on the basis of a"trans effect" due to increased intraneural concentrations ofsystem A-transported amino acids, or by alterations in AIBefflux since sodium-dependent AIB uptake was essentially uni-directional during the brief experimental periods employed inthese studies. Neither hyperglycemic concentrations of glucosenor physiologic or supraphysiologic concentrations of insulinacutely affect AIB uptake in vitro in nondiabetic endoneurial
1662 D. A. Greene, S. A. Lattimer, P. B. Carroll, J. D. Fernstrom, and D. N. Finegold

preparations at the K,. Administration of an aldose reductaseinhibitor that normalizes nerve MI levels corrects the reduc-tion in sodium-gradient dependent AIB uptake after 2 wk ofexperimental diabetes. MI supplementation (that in the dia-betic rat does not alter nerve polyol pathway intermediates[29]) also corrects sodium-gradient dependent AIB uptake,implicating MI depletion in this transport defect in diabeticperipheral nerve. However, the MI supplementation that re-produced the effect of sorbinil on sodium-dependent AIB up-take did not significantly raise the overall tissue MI content ofdiabetic rabbit sciatic nerve. As discussed below, this apparentparadox is consistent with the concept of Winegrad and co-workers who postulate that depletion or repletion of a specificsmall metabolically labile pool of tissue MI which is poorlyreflected by changes in overall tissue MI content may be par-ticularly relevant to abnormalities in membrane transport indiabetic tissues (8, 26). This concept was recently extended torenal tubular transport by the suggestion that impaired renaltubular sodium-dependent ascorbate transport in the strepto-zotocin-diabetic rat is corrected by dietary MI supplementa-tion without any concomitant change in renal cortical or med-ullary MI content (34).
Diabetes raises plasma BCAA levels in the rat (35), man(36), and the rabbit. The parallel increases in plasma and brainBCAA in the diabetic rat (35) contrast with the paradoxicaldecrease in diabetic rabbit peripheral nerve BCAA level,though this difference did not attain statistical significance.While species effects cannot be excluded, these divergent re-sponses in brain and peripheral nerve BCAA's to the raisedplasma BCAAlevels in diabetes more likely reflects a funda-mental differences in brain and nerve amino acid transport:system A transport occurs in peripheral nerve but not at theblood-brain barrier (37) where sodium-independent bidirec-tional system L would facilitate equilibration of brain andplasma BCAA levels, while the uptake of other large neutralamino acids would be reduced in diabetes due to competitiveinhibition by BCAA's (37). In peripheral nerve, active system-A uptake would greatly exceed influx via the bidirectionalsystem L, which would promote efflux of amino acids trans-ported in common and competitively by both systems (38).(The failure to detect a fall in system A amino acid contentdespite diminished AIB uptake in diabetic nerve may reflectthe large tissue pool of system A amino acids [38].) System Lamino acids (BCAA's, tyrosine and phenylalanine) tended tobe lower in diabetic nerve, though not statistically significantlyso, consistent with a reduction in system A amino acid trans-port and the much smaller tissue pool size of system L aminoacids. Thus the observed effects of diabetes on amino acidcontent in diabetic rabbit nerve are not inconsistent with asignificant reduction in vivo in sodium-dependent system Aamino acid transport in diabetic nerve.
MI regulates intracellular metabolism primarily through itsincorporation into inositol-containing membrane phospho-lipids that serve as precursors for phospholipase-C-generatedinositol-phosphate and diacylglycerol second messengers (39).The known relationship(s) between phosphoinositide signalingand membrane transport are widespread and complex, in-volving highly regulated inositol-1,4,5-trisphosphate- and ino-sitol- 1,4,5-tetrakisphosphate-mediated intracellular (40, 41)and transplasmalemmal (40) calcium fluxes, and diacyl-glyc-erol-mediated stimulation of phospholipid-and-calcium-de-pendent protein kinase C (39). Protein kinase C agonists affect
growth and differentiation in some cells, and have multipleeffects on membrane transport such as the induction of theglucose transporter gene (42). In LLC-PK' cells, PKC-me-diated activation of system A sodium-dependent amino acidtransport is demonstrable primarily in confluent cells withconstitutively low amino acid uptake but not in dividing cellswith constitutively high amino acid transport (43). The sor-binil- and MI-sensitive defect in amino acid transport in dia-betic peripheral nerve may be a direct consequence of theestimated fourfold rise in intraaxonal sodium (if the AIB trans-port defect resides in the axonal compartment), or a similarreduction in the transmembrane electrochemical sodium-gra-dient in the Schwann cell, both of which may reflect impairedsodium-potassium ATPase activity (3). Alternatively, MI de-pletion in diabetic nerve may independently reduce sodium-potassium ATPase activity and sodium-dependent AIB up-take, the latter possibly via an indirect mechanism analogousto that described in quiescent LLC-PK' cells (43). The fact thatPMA, a protein kinase C agonist that rapidly corrects the MI-related reduction in sodium-potassium ATPase in diabeticrabbit and rat nerve (4-6), does not acutely improve sodium-gradient dependent AIB uptake in vitro in endoneurial prepa-rations from diabetic rabbits implies that these two processesare independent, or that the reestablishment of the normalelectrochemical sodium gradient is delayed once sodium-po-tassium ATPase activity is restored. A third possibility, thatimpaired sodium-gradient dependent AIB uptake in diabeticperipheral nerve represents a consequence of MI depletion atsome distant site (perhaps the anterior horn or dorsal rootganglia neuronal cell body) that is fully corrected by MI-sup-plementation, cannot be rigorously excluded. In any case, bothsodium-dependent AIB uptake and sodium-potassium ATP-ase activity appear to be reduced in diabetic nerve as a result ofunderlying MI depletion. These two defects may be linkedserially or in parallel, and may involve one or more metabolicpools of MI, and/or distinct but phosphoinositide-related pro-cesses (39). All of these possibilities imply that metabolism of aspecific MI pool or pools rather than the total tissue MI con-tent mediate some of the effects of MI depletion and repletionon nerve metabolism such as the defect in system A aminotransport, as has been suggested by Winegrad and his col-leagues (8, 26).
Although rapidly reversible slowing of nerve conduction inhuman (9, 44) and animal (45) diabetes theoretically may beattributable in part to acute alterations in nerve MI metabo-lism, most but not all slowing of nerve conduction in estab-lished diabetes and diabetic neuropathy is thought to reflectnerve fiber loss, atrophy, and/or demyelination (46). Reducedleucine incorporation into myelin in diabetic rodent nerve hasbeen postulated to be a contributing factor to peripheral nervehypomyelination in diabetes (47, 48). This defect may reflectimpaired amino acid uptake by diabetic peripheral nerve invitro (while this defect appeared to be partially insulin sensi-tive, pharmacological concentrations of insulin far in excess ofthose employed in the present report were required (46)). Re-duced amino acid uptake by dorsal root ganglia from thestreptozotocin diabetic rat is associated with diminished so-dium-potassium ATPase activity, and has been invoked in theaccompanying axonal "dwindling" or "atrophy" (49, 50).Hence chronically reduced MI- and sorbinil-responsive so-dium-dependent amino acid uptake in axons and/or Schwanncells may be important links between acute metabolic derange-
Amino Acid Uptake in Diabetic Nerve 1663

ments and chronic structural defects in diabetic nerve, therebyproviding a plausible mechanism by which longstanding alter-ations in nerve MI metabolism induced by hyperglycemiamight contribute to the pathogenesis of the structural hall-marks of chronic diabetic neuropathy.
In summary, important polyol-pathway related abnormali-ties in MI metabolism in diabetic peripheral nerve, such as thatresponsible for impaired sodium-amino acid cotransport, maybe corrected by MI supplementation that fails to detectablyalter the concurrent decrease in whole-nerve MI content. Thismost likely reflects the fact that the metabolically active, physi-ologically important MI pools in the peripheral nervous sys-tem may represent only a small fraction of, and/or be poorlyreflected by, the composite tissue MI content of peripheralnerve. Therefore, the presence or absence of alterations inwhole tissue MI content in response to diabetes or metabolicintervention are poor indicators of the presence or absence ofphysiologically and perhaps pathogenetically important ab-normalities in MI or phosphoinositide metabolism.
Acknowledgments
The authors gratefully acknowledge the technical assistance of CarolKorbanic, Barbara Thornton, and Lorraine Weber, and advice of theBiostatistics Core of the Michigan Diabetes Research and TrainingCenter. This research was supported in part by the U. S. Public HealthService (RO1-DK26862 and DK38304 [Dr. Greene], and HD-24730[Dr. Fernstrom]), the American Diabetes Association (Dr. Finegold),and the Harry Soffer Memorial Research Fund of the University ofPittsburgh (Dr. Greene).
References
1. Committee on Health Care Issues, American Neurological Asso-ciation. 1986. Does improved control of glycemia prevent or amelio-rate diabetic neuropathy? Ann. Neurol. 19:288-290.
2. Greene, D. A., S. A. Lattimer, and A. A. F. Sima. 1988. Patho-genesis and prevention of diabetic neuropathy. Diabetes/MetabolismRev. 4:201-221.
3. Brismar, T., A. A. F. Sima, and D. A. Greene. 1987. Reversibleand irreversible nodal dysfunction in diabetic neuropathy. Ann.Neurol. 21:504-507.
4. Kim, J., and D; A. Greene. 1987. Correction of the myo-inosi-tol-related Na/K-ATPase defect in axolemmal-enriched, protein ki-nase C-containing isolated membranes from diabetic rat peripheralnerve by phorbol myristate. Clin. Res. 35:624A.
5. Lattimer, S. A., and D. A. Greene. 1987. Protein kinase C ago-nists rapidly correct impaired Na/K-ATPase in diabetic nerve in vitro.Diabetes. 36:75A. (Abstr.)
6. Lattimer, S. A., A. A. F. Sima, and D. A. Greene. 1989. Rapid invitro correction of impaired (Na,K)-ATPase in diabetic peripheralnerve by protein kinase-C agonists: suggested phosphoinositide regula-tion and integration of neural (Na,K)-ATPase. Am. J. Physiol. 256(Endocrinol. Metab. 19): E264-E269.
7. Greene, D. A., S. A. Lattimer, and A. A. F. Sima. 1987. Sorbitol,phosphoinositides and the sodium-potassium ATPase in the pathogen-esis of diabetic complications. N. Engl. J. Med. 316:599-606.
8. Winegrad, A. I. 1987. Banting lecture 1986. Does a commonmechanism induce the diverse complications of diabetes? Diabetes.36:396-406.
9. Judzewitsch, R. G., J. B. Jaspan, K. S. Polonsky, C. R. Weinberg,J. B. Halter, E. Halar, M. A. Pfeifer, C. Vukadinovic, L. Bernstein, M.Schneider, et al. 1983. Aldose reductase inhibition improves nerveconduction velocity in diabetic patients. N. Engl. J. Med. 308:119-125.
10. Sima, A. A. F., V. Bril, V. Nathaniel, T. A. J. McEwen, M. B.Brown, S. A. Lattimer, and D. A. Greene. 1988. Regeneration andrepair of myelinated fibers in sural nerve biopsies from patients withdiabetic neuropathy treated with sorbinil, an investigational aldosereductase inhibitor. N. Engl. J. Med. 319(9):548-555.
11. Dyck, P. J., B. R. Zimmerman, T. H. Vilen, S. R. Minnerath,J. L. Karnes, J. K. Yao, and J. F. Poduslo. 1988. Nerve glucose,fructose, sorbitol, myo-inositol, and fiber degeneration and regenera-tion in diabetic neuropathy. 319:542-548.
12. Salway, J. G., J. A. Finnegan, D. Barnett, L. Whitehead, A.Karunanayka, and R. B. Payne. 1978. Effect of myoinositol on periph-eral nerve function in diabetes. Lancet. ii: 1282-1294.
13. Clements, R. S., Jr., B. Vourganti, T. Kuba, S. J. Oh, and B.Darnell. 1979. Dietary myo-inositol intake and peripheral nerve func-tion in diabetic neuropathy. Metab. Clin. Exp. 28:477-483.
14. Greene, D. A., M. J. Brown, S. N. Braunstein, S. S. Schwartz,A. K. Asbury, and A. I. Winegrad. 1981. Comparison of clinical courseand sequential electrophysiological tests in diabetics with symptomaticpolyneuropathy and its implications for clinical trials. Diabetes.30:139-147.
15. Clements, R. S., Jr. 1985. Review of myoinositol and sorbinilstudies. Clin. Physiol. 5(Suppl. 5):90-93.
16. Gregerson, G., H. Borsting, P. Theil, and C. Servo. 1978.Myoinositol and function of peripheral nerve in human diabetics: acontrolled clinical trial. Acta Neurol. Scand. 58:241-248.
17. Gregersen, G., B. Bertelsen, H. Harbo, E. Larsen, J. R. Ander-son, A. Helles, M. Schmiegelow, and J. E. J. Christensen. 1983. Oralsupplementation of myo-inositol: effects on peripheral nerve functionin human diabetics and on the concentration in plasma, erythrocytes,urine and muscle tissue in human diabetics and normals. Acta Neurol.Scand. 67:164-172.
18. Ward, J. D. 1973. The polyol pathway in the neuropathy ofearly diabetes. In Early Diabetes. R. A. Camerini-Davalos and H. S.Cole, editors. Academic Press, NewYork. 425-432.
19. Mayhew, J. A., K. R. W. Gillon, and J. N. Hawthorne. 1983.Free and lipid inositol, sorbitol and sugars obtained post mortem fromdiabetic and control subjects. Diabetologia. 24:13-15.
20. Dyck, P. J., W. R. Sherman, L. M. Hallcher, F. J. Service, P. C.O'Brien, L. A. Grina, P. J. Palumbo, and C. J. Swanson. 1980. Humandiabetic endoneurial sorbitol, fructose and myo-inositol related tosural nerve morphometry. Ann. Neurol. 8:590-596.
2 1. Greene, D. A., V. Bril, S. A. Lattimer, and A. A. F. Sima. 1987.Correction of myo-inositol depletion in diabetic human sural nerve bytreatment with an aldose reductase inhibitor. Diabetes. 36(Suppl.1):86A. (Abstr.)
22. Asbury, A. K. 1988. Understanding diabetic neuropathy. Edi-torial. N. Engl. J. Med. 319:577-578.
23. Dyck, P. J., B. R. Zimmerman, T. H. Vilen, S. R. Minnerath,J. L. Karnes, J. K. Yao, and J. F. Poduslo. 1988. Letter to the Editor. N.Engl. J. Med. 320:59.
24. Winegrad, A. I., D. A. Simmons, and D. B. Martin. 1988. Letterto the Editor. N. Engl. J. Med. 320:57-58.
25. Sima, A. A. F., and D. A. Greene. 1988. Letter to the Editor. N.Engl. J. Med. 320:58-60.
26. Simmons, D. A., E. F. 0. Kern, A. I. Winegrad, and D. B.Martin. 1986. Basal phosphatidylinositol turnover controls aorticNa+/K+-ATPase. J. Clin. Invest. 77:503-513.
27. Greene, D. A., and Winegrad, A. I. 1981. Effects of acuteexperimental diabetes on composite energy metabolism in peripheralnerve axons and Schwann cells. Diabetes. 30:967-974.
28. Greene, D. A., A. I. Winegrad, J. L. Carpentier, M. J. Brown,M. Fukuma, and L. Orci. 1979. Rabbit sciatic nerve fascicle and 'en-doneurial' preparations for in vitro studies of peripheral nerve glucosemetabolism. J. Neurochem. 33:1007-1018.
29. Greene, D. A., P. V. DeJesus, and A. I. Winegrad. 1975. Effectof insulin and dietary myoinositol on impaired peripheral motor nerve
conduction velocity in acute streptozotocin diabetes. J. Clin. Invest.55:1326-1336.
1664 D. A. Greene, S. A. Lattimer, P. B. Carroll, J. D. Fernstrom, and D. N. Finegold

30. Fernstrom, J. D., M. S. Fernstrom, P. E. Grubb, and E. A. Volk.1985. Absence of chronic effects of dietary protein content on braintryptophane concentrations in rats. J. Nutr. 115:1337-1444.
31. Shotwell, M. A., M. S. Kilberg, and D. L. Oxender. 1983. Theregulation of neutral amino acid transport in mammalian cells. Bio-chem. Biophys. Acta. 737:267-284.
32. Kilberg, M., M. E. Handlogten, and H. N. Christensen. 1980.Characteristics of an amino acid transport system in rat liver for gluta-mine, asparagine, histidine and closely related analogs. J. Biol. Chem.255:4011-4019.
33. Wheeler, D. D. 1975. Amino acid transport in peripheral nerve:specificity of uptake. J. Neurochem. 24:97-104.
34. Yue, D. K., S. McLennan, E. Fisher, S. Heffernan, C. Capo-greco, G. R. Ross, and J. R. Turtle. 1989. Ascorbic acid metabolismand polyol pathway in diabetes. Diabetes. 38:257-261.
35. Crandall, E. A., and J. D. Fernstrom. 1983. Effect of experi-mental diabetes on the levels of aromatic and branched-chain aminoacids in rat blood and brain. Diabetes. 32:222-230.
36. Felig, P. 1975. Amino acid metabolism in man. Annu. Rev.Biochem. 44:933-955.
37. Pardridge, W. M. 1977. Regulation of amino acid availability tothe brain. In Nutrition and the Brain. Vol. 1. R. J. Wurtman and J. J.Wurtman, editor. Raven Press, NewYork. 141-204.
38. Christensen, H. N. 1987. Role of membrane transport in inter-organ amino acid flows: where do the depleted amino acids go inphenylketonuria? In Amino Acids in Health and Disease: New Per-spectives. S. Kaufman, editor. Alan R. Liss, NewYork. 1-17.
39. Hawthorne, J. N. 1988. Phosphoinositides and metabolic con-trol: how many messengers? Biochem. Soc. Trans. 16:657-660.
40. Berridge, M. J. 1988. Inositol lipids and calcium signalling.Proc. R. Soc. Lond. B. 234:359-378.
41. Danoff, S. K., S. Supattapone, and S. H. Synder. 1988. Charac-terization of a membrane protein from brain mediating the inhibitionof inositol 1,4,5-trisphosphate receptor binding by calcium. Biochem.J. 254:701-705.
42. Hiraki, Y., 0. M. Rosen, and M. J. Birnbaum. 1988. Growthfactors rapidly induce expression of the glucose transporter gene. J.Biol. Chem. 263:13655-13662.
43. Dawson, W., and J. S. Cook. 1987. Parallel changes in aminoacid transport and protein kinase C localization in LLC-PKI cellstreated with TPA or diradylglycerols. J. Cell. Physiol. 132:104-110.
44. Troni, W., Q. Carta, R. Cantello, M. T. Caselle, and I. Rainero.1984. Peripheral nerve function and metabolic control in diabetesmellitus. Ann. Neurol. 16:178-183.
45. Greene, D. A., S. Yagihashi, S. A. Lattimer, and A. A. F. Sima.1984. Nerve Na+-K+-ATPase, conduction and myo-inositol in theinsulin deficient BB rat. Am. J. Physiol. 247:E534-E539.
46. Behse, F., F. Buchthal, and F. Carlsen. 1977. Nerve biopsy andconduction studies in diabetic neuropathy. J. Neurol. Neurosurg. Psy-chiatry. 40:1072-1082.
47. Spritz, N., H. Singh, and B. Marman. 1975. Metabolism ofperipheral nerve myelin in experimental diabetes. J. Clin. Invest.55:1049-1056.
48. Spritz, N., H. Singh, and B. Marman. 1975. Decrease in myelincontent of rabbit sciatic nerve with aging and diabetes. Diabetes.24:680-683.
49. Thomas, P. K., D. W. Wright, and E. Tzebelikos. 1984. Aminoacid uptake by dorsal root ganglia from streptozotocin-diabetic rats. J.Neurol. Neurosurg. Psychiatr. 47:912-916.
50. Green, R. J., R. H. M. King, P. K. Thomas, and D. N. Baron.1985. Sodium-potassium ATPase activity in the dorsal root ganglia ofrats with streptozotocin-induced diabetes. Diabetologia. 28:104-107.
Amino Acid Uptake in Diabetic Nerve 1665





























